
Submitted:
31 October 2023
Posted:
01 November 2023
Read the latest preprint version here
Abstract
Herpesvirus is a prevalent pathogen that primarily infects human epithelial cells and has the ability to reside in neurons. In the field of otolaryngology, herpesvirus infection primarily leads to hearing loss and vestibular neuritis, and is considered the primary hypothesis regarding the pathogenesis of vestibular neuritis. Individuals afflicted with vestibular neuritis experience diz-ziness, which significantly impacts their daily lives. The impact of herpes virus infection on host cell processes and the targeted clearance of infected host cells by immune cells are likely the primary pathogenic mechanisms underlying vestibular neuritis. In this review, we provide a summary of the effects of herpes virus on cellular processes in both host cells and immune cells, with a focus on HSV-1 and HCMV as illustrative examples.
Keywords:
Keywords: Herpesvirus
; Herpes Simplex virus Type 1
; Human cytomegalovirus
; Vestibular neuritis
; Celler processes
1. Introduction
Human herpesviruses are large, enveloped, double-stranded DNA viruses that cause a variety of diseases and establish lifelong latent infections in the majority of the global population. The Herpesviridae family comprises nine viruses that are capable of causing human infections and is divided into three subfamilies: Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae[1]. Alphaherpesvirinae consists of Herpes simplex virus I (HSV-1), HSV-2, and Varicella zoster virus (VZV). Betaherpesvirinae includes Human cytomegalovirus (HCMV), Human herpesvirus 6A (HHV-6A), HHV-6B, and HHV-7. Epstein–Barr virus (EBV) and Kaposi's sarcoma herpesvirus (KSHV) belong to Gammaherpesvirinae.
Vestibular neuritis (VN) is a clinical condition in the field of otolaryngology characterized by acute and persistent peripheral vertigo caused by unilateral vestibular afferent nerve block. The cause of VN is still unknown, but the most common hypothesis is viral infection or reactivation, particularly by HSV (Table 1). Several researchers have reported herpes virus infection in the vestibular ganglion of patients with vestibular neuritis[2,3,4,5,6,7]. Arbusow reported the presence of HSV-1 DNA in both the human vestibular ganglion and vestibular nuclei[8], suggesting the potential migration of the virus to the human vestibular labyrinth[5]. Furthermore, HSV-1 DNA or HSV latency-related transcripts have been detected in vestibular ganglia removed during surgery in patients with Meniere's disease[4,9]. Herpesviruses have a tendency to invade sensory neurons, establish a latency period, and can be reactivated to cause disease. The initial infection or reactivation of the herpesvirus can profoundly affect cellular processes in the host. Mice inoculated with HSV-1 and HSV-2 into the middle ear exhibited hearing loss and vestibular dysfunction. HSV infection was observed in columnar epithelial cells of the infected mice in the stria vascularis, leading to apoptosis in a portion of the infected cells, while many uninfected cells in the spiral organ of Corti also underwent apoptosis. While vestibular ganglion cells did not undergo apoptosis, some of the cells experienced functional loss[10]. Mice inoculated with HSV-1 after auricle also exhibited vestibular dysfunction along with the death of vestibular ganglion cells[11]. In addition to the damage caused by virus infection to cells, the killing of host cells by immune cells may also contribute to the pathogenesis of vestibular neuritis. The coexistence of CD8+ cells and HSV-1 has been observed in the vestibular ganglion cells of patients with vestibular neuritis.
This article provides a summary of the alterations in cellular processes post-infection, using HSV-1 and HCMV as exemplars, with the aspiration that this knowledge will aid in the treatment of VN and other diseases instigated by human herpesviruses.
2. Herpes Simplex virus Type 1
2.1. The process of HSV-1 entering host cells, replication and assembly
HSV-1 has a spherical shape. The complete HSV-1 virus comprises double-stranded DNA, a nucleocapsid, teguments, envelope proteins, and a lipid envelope (Figure 1A)[12]. The nucleocapsid shell exhibits a symmetrical three-dimensional icosahedral structure. HSV-1 is primarily transmitted through close contact[13]. The entry of herpesvirus into human cells for receptor binding and membrane fusion requires the involvement of multifunctional viral glycoproteins on its surface[14,15,16]. HSV-1 carries a minimum of 12 different glycoproteins. The viral fusion protein glycoprotein B (gB) and the hetero-oligomers glycoprotein H/glycoprotein L (gH/gL) constitute the core entry glycoproteins of the herpesvirus[17]. Briefly, when HSV is adsorbed on the cell membrane surface, the initial non-specific binding between glycoprotein gC and/or gB and the heparan sulfate mucin (HSPG) on the cell surface reduces the spatial distance between the viral envelope and the cell membrane. gD can specifically bind to Herpes virus entry mediator (HVEM), nectin-1, nectin-2, or 3-O-sulfated heparan sulfate (3-OS-HS). This binding further initiates gH and gL. Subsequently, gH-gL transmits signals to gB [18]. gB undergoes a conformational change, inserts into the host cell membrane, and then refolds to fuse the cellular and viral membranes together (Figure 1B). The refolding of multiple gB trimers creates pores in the membrane, initiating the fusion process between the viral envelope and cell membrane. This process may enable the viral nucleocapsid and DNA to enter the cytoplasm and translocate to the nucleus[19,20].
