
Submitted:
28 October 2023
Posted:
31 October 2023
You are already at the latest version
Abstract
Viral infection activates the transcription factors IRF3 and NF-κB, which synergistically induce type I interferons (IFNs). Here, we identify the E3 ubiquitin ligase RNF138 as an important negative regulator of virus-triggered IRF3 activation and IFN-β induction. Overexpression of RNF138 inhibited virus-induced activation of IRF3 and transcription of the IFNB1 gene, whereas knockout of RNF138 promoted virus-induced activation of IRF3, and transcription of the IFNB1 gene. We further found that RNF138 promotes ubiquitination of PTEN and subsequently inhibits PTEN interactions with IRF3, which is essential for PTEN-mediated nuclear translocation of IRF3, thereby inhibiting IRF3 import into the nucleus. Our findings suggest that RNF138 negatively regulates virus-triggered signaling by inhibiting the interaction of PTEN with IRF3, and these data provide new insights into the molecular mechanisms of cellular antiviral responses.
Keywords:
Antiviral natural immunity
; RNF138
; IRF3
; PTEN
1. Introduction
The innate immune system is the host’s first line of defense against infection by pathogenic microorganisms[1]. Following viral infection, host pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) and activate a series of downstream signaling cascades that induce the production of type I interferons and pro-inflammatory cytokines[1,2,3]. Double-stranded RNA (dsRNA) or single-stranded RNA (ssRNA) generated during viral infection and replication are classical PAMPs that are detected by various PRRs including RIG-I-like receptors (RIG-I and MDA5) and Toll-like receptors (TLR3)[4,5], which then activates the adaptor protein MAVS (also known as IPS-1, VISA, and Cardif) and TRAF3, and then leads to phosphorylation of the downstream kinase TBK1, which promotes the activation of the transcription factors IRF3 and NF-κB, and thus induces the transcription and expression of downstream antiviral genes[6,7,8,9,10,11,12,13].
IRF3, the most critical transcription factor in the IFN-β induction pathway, is important in the antiviral innate immune response process[14,15]. Upon recognition of the pathogen by the cell, IRF3 is first phosphorylated and then undergoes a conformational change. IRF3 forms a homodimerization and undergoes nuclear translocation to bind to the promoter sequence of the target gene, the interferon-stimulated response element (ISRE), and induces antiviral gene expression[16,17]. IRF3 activation is finely regulated by post-translational modifications (PTMs) as it undergoes phosphorylation, dimerization, and nuclear translocation[18]. IRF3 is stably and continuously expressed in various cells, and in the resting state, IRF3 is located in the cytoplasm as a non-activated monomer. When the virus infects the cell, it causes the C-terminus of IRF3 to be phosphorylated, and the conformation changes to form a homodimer[19,20,21]. In addition, IRF3 acts as a pro-apoptotic factor in the late stage of viral infection, triggering the RLR-induced IRF-3-mediated pathway of apoptosis (RIPA) to eliminate infected cells and invading microorganisms[22]. The important role of IRF3 in the induction of the gene encoding IFN-β suggests that it must be precisely regulated to determine the appropriate immune response to invading viruses[19,23,24,25,26].
Nuclear translocation of IRF3 is tightly controlled by the nuclear export sequence (NES) and NLS[27], which are essential for its activation. Recent studies have reported that USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating input protein KPNA2[28]. In addition, the Ser97 phosphorylation site of IRF3 is crucial for its entry into the nucleus because our previous results revealed that PTEN exercises a dephosphorylation function on the Ser97 phosphorylation site of IRF3 through its phosphatase activity, thus promoting IRF3 import into the nucleus and activating the expression of type I interferons[29]. However, regulating nuclear translocation of IRF3 is a complex and dynamic process, and whether other negative regulators are involved in PTEN-mediated nuclear translocation of IRF3 still requires in-depth study.
RNF138 acts as a ubiquitin-E3 ligase, promotes cell survival by counteracting apoptotic signals as well as directly engaging in apoptosis, and is also involved in DNA damage response, tumourigenesis, neurodegenerative diseases, and chronic inflammatory processes[30,31,32,33,34,35,36]. However, it has been less studied in antiviral innate immunity. It was shown that the African swine fever virus pI215L negatively regulates the cGAS-STING signaling pathway by recruiting RNF138 to inhibit K63 ubiquitination of TBK1[37]. Recent studies have shown that the nuclear E3 ubiquitin ligase RNF138, a negative regulator in the inflammatory innate response, represses LPS-triggered transcription of late inflammatory genes by degrading SMARCC1 of the SWI/SNF complex[38]. However, whether RNF138 inhibits virus-triggered IRF3 activation and IFN-β induction is unclear.
This study found that RNF138 plays an important cytoplasmic role in virus-triggered IRF3 nuclear translocation and cellular antiviral responses. Further studies showed that RNF138 interacts with IRF3 and PTEN. RNF138 inhibits the interaction between PTEN and IRF3 by ubiquitinating PTEN after viral infection, which inhibits PTEN-mediated IRF3 nuclear translocation, thereby inhibiting virus-triggered IRF3 nuclear translocation and subsequent expression of downstream genes. Collectively, these findings define a previously unknown function of cytoplasmic RNF138 in antiviral innate immunity and establish a mechanistic link between the triad of RNF138, PTEN, and IRF3 nuclear translocation, thus providing new insights into the molecular mechanisms by which viral infection triggers IRF3 nuclear translocation.
2. Results
2.1. RNF138 negatively regulates viral RNA-triggered signaling
The E3 ubiquitin ligase RNF138 is a negative regulator in inflammatory responses, suppressing LPS-triggered transcription of late inflammatory genes by degradation of SMARCC1 of the SWI/SNF complex [38]. However, whether RNF138 plays a role in antiviral innate immunity is unknown. To investigate whether cytoplasmic RNF138 is also involved in antiviral innate immune signaling, we first explored the effect of RNF138 on SeV (Sendai Virus)-induced reporter gene activation. The reporter assays indicated that overexpression of RNF138 in HEK293T cells inhibited SeV-induced activation of the IFNβ, ISRE, and NF-κB promoters (Figure 1A). Quantitative PCR (qPCR) experiments indicated that overexpression of RNF138 inhibited SeV-induced transcription of downstream genes, including IFNB1 CXCL10 (Figure 1B). According to the literature, type I interferon induction requires activation of the transcription factor IRF3[39]. Overexpression of RNF138 inhibited SeV-induced phosphorylation and nuclear translocation of IRF3 (Figure 1C-D). These results suggest that RNF138 inhibits virus-triggered IRF3 activation and IFNB1 gene transcription.
2.2. knockout of RNF138 potentiates virus-induced IRF3 activation and IFNB1 transcription
Next, to explore whether endogenous RNF138 is required for virus-induced innate immunity signaling (in particular virus-triggered IRF3 activation) under physiological conditions, we generated the RNF138 knockout HEK293T cells by CRISPR-Cas9 and three RNF138 shRNA expression plasmids (#1, #2 and #3) which target different sites in RNF138 mRNA. The immunoblotting results showed that RNF138-KO#1 was more effective in knocking out RNF138 at the protein level (Figure 2A upper panel), which was used for all of the following experiments, whereas shRNF138#1 and #2 were effective in downregulating RNF138 expression at the protein level (Figure 2A bottom panel). Next, we also observed the effect of shRNF138 on SeV-induced ISRE activation. Reporter assays indicated that RNF138 knockdown enhanced SeV-induced ISRE activation in 293T cells compared to controls (Figure 2B).
To further assess the effect of RNF138 knockout on the expression of endogenous genes of IFNB1 and ISGs, RNF138 knockout HEK293T cells were infected with SeV. qPCR experiments indicated that mRNA level of IFNB1, CXCL10, ISG15, and IL-6 were significantly higher in RNF138 knockout HEK293T cells in SeV infection (Figure 2C). Further, we found that RNF138 knockdown significantly enhanced SeV-induced IRF3 phosphorylation, dimerization, and nuclear translocation (Figure 2D-F). These results suggest that RNF138 is a negative regulator of virus-triggered IRF3 activation and IFNβ transcription.
