
Submitted:
31 October 2023
Posted:
31 October 2023
You are already at the latest version
Abstract
Sterol regulatory element-binding proteins (SREBPs) are transcription factors that regulate genes involved in the biogenesis of cholesterol, fatty acids, and triglycerides. SREBPs are involved in the pathogenesis of non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), fibrosis, and hepatocellular carcinoma (HCC). We review the mechanisms by which SREBPs regulate lipogenesis, the alterations in these processes associated with liver diseases, and therapeutic strategies to target these pathways.
Keywords:
SREBPs
; NAFLD
; NASH
; fibrosis
; hepatocellular carcinoma (HCC)
; lipogenesis
; endoplasmic reticulum (ER) stress
; therapeutic target
Introduction
The global burden of nonalcoholic fatty liver disease (NAFLD), now termed metabolic dysfunction-associated liver disease (MASLD) is increasing worldwide in parallel with that of obesity; the prevalence of NAFLD in the overweight population is 79%[1]. NAFLD is characterized by pathologic liver features ranging from steatosis to non-alcoholic steatohepatitis (NASH, now called metabolic dysfunction-associated steatohepatitis, or MASH), characterized by steatosis, inflammation, and fibrosis. Between 20% and 30% of NAFLD cases progress to NASH, and 10%–20% of NASH cases progress to cirrhosis (fibrosis stage 4) [2,3]. NAFLD, NASH, and cirrhosis are associated with increased risks of hepatocellular carcinoma (HCC) and liver transplantation [4]. Many lines of evidence have revealed that hepatocyte cell death, inflammation, oxidative stress, endoplasmic reticulum (ER) stress, and lipotoxicity, which promote MASLD to progress into hepatocarcinogenesis.
Compared with livers of healthy subjects, de novo lipogenesis is increased by 26% in livers of patients with NAFLD or NASH [5,6,7,8,9]. The alterations in lipid metabolism leads to the lipotoxicity and hepatocyte cell death that contribute to HCC development.
Sterol regulatory element-binding proteins (SREBPs) are transcription factors that regulate lipid lipogenesis; alterations in this process have been associated with pathogenesis of NAFLD, NASH, and eventually HCC. SREBPs regulate transcription of genes that control biogenesis of triglycerides (TG), fatty acids (FAs), and cholesterol in mammals [10].
Mammalian SREBPs are encoded by the genes SREBF1 and SREBF2. SREBP1 expresses 2 isoforms: SREBP-1a and SREBP-1c, which differ in their first exons due to different transcriptional start sites. SREBP1 isoforms are abundant in liver, white adipose tissue, adrenal glands, skeletal muscle, and brains of mice and humans, while SREBP2 is expressed ubiquitously [11]. SREBP-1c regulates transcription of genes involved in FA and triacylglycerol synthesis, whereas SREBP-1a stimulates FA and cholesterol synthesis. SREBP-2 regulates transcription of genes that control cholesterol synthesis; all these pathways interact and overlap in a complicated fashion [12,13].
The transcription activation domain of SREBPs is located at the N-terminus. SREBPs are expressed as precursors that are cleaved to release the N-terminal domain, which can then enter the nucleus to stimulate the transcription of target genes. An escort protein, SREBP cleavage-activating protein (SCAP), transports SREBP precursors to the Golgi apparatus, where the active forms are generated by 2 proteases (site 1 protease and site 2 protease) and an anchoring protein. Insulin induced genes (INSIGS), ER membrane proteins with 6 transmembrane helices, bind the sterol-sensing domains of SCAP, causing the retention of the SCAP–SREBP complex based on sterol levels [14].
During development of liver diseases, SREBP activation or repression alters lipid profiles, contributing to metabolic disorders and cancer. Signaling pathways, such as the phosphatidylinositol 3-kinase (PI3K)–protein kinase B (PKB, Akt)–mammalian target of rapamycin (mTOR) pathway, regulate SREBP activation and are altered under different physiologic conditions [15,16]. Strategies to target the SREBP pathways have therefore been studied and developed. We review the roles and mechanism of SREBPs in healthy vs diseased liver and potential therapies to target these pathways.
Multifunctional role of SREBPs in NAFLD and NASH
Because SREBPs have multifunctional roles in lipid metabolism, many metabolic disorders are related to SREBP deregulation, such as type 2 diabetes, dyslipidemia, atherosclerosis, and hepatic steatosis [17,18]. Variants of SREBP1 have been associated with increased risk for NAFLD [19]. Transgenic overexpression of SREBP-1c in in mouse liver led to increased lipogenesis and hepatic steatosis [20]. On the other hand, deletion of the gene encoding SREBP-1c in livers of ob/ob mice, which are insulin resistant, results in an approximate 50% reduction of hepatic triglycerides (TGs), indicating the role of SREBP-1c in the hepatic steatosis in ob/ob mice [21]. In ob/ob mice, diet-induced NAFLD and steatosis were reversed by SREBP-1c antisense oligonucleotides, without improving insulin hepatic resistance [22].
HIV-patients with insulin-resistant lipodystrophia have altered hepatic expression of SREBP1 and peroxisome proliferator activated receptor gamma (PPARγ), compared with NAFLD or control subjects, which contributes to pathogenesis of steatosis and fibrosis [23]. Alterations in patatin-like phospholipase3 (PNPLA3) activity have also been associated with development of NASH [24,25]; polymorphisms in PNPLA3 have been associated with severity of NAFLD [26]. Expression of PNPL3 is regulated by SREBP-1c, which binds to the PNPLA3 promoter. Accumulation of PNPL3 on lipid droplets stimulates lipid accumulation in mouse hepatocytes [26,27].
Insulin mediates expression of the SREBP1, proteolytic processing of its products, and thereby hepatic lipogenesis. Insulin stimulates of SREBP-1c to active the PI3K–Akt and Akt–mTORC1 signaling pathways. Furthermore, AMPK, an energy sensor for cellular energy of homeostasis, inhibits cleavage and transcriptional activation of SREBP via phosphorylation. If AMPK activity is stimulated with metformin, SREBP-1c cleavage and nuclear translocation (via ser 372 phosphorylation) are suppressed, leading to attenuation of liver steatosis in mice deficient in low-density lipoprotein (LDL) with diet-induced insulin resistance [28].