The transportation of viral capsids and vesicles carrying viral glycoproteins in the cytoplasm is closely linked to microtubules, and their translocation along axons depends on microtubules[21,22,23,24]. HSV-I utilizes microtubules and actin for retrograde entry into cells along axons, as well as for retrograde transport during virus assembly and exit[25]. There are two types of axonal transport: fast and slow[26,27]. Fast axonal transport occurs in both cis and retrograde directions, transporting mitochondria, neurotransmitters, channel proteins, and more. In contrast, slow axonal transport occurs in a paracrine direction, transporting cytoskeletal components such as neurofilaments, microtubule proteins, and actin[26,28]. HSV-1 is actively directed to spread from neurons through the axonal cytoskeleton and molecular motors. Studies using time-lapse microscopy have shown that HSV-1 undergoes rapid axonal flow in both directions[29,30]. After the nucleocapsid is transported to the surrounding area of the nucleus, it can interact with the nuclear pore complex, and then dsDNA is injected into the nucleus through the nuclear pore[31].
Figure 2.
Schematic diagram of intracellular replication and assembly of herpesviruses. Once the HSV-1 nuclear capsid enters the cell, it binds to the motor protein associated with the microtubule. Subsequently, the nucleocapsid is transported towards the nucleus via microtubules. Upon reaching the vicinity of the nucleus, the HSV-1 capsid injects its genomic DNA into the nucleus through the nuclear pore complex. The injected viral genomic DNA is targeted to the PML body within the nucleus. The replicating viral genomic DNA assembles with a nucleocapsid that consists of early proteins synthesized in the nucleus and late proteins synthesized in the cytoplasm. Subsequently, it crosses the nuclear membrane and enters the cytoplasm. During this process, the virus is initially coated with an envelope, which may originate from the inner membrane of the nuclear envelope. Subsequently, the viruses lose the initial envelope through fusion with the outer nuclear membrane and are released into the cytoplasm without an envelope. Upon arrival in the cytoplasm, the capsid is subsequently reenveloped within an intracellular organelle, where it acquires its mature envelope and completes tegumentation. The capsid undergoes secondary envelopment before being released from the cell. During this process, the nucleocapsid, which is now associated with tegument proteins, buds into the membrane of a cytoplasmic organelle, resulting in the formation of an enveloped virion inside a vesicle. The origin of organelle membranes in the secondary envelope is still controversial, with some suggesting that they may originate from membrane tubes derived from recycled endosomes or vesicles from the trans Golgi network. Viruses that have completed the secondary envelopment are released through exocytosis.
Figure 2.
Schematic diagram of intracellular replication and assembly of herpesviruses. Once the HSV-1 nuclear capsid enters the cell, it binds to the motor protein associated with the microtubule. Subsequently, the nucleocapsid is transported towards the nucleus via microtubules. Upon reaching the vicinity of the nucleus, the HSV-1 capsid injects its genomic DNA into the nucleus through the nuclear pore complex. The injected viral genomic DNA is targeted to the PML body within the nucleus. The replicating viral genomic DNA assembles with a nucleocapsid that consists of early proteins synthesized in the nucleus and late proteins synthesized in the cytoplasm. Subsequently, it crosses the nuclear membrane and enters the cytoplasm. During this process, the virus is initially coated with an envelope, which may originate from the inner membrane of the nuclear envelope. Subsequently, the viruses lose the initial envelope through fusion with the outer nuclear membrane and are released into the cytoplasm without an envelope. Upon arrival in the cytoplasm, the capsid is subsequently reenveloped within an intracellular organelle, where it acquires its mature envelope and completes tegumentation. The capsid undergoes secondary envelopment before being released from the cell. During this process, the nucleocapsid, which is now associated with tegument proteins, buds into the membrane of a cytoplasmic organelle, resulting in the formation of an enveloped virion inside a vesicle. The origin of organelle membranes in the secondary envelope is still controversial, with some suggesting that they may originate from membrane tubes derived from recycled endosomes or vesicles from the trans Golgi network. Viruses that have completed the secondary envelopment are released through exocytosis.
