
Submitted:
31 October 2023
Posted:
31 October 2023
You are already at the latest version
Abstract
Anti-DNA antibodies are hallmark autoantibodies produced in systemic lupus erythematosus (SLE), but their pathogenetic roll is not fully understood. Accumulating evidence suggests that some anti-DNA antibodies enter different types of live cells and affect the pathophysiology of SLE by stimulating or impairing these cells. Circulating neutrophils in SLE are activated by type I interferon or other stimuli and primed to release neutrophil extracellular traps (NETs) on additional stimulation. Anti-DNA antibodies are also involved in this process and may induce NET release. Thereafter, they bind and protect extracellular DNA in the NETs from digestion by nucleases, resulting in increased NET immunogenicity. This review discusses the pathogenetic role of anti-DNA antibodies in SLE, mainly focusing on recent progress in the two research fields concerning antibody penetration into live cells and NETosis.
Keywords:
anti-DNA antibodies
; systemic lupus erythematosus (SLE)
; penetration
; endocytosis
; neutrophil extracellular traps (NETs)
; NETosis
; pathogenesis
1. Introduction
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease which preferentially affects women 20-40 years of age. Although clinical manifestations are varied, ranging from mild to severe, SLE often begins with fever, skin rash, or arthritis, and develops organ lesions such as serositis and glomerulonephritis, or neuropsychiatric symptoms (NPSLE) [1,2]. Were it not for appropriate diagnosis and treatment, the organ lesions could leave patients severely disabled. Multiple genetic susceptibility and environmental factors are thought to lead to a breakdown of immunological self-tolerance, and different autoantibodies against nuclear antigens are detected in the serum [3]. Many SLE-susceptibility genes have been linked to type I interferon (IFN) production or responses, and therefore numerous studies have been carried out to understand the “IFN signature” in SLE. So far, type I IFNs have been implicated in loss of tolerance, activation of neutrophils and release of neutrophil extracellular traps (NETs), production of B-cell activating factor (BAFF), and other events; nevertheless, our understanding of the pathophysiology of SLE is still incomplete [4].
Among the antinuclear antibodies (ANA), those reactive with double-stranded (ds)DNA and Sm nucleoprotein are relatively specific for SLE and included in the classification criteria for this disease proposed by the European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) [5]. According to the current criteria, detection of ANA at a titer of 1:80 or higher on HEp-2 cells is adopted as an entry criterion, and the presence of anti-dsDNA antibody or anti-Sm antibody is weighted heavily in the additive immunology domain criteria. In typical cases, serum titers of anti-DNA antibodies correlate with disease activity and they are regularly monitored over clinical follow-up. However, despite many efforts, our understanding of the pathogenetic role of these anti-DNA antibodies in SLE remains incomplete. This review discusses how anti-DNA antibodies are involved in lupus pathogenesis mainly focusing on two issues: antibody penetration into live cells, and relevance to NETosis, both issues that have been intensively studied recently.
2. Generation of anti-DNA antibodies
As native DNA itself is not immunogenic, how and why patients with SLE consistently produce anti-DNA antibodies remains an open question. In one study, DNA exogenously added to cultures of HEK 293T cells which had been transfected with the gene for the SLE susceptibility allele HLA-DR15 was internalized and then expressed on the cell surface together with this MHC class II molecule [6]. These investigators created NFAT-GFP reporter cells that were transfected with anti-DNA B cell receptors, and which expressed GFP and IL-2 upon crosslinking of the receptors. When cocultured with the above-mentioned DNA presenting cells, the reporter cells were activated to produce GFP and IL-2. MHC class II molecules are generally present peptide antigens to helper T cells, but this study proposes an unexpected role of MHC molecules in activation of DNA-reactive B cells.
The generation of monoclonal antibody-producing hybridomas using human peripheral blood lymphocytes is difficult and usually yields solely low affinity IgM antibodies. However, recent advances in molecular technologies have facilitated the production of human monoclonal anti-DNA antibody-like proteins by transfection of HEK 293T cells with the immunoglobulin heavy chain genes identified from a single B cell from the peripheral blood of a patient with SLE [7]. Applying this technique to analyze the variable region gene usage of anti-DNase1L3 neutralizing antibodies, interestingly, some were found to have been derived from anti-DNase1L3 germline-encoded precursors which had acquired cross-reactivity to dsDNA following somatic hypermutation [8]. Another study reported that some mouse anti-dsDNA monoclonal antibodies were cross-reactive with spermatid nuclear transition protein 1 [9]. These studies suggest the possibility that anti-DNA antibodies might initially be produced in response to unexpected DNA-binding protein antigens. Additionally, as discussed later, the release of NETs is increased in SLE; it is possible that oxidized DNA present in NETs [10], which is known to be immunogenic [11], acts as a primary antigen triggering the production of antibodies cross-reactive to native DNA.
3. Penetration of anti-DNA antibodies into live cells (Figure 1)
The ability of ANA to enter the nucleus of live cells was initially reported by Alarcón-Segovia et al. in 1978 [12]. By a direct immunostaining method without using a second antibody, they documented internalization of anti-RNP antibodies obtained from a patient with mixed connective tissue disease into normal peripheral blood mononuclear cells (PBMCs). Soon after, they reported similar findings with anti-DNA antibodies as well [13]. Initially, these findings met with skepticism, but gradually many studies have confirmed this phenomenon [14,15,16]. Mechanisms responsible for internalization are multifarious. Some anti-DNA antibodies enter cells via Fc-receptor mediated endocytosis, but there are examples showing that recombinant single chain fragments of the variable chains (scFv) lacking the Fc region can still enter cells [17,18]. Some anti-DNA antibodies enter the nucleus and bind chromatin DNA while others remain in the cytoplasm, but which factors determine such movement remains unidentified.