We also generated RNF138 knockout RAW264.7 and L929 cells by CRISPR-Cas9. qPCR experiments indicated that mRNA levels of IFNB1 and IL-6 were significantly higher in RNF138 knockout RAW264.7 cells in SeV or VSV (Vesicular Stomatitis Virus) infection (Figure 3A). Consistent with the gene induction assays, SeV or VSV-induced IRF3 phosphorylation was enhanced in RNF138 knockout RAW264.7 and L929 cells (Figure 3B-C). Thus, RNF138 is important in virus-triggered IRF3 activation in mouse cells.
2.3. RNF138 negatively regulates virus-triggered signaling by Targeting IRF3
As mentioned above, overexpression and endogenous experiments indicate that RNF138 acts as a negative regulator of innate immune signaling for virus-triggered IRF3 activation. Next, to find the level at which RNF138 regulates the virus-induced activation of the IRF3 activation pathway. HEK293T cells were co-transfected with plasmids expressing RNF138 and the other signal components. RNF138 inhibited ISRE activation mediated by all upstream activators (RIG-1, MAVS, TBK1, IRF3), including the constitutively active phosphorylation mimetic IRF3-5D (Figure 4A). The result suggests that RNF138 functions at the level of IRFThen, we checked the interaction between RNF138 and IRFCo-immunoprecipitation experiments showed that RNF138 was associated with IRF3 (Figure 4B-C). Importantly, RNF138 binds to IRF3 under physiological conditions, while the interaction between RNF138 and IRF3 is enhanced in the early stage of SeV infection but weakened in the late stage of infection (Figure 4D). These results indicated that RNF138 may act as a virus-triggered innate immunity signal at the level of IRF3.
RNF138 contains a domain located at the N-terminal end (RING), a ZNF domain in the middle, and a C-terminal domain (UIM), and IRF3 contains a DBD, TAD transcriptional activation, and RD nuclear response domain[29,30]. To determine which domain of RNF138 interacts with IRF3, the different domain mutants of RNF138 or IRF3 were constructed (Figure 4E-F). Co-immunoprecipitation experiments showed that the RING domain of RNF138 interacts with IRF3 (Figure 4E), whereas the transcriptional activation domain IAD of IRF3 interacts with RNF138 (Figure 4F). These results suggest that RNF138 binds to the IAD domain of IRF3 via the RING domain.
RNF138 is an E3 ubiquitin ligase. To determine whether RNF138 could ubiquitinate IRF3 and target IRF3 for degradation. Reporter assays indicated that overexpression of RNF138 in HEK293T cells inhibited SeV-induced (left) and IRF3-mediated (right) ISRE activation, but the inhibitory effect was lost in the RNF138 enzyme inactivation mutant (C18A/C54A) (Figure 4G). This result implied that RFN138 depended on its enzymatic activity to participate in the antiviral signaling. Interestingly, we found that overexpression of RNF138-WT and -CA had little effect on the polyubiquitination of IRF3 (Figure 4H) and the stability of IRF3 (Figure 4I). These results suggested that IRF3 activation regulation by RNF138 may be affected by the IRF3-interacting protein ubiquitination.
2.4. RNF138 inhibits IRF3 activation by ubiquitinating PTEN
Next, we sought to find regulators that interact with IRFOur previous findings revealed that the tumor suppressor PTEN could release the negatively regulated phosphorylation of IRF3 at the Ser97 site upon viral infection, thereby promoting IRF3 nuclear translocation[29]. To determine whether RNF138 interacts with PTEN, co-immunoprecipitation experiments showed that RNF138 is associated with PTEN (Figure 5A). Interestingly, overexpression of RNF138 inhibited PTEN interactions with IRF3 (Figure 5B), while knockout of RNF138 had the opposite effect (Figure 5C). To further determine whether RNF138 affects PTEN ubiquitination and co-immunoprecipitation showed that overexpression of RNF138 significantly promoted polyubiquitination of PTEN (Figure 5D). qPCR experiments indicated that PTEN overexpression promoted SeV-induced IFNB1 mRNA expression, significantly higher in RNF138 knockout HEK293T cells (Figure 5E). Furthermore, RNF138 overexpression inhibited PTEN-mediated nuclear translocation of IRF3 (Figure 5F). These results suggest that RNF138 may affect PTEN function by promoting PTEN ubiquitination, inhibiting PTEN-IRF3 interactions, and thus inhibiting PTEN-mediated IRF3 nuclear translocation, suppressing IRF3 activation and antiviral immune responses.
3. Discussion
Based on the fact that RNF138 is an E3 ubiquitin ligase and is predominantly distributed in the nucleus. At the same time, there is growing evidence that RNF138 protects genomic stability [31,32,40,41] and in cancer development[42,43,44]. For example, RNF138 drives NF-κB activation and lymphomagenesis by destabilizing MYD88 L265P through ubiquitination [33]; similarly, it has been shown that RNF138 inhibits colorectal cancer (CRC) by preventing hyperactivation of NF-κB signaling [44]. However, there are few studies on the role of RNF138 in antiviral innate immunity. Recent literature reports that RNF138 inhibits LPS-triggered late inflammatory gene transcription[38]. However, whether cytoplasmic RNF138 inhibits virus-triggered IRF3 activation and IFN-β induction is unclear.
In this study, we investigated the role of RNF138 in IRF3-mediated signaling. Overexpression of RNF138 significantly inhibited SeV-induced activation of ISRE, NF-κB, and IFN-β promoters, as well as SeV-induced phosphorylation, dimerization, and translocation into the nucleus of IRF3, whereas knockout of RNF138 had the opposite effect. Mouse cell knockout indicates that RNF138 deficiency promotes SeV or VSV-induced phosphorylation of IRFThese findings establish a critical role for RNF138 in the innate immune response to SeV or VSV. Although RNF138 had little effect on IRF3 ubiquitination and RNF138 did not affect the stability of the IRF3 protein, in combination with reporter assays, the RNF138-CA enzyme inactivation mutant lost the ability to inhibit both SeV-induced and IRF3-mediated ISRE activation, so we suggest that IRF3 activation regulation by RNF138 may be affected by the IRF3-interacting protein ubiquitination.
It is well known that the activation of IRF3 prior to import into the nucleus involves two basic steps: phosphorylation and dimerization. However, our previous results revealed that PTEN exercises a dephosphorylation function on the Ser97 phosphorylation site of IRF3 through its phosphatase activity in the type I interferon-inducible pathway, thereby promoting the nuclear translocation of IRF3 and activating the expression of type I interferon[29]. Therefore, our laboratory proposes that the activation of IRF3 and its import into the nucleus may be determined by at least three steps: carboxy-terminal phosphorylation, dimerization, and amino-terminal dephosphorylation. Thus, positive regulation of IRF3 activation by PTEN-mediated dephosphorylation controls nuclear translocation of IRFIn our study, although RNF138 did not affect the ubiquitination of IRF3 protein, RNF138 affected the ubiquitination of PTEN, which was ubiquitinated and likely inhibited the interaction of IRF3 with PTEN and consequently inhibited the import of IRF3 into the nucleus. We have seen that the interactions between IRF3 and PTEN were enhanced after RNF138 knockout, suggesting that the ubiquitination of PTEN may inhibit the interactions between IRF3 and PTEN. However, the mechanism of ubiquitination of PTEN by RNF138 and its detailed mechanism for IRF3 activation remain to be further investigated.
We also found that, unlike knockout of PTEN, which did not affect the phosphorylation and dimerization of IRF3, knockout of RNF138 promoted the phosphorylation and dimerization of IRF3 (Figure 2D-E), suggesting that RNF138 may also inhibit the phosphorylation and dimerization process of IRF3 by affecting the other IRF3-interacting protein. That is, RNF138-mediated regulation of innate immunity also has complexity and diversity.
