
Submitted:
27 October 2023
Posted:
30 October 2023
You are already at the latest version
Abstract
A healthy cornea is a nonvascularized, transparent tissue. It is nourished by diffusion through the tear film, aqueous fluid and neurotrophins supplied by the corneal innervation. The cornea is devoid of both blood and lymphatic vessels, which makes it an immunologi-cally privileged tissue. Corneal neovascularization develops due to an imbalance between angiogenic and anti-angiogenic factors and can lead to a significant decrease in visual acui-ty. It may cause scarring, lipid keratopathy, and corneal edema. The resulting pathological vascularization of the cornea reduces its immune privilege and significantly worsens the prognosis after subsequent penetrating keratoplasty, increasing the risk of rejection of the transplanted flap. Neovascularization is favored by many disease processes, including auto-immune diseases, viral inflammations, mainly herpes and varicella-zoster, and blepharitis [1]. There are several options to treat corneal neovascularization, both pharmacological and surgical. Angiostatic treatment involves inhibiting the growth of new vessels by suppressing inflammatory responses. VEGF inhibitors, steroids, cyclosporine A, and amniotic membrane grafts secured to the cornea work this way. VEGF inhibitors are the agents with the direct and most potent angiostatic effect. Angioregressive treatment, on the other hand, results in the regression of already existing pathological vessels. This therapy is also based on VEGF inhibitors, which, however, have the most potent effect on newly formed and small vessels but are less effective in reducing large-caliber and mature vessels. A more effective method for such vessels is ablation, which involves surgical closure. The most commonly used an-gio-occlusive method is needle diathermy and argon or yellow laser therapy. In advanced corneal neovascularization, corneal transplant surgery may be necessary. It is best performed after pretreatment with anti-VEGF preparations, often continued after corneal transplanta-tion to reduce the risk of transplant rejection [2]. This paper discusses the pathogenesis of corneal neovascularization and the current and future trends in its treatment.
Keywords:
cornea
; neovascularization
; steroids
; anti-VEGF drugs
; keratoplasty
; angiogenic factors
; anti-angiogenic factors
Introduction
Corneal diseases are one of the most common causes of blindness worldwide, along with age-related macular degeneration, diabetic retinopathy, and glaucoma. A normal cornea is characterized by transparency and a normal refractive surface. During various corneal diseases, neovascularization often develops; it is estimated to occur in about 1.4 million people annually, with vision loss in about 12% [1].
Corneal neovascularization is a disease process caused by corneal hypoxia, associated with an imbalance between angiogenic and anti-angiogenic factors. It is characterized by the formation of new blood vessels growing in from the corneal stroma and developing from pre-existing pericorneal vascular structures as a result of the proliferation and migration of vascular endothelial cells into the corneal stroma [1,2]. Pathological blood vessels are immature and lack structural integrity. When evaluating corneal neovascularization, attention should be paid to the source of vascularization (conjunctival, stromal, iridal), vessel penetration depth, branching, and leakage, causing lipid exudates and corneal edema. Clinically, the afferent vessels are narrower, straighter, located in the deeper layers of the cornea, and bright red. Drainage, i.e., efferent vessels, are of large caliber and dark red. Untreated corneal neovascularization predisposes to lipid keratopathy due to protein and lipid deposition in the stroma, chronic inflammation and corneal ulceration, corneal edema, and scar formation. Risk factors for corneal neovascularization include ocular surface diseases, allergies, autoimmune diseases, degeneration, trauma, chemical and thermal burns, past surgeries, keratitis (most commonly herpetic or chlamydial), onchocerciasis, bacterial and fungal inflammation, recurrent uveitis and corneal hypoxia caused by, for example, wearing contact lenses, especially soft hydrogel lenses, as well as congenital and acquired corneal diseases, corneal transplant rejection or limbal stem cell deficiency (LSCD).Some systemic conditions, such as diabetes or collagen-related diseases, can indirectly lead to corneal neovascularization. The consequence of these conditions is the loss of corneal stem cells, resulting in loss of transparency and significant deterioration of visual acuity up to and including blindness [1,2,3]. The formation of pathological corneal vessels is also associated with concomitant corneal lymphangiogenesis, which involves an immune response consisting of the presentation of antigens and antigen-presenting cells in the surrounding lymph nodes, which favors the rejection of corneal grafts [4] (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5).
Treatment of corneal neovascularization
Treatment of corneal neovascularization is either topical treatment with steroid and non-steroidal anti-inflammatory drugs, cyclosporine A, anti-VEGF drops, or surgical treatment, such as laser photocoagulation, diathermy/fine needle cauterization, superficial keratectomy, subconjunctival injections of anti-VEGF drugs, amniotic membrane transplantation, and corneal transplantation. Treatment of corneal neovascularization is not always effective and can produce side effects.
Topical treatment
Steroids
Corneal neovascularization is a condition that is usually accompanied by an inflammatory process, so steroidal anti-inflammatory drugs, especially corticosteroids, which have anti-inflammatory and immunosuppressive properties, play an important role. Applied in the form of drops into the conjunctival sac, they can help reduce the local immune response and inhibit the growth of new blood vessels. In some cases, subconjunctival injections of corticosteroids can be considered as they provide higher drug concentrations directly to the affected area, potentially leading to better outcomes with fewer systemic side effects. However, long-term use of topical steroids can cause side effects, which include increased intraocular pressure (glaucoma), cataract formation, and bacterial and fungal superinfections. In addition, steroids have limited anti-angiogenic impact, as they do not reduce mature corneal neovascularization [5].
A potent steroid drug is dexamethasone suspension at a concentration of 0.1%, 1-2 drops into the conjunctival sac up to five times daily. The frequency of administration can be increased, depending on the severity of the condition and response to treatment, up to 1-2 drops into the conjunctival sac every 30-60 minutes for the first few days until improvement occurs. If there is no improvement, subconjunctival or systemic steroid treatment should be considered. Dexamethasone is a synthetic glucocorticosteroid with potent and long-lasting anti-inflammatory, anti-edema, anti-allergic and immunosuppressive effects, which affects all phases of the inflammatory process, inhibits leukocyte migration, blocks IgE-dependent secretion of histamine and leukotrienes, inhibits the synthesis and release of cytokines, inhibits the activity of phospholipase A2 and prevents the release of arachidonic acid and the synthesis of inflammatory mediators (leukotrienes and prostaglandins), inhibits the migration of phagocytic cells in response to an inflammatory stimulus, inhibits the release of kinins and the production of antibodies, reduces capillary permeability and edema [6,7]. Dexamethasone 0.1%, administered into the conjunctival sac, is absorbed into the aqueous fluid, cornea, iris, choroid, ciliary body, and retina. Absorption of the active ingredient from the conjunctival sac into the general circulation is so minimal that pharmacokinetics and systemic effects are clinically insignificant. Steroid drugs also have adverse effects and can contribute to the development of infections, steroid glaucoma, and posterior chamber cataracts; they can also promote the recurrence of Herpes simplex [8].
The group of topically administered steroid drugs also includes loteprednol etabonate 0.5%, 1-2 drops up to four times a day. Loteprednol etabonate is synthesized by structural modification of prednisolone derivative compounds and belongs to the topical corticosteroids, which are transformed into inactive carboxylic acid derivatives. It shows a potent anti-inflammatory effect but has little effect on intraocular pressure and the development of steroid glaucoma. The increase in pressure occurs after a much longer period of administration compared to prednisolone acetate. In clinical trials, only 1.7% of patients experienced increased intraocular pressure (≥10 mm Hg). Few patients showed significant intraocular pressure elevation, but this quickly returned to normal after discontinuing the drug [9].
The 0.1% aqueous solution of betamethasone acetate is used topically in a dosage regimen of four to eight times daily. The higher concentration, i.e., 0.2%, has a potency similar to dexamethasone, a better safety profile, and requires less frequent administration. Its effect persists for several hours.
Fluorometholone, 0.1% suspension, is applied two to four times daily, one drop into the conjunctival sac. During the first 24 to 48 hours, the dosage can be increased by administering one drop every four hours.
Prednisolone Sodium Phosphate 0.5% and 1.0%; at the lower concentration, it can be used every 1-2 hours, 1-2 drops into the conjunctival sac, and at the higher concentration, even every hour for the first two days.
Other topical corticosteroids include difluprednate 0.05%, approved for use four times daily, loteprednol etabonate 1.0% suspension used twice daily, and loteprednol etabonate ophthalmic gel 0.38% used three times daily [10]. The latter reduces the number of inflammatory cells in the anterior chamber and significantly alleviates ocular pain (74.4%) with only minimal intraocular pressure elevation [11].
In more severe cases of corneal neovascularization associated with underlying retinal diseases, intravitreal steroid injections may be an option. This therapeutic approach is less commonly used, but it can indirectly inhibit the corneal neovascularization process in some severe cases.
Triamcinolone acetonide 4% applied to the vitreous body is an option for the treatment of neovascular lesions, both anterior and posterior, and vitreoretinal proliferation, often associated with macular edema. The most common complications of intravitreal triamcinolone include secondary ocular hypertension in about 40% of eyes, glaucoma, and cataracts. The duration of action of a 20 mg dose of triamcinolone administered to non-vitrectomized eyes is about 6-9 months. Intravitreal triamcinolone acetonide can also be an adjunctive treatment for neovascular and edematous disorders [12].
Sustained-release dexamethasone, a biodegradable intravitreal implant measuring approximately 0.46 mm in diameter and 6 mm in length, is injected into the vitreous chamber at a single dose of 0.7 mg. Following application, the active ingredient gradually diffuses from the implant, providing therapeutic concentrations for 5-6 months. Sustained-release dexamethasone therapy can be repeated; recurrence of macular edema is an indication for re-treatment. The implant matrix comprises a polylactide polymer and poly-glycolic acid, which biodegrades by hydrolysis to lactic and glycolic acid and subsequently to carbon dioxide and water [13].
