
Submitted:
26 October 2023
Posted:
27 October 2023
You are already at the latest version
Abstract
Arginine shows Jekyll and Hyde behavior in several respects. It participates in protein folding via ionic and H-bonds and cation-pi interactions; the charge and hydrophobicity of its side chain makes it a disorder-promoting amino acid. Its methylation in histones; RNA binding proteins; chaperones regulates several cellular processes. The arginine-centric modifications are important in oncogenesis and as biomarkers in several cardiovascular diseases. The cross-links involving arginine in collagen and cornea are involved in pathogenesis of tissues but have also been useful in tissue engineering and wound-dressing materials. Arginine is a part of active site of several enzymes such as GTPases, peroxidases, and sulfotransferases. Its metabolic importance is obvious as it is involved in production of urea, NO, ornithine and citrulline. It can form unusual functional structures such as molecular tweezers in vitro and sprockets which engage DNA chains as part of histones in vivo. It has been used in design of cell-penetrating peptides as drugs. Arginine has been used as an excipient in both solid and injectable drug formulations; its role in suppressing opalescence due to liquid-liquid phase separation is particularly very promising. It has been known as a suppressor of protein aggregation during protein refolding. It has proved its usefulness in protein bioseparation processes like ion-exchange, hydrophobic and affinity chromatographies. Arginine is an amino acid, whose importance in biological sciences and biotechnology continues to grow in diverse ways.
Keywords:
molecular tweezers
; cation-pi interactions
; intrinsic disorder
; protein-protein interactions
; hydrophobic interaction chromatography
; monoclonal antibody production
; protein aggregation
; protein refolding
; arginine methyltransferases
; post-translational modification
1. Introduction
Each of the amino acids present in proteins contribute in a different way to the structure and function of the proteins. This contribution due to their distinct side chains, of course, is modulated by the place of that amino acid in the primary sequence and the milieu around the protein. Arginine, with a guanidino group in its side chain and the highest pKa value (13.8) of all amino acids [1], is perhaps “more unequal among unequals”. In line with these considerations, Arakawa et al. (2007) wrote “there is insufficient information on arginine to elucidate its unique properties” [2]. While the subsequent period has seen some more insights into the behavior of this intriguing amino acid being gained; its footprint in terms of its role in protein structure (and intrinsic disorder], biological processes, and many biotechnological applications seem to be expanding as an unfinished story.
Aside from its role in shaping structure or disorder in proteins (see below), the way arginine acts in several applications continues to be debatable. These applications are diverse and include its function in protein refolding, protein stabilization, as additive in freeze-drying of biologically active proteins such as enzymes, hormones, and protein pharmaceuticals. This review is aimed at covering at a single place what we know about arginine functioning in different contexts. Apart from updating information about the intriguing behavior of this most basic amino acid, this should stimulate cross-fertilization of ideas from different sectors, which happen to be an eclectic mix.
2. Arginine in Protein Structure and Intrinsic Disorder
It has been reported that “arginine is an abundant (5.1%) amino acid… second most enriched amino acid in protein-protein interactions (after tryptophan)” [3]. In fact, as per Composition Profiler portal (http://www.cprofiler.org/help.html) [4], where amino acid compositions of the standard datasets have been pre-computed, as means and standard deviations over 100,000 bootstrap iterations, arginine accounts for 5.40 ± 0.04%, 4.93 ± 0.06%, and 6.56 ± 0.13% of residues in SwissProt 51 [5], PDB Select 25 [6], and surface residues determined by the Molecular Surface Package over a sample of PDB structures of monomeric proteins, suitable for analyzing phenomena on protein surfaces, such as binding [4], respectively. Among all amino acids, arginine was shown to have the highest mutability in the case of missense mutations linked to the genetic disorders [7]. This is in spite of the fact that this amino acid can be encoded by 6 codons.
Structurally, lysine is closest to arginine and has the highest frequency among amino acids in replacing arginine in the primary sequences of the same protein from different organisms. However, that number is merely 48, as compared to frequency of 83 for glutamic acid being replaced by aspartic acid [8]. At all physiologically relevant pH, arginine remains protonated. Unlike lysine, irrespective of pH, arginine serves exclusively as an H-donor [9]. In line with these considerations, Harms et al. (2011) conducted a systematic study of the ionizable groups buried in the hydrophobic interior of proteins using staphylococcal nuclease as a model [10]. This analysis revealed that lysine, aspartic acid, and glutamic acid residues at 25 internal positions in this protein can have highly anomalous pKa values, with some being shifted by as many as 5.7 pH units relative to normal pKa values in water [10]. On the contrary, arginine residues at the same internal positions exhibit no detectable shifts in pKa, all being charged at pH ≤ 10 [10]. It was also emphasized that the remarkable potential of arginine residues to remain protonated in environments otherwise incompatible with charges is determined by the capability of the guanidinium moiety of the arginine side chain to be he effectively neutralized via multiple hydrogen bonds to protein polar atoms and to site-bound water molecules [10]. The authors argued that “this unique capacity of Arg side chains to retain their charge in dehydrated environments likely contributes toward the important functional roles of internal Arg residues in situations where a charge is needed in the interior of a protein, in a lipid bilayer, or in similarly hydrophobic environments” [10].
Arginine, along with histidine and methionine, facilitates compactness of the protein tertiary structure by bringing together various secondary structure elements. These amino acids, thus also contribute to the enhanced stability of proteins from thermophiles [11]. The relative hydrophobicity of arginine is lower than that of lysine, being slightly higher than that of the glutamic acid and close to the hydrophobicity of alanine [12]. While arginine shows no preference in its positioning in α-helices, β-sheets or β-turns, among α-helices, it is more often found (along with glutamic acid, glutamine, and lysine) in α-helices near the surface of the globular proteins [13]. In terms of the propensity to form α-helix, arginine is better than lysine and glutamic acid; whereas both alanine and leucine are better than it [14]:
Ala > Leu > Arg > Met > Lys > Gln > Glu
Based on the analysis of the stability and the folding and unfolding rates of 12 alanine-based α-helical peptides with a nearly identical composition and containing three pairs of positively and negatively charged residues (either Glu−/Arg+, Asp−/Arg+, or Glu−/Lys+), Meuzelaar et al. (2016) reported that the Glu−/Arg+ salt bridge promoted α-helix content and stability at neutral pH, with the relative helicity and thermodynamic stability of the Glu−/Arg+ peptides following the trend (i + 4) Glu−/Arg+ > (i + 3) Glu−/Arg+ ≈ (i + 4) Arg+/Glu− > (i + 3) Arg+/Glu− [15]. These observations were in line with previous studies [16,17,18] and confirmed that a Glu−/Arg+-oriented salt bridge with Glu− and Arg+ spaced four peptide units apart is most favorable for the folded α-helical conformation [15]. Furthermore, Meuzelaar et al. (2016) showed that the optimized Glu−/Arg+-oriented salt bridge noticeably accelerated α-helix formation, and slowed down the unfolding of the α-helix, acting in these respects much better than the optimized Asp−/Arg+ and Glu−/Lys+ salt bridges [15]. However, these authors also found that the correlation between thermodynamic and kinetic effects of salt bridges is not a general phenomenon, as although the rates of α-helix formation at neutral pH follow the order (i + 4) Glu−/Arg+ > (i + 4) Asp−/Arg+ ≫ (i + 4) Glu−/Lys+, the conformational stability forms a different order, (i + 4) Glu−/Arg+ > (i + 4) Asp−/Arg+ ≈ (i + 4) Glu−/Lys+, suggesting that the salt bridges, which do not contribute positively to thermodynamic stability may still play a kinetic role in formation or unfolding of secondary structures in proteins [15].
One of the important paradigm shifts in our understanding of proteins has been the discovery of intrinsic disorder in proteins. It is now clear that disorder (or unstructure, as some people prefer to call it), just like structure, plays a number of crucial roles in function of proteins [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48]. Since disordered proteins do not fold at physiological conditions, there are noticeable differences between the ordered and disordered proteins at the level of their amino acid sequences in terms of their amino acid compositions, charge, flexibility, hydrophobicity, sequence complexity, and type and rate of amino acid substitutions over evolutionary time. In fact, intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) are significantly depleted in order-promoting residues Asn, Cys, His, Ile, Leu, Met, Phe, Trp, Tyr, and Val, being substantially enriched in disorder-promoting residues Ala, Arg, Gln, Glu, Gly, Lys, Pro, Ser, and Thr [4,19,20,21]. Therefore, Arg is among the disorder-promoting amino acid. The charge combined with hydrophobicity and side chain flexibility are deciding factors for this propensity [22].
It was pointed out that the arginine-rich motifs (ARMs), being among the common RNA binding domains in proteins, can function as independent recognition units, separated from the protein in which they are found [10,23]. Although ARMs in different proteins are all characterized by a preponderance of arginine residues, they have low overall sequence similarity and appear to have arisen independently throughout phylogeny [10]. It was pointed out that since ARMs are intrinsically disordered, “individual arginine residues govern binding to an RNA ligand, and the inherent flexibility of the peptide backbone may make it possible for “semi-specific” recognition of a discrete set of RNAs by a discrete set of ARM peptides and proteins” [10].
Among the proteins with the extremely high arginine content a special place is taken by protamines, which are small, most abundant sperm nuclear proteins in many species [24], which were isolated from the sperm by Friedrich Miescher almost 150 years ago [25]. In fact, 48% residues in the amino acid sequences of human protamines are arginines [24], and the true protamines can contain up to 70% arginine [26]. In line with these observations, Figure 1A shows that as a protein family, protamines are characterized by the exceptionally high arginine content. Based on their highly biased amino acid composition (see Figure 1A), it is not surprising to find that almost all members of the protamine family are intrinsically disordered (see Figure 1B,C). This is agreement with rather limited experimental data showing that the free protamine is unstructured in solution [27].
Being synthesized in the late-stage spermatids of many animals and plants protamines are responsible for condensation the spermatid genome into a genetically inactive state. In addition, at least seven major functions were ascribed to protamines, such as tight packaging and condensation of a paternal genome into a compact and hydrodynamic nucleus required for the fast movements of spermatozoa; protecting the paternal genetic message by making it inaccessible to nucleases or mutagens; removal of transcription factors and other proteins leading to a blank paternal genetic message that devoid of epigenetic information; the imprinting of the paternal genome during spermatogenesis; acting as an epigenetic mark on some regions of the sperm genome; acting as a checkpoint during spermiogenesis, and potentially playing some specific roles in the fertilized eggs [24,35]. All vertebrate protamines contain a set of small “anchoring” domains containing multiple arginine or lysine amino acids and utilized in DNA binding, and also have multiple serine and threonine residues often targeted for phosphorylation [26].
Protamine binding to DNA generates a remarkably stable complex, where a protamine wraps around the DNA helix in the major groove [36], and were tight binding of one protamine molecule per turn of DNA helix [37] is achieved via “he combination of hydrogen bonds and electrostatic bonds that form between the guanidinium groups of each arginine residue in the anchoring domains of the protamine and the phosphate groups in both DNA strands” [26].
3. Cation-π Interactions Often Involve Arginine
Apart from forming ionic bonds and H-bonds, the cation-π interactions of arginine with aromatic ring containing amino acids and ligands (of proteins containing arginines) are rather important in structural biology. The cation-π interactions originates because “electron-rich π system above and below the benzene ring is partially negative and this negatively charged region of the quadrupole interacts with positively charged amino acids” [38,39]. Such cation-π interactions have long ago emerged as an important type of non-covalent bonds, which play a number of crucial roles in function of both structured and intrinsically disordered proteins. This includes their involvement in molecular recognition of ligands by proteins as evidenced by cation-π interaction between the ammonium ion of choline with the indole group of tryptophan in cholinesterase [40]. Similarly, the recognition of Rev peptide by the HIV Rev-responsive element was found to have a significant contribution of cation-π interactions involving arginine residues [41]. In proteins, phenylalanine, tyrosine and tryptophan can act as the π-systems, and lysine and arginine can be the cations, whereas histidine represents a unique system, as it can be a cation as well as a π-system. Hohlweg et al. (2018) discussed how the cation-π interaction facilitates placement of arginine in the transmembrane helical environment in an ATPase [42]. More important, arginine has a special role in the proton translocation by the ATPase [42].
Early studies focused on just geometrical criterion in identifying the occurrence of the cation-π interaction in protein structures. Justin P. Gallivan and Dennis A. Dougherty [43] discussed why that is problematic and factored in energetic considerations. Their estimates showed that >25% of tryptopan residues in PDB can form cation-π interactions, and arginine is more probable than lysine in these interactions [43]. Kumar et al. (2018) explained why cation-π interactions maybe more common with arginine as compared to lysine [44]. In the case of lysine, the interaction has a much higher contribution from electrostatic forces, whereas in the case of arginine, there is about an equal contribution from electrostatic and dispersion forces. This means that the arginine-π interactions are not that much affected by the polar environments [44]. A recent review provides an update on importance of cation-π interactions in diverse areas including healthcare [45]. The authors pointed out that stabilization due to the cation-π interaction is higher at higher temperature. This is seen to be exploited in enzymes from thermophiles, which have more cation-π interactions than their mesophilic counterparts [45].
Also, cation-π interactions play an important role in protein-nucleic acid interactions. For example, it is recognized that 72–87% of known protein-RNA interfaces contain arginine [46,47]. This enrichment of the protein-RNA interfaces in arginine is not random, as substitution of arginine with lysine often decreases the specificity of protein-RNA complexes [48]. Furthermore, although for a long time, it was known that interaction of arginine-containing proteins and RNA can be described within the frames of an arginine-fork model [48], where a single arginine forms four complementary contacts with nearby phosphates, yielding a two-pronged backbone readout, recently, it was established that in addition to arginine interactions with the phosphate backbone and the major-groove edge of guanine, there are also simultaneous cation-π contacts between the guanidinium group of arginine and flanking nucleobases [49]. This is illustrated by Figure 1 representing different modes of the two-pronged arginine interaction with RNA [49]. Here, Figure 2A shows arginine-fork model proposed for the explanation of the extraordinary specificity of arginine for interaction between the HIV-1 Tat protein and TAR RNA [48]. Figure 2B underscores the possibility of the guanidinium interactions with both phosphate and nucleobases as evidenced by the results of computational modeling of arginine bound to TAR [50]. Finally, Figure 2C shows the related details of TAR in complex with TBP6.7, where “R47 salt-bridges to the Uri23 phosphate with simultaneous cation-π interactions between the guanidinium and nucleobases Ade22 and Uri23” [51].
Involvement of cation-π interactions between aromatic amino acids, such as tyrosine, and basic residues, such as arginine, in liquid-liquid phase separation (LLPS) of several intrinsically disordered proteins has been reported [52,53,54]. Recently, it was shown that the multiple cation-π interactions are responsible for LLPS of the complex between synaptophysin and synapsin [39]. Here, the C-terminal region of synaptophysin (residues 219-308) contains 100 repeat regions, 9 of which start with tyrosine (Y-G-P/Q-Q-G) [55] and therefore obviously acts as a donor of π systems. On the other hand, synapsin contains 85 positively charged amino acids and has a polybasic C-terminal intrinsically disordered region that contains 31 positively charged residues, most of whsich are arginine (21/31) [56,57], thereby acting as a source of cations, many of which are arginine residues. The crucial role of the multiple cation-π interactions in the synaptophysin-synapsin coacervation was supported by the observation that the synaptophysin mutant form, where all nine tyrosine residues (Y245, Y250, Y257, Y263, Y269, Y273, Y284, Y290, and Y295) were replaced with serine failed to coacervate synapsin despite the fact this mutant form retains the negative charge of synaptophysin (−8.3) [39]. Based on these findings, the authors concluded “these results are consistent with the possibility that multivalent electrostatic π–cation interactions rather than simple negative–positive charge interactions mainly govern the coacervation between synaptophysin and synapsin in living cells” [39].
In the same vein, the phase separation of fused in sarcoma (FUS) protein was shown to be regulated by cation-π interaction between arginine and tyrosine residues [54]. The methylation of arginine affected this cation-π interaction and can prevent the phase separation [45] (please see the section on posttranslational modifications below, wherein arginine methyltransferases have been discussed). Furthermore, a novel arginine-based interactions, arginine π-stacking, was recently described [58]. This interaction mode involves arginine’s own π-cloud in the guanindino group. The tau protein aggregation leading to the formation of amyloid-like fibrils represents the basis of the pathogenesis of various taupathies including Alzhiemer’s disease [58]. It was proposed that π-stacking by arginine residues can promote aberrant fibril interactions and also can drive the binding of other proteins to tau fibrils, thereby contributing to the formation of toxic aggregates [58].
