
Submitted:
27 October 2023
Posted:
30 October 2023
You are already at the latest version
Abstract
Valsartan (Val) is an important antihypertensive medication with side effects such as dizziness and angioedema. These constraints are due to the low oral bioavailability and relatively short half-life. The goal here is to incorporate Val into a mixed micelles system that can enhance transdermal delivery. Thin-film hydration and micro-phase separation were used to produce Val-loaded mixed micelle systems. A variety of factors, including surfactant type and drug-to-surfactant ratio, were optimized to produce micelles with low size and high Val entrapment efficiency (EE). The size, polydispersity index (PDI), zeta-potential, and drug entrapment efficiency of the prepared micelles were all measured. The drug release profiles in vitro were assessed using dialysis bags, and the permeation through abdominal rat skin was assessed using a Franz diffusion cell. All formulations had high EE levels exceeding 90% and low particle charges. The micellar sizes ranged from 107.6 to 191.7nm, with average PDI values of 0.3. The in vitro release demonstrated a uniform slow rate that lasted one week with varying extents. F7 demonstrated a significant (P < 0.01) transdermal efflux of 68.84 ± 3.96 µg/cm2/h through rat skin when compared to the control. As a result, the enhancement factor was 16.57. Val-loaded mixed micelles were successfully prepared using two simple methods with high reproducibility, and extensive transdermal delivery was demonstrated in the absence of any aggressive skin modifying enhancers.
Keywords:
Valsartan
; Thin-film Hydration
; Franz Diffusion Cell
; Tween 80
1. Introduction
Valsartan (Val) is a popular antihypertensive medication that belongs to the angiotensin II receptor antagonist class. It was listed as one of the top 200 prescribed medications in the United State [1]. The low oral bioavailability (23%) due to its poor aqueous solubility, as well as the relatively short elimination half-life (7.5 hours), are drawbacks for its therapeutic efficacy, resulting in more severe adverse effects such as dizziness, joint pain, and angioedema [2,3]. This has prompted many researchers to work on improving Val's overall therapeutic performance by modifying the drug's pharmacokinetic properties. In the treatment of chronic diseases such as hypertension, transdermal drug delivery demonstrated significant advantages over the oral route [4]. These include the highly controlled blood levels of drugs, comparable to iv infusion treatment, avoiding GIT-related side effects, and minimizing the drug's systemic adverse effects, as well as the possibility of reducing the frequency of administration, which leads to higher patient compliance.
Aside from the low oral bioavailability, the physico-chemical properties of Val, such as its low molecular weight (435.5 D), partition coefficient (log p = 4.5), and pKa value of 4.75, make it a viable candidate for transdermal drug delivery [5,6,7]. Manipulation of the skin's protective impermeable stratum corneum layer to allow drug efflux into the cutaneous layer is regarded as critical for the successful design of a transdermal delivery system. These substances are known as skin penetration enhancers. They can disrupt the stratum corneum's integrity through a variety of mechanisms, including changing the configuration order and/or partial extraction of intercellular lipids, resulting in mobilization, and adjusting the drug partition coefficient to increase its diffusion through the skin [8,9,10]. Many substances, including solvents such as alcohols and dimethyl sulfoxide (DMSO), essential oils and fatty acids such as terpenes, linoleic and oleic acids, and azones and surfactants, have been shown to increase skin permeability to many drugs [11,12,13,14].
Drug incorporation in nano-drug delivery systems has been used to improve cellular uptake and absorption [15,16], aqueous solubility of poorly soluble drugs [17,18], and drug residence time in the body [19]. Many nano-particulate systems have been used to improve drug skin permeability. Liposomes, solid lipid nanoparticles, transfersomes, niosomes, nanoemulsions, and mixed micelles have been successfully used to improve transdermal penetration for many drugs [20,21,22,23,24].
Micelles are association colloids that are formed by aggregation of amphiphilic molecules, containing polar heads and non-polar tails. They associate in water to form spherical shape particles in which non-polar tails hide on the inside [19]. Thus, hydrophobic drugs can be incorporated in the hydrophobic core of micelles. The hydrophilic surface of such micellar systems has the advantage of being intrinsically protected against removal in the systemic circulation by the phagocytic immune mechanism without the need for additional modification. As a result, they have a longer in vivo residence time [25,26,27]. They also comprise additional advantages including their simple production by self-assembly methods, hold a highly promising option for the delivery of poorly soluble drugs and enhancing their solubility and bioavailability [28]. The size of a micelle is highly dependent on the molecular size and configuration of the amphiphile [29]. Micelles have been extensively applied for enhancing the solubility and bioavailability of many drugs [29,30]. Polymeric micelles are unique type of micelles that are formed by amphiphilic block co-polymers composed of alternating hydrophilic and hydrophobic segments [31]. They can form micelles by a molecular plumbing out to hide the hydrophobic segment toward the core and the hydrophilic segment on the shell [32]. These are also known as unimer micelles and characterized by higher stability compared with conventional micelles [33]. Mixed micelles are micelles formed by association of two or more species of amphiphilic compounds. Their smaller sizes (below 60 nm) and simple method of production are important advantages over vesicular bilayer systems for the delivery of poorly soluble drugs, especially through the parenteral route [34].
Micellar systems have been widely used to improve the delivery of anticancer drugs such as paclitaxel [35], doxorubicin [36,37], and camptothecin [38]. Song and colleagues [39] discovered that TPGS/Phospholipids mixed micelles significantly increased icariside II anticancer activity in multi-resistant breast cancer cell lines. Ould-Ouali and colleagues [40] demonstrated that incorporating a poorly water-soluble drug, risperidone, into a polymeric micelle system improved its solubility. Mixed micelles were successfully used as a carrier for hydrophobic drugs such as Curcumin, and the drug absorption rate was increased [41].
The goal of this study was to optimize a mixed micelle system for transdermal delivery of Val in order to improve therapeutic performance by providing prolonged uniform drug levels while minimizing drug side effects.
2. Results
2.1. Formulation Factors:
Eight formulations were suggested to compare number of factors including two method of preparation; micro-phase separation and thin-film hydration, the type of co-surfactants, and the ratio of drug to surfactants. Full description of the exact composition of each formulation is presented in Table 1. All formulations contained Tween 80 as main surfactant in combination of either sodium dioxycholate (SDC) or Span 80 as co-surfactant.
2.2. Characterization of the mixed micelles formulations
2.2.1. Particle Size, Polydispersity Index, Zeta Potential and Structural Morphology
The average particle sizes for all the prepared formulations were in the nano-range lying between 107.6 nm to 191.7nm. Figure 1 and Table 2 depicts the mean particle sizes of all the prepared formulations. In order to explore the impact of the method of preparation on the micellar mean particular sizes, the results of formulation with exact same composition such as F1 with F6, F3 with F7, and F4 with F8 formulations were compared. There was no significant difference between both F3, F7 and F4, F8 pairs while a significant difference is observed between F1 and F6. This shows that there is no trend leading to a conclusive evidence that the particle sizes were affected by the method of preparation. Comparing the particle sizes of F5 with F7, F6 with F8, and F2 with F3 who differ only in the surfactants: drug ratio, it is notable that the sizes are significantly lower with higher ratios (P < 0.05). The effect of using SDC versus Span 80 as a second surfactant with Tween 80 was found to be insignificant on the sizes of micelles. F3 formulation (containing 500 mg Span) exhibited a significantly lower particle size (P < 0.05) than F2 formulation (containing 250mg Span); 112.74±1.73 nm and 137.03±3.42 nm, respectively. This indicates that the ratio of Span 80 is critical for the micelle size. The difference in the type of surfactant from Span 80 (F3 formulation) to SDC (F4 formulation) did not affect the particle size significantly (P > 0.05).
The polydispersity indices for all the prepared formulations were demonstrated in Figure 2 and values are presented in Table 2. Polydispersity index values ranged from 0.24 to 0.39. A cut-off values of 0.3 has been commonly accepted in the literature as maximum for good size uniformity among a single nanoparticle formulation [42]. Therefore, only formulations F5, F6, F7, and F8 are considered to have narrow micelle size distribution. It is also conclusive that the thin-film hydration method is superior over the micro-phase separation method regarding the uniformity of the micelle sizes.

Table 2.
Mean particle size, zeta potential, percent entrapment efficiency, and percent drug loading measurements for all formulations.
Table 2.
