
Submitted:
27 October 2023
Posted:
30 October 2023
You are already at the latest version
Abstract
With nearly 1,700 species, Microsporidia represent a group of obligate intracellular eukaryotes with veterinary, economic and medical impacts. To help in understanding the biological functions of these microorganisms, complete genome sequencing is used routinely. Nevertheless, the proper prediction of their gene catalogue is challenging due to taxon-specific evolutionary features. As innovative genome annotation strategies are needed to obtain a representative snapshot of the overall lifestyle of these parasites, the MicroAnnot tool, a dedicated workflow for microsporidian sequence annotation using data from curated databases of accurately annotated microsporidian genes, has been developed. Furthermore, specific modules have been implemented to perform small genes (< 300bp) and transposable element identification. Finally, functional annotation was performed using the signature-based InterProScan software. MicroAnnot’s accuracy has been verified by the re-annotation of four microsporidian genomes for which structural annotation had previously been validated. With its comparative approach and transcriptional signal identification method, MicroAnnot provides accurate prediction of translation initiation sites, efficient identification of transposable elements, as well as high specificity and sensitivity for microsporidian genes including those under 300 bp. Thanks to its web interface (https://microannot.org), MicroAnnot is available to the community to ensure high quality annotation of microsporidian genomes.
Keywords:
Microsporidia
; Structural annotation
; dedicated workflow
; high quality annotation.
1. Introduction
The microsporidian phylum, an evolved branch of the rozellids [1] includes over 1,700 species divided into more than 220 genera infecting an extremely diverse range of hosts protists to all major animal phyla [2]. Microsporidia are vastly represented in aquatic environments and food webs. Numerous species can also be found infecting animals of veterinary and economic importance such as bees or silkworms [3]. In addition, some microsporidian species may be involved in human diseases with 17 species belonging to 10 genus [4] that have been described as leading to severe syndromes predominantly in immunocompromised patients. This includes acquired immunodeficiency syndrome (AIDS) patients but also patients that have undergone organ transplants and been treated with immunosuppressive drugs [4,5,6,7].
With the advent of next-generation sequencing (NGS) technologies, the systematic sequencing of microsporidian genomes has been undertaken and over the last few decades, genomic data has rapidly been accumulating with more than 50 genomes now available [8]. The study of these genomes has allowed researchers to highlight the consequences of the distinct evolutionary patterns in this parasitic group. Indeed, due to their adaptation to obligate intracellular parasitism, « the genomes of Microsporidia are under strong selective pressures which conduct them to present specific characteristics. Thus, microsporidian genome sizes are highly reduced, with some species having reduced their genomic content to potentially the lowest limit required for life [9]. The human infecting species Encephalitozoon intestinalis with 2.3 Mbp harbors the smallest microsporidian genome sequenced [10]. The genomic reduction in microsporidia is not just limited to a massive loss of genes as it also affects the gene length. For example, Encephalitozoon cuniculi Coding DNA Sequences (CDSs) are on average 15% shorter than their yeast orthologs [11]. This CDS size reduction also leads to the presence of around 8.5% of the CDSs with a size under 300 nucleotides [12,13]. Another consequence of microsporidian gene compaction is the removal of intronic sequences. Introns with reduced size may remain in a small number of genes however, but this is not common to all species thus making their prediction more difficult [13,14]. Microsporidian genes also present a strong compaction of the 5' and 3'UTRs (UnTranslated Regions). In some cases, the 5’UTR can even be absent, and the transcribed mRNAs begin with the translation initiation codon [12,15,16,17,18,19]. This high reduction of 5’UTR length seems to be an advantage for the identification of transcriptional regulation signals that are therefore localized near the translation initiation codon. Numerous studies have shown that these signals are highly conserved amongst microsporidian species and consensus sequences have been defined [12,13,18]. Thus, CCC-like or GGG-like signals are located upstream of the Translation Initiation Site (TIS). In genomes with a low G+C content, these signals can often be replaced by a strong A+T-bias (more than 90%) near to the TIS. A final feature affecting the size of microsporidian genomes is the shortening of intergenic regions which are essential for transcription as they contain promoters and enhancers (Jespersen et al 2022).
Microsporidian genomes are characterized by a high rate of sequence evolution which has induced difficulties in positioning these microorganisms in the phylogenetic eukaryotic tree [1]. The identification of orthologous genes between microsporidian species has also proved to be challenging even for genes crucial to their infectious process [20] or DNA repair [21]. While genomes tend to present extreme reduction, one exceptionally large microsporidian genome of 51 Mbp has been reported in the mosquito parasite Edhazardia aedis. This is mainly due to the expansion of Transposable Elements (TE) families in the genome of this microsporidian species. The high rate of sequence evolution in the microsporidian phylum also concerns TEs, making them hard to identify using comparative approaches alone with sequences from other organisms [22,23].
Rapid and cost-effective next-generation sequencing (NGS) technologies have produced, and are still producing, numerous new microsporidian genome sequences. After genome assembly, efficient genome annotation represents a crucial step to point out all biological processes that govern the life of these microorganisms. Computational genome annotation has become one of the principal research areas in computational biology [24]. However, the microsporidian genome features previously described turn out to be the pitfall of classical methods for producing accurate prediction of complete gene repertoires. Until now, microsporidian genomes have been annotated using ab initio protein predictions that were based primarily on the detection of Open Reading Frames (ORF) using various generalist software such as GeneMarkES, Augustus [25,26] or Glimmer, that could be combined with the detection of CCC- and GGG-like motifs found in close proximity of microsporidian TISs [13,27,28]. Such signals significantly improve prediction of translational initiation codons which are otherwise defined as the first AUG codon of the studied ORF. In addition, extrinsic data, such as the one available from orthologous gene sequences has also been intensely used to carry out structural annotation [29]. Due to the high rate of sequence evolution in the microsporidian phylum, such approaches require an optimization of the comparison tool parameters (e.g. with BLAST). These parameters also need to ensure unambiguous prediction of TEs as much as possible. Finally, small protein-coding genes are often overlooked during structural annotation due to their shortness, lack of sequence conservation, and/or lack of known functions [30].
To address the challenging question of microsporidian genome annotation, we developed a dedicated annotation pipeline called MicroAnnot. MicroAnnot ensures gene prediction as well as structural and functional annotation. Firstly, using curated databases of validated proteomes from four microsporidian species, an extrinsic approach has been implemented using the BLAST tools [31] with optimized parameters to identify divergent or small orthologous sequences. The results were also exploited to predict potential translation initiation codons that were further validated using upstream transcriptional signals. Secondly, using these newly predicted genes as training set for the Glimmer tool [32], an ab initio sequence annotation was carried out and the potential translation initiation codons for the newly identified sequences were once again validated using transcriptional signals. All predicted CDSs were then used as queries against a microsporidian specific TE database. The identification of rRNAs was also done by a comparative approach using a database containing small subunit (SSU) rRNA sequences representative of all the microsporidian phylogenetic diversity and tRNA detection done using tRNAScan-SE [33]. MicroAnnot has been tested on four microsporidian genomes and it yielded an annotation of higher quality on all the evaluated criteria (specificity, sensitivity, translational initiation codon prediction, small gene characterization and TE identification). Furthermore, functional annotation using the InterProScan tool [34] can also be included in the result files as either GENBANK, EMBL or GFF annotation files.
2. Results
2.1. MicroAnnot Usage
By selecting “New analysis” in the right web banner (Figure 1A), the MicroAnnot tool will display different parameters that can be modulated (Figure 1B). The software takes as an input a flat file containing microsporidian genome FASTA sequence(s) to be annotated (maximum size data 100 Mbytes). The web interface provides the user the possibility to adjust the threshold values of the different approaches and softwares used. The user may also select the training dataset for the Glimmer software when less than 50 CDS are identified by the comparative search approach. Functional annotation using the InterProScan software is disabled by default but can be enabled by clicking on the “activate” box.
When the input file is uploaded, the analysis parameters defined and the databases selected, the analysis can be launched by clicking on the "Submit" button. Once the job submission is done, the user can follow its execution (Figure 2A and 2B). Once the job is finished, several compressed archives can be directly downloaded. The annotation data can be picked up in GENBANK, EMBL and GFF formats (Figure 2C). In addition, an archive is also generated containing uncertain annotations that must be manually validated (see algorithm description below).