Fluocinolone Acetonide (FAc) intravitreal implant 0.19 mg is a non-biodegradable, injectable corticosteroid microimplant that releases fluocinolone acetonide at an initial rate of 0.25 µg/day (average dose 0.2 µg/day) and remains effective for 36 months. Fluocinolone acetonide is a medium potency glucocorticoid receptor agonist and does not exhibit mineralocorticoid activity. The primary mechanism of action, as with other corticosteroids, is to stimulate an increase in lipocortin synthesis, especially phospholipase A2, which prevents the formation of prostaglandins and leukotrienes, potent mediators of inflammation, by inhibiting the release of their common precursor, arachidonic acid, from the phospholipid membrane. In addition to their anti-inflammatory effects, steroids injected into the vitreous chamber reduce intravitreal levels of vascular endothelial growth factor (VEGF), resulting in regression of active neovascularization. FAc has been shown to inhibit leukocyte migration, the release of heparin, growth and angiogenic factors, and the secretion of pro-inflammatory cytokines that stimulate VEGF production. As with other corticosteroids, the most common adverse events associated with fluocinolone acetonide intravitreal implants are cataracts and elevated intraocular pressure [14]. The Fluolocinolone Acetonide implant is registered for treating diabetic macular edema and non-infectious uveitis, and other uses are off-label [15].
Systemic corticosteroids taken orally can be used in cases where corneal neovascularization is associated with systemic inflammation or autoimmune diseases. However, systemic steroid use is associated with several potential side effects, so careful consideration of the benefit-risk balance is necessary. The most commonly used oral steroids for treating ophthalmic conditions are prednisone and methylprednisolone.
Prednisone is a synthetic glucocorticosteroid, a derivative of cortisol, an inactive compound metabolized in the liver to active prednisolone, with potent anti-inflammatory effects. It is assumed that 5 mg of prednisone has an anti-inflammatory effect equivalent to 4 mg of methylprednisolone or triamcinolone, 0.75 mg of dexamethasone, 0.6 mg of betamethasone, and 20 mg of hydrocortisone. The usual doses range from 5-60 mg of prednisone per day. The dose should be determined individually depending on the disease type and treatment response. Before planned discontinuation of the drug, e.g., when the desired therapeutic effect has been achieved, it is advisable to gradually reduce the dose by 2-5 mg every 2-7 days to the lowest effective dose or until complete discontinuation.
Prednisone reduces the accumulation of leukocytes and their adhesion to the endothelium of capillary vessels, inhibits phagocytosis and lysosome breakdown, reduces the number of lymphocytes, eosinophils, monocytes, blocks IgE-dependent secretion of histamine and leukotrienes. In addition, it inhibits the synthesis and release of cytokines: interferon γ, interleukins IL-1, IL-2, IL-3, IL-6, TNF-α, and GM-CSF. By inhibiting the activity of phospholipase A2 via lipocortin, prednisone prevents the release of arachidonic acid and the synthesis of inflammatory mediators (leukotrienes and prostaglandins). By reducing capillary permeability, it decreases tissue edema.
Prednisone also exhibits immunosuppressive effects. The mechanisms of immunosuppressive action have yet to be entirely understood. Still, it is known that prednisolone can prevent or inhibit cellular immune responses, as well as specific mechanisms related to the immune response. It reduces the number of T lymphocytes, monocytes, and acidophilic granulocytes. It also reduces the binding of immunoglobulins to receptors on the cell surface. Prednisone inhibits the synthesis or release of interleukins by decreasing T-lymphocyte blastogenesis and reducing the severity of the early immune response. It can also impede the penetration of immune complexes across basement membranes and reduce the concentration of complement components and immunoglobulins.
Complications of prednisone use include corneal deposits in the form of multicolored crystals, highly refractive, located mainly subepithelially and in the anterior corneal stroma [16].
Methylprednisolone aceponate produces a greater anti-inflammatory effect, causes less sodium and water retention than prednisolone, and is at least four times more potent than hydrocortisone. It acts mainly intracellularly at the DNA level, so its action is delayed after administration. Methylprednisolone reduces the accumulation of leukocytes and their adhesion to the endothelium, inhibits phagocytosis and lysosome breakdown, reduces the number of lymphocytes, eosinophils, and monocytes, blocks IgE-dependent histamine and leukotriene secretion, inhibits the synthesis and release of cytokines such as interferon-gamma, interleukins IL-1, IL-2, IL-3, IL-6, TNF-α, inhibits the activity of phospholipase A2, prevents the release of arachidonic acid, and, consequently, the synthesis of inflammatory mediators. Methylprednisolone aceponate also inhibits capillary permeability and reduces edema.
The dosage of methylprednisolone depends on the type and severity of the disease and the patient's response to the drug. As a rule, the daily dose should be taken in the morning, between 6 a.m. and 8 a.m., so as not to disrupt the hypothalamic-pituitary-adrenal axis. After long-term treatment, especially with relatively high doses, methylprednisolone should not be abruptly discontinued, but its dose should be gradually reduced. Usually, relatively high initial doses are used, i.e., up to 48 mg daily, and sometimes even higher in acute conditions. Depending on the course of the disease and response to treatment, the dose can be reduced to as low a maintenance dose as possible, generally to 4-12 mg of methylprednisolone per day. Long-term use with low maintenance doses is often necessary in treating chronic diseases. In cases where methylprednisolone doses need to be discontinued or reduced, daily doses above 12 mg should be reduced by 4 mg every day or every few days, and daily doses up to 12 mg are reduced by 2 mg every 2 to 3 days or by 4 mg every 4 to 6 days. In the last week, 2 mg of methylprednisolone should be taken every other day. Intermittent treatment consists of a double daily dose used every other morning. Such a regimen aims to ensure a favorable profile of glucocorticosteroids and minimize adverse effects, such as hypothalamic-pituitary-adrenal axis inhibition or Cushing's syndrome. Long-term use of corticosteroids can cause posterior subcapsular and nuclear cataracts, especially in children, increased intraocular pressure, and glaucoma. In most studies reviewed, the development of ocular hypertension in children occurred within a month of starting steroid treatment [17]. With prolonged steroid therapy, the prevalence of secondary fungal and viral eye infections increases, and central serous chorioretinopathy (CSR) may develop.
Steroids can be combined with other treatments, such as anti-VEGF treatment, whose direct therapeutic goal is to inhibit the growth of new blood vessels. Such combination therapy aims to reduce the inflammatory process and pathological vessel growth.
Nonsteroidal anti-inflammatory drugs (NSAIDs)
Nonsteroidal anti-inflammatory drugs (NSAIDs) have limited effects on corneal neovascularization. Some evidence suggests that NSAIDs may help inhibit the angiogenic process associated with corneal neovascularization by reducing proinflammatory molecules that contribute to developing abnormal blood vessels in the cornea. However, the effectiveness of NSAIDs in treating corneal neovascularization can vary, and their use does not always lead to complete regression of abnormal blood vessels. Non-steroidal anti-inflammatory drugs (NSAIDs) should be used with caution, as they can cause corneal ulceration and melting [18] and neurotrophic keratitis [19].
Of the many topical NSAIDs, 0.09% bromfenac sodium ophthalmic solution is clinically useful. One drop is instilled in the treated eye twice daily for two weeks. The duration of treatment should be at most two weeks due to the lack of safety data for more extended use. Bromfenac exerts anti-inflammatory effects by inhibiting cyclooxygenase 2 (COX-2) and blocking prostaglandin synthesis. Bromfenac likely inhibits the formation of corneal neovascularization and reduces the expression of VEGF and COX-2 in corneal tissue after alkali burns [20]. In addition, bromfenac also significantly decreased vascular endothelial growth factor (VEGF) level and monocyte chemoattractant protein-1 level [21].
Another topical NSAID is nepafenac at a concentration of 0.1% and 0.3%. The recommended dose is 1 drop three times daily at a concentration of 0.1% or 1 drop once daily at 0.3% into the conjunctival sac for two weeks. The administration period can be extended to three weeks. Nepafenac is a pro-drug of amphenac. After topical administration into the eye, nepafenac penetrates the cornea and undergoes rapid bioactivation to amfenac, a potent non-steroidal anti-inflammatory drug. Like other NSAIDs, its anti-inflammatory effect consists of inhibiting cyclooxygenase 2 (COX-2) and blocking prostaglandin synthesis. Chronic use of nepafenac can lead to corneal epithelial damage, thinning, ulceration, or corneal perforation [22].
The most widely used NSAID is diclofenac sodium, an aminophenylacetic acid derivative with potent anti-inflammatory, analgesic, and antipyretic effects. It is used as 0.1% drops three to five times daily for as long as necessary, but the treatment should not exceed four weeks. The drug's action is mainly based on the inhibition of cyclooxygenases involved in the synthesis of prostaglandins, which act as mediators of inflammation. However, diclofenac has a greater inhibitory effect on cyclooxygenase-1, a constitutive enzyme engaged in the synthesis of prostaglandins with physiological functions, than on inducible cyclooxygenase-2, responsible for the synthesis of pro-inflammatory prostaglandins at the site of inflammation. In addition, it reversibly inhibits platelet aggregation stimulated by adenosine diphosphate (ADP) and collagen. The most common adverse effect is a transient burning sensation in the eyes of mild to moderate intensity. Damage to the corneal epithelium and the development of punctate keratitis may occur during long-term use of diclofenac eye drops. A hydrogel combining diclofenac sodium with bevacizumab has been shown to have better anti-angiogenic effects than a drug containing bevacizumab alone due to its synergistic anti-VEGF and anti-inflammatory effects [23]. NSAIDs are thus an effective adjunct to anti-angiogenic treatment for corneal neovascularization.