Mean particle size, zeta potential, percent entrapment efficiency, and percent drug loading measurements for all formulations.
| Formulation | Particle size (nm) | Poly-dispersity index | Zeta-potential (mV) | EE% |
| F1 | 107.6±0.6* | 0.33±0.01 | -0.11±0.41 | 88.1±4.4 |
| F2 | 137.0±3.4 | 0.37±0.02 | 3.74±6.93 | 82.7±3.3 |
| F3 | 112.7±1.7 | 0.31±0.01 | -0.67±4.87 | 95.3±5.7 |
| F4 | 117.7±7.4 | 0.39±0.03 | 0.92±5.90 | 91.8±4.1 |
| F5 | 140.4±1.0 | 0.25±0.00 | 0.26±7.10 | 87.3±4.4 |
| F6 | 191.7±8.5 | 0.27±0.05 | 5.93±5.48 | 86.4±3.1 |
| F7 | 112.6±0.4 | 0.24±0.01 | -4.93±3.10 | 96.2±6.7 |
| F8 | 119.7±0.5 | 0.24±0.02 | 3.85±6.34 | 94.9±2.8 |
* Mean ± standard deviation
The zeta-potential values for each formulation shown in Figure 3 and Table 2 can be used to estimate the surface charge density of the prepared micelles. The results showed that all formulations had low zeta-potential values, despite differences in charge nature. The F6 formulation achieved the highest value (+5.93 mV). Having zeta-potential values above 30mV has been widely accepted in the literature as a measure of colloidal stability [43]. Other factors, such as the required hydrophilic lipophilic balance (RHLB), interfacial tension, and the concentration of the surfactant(s), are more influential on the stability of association colloids such as micelles. Micellar systems are lyophilic (solvent-like) colloids that are stabilized primarily by the formation of a protective solvent sheath rather than by high charge density [44,45].
Figure 1.
Histogram of particle characteristics all the prepared mixed micelles formulations.

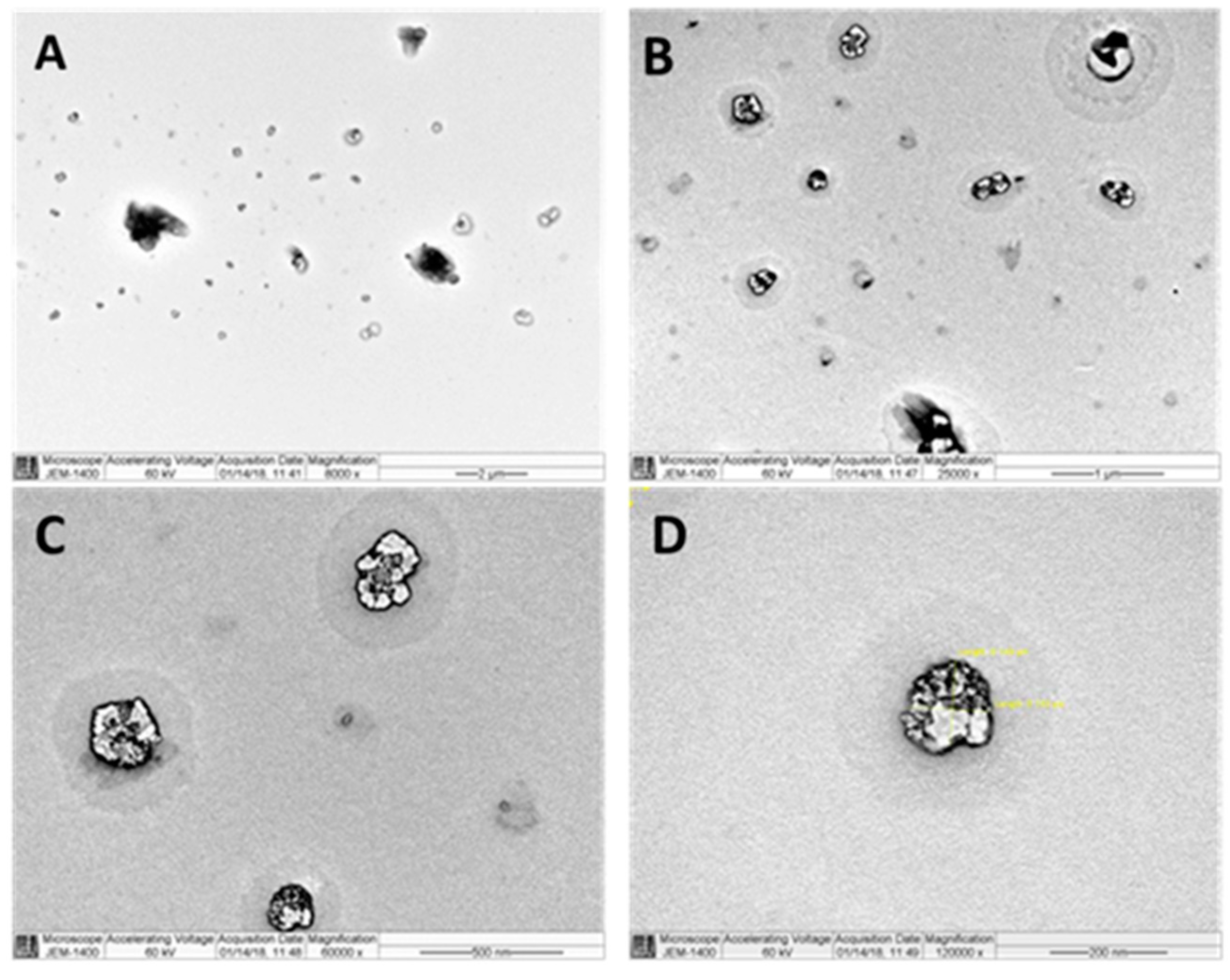
Figure 2 shows TEM images of various micellar samples at various magnification powers. The majority of the particles in Image A had particle sizes of around 100 nm. The presence of multiple dark spots within partiles in Figure 2 images B, C, and D clearly demonstrates drug encapsulation in the core of the particle. The presence of a transparent layer surrounding the micelles is attributed to the presence of a sovent stagnant layer.
Figure 2.
TEM micrograph of Val-loaded mixed micelles.
The SEM image in Figure 3 confirmed the 100 nm average particle size shown in the TEM micrographs. They indicated that the particle shape was spherical. The resistance to degradation by the applied high energy, 100 KV, indicated the prepared micelles' high stability and robust nature, which is unusual for such vesicular particles.
Figure 3.
SEM micrograph of Val-loaded mixed micelles.

2.2.2. Drug entrapment efficiency
All of the prepared formulations had EE% values greater than 80%, which is an advantage of micellar systems. The formulations with a higher surfactant to drug ratio clearly had a higher EE%. Table 2 shows that formulations F3, F4, F7, and F8 with a surfactant-to-drug ratio of 100:1 had significantly higher EE% (P < 0.05), with an average of 94%, than formulations with surfactant ratios of 1:45 (F2) and 1:50 (F1, F5, and F6), with an average of 86%.
2.2.3. In vitro release:
Figure 4 depicts the Val release profile from micellar formulations prepared using the microphase separation method (F1 to F4) for a period of one 7 days. It is clear that the rate of release from F1 and F2 is very similar, with no significant difference at any point (P < 0.05), while F3 and F4 showed a significantly slower rate. This indicates that the incorporation of a higher Surfactant to Val ratio is the key parameter in delaying the release rate, and the type of co-surfactant had no effect on the Val release from mixed micelles prepared by the microphase separation method. The change in the Val:Span 80 ratio from 1:5 (F2) to 1:20 (F3) caused a significant delay in the release rate. The same pattern was observed with SDC formulations, as F4 containing 1:20 had a significantly slower release rate than F1 containing 1:10.
Figure 4.
Cumulative percentage (%) release of Valsartan from four mixed micelles formulations prepared by micro-phase separation method against time.
Figure 4.
Cumulative percentage (%) release of Valsartan from four mixed micelles formulations prepared by micro-phase separation method against time.