2.2. MicroAnnot Algorithm
The details of the MicroAnnot algorithm are shown in Figure 3. Briefly, the complete set of Open Reading Frames (ORFs) of a given minimum size (default value 240 nt) is extracted from input FASTA sequence(s) and after translation is used as a query to perform an initial BLASTP analysis against well annotated microsporidian proteomes incorporated in MicroAnnot, namely Encephalitozoon cuniculi, Nosema ceranae, Enterocytozoon bieneusi and Anncaliia algerae [13]. As some microsporidian genes exhibit drastic size reduction, a second BLASTP analysis is performed after extracting all CDS sequences coding for short proteins (less than 80 amino acids, Figure 3 left part). For this second analysis, BLASTP parameters are optimized to identify sequence similarity between short peptide sequences and a specific database constructed from biologically and/or manually validated small gene sequences [12,13]. The significant results of the two BLASTP analyses are processed to optimize identification of TISs either by considering the alignment of the N-terminal part of the proteins or secondly by the presence of transcriptional signals in proximity of the start initiation codon when the N-terminal alignment is not available after local BLASTP alignment.
Figure 3.
An overview of the workflow supported by the MicroAnnot pipeline. The different modules using different bioinformatics programs and databanks are displayed. The detailed description of each panel and module is provided in the text. Functional analysis done by InterProScan in orange (optional), need to be selected in the first page of the interface (Figure 1B).
Figure 3.
An overview of the workflow supported by the MicroAnnot pipeline. The different modules using different bioinformatics programs and databanks are displayed. The detailed description of each panel and module is provided in the text. Functional analysis done by InterProScan in orange (optional), need to be selected in the first page of the interface (Figure 1B).
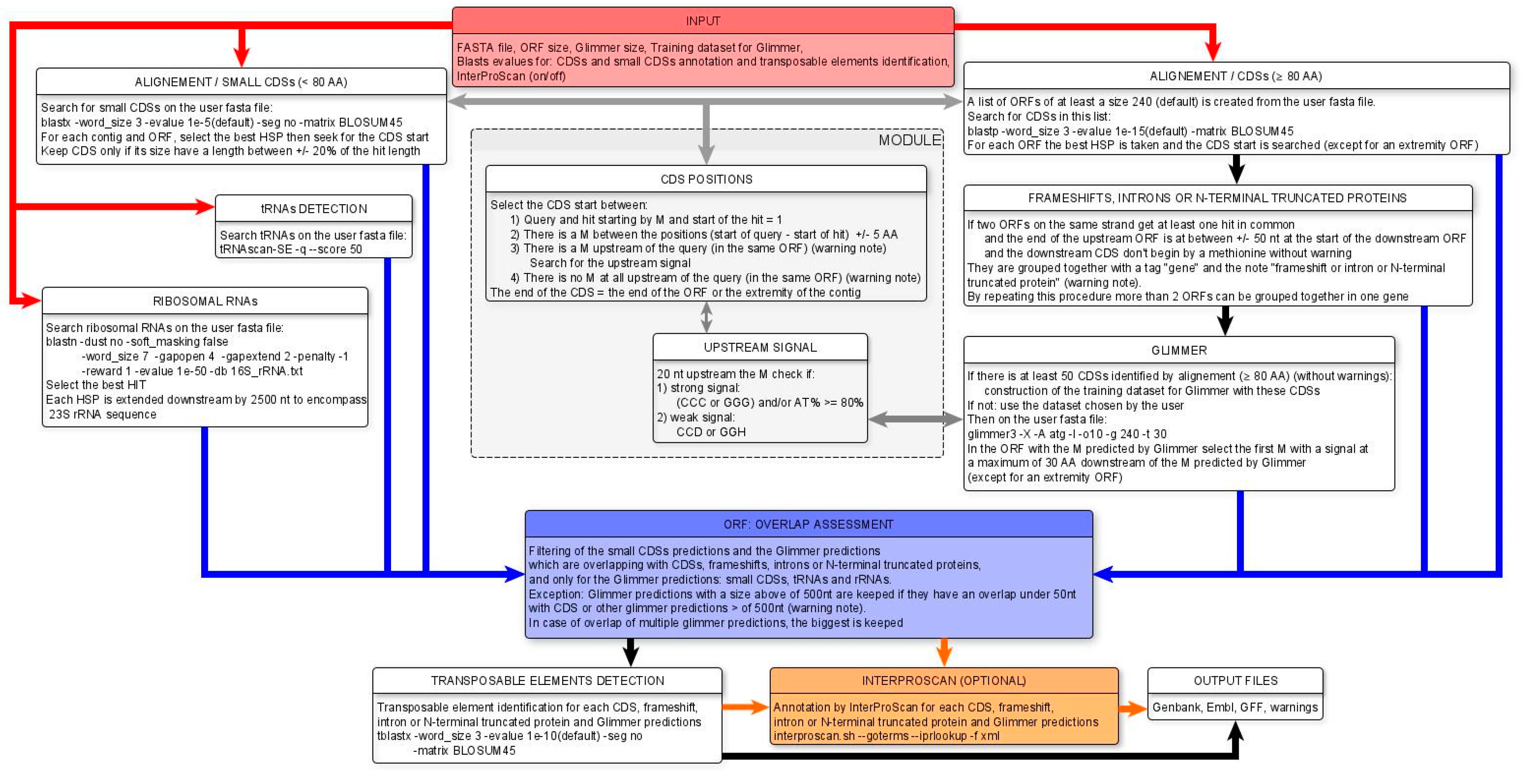
Results of the first BLASTP analysis are also exploited to identify potential i) frameshifts, ii) introns and iii) 5’ truncated CDSs. If more than 50 CDSs are well predicted by our first homology approach, their sequences are used to train the Glimmer model. Otherwise, the user has the possibility to use one of the Glimmer models defined from the four well predicted proteomes. TISs predicted by Glimmer annotation are then validated by identification of upstream transcriptional signals. If these signals are absent, the CDS sequence is scanned to highlight these signals upstream each potential translation initiation codon. As soon as an ATG codon is closely preceded by these signals, it is considered as the correct translation initiation codon and the predicted CDS is adjusted.
In parallel, MicroAnnot also implements non-coding features annotation. These are done using dedicated methods and tools. Transfer RNA (tRNA) annotation is conducted using the tRNAscanSE tool [33] directly embedded in the MicroAnnot pipeline. For the identification of rRNA-encoding genes (16S-23S rRNA units), a BLASTN analysis is conducted using the user’s input sequences as a query against a rRNA personal database including sequences representing the complete phylogenetical diversity of microsporidian rRNAs [35].
To avoid redundancy when compiling all generated annotation data, all Glimmer predicted CDSs overlapping with other predictions are eliminated. We consider homology driven annotation to be more reliable than Glimmer annotations and this step helps in keeping only the best annotations in MicroAnnot’s final output. We set an exception for Glimmer CDSs above 500 nucleotides which are retained if they have fewer than 50 nucleotides overlapping with other predictions.
After overlap curation, potential transposable element (TE) sequences are also identified by comparing all predicted CDSs to a microsporidian specific TE database using a TBLASTX approach. Finally, a functional annotation of all predicted CDSs is computed using InterProScan and the translated CDS sequences. This step is optional and deactivated by default (Figure 1B).
The final annotation results are available to the user In GENBANK, EMBL and GFF annotation formats. An additional text file called “warning” is also generated and contains all the annotations for which the MicroAnnot tool invites caution. This includes potential frameshifts, introns, 5’ truncated genes, TIS too far from the ORF start, and overlapping genes. In all these cases, the CDS feature created in the output files is replaced by the “gene” feature and a warning note is added.
2.3. MicroAnnot Validation
Comparative analysis of the annotations proposed by MicroAnnot and several previous annotations revealed higher sensitivity and specificity for MicroAnnot in regard to manually curated reference annotations [13] (Table 1 and Table 2, and supplementary data 1 to 4). Indeed, more than 85% of the genes that could previously only be identified by manual annotation are predicted, with a percentage rising to about 94% for the E. bieneusi species. It should also be noted that the MicroAnnot tool is efficient for the annotation of new genes, including 296 for N. ceranae. MicroAnnot reported some false positives, but their number remained relatively low. Furthermore, for the species harbouring the highest numbers of mis-predicted genes, a large number are proposed with a warning (27/35) to invite the user to control manually these predictions for N. ceranae species, and for the E. bieneusi species, 18 out of 25 falsely predicted genes correspond to low-complexity sequences mis-identified by the Glimmer tool (supplementary S4). For genes that had been mis-predicted initially within these genomes, The MicroAnnot tool also produced a significant improvement, with less than 25% false positives overall and even the detection of none of the genes incorrectly predicted in previous annotations of the E. cuniculi and E. intestinalis genomes. MicroAnnot's algorithm can also identify introns, frameshifts, sequencing errors and pseudogenes in sequences, and the comparative analysis reveals an efficiency of over 63% (E. intestinalis) going up to 100% (E. cuniculi) for intron identification, and between 39,5% (E. bieneusi) and 90% (E. intestinalis) for the detection of other non-canonical sequences and sequence features (frameshifts, sequencing errors, pseudogenes). For the determination of TISs, the MicroAnnot tool also displays conclusive results. Compared with the four other genome annotations and mis-predictions, MicroAnnot predicts the correct translation initiation codon in over 70% of the cases. Furthermore, additional TISs were corrected for the four species respectively. Finally, the TE prediction module of MicroAnnot proved particularly effective, enabling us to show that 693 CDSs actually correspond to such elements in N. ceranae.