Although both cationic arginine and lysine residues are commonly found in proteins capable of phase separation, arginine-rich proteins are observed to undergo LLPS more readily than lysine-rich proteins [54,59,60,61,62,63,64]. This observation supports the accepted notion that arginine, which is abundant in the RNA-binding proteins (RBPs), is considered as an important LLPS driver [54,60,61,62,63,64]. Although the difference between the lysine and arginine potentials to drive LLPS was ascribed to the fact that in addition to the ability to form cation−π interactions (with arginine forming stronger cation-π interactions with aromatic groups), arginine is capable of formingπ−π interactions [64,60,65,66,67,68,69], recently it was established that the reentrant phase behavior and tunable viscoelastic properties of the dense LLPS phase are determined by the arginine hydrophobicity [59].
An important case of the arginine-centric pathogenesis is given by a set of dipeptide repeat proteins generated as a result of a hexanucleotide repeat expansion (HRE) GGGGCC (G4C2) mutation in the 5′ non-coding region of the gene C9 open reading frame 72 (C9orf72) gene, which is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) [70,71,72,73]. Despite its intronic localization and lack of an ATG start codon, this expanded microsatellite region was shown to encode a series of dipeptide repeat (DPR) proteins, that can be produced by its sense (poly(Gly-Ala), poly(Gly-Pro), and poly(Gly-Arg)) [74,75] as well as antisense translation (poly(Pro-Ala), poly(Pro-Arg), and poly(Pro-Gly)) [76]. All six DPR species are produced form the expanded microsatellite region via repeat-associated non-ATG (RAN) translation [76] and can be found in CNS tissue from the ALS and FTLD patients [74,75,76]. Since the length of the pathogenic expanded repeat region of C9orf72 can range from 45 to several thousand units [73], the resulting DPR polypeptides can be very long. It was indicated that the highly charged arginine-rich polypeptides poly(Gly-Arg) and poly(ProArg) are the most toxic species in Drosophila, yeast, and mammalian primary neurons [77,78,79,80,81]. Furthermore, it was shown that both sense and anti-sense RNA foci from C9orf72 expansions can be found in the same cell indicating that multiple DPRs can be translated simultaneously [82,83] and therefore can have the complex biological interactions originating from simultaneous expression of multiple DPR variants [84].
In cellular model studies, the arginine-rich DPRs were found in nucleolus causing impaired rRNA synthesis and ribosome biogenesis [85,86]. Furthermore, the majority of the binding partners found the overlapping interactomes of the poly(Gly-Arg) and poly(ProArg) were reported as proteins with low-complexity domains (LCDs) and ribosomal proteins [87,88,89]. Based on these and similar observations, Hana M. Odeh and Shorter J. Shorter concluded that one of the important mechanisms of high toxicity of the arginine-rich DPRs can be associated with the capability of these polypeptides to cause aberrations in cellular LLPS processes [90]. Furthermore, poly(Gly-Arg) was shown to form cytoplasmic inclusions that sequester RNA and RNA-binding proteins including stress granule (SG) proteins, including the key driver of the SG assembly, Ras GTPase-activating protein-binding protein 1 (G3BP1), as well as YTH domain-containing family (YTHDF) proteins capable of binding of the N6-methyladenosine (m6A)-modified mRNAs [91]. Figure 3 represents this pathological mechanism, showing an importance of the interplay between poly(Gly-Arg), RNA, RNA-binding proteins (RBPs), G3BP1, YTHDF, and m6A-mRNA in the development of the aforementioned cytoplasmic biomolecular condensates [91]. Therefore, the capability of arginine-rich DPR proteins to alter the LLPS and biogenesis of various MLOs represents an important mechanism contributing to the potential of these polypeptides to promote nucleolar toxicity, inhibit protein synthesis, impair ribosomal RNA processing and ribosome biogenesis, and interact with RNA-binding proteins [80,81,86,89,92,93,94,95,96].
4. Importance of Arginine in Post-Translational Modifications of Proteins
Post translational modifications (PTMs) “affect localization, interaction state, stability, and turnover of proteins” [97]. PTMs are extensively involved in regulation of biological activities of proteins and are part of the mechanisms by which signal transduction takes place [98,99]. For example, in moonlighting function, the PTM may be absent or different. PTMs are also involved in redox homeostasis [100]. IDPs and IDRs are more prone to PTMs than ordered proteins and domains [19,97,98,99,101,102,103,104,105,106].
Arginine, being a disorder-promoting amino acid, is also targeted by a few biologically important PTMs [107]. However, an important consequence of replacing amino group in the side chain (of lysine) with the guanidino group (in arginine) is the drastic reduction in the post-translational modifications of the side chain. Lysine is known to undergo many different kinds of acylation of the side chain amino groups in proteins. In fact, more than 100,000 sites of Lys modifications in over 10,000 proteins have been mapped [108], and the list of the lysine-centric PTMs includes acetylation, methylation, ubiquitination, SUMOylation, NEEDylation, propionylation, butylation, crotonylation, malonylation, succinylation, glutarylation, β-hydroxybutylation, 2-hydroxyisobutyryation, lactylation, and benzoylation [109]. This wide range of reversible lysine-based PTMs are known to regulate enzyme activities, chromatin structure, protein-protein interactions, protein stability, and cellular localization. In fact, acetylation alone regulates a large set of biological functions, such as epigenetics, homeostasis, metabolism, signal transduction, cell cycle, DNA repair, transcription, development, and aging [110]. The biological importance of the PTMs of lysine residues is borne out by the fact that there are enzymes which are called writers, which introduce these PTMs, erasers, which reverse these PTMs, and readers, which are responsible for the downstream outputs of these PTMs [109]. On the other hand, only two kinds of PTMs were reported for arginine, methylation and citrullination, which are catalyzed by arginine methyltransferases and arginine deiminases, respectively [107]. Citrulination leads to the production of auto-antibodies in rheumatoid pathogenesis [111].
Both lysine and arginine undergo methylation. Methylation of histones and other regulatory proteins involved in RNA binding, transcription, translation, chaperone activity, etc. is a key regulatory mechanism of cellular metabolism. In fact, protein methylation is a PTM involved in a vast number of processes [112] and “1% of the functional genome encodes for the enzymes catalyzing protein methylation” [112,113]. Methylation of proteins involves the transfer of a methyl group (CH3) onto either an arginine or lysine residue, with the arginine methylation being far more common on the proteomic scale than lysine methylation [114,115]. Both mono- and di-methylarginines are formed, the latter in either asymmetric or symmetric forms Arginine methylation is catalyzed by protein arginine methyltransferases (PRMTs) [116], which are “ubiquitously expressed both at the cellular compartmentalization, and tissue expression levels” [112]. This family of proteins includes “three types of that catalyse this reaction, each responsible for a different ArgMe end-product: Type I PRMTs (PRMT1, -2, -3, -4, -6 and -8) lead to asymmetric dimethylarginine (ADMA); Type II PRMTs (PRMT5 and -9) produce symmetric dimethylarginine (SDMA); and Type III PRMT (PRMT7) forms monomethyl arginine (MMA) only” [112] (see Figure 4). ADMA and SDMA are both reported to inhibit nitric oxide synthase [107].
Samuel et al. (2021) have discussed inhibitors of protein methyltransferases as they have shown promise as new therapies for certain types of brain cancers [112]. Figure 5 outlines how arginine methylation is involved in oncogenesis. Arginine methyltransferases, apart from being inhibited by synthetic small molecular weight inhibitors, are also controlled by alternative splicing, PTMs, miRNA, and via interactions with other proteins [112].
The importance of arginine methylation in renal transplants has attracted considerable attention [117,118,119,120], as circulatory ADMA, which serves as the endogenous nitric oxide (NO) synthase inhibitor is involved in progression of kidney disease, being associated with mortality in renal transplant recipients (RTR) and increased risk of end-stage renal disease in chronic kidney disease (CKD) populations [121]. Furthermore, hypertension was also shown to be associated with elevated circulating ADMA concentrations [122], whereas ADMA and SDMA are considered as cardiovascular risk factors and have emerged as predictors of cardiovascular events and death in a range of pathologies [118,119,120,123,124]. Recently, arginine methylation has been shown to be useful as a biomarker of cardiovascular diseases [125].
5. Crosslinks of Arginines and Their Biological Importance
Apart from the aforementioned PTMs of arginine, its participation in the crosslink formation and implications of such crosslinks have been a focus of attention for a number of years now. Crosslinking occurs both in vivo and in vitro [126]. Although chemical crosslinking often links amino group of the lysine or -SH group of the cysteine, the crosslinks involving arginine have also been reported to form in vitro [127]. Collier et al. (2016) described formation of four types of lysine-arginine crosslinks in collagen present in bones, tendons, ligaments, and dermis as a part of the non-enzymatic glycation [128]. These advanced glycation end products (AGEs) are formed in diabetes and other age related diseases [128]. Because of type I collagen has long half-life, which can be up to 200 years in tendon [129], this protein is particularly prone to AGE cross-linking in a number of different tissues [128]. Depending on the reactive dicarbonyl donors, such as glucose, deoxyglucosone, methylglyoxal, and glyoxal, four lysine-arginine AGE cross-links, glucosepane, 3-deoxyglucosone-derived imidazolium crosslink (DOGDIC), methylglyoxal-derived imidazolium crosslink (MODIC), and glyoxal-derived imidazolium crosslink (GODIC), are commonly formed [128]. It was also reported that these AGE cross-links can occur in human lens protein, where they can be found at concentrations of 132.3–241.7 pmol/mg of protein for glucosepane, 1.3–8.0 pmol/mg of protein for DOGDIC, 40.7–97.2 pmol/mg of protein for MODIC and concentrations below the quantifiable level of the instrument for GODIC [130].
Photosensitized crosslinking of cornea has many clinical applications that include stromal stiffening for treatment of ectatic diseases, such as keratoconus, photobonding of LASIK flaps to the corneal stroma, and sealing wounds and lacerations [131,132,133,134,135]. While riboflavin had been used earlier for the photosensitized crosslinking, its replacement by rose bengal has several advantages [136], including the important capability of rose bengal molecules to remain in a ∼100 µm layer of stroma near the epithelial surface rather than diffusing throughout as occurs for riboflavin [137,138]. The protocols involving presence of oxygen or its absence have been described [136]. Wertheimer et al. (2020) reported that arginine acting as an electron donor promoted the corneal photosensitized crosslinking by rose bengal even in the absence of oxygen [136].
A collagen-chitosan 3D-hybrid scaffold was reported to be “cross-linked by arginine” to improve stability of this scaffold for tissue engineering [139]. No explanation of how arginine acts as a cross-linker was given; even more intriguing was the statement that crosslinking could also be carried out in the absence of arginine. The protocol for preparation of the scaffold involved freeze-drying. It is likely that arginine helped the scaffold structure during freezing or drying stage [139].
An unusual catalytic reaction was recently described for one of the radical S-adenosylmethionine (RaS) enzymes (a diverse protein superfamily capable of catalyzing chemically difficult transformations), which led to the formation of a crosslink between arginine and tyrosine during the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs that represent natural products with diverse structures and functions) [140]. This arginine-tyrosine crosslinking resulted in the “installation of a macrocyclic carbon–carbon bond that links the unactivated δ-carbon of an arginine side chain to the ortho-position of a tyrosine-phenol” thereby generating a unique macrocyclization motif [140].
DNA-binding proteins frequently have arginine residues that are used for interaction with DNA [141]. This observation was used to construct reactive DNA probes for the proximity labeling of DNA-binding [142]. For example, 1,3-diketone-modified nucleotides and DNA capable of cross-linking with arginine-containing peptides and protein were synthesized as a probe to identify binding regions in such proteins including histones [142]. It should be mentioned here that the chemical modification of guanidino group has been mostly restricted to the reaction with diketones, such as phenylglyoxal, 1,2-cyclohexanedione and a trimer of 2,3-butanedione [143,144,145].
Jones et al. (2019) have described the synthesis of two crosslinkers, one homobifunctional (based upon aromatic glyoxal moieties) for forming arginine-arginine crosslinks; and another heterobifunctioal (based upon diketone and NHS moieties) for forming lysine-arginine crosslinks. In these designs, attention was paid to typical distances found in protein-protein interactions involving lysine and arginine groups [3]. This was factored in deciding the span between the two reactive ends of the cross-linkers [126].
Because of arginine was shown to act as a very useful bioactive component due to its excellent biosafety, antimicrobial properties, and therapeutic effects on wound healing, and because it can also be used for treatment of specific pathological conditions, such as diabetes and trauma/hemorrhagic shock, there are multiple forms of arginine-based therapy [146]. The usefulness of arginine for wound healing is known for decades, and there are multiple arginine-based systems for the application in wound healing that can be classified as direct supplemental approaches of free arginine and indirect approaches based on the arginine derivatives, where modified arginine can be released after biodegradation, e.g., from wound-healing dressings [146]. Among various means for arginine incorporation into the wound-dressing material are electrostatic attachment to high-molecular-weight hyaluronic acid (HA, which is one of the most common extracellular matrix biomacromolecules) [147], or to the lignin nanofibrils [148], or to silicon and inositol to form arginine silicate inositol (ASI) complex [149]. Arginine can also be covalently attached to scaffold molecular chains via imine-type bonds or conjugate to polymeric skeletons [146], with the characteristic examples given by the composite hydrogels of poly(vinyl alcohol) (PVA) and oxidized polysaccharides [150], arginine-crafted chitosan [151], and arginine-based poly(ester amides) (Arg-PEAs), which represent a family of biodegradable and biocompatible synthetic polymers consisting of three nontoxic building blocks, L-arginine, diols, and dicarboxylic acids [152].
6. Arginine in the Active Sites of Enzymes
Given the positive charge on its side chain, which enable both electrostatic and cation-π interactions, it is not surprising that arginine plays an important role in the active site of enzymes, quite often directly participating in the binding of a substrate. Cotton et al. (1977) had studied complex formation between gunidine hydrochloride and p-nitrophenyl phosphate dianion and suggested that gunidino side chain of arginine orients the phosphate group correctly during enzymatic hydrolysis of phosphate compounds [153]. The involvement of arginine in binding of iodine in the active site of horse radish peroxidase was established by chemical modification studies with phenylglyoxal [154]. Even much earlier, chemical modification with diones was used for identifying arginine as an important active site residue of the avian liver phosphoenolpyruvate carboxykinase, an enzyme which is a part of the central metabolic pathway associated with the gluconeogenesis [155]. The kinetic data indicated involvement of the active site arginine in the carbon dioxide binding and activation [155]. Another example with important active site arginine is given by the argininosuccinate synthase, a key enzyme in urea synthesis, deficiency of which is associated with hyperammonemia [156]. The participation of this arginine in the ATP binding during the catalysis was also identified by chemical modification studies [157].
It is important to emphasize here that the role of arginine in the enzyme active sites is not limited to binding of the substrates/ligands, as this residue is also known to have a catalytic role in several cases. For example, arginine has been shown to act as a general acid catalyst in DNA cleavage by a site specific serine recombinase [158]. In xanthine dehydrogenase also, arginine actively participates in both binding and catalytic steps [159]. Ypt/Rab proteins are monomeric GTPases and were found to have 5 invariant arginine residues (dubbed as arginine finger) in their catalytic domain, with substitution of only one of them rendering the GAPs almost completely inactive [160].
Sulphotranferases have broad specificity towards -OH containing substrates. Arginine residues were found to be critical for binding of the coenzyme in sulphation of simple phenols by human phenol sulphotransferase, an important detoxification enzyme with broad substrate specificity and lack of endogenous substrates [161]. Recently, a volume on the sulfurtransferases represented studies focused on the use of the site directed mutagenesis to establish the role of arginine residue in the catalysis by thiosulphate sulfurtransferases (TST) containing R-K-G-V-T-A motif [162]. TSTs, which are also known as rhodanases, find wide applications in medicine and biotechnology. TST also produces hydrogen sulfide which has emerged as an important player in both intra- and intercellular signaling [162].
Human type I D-myo-Inositol 1,4,5-trisphosphate 5-phosphatase, an enzyme involved in generating calcium signal, which in turn regulates several cellular process, has two reactive arginine residues in its active site that play crucial role in the enzymatic activity of this protein [163]. These arginine residues are part of the 10 amino acid-long sequence M-N-T-R-C-P-A-W-C-D-R-I-L, which is conserved and is involved in substrate recognition [163]. Analogously, the triad of arginine residues in the anionic binding pocket of the ArsC arsenate reductase of plasmid R773 that catalyzes reduction of arsenate in Escherichia coli, was shown to be in arsenate binding and transition-state stabilization [164]. In the same vein, in sulfite oxidising enzymes, the active site arginine has been shown to be critical for electron transfer from Mo to heme redox center [165]. This arginine residue is conserved in these Mo-containing enzymes and is associated with a clinical mutation leading to sulfite oxidase deficiency [165].