Cyclosporin A
Cyclosporin A is a cyclic immunomodulatory polypeptide with immunosuppressive activity. Cyclosporin is usually used at a concentration of 0.1%, 1 drop, once a day before bedtime. It is recommended to use nasolacrimal duct occlusion and close the eyelids for 2 minutes after administration to reduce systemic absorption of the drug. Occasionally, prescription drops are prepared at a higher concentration of 0.5-2% when the corneal disease process is severe or 0.05%-0.1% when there are clinical indications for lower concentrations. Cyclosporin A inhibits the production and/or release of pro-inflammatory cytokines, including interleukin 2 (IL-2), also known as T-cell growth factor (TCGF). It also specifically and reversibly inhibits lymphocytes in the G0 or G1 phase of the cell cycle. After administration into the conjunctival sac, cyclosporin A is passively absorbed by T lymphocytes infiltrating the cornea and conjunctiva, where it inactivates calcineurin phosphatase. Inactivation of calcineurin inhibits the dephosphorylation of the transcription factor NF-AT and prevents the translocation of NF-AT into the cell nucleus, thereby blocking the release of pro-inflammatory cytokines such as IL-2. Hence, cyclosporin A is an immunosuppressive drug that inhibits the immune system response, particularly the activation of T lymphocytes. In the context of corneal neovascularization, the anti-inflammatory and immunosuppressive properties of cyclosporine may help control the angiogenic process. Eye drops containing cyclosporine allow direct application of the drug to the affected area to inhibit the growth of new blood vessels and reduce inflammation in the cornea. Sonmez, Beden, and Erkan showed that topical cyclosporine 0.05% could cause regression of corneal stromal neovascularization and, in selected cases, reduce the risk of corneal graft rejection [24]. In contrast, topical treatment with cyclosporine 0.1% appears to be more effective in inhibiting newly formed corneal neovascularization than bevacizumab eye drops applied to the conjunctival sac [25]. It should be noted that using cyclosporine to treat corneal neovascularization is not a first-line treatment. Depending on the cause of the neovascularization and the patient's overall condition, other therapeutic options such as corticosteroids, anti-VEGF preparations, and even surgical interventions should be considered [25,26].
Inhibitors of vascular endothelial growth factor (anti-VEGF)
Inhibitors of vascular endothelial growth factor (anti-VEGF) play a vital role in the process of angiogenesis, the growth of new blood vessels in various tissues, including the cornea. Anti-VEGF preparations, used as drops or subconjunctival injections, such as bevacizumab, aflibercept, and ranibizumab, bind to VEGF and prevent interaction with its receptors, thereby reducing the angiogenic response. Newly formed vessels show a good response to treatment with anti-VEGF agents, in contrast to mature vessels in chronic neovascularization, when these drugs are less effective due to abundant pericyte recruitment to the sites of angiogenesis. Therefore, anti-VEGF therapy targets new blood vessels, is a symptomatic treatment, and requires repetition to maintain efficacy over time [27]. Currently, there are several commonly used anti-VEGF drugs.
Ranibizumab is a fragment of a recombinant humanized monoclonal antibody produced in Escherichia coli cells using recombinant DNA technology, with a molecular weight of 48 kDa and high affinity to human vascular endothelial growth factor type A (VGEF-A). Ranibizumab binds to and therefore blocks all VEGF-A isoforms (e.g., VEGF110, VEGF112, and VEGF165), thereby preventing VEGF-A from binding to its receptors VEGFR-1 and VEGFR2. This is important because the binding of VEGF-A to receptors leads to the proliferation of endothelial cells and the formation of new vessels. Since ranibizumab is an antigen-binding Fab fragment without an Fc domain, its size is approximately one-third that of bevacizumab. Therefore, ranibizumab may have better penetration into the cornea than bevacizumab [28]. Ranibizumab exhibits anti-angiogenic properties by simultaneously suppressing the growth of blood and lymphatic vessels, which accentuates its therapeutic potential in the treatment of corneal neovascularization [29]. Topical 1% ranibizumab, in drop form, reduces the area of stable neovascularization and decreases vessel diameter, while no reduction in vessel length was observed [30]. In addition, ranibizumab administered subconjunctivally significantly decreases the level of VEGF in the aqueous fluid, reducing the area of neovascularization at the iridocorneal angle and iris. Therefore, ranibizumab might also be applied to treat neovascular glaucoma [31]. Subconjunctival ranibizumab injections at 0.5 mg/0.05 ml doses are administered once or repeated after 1 to 2 months, depending on the clinical effect.
Aflibercept, referred to in the literature as "VEGF Trap," is a recombinant fusion protein consisting of the extracellular components of VEGFR-1 and VEGFR-2 fused to the Fc fragment of human IgG1. Aflibercept is produced by recombinant DNA technology; it is a glycoprotein dimer with a molecular weight of 115 kDa. Aflibercept binds to circulating VEGF, acting as a "VEGF trap." Thus, it inhibits the activity of the vascular endothelial growth factor subtypes, i.e., VEGF-A and VEGF-B, and placental growth factor (PGF), suppressing the growth of new blood vessels. Aflibercept inhibits bFGF-induced corneal neovascularization and can be used in eyes previously treated with bevacizumab or ranibizumab [32]. A comparative analysis of the cytotoxic effects of bevacizumab, ranibizumab, and aflibercept showed that ranibizumab and aflibercept caused less corneal epithelial damage in patients with pre-existing corneal epithelial lesions [33,34]. Aflibercept can be administered as conjunctival drops three times daily at a dose of 2mg/0.05 ml [35] or subconjunctivally at a dose of 2mg/0.05 ml at a time. Subconjunctival injection can be repeated after 1 to 2 months, depending on the clinical effect.
Brolucizumab is a humanized monoclonal single-chain antibody fragment (scFv) produced by recombinant DNA synthesis in Escherichia coli cytoplasm. It acts as a VEGF inhibitor. The US FDA approved the drug for treating wAMD in 2019. Reports on the use of brolucizumab eye drops or subconjunctival injections for treating corneal neovascularization are yet to be published.
Bevacizumab is a recombinant humanized monoclonal antibody produced using DNA technology. Topical, subconjunctival, and intraocular application of bevacizumab can reduce corneal neovascularization and improve corneal transparency. Vascular diameter and neovascularization area are reduced by 24% and 61%, respectively [36]. Maximum effects are observed with early topical administration of bevacizumab. Subconjunctival bevacizumab injections are also effective in treating corneal neovascularization [37]. A comparison between subconjunctival administration and topical application of bevacizumab has shown that both methods effectively inhibit corneal angiogenesis and reduce inflammation. However, there is concern that topical but not subconjunctival bevacizumab may weaken corneal epithelial adhesion to the basement membrane, causing delayed wound healing and thinning of the corneal stroma. These adverse effects increase with higher doses (>1.0%) and longer treatment duration (> one month) [38,39]. Topical 1.25% bevacizumab can cause epitheliopathy in the second month of treatment, but at a concentration of 0.5%, the prevalence of corneal epithelial defects is low [40]. Bevacizumab can be administered subconjunctivally at a dose of 1.25mg/0.05ml; the injection can be repeated after 1 to 2 months, depending on the clinical effect. Higher concentrations of bevacizumab administered subconjunctivally, i.e., 2.5mg/0.1ml, have also been reported [41].
Despite the extensive literature confirming the effectiveness of treating corneal neovascularization with subconjunctival injections or drops of anti-VEGF drugs, this is an off-label treatment. The patient is required to sign an informed consent before treatment.
Tissue inhibitors of metalloproteinases(TIMP)
Tissue inhibitors of metalloproteinases (TIMPs) are endogenous inhibitors produced by cells to regulate MMP activity. TIMPs bind to active MMPs and inhibit their enzymatic function. There are four known TIMPs, i.e., TIMP-1, TIMP-2, TIMP-3, and TIMP-4. All are similar in structure but differ in their degree of affinity and expression profile. Their activity is regulated by cytokines and growth factors. TIMP-2, -3, and -4 inhibit all known human extracellular matrix proteases. A non-selective inhibitor of metalloproteinases is a tetracycline group antibiotic, doxycycline, which inhibits metalloproteinase activity by chelating zinc and calcium ions. In inflammatory conditions, suppression of enzymes that compromise the cornea's structural integrity can block corneal neovascularization. Oral doxycycline is used at a dose of 100 mg twice daily on the first day of treatment, followed by a maintenance dose of 100 mg once daily. Combining orally administered doxycycline with topical corticosteroids has inhibited neovascularization [42].
Other drugs
Rapamycin (sirolimus), a macrolide antibiotic immunosuppressant, effectively reduces angiogenesis, probably by inhibiting proinflammatory cytokines. For this reason, rapamycin could be used as a regulator of angiogenesis in treating corneal diseases manifested by neovascularization. Rapamycin has been effective in Herpes-1 virus (HSV-1)-induced interstitial keratitis[43]. Rapamycin effectively down-regulates mRNA expression levels of tumor necrosis factor-α (TNF-α) and interleukin-1beta (IL-1β). It also inhibits neutrophil and macrophage infiltration and suppresses inflammation-related angiogenesis mediated by matrix metalloproteinase-2 (MMP-2). Besides, rapamycin inhibits inflammation induced by corneal alkali burns by regulating angiogenesis through HIF-1α/VEGF, serum cytokines TNF-α, IL-6, interferon-gamma (IFN-γ), and granulocyte-macrophage colony-stimulating factor (GM- CSF) [44]. The initial dose is 2 mg daily but can be modified depending on the clinical effect, no more often than every 7-14 days.
Tacrolimus is used subconjunctivally at 0.25 mg/0.05 ml and as conjunctival drops at 5 mg/5 ml four times daily [45]. Topical tacrolimus, applied as an ointment at a dose of 0.2 mg/g twice daily to inhibit corneal graft rejection due to neovascularization, is safe and effective in improving graft survival in high-risk patients [46]. Tacrolimus eye drops, a concentration of 0.03%, have effectively prevented irreversible corneal graft rejection in high-risk patients [47].
Other potential therapeutic targets
Interleukin-1 receptor antagonist (IL-1 Ra) is a naturally occurring protein that is crucial in regulating the inflammatory response in various tissues. It seems to show promise in the treatment of corneal neovascularization. IL-1 Ra belongs to the interleukin-1 (IL-1) family of proteins, along with IL-1α, IL-1β, interleukin-18. Recombinant IL-1 Ra is currently used in rheumatoid conditions as an anti-inflammatory drug, inhibiting the biological activity of IL-1α and IL-1β. IL-1 antagonist (IL-1 Ra) is produced under the influence of cytokines, viral antigens, and acute phase proteins, and is a limiting factor for IL-1β in the chronic phase of inflammation. There are two types of receptors for IL-1, i.e., IL-1 type 1 receptor (IL-1RI) and IL-1 type 2 receptor (IL-1 RII). Stimulation of IL-1RI involving an additional IL-1RAcP protein induces signal transduction and action depending on the type of cell undergoing activation. Binding to IL-1RII does not lead to cell signaling. IL-1Ra binds to both receptors I and II [48]. IL-1Ra has been studied for its potential role in treating corneal neovascularization due to its ability to modulate the inflammatory response. It works by binding to the IL-1 receptor and preventing the proinflammatory effects of IL-1, including the expression of proangiogenic molecules such as VEGF and iNOS. By blocking the action of IL-1 (IL-1α and IL-1β), IL-1 Ra can help reduce inflammation and potentially inhibit the growth of new blood vessels in the cornea [1,49]. Interleukin-1 receptor antagonist eye drops, at a concentration of 2.5%, were effective and well tolerated in treating epitheliopathy [50].