3. Discussion
Although the correct annotation of genomes over the last decade has led to the development of increasingly innovative and specific approaches that consider the characteristics of each studied genome, this initial step in the in silico exploration of genomes is still often a source of error. Indeed, gene prediction is a complex process, especially in eukaryotes, and the prediction of all the genes in an organism is never 100% correct. A benchmark study of ab initio gene prediction methods in diverse eukaryotes using the most widely used gene prediction programs has shown that all these programs harbour numerous strengths but also various weaknesses [36]. These authors also concluded that ab initio gene structure prediction is a very challenging task, which should be further investigated [36]. For prokaryotic species, whose genome organization is close to that of Microsporidia, many common CDS predictors failed to identify the complete gene catalogue because some genes features fell outside the defined rules such as non-standard codon usage, overlapping genes and small genes [37].
Due to their characteristics, the structural annotation of microsporidian genomes to define their genetic potential can quickly prove to be a real challenge. To take these constraints into consideration, we have developed a tool dedicated to the annotation of these particular genomes. The annotation carried out using the MicroAnnot tool on the four benchmark genomes gives particularly conclusive results in terms of specificity and sensitivity, while conventional softwares used for the annotation of different microsporidian genomes (E. cuniculi ; Glimmer prediction [11], E. intestinalis, BLAST procedures [10], N. ceranae ; Glimmer [38] and E. bieneusi ; FunGene and Glimmer3 [39]) do not offer predictions of the same quality given several badly predicted or even non predicted genes which can add up to as much as 10% of the total genes of the studied species [13].
Achieving complete genome annotation based on ab initio predictions can be particularly effective when it comes to model organisms but the results in terms of sensitivity and specificity can drop for non-model species [36,40]. Annotations obtained using comparative methods such as the alignment of protein sequences against predicted CDS, give precise and reliable results [41]. However, due to high rates of sequence evolution in microsporidian sequences [42], comparative approaches are difficult to implement with these species: the correct alignment of orthologous sequences from different microsporidian species and the parameters of the comparative tools all need to be optimised. To provide high quality alignments with the proteome sequences but also with the small gene sequences, the parameters of the BLAST software were modified, notably with the use of the BLOSUM45 matrix. This « deeper » matrix provides very sensitive similarity searches but also produces alignment overextension into less homologous regions such as N-terminal regions [43]. The alignment of the N-terminal regions has been used by MicroAnnot to unambiguously determine gene TISs. Indeed, correct recognition of this initiation codon is crucial in gene prediction to highlight gene structure and its product [44]. Nevertheless, due to high rate of sequence evolution, local alignment with BLAST software may stop before reaching the methionine defining the N-terminal end of the sequence. In this case, MicroAnnot considers the length of the homologous database sequence and the position of the BLAST local alignment to propose a potential translation initiation site in the query sequence. Validation of predicted TISs can be achieved by evaluation of the ATG context [45]. The search for the Kozak sequence cannot be applied to microsporidian genes because the mRNAs are characterized by highly reduced or even absent 5’ untranslated regions (UTR) and only a bias in +4 position for an adenine or a guanine residue has been described [18]. However, 5’ UTR size reduction represents an advantage because the transcription regulatory signals are in close proximity to the translation initiation codon. Characterized CCC-like or GGG-like signals, or a strong adenine/thymine-rich sequence (approximately 90%) upstream of TISs are conserved within all microsporidian genomes [13,18,46,47] and their identification allows to unambiguously support TIS prediction. The search for these signals using the MicroAnnot tool proved particularly relevant by ensuring the correct prediction of more than 70% of the previously mis-predicted TISs. In the case of E. cuniculi, more than 23% of TISs [12,13,18] had been badly predicted during the first annotation of this genome not using detection of such signals [11]. The annotation obtained using MicroAnnot, without considering the reference Encephalitozoon species, presents only around 20% of mis-predicted TISs (Table 1). This value drops to less than 2.5% for the annotation of the E. intestinalis genome, carried out using the E. cuniculi’s proteome as a reference. It should also be noted that of the 32 badly predicted TISs in this species, 12 are contiguous to the putative correct ones (supplementary data 2). This comparative approach coupled with the identification of the correct translation initiation codon also proves relevant for identifying potential short introns found mainly within genes coding for ribosomal proteins [14,18,48].
The CCC- and GGG-like signals have been successfully used for the annotation of the gene TISs in different microsporidian species such as Ordospora colligata [47] or N. ceranae [28]. These signals also proved relevant to ensure the characterization of small microsporidian genes (CDS size < 300 nt) which are relatively frequent due to the general reduction of CDS sizes in microsporidia when compared to their orthologs in other fungal species [11]. Despite their number, these small genes are often misreported by generalist gene predictors. Advances in high-throughput technologies have highlighted an emerging world of proteins composed by small open reading frame-encoded micro-peptides [49]. Based on comparative approaches, the MicroAnnot tool proved particularly effective in ensuring the annotation of genes that had been ignored during the initial annotation of the four genomes studied in this work. These missing genes mostly correspond to small CDSs. For N. ceranae, 296 new genes have been identified and 184 of them harbour a CDS smaller than 300 nt in length. Most of these new genes were previously highlighted by Pelin et al. who used an in-house script that combines Glimmer's ab initio gene prediction algorithm, and CCC- and GGG-like motifs found in close proximity to microsporidian transcription initiation sites [28] reinforcing the relevance of our approach. Differences in genome sizes between microsporidian species are essentially due to the presence of TEs [22,50]. Unfortunately, gene predictors can predict CDS in these TEs which can thus lead to an incorrect estimation of the number of genes in microsporidia [13]. The first step in structural annotation involving exhaustive identification of repetitive elements is still challenging [24]. Despite their prevalence and importance, TE sequences remain poorly annotated and studied in almost all model systems [51]. Functional annotation of many microsporidian CDSs revealed that they contained specific TE ORF domains and motifs (see for example product description of Dictyocoela muelleri species [52] in MicrosporidiaDB [53]. In addition, the identification of large multigene families among which some members have a low percentage of similarity with TE sequences show difficulties to identify certain TE families within microsporidian genomes [13]. Thus, MicroAnnot includes a specific module based on a comparative approach with well predicted TEs to scan all predicted CDSs. This method allows a fine TE detection and finally 693 predicted CDS were in fact TEs for the N. ceranae species.
Although structural annotation methods based on sequence homology are very effective, they are closely dependent on the presence of orthologous genes in the queried databases used for annotation. This is not a problem if we consider the pangenome but it is more problematic for the core genome [54] especially for organisms such as microsporidia that can be found in multiple ecological niches and therefore present variable gene contents [23,55]. So, to ensure the annotation of new genes, the implementation of an ab initio method is needed and the Glimmer tool which was used for the annotation of several microsporidian genomes was selected [27,28,56,57]. For the best possible prediction, the dataset used for the construction of the Glimmer model must be perfectly reliable. The sequences included in the composition of this dataset are produced during the comparative annotation approach carried out by the MicroAnnot tool. This approach uses as references, genes whose annotation has been validated manually and, in some cases, experimentally [12,13]. Hence, these sequences ensure the extraction of unambiguous CDSs with translational start sites well defined and validated by the presence of transcriptional regulatory signals in the upstream region. Once the CDSs are predicted by Glimmer, their position in the genome is evaluated and it makes it possible to eliminate wrongly predicted genes by overlap search. This overlap is notably responsible for poor prediction of 20% and 28% of genes in E. bieneusi and N. ceranae respectively [13]. Incorrectly predicted genes may also result from sequencing errors leading to frameshifts. The comparative approaches utilized by MicroAnnot limit these erroneous gene predictions because the potential frameshifts are also evaluated. Frameshift characterization can only be done during the comparative approach step, and their identification is directly correlated to the reference proteomes available to the MicroAnnot tool and highlights the importance of integrating additional reference genomes more representative of the phylogenetic diversity of microsporidia for better identification (see below). This type of errors is however less frequent with the improvement of sequencing approaches, base calling algorithms [58], and third generation techniques [59,60]. The comparative analysis implemented in MicroAnnot also allows the identification of pseudogenes that may be present in microsporidian genomes [61]. The MicroAnnot tool also mis-predicted some genes. All these genes however, were predicted during the ab initio annotation step with the Glimmer tool. This is likely linked to Glimmer specificity concerns previously described [36]. However, for N. ceranae, 71% of incorrectly predicted genes (27 out of 38) harbour a “warning” giving the possibility to the user to invalidate these predictions. As for the initial annotation of the E. bieneusi genome, sequences of low complexity are annotated as CDSs during the ab initio approach, but this number drops from 89 to 18 with the MicroAnnot tool while these annotations are carried out with the Glimmer tool in both cases.