An interesting role of arginine has been observed in the peroxisomal enzyme human D-amino acid oxidase (hDAAO) [166]. The enzyme is involved in the degradation of D-serine, which is the main co-agonist of N-methyl D-aspartate receptors in brain and hence is involved in brain function and some of its diseases. An arginine residue present on the monomer-monomer interface and located 20 Å from the assumed second ligand-binding site was shown to be responsible for FAD binding. The mutation of this arginine resulted in increasing innate mistargetting of the enzyme to the nucleus [166]. Heme nitrite reductase produces NO and ammonium ion and is a key enzyme of nitrogen cycle. Recently, an arginine residue was reported to assist substrate binding and donate a proton during the catalysis [167].
7. Metabolic Importance of Arginine
Arginine is involved in production of urea, creatine phosphate, polyamines, nitric oxide, ornithine and citrulline. Most of the physiological effects of arginine result from its production of NO by nitric oxide synthase [168,169]. Arginine is a conditional essential amino acid and lack of its adequate bioavailability plays “pivotal role in the pathogenesis of a growing number of varied diseases, including sickle cell disease, thalassemia, malaria, acute asthma, cystic fibrosis, pulmonary hypertension, cardiovascular disease, certain cancers, and trauma, among others” [170].
Hristina et al. (2014) have reviewed the role of arginine in energy metabolism [171]. These authors emphasized that despite the possibility of the body to synthesize L-arginine, burns, infections, insufficient circulation, intensive physical activity, severe wounds, or sterility require exogenous supplementation of this amino acid [171]. Its exogenous supplementation under intensive sport activity helps in generation of ATP via AMP kinase pathway [171]. In a view of arginine involvement in energy production, it is not surprising that its metabolism is also affected during bacterial infection and cancer, with the two key enzymes in this context being arginine deiminase and arginase [172]. Although arginine is necessary for function of neuronal cells, it also causes neuroinflammation and NO production. A consequence of latter is that arginine depletion actually helps spinal cord regeneration after an injury [173]. Furthermore, recent metabonomic-based analysis identified arginine in plasma as a key biomarker among the 85 differentials metabolites identified between the chronic obstructive pulmonary disease (COPD) patients and healthy people [174].
8. Arginine in Protein Interactions
8.1. Arginine Tweezers
Jeanne Leblond and Anne Petitjean (2011) provided a good introduction to molecular tweezers as a class supramolecular systems, which now have footprints in diverse areas [175]. Two interacting moieties connected by a span shape the tweezers. Rigidity or flexibility of the span decides how specific the tweezers would be in recognizing the molecules. Smart tweezers, which are sensitive to the ionic, light, and redox stimuli are also known [175].
Kaiser et al. (2018) have investigated the evolution of amino acyl tRNA synthtases since the pre-biotic era [176]. These enzymes have two classes. In both, activation of the amino acid is the most conserved feature. The authors described how a conserved pair of arginine residues form the functional ends of tweezers to hold the adenosine phosphate in class II enzymes (see Figure 6) and also established that this structural motif (together with the Backbone Brackets motif found in the class I enzymes) can be traced back to the primordial forms of Protozymes of the aminoacyl tRNA synthetases and their more efficient successors, the Urzymes [176].
In yet another powerful application of arginine-based molecular tweezers, these were shown to interfere with the pathogenic aggregation of abnormal Aβ, α-synuclein, and TTR to redirect this process to the formation of non-toxic amorphous aggregates that could be degraded and cleared [177]. A good review of emerging importance of molecular tweezers in general in the drug discovery programs is available [178]. An important review focusing on applications of tweezers based upon basic amino acids lysine and arginine and their applications as drugs for inhibiting toxic aggregates is also available [179].
8.2. Sprocket Arginine Residues in Histones
Hodges et al. (2015) discussed another interesting arginine-based functionality, the DNA minor groove binding of the histone octamer in a sprocket-like manner [180]. As the authors explained, “the intermolecular contacts that bind DNA to the histone octamer is the series of histone arginine residues that insert into the DNA minor groove at each superhelical location where the minor groove faces the histone octamer…” [180]. These arginine residues that insert into the DNA minor groove are referred to as “sprocket” arginines, “since they engage the DNA “chain” like the teeth of a bicycle sprocket wheel” [180]. They are highly conserved among eukaryotic species and have important roles in DNA binding. In fact, the nucleosome core particle is formed via the high affinity binding of the histone octamer containing two copies each of histones H2A, H2B, H3, and H4 to ~147 bp of DNA. The corresponding histone-DNA binding is facilitated by more than 100 direct interactions of the histone main-chain and side-chain groups with the DNA sugar-phosphate backbone and a similar number of indirect water-mediated interactions [181,182]. In the nucleosome structure, the DNA minor groove faces the histone octamer at 14 locations [183], each containing a “sprocket” arginine, the side chain of which extends into the DNA minor groove and makes extensive contacts with the DNA backbone [180], constituting a significant fraction of the solvent accessible surface area that is buried upon histone-DNA binding [182]. In Saccharomyces cerevisiae (budding yeast), mutations of the individual “sprocket” arginine residues, which are histone H4 R45A, H3 R63A, H3 R83A, H2A R43A, H2B R36A, H2A R78A, and H3 R49A, compromised repair of UV-induced DNA lesions and affected gene expression and cryptic transcription [180].
8.3. Rings, Strings, and Stacks Made of the Arginine Residues at Protein-Protein Interfaces
Although the most obvious role of positively charged arginines (which are overrepresented at the oligomerization interfaces) in protein-protein interactions is the formation electrostatic contacts with oppositely charged residues, in a solvated environment, arginines are capable of formation of stable, short-range interactions via their guanidinium groups, with water molecules stabilizing the positively charged guanidinium groups and posessing a bridging effect on the arginines pairs [184]. Based on the analysis of over 70,000 protein structures in PDB, Neves et al. (2012) have found that “Clusters of four to eight arginine residues with Cζ-Cζ distances < 5 Å, organized as rings with 4 to 8 members, stacks of two arginines, and strings of stacked arginines, are commonly located at the interfaces of oligomeric proteins” [185]. The authors also revealed the presence of the planar stacking of guanidinium groups of arginines, bridged by hydrogen bonds and interactions with water molecules and showed that “guanidinium groups are commonly involved in 5 hydrogen bonds with water molecules and acceptor groups from surrounding amino acids” [185]. These are important observations indicating the prevalence of arginine clusters “that are exposed to interact with and potentially be controlled or switched by charged metabolites, membrane lipids, nucleic acids or side chains of other proteins” [185].
9. Arginine Applications in Biotechnology
So far, we have focused on arginine as a part of proteins. Its diverse applications as a free amino acid are also very important. There are some other applications as well, wherein arginine is a part of peptides. We briefly review such applications in different contexts in biotechnology.
9.1. Arginine as an Excipient in Drug Formulations
Excipients are added to enhance protein stability, ensure tonicity and enable efficient drug delivery. In a series of reports, Wang has comprehensively reviewed the use of excipients for oral drug delivery and in liquid and solid formulations [186,187,188]. Arginine is among the few amino acids, which have been used as excipients. While it has been used in solid drug formulations, its usefulness in protein drug injectable formulations is even more interesting.
Administration by injections, by far, has been the most frequently utilized route of administration of protein drugs. Several recent comprehensive reviews on proteins as injectables are available [189,190,191]. One major problem is the formation of opalescence in protein solutions as injectables. Opalescence in protein solutions is generally associated with aggregation and often precedes phase separation. In recent years, phase separation (discussed earlier) in cells has attracted considerable attention [52,63,192,193,194,195,196,197,198,199,200,201,202,203,204]. The biomolecular condensates formed as a result of this liquid-liquid phase separation (LLPS) are known as membrane-less organelles or biomolecular condensates. Weak interactions of all kinds, such as electrostatic, hydrophobic, π-π, and cation-π are implicated in LLPS. Proteins with intrinsic disorder and prions are more prone to participate in LLPS [205,206,207,208]. In the case of antibodies (including monoclonal antibodies), high concentrations (about 100 mg/ml) are commonly used in therapeutic formulations. Under these conditions, aggregation leading to cloudy appearance (opalescence) followed by crystallization or LLPS has been observed. Ashlesha S. Raut and Devendra S. Kalonia have given a good overview of the theoretical framework concerning formation of this cloudy appearance [209]. While pH, ionic strength, temperature obviously are important parameters, a useful survey of excipients which prevent this phenomenon has been provided.
Oki et al. reported that of the various excipients tested, arginine was the most effective in preventing formation of opalescence and LLPS [210]. Banks and Cordia screened sucrose, proline, sorbitol, glycerol, arginine and urea for preventing phase separation at different concentrations [211]. They observed that “…. all six were found to be preferentially excluded from the native state monomer by vapor pressure osmometry, and no apparent correlations to the excipient dependence of mAb-B melting temperatures were observed. These results and those of the effects of solution pH, addition of salt, and impact of a small number of charge mutations were most consistent with a mechanism of local excipient accumulation, to an extent dependent on their type, with the specific residues that mediate mAb-B electrostatic protein–protein interactions” [211].
Pyne and Mitra tracked the hydration of lysozyme by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) in the presence of sucrose, arginine, BSA, and ubiquitin [212]. They found that hydration level changes can be correlated with the extent of LLPS. Sucrose promoted LLPS; BSA diminished it. Arginine had a “subtle” effect, while the effect of ubiquitin was concentration dependent [212].
It is notable that arginine has been an excipient in the FDA approved drugs TNKase® (tenecteplase) and Activase® (alteplase), which are formulations of the biosynthetic forms of human tissue-type plasminogen activator produced by Genentech [2].
9.2. Arg as a Suppressor of Protein Aggregation during Protein Refolding
Among amino acids, interactions of free arginine with proteins are more complex than often realized. It has attracted significant attention from researchers in the areas of protein refolding.
Arakawa’s group [2,213,214] pointed out that arginine helps in protein refolding by reducing aggregation but has no effect in increasing the protein stability under thermal stress. At the concentration range of 0.5-2 M, arginine could help in extracting the active proteins from the inclusion bodies, thereby indicating that it enhances protein solubility. These researchers suggested that arginine prevents aggregation because of π electron-cation interaction between the guanidino side chain of arginine and tryptophan residues of the proteins [2,213,214].
Baynes et al. (2005) suggested a different and more complex hypothesis about prevention of aggregation by arginine [215]. They worked with association of insulin and its antibody and with refolding of carbonic anhydrase. According to these workers, arg acts as a neutral crowder. Their coining this term is an unfortunate description [215]. Protein crowders with large diversity of structures have been described, including those which carry no charge; PEG is one such example and has been frequently used as a protein crowder [216]. Unlike these conventional crowders, which generally facilitate association, a “neutral crowder” as described by Baynes et al. (2005) “slows protein association and may either speed or slow dissociation” [215]. Arginine as per their observations slowed down association and speeded up dissociation; on the other hand, guanidium salt had no effect on association, but accelerates dissociation of protein-protein complexes [215]. The authors explain these effects in terms of the transition state theory in the context of protein-protein interactions [215].
Working with refolding of recombinant consensus interferon, Liu et al. (2007) observed that arginine prevented the formation of insoluble aggregates but soluble oligomers were still formed [217]. These authors suggested that arginine stabilized unfolded structures giving them more time to refold correctly. At the same time, arginine beyond 0.5 M concentration stabilized these unfolded species so much that they did not fold correctly but oligomerize (explaining why refolding yield decreased when arginine concentration was increased beyond 0.5 M). While their results about concentration dependence of the yield of refolded interferon are interesting, the explanations are far from convincing [217]. The same group, investigating the effect of arginine on refolding on different proteins, later observed that while carbonic anhydrase (with no cysteine) could be refolded to 100% yield; GFP (with 2 cysteine residues) formed soluble oligomers [218]. Recombinant human colony stimulating factor (with 5 cysteines) formed significant amount of insoluble aggregates. It is known that correct pairing of cysteines to form disulphide bonds requires a separate strategy necessitating chemicals or enzymatic interventions [219]. Thetefore, mixing up the role of arginine with disulphide bonds was not a good idea.
Around the same time, Arakawa’s group in collaboration with Timsheff further refined their ideas on how arginine suppresses protein-protein interactions [220]. Their discussion involved comparing arginine with GdnHCl (a denaturant) and betaine (an osmolyte known to stabilize proteins). They noted that protein stabilizers like betaine are excluded from the protein surface (preferential exclusion) allowing proteins to get preferentially hydrated, whereas the denaturants bind to the protein surface [220]. Arginine was show to behave in a more complex way, which is in between the behavior of the denaturants and stabilizers. It binds to proteins weakly requiring high concentration of around 2M to elute antibodies from protein A columns. Unlike what is expected from an aggregation suppressor, arginine increases the surface tension of water, in fact more strongly than even GdnHCl. At the same time, it is chaotropic (breaks the water structure including in the hydration shell of water); i.e., not forming H-bonds with water and directly binding to the protein surface. As chaotopes increase the solubility of hydrophobic moieties, they are aggregate suppressors for proteins but can unfold the proteins (by interacting with hydrophobic side chains in proteins); this property is dependent on temperature and concentration [220]. It was also observed that arginine interacts well with tyrosine and tryptophan, moderately with hydrophilic amino acids and least with hydrophobic amino acids like leucine, isoleucine, and valine [220]. Arginine also binds to peptide bonds. This is the complex nature of the arginine binding to the protein surface. This overcomes the surface tension effects and exclusion from the protein surface as per preferential interaction theory. This leads to arginine suppressing protein aggregation but not destabilizing proteins either [220].
A major insight into arginine function was provided by Das et al. (2007), and their results deserve more attention [221]. Working with the Aβ1-42 peptide, these authors observed that it is the cluster of arginines, which bind to the peptide and suppresses its aggregation [221]. Light scattering increased with arginine concentration but reached a plateau beyond 0.5 M. They believe that this clustering aligns methylene groups of arginine to create a hydrophobic surface, thereby suggesting that the hydrophobic interactions are the cause of the aggregation suppression in the amyloid peptide [221].
While highlighting the involvement of hydrophobic interaction per se is not novel, the molecular size of arginine has been an important consideration in the discussion of preferential exclusion in the incisive analysis by Arakawa et al. [220]. Clustering by arginines, if taken into consideration, would make a considerable difference to that quantitative analysis.
Finally, mention should be made of the caution by Kim et al. (2016) that arginine under oxidizing conditions can produce oxides of nitrogen which can “fragment” the proteins [222].
All in all, one needs to test an additive like arginine with the specific pharmaceutical protein under all possible exposure conditions before using it.
9.3. Arginine Containing Cationic Cell-Penetrating Peptides in Drug Design
Cell-penetrating peptides (CCPs) are short peptides which can cross cell membranes. The CCPs also enable other molecules (bound or linked to them) to enter into cells and hence have shown considerable promise in drug design and even possibility of engineering of mammalian genome [223,224,225]. Cationic CCPs include arginine-based peptides of essentially three kinds: polyarginine (having 6 or 8 consecutive arginine residues); Tat peptides (such as Tat 49-57 derived from Tat protein of HIV, in which 6 arginine residues are present), and four arginine residues are linked on both ends of 6-amniohexanoic acid. Recent work using variants of TAT-based CPP variants suggest that hydrophobicity correlates positively with the cell-penetrating ability of these CPPs [226].
9.4. Arginine in Ion-Exchange, Hydrophobic Interaction, and Affinity Chromatography
Arakawa et al. (2007) presented a useful review covering usefulness of arginine in protein purification with various separation strategies [2]. It was found that the presence of arginine decreases the non-specific protein binding to the media during gel filtration. In ion-exchange chromatography of antibodies and human interleukin-6, protein recovery was better if arginine was present in the loading buffer. In case of hydrophobic interaction chromatography (HIC), arginine presence in eluting buffer improved recovery of proteins. It also led to better recovery of antibodies from Protein A column [227]. More recent work showed that arginine in elution buffer reduced the presence of host proteins and soluble aggregates while purifying bi-specific antibodies and mAb on HIC columns [228].
The positive charge of arginine enables its interaction with nucleic acids. Arginine as a ligand has been quite extensively used for purification of nucleic acids. Notable are the protocols for purifying supercoiled DNA plasmids [229] and DNA-based vaccine [230]. Meanwhile, 3-D printed chromatographic supports linked to arginine have been described which bind to pDNA [231].