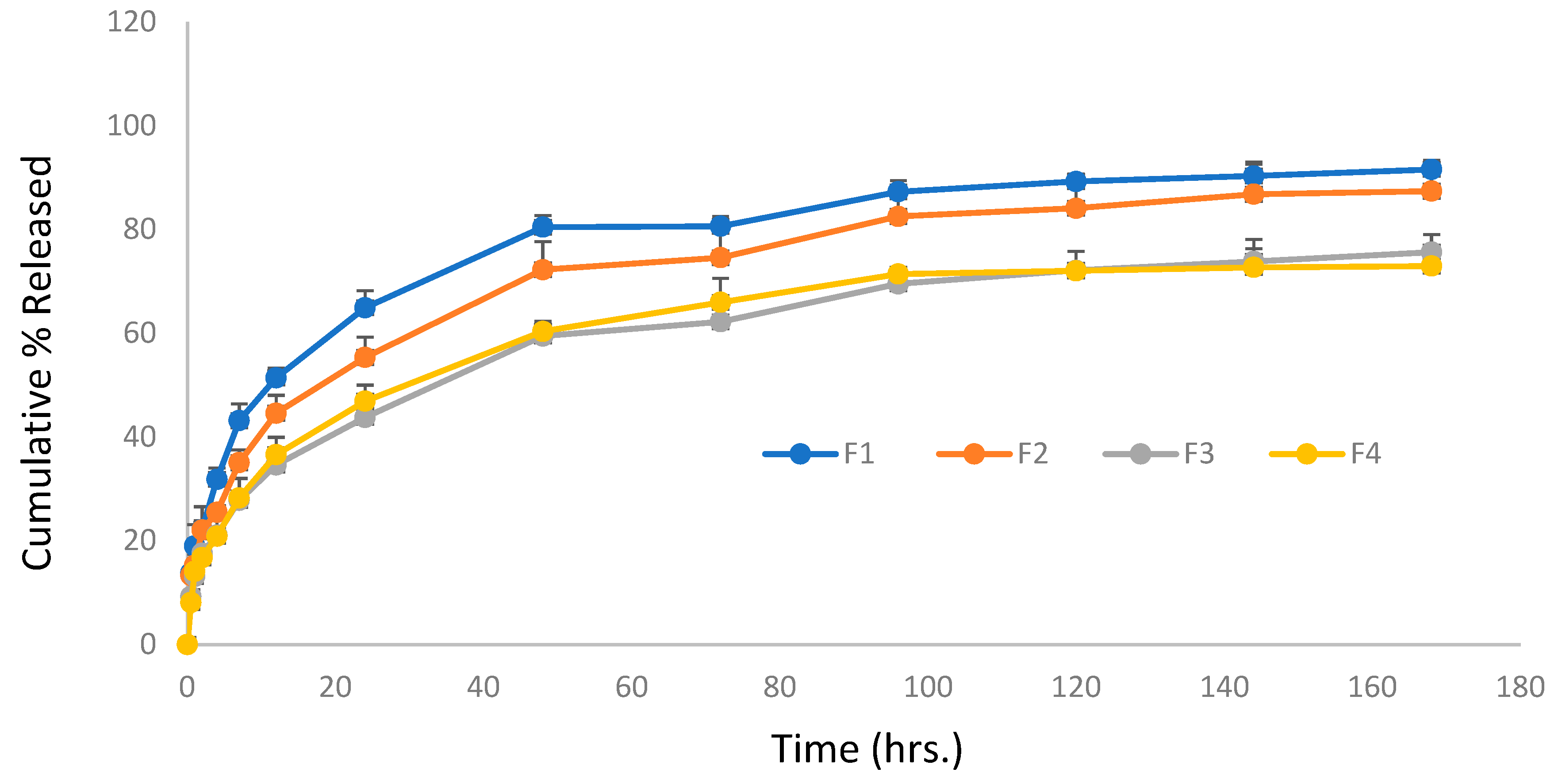
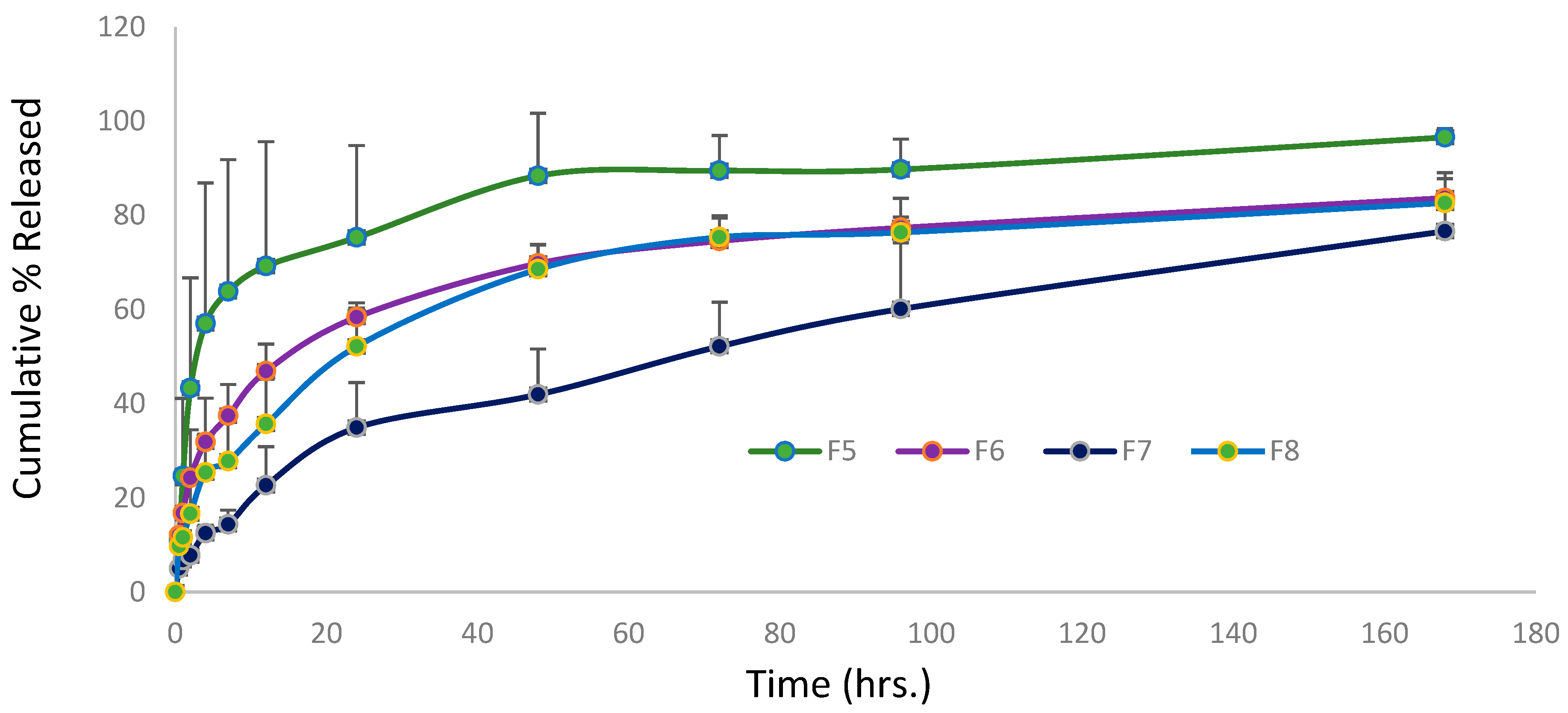
Figure 5 illustrates the val-release profile from formulations F5 to F8 over a 7-day period. This result shows that the F5-F8 formulations had controlled release at different rates. After 24 hours, the cumulative val% released was 75% for F5, 58% for F6, 52% for F8, and only 34% for
F7 formulation. According to the findings of this study, a lower drug-to-surfactant ratio slowed drug release from mixed micelle formulations. F5 had the fastest val-release among every formulation during the first 12 hours, making it suitable for per-oral controlled release because it allows for a gradual release of around 70% of Val within 12 hours, enabling a once-daily administration frequency with the maximum fraction of the dose absorbed. During the first 48 hours, F6 showed a faster rate of val release than F8, but the rate of release from both formulations nearly coincided until the end of the study. This is a consequence of the similar surfactant/co-surfactant composition of both formulations and the comparable quantity of drug remaining in both formulations after 48 hours. This pattern of resemblance was not observed in formulations containing Span 80 (F5 and F7). This can be explained by the higher hydrophilic nature of SDC-containing formulations when compared to Span 80-containing formulations. The F7 formulation demonstrated an almost constant rate of release, reaching a total of 42% after 2 days and gradually increasing to 76% after 7 days. This slow profile may be advantageous for a variety of delivery systems, including transdermal, long acting parenteral, and implantable systems. For the first time, our study reports an extremely slow drug release profile for polymeric or small molecule micellar systems. In a recent study, curcumin was combined with cholesterol [46] in an optimal niosome composed of a 7:3 ratio of Span 80: Tween 80. Within 24 hours, 75% of the curcumin had been released, according to the researchers. Aboud et al. [47] investigated val release from mixed micelles composed of Pluronic F127 and Tween 80 in varying ratios for 12 hours. They reported that the cumulative % of drug released from 9 formulations ranged from 25 to 60%.