Nitric oxide synthase (NOS)
There are three isoforms of NOS: endothelial NOS (eNOS or NOS1), neuronal NOS (nNOS or NOS2), and inducible NOS (iNOS or NOS3). The role of nitric oxide synthase in corneal neovascularization is complex. Endothelium-derived NOS is found in vascular endothelial cells and is critical in regulating vascular tone and blood flow. It can promote angiogenesis by stimulating the proliferation and migration of endothelial cells, which are essential for forming new blood vessels. On the other hand, it may also have anti-angiogenic effects by inhibiting signaling mediated by VEFG, a major driver of angiogenesis.
Neuronal NOS is mainly found in nerve cells and is involved in neurotransmission. Its role in corneal neovascularization is unclear, and its involvement may be limited compared to other isoforms.
Inducible NOS is typically not expressed in healthy tissues but can be induced by various pro-inflammatory factors such as cytokines and bacterial endotoxins. iNOS-derived nitric oxide often contributes to inflammation and immune responses. iNOS expression may promote angiogenesis by recruiting immune cells and facilitating tissue remodeling.
Interactions between NOS isoforms and their impact on corneal neovascularization may vary depending on the underlying causes of neovascularization and the overall inflammatory microenvironment. In addition, the balance between nitric oxide's proangiogenic and anti-angiogenic effects complicates understanding its role. Research into the molecular processes involved in corneal neovascularization continues, and potential therapeutic strategies targeting NOS signaling are being developed [51].
Galectin-3 inhibitors
The therapeutic success of anti-VEGF drugs is undeniable; however, their effect is limited against mature neovascularization, whose vessels are covered with pericytes. Galectin-3 inhibitors appear promising, as they can attenuate the course of corneal neovascularization and affect fibrotic processes in the cornea [52].
Pigment Epithelium-Derived Factor (PEDF)
Pigment Epithelium-Derived Factor can inhibit angiogenesis via VEGF, bFGF, and interleukin-8 (IL-8/CXCL8) by inhibiting endothelial cell migration to areas of neovascularization and concomitantly inducing cell apoptosis [53]. It has been suggested that using exogenous PEDF may help inhibit the growth of new blood vessels and reduce corneal neovascularization [54].
PDGFreceptor inhibitor
Intraperitoneal injection of a PDGF receptor inhibitor has been shown to reduce pericapillary pericytes and reduce vascular density in advanced corneal neovascularization. PDGF is a protein that plays a significant role in cell growth and division. It is released by platelets, cells involved in blood clotting, wound healing, and tissue repair processes. PDGF has been shown to have both proangiogenic and mitogenic effects. PDGF can interact with other growth factors and cytokines. This interaction can enhance the angiogenic response and contribute to the development of new blood vessels [55].
Aganirsen, an experimental drug in the form of squalamine lactate eye drops, has been studied for its potential in treating corneal neovascularization. It dose-dependently inhibits IRS-1 expression and angiogenesis while, at the same time, reducing VEGF-A and the proinflammatory cytokine IL-1b.Topical application of aganirsen was well tolerated and showed no side effects. After 90 days, the area of corneal neovascularization was reduced by 26.20%; the improvement persisted after 180 days [56,57].
Surgical treatment
Fine needle diathermy
Fine needle diathermy (FND) is commonly used to occlude corneal vessels. One way to perform it is to use a 3/8 needle with a 10-0 nylon suture [58,59]. The procedure involves inserting the needle close to the limbus, parallel to the vessel, and at the same depth as the vessel to be closed. With larger vessels, it was possible to insert the needle tip into the vessel lumen. Diathermy is continued until mild whitening of the corneal stroma. This procedure is repeated in other areas affected by neovascularization. It can be performed simultaneously with corneal transplantation, thus reducing the risk of intraoperative bleeding and graft rejection[60]. Complications of needle diathermy are reversible and include intracorneal stromal hemorrhages, transient corneal opacity, transient corneal whitening in the stroma adjacent to the needle, which completely resolves after 24-48 hours, and microperforations, especially with thin corneas [2]. This method may have adverse effects through the impact of heat energy on limbal stem cells and endothelium. It may also secondarily lead to neovascularization due to the release of proangiogenic substances. To reduce FND-related complications, a procedure modification has been introduced, where diathermy is performed under fluorescein angiography guidance [60,61].
Laser therapy
Laser therapies, namely argon laser photocoagulation and the 577 nm yellow laser, can be used to obliterate abnormal blood vessels in the cornea, thus preventing the rejection of the transplanted flap, and are also used in the treatment of lipid keratopathy. Laser photocoagulation facilitates obliteration of efferent vessels with a large diameter and slower blood flow. In contrast, it is difficult to obliterate afferent vessels because they are thinner and more deeply located in the cornea and are characterized by rapid blood flow. In their case, laser therapy is ineffective, and when repeated, thermal damage may promote the recurrence of neovascularization [62,63].Complications of laser therapy can include corneal endothelial damage, corneal thinning, intraocular hemorrhage, pupillary displacement, iris atrophy, and corneal suture lysis after corneal transplantation [2].
Photodynamic therapy (PDT) safely removes corneal neovascularization. Intrastromal injections of the photosensitizing substance verteporfin selectively induce regression of lymphatic vessels[64,65]. In addition, PDT generates reactive oxygen species that destroy the endothelium and vascular basement membrane, causing local thrombosis and leading to vascular remodeling. This minimally invasive treatment leads to the eradication of the pathological corneal vascular network without damaging healthy tissue. Additionally, it has minimal systemic effects, making it safe when multiple sessions are needed [66,67].
Nd:YAG laser photocoagulation (532 nm) can reduce the area of corneal neovascularization without significant side effects. At three months after the completion of frequency-doubled Nd:YAG (532 nm) laser photocoagulation, the reduction in corneal opacity area and corneal neovascularization area was 7.01% and 44.08%, respectively [68].
A femtosecond laser also appears to be effective for treatment for corneal neovascularization. Until now, studies have only been conducted on an animal model. Due to the high equipment cost, this treatment method is not widely used [2].
Amniotic membrane transplantation
Treatment of corneal neovascularization may involve suturing the amniotic membrane, which has anti-inflammatory and anti-angiogenic effects [60]. The amniotic membrane consists of a monolayer of epithelium, a thick basement membrane, and a nonvascularized, hypocellular matrix. It releases TIMP2 proteins that significantly reduce basic fibroblast growth factor (bFGF) inhibitingthe proliferation of fibrovascular tissue and the differentiation of fibroblasts into myofibroblasts is also inhibited.
The amniotic membrane is ideal for supporting corneal epithelial proliferation. Laminin, fibronectin, type IV, V, VII collagen, and integrins 4 and 6, which are found in the amniotic membrane, can stimulate the differentiation and hyperplasia of corneal epithelial cells, increase their adhesion to the basement membrane and replenish limbal stem cells. The amniotic basement membrane, by its composition, is similar to the basement membrane of the conjunctiva and cornea, making it an almost ideal material for reconstructing the ocular surface [69,70].
Superficial keratectomy
Superficial keratectomy is a surgical procedure to remove the superficial layers of the cornea, i.e., the epithelium, Bowman's membrane, and sometimes the superficial layers of the corneal stroma. It can be used to treat a variety of corneal conditions, including neovascularization. The purpose of superficial keratectomy is to remove abnormal blood vessels that reduce corneal translucency and impair corneal function, leading to corneal scarring, inflammation, decreased visual acuity, and even vision loss. Intraoperatively, mitomycin C 0.02% can be used to reduce postoperative corneal haze or amniotic membrane transplantation to improve surgical outcomes. Superficial keratectomy eliminates the underlying cause, i.e., abnormal blood vessels, therefore stimulating the cornea's natural healing processes. After the procedure, the cornea begins to heal, and the surrounding healthy tissue regenerates. Superficial keratectomy is often combined with other treatments, such as topical or laser therapy. These additional treatments can help control the underlying condition that caused the neovascularization. It should be noted that although superficial keratectomy can be effective in treating corneal neovascularization, the success of the procedure depends on various factors, including the cause of the neovascularization, the degree of blood vessel growth, and the patient's overall eye condition. In addition, like any surgical procedure, superficial keratectomy may involve complications, such as infection, delayed healing, or recurrence of neovascularization [71].
Corneal transplantation
Corneal transplantation is performed in severe cases where neovascularization has led to significant impairment of visual acuity. It should be noted, though, that the ingrowth of new vessels causes a loss of immune preference and increases the risk of graft rejection [64,71]. Patients with corneal neovascularization are prepared for corneal or corneal limbus transplantation by subconjunctival injection of anti-VEGF solution. Anti-angiogenic treatment is also administered after corneal transplantation to prevent rejection of the transplanted flap and continues after corneal limbus transplantation until a stable epithelium is obtained [60]. For patients with repeated graft failure or rejection due to refractory central corneal neovascularization, placement of a keratoprosthesis may be the treatment of choice.
Combination therapies
Combination therapies show synergistic effects and translate into a better therapeutic outcome. Combining anti-inflammatory drugs and anti-angiogenic agents, e.g., dexamethasone with bevacizumab, is more effective for corneal neovascularization than monotherapy [72]. A marked decrease in corneal angiogenesis can be achieved with bevacizumab in combination with argon laser therapy. Argon laser photocoagulation provides treatment by closing abnormal corneal vessels, while bevacizumab works to prevent new angiogenesis [62,73].