Affinity chromatography is currently mostly used by implying an affinity matrix, which can selectively bind to a recombinant fusion protein consisting of a fusion tag attached to the target protein [232,233,234]. An “Argi” system has been described in which the affinity matrix is a biotinylated DNA aptamer linked to streptavidine-agarose support [235]. The matrix could selectively bind fusion proteins with fusion tags of cationic CPPs consisting of either polyarginine or Tat49-57 peptide. The bound fusion proteins could be eluted in fairly pure state by incorporating around 300 mM GuHCl; the yield was > 90% [235].
10. Conclusions and Future Perspective
The physical chemistry of arginine (a guanidino group and 3 methylene groups attached to the alpha carbon atom) leads to many interesting and intriguing functional consequences and a wide range of applications. For example, arginine is among the most abundant osmolyte in case of mollusks and crustaceans in shallow waters [236]. Its protective behavior during freezing and freeze-drying, as well its usefulness in protein folding have both led to considerable debate. But its usefulness in preventing opalescence in protein injectibles, which is likely to see much more work, is unquestionable. The finding that arginine self-assembles in aqueous medium in the form of clusters may possibly lead to some rethink about the exact mechanisms in its roles.
Recent work indicates that the design of biosensors based on arginine and for its determination is also likely to see more work in near future [237,238,239,240]. Another interesting futuristic direction is obtaining arginine from waste via production of arginine-rich proteins. Waste valorization is now is a part of the development of bioeconomy that shows immense potential in crafting sustainable technology [241]. Li et al. (2018) have mentioned how arginine indirectly obtained from waste can prove useful in this context [242]. Many other recent articles highlight this as an emerging strategy [243,244,245]. A comprehensive techno-economic analysis of this approach (though not just limited to arginine) has also appeared recently [245].
As a disorder promoting amino acid, it continues to find novel applications. A cluster of five arginine residues engineered into Fab fragment of an antibody against hen egg while lysozyme enhanced the association rate of Ag-Fab complex formation by increasing the conformational diversity of the antibody fragment [246]. As an affinity maturation strategy, this has tremendous value in therapeutics. Both protein injectables and antibodies are seeing an upsurge in drug discovery and design sector.
Molecular tweezers based upon arginine is another area where we are likely to see more work and more exciting applications. Same goes for CPPs based upon arginine in drug targeting. In general, while its role in defining protein structure, especially via cation-π interactions, would continue to be interesting; it is its function in IDPs/IDRs (such as being part of hot spots in protein-protein interactions involving regulatory proteins), which is more likely to see more innovative work and novel insights. Arginine can still throw up surprises as we further look at it in the context of structure-disorder-function continuum model [247].
Author Contributions
Both authors contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Fitch, C.A.; Platzer, G.; Okon, M.; Garcia-Moreno, B.E.; McIntosh, L.P. Arginine: Its pKa value revisited. Protein Sci. 2015, 24, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Tsumoto, K.; Kita, Y.; Chang, B.; Ejima, D. Biotechnology applications of amino acids in protein purification and formulations. Amino Acids 2007, 33, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.X.; Cao, Y.; Tang, Y.L.; Wang, J.H.; Ding, Y.H.; Tan, H.; Chen, Z.L.; Fang, R.Q.; Yin, J.; Chen, R.C.; et al. Improving mass spectrometry analysis of protein structures with arginine-selective chemical cross-linkers. Nat. Commun. 2019, 10, 3911. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct. Biol. 2007, 7, 56. [Google Scholar] [CrossRef]
- Petsko, G.A.; Ringe, D. Protein Structure and Function; New Science Press: 2004.
- Stryer, L. Biochemistry, 3rd ed.; W H Freeman: New York, 1988. [Google Scholar]
- Harms, M.J.; Schlessman, J.L.; Sue, G.R.; Garcia-Moreno, B. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. USA 2011, 108, 18954–18959. [Google Scholar] [CrossRef] [PubMed]
- Brinda, K.V.; Vishveshwara, S. A network representation of protein structures: Implications for protein stability. Biophys. J. 2005, 89, 4159–4170. [Google Scholar] [CrossRef]
- Chen, H.; Gu, F.; Huang, Z. Improved Chou-Fasman method for protein secondary structure prediction. BMC Bioinform. 2006, 7 (Suppl. 4), S14. [Google Scholar] [CrossRef]
- Kumar, S.; Bansal, M. Geometrical and sequence characteristics of alpha-helices in globular proteins. Biophys. J. 1998, 75, 1935–1944. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Meuzelaar, H.; Vreede, J.; Woutersen, S. Influence of Glu/Arg, Asp/Arg, and Glu/Lys Salt Bridges on alpha-Helical Stability and Folding Kinetics. Biophys. J. 2016, 110, 2328–2341. [Google Scholar] [CrossRef] [PubMed]
- Huyghues-Despointes, B.M.; Scholtz, J.M.; Baldwin, R.L. Helical peptides with three pairs of Asp-Arg and Glu-Arg residues in different orientations and spacings. Protein Sci. 1993, 2, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Munoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994, 1, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Meuzelaar, H.; Tros, M.; Huerta-Viga, A.; van Dijk, C.N.; Vreede, J.; Woutersen, S. Solvent-Exposed Salt Bridges Influence the Kinetics of alpha-Helix Folding and Unfolding. J. Phys. Chem. Lett. 2014, 5, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Obradovic, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 89–100. [Google Scholar]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef]
- Bhowmick, J.; Chandra, S.; Varadarajan, R. Deep mutational scanning to probe specificity determinants in proteins. In Structure and Intrinsic Disorder in Enzymology; Elsevier: 2023; pp. 31–71.
- Smith, C.A.; Chen, L.; Frankel, A.D. Using peptides as models of RNA-protein interactions. Methods Enzym. 2000, 318, 423–438. [Google Scholar] [CrossRef]
- Oliva, R. Protamines and male infertility. Hum. Reprod. Update 2006, 12, 417–435. [Google Scholar] [CrossRef]
- Miescher, F. Das Protamin, eine neue organische Base aus den Samenfäden des Rheinlachses. Berichte Der Dtsch. Chem. Ges. 1874, 7, 376–379. [Google Scholar] [CrossRef]
- Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol. 2007, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Hud, N.V.; Milanovich, F.P.; Balhorn, R. Evidence of novel secondary structure in DNA-bound protamine is revealed by Raman spectroscopy. Biochemistry 1994, 33, 7528–7535. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac. Symp. Biocomput. 2012, 128–139. [Google Scholar]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci. 2022, 31, e4496. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Uversky, V.N. Analyzing IDPs in interactomes. Intrinsically Disordered Proteins: Methods and Protocols.
- Oliva, R.; Dixon, G.H. Vertebrate protamine genes and the histone-to-protamine replacement reaction. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 25–94. [Google Scholar] [CrossRef]
- Balhorn, R.; Cosman, M.; Thornton, K.; Krishnan, V.; Corzett, M.; Bench, G.; Kramer, C.; Hud, N.; Allen, M.; Prieto, M. Protamine Mediated Condensation of DNA in Mammalian Sperm; Cache River Press: 1999.
- Bench, G.S.; Friz, A.M.; Corzett, M.H.; Morse, D.H.; Balhorn, R. DNA and total protamine masses in individual sperm from fertile mammalian subjects. Cytometry 1996, 23, 263–271. [Google Scholar] [CrossRef]
- Marshall, M.S.; Steele, R.P.; Thanthiriwatte, K.S.; Sherrill, C.D. Potential energy curves for cation-pi interactions: Off-axis configurations are also attractive. J. Phys. Chem. A 2009, 113, 13628–13632. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Lee, S.E.; Jeong, S.; Lee, J.; Park, D.; Chang, S. Multivalent electrostatic pi-cation interaction between synaptophysin and synapsin is responsible for the coacervation. Mol. Brain 2021, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef]
- Michael, L.A.; Chenault, J.A.; Miller, B.R., 3rd; Knolhoff, A.M.; Nagan, M.C. Water, shape recognition, salt bridges, and cation-pi interactions differentiate peptide recognition of the HIV rev-responsive element. J. Mol. Biol. 2009, 392, 774–786. [Google Scholar] [CrossRef]
- Hohlweg, W.; Wagner, G.E.; Hofbauer, H.F.; Sarkleti, F.; Setz, M.; Gubensak, N.; Lichtenegger, S.; Falsone, S.F.; Wolinski, H.; Kosol, S.; et al. A cation-pi interaction in a transmembrane helix of vacuolar ATPase retains the proton-transporting arginine in a hydrophobic environment. J. Biol. Chem. 2018, 293, 18977–18988. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation-pi interactions in protein-ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef]
- Bergsma, S.; Poulios, E.; Charalampogiannis, N.; Andraws, O.; Achinas, S. Cation-pi Interaction as a Key Player in Healthcare: A Mini-Review. Digit. Med. Healthc. Technol. 2022. [Google Scholar] [CrossRef]
- Kruger, D.M.; Neubacher, S.; Grossmann, T.N. Protein-RNA interactions: Structural characteristics and hotspot amino acids. RNA 2018, 24, 1457–1465. [Google Scholar] [CrossRef]
- Barik, A.C.N.; Pilla, S.P.; Bahadur, R.P. Molecular architecture of protein-RNA recognition sites. J. Biomol. Struct. Dyn. 2015, 33, 2738–2751. [Google Scholar] [CrossRef]
- Calnan, B.J.; Tidor, B.; Biancalana, S.; Hudson, D.; Frankel, A.D. Arginine-mediated RNA recognition: The arginine fork. Science 1991, 252, 1167–1171. [Google Scholar] [CrossRef]
- Chavali, S.S.; Cavender, C.E.; Mathews, D.H.; Wedekind, J.E. Arginine Forks Are a Widespread Motif to Recognize Phosphate Backbones and Guanine Nucleobases in the RNA Major Groove. J. Am. Chem. Soc. 2020, 142, 19835–19839. [Google Scholar] [CrossRef]
- Leclerc, F.; Karplus, M. MCSS-based predictions of RNA binding sites. Theor. Chem. Acc. 1999, 101, 131–137. [Google Scholar] [CrossRef]
- Belashov, I.A.; Crawford, D.W.; Cavender, C.E.; Dai, P.; Beardslee, P.C.; Mathews, D.H.; Pentelute, B.L.; McNaughton, B.R.; Wedekind, J.E. Structure of HIV TAR in complex with a Lab-Evolved RRM provides insight into duplex RNA recognition and synthesis of a constrained peptide that impairs transcription. Nucleic Acids Res. 2018, 46, 6401–6415. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef]
- Sudhof, T.C.; Lottspeich, F.; Greengard, P.; Mehl, E.; Jahn, R. A synaptic vesicle protein with a novel cytoplasmic domain and four transmembrane regions. Science 1987, 238, 1142–1144. [Google Scholar] [CrossRef]
- Park, D.; Wu, Y.; Lee, S.E.; Kim, G.; Jeong, S.; Milovanovic, D.; De Camilli, P.; Chang, S. Cooperative function of synaptophysin and synapsin in the generation of synaptic vesicle-like clusters in non-neuronal cells. Nat. Commun. 2021, 12, 263. [Google Scholar] [CrossRef]
- Ueda, T.; Greengard, P. Adenosine 3':5'-monophosphate-regulated phosphoprotein system of neuronal membranes. I. Solubilization, purification, and some properties of an endogenous phosphoprotein. J. Biol. Chem. 1977, 252, 5155–5163. [Google Scholar] [CrossRef]
- Ferrari, L.; Stucchi, R.; Konstantoulea, K.; van de Kamp, G.; Kos, R.; Geerts, W.J.C.; van Bezouwen, L.S.; Forster, F.G.; Altelaar, M.; Hoogenraad, C.C.; et al. Arginine pi-stacking drives binding to fibrils of the Alzheimer protein Tau. Nat. Commun. 2020, 11, 571. [Google Scholar] [CrossRef]
- Hong, Y.; Najafi, S.; Casey, T.; Shea, J.E.; Han, S.I.; Hwang, D.S. Hydrophobicity of arginine leads to reentrant liquid-liquid phase separation behaviors of arginine-rich proteins. Nat. Commun. 2022, 13, 7326. [Google Scholar] [CrossRef]
- Das, S.; Lin, Y.H.; Vernon, R.M.; Forman-Kay, J.D.; Chan, H.S. Comparative roles of charge, pi, and hydrophobic interactions in sequence-dependent phase separation of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 28795–28805. [Google Scholar] [CrossRef]
- Schuster, B.S.; Dignon, G.L.; Tang, W.S.; Kelley, F.M.; Ranganath, A.K.; Jahnke, C.N.; Simpkins, A.G.; Regy, R.M.; Hammer, D.A.; Good, M.C.; et al. Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 11421–11431. [Google Scholar] [CrossRef]
- Greig, J.A.; Nguyen, T.A.; Lee, M.; Holehouse, A.S.; Posey, A.E.; Pappu, R.V.; Jedd, G. Arginine-Enriched Mixed-Charge Domains Provide Cohesion for Nuclear Speckle Condensation. Mol. Cell 2020, 77, 1237–1250. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7. [Google Scholar] [CrossRef]
- Krainer, G.; Welsh, T.J.; Joseph, J.A.; Espinosa, J.R.; Wittmann, S.; de Csillery, E.; Sridhar, A.; Toprakcioglu, Z.; Gudiskyte, G.; Czekalska, M.A.; et al. Reentrant liquid condensate phase of proteins is stabilized by hydrophobic and non-ionic interactions. Nat. Commun. 2021, 12, 1085. [Google Scholar] [CrossRef]
- Prather, L.J.; Weerasekare, G.M.; Sima, M.; Quinn, C.; Stewart, R.J. Aqueous Liquid-Liquid Phase Separation of Natural and Synthetic Polyguanidiniums. Polymers 2019, 11. [Google Scholar] [CrossRef]
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P. Arginine “Magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc Chem. Res. 2018, 51, 1455–1464. [Google Scholar] [CrossRef]
- Gao, B.; Wyttenbach, T.; Bowers, M.T. Protonated arginine and protonated lysine: Hydration and its effect on the stability of salt-bridge structures. J. Phys. Chem. B 2009, 113, 9995–10000. [Google Scholar] [CrossRef]
- Vondrasek, J.; Mason, P.E.; Heyda, J.; Collins, K.D.; Jungwirth, P. The molecular origin of like-charge arginine-arginine pairing in water. J. Phys. Chem. B 2009, 113, 9041–9045. [Google Scholar] [CrossRef]
- Daoud, H.; Suhail, H.; Sabbagh, M.; Belzil, V.; Szuto, A.; Dionne-Laporte, A.; Khoris, J.; Camu, W.; Salachas, F.; Meininger, V.; et al. C9orf72 hexanucleotide repeat expansions as the causative mutation for chromosome 9p21-linked amyotrophic lateral sclerosis and frontotemporal dementia. Arch. Neurol. 2012, 69, 1159–1163. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Gijselinck, I.; Cruts, M.; Van Broeckhoven, C. The Genetics of C9orf72 Expansions. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., 3rd; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PR(n) poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef]
- Jovicic, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645. [Google Scholar] [CrossRef]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013, 126, 845–857. [Google Scholar] [CrossRef]
- Darling, A.L.; Breydo, L.; Rivas, E.G.; Gebru, N.T.; Zheng, D.; Baker, J.D.; Blair, L.J.; Dickey, C.A.; Koren, J., 3rd; Uversky, V.N. Repeated repeat problems: Combinatorial effect of C9orf72-derived dipeptide repeat proteins. Int. J. Biol. Macromol. 2019, 127, 136–145. [Google Scholar] [CrossRef]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.L.; Birsa, N.; Gittings, L.M.; Holbling, B.V.; Dyson, M.C.; Thoeng, A.; Neeves, J.; et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef]
- Odeh, H.M.; Shorter, J. Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg. Top. Life Sci. 2020, 4, 293–305. [Google Scholar] [CrossRef]
- Park, J.; Wu, Y.; Shao, W.; Gendron, T.F.; van der Spek, S.J.F.; Sultanakhmetov, G.; Basu, A.; Castellanos Otero, P.; Jones, C.J.; Jansen-West, K.; et al. Poly(GR) interacts with key stress granule factors promoting its assembly into cytoplasmic inclusions. Cell Rep. 2023, 42, 112822. [Google Scholar] [CrossRef]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- White, M.R.; Mitrea, D.M.; Zhang, P.; Stanley, C.B.; Cassidy, D.E.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Guo, L.; Gonzales, P.K.; Gendron, T.F.; Wu, Y.; Jansen-West, K.; O’Raw, A.D.; Pickles, S.R.; Prudencio, M.; Carlomagno, Y.; et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055. [Google Scholar] [CrossRef]
- Harrison, P.M. Intrinsic disorder and posttranslational modification: An evolutionary perspective. In Structure and Intrinsic Disorder in Enzymology; Elsevier: 2023; pp. 377–396.