Despite the progress made in developing increasingly efficient tools for genome annotation, the process requires manual curation to produce the most reliable results [24,62]. Furthermore, genome annotation is unfortunately not 100% accurate and needs to be updated regularly to take advantage of the new knowledge from comparative genomics, transcriptomics, proteomics, and metabolomics, continuously generated on the organisms under study, and more generally on all organisms, for annotation using sequence similarity search approaches for example. However, computer analysis methods have led to high levels of erroneous annotations which, when used, spread throughout international databases [63]. The MicroAnnot tool, while significantly increasing the sensitivity and specificity of predictions and reducing the number of incorrectly predicted TIS, is not yet 100% effective. For this reason, we plan to update it constantly, particularly in regard to the content of the databases used for comparative approaches. Today, the tool includes four reference proteomes, but this number will need to be increased by implementing others available and validated proteomes, thus enabling the representation of the entire microsporidian diversity. Meanwhile the sequencing of new microsporidian genomes, the annotation of genomes available in international databases and microsporidiaDB [53] for which transcriptomic data has also been produced to validate their annotation could rapidly be integrated in MicroAnnot for the annotation by the comparative approach. The annotation of these genomes would also provide new sequences for the databases used by the software. Following the integration of a new reference genome, a check of the existing data should be systematically carried out, as this may enable errors to be corrected. The objective is not to propagate annotation errors, but to correct them over time by adding new sequences. The annotation of each microsporidian genome is therefore no longer fixed but is a dynamic process enabling regular re-annotation [64].
Increasing the number of reference genomes would be crucial also for developing and integrating a specific module for the characterization of non-coding RNAs. High-throughput sequencing technologies such as RNA-seq have largely shed light on the world of ncRNA regulators [65], some of which have been systematically identified within microsporidian genomes using RNA-seq data [66,67,68,69]. Many methods predict ncRNA using sequence-derived features alone and they are difficult to apply to all species, especially microsporidia, which have a high rate of sequence evolution. To ensure the annotation of such ncRNAs, a synteny-driven “all-versus-all" BLASTN approach could be implemented following the addition of new genomes. This approach has previously been used to annotate the U1 small nuclear RNA [70], almost 15 years after the initial sequencing of the E.cuniculi genome [11].
4. Materials and Methods
4.1. Software Implementation
The software implementation utilized several tools and libraries for the analysis and processing of data. The following software components were employed: Perl (5.20.2), Bioperl (1.006924) with some modifications (a sort function was added on row 1264 in the genbank.pm file and on row 956 in the embl.pm file (usr/share/perl5/Bio/SeqIO), the operator 'eq' was used instead of '==' on row 350 in the Simple.pm file (usr/share/perl5/Bio/Location)), dos2unix (6.0.4), ncbi-blast (2.13.0+), tRNAscan-SE (2.0.7), Glimmer (3.02), and InterProScan (5.60-92.0).
4.2. Web Interface
A web interface was developed to facilitate data access and analysis. The following technologies were used for the web interface implementation: PHP (7.4.26), MySQL (14.14), HTML, CSS and JavaScript with thee libraries Bootstrap (4.3.1), dataTables (1.10.2), font-awesome (6.0.0-beta2), swiper (8.4.4), Modernizr (2.8.3), jquery (v2.1.0) and nanoScrollerJS (0.8.0). Analyses were conducted on a Linux-based web server with the following specifications: Debian 3.16.7. 100GB RAM Intel(R) Xeon(R) CPU E5-4620 0 @ 2.20GHz.
4.3. Databases
To ensure comparative annotation, the gene product sequences of the E. cuniculi, N. ceranae, A.algerae and E. bieneusi genes for which manually curated annotation had been performed and whose curation approach had been experimentally validated by 5'RACE PCR [13] were integrated into the MicroAnnot tool.
To enable the identification of the small genes (CDS size less than 300 nucleotides) using the comparative approach, a specific protein database was built from biologically and/or manually validated small gene sequences [12,13]. In addition, sequences in available microsporidian genomes that were orthologous to protein sequences of this database were added to the database. Some sequences annotated during the study of N. ceranae polyploidy [28] were also included in this database. Using all their respective CDS sequences, a Glimmer training dataset was built for each of the four reference genomes. To implement the TE database, all data from multiple published data sources were extracted [13,22,27,71]. These TE lists were completed by TE sequences identified thanks to the complete chromosome assembly of the A. algerae genome using PacBio Hifi sequencing technology (unpublished data). An exhaustive SSU rRNA database comprising the complete phylogenetical diversity of microsporidian rRNAs was built data from [35].
4.4. MicroAnnot Analysis of the Four Microsporidian Genomes
In order to validate the MicroAnnot tool, sequences from four genomes for which annotation had been carried out manually and published (Peyretaillade et al., 2012) were used as a query. They correspond to the sequences of E. cuniculi GB-M1 (GCA_000091225.2), E. intestinalis (GCA_000146465.1), N. ceranae BRL01 (GCA_000182985.1), and E. bieneusi H348 (GCA_000209485.1). For the E. cuniculi, N. ceranae and E. bieneusi genomes, annotations were carried out by selecting the four well annotated microsporidian proteomes incorporated in MicroAnnot, with the exception of those corresponding to the genome used as a query. For the annotation of the E. intestinalis genome, all four proteomes were selected. The results of this comparative study are presented in Table 1 and supplementary Tables S1 to S4. For all other parameters, default values were used.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Details of annotation results performed by MicroAnnot on Encephalitozoon cuniculi. All categories listed in Table 1 are presented in separated sheet; Table S2: Details of annotation results performed by MicroAnnot on Encephalitozoon intestinalis. All categories listed in Table 1 are presented in separated sheet. Table S3: Details of annotation results performed by MicroAnnot on Nosema ceranae. All categories listed in Table 1 are presented in separated sheet. Table S4: Details of annotation results performed by MicroAnnot on Enterocytozoon bieneusi. All categories listed in Table 1 are presented in separated sheet.
Author Contributions
J.T.: Programming, software development, Writing – review & editing; V.P.: Conceptualization, Validation, Writing – review & editing; I.W.: Conceptualization, Validation, Writing – review & editing; R.F.A.: Validation, Writing – review & editing; N.P.: Conceptualization, Software development, Review & editing; F.D.: review & editing; E.L: transposable element database building, review & editing; M.R.: Programming, software development; P.S.: Software development; E.P.: Conceptualization, Validation, Resources, Writing – review & editing, Supervision, Project administration.
Data Availability Statement
Not applicable
Acknowledgments
We acknowledge Dr Rimour-Laneury for their initial work on MicroAnnot. We are also grateful to Victor Berthod who participate on small gene database. The authors would like to thank the AuBi platform (https://www.france-bioinformatique.fr/fr/plateformes/aubi) and the Mésocentre of Clermont Auvergne University (https://mesocentre.uca.fr/).