Summary
Corneal neovascularization is a relatively common consequence of severe ocular surface and cornea pathologies, which can lead to significant visual impairment. Vascular neovascularization is a result of a complex mechanism that involves inflammation, hypoxia, innervation disorders, and limbal stem cell deficiency. Continuous advances in understanding new molecular pathways and diagnosing the severity of corneal neovascularization allow for novel and better therapeutic strategies. Treatment options for corneal neovascularization are still dependent on the maturity of the blood vessels. The primary therapy for corneal neovascularization includes anti-inflammatory and anti-VEGF drugs. In addition to VEGF, angiogenesis is also promoted by other mediators such as PDGF, bFGF, nitric oxide, and pro-inflammatory cytokines. Therefore, combination therapies acting on multiple angiogenic pathways may be more effective. In severe limbal stem cell deficiency with deep neovascularization, the best strategy is ocular surface stem cell transplantation (OSST), followed by keratoplasty. Patients with repeated graft failure or rejection may benefit from keratoprosthesis placement as the treatment of choice. Considerable hopes are associated with gene therapy, which may become a universal treatment for pathological corneal vascularization, regardless of the severity of the lesions.
References
- Feizi, S.; Azari, A.A.; Safapour, S. Therapeutic approaches for corneal neovascularization. Eye Vis. 2017, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Sharif, Z.; Sharif, W. Corneal neovascularization: updates on pathophysiology, investigations & management. Romanian J. Ophthalmol. 2019, 63, 15–22. [Google Scholar] [CrossRef]
- Gupta, D. , Illingworth, C. Treatments for corneal neovascularization: a review. Cornea 2011, 30, 927–938. [Google Scholar] [CrossRef]
- Cursiefen, C.; Chen, L.; Dana, M.R.; Streilein, J.W. Corneal lymphangiogenesis: evidence, mechanisms, and implications for corneal transplant immunology. Cornea 2003, 22, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Cursiefen, C.; Wenkel, H.; Martus, P.; Langenbucher, A.; Nguyen, N.X.; Seitz, B.; Kuchle, M.; Naumann, G.O. Impact of short-term versus long-term topical steroids on corneal neovascularization after non-high-risk keratoplasty. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 2001, 239, 514–521. [Google Scholar] [CrossRef]
- Gallemore, R.; Sharareh, B.; Wallsh, J. Therapeutic effect of dexamethasone implant in retinal vein occlusions resistant to anti-VEGF therapy. Clin. Ophthalmol. 2016, 10, 947–954. [Google Scholar] [CrossRef]
- Rezar-Dreindl, S.; Eibenberger, K.; Pollreisz, A.; Bühl, W.; Georgopoulos, M.; Krall, C.; Dunavölgyi, R.; Weigert, G.; Kroh, M.; Schmidt-Erfurth, U.; et al. Effect of intravitreal dexamethasone implant on intra-ocular cytokines and chemokines in eyes with retinal vein occlusion. Acta Ophthalmol. 2016, 95, e119–e127. [Google Scholar] [CrossRef] [PubMed]
- Mukwaya, A.; Mirabelli, P.; Lennikov, A.; Xeroudaki, M.; Schaupper, M.; Peebo, B.; Lagali, N. Genome-wide expression datasets of anti-VEGF and dexamethasone treatment of angiogenesis in the rat cornea. Sci. Data 2017, 4, 170111–170111. [Google Scholar] [CrossRef]
- Novack, G.D.; Howes, J.; Crockett, R.S.; Sherwood, M.B. Change in Intraocular Pressure During Long-Term Use of Loteprednol Etabonate. Eur. J. Gastroenterol. Hepatol. 1998, 7, 266–269. [Google Scholar] [CrossRef]
- Hosseini, K.; Gollamudi, S.; Reiser, H.; Walters, T.; Lindstrom, R.L. 0.2% Betamethasone Sodium Phosphate: A Multicenter, Randomized, Double-Masked Study to Compare Its Ocular Safety, Tolerability, and Efficacy to Vehicle in Cataract Surgery Subjects. Clin. Ophthalmol. 2023, ume 17, 2219–2230. [Google Scholar] [CrossRef]
- Fong, R.; Cavet, M.E.; DeCory, H.H.; Vittitow, J.L. Loteprednol etabonate (submicron) ophthalmic gel 0.38% dosed three times daily following cataract surgery: integrated analysis of two Phase III clinical studies. Clin. Ophthalmol. 2019, 13, 1427–1438. [Google Scholar] [CrossRef]
- Jonas, J.B.; Kreissig, I.; Degenring, R. Intravitreal triamcinolone acetonide for treatment of intraocular proliferative, exudative, and neovascular diseases. Prog. Retin. Eye Res. 2005, 24, 587–611. [Google Scholar] [CrossRef]
- Haller, J.A.; Bandello, F.; Belfort, R., Jr.; Blumenkranz, M.S.; Gillies, M.; Heier, J.; Loewenstein, A.; Yoon, Y.H.; Jiao, J.; Li, X.-Y.; et al. Dexamethasone Intravitreal Implant in Patients with Macular Edema Related to Branch or Central Retinal Vein Occlusion: Twelve-Month Study Results. Ophthalmology 2011, 118, 2453–2460. [Google Scholar] [CrossRef]
- Ayalasomayajula, S.P.; Ashton, P.; Kompella, U.B. Fluocinolone Inhibits VEGF Expression via Glucocorticoid Receptor in Human Retinal Pigment Epithelial (ARPE-19) Cells and TNF-α–Induced Angiogenesis in Chick Chorioallantoic Membrane (CAM). J. Ocul. Pharmacol. Ther. 2009, 25, 97–104. [Google Scholar] [CrossRef]
- Syed, Y.Y. Fluocinolone Acetonide Intravitreal Implant 0.19 mg (ILUVIEN((R))): A Review in Diabetic Macular Edema. Drugs 2017, 77, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.R.; Maguire, L.J.; Salomão, D.R. Bilateral Corneal Deposits 1 Week After Starting Oral Prednisone Therapy. JAMA Ophthalmol 2018, 136, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, U.; Ameen, B.S.; Nie, C.; Nechi, D.; Mazhar, I.J.; Yasir, M.; Sarfraz, S.; Shlaghya, G.; Narayana, S.H.; Khan, S. Association Between the Use of Systemic Steroids and Ocular Hypertension as a Side Effect in Pediatric Population: A Systematic Review. Cureus 2023, 15, e42112. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Nakagami, T.; Mochizuki, M.; Hata, N.; Tsuchiya, T.; Hotta, Y. Three Cases of Corneal Melting After Instillation of a New Nonsteroidal Anti-Inflammatory Drug. Cornea 2006, 25, 224–227. [Google Scholar] [CrossRef]
- Raj, N.; Panigrahi, A.; Alam, M.; Gupta, N. Bromfenac-induced neurotrophic keratitis in a corneal graft. BMJ Case Rep. 2022, 15, e249400. [Google Scholar] [CrossRef]
- Song, L.; Liu, X.; Chen, N.; Liu, J.; Nie, A.; Li, W. The Effect of Bromfenac Sodium Nanopolymer Used in Anterior Segment of the Eye on Corneal Neovascularization. Cell. Mol. Biol. 2022, 68, 330–338. [Google Scholar] [CrossRef]