- Uversky, V.N. Posttranslational Modifications. In Brenner's Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: London, UK; Walham, MA, USA; San Diego, CA, USA, 2013; Volume 5, pp. 425–430. [Google Scholar]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Gupta, M.N.; Uversky, V.N. Moonlighting enzymes: When cellular context defines specificity. Cell. Mol. Life Sci. 2023, 80, 130. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014, 23, 1077–1093. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef]
- Tsikas, D. Post-translational modifications (PTM): Analytical approaches, signaling, physiology and pathophysiology-part I. Amino Acids 2021, 53, 485–487. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.; Krishnamurthy, A.; Rethi, B. Current view on the pathogenic role of anti-citrullinated protein antibodies in rheumatoid arthritis. RMD Open 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.F.; Barry, A.; Greenman, J.; Beltran-Alvarez, P. Arginine methylation: The promise of a ‘silver bullet’ for brain tumours? Amino Acids 2021, 53, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.E.; Dlakic, M.; Clarke, S. Automated identification of putative methyltransferases from genomic open reading frames. Mol. Cell. Proteom. 2003, 2, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteom. 2014, 13, 372–387. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Post, A.; Bollenbach, A.; Bakker, S.J.L.; Tsikas, D. Whole-body arginine dimethylation is associated with all-cause mortality in adult renal transplant recipients. Amino Acids 2021, 53, 541–554. [Google Scholar] [CrossRef]
- Said, M.Y.; Bollenbach, A.; Minovic, I.; van Londen, M.; Frenay, A.R.; de Borst, M.H.; van den Berg, E.; Kayacelebi, A.A.; Tsikas, D.; van Goor, H.; et al. Plasma ADMA, urinary ADMA excretion, and late mortality in renal transplant recipients. Amino Acids 2019, 51, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Said, M.Y.; Douwes, R.M.; van Londen, M.; Minovic, I.; Frenay, A.R.; de Borst, M.H.; van den Berg, E.; Heiner-Fokkema, M.R.; Kayacelebi, A.A.; Bollenbach, A.; et al. Effect of renal function on homeostasis of asymmetric dimethylarginine (ADMA): Studies in donors and recipients of renal transplants. Amino Acids 2019, 51, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Frenay, A.R.; van den Berg, E.; de Borst, M.H.; Beckmann, B.; Tsikas, D.; Feelisch, M.; Navis, G.; Bakker, S.J.; van Goor, H. Plasma ADMA associates with all-cause mortality in renal transplant recipients. Amino Acids 2015, 47, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Kronenberg, F.; Kielstein, J.T.; Morath, C.; Bode-Boger, S.M.; Haller, H.; Ritz, E. Asymmetric dimethylarginine and progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol. 2005, 16, 2456–2461. [Google Scholar] [CrossRef] [PubMed]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arter. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins 2017, 9. [Google Scholar] [CrossRef]
- Busch, M.; Fleck, C.; Wolf, G.; Stein, G. Asymmetrical (ADMA) and symmetrical dimethylarginine (SDMA) as potential risk factors for cardiovascular and renal outcome in chronic kidney disease—Possible candidates for paradoxical epidemiology? Amino Acids 2006, 30, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Marsden, A.J.; Riley, D.R.J.; Birkett, S.; Rodriguez-Barucg, Q.; Guinn, B.A.; Carroll, S.; Ingle, L.; Sathyapalan, T.; Beltran-Alvarez, P. Love is in the hair: Arginine methylation of human hair proteins as novel cardiovascular biomarkers. Amino Acids 2022, 54, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.N.; Perwez, M.; Sardar, M. Protein crosslinking: Uses in chemistry, biology and biotechnology. Biocatal. Biotransform. 2020, 38, 178–201. [Google Scholar] [CrossRef]
- Mukherjee, J.; Majumder, A.B.; Gupta, M.N. Adding an appropriate amino acid during crosslinking results in more stable crosslinked enzyme aggregates. Anal. Biochem. 2016, 507, 27–32. [Google Scholar] [CrossRef]
- Collier, T.A.; Nash, A.; Birch, H.L.; de Leeuw, N.H. Intra-molecular lysine-arginine derived advanced glycation end-product cross-linking in Type I collagen: A molecular dynamics simulation study. Biophys. Chem. 2016, 218, 42–46. [Google Scholar] [CrossRef]
- Thorpe, C.T.; Streeter, I.; Pinchbeck, G.L.; Goodship, A.E.; Clegg, P.D.; Birch, H.L. Aspartic acid racemization and collagen degradation markers reveal an accumulation of damage in tendon collagen that is enhanced with aging. J. Biol. Chem. 2010, 285, 15674–15681. [Google Scholar] [CrossRef] [PubMed]
- Biemel, K.M.; Friedl, D.A.; Lederer, M.O. Identification and quantification of major maillard cross-links in human serum albumin and lens protein. Evidence for glucosepane as the dominant compound. J. Biol. Chem. 2002, 277, 24907–24915. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Martin, E.; Gallego-Munoz, P.; Ibares-Frias, L.; Marcos, S.; Perez-Merino, P.; Fernandez, I.; Kochevar, I.E.; Martinez-Garcia, M.C. Rose Bengal and Green Light Versus Riboflavin-UVA Cross-Linking: Corneal Wound Repair Response. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4821–4830. [Google Scholar] [CrossRef] [PubMed]
- Seiler, T.G.; Engler, M.; Beck, E.; Birngruber, R.; Kochevar, I.E. Interface Bonding With Corneal Crosslinking (CXL) After LASIK Ex Vivo. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6292–6298. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Munoz, P.; Ibares-Frias, L.; Lorenzo, E.; Marcos, S.; Perez-Merino, P.; Bekesi, N.; Kochevar, I.E.; Martinez-Garcia, M.C. Corneal Wound Repair After Rose Bengal and Green Light Crosslinking: Clinical and Histologic Study. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Alt, C.; Webb, R.H.; Melki, S.; Kochevar, I.E. Corneal Crosslinking With Rose Bengal and Green Light: Efficacy and Safety Evaluation. Cornea 2016, 35, 1234–1241. [Google Scholar] [CrossRef]
- Bekesi, N.; Kochevar, I.E.; Marcos, S. Corneal Biomechanical Response Following Collagen Cross-Linking with Rose Bengal-Green Light and Riboflavin-UVA. Investig. Ophthalmol. Vis. Sci. 2016, 57, 992–1001. [Google Scholar] [CrossRef]
- Wertheimer, C.M.; Mendes, B.; Pei, Q.; Brandt, K.; Kochevar, I.E. Arginine as an Enhancer in Rose Bengal Photosensitized Corneal Crosslinking. Transl. Vis. Sci. Technol. 2020, 9, 24. [Google Scholar] [CrossRef]
- Wertheimer, C.M.; Elhardt, C.; Kaminsky, S.M.; Pham, L.; Pei, Q.; Mendes, B.; Afshar, S.; Kochevar, I.E. Enhancing Rose Bengal-Photosensitized Protein Crosslinking in the Cornea. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1845–1852. [Google Scholar] [CrossRef]
- Cherfan, D.; Verter, E.E.; Melki, S.; Gisel, T.E.; Doyle, F.J., Jr.; Scarcelli, G.; Yun, S.H.; Redmond, R.W.; Kochevar, I.E. Collagen cross-linking using rose bengal and green light to increase corneal stiffness. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Udhayakumar, S.; Shankar, K.G.; Sowndarya, S.; Venkatesh, S.; Muralidharan, C.; Rose, C. L-arginine intercedes bio-crosslinking of a collagen–chitosan 3D-hybrid scaffold for tissue engineering and regeneration: In silico, in vitro, and in vivo studies. RSC Adv. 2017, 7, 25070–25088. [Google Scholar] [CrossRef]
- Caruso, A.; Martinie, R.J.; Bushin, L.B.; Seyedsayamdost, M.R. Macrocyclization via an arginine-tyrosine crosslink broadens the reaction scope of radical S-adenosylmethionine enzymes. J. Am. Chem. Soc. 2019, 141, 16610–16614. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.F. Arginyl residues and anion binding sites in proteins. Mol. Cell. Biochem. 1979, 26, 71–92. [Google Scholar] [CrossRef]
- Leone, D.L.; Hubalek, M.; Pohl, R.; Sykorova, V.; Hocek, M. 1,3-Diketone-Modified Nucleotides and DNA for Cross-Linking with Arginine-Containing Peptides and Proteins. Angew. Chem. Int. Ed. Engl. 2021, 60, 17383–17387. [Google Scholar] [CrossRef]
- Means, G.; Feeney, R. Chemical Modification of Proteins; Hoiden-Day Inc.: San Francisco, CA, USA, 1971. [Google Scholar]
- Tyagi, R.; Gupta, M.N. Chemical modification and chemical crosslinking for enhancing thermostability of enzymes. In Thermostability of Enzymes; Gupta, M.N., Ed.; Springer: Berlin, 1993; pp. 146–160. [Google Scholar]
- Tyagi, R.; Gupta, M.N. Chemical modification and chemical crosslinking of proteins/enzyme stabilization. Biochemistry 1998, 63, 395–407. [Google Scholar]
- Zhou, Y.; Liu, G.; Huang, H.; Wu, J. Advances and impact of arginine-based materials in wound healing. J. Mater. Chem. B 2021, 9, 6738–6750. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kuroyanagi, Y. Development of a wound dressing composed of hyaluronic acid sponge containing arginine and epidermal growth factor. J. Biomater. Sci. Polym. Ed. 2010, 21, 715–726. [Google Scholar] [CrossRef]
- Reesi, F.; Minaiyan, M.; Taheri, A. A novel lignin-based nanofibrous dressing containing arginine for wound-healing applications. Drug Deliv. Transl. Res. 2018, 8, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Durmus, A.S.; Tuzcu, M.; Ozdemir, O.; Orhan, C.; Sahin, N.; Ozercan, I.H.; Komorowski, J.R.; Ali, S.; Sahin, K. Arginine Silicate Inositol Complex Accelerates Cutaneous Wound Healing. Biol. Trace. Elem. Res. 2017, 177, 122–131. [Google Scholar] [CrossRef]
- Baron, R.I.; Culica, M.E.; Biliuta, G.; Bercea, M.; Gherman, S.; Zavastin, D.; Ochiuz, L.; Avadanei, M.; Coseri, S. Physical Hydrogels of Oxidized Polysaccharides and Poly(Vinyl Alcohol) for Wound Dressing Applications. Materials 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Antunes, B.P.; Moreira, A.F.; Gaspar, V.M.; Correia, I.J. Chitosan/arginine-chitosan polymer blends for assembly of nanofibrous membranes for wound regeneration. Carbohydr. Polym. 2015, 130, 104–112. [Google Scholar] [CrossRef]
- Wu, J.; Mutschler, M.A.; Chu, C.C. Synthesis and characterization of ionic charged water soluble arginine-based poly(ester amide). J. Mater. Sci. Mater. Med. 2011, 22, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; la Cour, T.; Hazen, E.E., Jr.; Legg, M.J. The role of arginine residues at enzyme active sites. The interaction between guanidinium ions and p-nitro-phenyl phosphate and its effect on the rate of hydrolysis of the ester. Biochim. Biophys. Acta 1977, 481, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Adak, S.; Bandyopadhyay, D.; Bandyopadhyay, U.; Banerjee, R.K. An essential role of active site arginine residue in iodide binding and histidine residue in electron transfer for iodide oxidation by horseradish peroxidase. Mol. Cell. Biochem. 2001, 218, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Nowak, T. Arginine residues at the active site of avian liver phosphoenolpyruvate carboxykinase. J. Biol. Chem. 1989, 264, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Walker, V. Ammonia metabolism and hyperammonemic disorders. Adv. Clin. Chem. 2014, 67, 73–150. [Google Scholar] [PubMed]
- Kumar, S.; Lennane, J.; Ratner, S. Argininosuccinate synthetase: Essential role of cysteine and arginine residues in relation to structure and mechanism of ATP activation. Proc. Natl. Acad. Sci. USA 1985, 82, 6745–6749. [Google Scholar] [CrossRef] [PubMed]
- Keenholtz, R.A.; Mouw, K.W.; Boocock, M.R.; Li, N.S.; Piccirilli, J.A.; Rice, P.A. Arginine as a general acid catalyst in serine recombinase-mediated DNA cleavage. J. Biol. Chem. 2013, 288, 29206–29214. [Google Scholar] [CrossRef]
- Pauff, J.M.; Hemann, C.F.; Junemann, N.; Leimkuhler, S.; Hille, R. The role of arginine 310 in catalysis and substrate specificity in xanthine dehydrogenase from Rhodobacter capsulatus. J. Biol. Chem. 2007, 282, 12785–12790. [Google Scholar] [CrossRef]
- Albert, S.; Will, E.; Gallwitz, D. Identification of the catalytic domains and their functionally critical arginine residues of two yeast GTPase-activating proteins specific for Ypt/Rab transport GTPases. EMBO J. 1999, 18, 5216–5225. [Google Scholar] [CrossRef]
- Chen, G.; Chen, X. Arginine residues in the active site of human phenol sulfotransferase (SULT1A1). J. Biol. Chem. 2003, 278, 36358–36364. [Google Scholar] [CrossRef]
- Sulfurtransferases: Essential Enzymes for Life; Nagahara, N. , Ed.; Elsevier: San Diego, CA, USA, 2023. [Google Scholar]
- Communi, D.; Lecocq, R.; Erneux, C. Arginine 343 and 350 are two active residues involved in substrate binding by human Type I D-myo-inositol 1,4,5,-trisphosphate 5-phosphatase. J. Biol. Chem. 1996, 271, 11676–11683. [Google Scholar] [CrossRef]
- Shi, J.; Mukhopadhyay, R.; Rosen, B.P. Identification of a triad of arginine residues in the active site of the ArsC arsenate reductase of plasmid R773. FEMS Microbiol. Lett. 2003, 227, 295–301. [Google Scholar] [CrossRef]
- Hsiao, J.C.; McGrath, A.P.; Kielmann, L.; Kalimuthu, P.; Darain, F.; Bernhardt, P.V.; Harmer, J.; Lee, M.; Meyers, K.; Maher, M.J.; et al. The central active site arginine in sulfite oxidizing enzymes alters kinetic properties by controlling electron transfer and redox interactions. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 19–27. [Google Scholar] [CrossRef]
- Murtas, G.; Sacchi, S.; Pollegioni, L. Substitution of Arginine 120 in Human D-Amino Acid Oxidase Favors FAD-Binding and Nuclear Mistargeting. Front. Mol. Biosci. 2019, 6, 125. [Google Scholar] [CrossRef]
- Sarkar, A.; Bhakta, S.; Chattopadhyay, S.; Dey, A. Role of distal arginine residue in the mechanism of heme nitrite reductases. Chem. Sci. 2023, 14, 7875–7886. [Google Scholar] [CrossRef]
- Morris, S.M., Jr. Arginine metabolism: Boundaries of our knowledge. J. Nutr. 2007, 137, 1602S–1609S. [Google Scholar] [CrossRef]
- Tong, B.C.; Barbul, A. Cellular and physiological effects of arginine. Mini Rev. Med. Chem. 2004, 4, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Hamilton-Reeves, J.; Martindale, R.G.; Sarav, M.; Ochoa Gautier, J.B. Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr. Clin. Pr. 2017, 32, 30S–47S. [Google Scholar] [CrossRef] [PubMed]
- Hristina, K.; Langerholc, T.; Trapecar, M. Novel metabolic roles of L-arginine in body energy metabolism and possible clinical applications. J. Nutr. Health Aging 2014, 18, 213–218. [Google Scholar] [CrossRef]
- Xiong, L.; Teng, J.L.; Botelho, M.G.; Lo, R.C.; Lau, S.K.; Woo, P.C. Arginine Metabolism in Bacterial Pathogenesis and Cancer Therapy. Int. J. Mol. Sci. 2016, 17, 363. [Google Scholar] [CrossRef]
- Erens, C.; Van Broeckhoven, J.; Bronckaers, A.; Lemmens, S.; Hendrix, S. The Dark Side of an Essential Amino Acid: L-Arginine in Spinal Cord Injury. J. Neurotrauma 2023, 40, 820–832. [Google Scholar] [CrossRef]
- Ma, C.; Liao, K.; Wang, J.; Li, T.; Liu, L. L-Arginine, as an essential amino acid, is a potential substitute for treating COPD via regulation of ROS/NLRP3/NF-kappaB signaling pathway. Cell Biosci. 2023, 13, 152. [Google Scholar] [CrossRef]
- Leblond, J.; Petitjean, A. Molecular tweezers: Concepts and applications. ChemPhysChem 2011, 12, 1043–1051. [Google Scholar] [CrossRef]
- Kaiser, F.; Bittrich, S.; Salentin, S.; Leberecht, C.; Haupt, V.J.; Krautwurst, S.; Schroeder, M.; Labudde, D. Backbone Brackets and Arginine Tweezers delineate Class I and Class II aminoacyl tRNA synthetases. PLoS Comput. Biol. 2018, 14, e1006101. [Google Scholar] [CrossRef]
- Schrader, T.; Bitan, G.; Klarner, F.G. Molecular tweezers for lysine and arginine—Powerful inhibitors of pathologic protein aggregation. Chem. Commun. 2016, 52, 11318–11334. [Google Scholar] [CrossRef]
- Mbarek, A.; Moussa, G.; Chain, J.L. Pharmaceutical Applications of Molecular Tweezers, Clefts and Clips. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Hadrovic, I.; Rebmann, P.; Klarner, F.G.; Bitan, G.; Schrader, T. Molecular Lysine Tweezers Counteract Aberrant Protein Aggregation. Front. Chem. 2019, 7, 657. [Google Scholar] [CrossRef]