In patients with NAFLD, treatment with antrodan, which is used to treat alcohol-associated steatohepatitis, alleviate fatty liver symptoms and the liver injuriesal, by altering AMPK–Sirt1–PPARγ–SREBP-1c signaling, although the mechanism not clear [29]. Similarly, xanthohumol inhibits the development of fatty liver in mice by impairing EF–Golgi translocation of the SCAP–SREBP complex and blocking incorporation into common coated protein II vesicles [30]. In patients with hepatic steatosis and lipodystrophy, leptin treatment improves insulin-stimulated hepatic and peripheral glucose metabolism, suggesting potential therapeutic approaches [31].
Micro-RNAs (miRNAs) have been identified that regulate lipid metabolism at the post-transcriptional level. The miRNAs can affect intracellular lipid levels, cholesterol transportation, and HDL formation by affecting SREBP expression [32,33]. Long non-coding RNAs (lncRNAs) also regulate hepatic lipogenesis, and alterations in lncRNAs have been associated with NASH. For example, lncARSR is upregulated in serum and liver of NAFLD patients and mouse models by methionine-choline deficient (MCD) diet feeding. lncARSR mediates lipogenesis via the Akt–SREBP-1c pathway [34]. miRNAs and lncRNAs also regulate lipid metabolism synthetically. In cell lines and mouse models of NAFLD, abundant transcript 1 (NEAT1) binds miR-140 to exacerbate the progression, through inactivating the AMPK–SREBP-1 pathway, which presenting a new therapeutic strategy[35].
Studies support the roles of lncRNAs in hepatic lipid accumulation. For example, the lncRNA Gm15622 is highly expressed in livers of mice with high-fat diet (HFD)-induced obesity. Gm15622 acts as a sponge for the miRNA miR-742-3p, thereby increasing expression of SREBP-1c to promote lipid accumulation. These findings have increased our understanding of how lipid metabolism and accumulation are altered during development of NAFLD [36].
SREBPs may mediate Liver Fibrosis via TGF-β Signaling during Chronic liver disease progressing to HCC
Liver fibrosis develops with chronic liver disease or injury, such as in NAFLD, hepatitis virus infections, or alcohol-induced steatohepatitis, leading to HCC and liver failure [47]. Liver fibrosis is characterized by an excess deposition of extracellular matrix (ECM) in the perisinusoidal space (between hepatocytes and the sinusoids), distorting liver architecture and causing liver stiffness [47,48]. The ECM, a network of extracellular macromolecules and minerals, contains enzymes, glycoproteins, and proteins such as collagen that provide structural and biochemical support to surrounding cells. Collagen, the main structural protein in connective tissues, is produced by myofibroblasts (hepatic stellate cells in liver).
Transforming growth factor beta (TGF-β) activates signaling pathways that lead to liver fibrosis. TGF-β signaling, via TβRII and SMADs, causes quiescent HSCs (5%–8% of liver cells) to transdifferentiate into activated HSCs, which are highly proliferative, contractile, and fibrogenic. Activated HSCs produce ECM proteins and promote not only fibrogenesis but hepatic inflammation. TGF-β signaling pathways interact with MAPK, mTOR, PI3K/AKT, and Rho/GTPase pathways [49], and are regulated by SREBP-1c, which inhibits HSC.
In rat and mouse HSCs, leptin-induced β-catenin signaling reduces levels of SREBP-1c protein and activity, independent of conventional key regulators of SREBP-1c activity, such as SCAP, INSIG, and other proteins [50]. This observation provides a potential mechanisms of the liver fibrogenesis associated with increased levels of leptin in obese individuals, and indicates the important function of SREBP signaling in HSCs.
Further investigation into the mechanisms by which SREBP1c regulates HSCs and liver fibrosis[51] demonstrated that overexpression of SREBP1c inhibited liver fibrosis in mice, by reducing levels of TGFβ1 and signaling via SMAD3 and Akt1/2/3[51]. In activated HSCs, SREBP1c was reported to regulate epigenetic factors such as bromodomain-containing chromatin-modifying factor bromodomain protein 4 (BrD4) and methionine adenosyltransferase 2B (MAT2B) to prevent liver fibrogenesis. Interestingly, PPARγ, a transcription factor that regulates activation of HSCs, increases with levels of SREBP1c and together these proteins inhibits HSC activation.
SREBP2 has been implicated in liver fibrosis via regulation of cholesterol levels in HSCs. In mouse models of NASH and primary mouse HSCs, the nuclear form of SREBP2 increases with HSC activation. Addition of 25-hydroxycholesterol or LDL, which promotes the formation of the SCAP–INSIG complex, decreased the nuclear form of SREBP2 in quiescent HSCs, but not in activated HSCs; this difference is attributed to differences in levels of INSIG-1 and INSIG-2 in cells. Activated HSCs have nearly undetectable levels of INSIG-1 and INSIG-2, resulting in the constitutive processing of SREBP when excess free cholesterol accumulates. The feedback regulation of cholesterol homeostasis mediated by SREBP2 is disrupted in HSCs. This disruption affects the sensitivity of HSCs to activation by TGF-ß, resulting in accumulation of free cholesterol and contributing to the vicious cycle of liver fibrosis [52]. Therefore, the SREBP pathway has a crucial role in the process of liver fibrosis and requires careful characterization.
SREBPs are defined as pro-fibrotic mediators because they activate TGF-β in lipotoxicity-induced fibrosis development. There are also investigations into SREBP mediation of kidney fibrosis, via a lipid-independent pathways [53]. SREBPs also regulate cell functions such as autophagy, metabolic circadian rhythm, and other process that affect expression of non-lipogenic genes in different organs or cell lines during fibrosis development. In light of the similarities among cell and tissue types in fibrosis signaling, it is worthwhile to address the versatility and complexity of SREBPs in mediating liver fibrosis, and hopefully identify inhibitors that can be used to treat liver and other diseases.
Role of SREBPs in HCC
HCC is the second-leading cause of cancer-related death worldwide. Studies have shown that alterations in lipid metabolism and accumulation of lipid metabolic products contributes to hepatocarcinogenesis. HCCs of high histologic grade, advanced, and metastatic tumors overexpress SREBP-1. On the other hand, downregulation of SREBP1 inhibits proliferation and apoptosis in HCC cell lines, such as HepG2 [54]. Tissue microarray analysis showed that high expression of spindlin 1 (SPIN1), associated with HCC malignancy, is co-activated with SREBP-1c and that the proteins cooperate to increase intracellular TGs, cholesterols, and lipid droplets, which promote progression of HCC [55].