Anti-DNA antibodies form immune complexes with DNA in vivo in the plasma or in vitro in culture medium. Although these immune complexes would be trimmed by DNase, when researchers use pure antibodies, they must be washed thoroughly in high salt buffer and/or alkaline buffer [19,20]. Because the ratio of absorbance at 260 nm and 280 nm changes only slightly, but significantly, before and after washing, it is speculated that, for example, without sufficiently thorough washing, short oligonucleotides remain attached to the antigen-binding cleft of the antibodies purified by protein G column. Even after preparation of ultra purified antibodies, however, they would still again bind to DNA in the medium or on the cell surface when added to cell cultures. Therefore, even very highly purified anti-DNA antibodies should be considered as immune complexes in most studies even without addition of exogenous DNA.
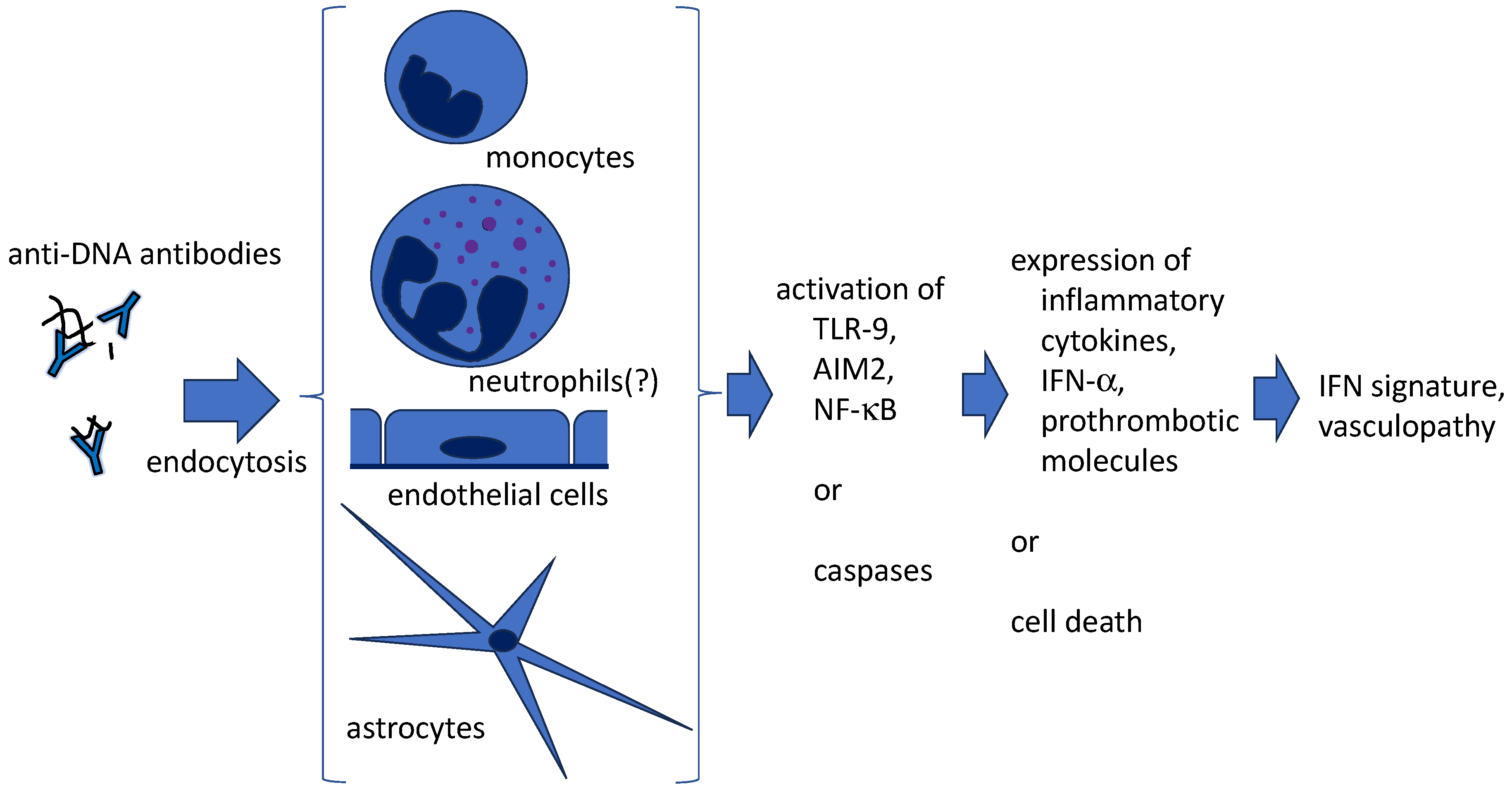
Figure 1.
Internalization of anti-DNA antibodies by living cells may affect the pathophysiology of SLE. Apart from immortalized or genetically modified cell lines, anti-DNA antibodies are also demonstrated to enter several normal cell types, accompanied by DNA. As a result, cells would be activated to produce lupus-prone cytokines, prothrombotic molecules, or might be impaired.
Figure 1.
Internalization of anti-DNA antibodies by living cells may affect the pathophysiology of SLE. Apart from immortalized or genetically modified cell lines, anti-DNA antibodies are also demonstrated to enter several normal cell types, accompanied by DNA. As a result, cells would be activated to produce lupus-prone cytokines, prothrombotic molecules, or might be impaired.
In parallel with the discovery of various different intracellular nucleic acid sensors, it has been suggested that the DNA which enters the cells accompanying anti-DNA antibodies stimulates Toll-like receptors (TLRs) or other nucleic acid sensors expressed in the endosome or in the cytosol, leading to production of cytokines relevant to lupus pathogenesis [21,22]. In line with this, our laboratory showed that the mouse monoclonal antibody 2C10, which specifically recognizes dsDNA and does not bind single-stranded (ss)DNA, enters the nucleus of PBMCs from healthy subjects and induces expression of cytokines commonly implicated in lupus, including IFN-a, IFN-b, TNF-a, IL-1b, and MCP-1 [23]. Internalization of 2C10 was significantly inhibited by the macropinocytosis inhibitor cytochalasin D, but not by an Fcg-receptor blocker. Cytokine expression was suppressed by cytochalasin D and the TLR-9 inhibitor chloroquine. In addition, the NLRP3 inhibitor shikonin suppressed the secretion of certain cytokines, including IL-1b. These results suggest that 2C10 was endocytosed mainly by monocytes via macropinocytosis, and the accompanying DNA ligated TLR-9 in the endosome, and after leaking into the cytosol, stimulated AIM-2. Another monoclonal anti-DNA antibody, WB-6, which is cross-reactive with dsDNA, ssDNA and cardiolipin-b2GPI, was observed to enter normal monocytes and induce tissue factor expression [20,24].