Conflicts of Interest
The authors declare that they have no conflict of interest
References
- Corsaro, D. Insights into Microsporidia Evolution from Early Diverging Microsporidia. Exp Suppl 2022, 114, 71–90. [Google Scholar] [CrossRef]
- Han, B.; Weiss, L.M. Microsporidia: Obligate Intracellular Pathogens Within the Fungal Kingdom. Microbiol Spectr 2017, 5. [Google Scholar] [CrossRef]
- Stentiford, G.D.; Becnel, -J. J.; Weiss, L.M.; Keeling, P.J.; Didier, E.S.; Williams, B. -a. P.; Bjornson, S.; Kent, M.-L.; Freeman, M.A.; Brown, M.J.F.; et al. Microsporidia - Emergent Pathogens in the Global Food Chain. Trends Parasitol 2016, 32, 336–348. [Google Scholar] [CrossRef]
- Han, B.; Pan, G.; Weiss, L.M. Microsporidiosis in Humans. Clin Microbiol Rev 2021, 34, e00010-20. [Google Scholar] [CrossRef]
- Ziad, F.; Robertson, T.; Watts, M.R.; Copeland, J.; Chiu, G.; Wang, D.; Stark, D.; Graham, L.; Turner, C.; Newbury, R. Fatal Disseminated Anncaliia Algerae Myositis Mimicking Polymyositis in an Immunocompromised Patient. Neuromuscular Disorders 2021, 31, 877–880. [Google Scholar] [CrossRef]
- Coyle, C.M.; Weiss, L.M.; Rhodes, L.V.; Cali, A.; Takvorian, P.M.; Brown, D.F.; Visvesvara, G.S.; Xiao, L.; Naktin, J.; Young, E.; et al. Fatal Myositis Due to the Microsporidian Brachiola Algerae, a Mosquito Pathogen. New England Journal of Medicine 2004, 351, 42–47. [Google Scholar] [CrossRef]
- Anderson, N.W.; Muehlenbachs, A.; Arif, S.; Bruminhent, J.; Deziel, P.J.; Razonable, R.R.; Wilhelm, M.P.; Metcalfe, M.G.; Qvarnstrom, Y.; Pritt, B.S. A Fatal Case of Disseminated Microsporidiosis Due to Anncaliia Algerae in a Renal and Pancreas Allograft Recipient. Open Forum Infect Dis 2019, 6, ofz285. [Google Scholar] [CrossRef]
- Williams, B.A.P.; Williams, T.A.; Trew, J. Comparative Genomics of Microsporidia. Exp Suppl 2022, 114, 43–69. [Google Scholar] [CrossRef]
- Jespersen, N.; Monrroy, L.; Barandun, J. Impact of Genome Reduction in Microsporidia. Exp Suppl 2022, 114, 1–42. [Google Scholar] [CrossRef]
- Corradi, N.; Pombert, J.-F.; Farinelli, L.; Didier, E.S.; Keeling, P.J. The Complete Sequence of the Smallest Known Nuclear Genome from the Microsporidian Encephalitozoon Intestinalis. Nat Commun 2010, 1, 77. [Google Scholar] [CrossRef]
- Katinka, M.D.; Duprat, S.; Cornillot, E.; Méténier, G.; Thomarat, F.; Prensier, G.; Barbe, V.; Peyretaillade, E.; Brottier, P.; Wincker, P.; et al. Genome Sequence and Gene Compaction of the Eukaryote Parasite Encephalitozoon Cuniculi. Nature 2001, 414, 450–453. [Google Scholar] [CrossRef]
- Belkorchia, A.; Gasc, C.; Polonais, V.; Parisot, N.; Gallois, N.; Ribière, C.; Lerat, E.; Gaspin, C.; Pombert, J.-F.; Peyret, P.; et al. The Prediction and Validation of Small CDSs Expand the Gene Repertoire of the Smallest Known Eukaryotic Genomes. PLoS ONE 2015, 10, e0139075. [Google Scholar] [CrossRef]
- Peyretaillade, E.; Parisot, N.; Polonais, V.; Terrat, S.; Denonfoux, J.; Dugat-Bony, E.; Wawrzyniak, I.; Biderre-Petit, C.; Mahul, A.; Rimour, S.; et al. Annotation of Microsporidian Genomes Using Transcriptional Signals. Nat Commun 2012, 3, 1137. [Google Scholar] [CrossRef]
- Biderre, C.; Méténier, G.; Vivarès, C.P. A Small Spliceosomal-Type Intron Occurs in a Ribosomal Protein Gene of the Microsporidian Encephalitozoon Cuniculi. Mol Biochem Parasitol 1998, 94, 283–286. [Google Scholar] [CrossRef]
- Corradi, N.; Gangaeva, A.; Keeling, P.J. Comparative Profiling of Overlapping Transcription in the Compacted Genomes of Microsporidia Antonospora Locustae and Encephalitozoon Cuniculi. Genomics 2008, 91, 388–393. [Google Scholar] [CrossRef]
- Gill, E.E.; Lee, R.C.H.; Corradi, N.; Grisdale, C.J.; Limpright, V.O.; Keeling, P.J.; Fast, N.M. Splicing and Transcription Differ between Spore and Intracellular Life Stages in the Parasitic Microsporidia. Mol Biol Evol 2010, 27, 1579–1584. [Google Scholar] [CrossRef]
- Heinz, E.; Williams, T.A.; Nakjang, S.; Noël, C.J.; Swan, D.C.; Goldberg, A.V.; Harris, S.R.; Weinmaier, T.; Markert, S.; Becher, D.; et al. The Genome of the Obligate Intracellular Parasite Trachipleistophora Hominis: New Insights into Microsporidian Genome Dynamics and Reductive Evolution. PLoS Pathog 2012, 8, e1002979. [Google Scholar] [CrossRef]
- Peyretaillade, E.; Gonçalves, O.; Terrat, S.; Dugat-Bony, E.; Wincker, P.; Cornman, R.S.; Evans, J.D.; Delbac, F.; Peyret, P. Identification of Transcriptional Signals in Encephalitozoon Cuniculi Widespread among Microsporidia Phylum: Support for Accurate Structural Genome Annotation. BMC Genomics 2009, 10, 607. [Google Scholar] [CrossRef]
- Williams, B.A.P.; Slamovits, C.H.; Patron, N.J.; Fast, N.M.; Keeling, P.J. A High Frequency of Overlapping Gene Expression in Compacted Eukaryotic Genomes. Proc Natl Acad Sci U S A 2005, 102, 10936–10941. [Google Scholar] [CrossRef]
- Polonais, V.; Prensier, G.; Méténier, G.; Vivarès, C.P.; Delbac, F. Microsporidian Polar Tube Proteins: Highly Divergent but Closely Linked Genes Encode PTP1 and PTP2 in Members of the Evolutionarily Distant Antonospora and Encephalitozoon Groups. Fungal Genetics and Biology 2005, 42, 791–803. [Google Scholar] [CrossRef]
- Mascarenhas dos Santos, A.C.; Julian, A.T.; Pombert, J.-F. The Rad9–Rad1–Hus1 DNA Repair Clamp Is Found in Microsporidia. Genome Biol Evol 2022, 14, evac053. [Google Scholar] [CrossRef]
- Parisot, N.; Pelin, A.; Gasc, C.; Polonais, V.; Belkorchia, A.; Panek, J.; El Alaoui, H.; Biron, D.G.; Brasset, E.; Vaury, C.; et al. Microsporidian Genomes Harbor a Diverse Array of Transposable Elements That Demonstrate an Ancestry of Horizontal Exchange with Metazoans. Genome Biol Evol 2014, 6, 2289–2300. [Google Scholar] [CrossRef]
- Peyretaillade, E.; Boucher, D.; Parisot, N.; Gasc, C.; Butler, R.; Pombert, J.-F.; Lerat, E.; Peyret, P. Exploiting the Architecture and the Features of the Microsporidian Genomes to Investigate Diversity and Impact of These Parasites on Ecosystems. Heredity (Edinb) 2015, 114, 441–449. [Google Scholar] [CrossRef]
- Ejigu, G.F.; Jung, J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology (Basel) 2020, 9, 295. [Google Scholar] [CrossRef]