- Matsumura, T.; Iwasaki, K.; Arimura, S.; Takeda, R.; Takamura, Y.; Inatani, M. Topical bromfenac reduces multiple inflammatory cytokines in the aqueous humour of pseudophakic patients. Sci. Rep. 2021, 11, 6018. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Noriega, K.; Butrón-Valdez, K.; Vazquez-Galvan, J.; Mohamed-Noriega, J.; Cavazos-Adame, H.; Mohamed-Hamsho, J. Corneal Melting after Collagen Cross-Linking for Keratoconus in a Thin Cornea of a Diabetic Patient Treated with Topical Nepafenac: A Case Report with a Literature Review. Case Rep. Ophthalmol. 2016, 7, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhu, Y.; Xing, C.; Li, S.; Bao, Z.; Lei, L.; Lin, D.; Wang, Y.; Chen, H.; Xu, X. An injectable thermosensitive hydrogel for dual delivery of diclofenac and Avastin(R) to effectively suppress inflammatory corneal neovascularization. International journal of pharmaceutics 2022, 625, 122081. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, B.; Beden, U.; Erkan, D. Regression of severe corneal stromal neovascularization with topical cyclosporine 0.05% after penetrating keratoplasty for fungal corneal ulcer. Int. Ophthalmol. 2007, 29, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, D.M.; Kahraman, N.; Balcioglu, E.; Duru, Z. Comparison of the inhibitory effect of topical cyclosporine A 0.1% and topical anti-VEGF application in an experimental model of corneal neovascularization. Arq. Bras. de Oftalmol. 2021, 85, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Cejka, C.; Cejkova, J.; Trosan, P.; Zajicova, A.; Sykova, E.; Holan, V. Transfer of mesenchymal stem cells and cyclosporine A on alkali-injured rabbit cornea using nanofiber scaffolds strongly reduces corneal neovascularization and scar formation. Histology and histopathology 2016, 31, 969–980. [Google Scholar] [PubMed]
- Krizova, D.; Vokrojova, M.; Liehneova, K.; Studeny, P. Treatment of Corneal Neovascularization Using Anti-VEGF Bevacizumab. J. Ophthalmol. 2014, 2014, 178132. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, W.; Cheng, S.-F.; Dastjerdi, M.H.; Ferrari, G.; Dana, R. Corneal Neovascularization and the Utility of Topical VEGF Inhibition: Ranibizumab (Lucentis) Vs Bevacizumab (Avastin). Ocul. Surf. 2012, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Bucher, F.; Parthasarathy, A.; Bergua, A.; Onderka, J.; Regenfuß, B.; Cursiefen, C.; Bock, F. Topical Ranibizumab inhibits inflammatory corneal hem- and lymphangiogenesis. Acta Ophthalmol. 2012, 92, 143–148. [Google Scholar] [CrossRef]
- Ferrari, G.; Dastjerdi, M.H.; Okanobo, A.; Cheng, S.-F.; Amparo, F.; Nallasamy, N.; Dana, R.M. Topical Ranibizumab as a Treatment of Corneal Neovascularization. Cornea 2013, 32, 992–997. [Google Scholar] [CrossRef]
- Liarakos, V.S.; Papaconstantinou, D.; Vergados, I.; Douvali, M.; Theodossiadis, P.G. The Effect of Subconjunctival Ranibizumab on Corneal and Anterior Segment Neovascularization: Study on an Animal Model. Eur. J. Ophthalmol. 2014, 24, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.B.; Sakimoto, T.; Javier, J.A.; Azar, D.T.; Wiegand, S.J.; Jain, S.; Chang, J.H. VEGF Trap(R1R2) suppresses experimental corneal angiogenesis. European journal of ophthalmology 2010, 20, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Choi, H.; Rho, C.R. Differential Effects of Bevacizumab, Ranibizumab, and Aflibercept on the Viability and Wound Healing of Corneal Epithelial Cells. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics 2016, 32, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Sella, R.; Ben Ishai, M.; Livny, E.; Nahum, Y.; Bahar, I. Subconjunctival Aflibercept for the Treatment of Formed Corneal Neovascularization. Eye Contact Lens: Sci. Clin. Pr. 2020, 47, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S. Treatment of corneal neovascularization with topical aflibercept in a case of exposure keratopathy following cerebellar astrocytoma surgery. Indian J. Ophthalmol. 2019, 67, 145–147. [Google Scholar] [CrossRef]
- Koenig, Y.; Bock, F.; Horn, F.; Kruse, F.; Straub, K.; Cursiefen, C. Short- and long-term safety profile and efficacy of topical bevacizumab (Avastin) eye drops against corneal neovascularization. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 2009, 247, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.-S.; Hu, F.-R.; Yang, C.-M.; Yeh, P.-T.; Chen, Y.-M.; Hou, Y.-C.; Chen, W.-L. Subconjunctival Injection of Bevacizumab in the Treatment of Corneal Neovascularization Associated With Lipid Deposition. Cornea 2011, 30, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-I.; Chung, J.L.; Hong, J.P.; Min, K.; Seo, K.Y.; Kim, E.K. Bevacizumab Application Delays Epithelial Healing in Rabbit Cornea. Investig. Opthalmology Vis. Sci. 2009, 50, 4653–4659. [Google Scholar] [CrossRef]
- de Redín, I.L.; Boiero, C.; Recalde, S.; Agüeros, M.; Allemandi, D.; Llabot, J.M.; García-Layana, A.; Irache, J.M. In vivo effect of bevacizumab-loaded albumin nanoparticles in the treatment of corneal neovascularization. Exp. Eye Res. 2019, 185, 107697. [Google Scholar] [CrossRef]
- Al-Debasi, T.; Al-Bekairy, A.; Al-Katheri, A.; Al Harbi, S.; Mansour, M. Topical versus subconjunctival anti-vascular endothelial growth factor therapy (Bevacizumab, Ranibizumab and Aflibercept) for treatment of corneal neovascularization. Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological Society 2017, 31, 99–105. [Google Scholar] [CrossRef]
- Bhatti, N.; Qidwai, U.; Hussain, M.; Kazi, A. Efficacy of sub-conjunctival and topical bevacizumab in high-risk corneal transplant survival. JPMA The Journal of the Pakistan Medical Association 2013, 63, 1256–1259. [Google Scholar] [PubMed]
- Homer, H.C.; Houman, M.S.; Hemmati, D. Treatment of Corneal Neovascularization. EyeNet Magazine October. 2013. [Google Scholar]
- Zapata, G.; Racca, L.; Tau, J.; Berra, A. Topical Use of Rapamycin in Herpetic Stromal Keratitis. Ocul. Immunol. Inflamm. 2012, 20, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, J.; Shi, Y.; Liu, M. Rapamycin inhibits corneal inflammatory response and neovascularization in a mouse model of corneal alkali burn. Exp. Eye Res. 2023, 233, 109539. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Joo, C.-K.; Chung, S.K. Comparative Study of Tacrolimus and Bevacizumab on Corneal Neovascularization in Rabbits. Cornea 2015, 34, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, L.; Gil, J.; Costa, E.; Tavares, C.; Rosa, A.; Quadrado, M.J.; Murta, J. Topical tacrolimus in high-risk corneal transplants. Eur. J. Ophthalmol. 2023, 34, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, O.A.; Marinho, D.R.; Kwitko, S. Topical 0.03% tacrolimus preventing rejection in high-risk corneal transplantation: a cohort study. Br. J. Ophthalmol. 2013, 97, 1395–1398. [Google Scholar] [CrossRef]