Clinical phenotypes of NPSLE are diverse and are classified into neurological syndromes (including headache, seizure disorders, and cerebrovascular disease) and diffuse psychiatric or neuropsychological syndromes (including cognitive impairment, mood disorder, anxiety disorder, and psychosis) [25]. At least some neurological symptoms are ascribed to the pathological effects of antiphospholipid antibodies (aPL) on the vascular system. Although it is hypothesized that some autoantibodies are involved, the pathogenetic role of autoantibodies in diffuse psychiatric or neuropsychological syndromes remains undefined [26]. In addition to the blood-brain barrier, however, several other interfaces may serve as sites of antibody transfer into the central nervous system (CNS), such as the meningeal barrier, the glymphatic pathway and the blood-cerebrospinal fluid barrier; the permeability of these barriers is considered to be increased under pathological conditions [25]. It is noteworthy that Stamou et al. [27] have documented the internalization of IgG-anti-IgG immune complexes by newborn rat hippocampal cells via Fcg receptors. Based on these findings, we tested whether 2C10 enters cells of the CNS, and found that it indeed enter the nucleus of rat astrocytes, but not neurons, in in vitro cultures [28]. The effects of 2C10 internalization on the function of astrocytes have not yet been determined, but given the pivotal role of astrocytes in regulating brain activity [29], they might be relevant to the pathogenesis of diffuse psychiatric or neuropsychological syndromes in NPSLE.
Using the well-studied mouse monoclonal anti-DNA antibodies 3D8 and 3E10, molecular mechanisms of cell-penetration have been explored and reviewed in detail [30]. Briefly, following the binding to cell surface heparan sulfate proteoglycan, 3D8 is engulfed into early endosomes, then dissociates from the heparan sulfate, changes its conformation, and escapes into the cytosol. In contrast, 3E10 is proposed to enter cells via a mechanism not involving endocytosis, but in a manner dependent on equilibrative nucleoside transporter 2 (ENT2). ENT2 is an integral membrane protein widely expressed in most cell types, playing a role in transporting nucleosides. Knockdown of ENT2 or adding the ENT2 inhibitor dipyridamole reduces the penetration of 3E10 into the cell. Further, 3E10 traffics to the nucleus via an uncertain mechanism. It is noteworthy that in a mouse model, a dimeric scFv structural 3E10 variant (designated DX1) was suggested to be transcytosed through brain endothelial cells and thus cross the blood-brain barrier [31]. Dipyridamole reduced the transfer of DX1. That study aimed to develop antibody-based immunotherapy for brain tumors, but could also be relevant to the pathological mechanism of NPSLE. It would be intriguing to explore the molecular mechanisms of how DX1 interacts with ENT2, enters and exits brain endothelial cells.
4. Anti-DNA antibodies and NETs
4.1. What are NETs?
Eight years after Takei et al. [32] described characteristic morphological changes of neutrophils stimulated by phorbol 12-myristate 13-acetate, Brinkmann et al. [33] described the basic structure and antimicrobial function of NETs by impressive electron microscopy. Since these publications, NET release has been recognized as a new type of cell death by neutrophils mediating antimicrobial suicide attacks [34]; this process has been designated NETosis. NETs are web-like structures released by neutrophils triggered by different stimuli; they are composed of DNA originating from decondensed chromatin or mitochondria, decorated by histones, HMGB1 and various neutrophil granular antimicrobial proteins or peptides including myeloperoxidase (MPO) and LL37 (37 amino acid residues of the C-terminal region of a human cationic antimicrobial protein, hCAP). Although similar extracellular trap (ET) formation has also been observed in mast cells [35] and eosinophils [36] playing a role in innate self-defense mechanisms, NETs have been most intensively studied recently in the context of autoimmunity.
4.2. NETs in autoimmune diseasses
As well as in infectious diseases, including most recently in COVID-19 [37], NETs are known to be triggered by a variety of sterile stimuli and are involved in autoimmune diseases including SLE, antiphospholipid syndrome (APS), anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), and rheumatoid arthritis [38].
4.2.1. SLE
Peripheral blood neutrophils obtained from SLE patients release more NETs than those from healthy donors triggered by different stimuli or even spontaneously in ex vivo experiments. Lande and colleagues [39] showed that NETs in sera obtained from SLE contain DNA, anti-DNA antibodies, and antimicrobial peptides LL37 and HNPs (human neutrophil peptides belonging to the a-defensin family). These complexes of DNA, anti-DNA and peptides stimulated normal plasmacytoid dendritic cells (pDCs) to produce IFN-a through the TLR-9 pathway. Interestingly, DNA-anti-DNA immune complexes alone did not stimulate pDCs, and LL37 and/or HNPs were necessary to activate them. It was suggested that LL37 induced aggregation of DNA fragments to form insoluble particles which were resistant to nuclease digestion, and enabled the DNA to enter the intracellular TLR-9-containing compartments of pDCs. In the same issue of that journal, another study focusing on pediatric SLE found that healthy neutrophils show increased levels of TLR-7 mRNA after exposure to SLE sera or IFN-a [40]. Accordingly, SLE neutrophils produced significantly high levels of IL-8 in response to a TLR-7 agonist. These observations prompted the researchers to assess the effect of anti-RNP antibodies. They found that SLE neutrophils, presumably primed in vivo by IFN-a, showed NETosis after 3 hours of culture with IgG anti-RNP antibodies purified from SLE serum in an FcgRIIa-, NADPH-, and TLR-7-dependent manner.
To prevent the NET release in SLE, a study tested the effect of ligation of one of the negative regulators of neutrophil function, the signal inhibitory receptor on leukocytes-1 (SIRL-1) [41]. By ligation with anti-SIRL-1 antibodies, spontaneous and anti-LL37 antibody-induced NET release by SLE neutrophils was significantly suppressed. These results suggest that NET release could be a strategically important therapeutic target in SLE.