SREBP-1 affects HCC development via different signaling pathways. Hepatoma-derived growth factor (HDGF) is co-expressed with SREBP-1 in HCCs, and is closely associated with HCC prognosis [56]. Similarly, apoptosis-antagonizing transcription factors (AATFs) are highly expressed in HCC tissues and cell lines; AATFs bind SREBP-1c to regulate proliferation, migration, colony formation, and tumor growth [57].
An HCC-associated tumor suppressor, zinc fingers and homeoboxes2 (ZHX2), inhibits SREBP-1c-regulated lipogenesis in cell lines and human specimens. This regulation pathway induces SREBP-1c degradation, by increasing transcription of miRNA-24-3P, to suppress HCC development [58]. Acyl CoA synthetase 4 (ACSL4), and oncogene and marker of the alpha-fetoprotein-high subtype of HCC, upregulates expression of SREBP-1 and downstream lipogenic enzymes to reprogram fatty acid metabolism through c-myc, which facilitates HCC progression [59]. In mice with diet-induces obesity that develop steatohepatitis and liver tumors, histone deacetylase 8 (HDAC8) and SREBP-1 are co-expressed in tumor tissues, and HDAC8 is directly upregulated by SREBP-1. Additionally, HDAC8 promotes cell proliferation by coordinating with the chromatin modifier EZH2 to epigenetically repress Wnt antagonists. This repression of Wnt antagonists inhibits cell death through the p53–p21 pathway, leading to G2/M cell cycle arrest. Notably, this regulatory pathway is also perturbed in human NAFLD-associated HCC tissues, in which HDAC8 is aberrantly upregulated by SREBP-1. [60].
When signal transducers and activators of transcription 5 (STAT5) are deficient, SREBP-1 and peroxisome PPARγ signaling is upregulated, activating tumor necrosis factor, reactive oxygen species, and STAT3, which contribute to hepatocarcinogenesis [61]. Expression analysis of human HCC specimens showed that SREBP-1 expression correlated with expression of UBC12, which contributes to HCC aggressiveness. If the NEDD8-activating enzyme E1, MLN4924, is incorporated in tumor xenograft mice, HCC progression is blocked [62].
mTOR signaling can activate SREBPs. One of the genes regulated by SREBP encodes enzyme fatty acid desaturase 2 (FADS2), which is upregulated in HCC cells [63]. The transcription factor Krüppel-like factor 10 (KLF10) is phosphorylated by AMPK, which then represses SREBP-1c and thereby the lipogenesis pathways in liver tumor cells [64].
Collectively, multiple pathways regulate SREBP-1 expression, activation, and stability to promote the proliferation, invasion, and migration of HCC cells, leading to tumor growth and metastasis. Moreover, genetic disruption or pharmaceutical blocking of regulators of SREBP pathways, such as with SCAP or gp78, inhibits lipid synthesis-related gene expression and reduces HCC progression in chow diet mouse models [65]. In addition to SCAP-dependent SREBP activation during the progression of hepatocellular carcinoma (HCC), SCAP-independent regulation also plays a role. Our research has demonstrated that chronic alcohol consumption-induced HCC upregulates IL-17A signaling, which further increases cholesterol and fatty acid synthesis via TNFR1-caspase-2-dependent activation of SREBP1/2 in both mouse and human steatotic hepatocytes [66]. Deletion of the IL-17A receptor results in significant downregulation of SREBP1/2 proteins and less lipid accumulation in ethanol and HFD fed mice. Therefore, targeting the IL-17A-SREBP signaling pathway may be a potential therapeutic strategy for patients with alcohol-induced HCC.
SREBP-2 regulates cholesterol synthesis, and SREBP-2 activation promotes cholesterol accumulation that contributes to liver tumor progression. P53 induces the mevalonate pathway through SREBP-2 maturation and induces expression of the cholesterol transporter gene ABCA1. A p53 activator, haploinsufficient tumor suppressor ASPP2, interacts with SREBP-2 and negatively regulates the mevalonate pathway to inhibit growth of HCC cells, . [67]Interaction of the oncogene staphylococcal nuclease with tudor domain containing-1 (SND-1) leads to altered activation of the sterol regulatory element-binding protein SREBP2 in hepatocellular carcinoma, promoting accumulation of cellular cholesteryl esters[68]. In FASN-knockout mice, which develop liver tumors, levels of HMGCR cholesterol synthesis and nuclear SREBP-2 are increased [69]. If FASN ubiquitination is blocked, the SREBP-1 and SREBP-2 degradation complexes are disrupted, preventing development of liver tumors [70].
Targeting SREBPs for treating HCC and chronic liver disease
Targeting SREBPs, by modulating the level of expression or activation process, small molecules or natural products as potential therapeutics for preventing or treating liver disease have been thoroughly investigated (Table 1). Song et al. [63] reported that a small molecule, betulin, specifically inhibits the maturation of SREBP by inducing interaction of SCAP with INSIGS, which could improve hyperlipidemia. The function of betulin in treating liver diseases has been studied in HCC cells. In mice with diethylnitrosamine-induced liver tumors, betulin suppressed tumor progression by reducing inflammation [64].
Sorafenib, a multi-kinase inhibitor of RAS–MEK–ERK signaling, VEGFR, and PDGFR, is a first-line treatment for HCC. Sorafenib reduces expression of SCD-1 to decrease synthesis of monounsaturated fatty acids, and activates AMPK to reduce levels of SREBP-1 and phosphorylate mTOR, which suppresses liver cancer [71]. Sorafenib is not an effective treatment for HCC, and high levels of SREBP-1 in tumors correlate with shorter survival. Administration of betulin increases the ability of sorafenib to kill HCC cells and slow growth of xenograft tumors by suppressing cellular glucose metabolism and reducing glycolytic activity [72].
Radiofrequency ablation is an important strategy for treatment of advanced HCC. After undergoing RFA, tumors in some patients have high protein levels of SREBP-1, which correlates with reduced survival. A small-molecule inhibitor of SREBP-1, SI-1, 1-(4-bromophenyl)-3-(pyridin-3-yl) urea, inhibits aerobic glycolysis and increases killing HCC cells by radiofrequency ablation and slows growth of xenograft tumors. SI-1, 1-(4-bromophenyl)-3-(pyridin-3-yl) urea therefore could be a promising approach for treatment of HCC [73].