Collectively, these findings define a previously unknown function of cytoplasmic RNF138 in antiviral innate immunity. We confirm the involvement of RNF138 in virus-triggered IRF3 nuclear translocation and how it regulates this process, and establish a mechanistic link between the triad of RNF138, PTEN, and IRF3 nuclear translocation, thus providing new insights into the molecular mechanisms by which viral infections trigger IRF3 nuclear translocation (Figure 6). In conclusion, our findings suggest that RNF138 ubiquitinates PTEN and subsequently inhibits PTEN interactions with IRF3, thereby inhibiting nuclear translocation of IRF3, which provides new insights into the mechanisms that control excessive cellular antiviral responses.
4. Materials and Methods
4.1. Reagents
This study utilized antibodies against IRF3 (#11312-1-AP), GFP (#50430-2-AP), GAPDH (#10494-1-AP), Lamin B (#12987-1-AP), Actin (#66009-1-Ig), HA (#51064-2-AP), Flag (#20543-1-AP), Myc (#16286-1-AP, all from Proteintch), RNF138 (#A10304, ABclonal), p-IRF3(Ser386) (#ET1608-22, Huabio), p-IRF3(Ser396) (#29047, CST), HRP-conjugated Mouse Anti-Rabbit IgG Light Chain (#AS061, ABclonal). 2×SYBR qPCR Mix (Thermo Fisher) were purchased from the indicated companies. Sendai virus (SeV) and Vesicular Stomatitis Virus (VSV) were kindly provided by Dr. Hong-Bing Shu of Wuhan University. The TCID50 of SeV is 10^-3.9/0.1mL. The MOI of VSV is 0.After virus infection, the HEK293T, RAW264.7 or L929 cells were cultured in an incubator at 37°C and 5% carbon dioxide for different times.
4.2. Constructs
The cDNAs encoding human RNF138 was amplified and cloned into the pEF vector. The ISRE, IFN-β, NF-κB promoter luciferase reporter plasmids, and mammalian expression plasmids for TBK1, IRF3, IRF3-5D, RIG-Ⅰ, MAVS, HA-Ub and PTEN were kindly provided by Dr. Hong-Bing Shu. Mammalian expression plasmids and prokaryotic expression plasmids for RNF138 was constructed by standard molecular biology techniques.
4.3. Transfection and Reporter Gene Assays
The HEK293T cells were seeded in 24-well plates and then transfected with IFN-β-luc, ISRE-luc or NF-κB-luc, HA-IRF3, shRNF138, RNF138 or its mutant plasmids or empty control vector by PEI transfection reagent, together with pRL-TK (Renilla luciferase) as a control. Twenty-four hours (for RNF138 overexpression) or thirty-six hours (for RNF138 knockdown) after transfection, the cells were stimulated with SeV for another 12 h in certain experiments; or the cells were harvested. The cells were lysed with 100μl passive lysis buffer for 30 min and subjected to measurements of dual-luciferase activity with a dual-specific luciferase assay kit and Luciferase Reporter Assay System (Promega). The firefly luciferase activity was normalized to the Renilla luciferase activity.
4.4. PCR
Total RNA was extracted from cells using TRIzol (BIOLOGY). A reverse transcription system (Takara) was used to synthesize cDNA. 2× qPCR SYBR Green Master Mix (Yeasen Biotechnology Co.,Ltd.) and a Bio Rad CFX Connect system were used for qPCR. The mRNA results were normalized to GAPDH expression. The qPCR primers sequences are listed in Supplementary information Table S1.
4.5. shRNA
The shRNAs targeting RNF138 were constructed by plasmid pLKO.1-TCR Vector and transfected by PEI into HEK293T cells followed by immunoblot analysis. The shRNA primer sequences in this study are listed in Supplementary information Table S2.
4.6. Generation of knockout cells by CRISPR/Cas9 technology
HEK293T or RAW264.7 or L929 cells with the knockout of RNF138 were generated by a Lenti-CRISPR/Cas9-v2 system. HEK293T cells were seeded in 60 mm dishes and transfected with lenti-CRISPR-RNF138-sgRNA#1, sgRNA#2 or lenti-CRISPR-Rnf138-sgRNA along with the packaging plasmids psPAX2 and pMD2.G by PEI transfection reagent. The medium was changed 6h after transfection. The supernatants containing lentivirus were harvested 48 h after infection, and were filtered through a 0.22 μm filter. HEK293T, RAW264.7 or L929 cells were incubated with the lentivirus for 36 h, and then were selected with puromycin for 7 days. The isolated single clonal knockout cells were confirmed with western blotting. The sgRNA primer sequences in this study are listed in Supplementary information Table S3.
4.7. Co-immunoprecipitation and Immunoblotting Analyses
For the co-immunoprecipitation experiments, the HEK293T cells were transfected with the indicated plasmid. After 24 h, the cells were harvested and lysed in 0.8 ml of lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% TritonX-100, sodium pyrophoshate, β-glycerophosphoate, 1 mM EDTA, Na3VO4, 10 μg/ml leupeptin). For immunoprecipitation reaction, 0.7 ml of cell lysate was incubated with Flag-beads or the indicated antibody and 40 μl of protein A/G agarose beads at 4°C. After overnight incubation, the beads were washed three times with 1 ml of Co-IP washing buffer (50 mM NaCl and 150 mM Tris, pH 7.5). The immunoprecipitates and whole cell lysate were subjected to SDS-PAGE, transferred onto nitrocellulose membranes and blotted as described previously[29]. For the endogenous immunoprecipitation experiments, HEK293T cells were seeded in 100 mm dishes and treated with SeV for the indicated times after 16 h. The subsequent procedures were performed as described above.
4.8. Ubiquitination assays
The HEK293T cells were transfected with the indicated plasmid. After 24 h, the cells were harvested and lysed on ice in 1 ml of lysis buffer as described above (100ul, in the presence of 1% SDS). Next, the cell lysates were denatured at 100°C for 5 min and then diluted to 10 volumes lysis buffer (without SDS) before incubation with anti-Flag beads at 4°C for over night. The beads were washed three times with 1 ml of Co-IP washing buffer and analyzed by immunoblotting.
4.9. Native PAGE
The HEK293T cells were harvested and lysed with ice-cold lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% TritonX-100, sodium pyrophoshate, β-glycerophosphoate, 1 mM EDTA, Na3VO4, 10 μg/ml leupeptin, without SDS). Next, the cell lysates were diluted with 2× loading buffer (50 mM Tris-HCl, pH 6.8, 30% glycerol and bromophenol blue, without SDS). Finally, the samples were loaded onto 8% native gels and separated at 20 mA for 30 minutes and 35 mA for 90 minutes on ice by electrophoresis, followed by immunoblot analysis.
4.Subcellular fractionation
Nuclear and cytoplasmic extracts were prepared with a nuclear-cytoplasmic extraction kit (Wuhan KeRui Biotechnology Co., Ltd.) according to the manufacturer’s instructions.
4.10. Statistical Analysis
All data were performed for at least two experiments with similar results, data were processed Microsofe Excel and Origin 6.0 and are presented as mean ± SD of one representative experiment, differences between control and experimental groups were determined by Student’s t-test. P values < 0.05 were considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, G.S., X.Z.; methodology, X.Z., C.L., J.F., J.Z.; software, X.Z.; validation, X.Z.; formal analysis, G.S., X.Z.; investigation, X.Z., C.L., J.F., J.Z.; data curation, X.Z.; writing—original draft preparation, X.Z.; writing—review and editing, G.S., X.Z., C.F.; supervision, G.S., M.G.; project administration, G.S., M.G.. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grants No. 31370187, 30870113, 81572447, 31871427, and 32260173), Zhongnan Hospital - Taikang Medical School (School of Basic Medical Sciences) of Wuhan University Joint Foundation (Grant No. JCZN2022010), and the Central Guidance on Local Science and Technology Development Fund of Tibet (Grant No. XZ202301YD0040C).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are provided in the article and its Supplementary file, or by the corresponding author upon reasonable request.