- Hodges, A.J.; Gallegos, I.J.; Laughery, M.F.; Meas, R.; Tran, L.; Wyrick, J.J. Histone Sprocket Arginine Residues Are Important for Gene Expression, DNA Repair, and Cell Viability in Saccharomyces cerevisiae. Genetics 2015, 200, 795–806. [Google Scholar] [CrossRef]
- Muthurajan, U.M.; Park, Y.J.; Edayathumangalam, R.S.; Suto, R.K.; Chakravarthy, S.; Dyer, P.N.; Luger, K. Structure and dynamics of nucleosomal DNA. Biopolymers 2003, 68, 547–556. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Magalhaes, A.; Maigret, B.; Hoflack, J.; Gomes, J.N.; Scheraga, H.A. Contribution of unusual arginine-arginine short-range interactions to stabilization and recognition in proteins. J. Protein Chem. 1994, 13, 195–215. [Google Scholar] [CrossRef]
- Neves, M.A.; Yeager, M.; Abagyan, R. Unusual arginine formations in protein function and assembly: Rings, strings, and stacks. J. Phys. Chem. B 2012, 116, 7006–7013. [Google Scholar] [CrossRef]
- Wang, W. Oral protein drug delivery. J. Drug Target 1996, 4, 195–232. [Google Scholar] [CrossRef]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Garcia Garcia, C.; Patkar, S.S.; Wang, B.; Abouomar, R.; Kiick, K.L. Recombinant protein-based injectable materials for biomedical applications. Adv. Drug Deliv. Rev. 2023, 193, 114673. [Google Scholar] [CrossRef]
- Ryan, S.; Shortall, K.; Dully, M.; Djehedar, A.; Murray, D.; Butler, J.; Neilan, J.; Soulimane, T.; Hudson, S.P. Long acting injectables for therapeutic proteins. Colloids Surf. B Biointerfaces 2022, 217, 112644. [Google Scholar] [CrossRef]
- Ibeanu, N.; Egbu, R.; Onyekuru, L.; Javaheri, H.; Khaw, P.T.; Williams, G.R.; Brocchini, S.; Awwad, S. Injectables and Depots to Prolong Drug Action of Proteins and Peptides. Pharmaceutics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013, 203, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015, 589, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Dundr, M.; Misteli, T. Biogenesis of nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000711. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Brangwynne, C.P. Nuclear bodies: The emerging biophysics of nucleoplasmic phases. Curr. Opin. Cell Biol. 2015, 34, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv. Colloid Interface Sci. 2017, 239, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, S.; Huang, Y.; He, X.; Cui, H.; Zhu, X.; Zheng, Y. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell 2015, 163, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: Overview of the evolution of the cell compartmentalization concept. Cell. Mol. Life Sci. 2022, 79, 251. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Nguemaha, V.; Mazarakos, K.; Qin, S. Why Do Disordered and Structured Proteins Behave Differently in Phase Separation? Trends Biochem. Sci. 2018, 43, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem. Sci. 2019, 44, 716–728. [Google Scholar] [CrossRef]
- Fonin, A.V.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B.Y.; Kulkarni, P.; Uversky, V.N. Biological soft matter: Intrinsically disordered proteins in liquid-liquid phase separation and biomolecular condensates. Essays Biochem. 2022, 66, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Raut, A.S.; Kalonia, D.S. Opalescence in monoclonal antibody solutions and its correlation with intermolecular interactions in dilute and concentrated solutions. J. Pharm. Sci. 2015, 104, 1263–1274. [Google Scholar] [CrossRef]
- Oki, S.; Nishinami, S.; Shiraki, K. Arginine suppresses opalescence and liquid-liquid phase separation in IgG solutions. Int. J. Biol. Macromol. 2018, 118, 1708–1712. [Google Scholar] [CrossRef]
- Banks, D.D.; Cordia, J.F. Suppression of Electrostatic Mediated Antibody Liquid-Liquid Phase Separation by Charged and Noncharged Preferentially Excluded Excipients. Mol. Pharm. 2021, 18, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Pyne, P.; Mitra, R.K. Excipients Do Regulate Phase Separation in Lysozyme and Thus Also Its Hydration. J. Phys. Chem. Lett. 2022, 13, 931–938. [Google Scholar] [CrossRef]
- Tsumoto, K.; Umetsu, M.; Kumagai, I.; Ejima, D.; Philo, J.S.; Arakawa, T. Role of arginine in protein refolding, solubilization, and purification. Biotechnol. Prog. 2004, 20, 1301–1308. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K. The effects of arginine on refolding of aggregated proteins: Not facilitate refolding, but suppress aggregation. Biochem. Biophys. Res. Commun. 2003, 304, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Baynes, B.M.; Wang, D.I.; Trout, B.L. Role of arginine in the stabilization of proteins against aggregation. Biochemistry 2005, 44, 4919–4925. [Google Scholar] [CrossRef]
- Gupta, M.N.; Uversky, V.N. Macromolecular crowding: How it affects protein structure, disorder, and catalysis. In Structure and Intrinsic Disorder in Enzymology; Elsevier: 2023; pp. 353–376.
- Liu, Y.D.; Li, J.J.; Wang, F.W.; Chen, J.; Li, P.; Su, Z.G. A newly proposed mechanism for arginine-assisted protein refolding--not inhibiting soluble oligomers although promoting a correct structure. Protein Expr. Purif. 2007, 51, 235–242. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Wang, Y.; Ding, H.; Su, Z. Different effects of l-arginine on protein refolding: Suppressing aggregates of hydrophobic interaction, not covalent binding. Biotechnol. Prog. 2008, 24, 1365–1372. [Google Scholar] [CrossRef]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 2018, 118, 1169–1198. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Ejima, D.; Tsumoto, K.; Obeyama, N.; Tanaka, Y.; Kita, Y.; Timasheff, S.N. Suppression of protein interactions by arginine: A proposed mechanism of the arginine effects. Biophys. Chem. 2007, 127, 1–8. [Google Scholar] [CrossRef]
- Das, U.; Hariprasad, G.; Ethayathulla, A.S.; Manral, P.; Das, T.K.; Pasha, S.; Mann, A.; Ganguli, M.; Verma, A.K.; Bhat, R.; et al. Inhibition of protein aggregation: Supramolecular assemblies of arginine hold the key. PLoS ONE 2007, 2, e1176. [Google Scholar] [CrossRef]
- Kim, N.A.; Hada, S.; Thapa, R.; Jeong, S.H. Arginine as a protein stabilizer and destabilizer in liquid formulations. Int. J. Pharm. 2016, 513, 26–37. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control Release 2014, 174, 126–136. [Google Scholar] [CrossRef]
- Peitz, M.; Pfannkuche, K.; Rajewsky, K.; Edenhofer, F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: A tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 4489–4494. [Google Scholar] [CrossRef]
- Allen, J.; Pellois, J.P. Hydrophobicity is a key determinant in the activity of arginine-rich cell penetrating peptides. Sci. Rep. 2022, 12, 15981. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K.; Nagase, K.; Ejima, D. The effects of arginine on protein binding and elution in hydrophobic interaction and ion-exchange chromatography. Protein Expr. Purif. 2007, 54, 110–116. [Google Scholar] [CrossRef]
- Hall, T.; Kelly, G.M.; Emery, W.R. Use of mobile phase additives for the elution of bispecific and monoclonal antibodies from phenyl based hydrophobic interaction chromatography resins. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1096, 20–30. [Google Scholar] [CrossRef]
- Valente, J.F.A.; Sousa, A.; Azevedo, G.A.; Queiroz, J.A.; Sousa, F. Purification of supercoiled p53-encoding plasmid using an arginine-modified macroporous support. J. Chromatogr. A 2020, 1618, 460890. [Google Scholar] [CrossRef]
- Soares, A.; Queiroz, J.A.; Sousa, F.; Sousa, A. Purification of human papillomavirus 16 E6/E7 plasmid deoxyribonucleic acid-based vaccine using an arginine modified monolithic support. J. Chromatogr. A 2013, 1320, 72–79. [Google Scholar] [CrossRef]
- Valente, J.F.A.; Carreira, T.S.; Dias, J.R.; Sousa, F.; Alves, N. Arginine-Modified 3D-Printed Chromatographic Supports. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Gupta, M.N. Methods for Affinity-Based Separations of Enzymes and Proteins; Springer Science & Business Media: 2002.
- Roy, I.; Gupta, M.N. Current trends in affinity-based separations of proteins/enzymes. Curr. Sci. 2000, 587–591. [Google Scholar]
- Mondal, K.; Gupta, M.N. The affinity concept in bioseparation: Evolving paradigms and expanding range of applications. Biomol. Eng. 2006, 23, 59–76. [Google Scholar] [CrossRef]
- Bartnicki, F.; Bonarek, P.; Kowalska, E.; Strzalka, W. The Argi system: One-step purification of proteins tagged with arginine-rich cell-penetrating peptides. Sci. Rep. 2017, 7, 2619. [Google Scholar] [CrossRef]
- Burg, M.B.; Ferraris, J.D. Intracellular organic osmolytes: Function and regulation. J. Biol. Chem. 2008, 283, 7309–7313. [Google Scholar] [CrossRef]
- Xu, M.; Liu, P.; Chen, J.; Peng, A.; Yang, T.; Zhang, X.; Xu, Z.; Rao, Z. Development of a Novel Biosensor-Driven Mutation and Selection System via in situ Growth of Corynebacterium crenatum for the Production of L-Arginine. Front. Bioeng. Biotechnol. 2020, 8, 175. [Google Scholar] [CrossRef]
- Stasyuk, N.; Gayda, G.; Demkiv, O.; Darmohray, L.; Gonchar, M.; Nisnevitch, M. Amperometric biosensors for L-arginine determination based on L-arginine oxidase and peroxidase-like nanozymes. Appl. Sci. 2021, 11, 7024. [Google Scholar] [CrossRef]
- Berketa, K.; Saiapina, O.; Fayura, L.; Sibirny, A.; Dzyadevych, S.; Soldatkin, O. Novel highly sensitive conductometric biosensor based on arginine deiminase from Mycoplasma hominis for determination of arginine. Sens. Actuators B Chem. 2022, 367, 132023. [Google Scholar] [CrossRef]
- Rahman, M.; Niu, J.; Cui, X.; Zhou, C.; Tang, N.; Jin, H.; Cui, D. Electrochemical Biosensor Based on l-Arginine and rGO-AuNSs Deposited on the Electrode Combined with DNA Probes for Ultrasensitive Detection of the Gastric Cancer-Related PIK3CA Gene of ctDNA. ACS Appl. Bio Mater. 2022, 5, 5094–5103. [Google Scholar] [CrossRef]
- Roy, I.; Gupta, M.N. White & grey biotechnologies for shaping a sustainable future. RSC Sustain. 2023, 1, 1722–1736. [Google Scholar]
- Li, S.Y.; Ng, I.S.; Chen, P.T.; Chiang, C.J.; Chao, Y.P. Biorefining of protein waste for production of sustainable fuels and chemicals. Biotechnol. Biofuels 2018, 11, 256. [Google Scholar] [CrossRef]
- De Schouwer, F.; Claes, L.; Vandekerkhove, A.; Verduyckt, J.; De Vos, D.E. Protein-rich biomass waste as a resource for future biorefineries: State of the art, challenges, and opportunities. ChemSusChem 2019, 12, 1272–1303. [Google Scholar] [CrossRef] [PubMed]
- Peydayesh, M.; Bagnani, M.; Soon, W.L.; Mezzenga, R. Turning Food Protein Waste into Sustainable Technologies. Chem. Rev. 2023, 123, 2112–2154. [Google Scholar] [CrossRef] [PubMed]
- Piercy, E.; Verstraete, W.; Ellis, P.R.; Banks, M.; Rockström, J.; Smith, P.; Witard, O.C.; Hallett, J.; Hogstrand, C.; Knott, G. A sustainable waste-to-protein system to maximise waste resource utilisation for developing food-and feed-grade protein solutions. Green Chem. 2023, 25, 808–832. [Google Scholar] [CrossRef]
- Maeta, S.; Nakakido, M.; Matsuura, H.; Sakai, N.; Hirata, K.; Kuroda, D.; Fukunaga, A.; Tsumoto, K. Arginine cluster introduction on framework region in anti-lysozyme antibody improved association rate constant by changing conformational diversity of CDR loops. Protein Sci. 2023, 32, e4745. [Google Scholar] [CrossRef]
- Gupta, M.N.; Uversky, V.N. Structure and Intrinsic Disorder in Enzymology; Elsevier: 2022.
Figure 1.
Characteristic sequence features of a protamine family. (A) Amino acid composition profile of 1880 protamines generatd by Composition Profiler (http://www.cprofiler.org/) [4]. The fractional difference is calculated as , where Cx is the content of a given amino acid in the query set (1880 protamines or known intrinsically disordered proteins), and Corder is the content of a given amino acid in the background set (Protein Data Bank Select 25). The amino acid residues are ranked from most order-promoting residue to most disorder-promoting residue. Positive values indicate enrichment, and negative values indicate depletion of a particular amino acid. The composition profile generated for experimentally validated disordered proteins from the DisProt database (black bars) is shown for comparison. In both cases, error bars correspond to the standard deviations over 10,000 bootstrap iterations. The composition profile analysis showed that 8 out of 10 order-promoting residues (W, I, Y, F, L, V, N, and M) were statistically depleted in the protamines (p-value ≤ 0.05), and 6 out of 10 disorder-promoting residue (R, A, Q, S, E, and P) were significantly enriched in these proteins. Note an extremely high relative content of arginine residues in protamines. (B) Charge-hydropathy and cumulative distribution function (CH-CDF) plot for 1880 protamines. The Y-coordinate is calculated as the distance of the corresponding protein from the boundary in the CH plot [28,29]. The X-coordinate is calculated as the average distance of the corresponding protein’s CDF curve from the CDF boundary [29]. The resulting CH-CDF plot allows distinguishing hybrid proteins containing moderate order and disorder from entirely ordered or entirely disordered proteins based on their position within the CH-CDF phase space, with the quadrant where the proteins are located determines their classification [30,31]. Q1, 14 proteins (0.74%) predicted to be ordered by CDF and compact/ordered by CH-plot. Q2, 153 proteins (8.14%) predicted to be ordered/compact by CH-plot and disordered by CDF (native molten globules or hibrid proteins containing ordered domains and high levels of intrinsic disorder). Q3, 1705 proteins (90.69%) predicted to be disordered by CH-plot and CDF (native coils and pre-moletn globules). Q4, 8 proteins (0.44%) predicted to be disordered by CH-plot and ordered by CDF. Herein, we utilized web platform Rapid Intrinsic Disorder Analysis Online (RIDAO) [32] to generate data for the CH-CDF plot. (C) PONDR® VSL2 output for 1880 protamines. PONDR® VSL2 score is the mean disorder score (MDS) for a protein, which is calculated as a sequence-length normalized sum of its per-residue scores. PONDR® VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR); i.e., percent of residues with disorder scores above 0.5. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which only one of these criteria applies. The boundaries of the colored regions represent arbitrary and accepted cutoffs for MDS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). Generally, a PPIDR value of less than 10% is taken to correspond to a highly ordered protein, PPIDR between 10% and 30% is ascribed to moderately disordered protein, and PPIDR greater than 30% corresponds to a highly disordered protein [33,34]. Mean disorder score (MDS) was calculated for each query protein as a protein length-normalized sum of all the per-residue disorder scores. The per-residue disorder score ranges from 0 to 1, where a score of 0 indicates fully ordered residues and a score of 1 indicates fully disordered residues. Residues with scores above the threshold of 0.5 were considered disordered residues. Residues with disorder scores between 0.25 and 0.5 were categorized as highly flexible, while those with scores between 0.1 and 0.25 were classified as moderately flexible. Of the 1880 protamines, none are predicted to be highly ordered, only 51 (2.7%) demonstrate moderate disorder or conformational flexibility, wheras the vast majority of this set (1823 protein or 97.3%) are predicted to be highly disordered.