Other compounds, such as cinobufotalin and emodin, inhibit HCC cells via SREBP-1 and its downstream targets. Cinobufotalin, extracted from the skin secretion of the giant toad, promotes HCC cells apoptosis, induces cell cycle G2/M arrest, and inhibits cell proliferation. Cinobufotalin downregulates SREBP-1 expression and inhibits de novo lipid synthesis in HCC cells [74]. In addition, emodin, a component of a Chinese medicinal herb Moldenke, induces apoptosis and activates expression of intrinsic apoptosis signaling pathway-related proteins, such as caspase 9, Bax, and BCL-2, in HCC cells. This apoptotic process may or may not depend on the SREBP-1 pathway, [75]. Emodin inhibits SREBP-2 transcriptional activity to suppress cholesterol metabolism and Akt signaling, which sensitizes HCC cells to the anti-cancer effect of sorafenib in vitro and in xenograft tumors [76]. Moreover, ursolic acid, a natural pentacyclic terpenoid, activates SREBP-2 and increases the expression of cholesterol biosynthesis-related enzymes to induce cell cycle arrest and apoptosis in HCC cells [77].
Conclusions
We have reviewed our understanding of the role of SREBP-mediated lipid metabolism in development of liver diseases and potential therapeutic targets. Inhibition of SREBP-mediated pathways with small molecules might prevent or slow progression of HCC (Figure 1). Multiple signaling pathways and molecules regulate expression, stability, and activation of of SREBP-1 and SREBP-2, which control gene transcription to regulate liver cancer cell proliferation, apoptosis, endoplasmic reticulum stress, and metastasis (Figure 2). Small molecules, compounds, or herbal extracts (Table 1) that target the SREBP-1- or SREBP-2-regulated mevalonate pathway to repress lipid metabolism might be developed to inhibit liver tumor progression.
SREBP inhibition is not always beneficial for liver diseases. For example, liver-specific PTEN deficiency results in excess lipids and steatosis in mice, whereas when combined with SCAP deletion it reduces steatosis, exacerbates the later stage development of inflammation and liver injury, and accelerates the development of NASH and HCC [78]. Targeting lipogenesis via the SREBP pathway is not straightforward, and therefore, alternative approaches that consider lipid levels, based on the physiologic situation, should be explored. To effectively address liver pathology, it is crucial to target different stages of disease progression precisely. This approach ensures the maintenance of lipid homeostasis while intervening in lipid metabolism.
Funding
This work was sponsored by the National Natural Science Foundation of China (31900541, 32293230) and the Science and Shanghai Pujiang Program (2021PJD027) (NL), and National Institutes of Health USA DK099205, AA028550, DK101737, P50 AA011999, P30 DK120515, DK091183 (TK).
Acknowledgments
The authors are grateful to BioRender.com, which provided us with a good tool to create Figure 2.
References
- Quek, J.; Chan, K. E.; Wong, Z. Y.; Tan, C.; Tan, B.; Lim, W. H.; Tan, D. J. H.; Tang, A. S. P.; Tay, P.; Xiao, J.; Yong, J. N.; Zeng, R. W.; Chew, N. W. S.; Nah, B.; Kulkarni, A.; Siddiqui, M. S.; Dan, Y. Y.; Wong, V. W.; Sanyal, A. J.; Noureddin, M.; Muthiah, M.; Ng, C. H. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2023, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Wong, R. J.; Harrison, S. A. Nonalcoholic Fatty Liver Disease Review: Diagnosis, Treatment, and Outcomes. Clin Gastroenterol Hepatol 2015, 13, 2062–70. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z. M. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011, 34, 274–85. [Google Scholar] [CrossRef] [PubMed]
- Jinjuvadia, R.; Patel, S.; Liangpunsakul, S. The association between metabolic syndrome and hepatocellular carcinoma: systemic review and meta-analysis. Journal of clinical gastroenterology 2014, 48. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Enooku, K.; Nakagawa, H.; Fujiwara, N.; Kondo, M.; Minami, T.; Hoshida, Y.; Shibahara, J.; Tateishi, R.; Koike, K. Altered serum acylcarnitine profile is associated with the status of nonalcoholic fatty liver disease (NAFLD) and NAFLD-related hepatocellular carcinoma. Sci Rep 2019, 9, 10663. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M. A.; Seki, E.; Hidalgo, J.; Koike, K.; Kaufman, R. J.; Karin, M. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Donnelly, K. L.; Smith, C. I.; Schwarzenberg, S. J.; Jessurun, J.; Boldt, M. D.; Parks, E. J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005, 115, 1343–51. [Google Scholar] [CrossRef]
- Moon, Y. A. The SCAP/SREBP Pathway: A Mediator of Hepatic Steatosis. Endocrinol Metab (Seoul) 2017, 32, 6–10. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Sato, R. Sterol metabolism and SREBP activation. Arch Biochem Biophys 2010, 501, 177–81. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T. I.; Osborne, T. F. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 2012, 23, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Cortes, V. A.; Rashid, S.; Anderson, N. N.; McDonald, J. G.; Liang, G.; Moon, Y. A.; Hammer, R. E.; Horton, J. D. Expression of SREBP-1c Requires SREBP-2-mediated Generation of a Sterol Ligand for LXR in Livers of Mice. Elife 2017, 6. [Google Scholar]
- Gong, Y.; Lee, J. N.; Lee, P. C.; Goldstein, J. L.; Brown, M. S.; Ye, J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab 2006, 3, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A. N.; Soto, H.; Zhu, S.; Babic, I.; Tanaka, K.; Dang, J.; Iwanami, A.; Gini, B.; Dejesus, J.; Lisiero, D. D.; Huang, T. T.; Prins, R. M.; Wen, P. Y.; Robins, H. I.; Prados, M. D.; Deangelis, L. M.; Mellinghoff, I. K.; Mehta, M. P.; James, C. D.; Chakravarti, A.; Cloughesy, T. F.; Tontonoz, P.; Mischel, P. S. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov 2011, 1, 442–56. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Griffiths, B.; Chung, Y. L.; Delpuech, O.; Griffiths, J. R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–81. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M. M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J. Y.; Gao, B.; Wierzbicki, M.; Verbeuren, T. J.; Shaw, R. J.; Cohen, R. A.; Zang, M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Eberle, D.; Clement, K.; Meyre, D.; Sahbatou, M.; Vaxillaire, M.; Le Gall, A.; Ferre, P.; Basdevant, A.; Froguel, P.; Foufelle, F. SREBF-1 gene polymorphisms are associated with obesity and type 2 diabetes in French obese and diabetic cohorts. Diabetes 2004, 53, 2153–7. [Google Scholar] [CrossRef]
- Ponugoti, B.; Fang, S.; Kemper, J. K. Functional interaction of hepatic nuclear factor-4 and peroxisome proliferator-activated receptor-gamma coactivator 1alpha in CYP7A1 regulation is inhibited by a key lipogenic activator, sterol regulatory element-binding protein-1c. Mol Endocrinol 2007, 21, 2698–712. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J. D.; Shimomura, I.; Hammer, R. E.; Brown, M. S.; Goldstein, J. L. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997, 99, 846–54. [Google Scholar] [CrossRef]