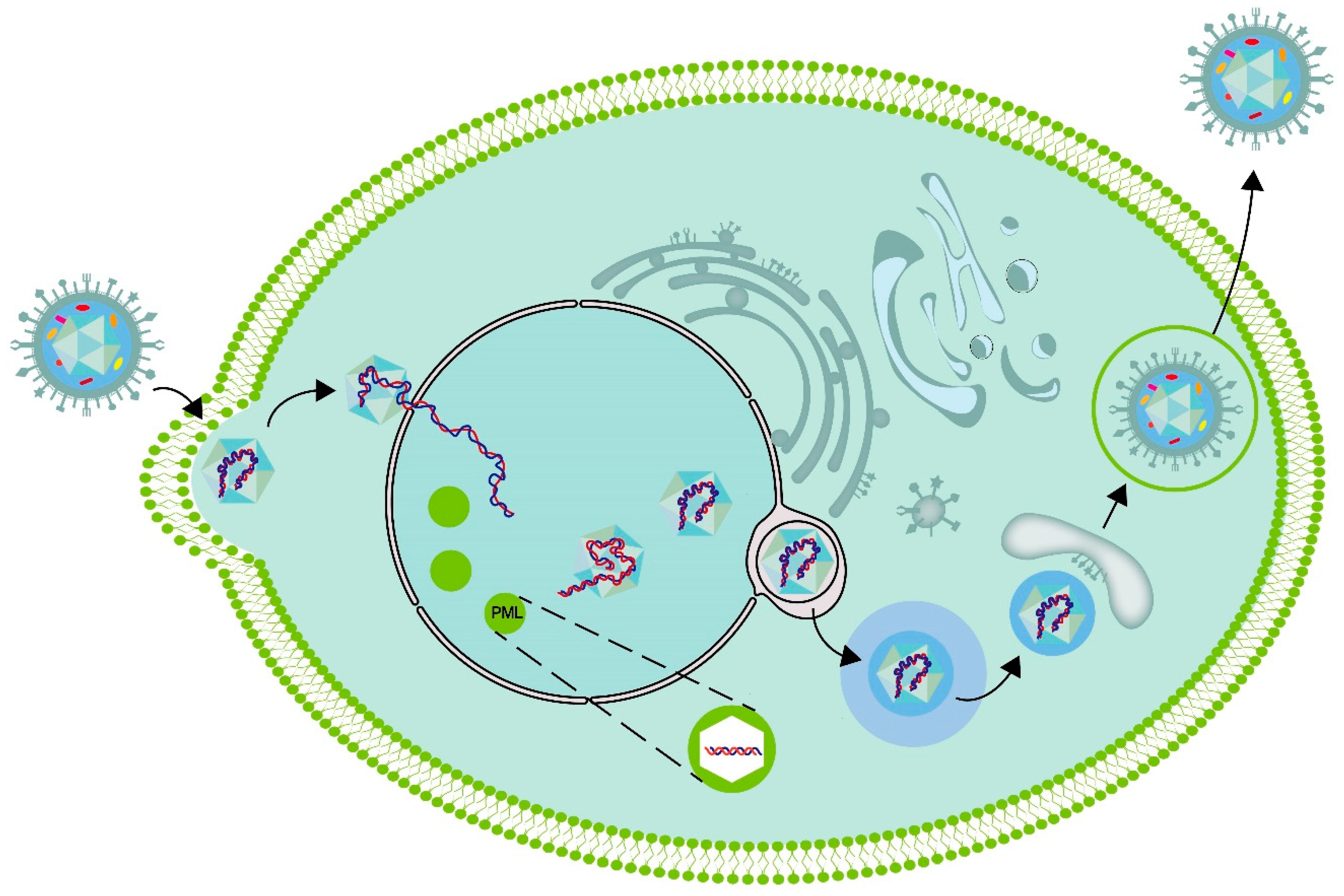
DNA viruses, such as herpesviruses, replicate in specific inclusions within the nucleus. These inclusions, referred to as viral replication compartments (VRCs), are the sites where viral DNA replication, viral transcription, and virion assembly take place [32,33,34]. Compartmentalization is an essential feature in living organisms. Cellular organisms typically utilize cell membranes to partition cells into compartments. Moreover, eukaryotic cells possess membrane-free compartments, such as stress granules and Pbodies[35,36]. Certain compartments exhibit liquid properties and are formed through a process known as liquid-liquid phase separation (LLPS), analogous to the formation of oil droplets in water[37]. There is a hypothesis suggesting that the nuclear viral replication compartments (VRCs) of DNA viruses, such as HSV-1, are also phase-separated condensates[33,34,38]. Michael Seyffert demonstrated that the HSV-1 transcription factor ICP4 has the ability to induce protein condensation, thereby imparting liquid-like properties to the VRC[39].