When comparing the val release profiles of formulations with the same exact composition prepared by different methods, such as F1 and F6, F3 and F7, and F4 and F8, it is also notable that there is very good similarity in the release rate for each pair, indicating that the preparation method has no impact and that both methods are suitable for drug incorporation into micelles.
Figure 5.
Cumulative percentage (%) release of Valsartan from four mixed micelles formulations, prepared by thin film hydration, method against time.
Figure 5.
Cumulative percentage (%) release of Valsartan from four mixed micelles formulations, prepared by thin film hydration, method against time.
The prepared micelles also demonstrated very good physical stability, as they remained integral while maintaining the slow drug release profile at 37°C and continuous shaking for 7 days. Similarly, self-assembled val-loaded polymeric micelles made of poly(D,L-lactide-co-glycolide)-poly(ethylene glycol) block copolymers revealed 9-day continuous val release with an average burst release of 20% [48]. Goo et al. [49] found that incorporating val into a solid self-dispersing micelle composed primarily of Tween 80 and Gelucire 44 increased its oral bioavailability in rats by more than twice.
The kinetics of val release from all micelle formulations were studied by fitting to zero order, Higuchi, and Hixon-Crowell equations, as well as determining the Peppas-Korsmeyer (n), and the results are shown in Table 3. The r2 values clearly indicated their best fit to the Higuchi equation. Korsmeyer et al. [50] and Peppas [51] proposed that n = 0.45 indicates Fickian diffusion, n = 0.46 to 0.88 indicates non-Fickian (anomalous) diffusion, n = 0.89 indicates case-II transport (erosion control and zero-order kinetics), and n = 0.90 indicates case-III transport (erosion control and zero-order kinetics). The calculated n values for all formulations were found to be between 0.385 and 0.442. The Fickian release kinetic model was suggested for the release of Val from the prepared mixed micells formulations based on the aforementioned criteria. This is consistent with several reports in the literature [52,53]. The absence of any burst release and the slow rate of drug diffussion that decreases with time were observed in all formulations to varying degrees. This demonstrates val incorporation in the micelle core and release dependence on concentration gradient.
Table 3.
The kinetic parameters of Val release as fitted by various model equations.
| Formulation code | Zero-order | Higushi- Diffusion |
Hixson-Crowell | Peppas-Korsmeyer exponent (n) |
| F1 | 0.947 | 0.993 | 0.970 | 0.409 |
| F2 | 0.948 | 0.995 | 0.977 | 0.385 |
| F3 | 0.935 | 0.990 | 0.956 | 0.399 |
| F4 | 0.945 | 0.994 | 0.968 | 0.423 |
| F5 | 0.854 | 0.935 | 0.894 | 0.418 |
| F6 | 0.937 | 0.990 | 0.961 | 0.407 |
| F7 | 0.965 | 0.996 | 0.980 | 0.524 |
| F8 | 0.962 | 0.997 | 0.984 | 0.438 |
2.2.4. In vitro skin permeation studies
In vitro skin permeation profile of valsartan from different micelles and control is shown in Figure 6. The skin permeation profile of valsartan from F7 composed of Tween 80/Span 80 micelles was obtained as significant compared to control micelles (P < 0.01).
Figure 6.
In vitro skin permeation profile of valsartan via rat abdominal skin from micelles and aqueous suspension of valsartan (control).
Figure 6.
In vitro skin permeation profile of valsartan via rat abdominal skin from micelles and aqueous suspension of valsartan (control).
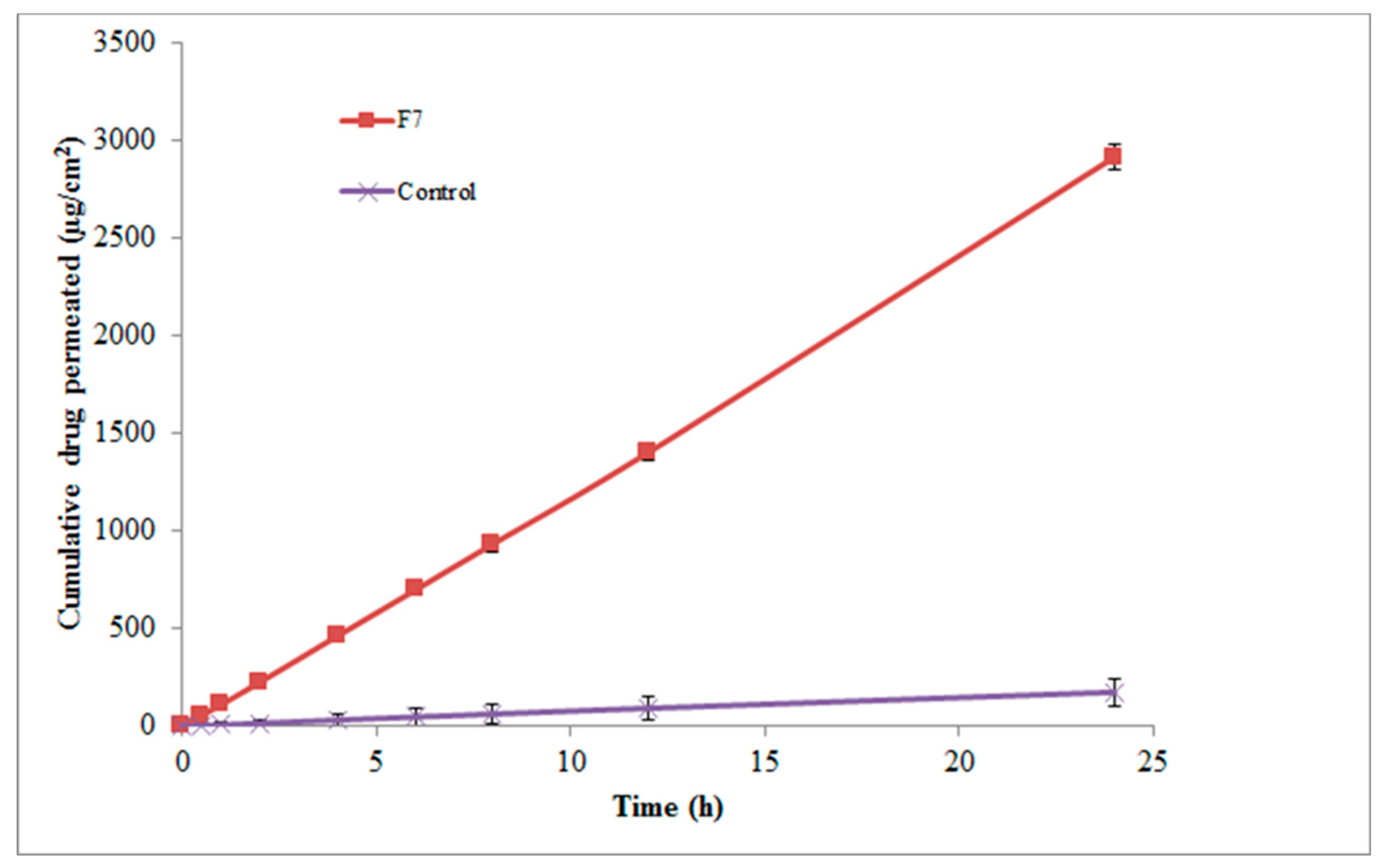
In this study, three distinct permeability parameters were predicted from permeation graphs plotted between cumulative valsartan permeated (g/cm2) and time (h) (Figure 6). These parameters were the rate of drug permeation via rat skin at steady state (Jss), the permeability coefficient (Kp), and the enhancement factor (Ef). Table 4 shows the Jss, Kp, and Ef values for the F7 micellar formulation and the control. F7 exhibited 16.57 Ef as well as Jss and Kp values equal to 68.84 ± 3.96 µg/cm2/h and 13.76± 0.064 × 10−3 cm/h, respectively, which were found to be significant when compared to the control (P < 0.01).
Table 4.
Permeability parameters of micelles and control.
| Formulation | JSS (µg/cm2/h)a | Kp (cm/h)a ×10-3 | Ef |
| Controlb | 4.15 ± 0.37 | 0.83 ± 0.016 | - |
| F7 (Span/Tween micelles) | 68.84 ± 3.96 | 13.76 ± 0.064 | 16.57 |
aMean ± SD, n = 3, baqueous suspension of valsartan was used as control.