Acknowledgments
We are grateful to Dr. Hong-Bing Shu for providing the virus and expression plasmids.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. International reviews of immunology 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, L.A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunological reviews 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Shu, H.B. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annual review of cell and developmental biology 2018, 34, 357–379. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Molecular cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunological reviews 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nature reviews. Microbiology 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 2001, 19, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 2008, 26, 535–584. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Molecular and cellular biology 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS letters 1998, 425, 112–116. [Google Scholar] [CrossRef]
- Qin, B.Y.; Liu, C.; Lam, S.S.; Srinath, H.; Delston, R.; Correia, J.J.; Derynck, R.; Lin, K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nature structural biology 2003, 10, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Kuzmanovic, T.; Zhang, Y.; Wetzel, J.L.; Sen, G.C. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016, 44, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol 2006, 7, 598–605. [Google Scholar] [CrossRef]
- Mori, M.; Yoneyama, M.; Ito, T.; Takahashi, K.; Inagaki, F.; Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 2004, 279, 9698–9702. [Google Scholar] [CrossRef]
- Servant, M.J.; Grandvaux, N.; tenOever, B.R.; Duguay, D.; Lin, R.; Hiscott, J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem 2003, 278, 9441–9447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.P.; McBride, K.M.; Weaver, B.K.; Dingwall, C.; Reich, N.C. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Molecular and cellular biology 2000, 20, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, M.X.; Tang, Z.; Zhang, Q.; Ye, J.; Xiong, T.C.; Zhang, Z.D.; Zhong, B. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J Exp Med 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.Y.; Chen, S.; Zhao, X.; Lei, C.Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat Immunol 2016, 17, 241–249. [Google Scholar] [CrossRef]
- Ismail, I.H.; Gagne, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat Cell Biol 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat Cell Biol 2015, 17, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lu, Y.; Han, D.; Yao, R.; Wang, H.; Zhong, S.; Luo, Y.; Han, R.; Li, K.; Fu, J.; et al. Rnf138 deficiency promotes apoptosis of spermatogonia in juvenile male mice. Cell Death Dis 2017, 8, e2795. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Liu, H.; Wang, X.; Hu, H.; Cao, Y.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; et al. MYD88 L265P elicits mutation-specific ubiquitination to drive NF-κB activation and lymphomagenesis. Blood 2021, 137, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Byun, K.; Hong, W.; Chuang, H.Y.; Pack, C.G.; Bayarsaikhan, E.; Paek, S.H.; Kim, H.; Shin, H.Y.; Ideker, T.; et al. Proteome-wide discovery of mislocated proteins in cancer. Genome research 2013, 23, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Long, P.; Samnakay, P.; Jenner, P.; Rose, S. A yeast two-hybrid screen reveals that osteopontin associates with MAP1A and MAP1B in addition to other proteins linked to microtubule stability, apoptosis and protein degradation in the human brain. Eur J Neurosci 2012, 36, 2733–2742. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Bjerrum, J.T.; Csillag, C.; Nielsen, F.C.; Olsen, J. Influence of smoking on colonic gene expression profile in Crohn's disease. PLoS One 2009, 4, e6210. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, W.; Liu, H.; Xue, M.; Liu, X.; Zhang, K.; Hu, L.; Li, J.; Liu, X.; Xiang, Z.; et al. Correction: African Swine Fever Virus pI215L Negatively Regulates cGAS-STING Signaling Pathway through Recruiting RNF138 to Inhibit K63-Linked Ubiquitination of TBK1. J Immunol 2022, 208, 1510–1511. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Z.; Liu, S.; Zhang, X.; Cao, X.; Jiang, M. RNF138 inhibits late inflammatory gene transcription through degradation of SMARCC1 of the SWI/SNF complex. Cell Rep 2023, 42, 112097. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 7945–7950. [Google Scholar] [CrossRef]
- Ismail, I.H.; Gagné, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat Cell Biol 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. RNF138 joins the HR team. Nat Cell Biol 2015, 17, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Han, D.; Liu, W.; Huang, R.; Ou, J.; Chen, X.; Zhang, X.; Wang, X.; Li, S.; Wang, L.; et al. RNF138 confers cisplatin resistance in gastric cancer cells via activating Chk1 signaling pathway. Cancer biology & therapy 2018, 19, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, X.; Feng, M.; Yao, L.; Deng, Z.; Zao, G.; Zhou, Y.; Chen, S.; Du, Z. Downregulation of RNF138 inhibits cellular proliferation, migration, invasion and EMT in glioma cells via suppression of the Erk signaling pathway. Oncology reports 2018, 40, 3285–3296. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, R.; Ying, J.; Li, X.; Jiao, T.; Guo, L.; Zhou, H.; Wang, H.; Tuersuntuoheti, A.; Liu, J.; et al. RING finger 138 deregulation distorts NF-кB signaling and facilities colitis switch to aggressive malignancy. Signal Transduction and Targeted Therapy 2022, 7, 185. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The overexpression of RNF138 inhibited the virus-triggered activations of IRF3 and IFNB1 gene transcription. (A) RNF138 inhibited the IFN-β promoter, ISRE, and NF-κB. HEK293T cells were transfected with the IFN-β, ISRE, NF-κB reporter, and control or RNF138 plasmid for 24 h and then infected with SeV for 12 h before luciferase assays. (B) Effects of RNF138 on SeV-induced transcription of downstream genes. HEK293T cells were transfected with control or RNF138 plasmid for 24 h and then infected with SeV for 8 h before qPCR analysis. (C) Effects of RNF138 on SeV-induced phosphorylation of IRF3 (Ser396, Ser386). HEK293T cells were transfected with control or myc-RNF138 plasmid for 24 h, then infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (D) Effects of RNF138 on SeV-induced nuclear translocation of IRFHEK293T cells were transfected with GFP or RNF138-GFP plasmid for 24 h, then infected with SeV for 8 h. Immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, B, D).
Figure 1.
The overexpression of RNF138 inhibited the virus-triggered activations of IRF3 and IFNB1 gene transcription. (A) RNF138 inhibited the IFN-β promoter, ISRE, and NF-κB. HEK293T cells were transfected with the IFN-β, ISRE, NF-κB reporter, and control or RNF138 plasmid for 24 h and then infected with SeV for 12 h before luciferase assays. (B) Effects of RNF138 on SeV-induced transcription of downstream genes. HEK293T cells were transfected with control or RNF138 plasmid for 24 h and then infected with SeV for 8 h before qPCR analysis. (C) Effects of RNF138 on SeV-induced phosphorylation of IRF3 (Ser396, Ser386). HEK293T cells were transfected with control or myc-RNF138 plasmid for 24 h, then infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (D) Effects of RNF138 on SeV-induced nuclear translocation of IRFHEK293T cells were transfected with GFP or RNF138-GFP plasmid for 24 h, then infected with SeV for 8 h. Immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, B, D).