- Huang, Q.; Wu, Z.H.; Li, W.F.; Guo, R.; Xu, J.S.; Dang, X.Q.; Ma, Z.G.; Chen, Y.P.; Evans, J.D. Genome and Evolutionary Analysis of Nosema Ceranae: A Microsporidian Parasite of Honey Bees. Front Microbiol 2021, 12, 645353. [Google Scholar] [CrossRef]
- Mikhailov, K.V.; Simdyanov, T.G.; Aleoshin, V.V. Genomic Survey of a Hyperparasitic Microsporidian Amphiamblys Sp. (Metchnikovellidae). Genome Biol Evol 2016, 9, 454–467. [Google Scholar] [CrossRef]
- Ndikumana, S.; Pelin, A.; Williot, A.; Sanders, J.L.; Kent, M.; Corradi, N. Genome Analysis of Pseudoloma Neurophilia: A Microsporidian Parasite of Zebrafish (Danio Rerio). J Eukaryot Microbiol 2017, 64, 18–30. [Google Scholar] [CrossRef]
- Pelin, A.; Selman, M.; Aris-Brosou, S.; Farinelli, L.; Corradi, N. Genome Analyses Suggest the Presence of Polyploidy and Recent Human-Driven Expansions in Eight Global Populations of the Honeybee Pathogen Nosema Ceranae. Environmental Microbiology 2015, 17, 4443–4458. [Google Scholar] [CrossRef]
- Ang’ang’o, L.M.; Herren, J.K.; Tastan Bishop, Ö. Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma Corneae ATCC 50505 Using in Silico Approaches. International Journal of Molecular Sciences 2023, 24, 3507. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Rothnagel, J.A. Emerging Evidence for Functional Peptides Encoded by Short Open Reading Frames. Nat Rev Genet 2014, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Dubuffet, A.; Chauvet, M.; Moné, A.; Debroas, D.; Lepère, C. A Phylogenetic Framework to Investigate the Microsporidian Communities through Metabarcoding and Its Application to Lake Ecosystems. Environ Microbiol 2021, 23, 4344–4359. [Google Scholar] [CrossRef] [PubMed]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A Benchmark Study of Ab Initio Gene Prediction Methods in Diverse Eukaryotic Organisms. BMC Genomics 2020, 21, 293. [Google Scholar] [CrossRef]
- Dimonaco, N.J.; Aubrey, W.; Kenobi, K.; Clare, A.; Creevey, C.J. No One Tool to Rule Them All: Prokaryotic Gene Prediction Tool Annotations Are Highly Dependent on the Organism of Study. Bioinformatics 2022, 38, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Chen, Y.P.; Schatz, M.C.; Street, C.; Zhao, Y.; Desany, B.; Egholm, M.; Hutchison, S.; Pettis, J.S.; Lipkin, W.I.; et al. Genomic Analyses of the Microsporidian Nosema Ceranae, an Emergent Pathogen of Honey Bees. PLoS Pathog 2009, 5, e1000466. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, D.E.; Morrison, H.G.; Lei, S.; Feng, X.; Zhang, Q.; Corradi, N.; Mayanja, H.; Tumwine, J.K.; Keeling, P.J.; Weiss, L.M.; et al. Genomic Survey of the Non-Cultivatable Opportunistic Human Pathogen, Enterocytozoon Bieneusi. PLoS Pathog 2009, 5, e1000261. [Google Scholar] [CrossRef]
- Baker, L.; David, C.; Jacobs, D.J. Ab Initio Gene Prediction for Protein-Coding Regions. Bioinformatics Advances 2023, 3, vbad105. [Google Scholar] [CrossRef]
- König, S.; Romoth, L.; Stanke, M. Comparative Genome Annotation. Methods Mol Biol 2018, 1704, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Marcet-Houben, M.; Gabaldón, T. Phylogenomics Supports Microsporidia as the Earliest Diverging Clade of Sequenced Fungi. BMC Biology 2012, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Selecting the Right Similarity-Scoring Matrix. Curr Protoc Bioinformatics 2013, 43, 3.5.1–3.5.9. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Singh, S.; Aseri, T.C. Global Sequence Features Based Translation Initiation Site Prediction in Human Genomic Sequences. Heliyon 2020, 6, e04825. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, H.; Jiang, T.; Zhang, L.; Zeng, J. TITER: Predicting Translation Initiation Sites by Deep Learning. Bioinformatics 2017, 33, i234–i242. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Fast, N.M.; Corradi, N. Microsporidian Genome Structure and Function. In Microsporidia; John Wiley & Sons, Ltd, 2014; pp. 221–229 ISBN 978-1-118-39526-4.
- Pombert, J.-F.; Haag, K.L.; Beidas, S.; Ebert, D.; Keeling, P.J. The Ordospora Colligata Genome: Evolution of Extreme Reduction in Microsporidia and Host-To-Parasite Horizontal Gene Transfer. mBio 2015, 6, 10.1128/mbio.02400-14. [Google Scholar] [CrossRef]
- Whelan, T.A.; Lee, N.T.; Lee, R.C.H.; Fast, N.M. Microsporidian Introns Retained against a Background of Genome Reduction: Characterization of an Unusual Set of Introns. Genome Biol Evol 2018, 11, 263–269. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, K.; Xun, C.; Chu, T.; Liang, S.; Zeng, Y.; Liu, Z. Small Open Reading Frame-Encoded Micro-Peptides: An Emerging Protein World. International Journal of Molecular Sciences 2023, 24, 10562. [Google Scholar] [CrossRef]
- de Albuquerque, N.R.M.; Ebert, D.; Haag, K.L. Transposable Element Abundance Correlates with Mode of Transmission in Microsporidian Parasites. Mobile DNA 2020, 11, 19. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol 2019, 20, 275. [Google Scholar] [CrossRef]
- Cormier, A.; Chebbi, M.A.; Giraud, I.; Wattier, R.; Teixeira, M.; Gilbert, C.; Rigaud, T.; Cordaux, R. Comparative Genomics of Strictly Vertically Transmitted, Feminizing Microsporidia Endosymbionts of Amphipod Crustaceans. Genome Biol Evol 2020, 13, evaa245. [Google Scholar] [CrossRef] [PubMed]
- Aurrecoechea, C.; Barreto, A.; Brestelli, J.; Brunk, B.P.; Caler, E.V.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; et al. AmoebaDB and MicrosporidiaDB: Functional Genomic Resources for Amoebozoa and Microsporidia Species. Nucleic Acids Res 2011, 39, D612–D619. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.S. A Review of Pangenome Tools and Recent Studies. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer: Cham (CH), 2020 ISBN 978-3-030-38280-3.
- Seatamanoch, N.; Kongdachalert, S.; Sunantaraporn, S.; Siriyasatien, P.; Brownell, N. Microsporidia, a Highly Adaptive Organism and Its Host Expansion to Humans. Front Cell Infect Microbiol 2022, 12, 924007. [Google Scholar] [CrossRef]