Our results were nearly four times higher than the enhancement ratio reported by Ahad et al. [5]. They found that a 15% ethanol carbopol gel formulation produced a 4.53 enhancement factor for val through rat skin. Their reported val transdermal efflux was 143.51 g/cm2/h, which is nearly twice our formulation's value. In another study, an optimized val-ethosome formula was tested ex vivo via rat abdominal skin and its antihypertensive effect was tested in vivo in Wistar rats. They discovered a significant increase in transdermal efflux, which was supported by a longer duration of blood pressure lowering action when compared to orally administered Val. [54]. Similarly, Ahad et al. [55] used the Box-Behnken experimental design to develop an optimized val ethosome formula containing 35% ethanol. They demonstrated an extremely high efflux through rat skin (801.36 ± 21.45 μg/cm2/h). In another study, the use of iso-eucalyptol has shown to enhance val skin penetration by a ratio of 7.4 and 3.6 via rat and human cadaver skin, respectively [56].
3. Discussion
The incorporation of Val into mixed micelles was intended to overcome the low bioavailability and allow the transdermal delivery of the drug to provide more uniform drug levels for prolonged period that consequently enhance the therapeutic outcomes. Micellar based systems have multiple advantages as a delivery moiety for drugs through the skin. This include their ability to emulsify wide range of lipophilic and hydrophilic drugs in hydro-dynamically stable nano-vesicles and their ability to modify the release rate of drugs in addition to the skin enhancing property of surfactants [57,58,59].
Mixed micelles have been successfully employed to enhance the transdermal delivery of many drugs including indirubin, arbutin, and diltiazem [60,61,62]. Seo et al. introduced a mixed micellar system composed of a combination of Kolliphor® EL, Tween 80, with PEG 400 as co-surfactant. They showed the effectiveness of their system in the transdermal delivery of an indirubin analog, KY19382 [60]. The dermal delivery of drugs for psoriasis treatment has been approached effectively via incorporation into mixed micelles [63,64]. Lapteva et al. [63] developed a polymeric micelle nano-system composed of a polylactide- methoxy-poly(ethylene glycol)-dihexyl co-polymer for the dermal delivery of tacrolimus for the treatment of psoriasis. The selective accumulation of micelles into hair follicles was visualized by confocal laser scanning microscopy images. In another study, resveratrol-loaded polymeric micelles was in vivo evaluated in psoriatic-like plaque mice model in a form of a gel and showed significant activity [64].
The selection of Tween 80 as the main surfactant together with either Span 80 or SDC as co-surfactant was based on their high biosafety, biodegradability and biocompatibility compared with large molecular weight polymeric surfactants, in addition to their wide spread use in food and pharmaceutical products [65,66]. They are approved by the US Food and Drug Administration for use in up to 1% in selected foods [67].
The formulations were divided to compare the efficiency of the thin-film hydration with the micro-phase separation methods for mixed micelles preparation. The results indicated that both methods are applicable however, thin-film hydration method showed more uniform particle size distribution and higher drug EE% while thee drug release was similar for both methods. This is in compliance with other reports that attribute the wide spread use of the thin-film method for its better applicability and higher drug entrapment efficiency, as well as minimal organic solvent residual traces [68,69].
The variation of drug release profile was dependent on the composition and allows for variable applications through different routes of administration. Regardless of their vesicular nature, our prepared micelles have shown a robust integrity and stability indicating from their ability to control the release of Val for a period of one week withstanding continuous shaking, multiple dilution and elevated temperature. Their resistance to depletion by the high energy (10 KV) employed for the development of SEM at high magnification (43,000x) is another evidence of their high stability. Such advantage is more common for cross-linked polymeric micelles. Xiong et al. [70] developed a novel mixed micelles composed of two co-polymers containing PCL cores that shown to have high integrity at very low concentrations and prolonged drug release for more than a week in addition to their stimulus triggered targeting ability. Stability is a main concern for the success of any micellar drug delivery system [71,72].
Beside the high integrity and robust properties, our prepared micelles showed a number of interesting attributes including low molecular size in the range of 100 nm, high entrapment efficiency, the uniform transdermal release rate over one day, and being entirely composed of safe materials.
4. Materials and Methods
4.1. Materials
Valsartan was kindly obtained as a gift from Riyadh Pharma Company (Riyadh, Saudi Arabia). Different kinds of surfactant such as sodium deoxycholate, Tween 80, and and Span 80 were purchased from Sigma–Aldrich Chemical Co. (St Louis, MO, USA). All other reagents and chemicals used were of either HPLC or analytical grade.
4.2. Preparation of Valsartan-loaded mixed micelles:
For the preparation of Val-loaded mixed micelles, two methods were employed; the micro-phase separation and the thin-film hydration.
4.2.1. The micro-phase separation method:
As previously described by Hu and his colleagues [73]. To summarize, Val and the surfactant mixture were dissolved in dichloromethane to form a true solution. The solution was then added drop by drop to an excess volume of distilled water while being stirred. The organic solvent was completely evaporated after three hours of stirring. To remove any unentrapped drug, the formed micellar dispersion was filtered through a 0.2 m membrane filter.
4.2.2. The thin-film hydration method:
Chen and his colleagues' [37] procedures were followed. In summary, dichloromethane was used to dissolve Val as well as the surfactants mixture. The solution was dried to form a thin film using a IKA rota-evaporator RV 10 V-C system (IKA-Werke GmbH & Co., Staufen, Germany) at 40°C + 0.5 and 100 rpm under a reduced 40 mbar vacuum pressure. The organic solvent was completely removed from the thin film by vacuuming it overnight. The dried film was hydrated with 50 mL of de-ionized water pre-heated to 40°C and stirred for 45 minutes at 40°C to form micellar drug dispersion. The dispersion was filtered through a 0.2 m membrane filter to remove any excess drug.
4.3. Particle size and zeta-potential:
Samples from each batch were diluted using distilled water to produce micellar concentration of ~ 0.1% before processing in a Brookhaven ZetaPALS (Brookhaven Instruments Corporation, Holtsville, NY, USA) to measure the mean particle size and polydispersity index of the size distribution. A 90° angle of detection was used for all measurements. The same instrument used to determine particle’s size were utilized for zeta-potential measurement by applying the Laser Doppler Velocimetry (LDV) mode on samples with the same concentration range at 25°C.
4.4. Drug entrapment efficiency and drug loading:
Val-loaded mixed micelles samples were filtered using 0.2 µm membrane filters and then diluted in the methanol. This process was repeated and drug concentration was determined using HPLC by the method previously described by Albekairy and colleagues [74]. Drug entrapment efficiency (%EE) was determined according to the Eq. 1.
The HPLC system consisted of Agilent 1200 series equipped with Photodiode Array Detector of 1260 series (Agilent, CA, USA). The separation and quantitative determination have been conducted utilizing an Eclipsed XBD column (Agilent -PN 993967) C18, 150 mm x 3.0 mm i.d. with particle size of 5 μm. The mobile phase was composed of 46 parts of phosphate buffer (pH 3.6 and 0.01 M), 44 parts of acetonitrile, 10 parts of methanol. Injection volume was adjusted to 20 μL and a constant flow-rate of 1 mL min-1 at an ambient temperature (25°C) was maintained along the analysis. Val peaks were detected at λ = 265 nm. The system integrated software Mass Hunter® was used to automatically calculate the peak areas. Val was eluted at 3.52 min.
4.5. Particles Morphology:
The morphological features of particles were examined by both Transmitted and scanning electron microscopy.
4.5.1. Transmission Electron Microscopy:
Transmission electron microscope (TEM) measurements were performed using a (JEM-1400 electron microscope; JEOL, Tokyo, Japan) operating at an acceleration voltage of 120 kV. Few drop of F3 formulation was placed on a 400-mesh carbon-coated copper grid. The samples were air dried at room temperature prior to measurement.
4.5.2. Scanning Electron Microscopy:
Scanning electron microscopy was used to examine the particle surface characteristics of the Val-loaded NLC (JSM-6360 LV, JEOL, Tokyo, Japan). Few drops from formulation F7 were mounted on carbon tape and sputter-coated with a thin gold layer in a high-vacuum evaporator using a gold sputter module (JFC-1100 fine coat ion sputter; JEOL). For scanning and producing photomicrographs of the coated samples, a 10KV acceleration voltage was used.
4.5.3. In-vitro release profile study:
The % of Val released from each mixed micelle formulation were determined by placing certain amount of a micellar dispersed in 1 ml of phosphate buffer pH 7 inside a dialysis tube (12 KDa cut- off) firmly tied from one end. Dialysis tube was closed then immersed in a vessel containing 50 ml of the same media and placed in a shaking water bath adjusted to 37±1°C and 80 rpm. 1 ml of each sample was withdrawn at pre-determined time intervals and replaced by fresh pre-heated medium to maintain sink condition. The released percentage of Val was determined in each sample using the same HPLC method.