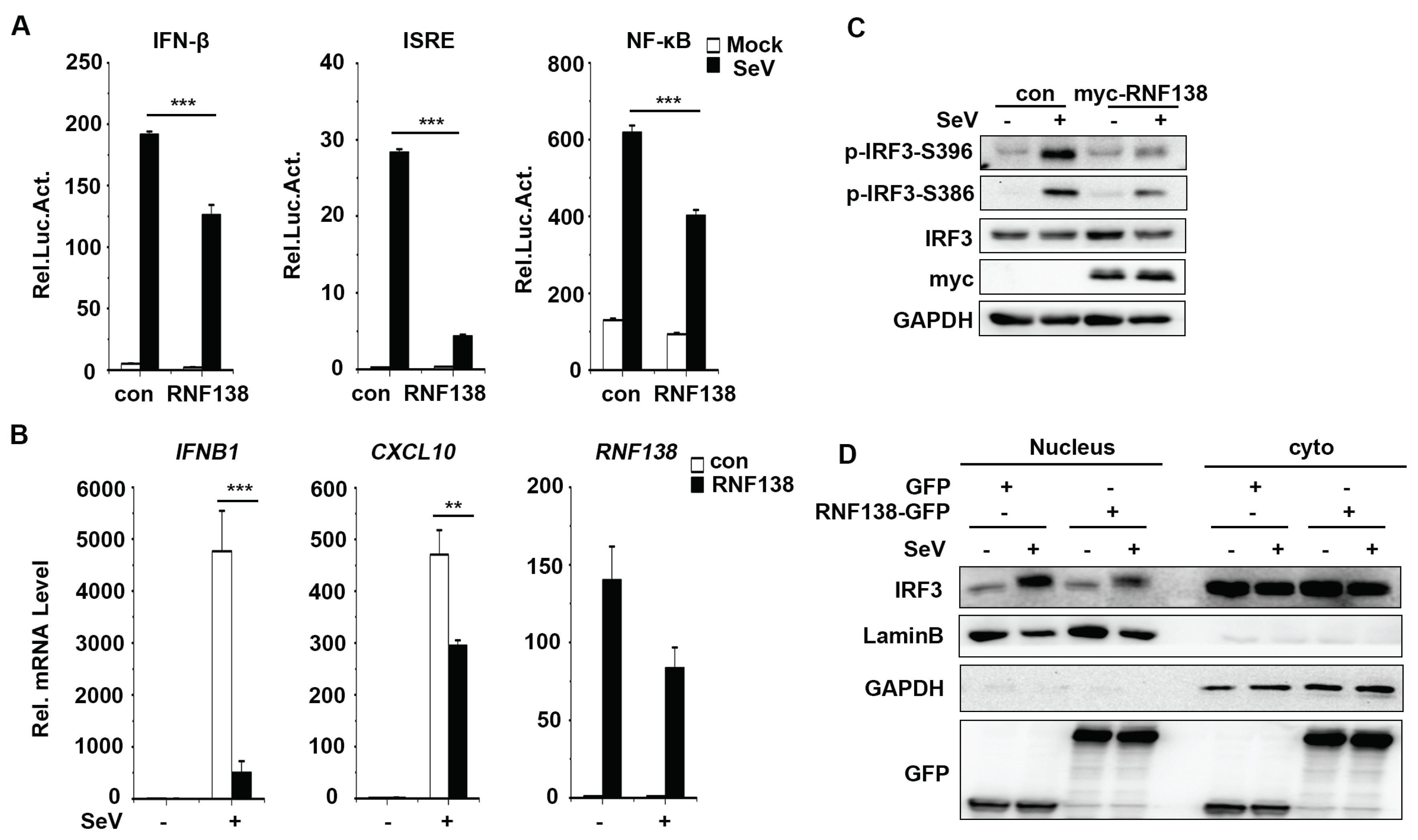
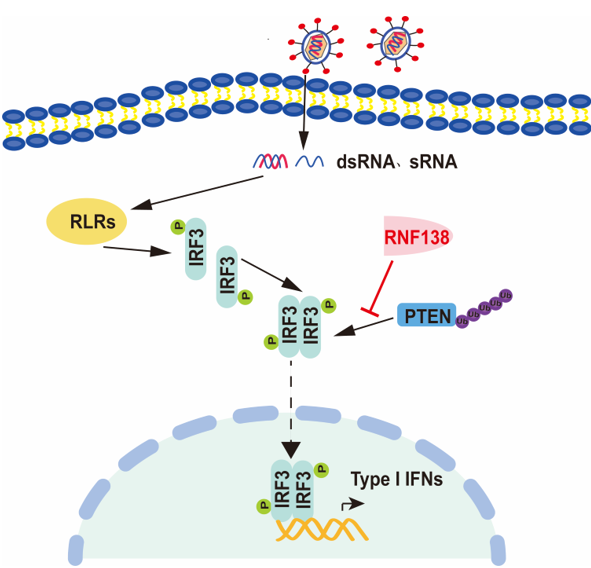
Figure 2.
Effects of knockdown and knockout of RNF138 on SeV-induced signaling and IRF3 activation. (A) RNF138-deficient (KO) HEK293T clones were generated by the CRISPR-Cas9 method (upper panel) and Effects of RNF138 RNAi on the expression of endogenous RNF138 (lower panel). Deficiency of RNF138 in the KO clones were confirmed by immunoblotting analysis with anti-RNFHEK293T cells were transfected with the indicated RNAi plasmids for 48 h before immunoblotting analysis with anti-RNF138. (B) Effects of RNF138 knockdown on Sev-induced activation of ISRE. HEK293T cells were transfected with the ISRE reporter, and control or RNF138 RNAi plasmids for 36 h and then infected with SeV for 12 h before luciferase assays. (C) Effects of RNF138 deficiency on SeV-induced transcription of downstream genes. RNF138-KO and control HEK293T cells were infected with SeV for the indicated times before qPCR analysis. (D) Effects of RNF138 deficiency on SeV-induced phosphorylation of IRF3 (Ser386, Ser396). RNF138-KO and control HEK293T cells were infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (E) Effects of RNF138 deficiency on SeV-induced dimerization of IRFRNF138-KO and control HEK293T cells were infected with SeV for 8 h, and then cell lysates were separated by native PAGE and analyzed by immunoblots with anti-IRF3. (F) Effects of RNF138 deficiency on SeV-induced nuclear translocation of IRFRNF138-KO and control HEK293T cells were infected with SeV for 8 h, and then immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in B, C, D).
Figure 2.
Effects of knockdown and knockout of RNF138 on SeV-induced signaling and IRF3 activation. (A) RNF138-deficient (KO) HEK293T clones were generated by the CRISPR-Cas9 method (upper panel) and Effects of RNF138 RNAi on the expression of endogenous RNF138 (lower panel). Deficiency of RNF138 in the KO clones were confirmed by immunoblotting analysis with anti-RNFHEK293T cells were transfected with the indicated RNAi plasmids for 48 h before immunoblotting analysis with anti-RNF138. (B) Effects of RNF138 knockdown on Sev-induced activation of ISRE. HEK293T cells were transfected with the ISRE reporter, and control or RNF138 RNAi plasmids for 36 h and then infected with SeV for 12 h before luciferase assays. (C) Effects of RNF138 deficiency on SeV-induced transcription of downstream genes. RNF138-KO and control HEK293T cells were infected with SeV for the indicated times before qPCR analysis. (D) Effects of RNF138 deficiency on SeV-induced phosphorylation of IRF3 (Ser386, Ser396). RNF138-KO and control HEK293T cells were infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (E) Effects of RNF138 deficiency on SeV-induced dimerization of IRFRNF138-KO and control HEK293T cells were infected with SeV for 8 h, and then cell lysates were separated by native PAGE and analyzed by immunoblots with anti-IRF3. (F) Effects of RNF138 deficiency on SeV-induced nuclear translocation of IRFRNF138-KO and control HEK293T cells were infected with SeV for 8 h, and then immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in B, C, D).