4.2.2. APS
NETs participate in a prothrombotic state by multiple mechanisms including inhibition of tissue factor pathway inhibitors, activation of platelets, activation of procoagulant factors, and induction of activated protein C (APC) resistance [42,43]. The frequency of aPL-positive patients in SLE is estimated to be 30-40% [44]. Not all of these patients exhibit aPL-related clinical manifestations, but they have a higher risk of vascular events than aPL-negative patients. In contrast, about half of the patients with APS are secondary APS, mostly associated with SLE. Thus, there is a significant overlap in the pathological condition in SLE and APS.
IgG purified from patients with primary APS and human IgG monoclonal anti-b2GPI antibodies both induced NET release from normal neutrophils in one study [45]. These investigators reported that anti-b2-GPI antibodies likely bind to the cell surface b2-GPI and thereby stimulate the cells. Further, in vivo testing of APS IgG in a mouse model resulted in exaggerated thrombosis with thrombi enriched for citrullinated histone H3 (a marker of NETs) [46]. Large amounts of human IgG were bound to the surface of the mouse neutrophils. Although endothelial cells, platelets and monocytes are the main players involved in APS pathogenesis, these reports revealed an important role for neutrophils as well. However, it remains uncertain how and to what extent b2-GPI is expressed on the normal neutrophil surface. In one study on large cohorts of patients with SLE, secondary APS associated with SLE, or primary APS, NET-release triggering activity of patient plasma samples on healthy neutrophils was compared [47]. The results showed that plasma samples collected from 60% of SLE, 61% of SLE + APS, and 45% of primary APS patients were able to induce NET release.
4.2.3. AAV
Different forms of NETosis are observed depending on how they are triggered. Two major types, late suicidal NETosis (also referred to as lytic NET formation) and early vital NETosis (rapid non-lytic NET formation) are reviewed elsewhere [48]. Late suicidal NETosis depends on production of reactive oxygen species by NADPH-oxidase and takes a few hours. Unfolded chromatin is released into the cytosol, decorated with granular and cytosolic proteins, and finally expelled by the plasma membrane disruption. In contrast, vital NETosis occurs within minutes of stimulation, independently of oxidants, and NETs are released by nuclear-envelope blebbing and vesicular export without rupture of the plasma membrane; thus the cells remain alive.
Characteristics of NETosis induced in healthy neutrophils by patient sera were compared in large cohorts of ANCA-positive AAV (n=80) and ANA-positive SLE (n=59). Interestingly, incubation of healthy neutrophils with AAV sera induced late suicidal NETosis, whereas SLE sera induced vital NETosis [49]. AAV-induced NETosis was triggered independently of IgG, whereas SLE-induced NETosis was dependent on Fcg receptor signaling. Soluble IgG isolated from SLE sera did not induce NETosis, but immobilized SLE-IgG, which mimics immune complexes, did. These results suggest that vital NETosis requires intensive cross-linking of Fcg receptors.
4.3. Quantification of NETs
The quantification of NETs is challenging due to their varied morphology, heterogeneous components, and especially their fragility. A possible simple approach to detect NETs in plasma or in culture medium is by using a sandwich ELISA with a capture antibody such as anti-MPO and a detection antibody such as anti-DNA. This may be satisfactory for certain assays, with accepted limitations, but developing a reliable, generally applicable ELISA system is not straightforward [50]. Recently, an improved highly sensitive ELISA protocol was proposed, using two different antibodies (anti-MPO and anti-citrullinated histone H3) for capture, and one (anti-DNA) for detection [51]. In parallel, a simple, inexpensive, immunofluorescence smear assay was developed, in which 1 µL of SLE plasma was fixed onto poly-L-lysine-coated glass slides, followed by staining extracellular DNA with SYTOX Green and DAPI. Fluorescence intensity was quantified using an image analysis software and assay results were shown to correlate well with the ELISA. An assay using a more sophisticated imaging system employing three-dimensional immunofluorescence confocal microscopy has been published as a video article; this technique can be utilized to observe the process of formation and degradation of NETs [52].
When we consider introducing an assessment of NETs into the clinical laboratory, it would be more practical to measure some NET-associated serological markers in place of the challenge of detecting the whole structure of NETs. One study has indicated that SLE patients have higher cell-free DNA, MPO activity, anti-MPO antibodies, DNase I concentration, and lower NETolytic activity compared to healthy controls [53]. These changes of NET-related parameters were shown to be correlated with disease activity.
4.4. Anti-NET antibodies
It is plausible that NETs are antigenic because molecules normally contained in the nucleus or granules are extruded, may be in a modified form, and exposed to the immune system for a long period due to decreased degradation activity. In fact, production of various autoantibodies reactive to NET components has been reported.
In one study, anti-NET antibodies were detected by indirect immunofluorescence in 10 of 19 patients with microscopic polyangiitis [54]; these antibodies were distinct from ANCA, but their target antigens were not determined. It is noteworthy that ANA was negative in all these anti-NET-positive patients, indicating that fundamental self-tolerance mechanisms were not entirely absent. In another study, IgG and IgM antibodies to NETs measured by ELISA were significantly elevated in patients with primary APS, and in SLE without aPL, relative to healthy controls [55]. In a recent larger cohort of primary APS, 45% of the aPL-positive patients had IgG and/or IgM anti-NET antibodies [56]. Importantly, analysis of associations of anti-NET antibodies with clinical manifestations revealed that IgG antibodies were associated with lesions affecting the white matter of the brain, while IgM antibodies tracked with complement consumption. Antigen specificities of these antibosies were analyzed using a 120-antigen microarray panel, and it was suggested that IgG antibodies to NETs were likely to be driven by reactivity with protein antigens in the NETs, while the IgM antibodies to NETs were likely to target DNA. In another study, anti-NET antibodies were detected in 35.7% of patients with SLE [57]. Interestingly, 37.0% of these patients were negative for anti-dsDNA antibodies, indicating that DNA is not necessarily a major antigen in the NETs, even in SLE.