- Moon, Y.-A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M. S.; Goldstein, J. L.; Horton, J. D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell metabolism 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Vitto, M.; Luz, G.; Luciano, T.; Marques, S.; Souza, D.; Pinho, R.; Lira, F.; Cintra, D.; De Souza, C. Reversion of steatosis by SREBP-1c antisense oligonucleotide did not improve hepatic insulin action in diet-induced obesity mice. Hormone and Metabolic Research 2012, 44, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.; Barbu, V.; Girard, P. M.; Kim, M.; Bastard, J.-P.; Wendum, D.; Paye, F.; Housset, C.; Capeau, J.; Serfaty, L. Altered hepatic expression of SREBP-1 and PPARγ is associated with liver injury in insulin-resistant lipodystrophic HIV-infected patients. Aids 2006, 20, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. C.; Horton, J. D.; Hobbs, H. H. Human fatty liver disease: old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R. M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World journal of gastroenterology 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C. J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Liang, J.; Ke, Y.; Li, C.; Cui, Y.; Shen, L.; Zhang, H.; Cui, A.; Liu, X.; Liu, C. Mouse patatin-like phospholipase domain-containing 3 influences systemic lipid and glucose homeostasis. Hepatology 2011, 54, 509–521. [Google Scholar] [CrossRef]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nature communications 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Chyau, C.-C.; Wang, H.-F.; Zhang, W.-J.; Chen, C.-C.; Huang, S.-H.; Chang, C.-C.; Peng, R. Y. Antrodan alleviates high-fat and high-fructose diet-induced fatty liver disease in C57BL/6 mice model via AMPK/Sirt1/SREBP-1c/PPARγ pathway. International journal of molecular sciences 2020, 21, 360. [Google Scholar] [CrossRef]
- Miyata, S.; Inoue, J.; Shimizu, M.; Sato, R. Xanthohumol improves diet-induced obesity and fatty liver by suppressing sterol regulatory element-binding protein (SREBP) activation. Journal of Biological Chemistry 2015, 290, 20565–20579. [Google Scholar] [CrossRef]
- Petersen, K. F.; Oral, E. A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G. W.; DePaoli, A. M.; Taylor, S. I.; Gorden, P. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. The Journal of clinical investigation 2002, 109, 1345–1350. [Google Scholar] [CrossRef]
- Moore, K. J.; Rayner, K. J.; Suárez, Y.; Fernández-Hernando, C., microRNAs and cholesterol metabolism. Trends in Endocrinology & Metabolism 2010, 21, 699-706. [CrossRef]
- Fernández-Hernando, C.; Suárez, Y.; Rayner, K. J.; Moore, K. J. MicroRNAs in lipid metabolism. Current opinion in lipidology 2011, 22, 86. [Google Scholar] [CrossRef]
- Zhang, M.; Chi, X.; Qu, N.; Wang, C. Long noncoding RNA lncARSR promotes hepatic lipogenesis via Akt/SREBP-1c pathway and contributes to the pathogenesis of nonalcoholic steatohepatitis. Biochemical and biophysical research communications 2018, 499, 66–70. [Google Scholar] [CrossRef]
- Sun, Y.; Song, Y.; Liu, C.; Geng, J. LncRNA NEAT1-MicroRNA-140 axis exacerbates nonalcoholic fatty liver through interrupting AMPK/SREBP-1 signaling. Biochem Biophys Res Commun 2019, 516, 584–590. [Google Scholar] [CrossRef]
- Ma, M.; Duan, R.; Shen, L.; Liu, M.; Ji, Y.; Zhou, H.; Li, C.; Liang, T.; Li, X.; Guo, L. The lncRNA Gm15622 stimulates SREBP-1c expression and hepatic lipid accumulation by sponging the miR-742-3p in mice [S]. Journal of lipid research 2020, 61, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, A.; Nabavizadeh, F.; Zekri, A.; Amiri, F. Naltrexone changes the expression of lipid metabolism-related proteins in the endoplasmic reticulum stress induced hepatic steatosis in mice. Clinical and experimental pharmacology and physiology 2017, 44, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, A.; Farahabadi, M.; Chavoshzadeh, S. A.; Barati, A.; Ababzadeh, S.; Mohammadbeigi, A., The effect of amygdalin on endoplasmic reticulum (ER) stress induced hepatic steatosis in mice. The Malaysian journal of medical sciences: MJMS 2018, 25, 16. [CrossRef]
- Kammoun, H. L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress–induced SREBP-1c activation and reduces hepatic steatosis in mice. The Journal of clinical investigation 2009, 119, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D. T.; Wu, J.; Back, S.-H.; Callaghan, M. U.; Ferris, S. P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J. R.; Fornek, J. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Developmental cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Harding, H. P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell metabolism 2008, 7, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R. A.; Wiest, M. M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M. J.; Sanyal, A. J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Shin, H.-S.; Choi, H. S.; Park, J.-W.; Jo, I.; Oh, E.-S.; Lee, K.-Y.; Lee, B.-H.; Johnson, R. J.; Kang, D.-H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Laboratory investigation 2014, 94, 1114–1125. [Google Scholar] [CrossRef]
- Kim, J. Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R. J.; Saltiel, A. R.; Karin, M., ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 2018, 175, 133-145. e15. [CrossRef]
- Basseri, S.; Austin, R. C. Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochemistry research international 2012, 2012. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M. H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A. H.; Park, W.-J., Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Experimental & molecular medicine 2019, 51, 1-16. [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A. N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8. [Google Scholar]
- Zhang, J.; Liu, Q.; He, J.; Li, Y. Novel Therapeutic Targets in Liver Fibrosis. Front Mol Biosci 2021, 8, 766855. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem 2016, 64, 157–67. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yan, K.; Fan, J.; Niu, M.; Zhou, Q.; Zhou, Y.; Chen, H.; Zhou, Y. The beta-catenin pathway contributes to the effects of leptin on SREBP-1c expression in rat hepatic stellate cells and liver fibrosis. Br J Pharmacol 2013, 169, 197–212. [Google Scholar] [CrossRef]
- Su, S.; Tian, H.; Jia, X.; Zhu, X.; Wu, J.; Zhang, Y.; Chen, Y.; Li, Z.; Zhou, Y. Mechanistic insights into the effects of SREBP1c on hepatic stellate cell and liver fibrosis. J Cell Mol Med 2020, 24, 10063–10074. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; Shimamura, K.; Usui, S.; Ebinuma, H.; Saito, H.; Watanabe, C.; Komoto, S.; Kawaguchi, A.; Nagao, S.; Sugiyama, K.; Hokari, R.; Kanai, T.; Miura, S.; Hibi, T. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–69. [Google Scholar] [CrossRef]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front Pharmacol 2020, 11, 265. [Google Scholar] [CrossRef]