Primary HSV-1 infections generally occur in the epithelial cells of oral and anal mucosa[25]. Following infections of the skin or mucosa innervated by sensory nerves, HSV-1 can undergo retrograde axonal transport to neuronal cell bodies. It can then establish lifelong latency within the dorsal root ganglia(DRG)[40,41] and trigeminal ganglia (TG) and can be reactivated, resulting in tissue damage. Clinical manifestations of HSV-1 infection are changeable, depending on host immune function and mode of viral transmission[42].
2.2. Host cell processes caused by HSV-1 infection
HSV-1 belongs to the lysogenic family of viruses, and its lytic replication results in the destruction of host cells. Cell aggregation is observed almost immediately after cells are infected with HSV-1, and the severity tends to increase with the number of infections [43]. According to Roizman et al [43], herpes simplex virus infection can lead to the production of multinucleated cells, which result from the fusion of functional cells with different phenotypic characteristics. In HSV-1-infected cells, Avitabile et.al [44] found that microtubules are partially broken, especially at the cell periphery, where the connection between the microtubule network and the plasma membrane appears to be lost. Subsequently, the microtubules form bundles around the nucleus, resulting in a near-spherical shape of the cells [44]. Heeg et al observed that infection with high doses of various strains of HSV-1 for two and a half hours resulted in cell rounding, accompanied by the breakdown of actin-containing microfilaments and the appearance of knob-like protuberances containing actin at the cell periphery[45]. Hampar et.al reported that HSV-1 infection of cells causes chromosome breaks, translocations, and fusions[46]. Roizman et al reported that protein synthesis must precede viral DNA synthesis in the early stages of HSV-infected cells[47]. Both functional and structural proteins required for viral proliferation are produced by the host cell's translation system. HSV has been observed to decrease protein synthesis and mRNA levels in host cells with the expression level of viral proteins rapidly increasing, accompanied by the rapid degradation of previously existing polyribosomes and some host cell mRNA[48]. Aubert and Blaho summarize that the manifestations of HSV-1 infection include (i) the loss of matrix binding proteins on the cell surface, leading to detachment; (ii) modifications of membranes; (iii) cytoskeletal destabilizations; (iv) nucleolar alterations; and (v) chromatin margination and aggregation or damage, as well as (vi) a decrease in cellular macromolecular synthesis[49].
Cellular autophagy, apoptosis, and necrosis pathways are crucial cellular processes that are interconnected to restrict the spread of pathogens by eliminating infected cells [50]. Viral proteins can interact with these signaling molecules, disrupting downstream signal transduction and promoting viral replication and spread. Dufour et al. demonstrated that the ribonucleotide reductase R1 subunit of HSV inhibits Caspase8, thereby protecting cells from apoptosis induced by tumor necrosis factor (TNF) α and Fas ligand [51]. Furthermore, the research group demonstrated that this HSV protein disrupts the structural domain interactions of the Toll interleukin (IL)-1 receptor, thereby inhibiting poly I:C-induced apoptosis in HeLa cells [52]. Moreover, in addition to inducing the formation of filopodia in infected cells to facilitate viral transmission through cell-to-cell contact [53], Us3 proteins disable Bad by inhibiting its phosphorylation [54], thereby safeguarding the cell against DNA fragmentation, nuclear disintegration, and apoptosis [53,55].
Autophagy is a crucial cellular process that involves the self-degradation and recycling of cellular components, including the cell membrane, cytoplasm, and organelles. It plays a role in eliminating misfolded proteins, damaged organelles, and intracellular pathogens. However, certain HSV proteins, such as US11 and ICP34.5, interfere with cellular autophagy. US11 is a ribosome-associated double-stranded RNA-binding protein that directly interacts with PKR [56]. On the other hand, ICP34.5 consists of a C-terminal structural domain and an N-terminal structural domain. The C-terminal domain recruits protein phosphatase 1 (PP-1) to inhibit PKR-mediated phosphorylation of eLF2α [57], while the N-terminal domain directly interacts with Beclin-1 to block autophagy [58]. In summary, there are multiple pathways through which the host induces apoptosis and autophagy in infected cells, and HSV employs its own proteins to interfere with certain steps in these pathways to protect the survival of infected cells and facilitate its own replication and dissemination.