Similarly to antibodies that bind to DNA- or RNA-binding proteins such as histone, Sm or RNP, SLE patients possess antibodies to antimicrobial DNA-binding peptides LL37 and HNPs in NETs [39]. In addition, DNA sensors AIM2 and IFI16, which are released from dying cells and are present in the plasma, bind extracellular DNA in NETs; antibodies against AIM2 and IFI16 are also produced in SLE [58]. Furthermore, extracellular DNA-MPO-AIM2/IFI16 complexes were detected in biopsy specimens of diffuse proliferative lupus nephritis, suggesting a distinct immunostimulatory role of AIM2 and IFI16 in the renal lesions.
Even though titers of anti-DNA antibodies correlate with such antibodies to NET components, there have been no reports which directly demonstrating that NETs induce production of anti-DNA antibodies. Rather, it is generally recognized that anti-DNA antibodies have been produced in SLE some time before the occurrence of pathological conditions with NET release. Thus, not all NET components induce autoantibody production. For example, SLE patients do not produce anti-MPO antibodies. Conversely, AAV patients do not produce anti-DNA antibodies. What controls the antigenicity of the NET components has not been clearly explained.
4.5. Amplification of SLE disease activity by anti-DNA antibodies and NETs (Figure 2)
As discussed above, NET formation is increased in SLE. In the following, representative findings informative for the mechanisms responsible for NET aggravation of the pathological condition in SLE are reviewed.
4.5.1. Aggravation of IFN signature
Culturing pDCs isolated from healthy donors with apoptotic or necrotic neutrophils does not result in their activation. In contrast, culturing with NETs induced production of IFNa by pDCs in a DNA- and TLR-9-dependent manner [39,40]. In another study, NETs were phagocytosed by macrophages and translocated from the phagosome to the cytosol, where they activated cGAS leading to production of type I interferon [59]. Furthermore, immune complexes of a panel of human monoclonal anti-DNA antibodies and NETs were suggested to be internalized by monocytes and endothelial cells in an Fc receptor-dependent manner, resulting in enhanced expression of type I IFN and NF-kB, respectively [60].
4.5.2. Protection of NETs from nuclease resistance by anti-DNA antibodies
DNA in the NETs is protected from nuclease digestion by various DNA-binding peptides and proteins, resulting in prolonged presence in the circulation and increased pathogenetic activity [39,58]. It has also been reported that a group of human monoclonal antibodies cross-reactive with dsDNA, Crithidia luciliae, histone, and apoptotic Jurkat cells, protected NETs from digestion by micrococcal nuclease or DNase I. Interestingly, another group of antibodies which were specific for dsDNA did not show significant protection [60]. In a different context, monoclonal antibodies to DNase1L3 protected chromatin from degradation by this enzyme [8]. DNase1L3 is a member of the DNase1 family, which is responsible for the DNase activity in plasma together with DNase1 itself. To make matters still more complicated, these anti-DNaseIL3 antibodies are cross-reactive with dsDNA, as described in Section 2 (Generation of anti-DNA antibodies).
4.5.3. Thrombogenic properties
Prognosis of SLE has improved and patients now enjoy nearly as long a life expectancy as the average in developed countries. Accordingly, it has become a problem that they suffer a higher cardiovascular disease risk than the general population. One of the causes of this may be the use of corticosteroids over extended periods. In addition, as discussed in Section 4.2.2. (APS) above, NETs lead to a prothrombotic and procoagulant state by multiple mechanisms. Even in patients with SLE that does not fulfil the criteria for APS, NET-related thrombogenic properties might be relevant to their prognosis. Also, anti-DNA antibodies may contribute directly to cardiovascular risk. Recent in vitro studies demonstrated that anti-dsDNA antibodies purified from SLE patients bind to the cell surface, and some of them enter the nucleus of healthy monocytes, leading to expression of proinflammatory and prothrombotic molecules including tissue factor [61]. These results suggest that anti-DNA antibodies as well as NETs play a role not only in the pathogenesis of SLE itself but also in associated cardiovascular disorders.
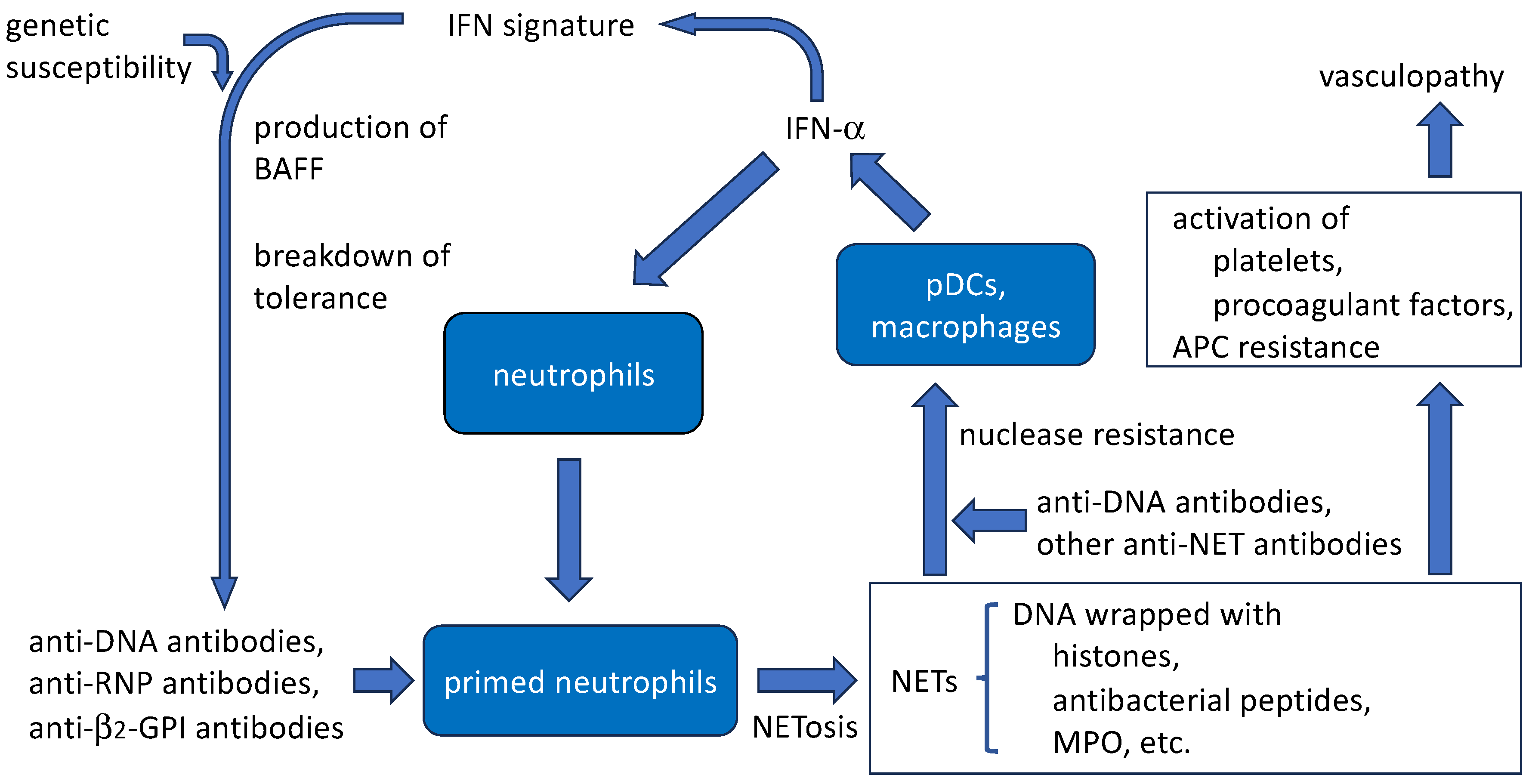
Figure 2.