4.5.4. Release kinetic analysis
The Val-release data were fitted to zero-order model (equation 2), Higushi diffusion model (equation 3), Hixson-Crowell model (equation 4) and Peppas and Korsmeyer model (equation 5).
where Q is the cumulative % Val released at time t; kz is the zero-order release rate constant, kh is the Higuchi diffusion release rate constant, Khc is the Hixson-Crowell release rate constant, Mt/M∞ is the fraction of drug released till time (t), and kp is the Peppas and Korsmeyer release rate constant. The exponent (n) in equation (5) is the slope of the line obtained by plotting log Mt/M∞ (up to 0.6) against log time t.
Q = Kz t
Q = kh t1/2
(100 –Q)1/3 = 1001/3 – Khc t
Mt/M∞ = kp tn
4.3. In vitro skin permeation studies
In vitro skin permeation profile of valsartan from different micelles in comparison to valsartan suspension (control) was studied using a Franz Diffusion Cell (FDC). The surface area and volume of FDC were 1.76 cm2 and 12 ml, respectively. The rat abdominal skin was utilized as a permeation membrane. Logan Transdermal Apparatus (SFDC6, Logan Instrument Corporation, Avalon, NJ, USA) was used to assess the skin permeation profile of valsartan. The skin was excised from the abdominal region of the rat and hair were removed using an electric clipper. The skin was prepared and stored as per the instructions specified in the literature [75,76]. On the day of experiment, the skin was mounted between the donor and receiver compartment of FDC and procedure was followed as reported in literature [75,76,77]. Initially, the donor compartment was kept empty and receiver compartment was filled with freshly prepared 12 ml of phosphate buffer (pH 7). The magnetic bar was included in FDC. The whole FDC assembly was placed in Logan Transdermal Apparatus. The receiver compartment fluid was rotated at 100 rpm and the temperature was fixed to 37 ± 0.5 ◦C using thermostat. The whole buffer was replaced at regular time interval of 30 minutes in order to stabilize the rat skin. It was found that the fluid in receiver compartment showed negligible HPLC response after 6 hours and beyond this time, indicating the complete stabilization of the skin. After stabilization of the skin, 1 ml of poloxamer micelles, Span/Tween micelles and control (each containing 5 mg of valsartan) was placed into each donor compartment. The donor compartment of each cell was sealed with paraffin film to provide occlusive environment. Aqueous suspension of the valsartan was used as the control for the determination of enhancement factor (Ef). Approximately 0.5 ml of aliquots from each formulation was carefully withdrawn and replaced with freshly produced phosphate buffer at regular intervals of 0, 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h, filtered using 0.45 µm membrane filter and analyzed for valsartan content using the same HPLC method.
Permeation data analysis:
The cumulative amount of valsartan permeated via rat skin (µg/cm2) was graphed as a function of time (h) for different micelles and control. The rate of drug permeation via rat skin at steady state (Jss) was determined by dividing the slope of the linear portion of the graph by the area of FDC. The values of permeability coefficient (Kp) and Ef were determined using equations (6) and (7), respectively [78,79]:
In which, C0 is the initial concentration of valsartan in the donor compartment.
5. Conclusions
Val-loaded mixed micelles were successfully prepared using two simple methods that were highly reproducible. The developed micelles have low micellar sizes with narrow size distribution, high drug entrapment efficiencies, high stability, and prolonged uniform release control with variable rates that can be tuned for multiple routes of administration. Without the use of any skin-modifying enhancers, the best formulation demonstrated extensive transdermal drug delivery through rat skin at a uniform slow rate for 24 hours. The prepared system is suitable for a wide range of drugs, particularly those in BCS classes II and IV.
Author Contributions
Conceptualization, Alaa Yassin (A.Y.) and Majed Halwani (M.H.); methodology, A.Y., Abdullah Alshwaimi (A.A.), Faiyaz Shakeel (F.S.), Raslan Kittaneh (R.K.), and Mostafa Omer (M.O.); software, Salam Massadeh (S.M.), and M.H.; validation, F.S., D.A., and Al Hassan Aodah (A.O.); formal analysis, Dilshad Ahmad (D.A.), M.O., A.O., and Prawez Alam (P.A.) ; investigation, A.A., S.M., R.K., and Saleh Alanazi (S.A.); resources, S.M., A.O., and A.Y.; R.K., A.A., and M.O.; writing—original draft preparation, A.Y., S.M., and F.S.; writing—review and editing, A.Y., and S.M.; visualization, A.A., R.K., P.A.; supervision, S.M., S.A., R.K., and A.Y.; project administration, R.K., S.A., and M.O.; funding acquisition, A.Y., M.O., and M.H.
Funding
This work was funded by a grant from KING ABDULLAH INTERNATIONAL RESEARCH CENTER (KAIMRC), National Guard Health Affairs, Riyadh, Saudi Arabia (Grant No. SP16/192/R). The funding agency had no role in the decision to publish or prepare the manuscript.
Institutional Review Board Statement
Not Applicable
Informed Consent Statement
Not Applicable
Data Availability Statement
Not Applicable
Acknowledgments
The Authors would like to thank the administration of College of Pharmacy, King Saud bin Abdulaziz University for Health Science for creating an excellent work environment for research and innovation.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- ClinCal.com. The top 200 of 2020, Version 2022.08. Available online: https://clincalc.com/DrugStats/Top200Drugs.aspx (accessed on 23 October 2023).
- Dixit, A.R.; Rajput, S.J.; Patel, S.G. Preparation and bioavailability assessment of SMEDDS containing valsartan. AAPS PharmSciTech 2010, 11, 314-321. [CrossRef]
- Ahad, A.; Aqil, M.; Kohli, K.; Sultana, Y.; Mujeeb, M. Ali, A. Role of novel terpenes in transcutaneous permeation of valsartan: effectiveness and mechanism of action. Drug Dev Ind Pharm 2011, 37:5, 583-596. [CrossRef]
- Aqil, M.; Sultana, Y.; Ali, A. Matrix type transdermal drug delivery systems of metoprolol tartrate: in vitro characterization. Acta Pharm 2003, 53, 119-125.
- Ahad, A.; Aqil, M.; Ali, A. Investigation of antihypertensive activity of carbopol valsartan transdermal gel containing 1,8-cineole. Int J Biol Macromol 2014, 64, 144-149. [CrossRef]
- Nishida, N.; Taniyama, K.; Sawabe, T.; Manome, Y. Development and evaluation of a monolithic drug-in-adhesive patch for valsartan. Int J Pharm 2010, 402, 103-109. [CrossRef]
- Ahad, A.; Aqil, M.; Ali, A. The application of anethole, menthone, and eugenol in transdermal penetration of valsartan: Enhancement and mechanistic investigation. Pharm Biol 2016, 54:6, 1042-1051. [CrossRef]
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv Drug Deliv Rev 2004, 56, 603-618. [CrossRef]
- Morteza-Semnani, K.; Saeedi, M.; Akbari, J.; Eghbali, M.; Babaei, A.; Hashemi, S.M.H.; Nokhodchi, A. Development of a novel nanoemulgel formulation containing cumin essential oil as skin permeation enhancer. Drug Deliv Transl Res 2022, 12, 1455-1465. [CrossRef]
- Thong, H.Y.; Zhai, H.; Maibach, H.I. Percutaneous penetration enhancers: an overview. Skin Pharmacol Physiol 2007, 20, 272-282. [CrossRef]
- Afouna, M.I.; Fincher, T.K.; Zaghloul, A.A.; Reddy, I.K. Effect of Azone upon the in vivo antiviral efficacy of cidofovir or acyclovir topical formulations in treatment/prevention of cutaneous HSV-1 infections and its correlation with skin target site free drug concentration in hairless mice. Int J Pharm 2003, 253, 159-168. [CrossRef]
- Herman, A.; Herman, A.P. Essential oils and their constituents as skin penetration enhancer for transdermal drug delivery: a review. J Pharm Pharmacol 2015, 67, 473-485. [CrossRef]
- Aungst, B.J. Structure/effect studies of fatty acid isomers as skin penetration enhancers and skin irritants. Pharm Res 1989, 6, 244-247. [CrossRef]
- Akbari, J.; Saeedi, M.; Farzin, D.; Morteza-Semnani, K.; Esmaili, Z. Transdermal absorption enhancing effect of the essential oil of Rosmarinus officinalis on percutaneous absorption of Na diclofenac from topical gel. Pharm Biol 2015, 53, 1442-1447. [CrossRef]
- Massadeh, S.; Omer, M. E.; Alterawi, A.; Ali, R.; Alanazi, F. H.; Almutairi, F.; Almotairi, W.; Alobaidi, F. F.; Alhelal, K.; Almutairi, M. S.; Almalik, A.; Obaidat, A.; Alaamery, M.; Yassin, A. E. Optimized Polyethylene Glycolylated Polymer-Lipid Hybrid Nanoparticles as a Potential Breast Cancer Treatment. Pharmaceutics 2020, 12(7), 666. [CrossRef]
- Singh S.; Pandey V.K.; Tewari R.P.; Agarwal V. Nanoparticle based drug delivery system: Advantages and applications. Ind J Sci Tech. 2011, 4, 177-184. [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.G.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res 2008, 14, 1310-1316. [CrossRef]
- Yassin A,; Alkatheri A.; Sharma R. Anticancer-loaded solid lipid nanoparticles: high potential advancement in chemotherapy. Dig. J. Nanomater. Bios. 2013, 8, 905-916.