2.3. Immune cell process caused by HSV-1 infection
The intrinsic and innate immune responses serve as the first line of defense against viral infections, including HSV. They work together to limit the spread of viral replication until the body develops an adaptive immune response. The intrinsic immune response is particularly effective during the initial HSV-1 infection and also contributes to the subsequent adaptive immune response [59].The innate immune response is initiated through the cellular expression of pattern recognition receptors (PRRs), which detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns [60,61]. This recognition stimulates the secretion of interferon (IFN) α, β, or γ, along with other cytokines [62]. These cytokines can act in an autocrine and paracrine manner and play a crucial role in controlling HSV infection and coordinating innate and adaptive immune responses.Among the PRRs, Toll-like receptors (TLRs) are involved in detecting HSV nucleic acids and proteins. TLRs 2, 3, and 9 are the major TLRs responsible for HSV detection [61,63]. Interaction between PAMPs and TLRs leads to IFN secretion [62]. TLR2 recognizes viral glycoproteins, TLR3 senses double-stranded RNA (dsRNA) produced during HSV replication, and TLR9 recognizes HSV DNA. TLR2 interacts with gH and gL on the viral envelope and signals through myeloid differentiation factor 88 (MyD88) [64,65]. TLR2 activation promotes the expression of pro-inflammatory cytokines, exerting antiviral effects. However, studies on TLR2-deficient mice infected with HSV have shown that these mice exhibit fewer symptoms and longer survival than wild-type mice, suggesting that TLR2 activation may have harmful effects on the host [66,67].TLR3 recognition of dsRNA plays a protective role against herpes simplex virus encephalitis (HSE) in children [68]. Defects in the TLR3 response in the central nervous system (CNS) have been observed in approximately 5% of children with HSE [69]. Mouse experiments suggest that astrocytes rely on TLR3 to mediate resistance to HSV infection [70]. However, another study demonstrated that TLR3-deficient neurons and oligodendrocytes were more susceptible to HSV-1 infection compared to control cells, indicating the importance of TLR3 in protecting neuronal cells from HSV infection [71].TLR9 is significant for certain cell types, such as plasmacytoid dendritic cells (pDC), where the absence of TLR9 results in impaired IFN responses [72,73].
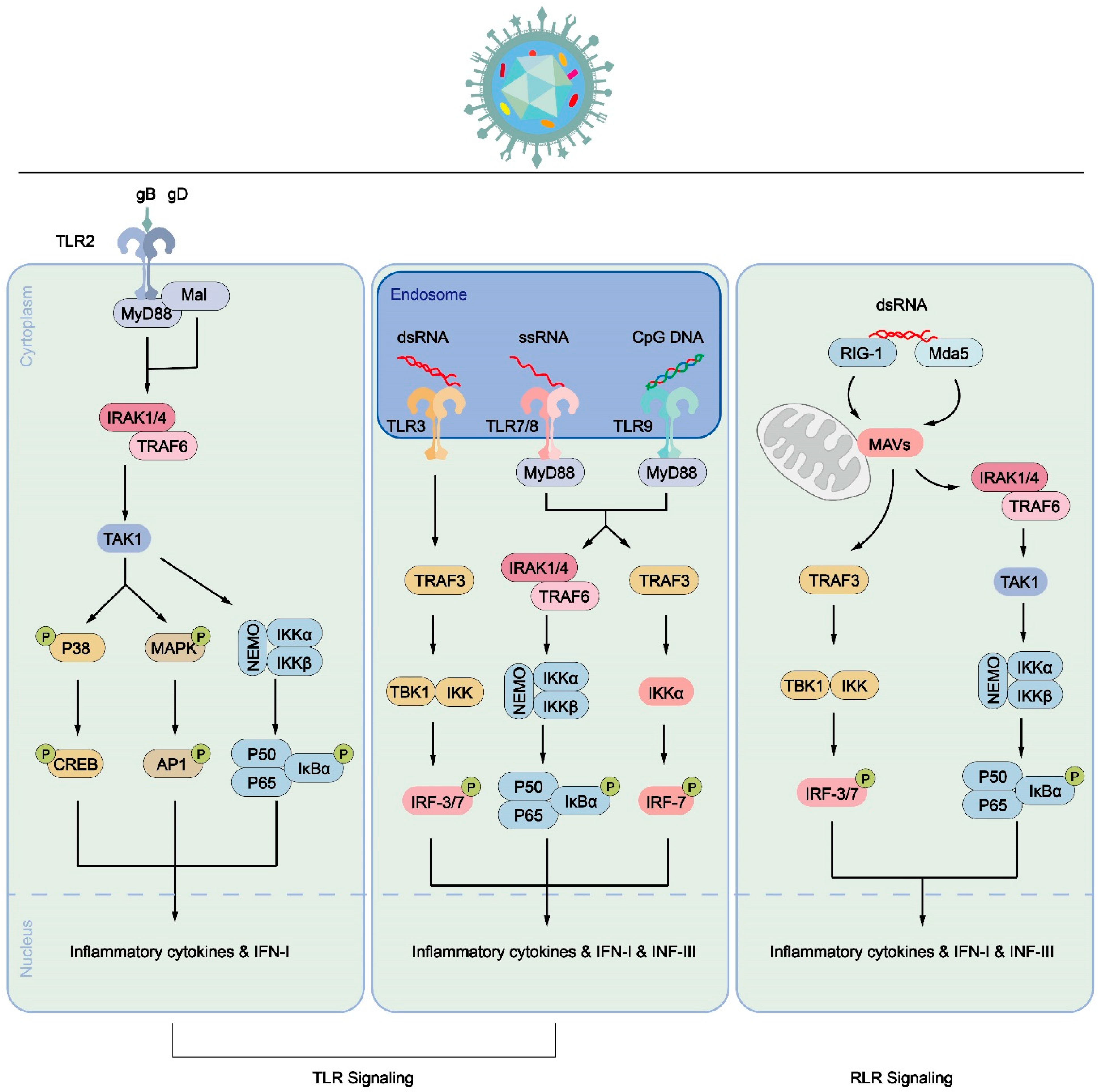
Figure 3.