Anti-DNA antibodies are involved in the process of NETosis. They likely to induce SLE neutrophils, which had been primed by IFN-a, to release NETs. They also bind to extracellular DNA in the NETs, which could enhance immunogenic activity of the NETs.
Figure 2.
Anti-DNA antibodies are involved in the process of NETosis. They likely to induce SLE neutrophils, which had been primed by IFN-a, to release NETs. They also bind to extracellular DNA in the NETs, which could enhance immunogenic activity of the NETs.
4.5.4. Induction of NET release by anti-DNA antibodies
Several studies have examined whether anti-DNA antibodies induce NET release from neutrophils. For example, mouse monoclonal anti-LL37 and anti-HNP antibodies could induce healthy human neutrophils to release NETs. F(ab’)2 fragments of anti-LL37 and anti-HNP also induced NET release, suggesting that these antibodies bind neutrophils not via Fc receptors but via cell surface antimicrobial peptides. However, a mouse monoclonal anti-DNA antibody H241 could not induce NET release in this study [39]. In another study, SLE plasma induced NET release by healthy neutrophils [41]. However, patient plasma contains many different antibodies, immune-complexes, cytokines and other factors. It was not determined which of those was responsible for the NET release. In a comparative study of plasma NET release activity in SLE and APS as described in Section 4.2.2. (APS) [47], increased levels of anti-dsDNA antibodies were associated with increased NET release, suggesting that anti-DNA antibodies may be responsible for NET release, but this was not determined, either. In a study of pediatric SLE described in Section 4.2.1. (SLE), anti-RNP antibodies induced patient neutrophils, but not healthy neutrophils, to die by releasing NETs [40]. Unfortunately, the effect of anti-DNA antibodies was not tested in that study.
In another study described in Section 4.2.3. (AAV), soluble IgG isolated from SLE patient serum did not induce NETosis, but immobilized SLE-IgG did so [49]. Recent observations by Patiño-Trives et al. [61] revealed that incubation of normal neutrophils for 6 hours long with IgG anti-DNA antibodies affinity purified from SLE sera induced NETosis. Thus, although evidence has been limited so far, induction of NETosis by anti-DNA antibodies is likely to be observed when assay conditions are appropriate.
5. Conclusions
Some, but not all, anti-DNA antibodies can enter live cells. Apart from immortalized cell lines, there have also been reports of the internalization of anti-DNA antibodies by different normal cell types including monocytes, vascular endothelial cells, and astrocytes. The mechanisms are multifarious, with some antibodies entering via Fc receptor-mediated endocytosis, but other mechanisms are also probable. Some of the antibody enters the nucleus, for which mechanisms remain to be elucidated. In any cases, such antibodies are thought to carry nucleotides which may stimulate the cells via TLR-9 or other nucleic acid sensors, resulting in cytokine production or sometimes apoptosis, and affecting the pathological condition of SLE.