- Li, C.; Yang, W.; Zhang, J.; Zheng, X.; Yao, Y.; Tu, K.; Liu, Q. SREBP-1 has a prognostic role and contributes to invasion and metastasis in human hepatocellular carcinoma. International journal of molecular sciences 2014, 15, 7124–7138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Bu, Y.; Feng, J.; Zhang, H.; Chen, Y.; Yang, G.; Liu, Z.; Yuan, H.; Yuan, Y.; Liu, L. SPIN1 triggers abnormal lipid metabolism and enhances tumor growth in liver cancer. Cancer Letters 2020, 470, 54–63. [Google Scholar] [CrossRef]
- Min, X.; Wen, J.; Zhao, L.; Wang, K.; Li, Q.; Huang, G.; Liu, J.; Zhao, X. Role of hepatoma-derived growth factor in promoting de novo lipogenesis and tumorigenesis in hepatocellular carcinoma. Molecular oncology 2018, 12, 1480–1497. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D. P.; Santhekadur, P. K.; Seneshaw, M.; Mirshahi, F.; Uram-Tuculescu, C.; Sanyal, A. J. A regulatory role of apoptosis antagonizing transcription factor in the pathogenesis of nonalcoholic fatty liver disease and hepatocellular carcinoma. Hepatology 2019, 69, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lin, Q.; Wu, Z.; Zhang, Y.; Wang, T.; Zhao, S.; Song, X.; Chen, C.; Wang, Z.; Xu, L. ZHX2 inhibits SREBP1c-mediated de novo lipogenesis in hepatocellular carcinoma via miR-24-3p. The Journal of Pathology 2020, 252, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer letters 2021, 502, 154–165. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.; Wong, G.; Yang, W.; Sun, H.; Shen, J.; Tong, J.; Go, M.; Cheung, Y. S.; Lai, P., Histone deacetylase HDAC8 promotes insulin resistance and-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res,(Received on July 24, 2015). [CrossRef]
- Mueller, K. M.; Kornfeld, J. W.; Friedbichler, K.; Blaas, L.; Egger, G.; Esterbauer, H.; Hasselblatt, P.; Schlederer, M.; Haindl, S.; Wagner, K. U. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology 2011, 54, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Heo, M. J.; Kang, S. H.; Kim, Y. S.; Lee, J. M.; Yu, J.; Kim, H. R.; Lim, H.; Kim, K. M.; Jung, J.; Jeong, L. S. UBC12-mediated SREBP-1 neddylation worsens metastatic tumor prognosis. International Journal of Cancer 2020, 147, 2550–2563. [Google Scholar] [CrossRef] [PubMed]
- Triki, M.; Rinaldi, G.; Planque, M.; Broekaert, D.; Winkelkotte, A. M.; Maier, C. R.; Raman, S. J.; Vandekeere, A.; Van Elsen, J.; Orth, M. F. mTOR signaling and SREBP activity increase FADS2 expression and can activate sapienate biosynthesis. Cell reports 2020, 31, 107806. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chen, R.-J.; Peng, S.-Y.; Yu, W. C.; Chang, V. H.-S. Therapeutic Targeting of Nonalcoholic Fatty Liver Disease by Downregulating SREBP-1C Expression via AMPK-KLF10 Axis. Frontiers in Molecular Biosciences 2021, 946. [Google Scholar] [CrossRef]
- Li, N.; Zhou, Z. S.; Shen, Y.; Xu, J.; Miao, H. H.; Xiong, Y.; Xu, F.; Li, B. L.; Luo, J.; Song, B. L. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 2017, 65, 1936–1947. [Google Scholar] [CrossRef]
- Ma, H. Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J. Y.; Alexandrov, L. B.; Koyama, Y.; Nishio, T.; Benner, C.; Heinz, S.; Rosenthal, S. B.; Liang, S.; Sun, M.; Karin, G.; Zhao, P.; Brodt, P.; McKillop, I. H.; Quehenberger, O.; Dennis, E.; Saltiel, A.; Tsukamoto, H.; Gao, B.; Karin, M.; Brenner, D. A.; Kisseleva, T. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol 2020, 72, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Chen, R.; Song, S.; Wang, H.; Sun, G.; Yang, H.; Jing, W.; Zhou, X.; Fu, Z.; Huang, G.; Zhao, J. ASPP2 inhibits tumor growth by repressing the mevalonate pathway in hepatocellular carcinoma. Cell Death Dis 2019, 10, 830. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Imaz, H.; Chico, Y.; Rueda, Y.; Fresnedo, O. Channeling of newly synthesized fatty acids to cholesterol esterification limits triglyceride synthesis in SND1-overexpressing hepatoma cells. Biochim Biophys Acta Mol Cell Biol Lipids 2019, 1864, 137–146. [Google Scholar] [CrossRef]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M. G.; Wang, J. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020, 69, 177–186. [Google Scholar] [CrossRef]
- Calvisi, D. F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S. A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J., Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071-1083. e5. [CrossRef]
- Liu, G.; Kuang, S.; Cao, R.; Wang, J.; Peng, Q.; Sun, C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. The FASEB Journal 2019, 33, 10089–10103. [Google Scholar] [CrossRef]
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y., SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell death & disease 2019, 10, 1-12. [CrossRef]
- Zou, X.-z.; Hao, J.-f.; Zhou, X.-h. Inhibition of SREBP-1 Activation by a Novel Small-Molecule Inhibitor Enhances the Sensitivity of Hepatocellular Carcinoma Tissue to Radiofrequency Ablation. Frontiers in Oncology 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Shen, M.; Li, J.; Zhang, R.; Li, X.; Zhao, L.; Huang, G.; Liu, J. Novel SREBP1 inhibitor cinobufotalin suppresses proliferation of hepatocellular carcinoma by targeting lipogenesis. European Journal of Pharmacology 2021, 906, 174280. [Google Scholar] [CrossRef]
- Yang, N.; Li, C.; Li, H.; Liu, M.; Cai, X.; Cao, F.; Feng, Y.; Li, M.; Wang, X. Emodin induced SREBP1-dependent and SREBP1-independent apoptosis in hepatocellular carcinoma cells. Frontiers in Pharmacology 2019, 10, 709. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Lee, Y.-M.; Oh, T.-I.; Shin, D. H.; Kim, G.-H.; Kan, S.-Y.; Kang, H.; Kim, J. H.; Kim, B. M.; Yim, W. J. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. International journal of molecular sciences 2018, 19, 3127. [Google Scholar] [CrossRef]
- Kim, G.-H.; Kan, S.-Y.; Kang, H.; Lee, S.; Ko, H. M.; Kim, J. H.; Lim, J.-H. Ursolic acid suppresses cholesterol biosynthesis and exerts anti-cancer effects in hepatocellular carcinoma cells. International journal of molecular sciences 2019, 20, 4767. [Google Scholar] [CrossRef]
- Kawamura, S.; Matsushita, Y.; Kurosaki, S.; Tange, M.; Fujiwara, N.; Hayata, Y.; Hayakawa, Y.; Suzuki, N.; Hata, M.; Tsuboi, M. Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. The Journal of Clinical Investigation 2022, 132. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Regulation of the SREBP Pathway in Liver Diseases. Hepatocarcinogenesis involves several stages, progressing through NAFL and NASH. Although progression is slow and partially reversible, the transition from NASH to HCC is irreversible. The SREBP pathway coordinates numerous signaling factors, such as the AKT–mTOR pathway and ER stress. These pathways regulate the different stages of cancer progression.