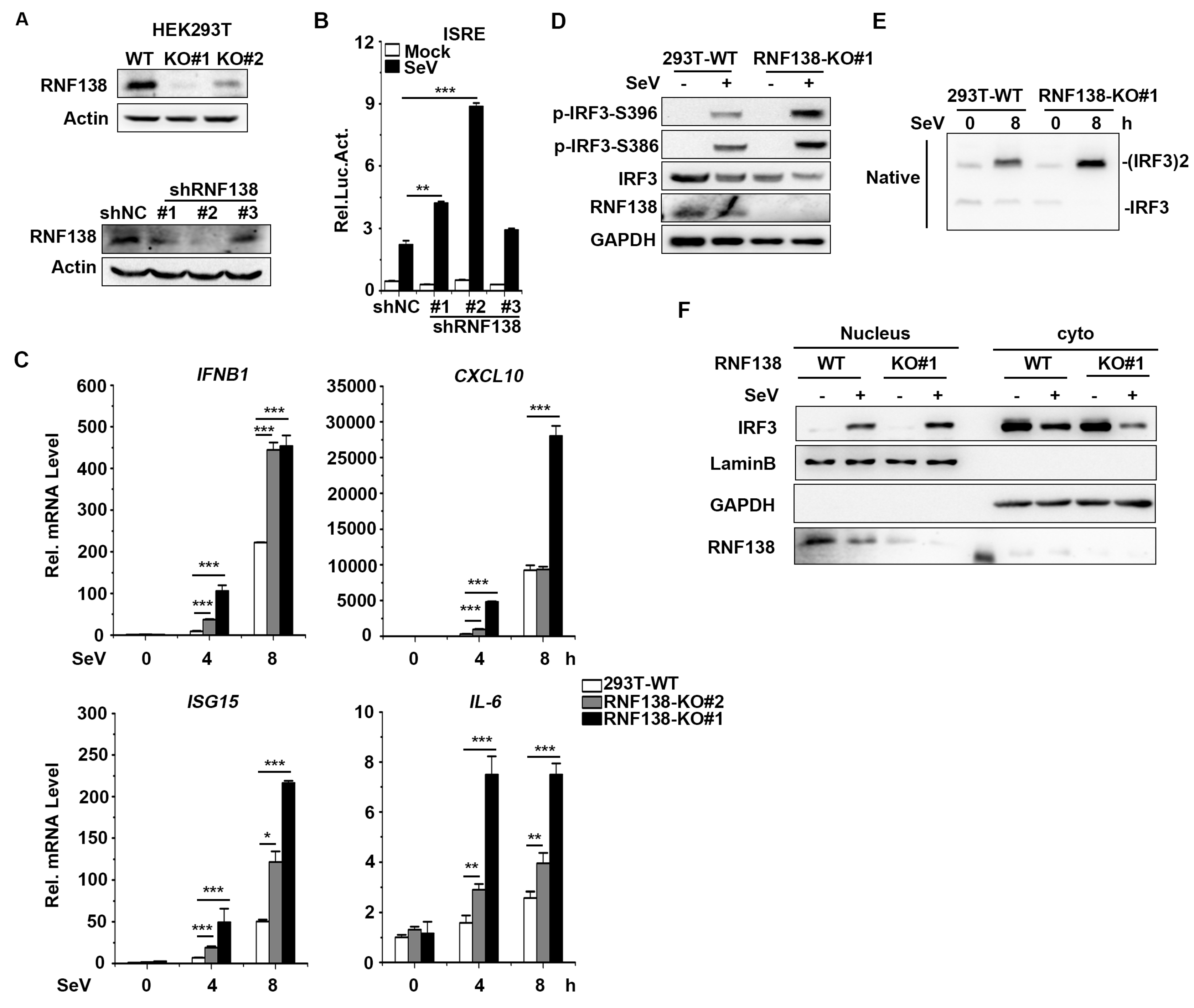
Figure 3.
Knockout of RNF138 in mouse cells promotes virus-induced IRF3 activation. (A) Effects of RNF138 deficiency on SeV or VSV-induced transcription of downstream genes. RNF138-KO and control RAW264.7 cells were infected with SeV or VSV for 8 h before qPCR analysis. (B) Effects of RNF138 deficiency on SeV-induced phosphorylation of IRF3 (Ser396). RNF138-deficient (KO) RAW264.7 or L929 clones were generated by the CRISPR-Cas9 method. RNF138-KO and control RAW264.7 or L929 cells were infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (C) Effects of RNF138 deficiency on VSV-induced phosphorylation of IRF3 (Ser396). RNF138-KO and control RAW264.7 or L929 cells were infected with VSV for 8 h before immunoblotting analysis with the indicated antibodies. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A).
Figure 3.
Knockout of RNF138 in mouse cells promotes virus-induced IRF3 activation. (A) Effects of RNF138 deficiency on SeV or VSV-induced transcription of downstream genes. RNF138-KO and control RAW264.7 cells were infected with SeV or VSV for 8 h before qPCR analysis. (B) Effects of RNF138 deficiency on SeV-induced phosphorylation of IRF3 (Ser396). RNF138-deficient (KO) RAW264.7 or L929 clones were generated by the CRISPR-Cas9 method. RNF138-KO and control RAW264.7 or L929 cells were infected with SeV for 8 h before immunoblotting analysis with the indicated antibodies. (C) Effects of RNF138 deficiency on VSV-induced phosphorylation of IRF3 (Ser396). RNF138-KO and control RAW264.7 or L929 cells were infected with VSV for 8 h before immunoblotting analysis with the indicated antibodies. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A).
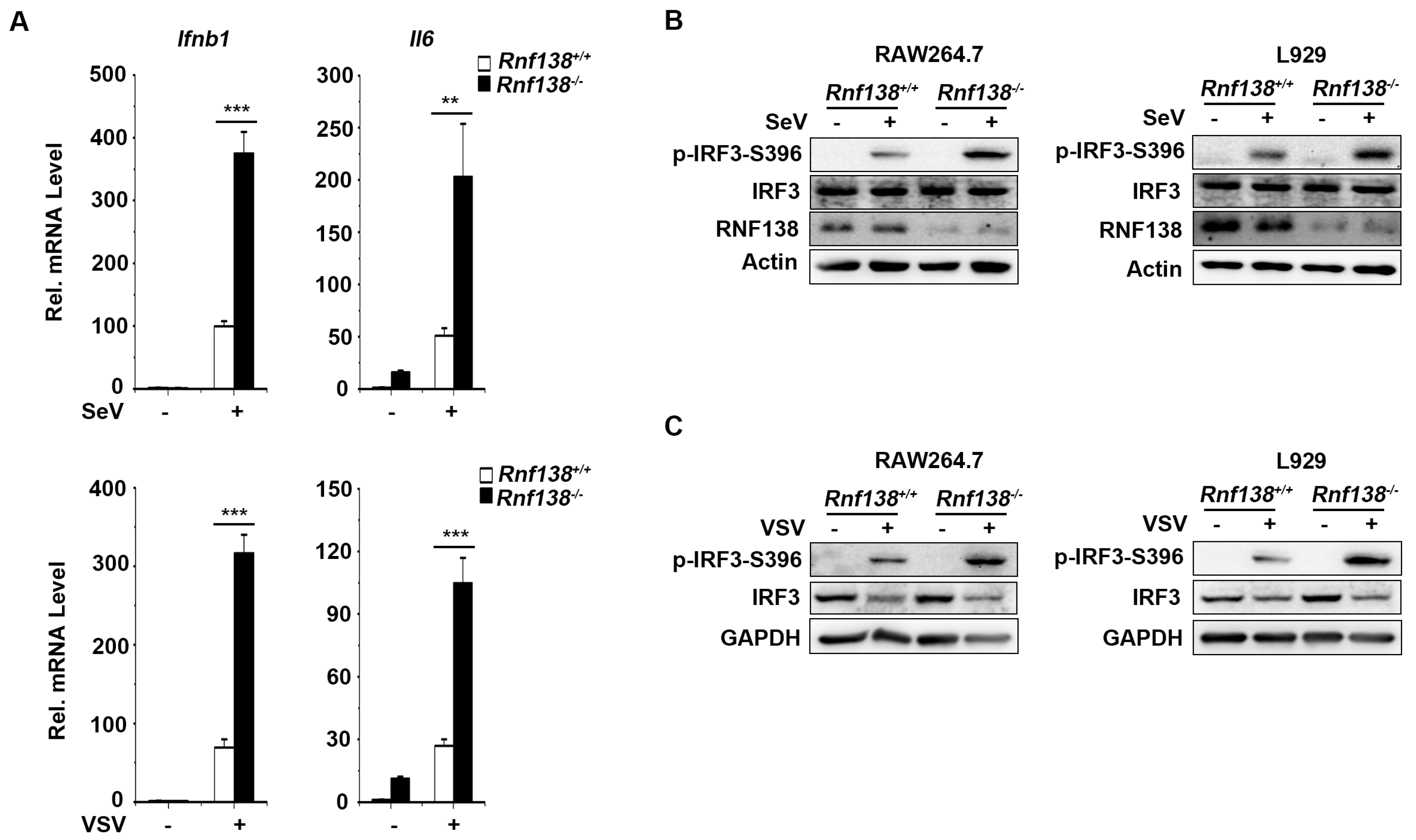
Figure 4.