In the circulation of patients with SLE, neutrophils are primed with IFN-a and other stimuli, and are prone to release NETs following additional triggers including DNA-anti-DNA immune complexes. NETs are protected from DNase digestion by different proteins, peptides, anti-DNA or other antibodies enveloping the DNA, and therefore persist for a long period. Such complexes of DNA and proteins/peptides are engulfed by pDCs and macrophages resulting in expression of type I IFN which plays a pivotal role in forming the IFN signature. Thus a vicious circle may be initiated. These accumulating findings indicate a need to formulate a new therapeutic approach targeting anti-DNA antibody production or NET release for the treatment of SLE.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, Ruiz-Irastorza G, Hughes G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Ameer MA, Chaudhry H, Mushtaq J, Khan OS, Babar M, Hashim T, Zeb S, Tariq MA, Patlolla SR, Ali J, et al. An overview of systemic lupus erythematosus (SLE) pathogenesis, classification, and management. Cureus 2022, 14, e30330. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky DS, Lipsky PE. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2020, 16, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Tsuji H, Ohmura K, Jin H, Naito R, Arase N, Kohyama M, Suenaga T, Sakakibara S, Kochi Y, Okada Y, et al. Anti-double-stranded DNA antibodies recognize DNA presented on HLA class II molecules of systemic lupus erythematosus risk alleles. Arthritis Rheumatol. 2022, 74, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Richardson C, Chida AS, Adlowitz D, Silver L, Fox E, Jenks SA, Palmer E, Wang Y, Heimburg-Molinaro J, Li QZ, et al. Molecular basis of 9G4 B cell autoreactivity in human systemic lupus erythematosus. J. Immunol. 2013, 191, 4926–4939. [Google Scholar] [CrossRef]
- Gomez-Bañuelos E, Yu Y, Li J, Cashman KS, Paz M, Trejo-Zambrano MI, Bugrovsky R, Wang Y, Chida AS, Sherman-Baust CA, et al. Affinity maturation generates pathogenic antibodies with dual reactivity to DNase1L3 and dsDNA in systemic lupus erythematosus. Nat. Commun. 2023, 14, 1388. [Google Scholar] [CrossRef]
- Uprety LP, Park YH, Jang YJ. Autoantigen spermatid nuclear transition protein I enhances pro-inflammatory cytokine production stimulated by anti-DNA autoantibodies in macrophages. Eur. J. Inflamm. 2022, 20, 1–9. [Google Scholar] [CrossRef]
- Hayden H, Klopf J, Ibrahim N, Knöbl V, Sotir A, Mekis R, Nowikovsky K, Eilenberg W, Neumayer C, Brostjan C. Quantitation of oxidized nuclear and mitochondrial DNA in plasma samples of patients with abdominal aortic aneurysm. Free Radic. Biol. Med. 2023, 206, 94–105. [Google Scholar] [CrossRef]
- Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J. Immunogenicity of DNA damaged by reactive oxygen species –– implications for anti-DNA antibodies in lupus. Free Radic. Biol. Med. 1997, 22, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Segovia D, Ruiz-Arguelles A, Fishbein E. Antibody to nuclear ribonucleoprotein penetrates live human mononuclear cells through Fc receptors. Nature 1978, 271, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Alarcón-Segovia D, Llorente L, Fishbein E, Díaz-Jouanen E. Abnormalities in the content of nucleic acids of peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Relationship to DNA antibodies. Arthritis Rheum. 1982, 23, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Vlahakos D, Foster MH, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Murine monoclonal anti-DNA antibodies penetrate cells, bind to nuclei, and induce glomerular proliferation and proteinuria in vivo. J. Am. Soc. Nephrol. 1992, 2, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Zannikou M, Bellou S, Eliades P, Hatzioannou A, Mantzaris MD, Carayanniotis G, Avrameas S, Lymberi P. DNA-histone complexes as ligands amplify cell penetration and nuclear targeting of anti-DNA antibodies via energy-independent mechanisms. Immunology 2015, 147, 73–81. [CrossRef] [PubMed]
- Park H, Kim M, Seo Y, Ham Y, Cho MY, Kwon MH. Cytosolic internalization of anti-DNA antibodies by human monocytes induces production of pro-inflammatory cytokines independently of the tripartite motif-containing 21 (TRIM21)-mediated pathway. Front. Immunol. 2019, 9. [CrossRef] [PubMed]
- Jang JY, Jeong JG, Jun HR, Lee SC, Kim JS, Kim YS, Kwon MH. A nucleic acid-hydrolyzing antibody penetrates into cells via caveolae-mediated endocytosis, localizes in the cytosol and exhibits cytotoxicity. Cell Mol. Life Sci. 2009, 66, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Im SR, Im SW, Chung HY, Pravinsagar P, Jang YJ. Cell- and nuclear-penetrating anti-dsDNA autoantibodies have multiple arginines in CDR3 of VH and increase cellular level of pERK and Bcl-2 in mesangial cells. Mol. Immunol. 2015, 67, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Fenton KA, Tømmerås B, Marion TN, Rekvig OP. Pure anti-dsDNA mAbs need chromatin structures to promote glomerular mesangial deposits in BALB/c mice. Autoimmunity 2010, 43, 179–188. [Google Scholar] [CrossRef]
- Saito M, Makino Y, Inoue K, Watanabe Y, Hoshi O, Kubota T. Anti-DNA antibodies cross-reactive with b2-glycoprotein I induce monocyte tissue factor through the TLR9 pathway. Immunol. Med. 2021, 44, 124–135. [Google Scholar] [CrossRef]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Inoue K, Ishizawa M, Kubota T. Monoclonal anti-dsDNA antibody 2C10 escorts DNA to intracellular DNA sensors in normal mononuclear cells and stimulates secretion of multiple cytokines implicated in lupus pathogenesis. Clin. Exp. Immunol. 2020, 199, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Virachith S, Saito M, Watanabe Y, Inoue K, Hoshi O, Kubota T. Anti-b2-glycoprotein I antibody with DNA binding activity enters living monocytes via cell surface DNA and induces tissue factor expression. Clin. Exp. Immunol. 2019, 195, 167–178. [Google Scholar] [CrossRef]
- Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Manca, E. Autoantibodies in neuropsychiatric systemic lupus erythematosus (NPSLE): Can they be used as biomarkers for the differential diagnosis of this disease? Clin. Rev. Allerg. Immunol. 2022, 63, 194–209. [Google Scholar] [CrossRef]
- Stamou M, Grodzki AC, van Oostrum M, Wollscheid B, Lein PJ. Fc gamma receptors are expressed in the developing rat brain and activate downstream signaling molecules upon cross-linking with immune complex. J. Neuroinflammation 2018, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Inoue K, Hoshi O, Kubota T. Internalization of anti-DNA antibodies by rat brain cells: a possible pathogenetic mechanism of neuropsychiatric lupus. Med. Res. Arch. 2023, 11. [CrossRef]