- Husseini, G.A.; Pitt, W.G. Micelles and nanoparticles for ultrasonic drug and gene delivery. Adv Drug Deliv Rev 2008, 60, 1137-1152. [CrossRef]
- Mao, Y.; Chen, X.; Xu, B.; Shen, Y.; Ye, Z.; Chaurasiya, B.; Liu, L.; Li, Y.; Xing, X.; Chen, D. Eprinomectin nanoemulgel for transdermal delivery against endoparasites and ectoparasites: preparation, in vitro and in vivo evaluation. Drug Deliv 2019, 26, 1104-1114. [CrossRef]
- Virani, A.; Puri, V.; Mohd, H.; Michniak-Kohn, B. Effect of Penetration Enhancers on Transdermal Delivery of Oxcarbazepine, an Antiepileptic Drug Using Microemulsions. Pharmaceutics 2023, 15. [CrossRef]
- Liu, D.; Ge, Y.; Tang, Y.; Yuan, Y.; Zhang, Q.; Li, R.; Xu, Q. Solid lipid nanoparticles for transdermal delivery of diclofenac sodium: preparation, characterization and in vitro studies. J Microencapsul 2010, 27, 726-734. [CrossRef]
- Parra, A.; Jarak, I.; Santos, A.; Veiga, F.; Figueiras, A. Polymeric Micelles: A Promising Pathway for Dermal Drug Delivery. Materials (Basel) 2021, 14. [CrossRef]
- Muzzalupo R.; Tavano L. Niosomal drug delivery for transdermal targeting: recent advances. Res. rep. transdermal drug deliv. 2015, 4, 23-33. [CrossRef]
- Atanase, L.I. Micellar Drug Delivery Systems Based on Natural Biopolymers. Polymers (Basel) 2021, 13. [CrossRef]
- Hussein, Y.H.A.; Youssry, M. Polymeric Micelles of Biodegradable Diblock Copolymers: Enhanced Encapsulation of Hydrophobic Drugs. Materials (Basel) 2018, 11. [CrossRef]
- Atanase, L.I.; Riess, G. Self-Assembly of Block and Graft Copolymers in Organic Solvents: An Overview of Recent Advances. Polymers (Basel) 2018, 10. [CrossRef]
- Deng C.; Jiang Y.; Cheng R.; Meng F.; Zhong Z. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: Promises, progress and prospects. Nano. Today 2012, 7, 467-480.
- Rangel-Yagui, C.O.; Pessoa, A., Jr.; Tavares, L.C. Micellar solubilization of drugs. J Pharm Pharm Sci 2005, 8, 147-165.
- Mall, S.; Buckton, G.; Rawlins, D.A. Dissolution behaviour of sulphonamides into sodium dodecyl sulfate micelles: a thermodynamic approach. J Pharm Sci 1996, 85, 75-78. [CrossRef]
- Aliabadi, H.M.; Lavasanifar, A. Polymeric micelles for drug delivery. Expert Opin Drug Deliv 2006, 3, 139-162. [CrossRef]
- Gothwal, A.; Khan, I.; Gupta, U. Polymeric Micelles: Recent Advancements in the Delivery of Anticancer Drugs. Pharm Res 2016, 33, 18-39. [CrossRef]
- FitzGerald, P.A.; Chatjaroenporn, K.; Warr, G.G. Structure and composition of mixed micelles of polymerized and monomeric surfactants. J Colloid Interface Sci 2015, 449, 377-382. [CrossRef]
- Rupp, C.; Steckel, H.; Müller, B.W. Mixed micelle formation with phosphatidylcholines: the influence of surfactants with different molecule structures. Int J Pharm 2010, 387, 120-128. [CrossRef]
- Zhao, Y.; Li, J.; Yu, H.; Wang, G.; Liu, W. Synthesis and characterization of a novel polydepsipeptide contained tri-block copolymer (mPEG-PLLA-PMMD) as self-assembly micelle delivery system for paclitaxel. Int J Pharm 2012, 430, 282-291. [CrossRef]
- Yoo, H.S.; Park, T.G. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J Control Release 2001, 70, 63-70. [CrossRef]
- Chen, Y.; Zhang, W.; Gu, J.; Ren, Q.; Fan, Z.; Zhong, W.; Fang, X.; Sha, X. Enhanced antitumor efficacy by methotrexate conjugated Pluronic mixed micelles against KBv multidrug resistant cancer. Int J Pharm 2013, 452, 421-433. [CrossRef]
- Opanasopit, P.; Yokoyama, M.; Watanabe, M.; Kawano, K.; Maitani, Y.; Okano, T. Block copolymer design for camptothecin incorporation into polymeric micelles for passive tumor targeting. Pharm Res 2004, 21, 2001-2008. [CrossRef]
- Song, J.; Huang, H.; Xia, Z.; Wei, Y.; Yao, N.; Zhang, L.; Yan, H.; Jia, X.; Zhang, Z. TPGS/Phospholipids Mixed Micelles for Delivery of Icariside II to Multidrug-Resistant Breast Cancer. Integr Cancer Ther 2016, 15, 390-399. [CrossRef]
- Ould-Ouali, L.; Noppe, M.; Langlois, X.; Willems, B.; Te Riele, P.; Timmerman, P.; Brewster, M.E.; Ariën, A.; Préat, V. Self-assembling PEG-p(CL-co-TMC) copolymers for oral delivery of poorly water-soluble drugs: a case study with risperidone. J Control Release 2005, 102, 657-668. [CrossRef]
- Patil, S.; Choudhary, B.; Rathore, A.; Roy, K.; Mahadik, K. Enhanced oral bioavailability and anticancer activity of novel curcumin loaded mixed micelles in human lung cancer cells. Phytomedicine 2015, 22, 1103-1111. [CrossRef]
- Kashanian, S.; Azandaryani, A.H.; Derakhshandeh, K. New surface-modified solid lipid nanoparticles using N-glutaryl phosphatidylethanolamine as the outer shell. Int J Nanomedicine 2011, 6, 2393-2401. [CrossRef]
- Thatipamula, R.; Palem, C.; Gannu, R.; Mudragada, S.; Yamsani, M. Formulation and in vitro characterization of domperidone loaded solid lipid nanoparticles and nanostructured lipid carriers. Daru 2011, 19, 23-32.
- Verwey, E.J.W. Theory of the Stability of Lyophobic Colloids. J. Phys. Colloid Chem. 1947, 51, 631-636. [CrossRef]
- Ding, B.; Ahmadi, S.H.; Babak, P.; Bryant, S.L.; Kantzas, A. On the Stability of Pickering and Classical Nanoemulsions: Theory and Experiments. Langmuir 2023, 39, 6975-6991. [CrossRef]
- Ghumman, S.A.; Ijaz, A.; Noreen, S.; Aslam, A.; Kausar, R.; Irfan, A.; Latif, S.; Shazly, G.A.; Shah, P.A.; Rana, M.; et al. Formulation and Characterization of Curcumin Niosomes: Antioxidant and Cytotoxicity Studies. Pharmaceuticals 2023, 16, 1406.
- Aboud, H.M.; Mahmoud, M.O.; Abdeltawab Mohammed, M.; Shafiq Awad, M.; Sabry, D. Preparation and appraisal of self-assembled valsartan-loaded amalgamated Pluronic F127/Tween 80 polymeric micelles: Boosted cardioprotection via regulation of Mhrt/Nrf2 and Trx1 pathways in cisplatin-induced cardiotoxicity. J Drug Target 2020, 28, 282-299. [CrossRef]
- Zhu, Q.; Zhang, B.; Wang, Y.; Liu, X.; Li, W.; Su, F.; Li, S. Self-assembled micelles prepared from poly(D,L-lactide-co-glycolide)-poly(ethylene glycol) block copolymers for sustained release of valsartan. Polym. Adv. Technol. 2021, 32, 1262-1271. [CrossRef]
- Goo, Y.T.; Park, S.Y.; Chae, B.R.; Yoon, H.Y.; Kim, C.H.; Choi, J.Y.; Song, S.H.; Choi, Y.W. Optimization of solid self-dispersing micelle for enhancing dissolution and oral bioavailability of valsartan using Box-Behnken design. Int J Pharm 2020, 585, 119483. [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. International Journal of Pharmaceutics 1983, 15, 25-35. [CrossRef]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 1985, 60, 110-111.