Figure 1.
Regulation of the SREBP Pathway in Liver Diseases. Hepatocarcinogenesis involves several stages, progressing through NAFL and NASH. Although progression is slow and partially reversible, the transition from NASH to HCC is irreversible. The SREBP pathway coordinates numerous signaling factors, such as the AKT–mTOR pathway and ER stress. These pathways regulate the different stages of cancer progression.
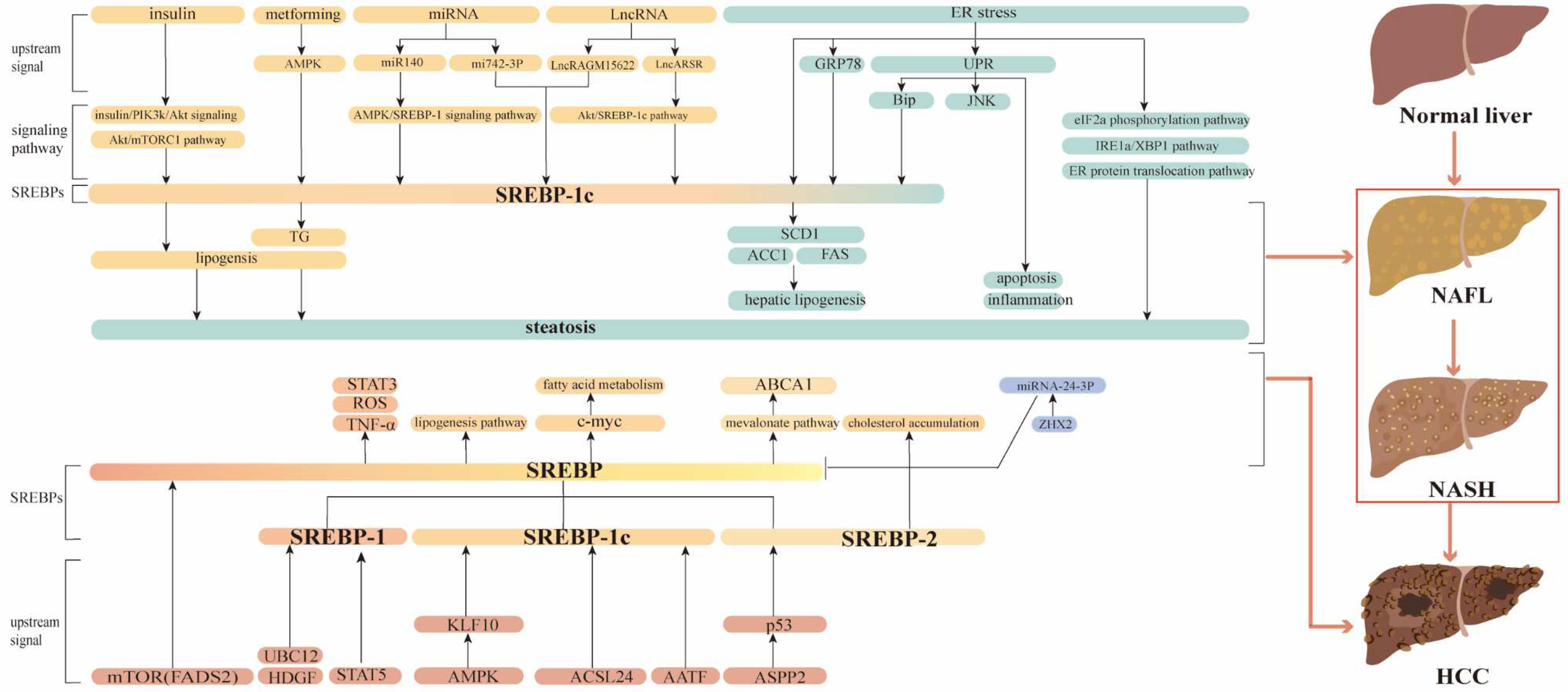
Figure 2.
Proper Processing of SREBP Maintains Lipid Homeostasis and Prevents Liver Disease. Multiple mediators regulate the nuclear activation of SREBP, which in turn affects lipogenesis, cholesterol levels, and other related lipid profiles. Alterations in lipid status can contribute to development of liver diseases. During the pathogenesis of liver diseases, altered functions of regulators such as transcription factors, miRNAs, and lcRNAs affect different steps of these pathways, in different cell types. The SREBP pathway is intricately linked with numerous signaling pathways, that regulate many cell and liver functions.
Figure 2.