- Ren, K.; Torres, R. Role of interleukin-1beta during pain and inflammation. Brain research reviews 2009, 60, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Li, L.; Liu, G.; Zhang, X.; Mukaida, N. Enhanced experimental corneal neovascularization along with aberrant angiogenic factor expression in the absence of IL-1 receptor antagonist. Investigative ophthalmology & visual science 2009, 50, 4761–4768. [Google Scholar]
- Amparo, F.; Dastjerdi, M.H.; Okanobo, A.; Ferrari, G.; Smaga, L.; Hamrah, P.; Jurkunas, U.; Schaumberg, D.A.; Dana, R. Topical interleukin 1 receptor antagonist for treatment of dry eye disease: a randomized clinical trial. JAMA ophthalmology 2013, 131, 715–723. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, G.-Q.; Lu, P.-R. Potential involvement of nitric oxide synthase but not inducible nitric oxide synthase in the development of experimental corneal neovascularization. JAMA ophthalmology 2011, 4, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-S.; Cao, Z.; Leffler, H.; Nilsson, U.J.; Panjwani, N. Galectin-3 Inhibition by a Small-Molecule Inhibitor Reduces Both Pathological Corneal Neovascularization and Fibrosis. Investig. Opthalmology Vis. Sci. 2017, 58, 9–20. [Google Scholar] [CrossRef]
- Mori, K.; Gehlbach, P.; Ando, A.; McVey, D.; Wei, L.; A Campochiaro, P. Regression of ocular neovascularization in response to increased expression of pigment epithelium-derived factor. Investigative ophthalmology & visual science 2002, 43, 2428–2434. [Google Scholar]
- Jin, J.; Ma, J.-X.; Guan, M.; Yao, K. Inhibition of Chemical Cautery–Induced Corneal Neovascularization by Topical Pigment Epithelium–Derived Factor Eyedrops. Cornea 2010, 29, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Dell, S.; Peters, S.; Mu¨ther, P.; Kociok, N.; Joussen, A.M. The Role of PDGF Receptor Inhibitors and PI3-Kinase Signaling in the Pathogenesis of Corneal Neovascularization. Investig. Opthalmology Vis. Sci. 2006, 47, 1928–1937. [Google Scholar] [CrossRef]
- Cursiefen, C.; Viaud, E.; Bock, F.; Geudelin, B.; Ferry, A.; Kadlecova, P.; Levy, M.; Al Mahmood, S.; Colin, S.; Thorin, E.; et al. Aganirsen antisense oligonucleotide eye drops inhibit keratitis-induced corneal neovascularization and reduce need for transplantation: the I-CAN study. Ophthalmology 2014, 121, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Cursiefen, C.; Bock, F.; Horn, F.K.; Kruse, F.E.; Seitz, B.; Borderie, V.; Früh, B.; Thiel, M.A.; Wilhelm, F.; Geudelin, B.; et al. GS-101 Antisense Oligonucleotide Eye Drops Inhibit Corneal Neovascularization: Interim Results of a Randomized Phase II Trial. Ophthalmology 2009, 116, 1630–1637. [Google Scholar] [CrossRef]
- Natarajan, R.; Ravichandran, S. Fine-needle diathermy for corneal vascularisation. Indian J. Ophthalmol. 2022, 70, 1868–1869. [Google Scholar] [CrossRef]
- Pillai, C.T.; Dua, H.S.; Hossain, P. Fine needle diathermy occlusion of corneal vessels. Investigative ophthalmology & visual science 2000, 41, 2148–2153. [Google Scholar]
- Drzyzga, Ł.M.-K.E. Angiogeneza w rogówce i strategie leczenia antyangiogennego. Okulistyka po Dyplomie 2013, 3. [Google Scholar]
- Le, V.N.H.; Schneider, A.-C.; Scholz, R.; Bock, F.; Cursiefen, C. Fine Needle-Diathermy Regresses Pathological Corneal (Lymph)Angiogenesis and Promotes High-Risk Corneal Transplant Survival. Sci. Rep. 2018, 8, 5707. [Google Scholar] [CrossRef] [PubMed]
- Gerten, G. Bevacizumab (avastin) and argon laser to treat neovascularization in corneal transplant surgery. Cornea 2008, 27, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.G.; Donato, E.; Cordeiro, M.d.C.; de Andrade, M.M.; de Freitas, J.A.H. Treating corneal neovascularization using a combination of anti-VEGF injection and argon laser photocoagulation application - case report. Romanian J. Ophthalmol. 2021, 65, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Bucher, F.; Bi, Y.; Gehlsen, U.; Hos, D.; Cursiefen, C.; Bock, F. Regression of mature lymphatic vessels in the cornea by photodynamic therapy. Br. J. Ophthalmol. 2014, 98, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Kim, M.K.; Seo, K.Y.; Ueta, M.; Yoon, K.C. Effectiveness of photodynamic therapy with verteporfin combined with intrastromal bevacizumab for corneal neovascularization in Stevens–Johnson syndrome. Int. Ophthalmol. 2017, 39, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Al-Torbak, A.A. Photodynamic therapy with verteporfin for corneal neovascularization. Middle East African journal of ophthalmology 2012, 19, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.J.; Ambati, B.K.; Marcus, D.M.; Ratanasit, A. Photodynamic therapy for corneal neovascularisation and lipid degeneration. Br. J. Ophthalmol. 2004, 88, 840–840. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Gehra, A.; Sirohi, N. Role of Frequency Doubled Nd. Yag Laser in Treatment of Corneal Neovascularisation. Journal of clinical and diagnostic research: JCDR 2016, 10, NC01–NC04. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Pi, Y.-L. Effect of amnion membrane transplantation on corneal neovascularization in 10 patients with alkali burn. 4. [CrossRef]
- Yang, L.-L.; Zhou, Q.-J.; Wang, Y.; Gao, Y.; Wang, Y.-Q. Comparison of the therapeutic effects of extracts from Spirulina platensis and amnion membrane on inflammation-associated corneal neovascularization. International journal of ophthalmology 2012, 37, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Scorcia, V.; Giannaccare, G.; Lucisano, A.; Vaccaro, S.; Battaglia, C.; Yu, A.C.; Bovone, C.; Busin, M.; Spena, R. Corneal neovascularisation following deep anterior lamellar keratoplasty for corneal ectasia: incidence, timing and risk factors. Br. J. Ophthalmol. 2021, 106, 1363–1367. [Google Scholar] [CrossRef]
- Hoffart, L.; Matonti, F.; Conrath, J.; Daniel, L.; Ridings, B.; Masson, G.S.; Chavane, F. Inhibition of corneal neovascularization after alkali burn: comparison of different doses of bevacizumab in monotherapy or associated with dexamethasone. Clin. Exp. Ophthalmol. 2010, 38, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Reidy, J.J.; Riaz, K.M. Short-term regression of corneal neovascularization with combination therapy of argon green laser photocoagulation and subconjunctival bevacizumab. Int. Med Case Rep. J. 2019, ume 12, 89–92. [Google Scholar] [CrossRef]
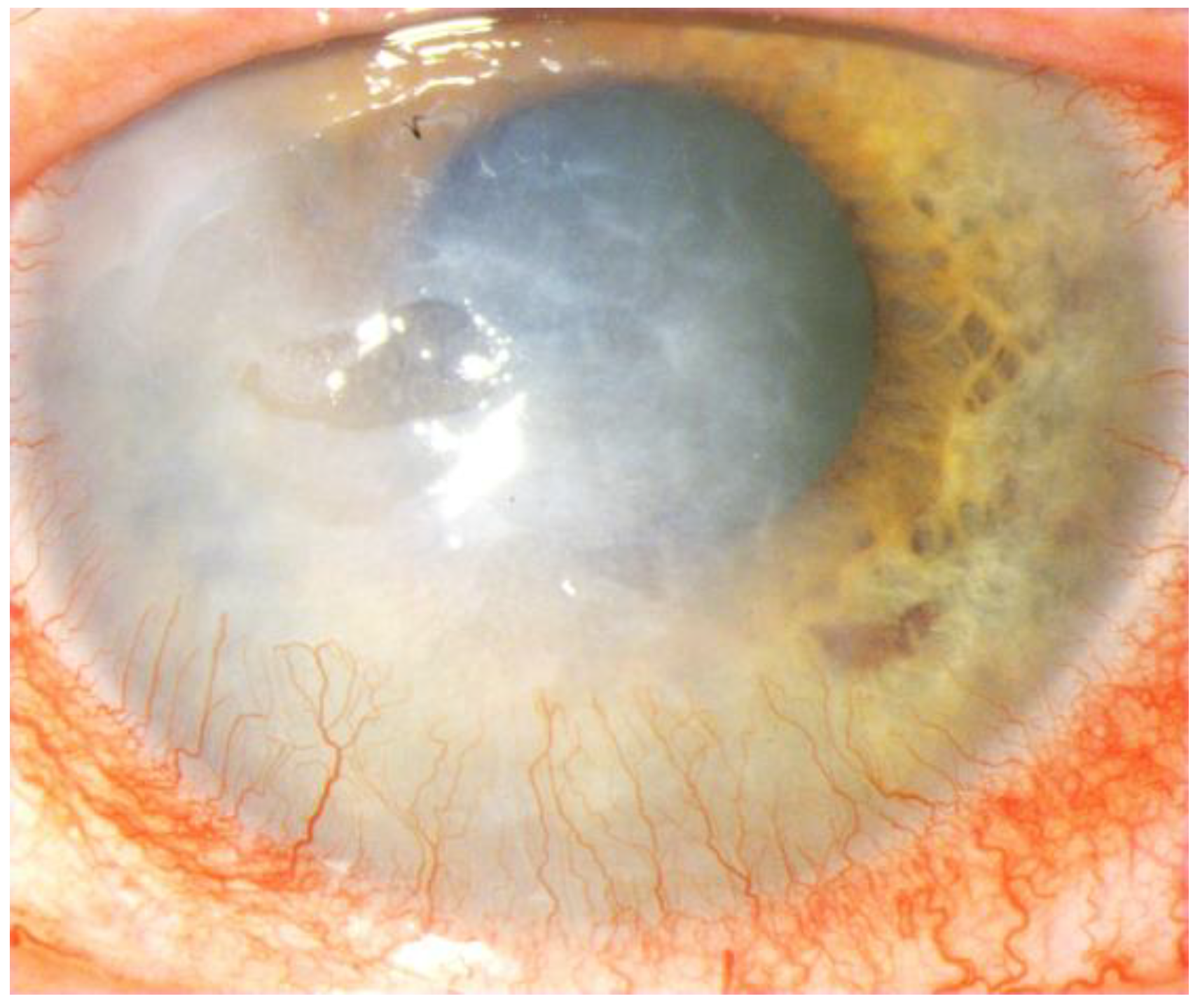
Figure 1.
Corneal neovascularization in autoimmune disease.
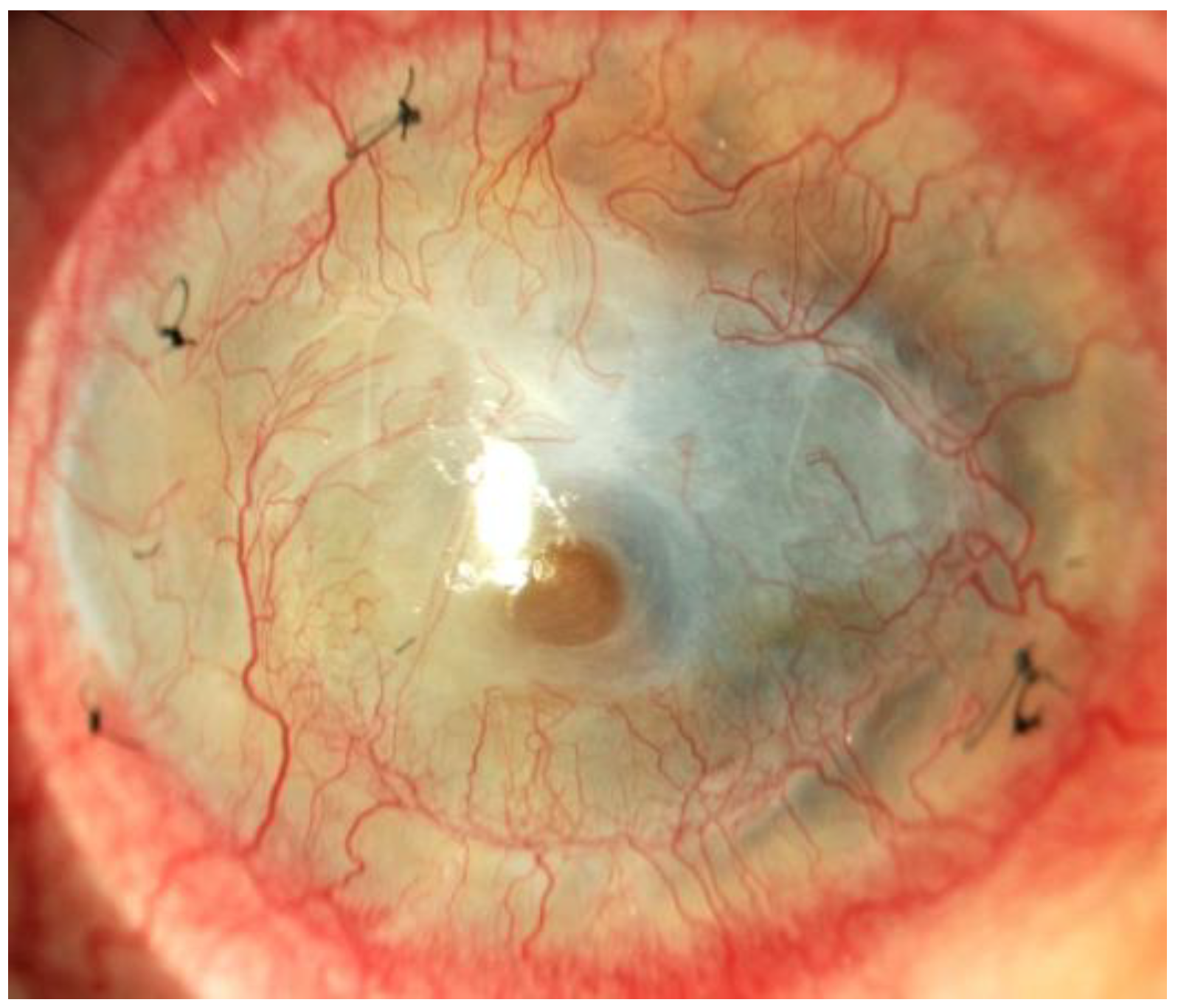
Figure 2.
Corneal neovascularization in crystalline keratopathy.