Pattern diagram of immune cell response process triggered by PRR signal triggered by HSV-1 infection. Inducing the secretion of inflammatory cytokines or IFN through the TLR signaling pathway (left, middle) or RLR signaling pathway (right). In the TLR signaling pathway, TLR2 recognizes signals induced by HSV-1 envelope proteins, such as gB or gD. The signal is transmitted to the cytoplasm, where MyD88 binds to the cytoplasmic domain of TLR2, leading to the activation of transcription factors like NF-κB. This activation promotes the translocation of P50/P65 into the nucleus and increases the expression of inflammatory cytokines and IFN-1. Additionally, TLR3, TLR7/8, and TLR9 signaling are activated by dsRNA, ssRNA, or CpG DNA, respectively, in endosomes. These signals activate IRF-3, IRF-7, and NF-κB, ultimately resulting in increased expression of inflammatory cytokines, IFN-1, IFN-III, and interferon-stimulated genes (ISGs). In the RLR signaling pathway, RIG-I and MDA5, which contain N-terminal caspase activation and recruitment domains, recruit and activate the mitochondrial antiviral signaling (MAVS) protein to mediate signal transduction. The activated MAVS protein further activates downstream signaling, promoting the expression of inflammatory cytokines and IFN.Both pathways contribute to the immune response against HSV-1 infection by triggering the production of inflammatory cytokines and interferons, which play crucial roles in controlling viral replication and coordinating innate and adaptive immune responses.
Figure 3.
Pattern diagram of immune cell response process triggered by PRR signal triggered by HSV-1 infection. Inducing the secretion of inflammatory cytokines or IFN through the TLR signaling pathway (left, middle) or RLR signaling pathway (right). In the TLR signaling pathway, TLR2 recognizes signals induced by HSV-1 envelope proteins, such as gB or gD. The signal is transmitted to the cytoplasm, where MyD88 binds to the cytoplasmic domain of TLR2, leading to the activation of transcription factors like NF-κB. This activation promotes the translocation of P50/P65 into the nucleus and increases the expression of inflammatory cytokines and IFN-1. Additionally, TLR3, TLR7/8, and TLR9 signaling are activated by dsRNA, ssRNA, or CpG DNA, respectively, in endosomes. These signals activate IRF-3, IRF-7, and NF-κB, ultimately resulting in increased expression of inflammatory cytokines, IFN-1, IFN-III, and interferon-stimulated genes (ISGs). In the RLR signaling pathway, RIG-I and MDA5, which contain N-terminal caspase activation and recruitment domains, recruit and activate the mitochondrial antiviral signaling (MAVS) protein to mediate signal transduction. The activated MAVS protein further activates downstream signaling, promoting the expression of inflammatory cytokines and IFN.Both pathways contribute to the immune response against HSV-1 infection by triggering the production of inflammatory cytokines and interferons, which play crucial roles in controlling viral replication and coordinating innate and adaptive immune responses.