- Wang, H.; Chen, H. Study of the drug release kinetics in nanoscale micelle to micelle system. In Proceedings of the 2010 3rd International Nanoelectronics Conference (INEC), 3-8 Jan. 2010, 2010; pp. 1331-1332.
- Tao, L.; Chan, J.W.; Uhrich, K.E. Drug loading and release kinetics in polymeric micelles: Comparing dynamic versus unimolecular sugar-based micelles for controlled release. Journal of Bioactive and Compatible Polymers 2016, 31, 227-241. [CrossRef]
- Bhosale, S.S.; Avachat, A.M. Design and development of ethosomal transdermal drug delivery system of valsartan with preclinical assessment in Wistar albino rats. J Liposome Res 2013, 23, 119-125. [CrossRef]
- Ahad, A.; Aqil, M.; Kohli, K.; Sultana, Y.; Mujeeb, M. Enhanced transdermal delivery of an anti-hypertensive agent via nanoethosomes: statistical optimization, characterization and pharmacokinetic assessment. Int J Pharm 2013, 443, 26-38. [CrossRef]
- Ahad, A.; Aqil, M.; Kohli, K.; Sultana, Y.; Mujeeb, M. Design, formulation and optimization of valsartan transdermal gel containing iso-eucalyptol as novel permeation enhancer: preclinical assessment of pharmacokinetics in Wistar albino rats. Expert Opin Drug Deliv 2014, 11, 1149-1162. [CrossRef]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-Microemulsifying Drug Delivery Systems: An Attractive Strategy for Enhanced Therapeutic Profile. Int Sch Res Notices 2014, 2014, 964051. [CrossRef]
- Alkilani, A.Z.; McCrudden, M.T.; Donnelly, R.F. Transdermal Drug Delivery: Innovative Pharmaceutical Developments Based on Disruption of the Barrier Properties of the stratum corneum. Pharmaceutics 2015, 7, 438-470. [CrossRef]
- Patel, P.V.; Patel, H.K.; Panchal, S.S.; Mehta, T.A. Self micro-emulsifying drug delivery system of tacrolimus: Formulation, in vitro evaluation and stability studies. Int J Pharm Investig 2013, 3, 95-104. [CrossRef]
- Seo, S.H.; Kim, E.; Joo, Y.; Lee, J.; Oh, K.T.; Hwang, S.J.; Choi, K.Y. A Mixed Micellar Formulation for the Transdermal Delivery of an Indirubin Analog. Pharmaceutics 2020, 12. [CrossRef]
- Liang, K.; Xu, K.; Bessarab, D.; Obaje, J.; Xu, C. Arbutin encapsulated micelles improved transdermal delivery and suppression of cellular melanin production. BMC Res Notes 2016, 9, 254. [CrossRef]
- Omray, K. Enhanced Transdermal Delivery of Diltiazem Hydrochloride Via Reverse Micellar Transformation Type Liquid Crystalline Gel. International Journal of Pharmaceutics and Drug Analysis 2014, 2, 225-228.
- Lapteva, M.; Mondon, K.; Möller, M.; Gurny, R.; Kalia, Y.N. Polymeric micelle nanocarriers for the cutaneous delivery of tacrolimus: a targeted approach for the treatment of psoriasis. Mol Pharm 2014, 11, 2989-3001. [CrossRef]
- Khurana, B.; Arora, D.; Narang, R.K. QbD based exploration of resveratrol loaded polymeric micelles based carbomer gel for topical treatment of plaque psoriasis: In vitro, ex vivo and in vivo studies. Journal of Drug Delivery Science and Technology 2020, 59, 101901. [CrossRef]
- Li, X.; Fan, R.; Wang, Y.; Wu, M.; Tong, A.; Shi, J.; Xiang, M.; Zhou, L.; Guo, G. In situ gel-forming dual drug delivery system for synergistic combination therapy of colorectal peritoneal carcinomatosis. RSC Advances 2015, 5, 101494-101506. [CrossRef]
- Mendonça, P.V.; Matos, A.; Sousa, A.F.; Serra, A.C.; Simões, S.; Coelho, J.F.J. Increasing the Bile Acid Sequestration Performance of Cationic Hydrogels by Using an Advanced/Controlled Polymerization Technique. Pharm Res 2017, 34, 1934-1943. [CrossRef]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92-96. [CrossRef]
- Ai, X.; Zhong, L.; Niu, H.; He, Z. Thin-film hydration preparation method and stability test of DOX-loaded disulfide-linked polyethylene glycol 5000-lysine-di-tocopherol succinate nanomicelles. Asian Journal of Pharmaceutical Sciences 2014, 9, 244-250. [CrossRef]
- El Zaafarany, G.M.; Awad, G.A.; Holayel, S.M.; Mortada, N.D. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm 2010, 397, 164-172. [CrossRef]
- Xiong, D.; Wen, L.; Peng, S.; Xu, J.; Zhang, L. Reversible Cross-Linked Mixed Micelles for pH Triggered Swelling and Redox Triggered Degradation for Enhanced and Controlled Drug Release. Pharmaceutics 2020, 12. [CrossRef]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res 2018, 11, 4985-4998. [CrossRef]
- Kim, S.; Shi, Y.; Kim, J.Y.; Park, K.; Cheng, J.-X. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle–cell interaction. Expert Opinion on Drug Delivery 2010, 7, 49-62. [CrossRef]
- Hu, K.; Li, J.; Shen, Y.; Lu, W.; Gao, X.; Zhang, Q.; Jiang, X. Lactoferrin-conjugated PEG-PLA nanoparticles with improved brain delivery: in vitro and in vivo evaluations. J Control Release 2009, 134, 55-61. [CrossRef]
- Albekery M.; Alharbi K.; Alarifi S.; Ahmad D.; Omer M.; Massadeh S.; Yassin A. Optimization of a nanostructured lipid carriers system for enhancing the biopharmaceutical properties of valsartan. Dig. J. Nanomater. Bios. 2017, 12, 381-389.
- Shakeel, F.; Baboota, S.; Ahuja, A.; Ali, J.; Aqil, M.; Shafiq, S. Nanoemulsions as vehicles for transdermal delivery of aceclofenac. AAPS PharmSciTech 2007, 8.
- Shakeel, F.; Haq, N.; Al-Dhfyan, A.; Alanazi, F.K.; Alsarra, I.A. Chemoprevention of skin cancer using low HLB surfactant nanoemulsion of 5-fluorouracil: a preliminary study. Drug Deliv 2015, 22, 573-580. [CrossRef]
- El Maghraby, G.M. Transdermal delivery of hydrocortisone from eucalyptus oil microemulsion: effects of cosurfactants. Int J Pharm 2008, 355, 285-292. [CrossRef]
- Shakeel, F.; Ramadan, W.; Ahmed, M.A. Investigation of true nanoemulsions for transdermal potential of indomethacin: characterization, rheological characteristics, and ex vivo skin permeation studies. J Drug Target 2009, 17, 435-441. [CrossRef]
- Shakeel, F.; Ramadan, W. Transdermal delivery of anticancer drug caffeine from water-in-oil nanoemulsions. Colloids Surf B Biointerfaces 2010, 75, 356-362. [CrossRef]
Table 1.
The composition and properties of each of the prepared valsartan mixed micelles formulation.
Table 1.
The composition and properties of each of the prepared valsartan mixed micelles formulation.
| Formulation | Val (mg) | Tween 80 (mg) |
Span 80 (mg) |
Sodium dioxy cholate (mg) | Method |
| F1 | 50 | 2000 | --- | 500 | Microphase separation (probe sonication) |
| F2 | 50 | 2000 | 250 | ---- | |
| F3 | 25 | 2000 | 500 | ---- | |
| F4 | 25 | 2000 | --- | 500 | |
| F5 | 50 | 2000 | 500 | --- | Thin-Film Hydration |
| F6 | 50 | 2000 | --- | 500 | |
| F7 | 25 | 2000 | 500 | --- | |
| F8 | 25 | 2000 | ---- | 500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