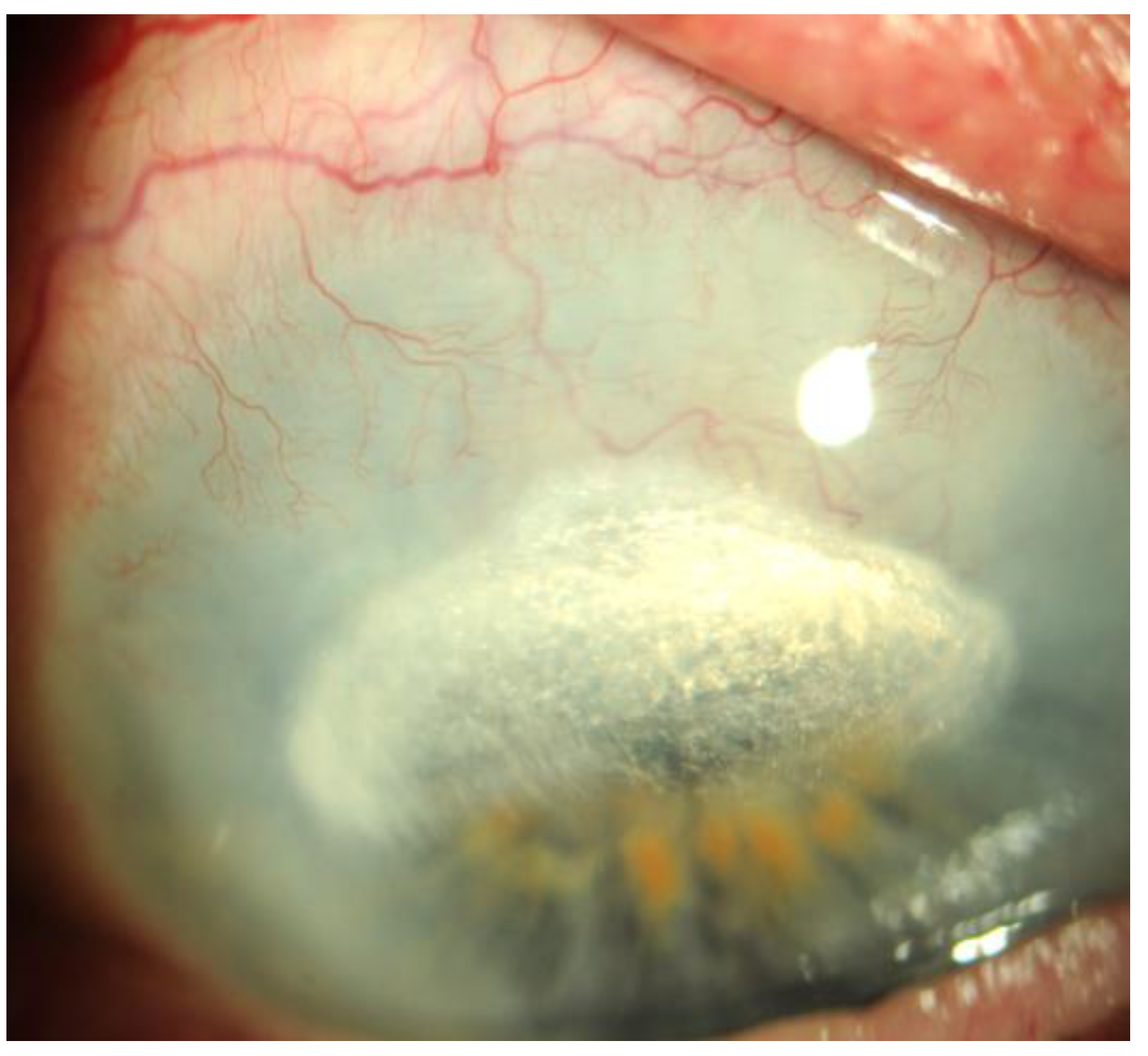
Figure 3.
Initial corneal neovascularization after penetrating keratoplasty .
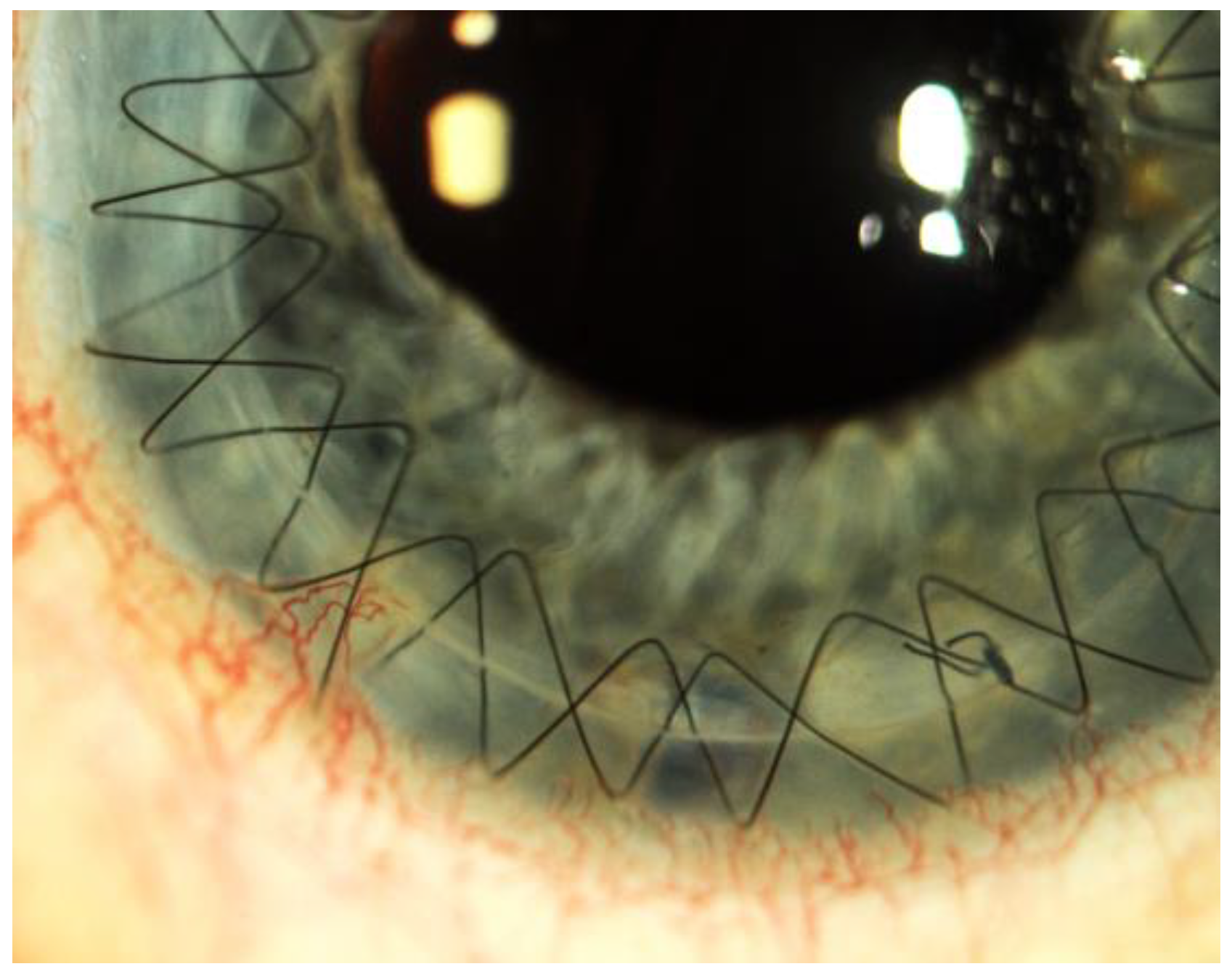
Figure 4.
Neovascularization after corneal transplantation.
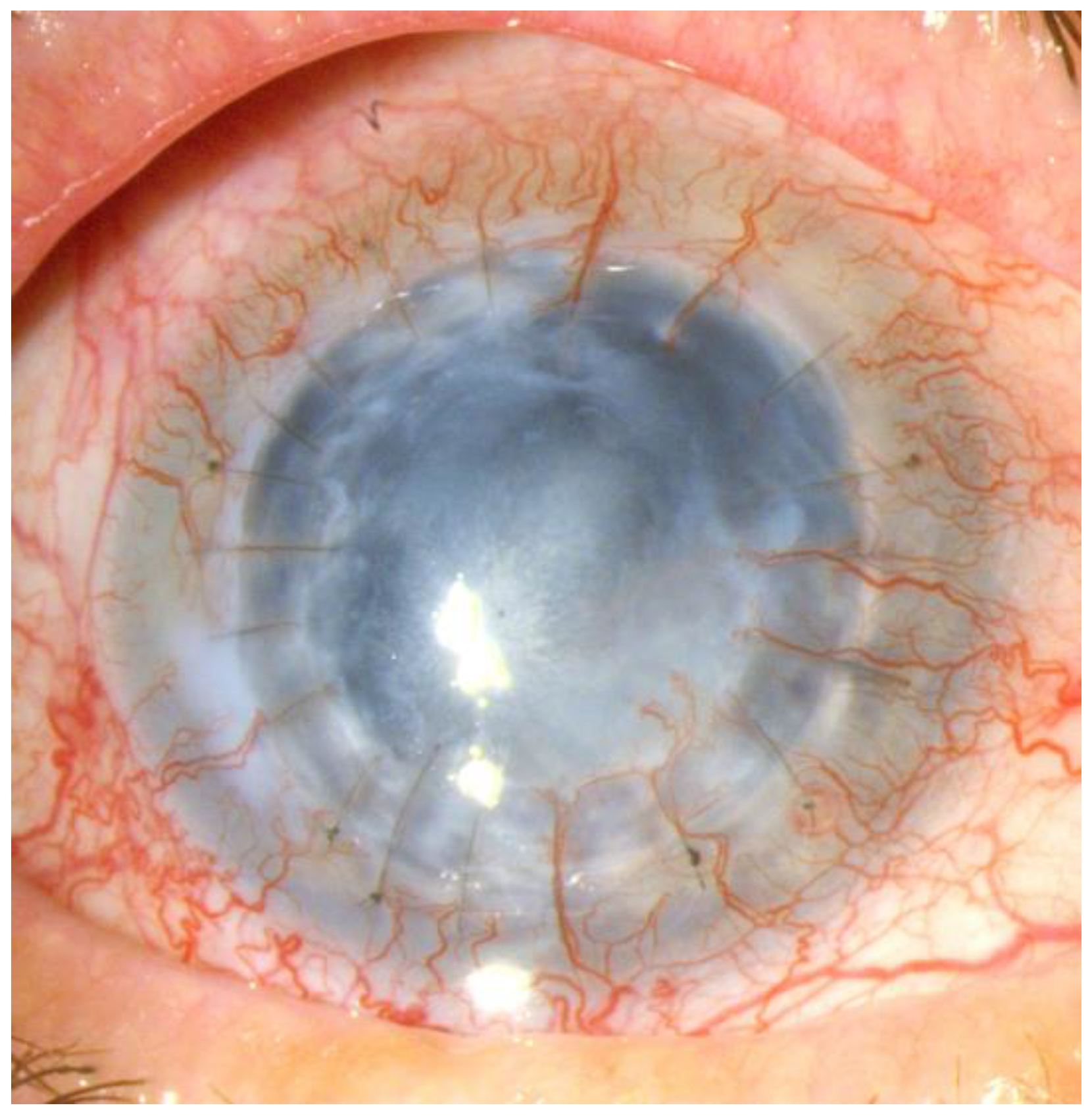
Figure 5.
Post-inflammatory corneal neovascularization.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