The adaptive immune response plays a crucial role in managing HSV infection and reactivation. Cell-mediated immunity, particularly involving T cells, is a key component of the adaptive immune response. After viral infection, cells present antigens to CD8+ T cells through surface major histocompatibility complex (MHC) class 1 molecules. This triggers the elimination of infected cells, limiting viral spread. T cells have been found to play a major role in the adaptive immune response to HSV. Specific T cells have been identified in sensory ganglia of infected individuals and in active and latent lesions of patients [74,75,76,77,78]. Following acute HSV infection, the percentage of blood-specific T cells is lower in infected individuals [79,80]. HSV-specific CD8+ T cells in the blood express high levels of cytolytic molecules when re-exposed to viral antigens [81]. CD4+ T cells recognize HSV-1 proteins and express cytokines associated with helper T cell type 1 (Th1)/Th0-like responses with cytolytic potential [80,82].
HSV-1 is capable of establishing a latency period in the dorsal root ganglia (DRG) of severely combined immunodeficient mice, even when CD8+ memory T cells are transplanted prior to infection. However, the presence of T cells reduces the number of infected DRG neurons, potentially limiting HSV-1 reactivation [83,84]. In mouse models, the rate of in vitro reactivation of trigeminal ganglia (TG) is directly correlated with viral ganglionic load, rather than the number of specific CD8+ T cells [85]. Specific CD8+ and CD4+ T cells are also present in the TG following human HSV-1 infection [74,75]. The infiltrating T cells in human infected TGs are characterized as memory effector T cells and surround the cell bodies and axons of neurons [74,86]. In mouse models, memory CD8+ T cells express interferon-gamma (IFN-γ), which prevents HSV replication in neurons and inhibits neuronal apoptosis, potentially promoting the survival of neurons and HSV-1 silencing and latency [87,88,89]. The mechanism of CD4+ and CD8+ T cell recognition of latently infected neurons is not fully understood. It is possible that there may be limited viral gene expression that can be recognized by T cells, allowing CD8+ T cell recognition and reactivation, along with potentially low levels of neuronal MHC class I molecule expression [90,91]. Additionally, satellite cells can act as antigen-presenting cells and express T-cell suppressor molecules to control HSV-1 latency without damaging neurons [92]. HSV also employs various strategies to inhibit antigen presentation and modulate adaptive immune responses. For example, the viral protein ICP47 blocks antigen presentation, and ICP34.5 inhibits autophagy, which is involved in antigen presentation [93]. Furthermore, HSV can inhibit antibody responses by interacting with antibodies and complement components, inhibiting antibody-dependent cell-mediated cytotoxicity [94]. These mechanisms suggest that HSV can modulate the adaptive immune response and influence the pathogenesis of the infection.
3. Human Cytomegalovirus
3.1. The process of HCMV entering host cells, replication and assembly
HCMV can be transmitted through various routes, including vertical transmission, contact transmission, and sexual transmission [95,96,97]. The virus infects different types of cells, such as leukocytes, fibroblasts, epithelial cells, and endothelial cells [98].HCMV entry into host cells involves several viral envelope glycoprotein complexes. Initially, three complexes were identified: gC-I, gC-II, and gC-III, which play a crucial role in viral entry [99]. gC-II is composed of glycoproteins M (gM) and N (gN), encoded by UL100 and UL73, respectively. It can attach to host cells through interaction with heparan sulfate proteoglycans on the cell surface [100,101]. The gH/gL complex, present in all herpesviruses, along with gB, forms the core membrane fusion machinery. gC-III is a heterotrimer consisting of the gH/gL complex linked to gO via a disulfide bond, commonly referred to as a trimer. Additionally, a pentameric complex consisting of gH/gL bound to three small glycoproteins encoded by UL128, UL130, and UL131A was discovered [103,104,105]. Binding of these complexes to the receptor transmits signals to gB, triggering membrane fusion [104,105]. gC-I is a homotrimer of gB, and different regions of gB are involved in fusion and/or interactions with proteins that trigger fusion [106,107]. After insertion into the target cell membrane, gB refolds into its post-fusion conformation, creating a fusion pore through which the viral capsid can enter the cell. Lateral interactions between gB trimers may contribute to expanding the fusion pore [17,108]. Several cell surface proteins, including platelet-derived growth factor receptor alpha (PDGFRα), epidermal growth factor receptor (EGFR), and integrins, have been reported to be associated with gB receptors [109,110,111,112].