- Chen, L.; Gao, X.; Li, R.; Zhang, L.; Huang, R.; Wang, L.; Song, Y.; Xing, Z.; Liu, T.; Nie, X.; et al. Complete Genome of a Unicellular Parasite (Antonospora Locustae) and Transcriptional Interactions with Its Host Locust. Microb Genom 2020, 6, mgen000421. [Google Scholar] [CrossRef] [PubMed]
- Polonais, V.; Niehus, S.; Wawrzyniak, I.; Franchet, A.; Gaspin, C.; Belkorchia, A.; Reichstadt, M.; Belser, C.; Labadie, K.; Couloux, A.; et al. Draft Genome Sequence of Tubulinosema Ratisbonensis, a Microsporidian Species Infecting the Model Organism Drosophila Melanogaster. Microbiology Resource Announcements 2019, 8, 10.1128/mra.00077-19. [Google Scholar] [CrossRef]
- Cacho, A.; Smirnova, E.; Huzurbazar, S.; Cui, X. A Comparison of Base-Calling Algorithms for Illumina Sequencing Technology. Brief Bioinform 2016, 17, 786–795. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.-C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci Data 2020, 7, 399. [Google Scholar] [CrossRef] [PubMed]
- Pagès-Gallego, M.; De Ridder, J. Comprehensive Benchmark and Architectural Analysis of Deep Learning Models for Nanopore Sequencing Basecalling. Genome Biol 2023, 24, 71. [Google Scholar] [CrossRef]
- Pombert, J.-F.; Selman, M.; Burki, F.; Bardell, F.T.; Farinelli, L.; Solter, L.F.; Whitman, D.W.; Weiss, L.M.; Corradi, N.; Keeling, P.J. Gain and Loss of Multiple Functionally Related, Horizontally Transferred Genes in the Reduced Genomes of Two Microsporidian Parasites. Proc. Natl. Acad. Sci. USA 2012, 109, 12638–12643. [Google Scholar] [CrossRef]
- Ouzounis, C.A.; Karp, P.D. The Past, Present and Future of Genome-Wide Re-Annotation. Genome Biol 2002, 3, comment2001.1–comment2001.6. [Google Scholar] [CrossRef]
- Goudey, B.; Geard, N.; Verspoor, K.; Zobel, J. Propagation, Detection and Correction of Errors Using the Sequence Database Network. Brief. Bioinform. 2022, 23, bbac416. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bencurova, E.; Srivastava, M.; Pahlavan, P.; Balkenhol, J.; Dandekar, T. Improving Re-Annotation of Annotated Eukaryotic Genomes. In Big Data Analytics in Genomics; Wong, K.-C., Ed.; Springer International Publishing: Cham, 2016; pp. 171–195. ISBN 978-3-319-41279-5. [Google Scholar]
- Fu, X.-D. Non-Coding RNA: A New Frontier in Regulatory Biology. Natl Sci Rev 2014, 1, 190–204. [Google Scholar] [CrossRef]
- Dong, Z.; Zheng, N.; Hu, C.; Deng, B.; Fang, W.; Wu, Q.; Chen, P.; Huang, X.; Gao, N.; Lu, C.; et al. Nosema Bombycis microRNA-like RNA 8 (Nb-milR8) Increases Fungal Pathogenicity by Modulating BmPEX16 Gene Expression in Its Host, Bombyx Mori. Microbiol Spectr 2021, 9, e0104821. [Google Scholar] [CrossRef]
- Guo, R.; Chen, D.; Chen, H.; Xiong, C.; Zheng, Y.; Hou, C.; Du, Y.; Geng, S.; Wang, H.; Dingding, Z.; et al. Genome-Wide Identification of Circular RNAs in Fungal Parasite Nosema Ceranae. Curr Microbiol 2018, 75, 1655–1660. [Google Scholar] [CrossRef]
- Shao, S.S.; Yan, W.Y.; Huang, Q. Identification of Novel miRNAs from the Microsporidian Parasite Nosema Ceranae. Infect Genet Evol 2021, 93, 104930. [Google Scholar] [CrossRef]
- Shen, Z.; Yang, Q.; Luo, L.; Li, T.; Ke, Z.; Li, T.; Chen, J.; Meng, X.; Xiang, H.; Li, C.; et al. Non-Coding RNAs Identification and Regulatory Networks in Pathogen-Host Interaction in the Microsporidia Congenital Infection. BMC Genomics 2023, 24, 420. [Google Scholar] [CrossRef]
- Belkorchia, A.; Pombert, J.-F.; Polonais, V.; Parisot, N.; Delbac, F.; Brugère, J.-F.; Peyret, P.; Gaspin, C.; Peyretaillade, E. Comparative Genomics of Microsporidian Genomes Reveals a Minimal Non-Coding RNA Set and New Insights for Transcription in Minimal Eukaryotic Genomes. DNA Res 2017, 24, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Tang, X.; Lan, L.; Zhang, X.; Zhang, X. The Genomic Survey of Tc1-like Elements in the Silkworm Microsporidia Nosema Bombycis. Acta Parasitol 2020, 65, 193–202. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Microannot interface. (A) Home page with the pipeline description. By clicking on help, the user is redirected to MicroAnnot scheme (see Figure 3). (B) The analysis section is also available on the home page. Sequences to annotate (fasta files) have to be downloaded from user files. Then the user can select the minimal size of ORF finding and the parameters used for Glimmer CDS prediction (minimal CDS size and training data set if less than 50 CDS are identified by homology). Here default parameters are presented for each section. Functional annotation with InterProScan is disabled by default but can be activated by checking (*).
Figure 1.
Microannot interface. (A) Home page with the pipeline description. By clicking on help, the user is redirected to MicroAnnot scheme (see Figure 3). (B) The analysis section is also available on the home page. Sequences to annotate (fasta files) have to be downloaded from user files. Then the user can select the minimal size of ORF finding and the parameters used for Glimmer CDS prediction (minimal CDS size and training data set if less than 50 CDS are identified by homology). Here default parameters are presented for each section. Functional annotation with InterProScan is disabled by default but can be activated by checking (*).
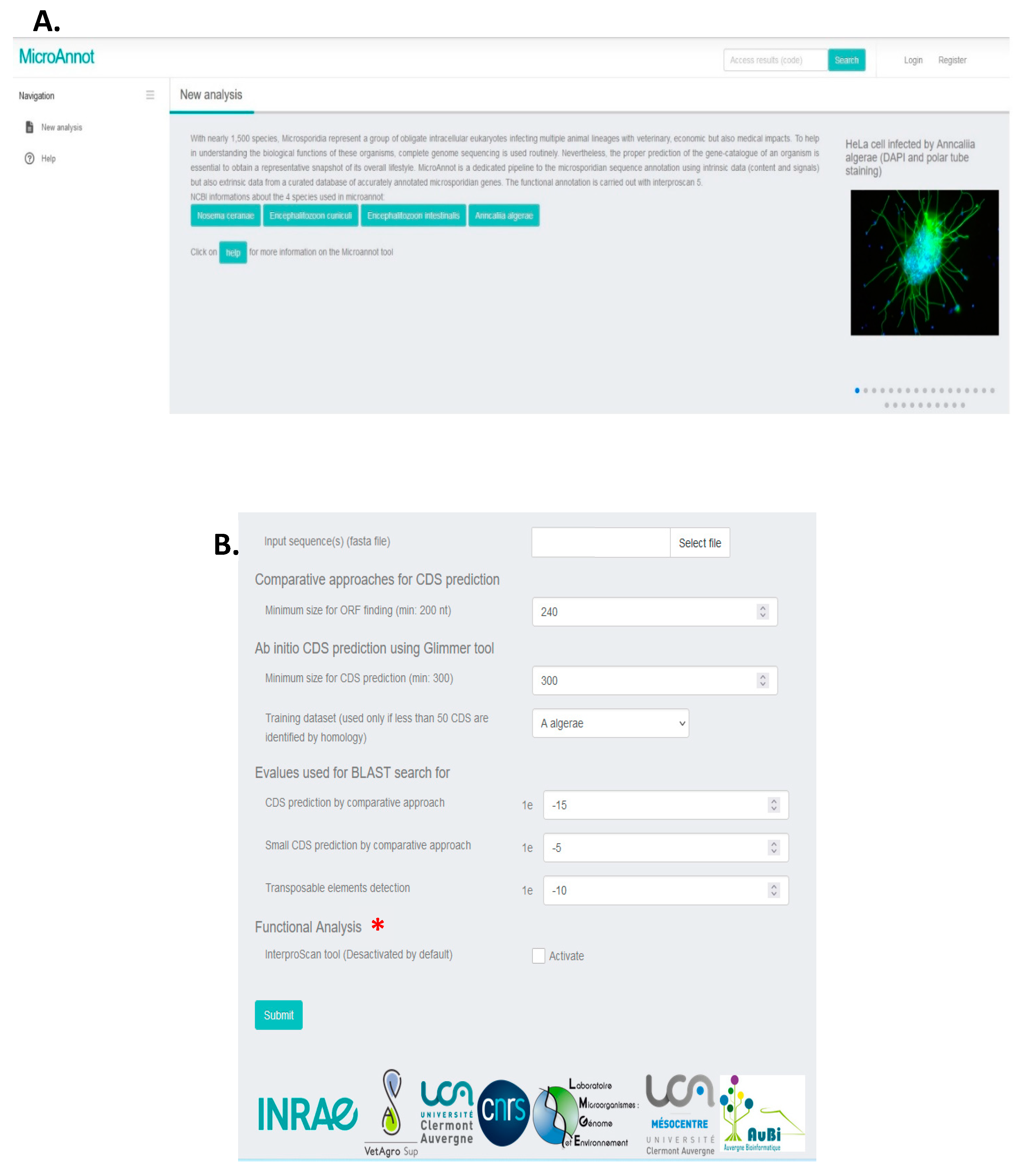
Figure 2.
Analysis seen by the user after job submission. (A) Pending, (B) Running. (C) When the analysis is finished, different files formats (EMBL, GenBank or GFF) are available for download as well as a warning text file grouping uncertain annotations that must be manually validated. At each step the sequence name (fasta file) is conserved along with all the analysis details in the information column.
Figure 2.
Analysis seen by the user after job submission. (A) Pending, (B) Running. (C) When the analysis is finished, different files formats (EMBL, GenBank or GFF) are available for download as well as a warning text file grouping uncertain annotations that must be manually validated. At each step the sequence name (fasta file) is conserved along with all the analysis details in the information column.