Figure 1.
Characteristic sequence features of a protamine family. (A) Amino acid composition profile of 1880 protamines generatd by Composition Profiler (http://www.cprofiler.org/) [4]. The fractional difference is calculated as , where Cx is the content of a given amino acid in the query set (1880 protamines or known intrinsically disordered proteins), and Corder is the content of a given amino acid in the background set (Protein Data Bank Select 25). The amino acid residues are ranked from most order-promoting residue to most disorder-promoting residue. Positive values indicate enrichment, and negative values indicate depletion of a particular amino acid. The composition profile generated for experimentally validated disordered proteins from the DisProt database (black bars) is shown for comparison. In both cases, error bars correspond to the standard deviations over 10,000 bootstrap iterations. The composition profile analysis showed that 8 out of 10 order-promoting residues (W, I, Y, F, L, V, N, and M) were statistically depleted in the protamines (p-value ≤ 0.05), and 6 out of 10 disorder-promoting residue (R, A, Q, S, E, and P) were significantly enriched in these proteins. Note an extremely high relative content of arginine residues in protamines. (B) Charge-hydropathy and cumulative distribution function (CH-CDF) plot for 1880 protamines. The Y-coordinate is calculated as the distance of the corresponding protein from the boundary in the CH plot [28,29]. The X-coordinate is calculated as the average distance of the corresponding protein’s CDF curve from the CDF boundary [29]. The resulting CH-CDF plot allows distinguishing hybrid proteins containing moderate order and disorder from entirely ordered or entirely disordered proteins based on their position within the CH-CDF phase space, with the quadrant where the proteins are located determines their classification [30,31]. Q1, 14 proteins (0.74%) predicted to be ordered by CDF and compact/ordered by CH-plot. Q2, 153 proteins (8.14%) predicted to be ordered/compact by CH-plot and disordered by CDF (native molten globules or hibrid proteins containing ordered domains and high levels of intrinsic disorder). Q3, 1705 proteins (90.69%) predicted to be disordered by CH-plot and CDF (native coils and pre-moletn globules). Q4, 8 proteins (0.44%) predicted to be disordered by CH-plot and ordered by CDF. Herein, we utilized web platform Rapid Intrinsic Disorder Analysis Online (RIDAO) [32] to generate data for the CH-CDF plot. (C) PONDR® VSL2 output for 1880 protamines. PONDR® VSL2 score is the mean disorder score (MDS) for a protein, which is calculated as a sequence-length normalized sum of its per-residue scores. PONDR® VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR); i.e., percent of residues with disorder scores above 0.5. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which only one of these criteria applies. The boundaries of the colored regions represent arbitrary and accepted cutoffs for MDS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). Generally, a PPIDR value of less than 10% is taken to correspond to a highly ordered protein, PPIDR between 10% and 30% is ascribed to moderately disordered protein, and PPIDR greater than 30% corresponds to a highly disordered protein [33,34]. Mean disorder score (MDS) was calculated for each query protein as a protein length-normalized sum of all the per-residue disorder scores. The per-residue disorder score ranges from 0 to 1, where a score of 0 indicates fully ordered residues and a score of 1 indicates fully disordered residues. Residues with scores above the threshold of 0.5 were considered disordered residues. Residues with disorder scores between 0.25 and 0.5 were categorized as highly flexible, while those with scores between 0.1 and 0.25 were classified as moderately flexible. Of the 1880 protamines, none are predicted to be highly ordered, only 51 (2.7%) demonstrate moderate disorder or conformational flexibility, wheras the vast majority of this set (1823 protein or 97.3%) are predicted to be highly disordered.
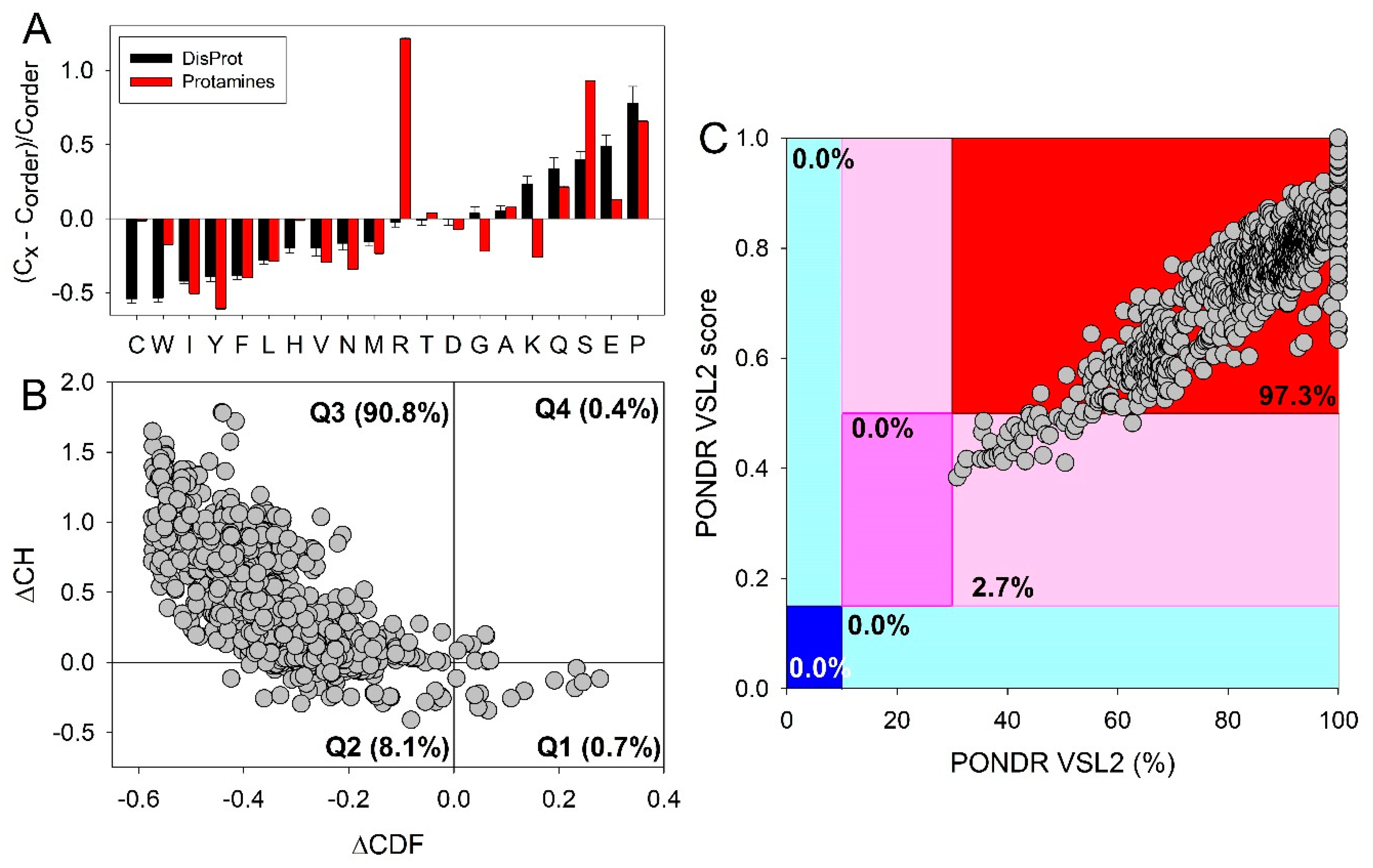
Figure 2.
Peculiarities of the arginine-based two-pronged arginine interactions with RNA. (A) Original arginine-fork model wherein the guanidinium group salt-bridges to two TAR phosphate groups [48]. (B) Computational modeling supports nucleobase and backbone interactions [50]. (C) TAR binding of R47 from TBP6.7 (PDB entry 6cmn [51]). Reprinted (adapted) with permission from S.S. Chavali, C.E. Cavender, D.H. Mathews, J.E. Wedekind, Arginine Forks Are a Widespread Motif to Recognize Phosphate Backbones and Guanine Nucleobases in the RNA Major Groove, J Am Chem Soc 142(47) (2020) 19835-19839. Copyright © 2020 American Chemical Society.
Figure 2.
Peculiarities of the arginine-based two-pronged arginine interactions with RNA. (A) Original arginine-fork model wherein the guanidinium group salt-bridges to two TAR phosphate groups [48]. (B) Computational modeling supports nucleobase and backbone interactions [50]. (C) TAR binding of R47 from TBP6.7 (PDB entry 6cmn [51]). Reprinted (adapted) with permission from S.S. Chavali, C.E. Cavender, D.H. Mathews, J.E. Wedekind, Arginine Forks Are a Widespread Motif to Recognize Phosphate Backbones and Guanine Nucleobases in the RNA Major Groove, J Am Chem Soc 142(47) (2020) 19835-19839. Copyright © 2020 American Chemical Society.
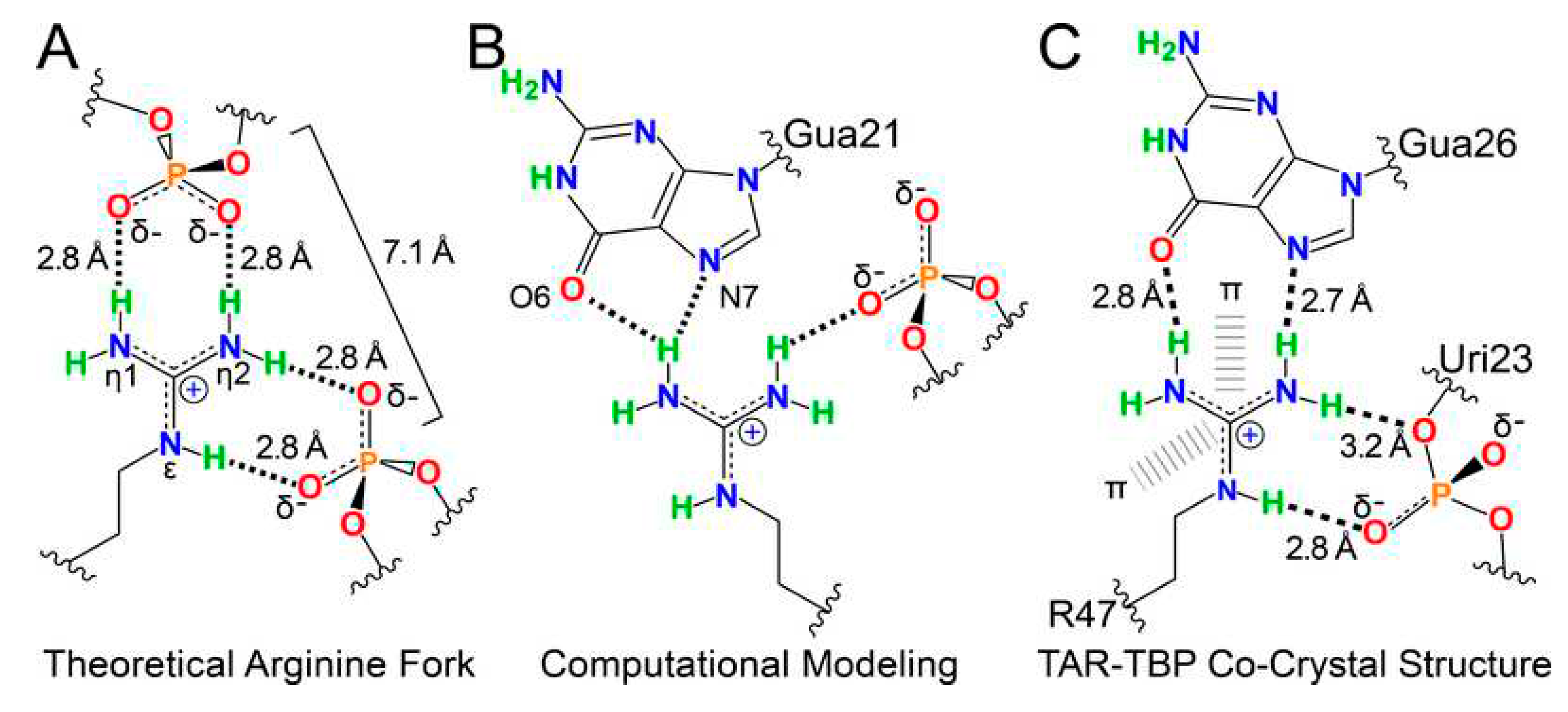
Figure 3.
Schematic representation of an interplay between the poly(Gly-Arg), RNA, RNA-binding proteins (RBPs), G3BP1, YTHDF, and m6A-mRNA leading to the development of the cytoplasmic poly(Gly-Arg) biomolecular condensates. Reprinted from Cell Reports, VOLUME 42, ISSUE 8, Jinyoung Park, Yanwei Wu, Wei Shao, Tania F. Gendron, Sophie J. F. van der Spek, Grigorii Sultanakhmetov, Avik Basu, Paula Castellanos Otero, Caroline J Jones, Karen Jansen-Wes1, Lillian M. Daughrity, Sadhna Phanse, Giulia Del Rosso, Jimei Tong, Monica Castanedes-Casey, Lulu Jiang, Jenna Libera, Björn Oskarsson, Dennis W. Dickson, David W. Sanders, Clifford P. Brangwynne, Andrew Emili, Benjamin Wolozin, Leonard Petrucelli, Yong-Jie Zhang. Poly(GR) interacts with key stress granule factors promoting its assembly into cytoplasmic inclusions, 112822 [91]. Copyright (2023), with permission from Elsevier.
Figure 3.
Schematic representation of an interplay between the poly(Gly-Arg), RNA, RNA-binding proteins (RBPs), G3BP1, YTHDF, and m6A-mRNA leading to the development of the cytoplasmic poly(Gly-Arg) biomolecular condensates. Reprinted from Cell Reports, VOLUME 42, ISSUE 8, Jinyoung Park, Yanwei Wu, Wei Shao, Tania F. Gendron, Sophie J. F. van der Spek, Grigorii Sultanakhmetov, Avik Basu, Paula Castellanos Otero, Caroline J Jones, Karen Jansen-Wes1, Lillian M. Daughrity, Sadhna Phanse, Giulia Del Rosso, Jimei Tong, Monica Castanedes-Casey, Lulu Jiang, Jenna Libera, Björn Oskarsson, Dennis W. Dickson, David W. Sanders, Clifford P. Brangwynne, Andrew Emili, Benjamin Wolozin, Leonard Petrucelli, Yong-Jie Zhang. Poly(GR) interacts with key stress granule factors promoting its assembly into cytoplasmic inclusions, 112822 [91]. Copyright (2023), with permission from Elsevier.