RNF138-mediated virus-triggered signaling at the level of IRF3. (A) The overexpression of RNF138 inhibited RIGI-, MAVS-, TBK1-, IRF3-, and IRF3-5D-mediated signaling. HEK293T cells were transfected with RNF138 or control plasmids together with ISRE reporter and the indicated plasmids. Luciferase assays were performed 24 h after transfection. (B) Co-immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells transfected with HA-IRF3 and RNF138-Flag or control vector for 24 h. (C) Co-immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells transfected with RNF138-Flag or control vector for 24 h. (D) Endogenous immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells infected with SeV for the indicated times. (E-F) Domain mapping of the interaction of RNF138 with IRFHEK293T cells were transfected with the indicated truncations before co-immunoprecipitation and immunoblotting analysis with the indicated antibodies. The schematic presentations of RNF138 and IRF3 truncations are shown at the top. (G) Effects of RNF138-WT and RNF138-CA (C18A/C54A) on SeV-induced or IRF3-mediated activation of ISRE. HEK293T cells were transfected with the ISRE reporter and a control, RNF138-WT, RNF138-CA plasmids for 24 h, and then infected with SeV for 12 h (left), or transfected with the ISRE reporter and HA-IRF3 together with a control, RNF138-WT, RNF138-CA plasmids for 24 h (right panel) before luciferase assays. (H) Effects of RNF138 and its CA mutants on polyubiquitination of IRFHEK293T cells were transfected with Flag-IRF3 and HA-Ub together with control, RNF138-WT, RNF138-CA plasmids for 24 h, followed by immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (I) Immunoblot analysis of protein level of IRF3 in HEK293T cells transfected with HA-IRF3 in fixed amounts and myc-RNF138 in different dosages. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, B, C, G).
Figure 4.
RNF138-mediated virus-triggered signaling at the level of IRF3. (A) The overexpression of RNF138 inhibited RIGI-, MAVS-, TBK1-, IRF3-, and IRF3-5D-mediated signaling. HEK293T cells were transfected with RNF138 or control plasmids together with ISRE reporter and the indicated plasmids. Luciferase assays were performed 24 h after transfection. (B) Co-immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells transfected with HA-IRF3 and RNF138-Flag or control vector for 24 h. (C) Co-immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells transfected with RNF138-Flag or control vector for 24 h. (D) Endogenous immunoprecipitation analysis of the interaction between RNF138 and IRF3 in HEK293T cells infected with SeV for the indicated times. (E-F) Domain mapping of the interaction of RNF138 with IRFHEK293T cells were transfected with the indicated truncations before co-immunoprecipitation and immunoblotting analysis with the indicated antibodies. The schematic presentations of RNF138 and IRF3 truncations are shown at the top. (G) Effects of RNF138-WT and RNF138-CA (C18A/C54A) on SeV-induced or IRF3-mediated activation of ISRE. HEK293T cells were transfected with the ISRE reporter and a control, RNF138-WT, RNF138-CA plasmids for 24 h, and then infected with SeV for 12 h (left), or transfected with the ISRE reporter and HA-IRF3 together with a control, RNF138-WT, RNF138-CA plasmids for 24 h (right panel) before luciferase assays. (H) Effects of RNF138 and its CA mutants on polyubiquitination of IRFHEK293T cells were transfected with Flag-IRF3 and HA-Ub together with control, RNF138-WT, RNF138-CA plasmids for 24 h, followed by immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (I) Immunoblot analysis of protein level of IRF3 in HEK293T cells transfected with HA-IRF3 in fixed amounts and myc-RNF138 in different dosages. **P < 0.01, ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, B, C, G).
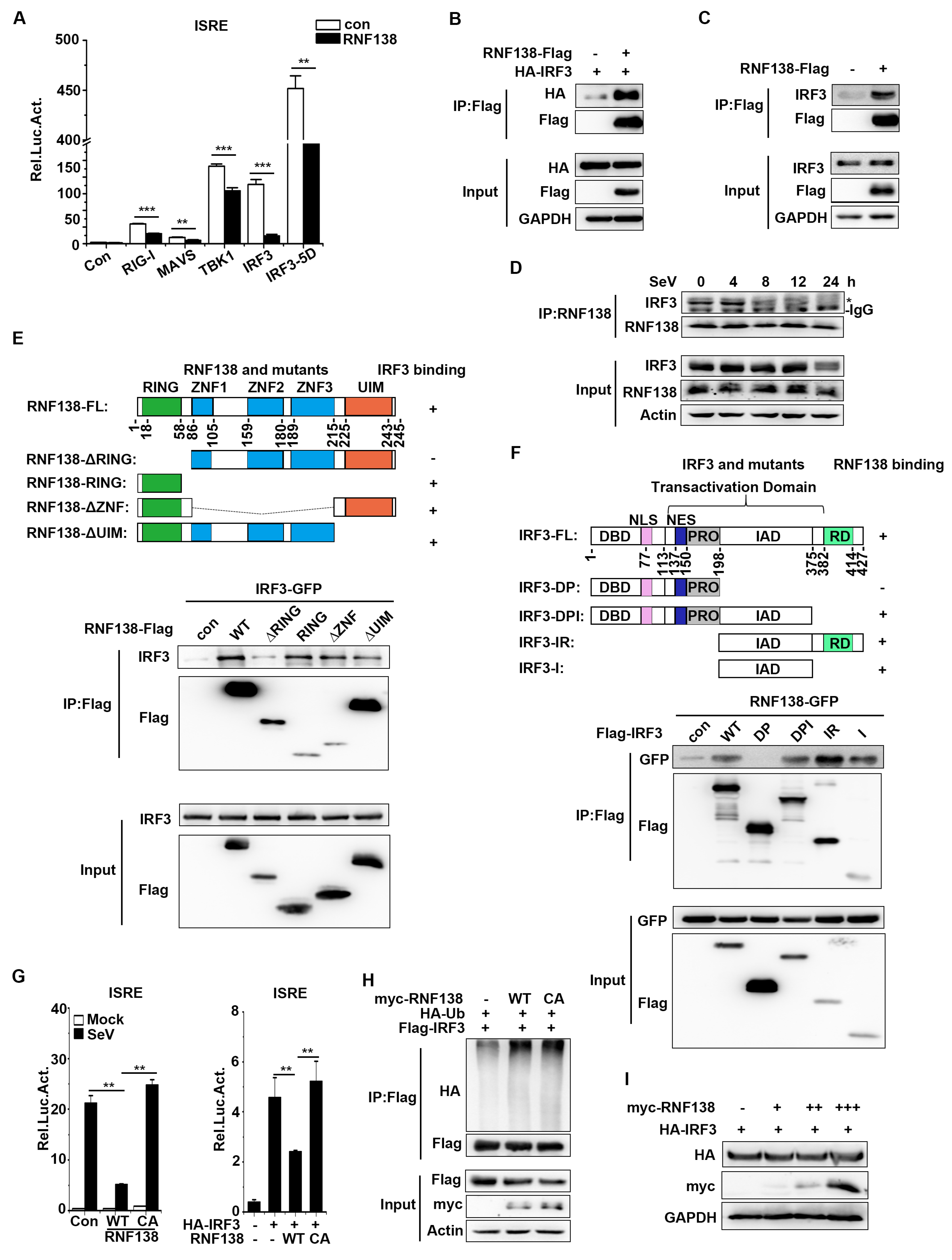
Figure 5.