Proper Processing of SREBP Maintains Lipid Homeostasis and Prevents Liver Disease. Multiple mediators regulate the nuclear activation of SREBP, which in turn affects lipogenesis, cholesterol levels, and other related lipid profiles. Alterations in lipid status can contribute to development of liver diseases. During the pathogenesis of liver diseases, altered functions of regulators such as transcription factors, miRNAs, and lcRNAs affect different steps of these pathways, in different cell types. The SREBP pathway is intricately linked with numerous signaling pathways, that regulate many cell and liver functions.
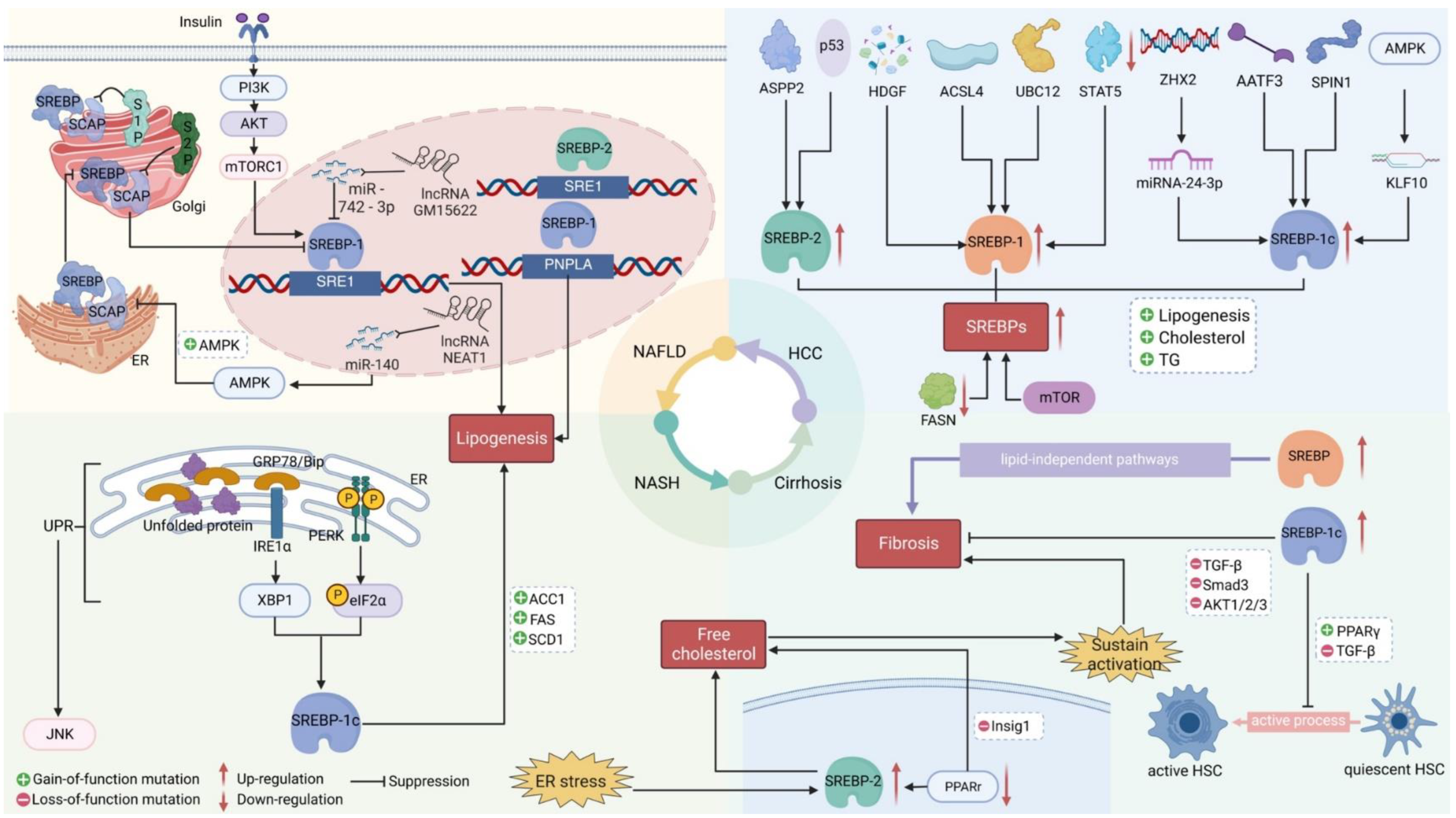

Table 1.
Agents That Target SREBP-mediated Lipogenesis.
| Drug | Disease | Mechanism | Targets | Cell Lines Tested | Mouse Models Tested (Dose) | Reference |
| Xanthohumol | Fatty liver | Impairs ER–Golgi translocation of the SCAP-SREBP complex by binding to SEC23 and SEC24 and blocking SCAP–SREBP incorporation into common coated protein II vesicles | SREBP1 | Huh-7 | HFD-induced fatty liver in male C57BL/6J mice (0.2% or 0.4%) | [30] |
| Antrodan | NAFLD | Reduces HFD-induced NAFLD via the AMPK–SREBP1c–PPARγ pathway | SREBP1 | none | (20-40 mg/kg) | [29] |
| Betulin | HCC | Inhibits cell glucose metabolism to prevent metastatic potential and facilitate inhibitory effect of sorafenib | SREBP1 | MHCC97-H | MHCC97-H xenograft tumors (2 mg/kg) | [72] |
| HCC | Inhibits ER–Golgi translocation of SREBPs | SREBP1 | none | Diethylnitrosamine-induced HCC in mice (50 mg/kg) | [65] | |
| Emodin | HCC | Induces apoptosis and reduces mitochondrial membrane potential; anticancer effects |
SREBP1 and its downstream targets, ACLY, ACCa, FASN, and SCD1 | Bel-7402 | none | [75-76] |
| Sorafenib | HCC | Reduces cell viability | SREBP1 and its target SCD1 | human Huh7.5 liver cancer cells | Huh7.5 xenograft tumors (20 mg/kg/d) | [71] |
| Cinobufotalin | HCC | Induces cell cycle G2–M arrest and apoptosis; inhibits cell proliferation by inhibiting d novo lipid synthesis |
SREBP1 | HepG2, SMMC-7721 | SMMC-7721 xenograft tumors (2.5 mg/kg, 5 mg/kg) | [74] |
| Ursolic acid | HCC | Activates SREBP2 and cholesterol biosynthesis-related genes and enzymes to lower cholesterol in cells | SREBP2 | SK-HEP-1, Hep3B | none | [77] |
| HFD, high-fat diet | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