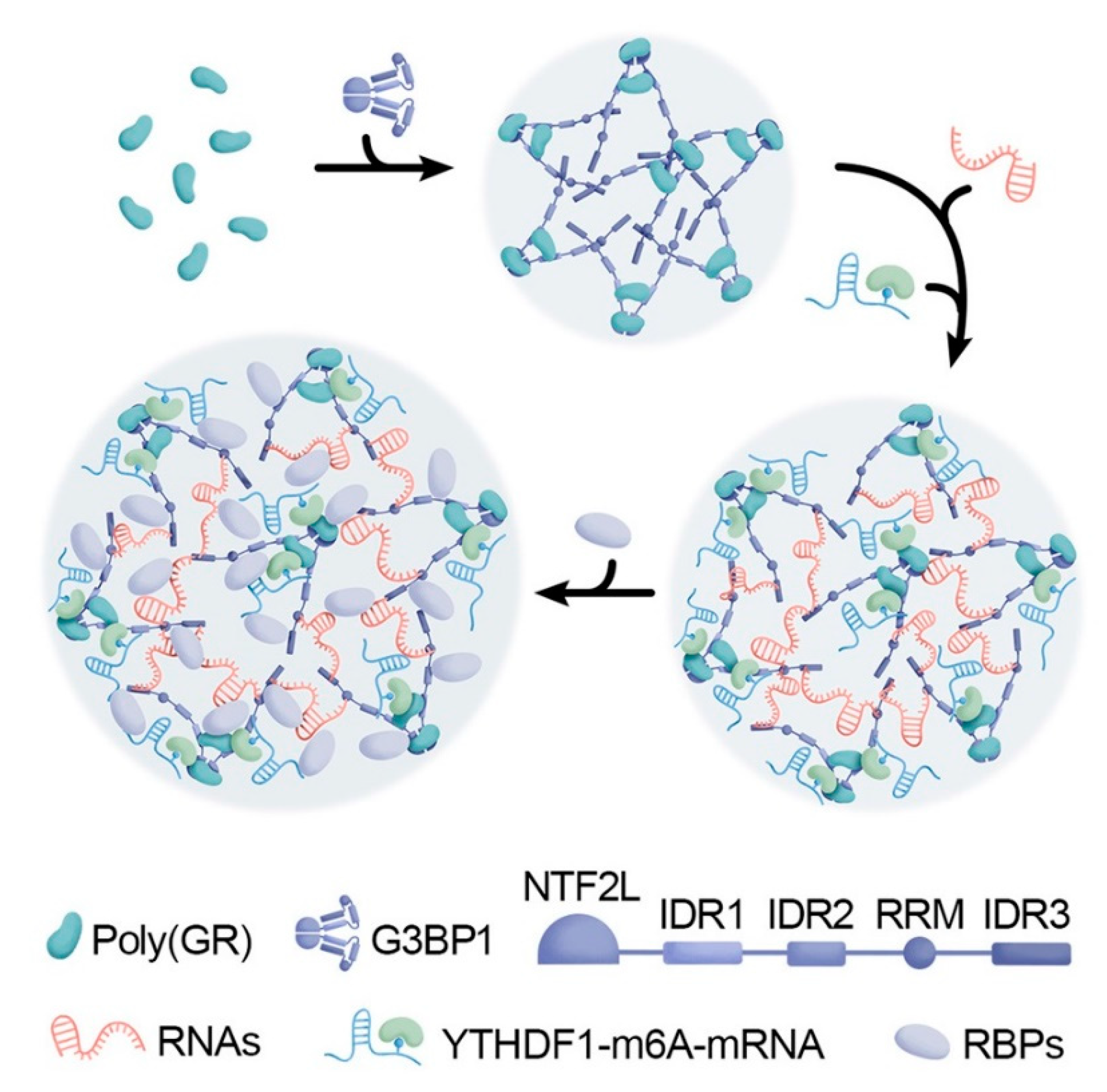
Figure 4.
Scheme showing the different enzymatic activities of type I, II and III protein arginine N-methyltransferases (PRMTs). Type I, II and III PRMT enzymes catalyze the transfer of a methyl group from S-adenosyl-l-methionine (SAM) to the side chain nitrogen on arginine residues on the target protein to produce monomethylarginine (MMA). Type I enzymes then catalyze the transfer of a further methyl group, asymmetrically onto the same nitrogen atom that results in asymmetric dimethylarginine (ADMA). Type II enzymes catalyze the transfer of a second methyl group symmetrically, onto the opposite nitrogen on the side chain of arginine (symmetric dimethylarginine, SDMA). Reproduced from [112] under CC Attribution license 4.0.
Figure 4.
Scheme showing the different enzymatic activities of type I, II and III protein arginine N-methyltransferases (PRMTs). Type I, II and III PRMT enzymes catalyze the transfer of a methyl group from S-adenosyl-l-methionine (SAM) to the side chain nitrogen on arginine residues on the target protein to produce monomethylarginine (MMA). Type I enzymes then catalyze the transfer of a further methyl group, asymmetrically onto the same nitrogen atom that results in asymmetric dimethylarginine (ADMA). Type II enzymes catalyze the transfer of a second methyl group symmetrically, onto the opposite nitrogen on the side chain of arginine (symmetric dimethylarginine, SDMA). Reproduced from [112] under CC Attribution license 4.0.
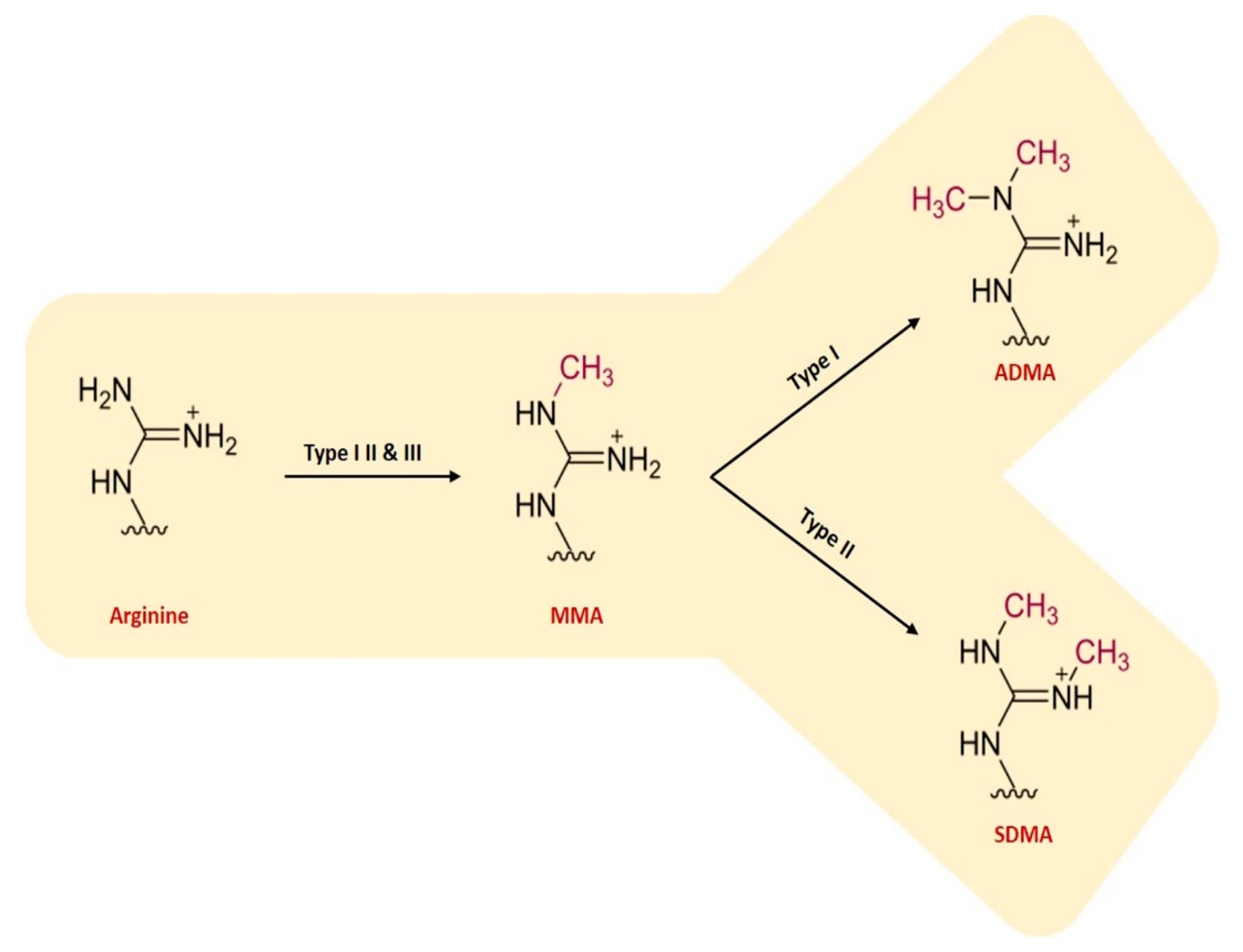
Figure 5.
Schematic representation of the mechanisms by which methylated arginine (ArgMe) might lead to oncogenesis. PRMTs and their alternatively spliced isoforms have diverse roles in transcriptional regulation, splicing and DNA damage repair, through MMA, SDMA and ADMA on specific targets. PRMTs methylate a combination of histone and non-histone targets. For example, PRMT2 methylates H3R8, inducing gene expression. PRMT1 methylates double strand break repair protein meiotic recombination 11 homolog (Mre11), anchoring it to the double strand break. PRMT5 methylates Sm ribonucleosomal proteins, promoting uridine-rich small nuclear ribonuclear protein (UsnRNPs) and survival motor neuron (SMN) spliceosomal complex assembly. Methylation of these targets results in the increased expression of oncogenic genes and the attenuation of tumour suppressive genes, either through promoter activation or repression by epigenetic regulation, alterations in splicing patterns or through an increase in genomic instability. PRMT activity has also been shown to promote tumor stem cell characteristics. Reproduced along with the legend from [112] under CC Attribution license 4.0.
Figure 5.
Schematic representation of the mechanisms by which methylated arginine (ArgMe) might lead to oncogenesis. PRMTs and their alternatively spliced isoforms have diverse roles in transcriptional regulation, splicing and DNA damage repair, through MMA, SDMA and ADMA on specific targets. PRMTs methylate a combination of histone and non-histone targets. For example, PRMT2 methylates H3R8, inducing gene expression. PRMT1 methylates double strand break repair protein meiotic recombination 11 homolog (Mre11), anchoring it to the double strand break. PRMT5 methylates Sm ribonucleosomal proteins, promoting uridine-rich small nuclear ribonuclear protein (UsnRNPs) and survival motor neuron (SMN) spliceosomal complex assembly. Methylation of these targets results in the increased expression of oncogenic genes and the attenuation of tumour suppressive genes, either through promoter activation or repression by epigenetic regulation, alterations in splicing patterns or through an increase in genomic instability. PRMT activity has also been shown to promote tumor stem cell characteristics. Reproduced along with the legend from [112] under CC Attribution license 4.0.
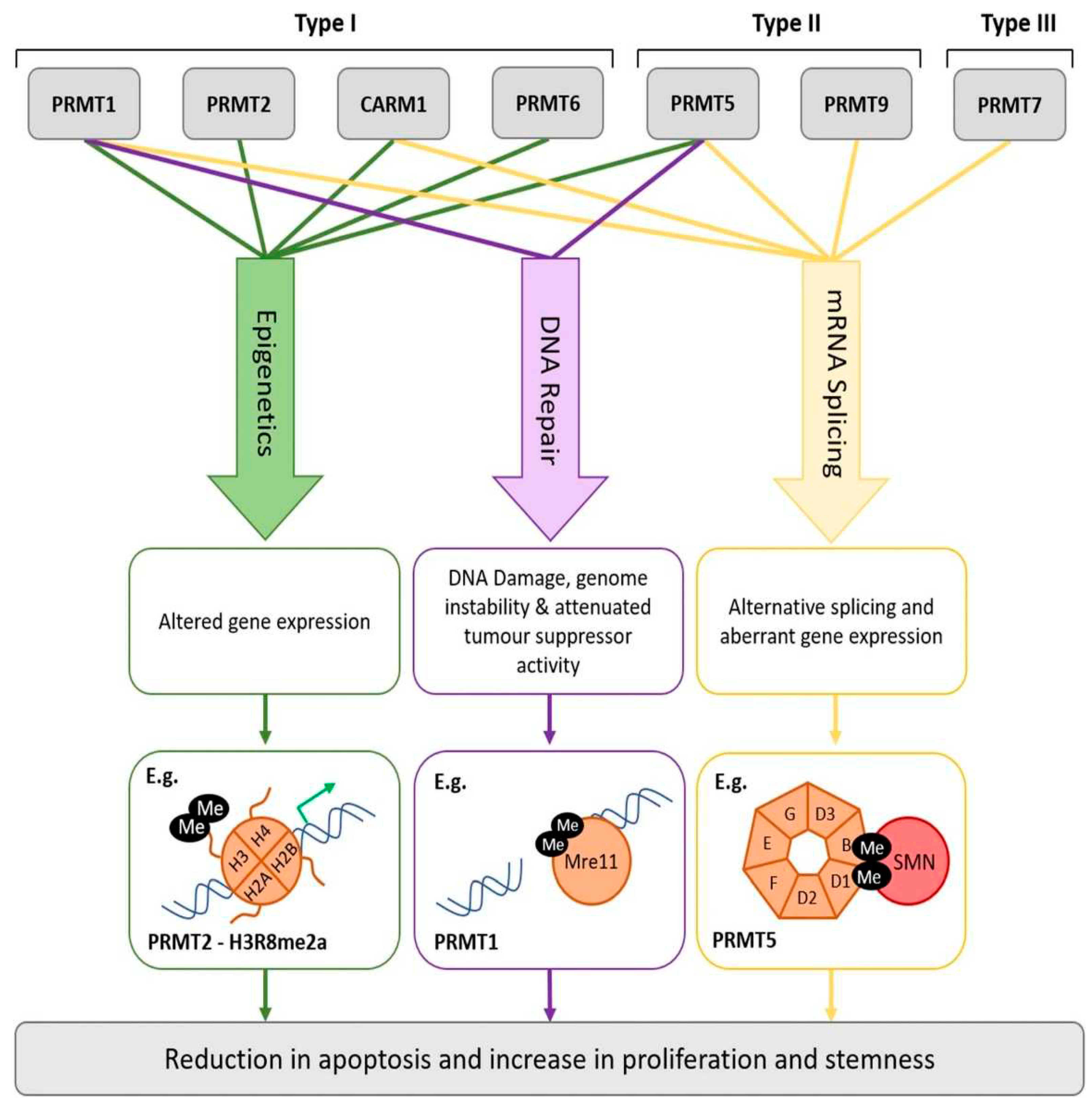
Figure 6.
Arginine Tweezers. Based on the analysis of 972 protein 3D structures (448 protein chains for Class I and 524 chains for Class II), Backbone Brackets and Arginine Tweezers were identified as structural motifs distinctive for binding to adenosine phosphate. Reproduced from Kaiser, F.; Bittrich, S.; Salentin, S.; Leberecht, C.; Haupt, V.J.; Krautwurst, S.; Schroeder, M.; Labudde, D. Backbone Brackets and Arginine Tweezers delineate Class I and Class II aminoacyl tRNA synthetases. PLoS Comput Biol 2018, 14, e1006101 [176] under CC Attribution license 4.0, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Figure 6.
Arginine Tweezers. Based on the analysis of 972 protein 3D structures (448 protein chains for Class I and 524 chains for Class II), Backbone Brackets and Arginine Tweezers were identified as structural motifs distinctive for binding to adenosine phosphate. Reproduced from Kaiser, F.; Bittrich, S.; Salentin, S.; Leberecht, C.; Haupt, V.J.; Krautwurst, S.; Schroeder, M.; Labudde, D. Backbone Brackets and Arginine Tweezers delineate Class I and Class II aminoacyl tRNA synthetases. PLoS Comput Biol 2018, 14, e1006101 [176] under CC Attribution license 4.0, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
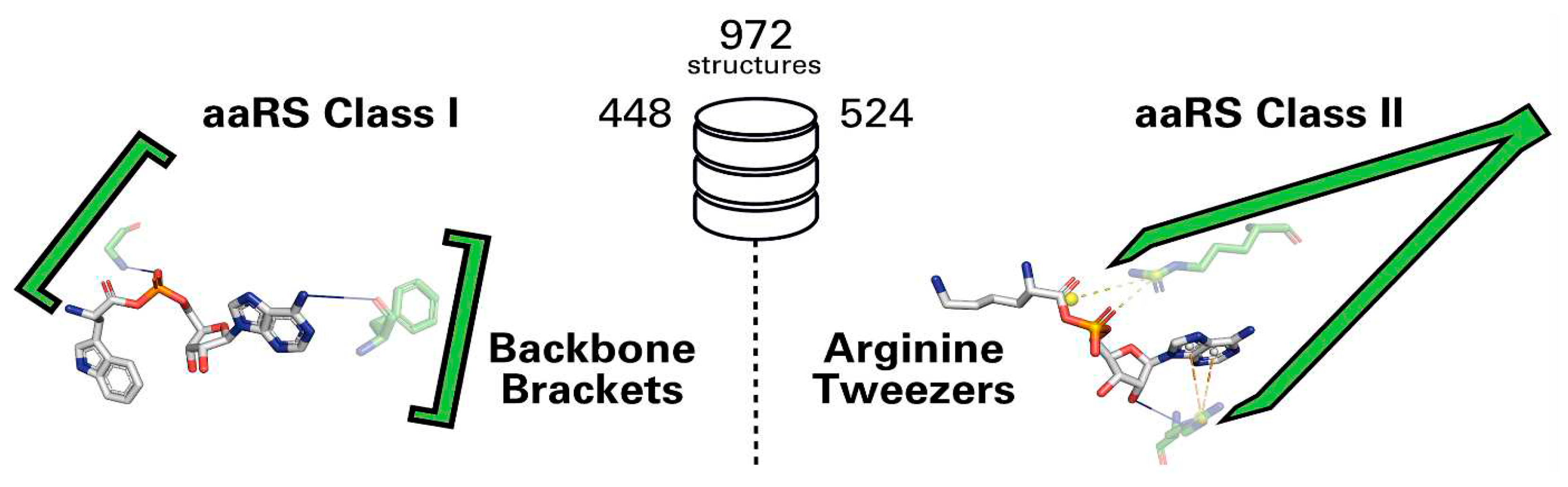
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