RNF138 inhibits IRF3 activation by ubiquitinating PTEN. (A) Co-immunoprecipitation analysis of the interaction between RNF138 and PTEN in HEK293T cells transfected with myc-RNF138 and PTEN-Flag or control vector for 24 h. (B) RNF138 inhibited the association of PTEN with IRFHEK293T cells were transfected with PTEN-Flag and HA-IRF3 with a control and myc-RNF138 plasmid for 24 h before immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (C) RNF138 deficiency promotes the association of PTEN with IRFRNF138-KO and control HEK293T cells were transfected with PTEN-Flag and HA-IRF3 for 24 h before immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (D) Effects of RNF138 on polyubiquitination of PTEN. HEK293T cells were transfected with PTEN-Flag and HA-Ub together with control or myc-RNF138 plasmid for 24 h, followed by immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (E) Effects of RNF138 deficiency on PTEN-mediated transcription of IFNB1 gene. RNF138-KO and control HEK293T cells were transfected with PTEN-Flag for 24 h and then infected with SeV for 8 h before qPCR analysis. (F) Effects of RNF138 on PTEN-mediated nuclear translocation of IRFHEK293T cells were transfected with control or PTEN-Flag plasmid together with GFP or RNF138-GFP plasmid for 24 h, then infected with SeV for 8 h. Immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, D, E).
Figure 5.
RNF138 inhibits IRF3 activation by ubiquitinating PTEN. (A) Co-immunoprecipitation analysis of the interaction between RNF138 and PTEN in HEK293T cells transfected with myc-RNF138 and PTEN-Flag or control vector for 24 h. (B) RNF138 inhibited the association of PTEN with IRFHEK293T cells were transfected with PTEN-Flag and HA-IRF3 with a control and myc-RNF138 plasmid for 24 h before immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (C) RNF138 deficiency promotes the association of PTEN with IRFRNF138-KO and control HEK293T cells were transfected with PTEN-Flag and HA-IRF3 for 24 h before immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (D) Effects of RNF138 on polyubiquitination of PTEN. HEK293T cells were transfected with PTEN-Flag and HA-Ub together with control or myc-RNF138 plasmid for 24 h, followed by immunoblotting and co-immunoprecipitation analysis with the indicated antibodies. (E) Effects of RNF138 deficiency on PTEN-mediated transcription of IFNB1 gene. RNF138-KO and control HEK293T cells were transfected with PTEN-Flag for 24 h and then infected with SeV for 8 h before qPCR analysis. (F) Effects of RNF138 on PTEN-mediated nuclear translocation of IRFHEK293T cells were transfected with control or PTEN-Flag plasmid together with GFP or RNF138-GFP plasmid for 24 h, then infected with SeV for 8 h. Immunoblot analysis of IRF3 in cytoplasmic (Cyto) and nucleus fractions with the indicated antibodies. ***P < 0.001 (unpaired t-test). Data represent at least two experiments with similar results (mean ± SD, n = 3 independent samples in A, D, E).
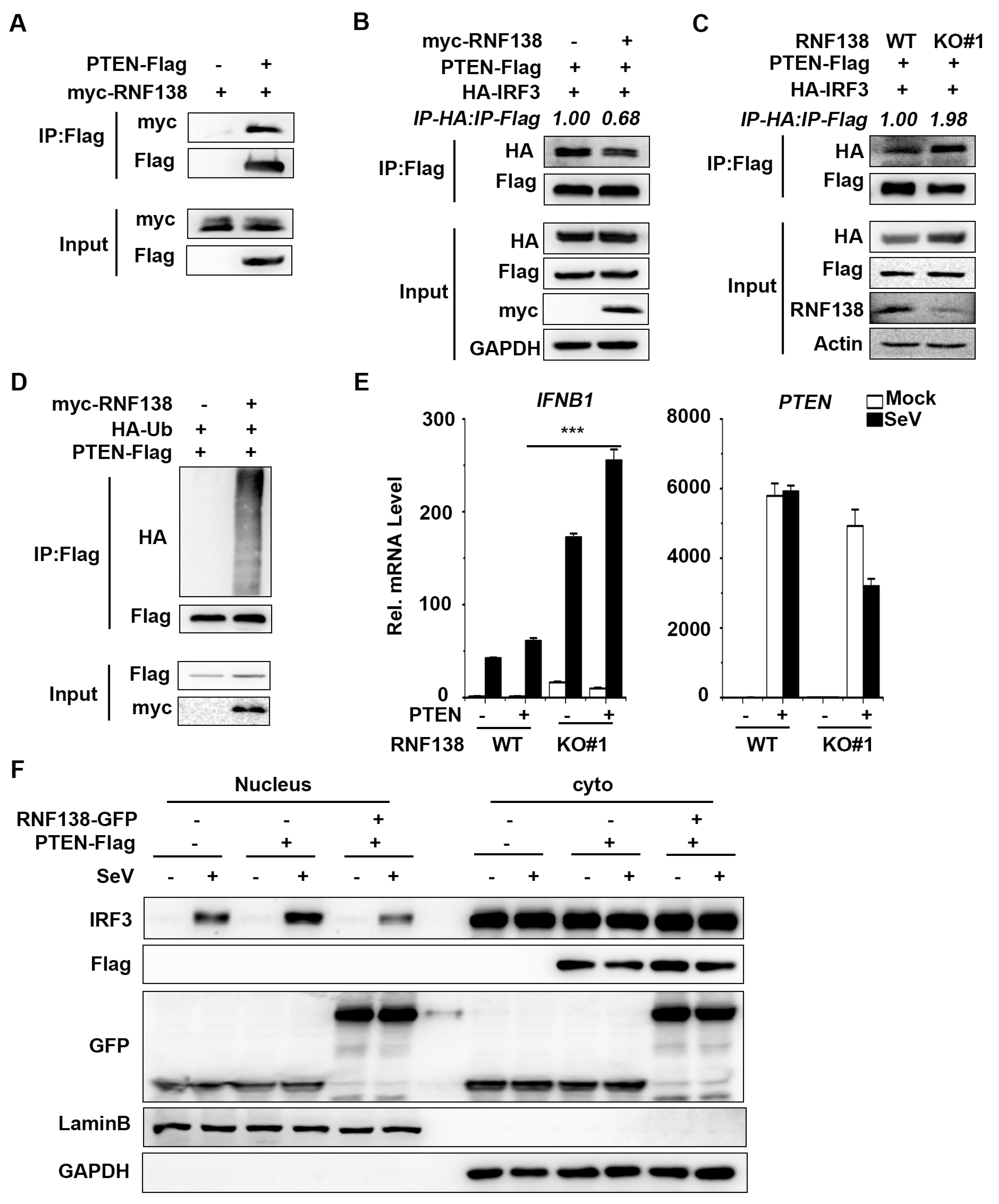
Figure 6.
The proposed working model of RNF138 in the regulation of antiviral innate immune response. RNF138 affects the ubiquitination of PTEN, which was ubiquitinated likely inhibited the interaction of IRF3 with PTEN, and consequently inhibited the import of IRF3 into the nucleus.
Figure 6.
The proposed working model of RNF138 in the regulation of antiviral innate immune response. RNF138 affects the ubiquitination of PTEN, which was ubiquitinated likely inhibited the interaction of IRF3 with PTEN, and consequently inhibited the import of IRF3 into the nucleus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