- Koob, AO. Astrocytes imagined. J. Integr. Neurosci. 2022, 21, 112. [Google Scholar] [CrossRef]
- Gordon RE, Nemeth JF, Singh S, Lingham RB, Grewal IS. Harnessing SLE autoantibodies for intracellular delivery of biologic therapeutics. Trends. Biotechnol. 2021, 39, 298–310. [Google Scholar] [CrossRef]
- Rattray Z, Deng G, Zhang S, Shirali A, May CK, Chen X, Cuffari BJ, Liu J, Zou P, Rattray NJW, et al. ENT2 facilitates brain endothelial cell penetration and blood-brain barrier transport by a tumor-targeting anti-DNA autoantibody. JCI Insight 2021, 6, e145875. [CrossRef] [PubMed]
- Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [CrossRef] [PubMed]
- von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [CrossRef] [PubMed]
- Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [CrossRef]
- Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stolzfus A, Borczuk AC, Loda M, et al. Neutrophil extracellular traps contribute to immmunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [CrossRef] [PubMed]
- Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed]
- Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [CrossRef]
- Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar]
- van Avondt K, Fritsch-Stork R, Derksen RHWM, Meyaard L. Ligation of signal inhibitory receptor on leukocytes-1 suppresses the release of neutrophil extracellular traps in systemic lupus erythematosus. PLoS ONE 2013, 8, e78459. [Google Scholar] [CrossRef] [PubMed]
- de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [CrossRef] [PubMed]
- Foret T, Dufrost V, du Mont LS, Costa P, Lakomy C, Lagrange J, Lacolley P, Regnault V, Zuily S, Wahl D. A new pro-thrombotic mechanism of neutrophil extracellular traps in antiphospholipid syndrome: impact on activated protein C resistance. Rheumatology 2022, 61, 2993–2998. [CrossRef] [PubMed]
- Ünlü O, Zuily S, Erkan D. The clinical significance of antiphospholipid antibodies in systemic lupus erythematosus. Eur. J. Rheumatol. 2016, 3, 75–84. [Google Scholar] [CrossRef]
- Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, Hernández-Ramírez D, Bockenstedt PL, Liaw PC, Cabral AR, et al. Release on neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies. A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, Pinsky DJ, Henke PK, Knight JS. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef]
- van der Linden M, van den Hoogen LL, Westerlaken GHA, Fritsch-Stork RDE, van Roon JAG, Radstake TRDJ, Meyaard L. Neutrophil extracellular trap release is associated with antinuclear antibodies in systemic lupus erythematosus and anti-phospholipid syndrome. Rheumatology 2018, 57, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- van Dam LS, Kraaij T, Kamerling SWA, Bakker JA, Scherer UH, Rabelink TJ, van Kooten C, Teng YKO. Intrinsically distinct role of neutrophil extracellular trap formation in antineutrophil cytoplasmic antibody-associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol. 2019, 71, 2047–2058. [Google Scholar] [CrossRef]
- Hayden H, Ibrahim N, Klopf J, Zagrapan B, Mauracher LM, Hell L, Hofbauer TM, Ondracek AS, Schoergenhofer C, Jilma B, et al. ELISA detection of MPO-DNA complexes in human plasma is error-prone and yields limited information on neutrophil extracellular traps formed in vivo. PLoS ONE 2021, 16, e0250265. [Google Scholar] [CrossRef]
- Matta B, Battaglia J, Barnes BJ. Detection of neutrophil extracellular traps in patient plasma: method development and validation in systemic lupus erythematosus and healthy donors that carry IRF5 genetic risk. Front. Immunol. 2022, 13, 951254. [Google Scholar] [CrossRef] [PubMed]
- Arends EJ, van Dam LS, Kraaij T, Kamerling SWA, Rabelink TJ, van Kooten C, Teng YKO. A high-throughput assay to assess and quantify neutrophil extracellular trap fprmation. J.Vis.Exp. 2019, 143, e59150. [CrossRef]
- Jeremic I, Djuric O, Nikolic M, Vlajnic M, Nikolic A, Radojkovic D, Bonaci-Nikolic B. Neutrophil extracellular traps-associated markers are elevated in patients with systemic lupus erythematosus. Rheumatol. Int. 2019, 39, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Hattanda F, Nakazawa D, Watanabe-Kusunoki K, Kusunoki Y, Shida H, Masuda S, Nishio S, Tomaru U, Atsumi T, Ishizu A. The presence of anti-neutrophil extracellular trap antibody in patients with microscopic polyangiitis. Rheumatology 2019, 58, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Zuo Y, Yalavarthi S, Gockman K, Madison JA, Gudjonsson JE, Kahlenberg JM, McCune WJ, Bockenstedt PL, Karp DR, Knight JS. Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheumatol. 2020, 72, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- Zuo Y, Navaz S, Tsodikov A, Kmetova K, Kluge L, Ambati A, Hoy CK, Yalavarthi S, de Andrade D, Tektonidou MG, et al. Anti-neutrophil extracellular trap antibodies in antiphospholipid antibody-positive patients: results from the Antiphospholipid Syndrome Alliance for Clinical Trials and InternatiOnal Networking clinical database and repository. Arthritis Rheumatol. 2023, 75, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhan M, Wang Z, Bao H, Di C, Xia C, Zhang X, Liu Y. Antibodies against neutrophil extracellular traps (NETs) potentiate clinical performance of anti-double-stranded DNA antibodies in systemic lupus erythematosus. Clin. Immunol. 2023, 249, 109297. [CrossRef] [PubMed]
- Antiochos B, Trejo-Zambrano D, Fenaroli P, Rosenberg A, Baer A, Garg A, Sohn J, Li J, Petri M, Goldman DW, et al. The DNA sensors AIM2 and IFI16 are SLE autoantigens that bind neutrophil extracellular traps. eLife 2022, 11, e72103. [Google Scholar] [CrossRef] [PubMed]
- Apel F, Andreeva L, Knackstedt LS, Streeck R, Frese CK, Goosmann C, Hopfner KP, Zychlinsky A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021, 14, eaax7942. [Google Scholar] [CrossRef]
- Lou H, Wojciak-Stothard B, Ruseva MM, Cook HT, Kelleher P, Pickering MC, Mongkolsapaya J, Screaton GR, Xu X. Autoantibody-dependent amplification of inflammation in SLE. Cell Death Dis. 2020, 11, 729. [Google Scholar] [CrossRef]
- Patiño-Trives AM, Pérez-Sánchez C, Pérez-Sánchez L, Luque-Tévar M, Ábalos-Aguilera MC, Alcaide-Ruggiero L, Arias-de la Rosa I, Román-Rodríguez C, Seguí P, Espinosa M, et al. Anti-dsDNA antibodies increase the cardiovascular risk in systemic lupus erythematosus promoting a distinctive immune and vascular activation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2417–2430. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