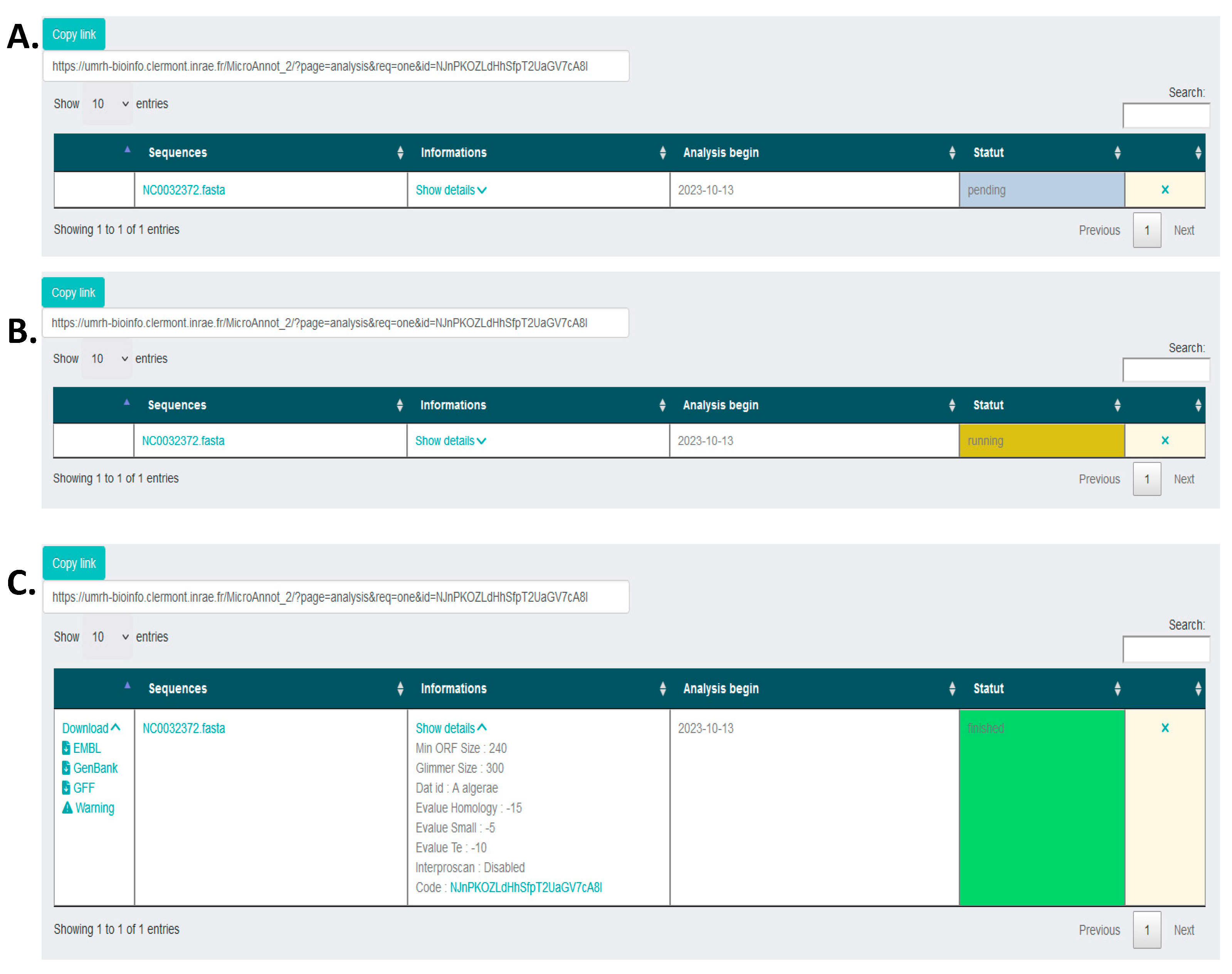

Table 1.
Evaluation of annotation results performed by MicroAnnot on four well annotated microsporidian genomes classified in three categories. i) annotation corrections proposed by MicroAnnot in comparison with annotation errors identified manually during different studies. Numbers into brackets correspond to the annotation corrections detected in this study by MicroAnnot; ii) additional annotation corrections identified with MicroAnnot and iii) additional annotation errors obtained with MicroAnnot. The number into brackets indicate the number of warning encouraging authors to check these annotations. Detailed information can be found in supplementary Table S1 to S4. TIS: Translational Initiation Site; TEs: Transposable Elements.
Table 1.
Evaluation of annotation results performed by MicroAnnot on four well annotated microsporidian genomes classified in three categories. i) annotation corrections proposed by MicroAnnot in comparison with annotation errors identified manually during different studies. Numbers into brackets correspond to the annotation corrections detected in this study by MicroAnnot; ii) additional annotation corrections identified with MicroAnnot and iii) additional annotation errors obtained with MicroAnnot. The number into brackets indicate the number of warning encouraging authors to check these annotations. Detailed information can be found in supplementary Table S1 to S4. TIS: Translational Initiation Site; TEs: Transposable Elements.
| E. cuniculi | E. intestinalis | N. ceranae | E. bieneusi | ||
|---|---|---|---|---|---|
| Annotation corrections | Falsely predicted TIS | 445 (299: 67.2%) | 199 (185: 93%) | 308 (232: 75.3) | 240 (172: 71.7%) |
| Falsely predicted gene | 11 (0: 100%) | 8 (0: 100%) | 76 (18: 76.3%) | 168 (4: 97.6%) | |
| Newly predicted introns | 3 (3:100%) | 19 (12: 63.2%) | 4 (3:75%) | - | |
| Unpredicted genes | 142 (128: 90.1%) | 115 (108: 93.9%) | 292 (250: 86.5%) | 70 (66: 94.3%) | |
| Unpredicted frameshift or sequencing error or pseudogene | - | 11(10: 90.9%) | 121 (74: 61.1%) | 43 (17: 39.5%) | |
| Additional annotation corrections | Corrected TIS | 77 | 18 | 15 | 26 |
| Falsely Predicted gene | 12 | _ | 3 | 8 | |
| Predicted introns | - | _ | 1 | _ | |
| Unpredicted genes | 23 | - | 296 | 15 | |
| Unpredicted frameshift or sequencing error or pseudogene | - | 1 | - | - | |
| TEs predicted as CDS | - | - | 42 | - | |
| Predicted TEs | - | - | 643 | - | |
| Additional annotation errors | Mispredicted TIS (warning) | 94 | 32 | 62 (49) | 68 (29) |
| Falsely Predicted gene (warning) | 1 | 1 | 11 (27) | 25 | |
| Mispredicted intron | 5 | 0 | _ | - | |
| Unpredicted genes | 47 | 4 | 22 | 33 | |
| Bad predicted TEs | 0 | 1 | - | - |
Table 2.
Comparisons of MicroAnnot performances with previous annotations. The specificity (Sp), the sensitivity (Sn) and TIS prediction of the genes are defined as: Sp=TP(TP+FP), Sn= TP(TP+FN), and TCP= TP(TP+FPT). TP: True positives FN: False negatives, FP: False positives and FPT: Falsely Predicted TIS. * true gene numbers obtained after compilation of all the reannotation studies [12,13,18] and present data corrected by MicroAnnot annotation. ** first annotation references [10,11,38,39] for E. bieneusi, N. ceranae, E. intestinalis and E. cuniculi genomes respectively.
Table 2.
Comparisons of MicroAnnot performances with previous annotations. The specificity (Sp), the sensitivity (Sn) and TIS prediction of the genes are defined as: Sp=TP(TP+FP), Sn= TP(TP+FN), and TCP= TP(TP+FPT). TP: True positives FN: False negatives, FP: False positives and FPT: Falsely Predicted TIS. * true gene numbers obtained after compilation of all the reannotation studies [12,13,18] and present data corrected by MicroAnnot annotation. ** first annotation references [10,11,38,39] for E. bieneusi, N. ceranae, E. intestinalis and E. cuniculi genomes respectively.
| E. cuniculi | E. intestinalis | N. ceranae | E. bieneusi | ||
|---|---|---|---|---|---|
| True gene numbers* | 2151 | 1940 | 2352 | 1770 | |
| Specificity (Sp) | 1st annotation** | 98.9% | 99.6% | 96.8% | 91% |
| MicroAnnot | 99.9% | 99.9% | 98.9% | 98.4% | |
| Sensibility (Sn) | 1st annotation** | 92.9% | 94.4% | 80% | 95.4% |
| MicroAnnot | 95.2% | 99.4% | 97.4% | 98% | |
| TIS Correctly predicted (TCP) | 1st annotation** | 80.5% | 90.0% | 88.0% | 87% |
| MicroAnnot | 90.0% | 97.8% | 94.5% | 93% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