HCMV entry into epithelial and endothelial cells occurs after endocytosis and requires binding to the pentameric complex neurofibrillary protein 2 (NRP 2) to trigger gB [113,114]. In fibroblasts, the trimers bind to PDGFRα and trigger gB at the plasma membrane [115,116,117].
After HCMV infection, the viral genome translocates to the nucleus. One of the first proteins expressed by HCMV is the Immediate Early 1 (IE 1) protein, which disrupts promyelocytic leukemia (PML) nuclear bodies and eliminates their inhibitory effect on viral gene transcription [118]. Replication and transcription of viral DNA occur in viral replication compartments (VRCs). The UL112-113 protein of HCMV plays a role in generating VRCs around the viral genome by liquid-liquid phase separation (LLPS), creating a pro-replicative environment [119]. Additionally, during the early stages of infection, most of the periplasmic proteins and glycoproteins are expressed in the cytoplasm and accumulate in specific cytoplasmic compartments called viral assembly compartments (AC) [120]. Cytoplasmic capsids are transported to the AC, where they form infectious mature particles.
3.2. Host cell processes caused by HCMV infection
HCMV infects organisms by entering through mucosal surfaces and subsequently disseminates to various tissues and organs within the body. Following invasion of the human body by HCMV, the majority of viral replication takes place locally. In 1975, Stagno initially reported a correlation between HCMV viral load and human disease, describing it as a threshold relationship. If the local viral load surpasses a critical threshold, the virus can disseminate via the bloodstream, resulting in damage to multiple target organs. The pathogenic risk associated with various target organs is intricately linked to their respective local viral loads. If the local immune response of a target organ fails to adequately control viral replication below a certain threshold, damage to the corresponding organ may ensue. Simultaneously, the local immune status also influences the threshold at which the virus becomes pathogenic. Strengthening the local immune status results in an increased threshold for viral pathogenicity, rendering the organ more resilient to the virus. Conversely, a weakened local immune status reduces the threshold for viral pathogenicity, making the local organ more susceptible to disease.
Primary infection of healthy individuals by HCMV is typically asymptomatic but can establish a lifelong latent infection in the host, which may lead to disease upon reactivation. The maintenance of latency and reactivation are regulated through the coordinated actions of both viral and cellular factors. HCMV establishes latency in hematopoietic stem cells and hematopoietic progenitor cells (HPCs) and persists in myeloid lineage cells. During latency, viral gene expression, including the major immediate early promoter (MIEP) that controls replication by regulating the IE gene, is reduced. The repression of viral genome transcription is also regulated through direct repression of IE gene expression and the expression of IE regulatory proteins. Buehler proposed a model in which pUL135 and pUL138, along with EGFR, form a molecular switch that regulates latency and replication status in HCMV infection. Upon viral entry and stimulation of the MEK/ERK signaling pathway, EGR-1 stimulates UL 138 expression, inhibiting viral replication to promote the establishment of latency. UL135 promotes EGFR recycling and counteracts the inhibitory effect of UL138, thereby promoting reactivation from latency and viral replication.
The regulation of latency also involves the modulation of host cell signaling. Recent studies have provided evidence that HCMV miRNAs play a role in modulating cellular signaling pathways and mediating latency in HPCs. For instance, Pan discovered miR-UL148, an HCMV-encoded miRNA, is expressed early during HPCs infection and can downregulate an IE activator, thereby promoting the establishment of latency. Diggins also found that miR-US25-1 can target the RhoA signaling axis to control cell proliferation. Moreover, HCMV protects HPCs from apoptosis and enhances HPCs survival through the synergistic effects of UL7 with miR-US5-1 and miR-UL112.
Virus reactivation can be triggered by various factors. For example, stimulation of stem cells moving to the periphery by granulocyte colony-stimulating factor can induce viral reactivation. Viral miRNAs, such as miR-US5-2 and miR-US22, as well as viral proteins, including UL135 and UL138, can also regulate reactivation by controlling EGFR. Additionally, the viral protein UL7 acts as a ligand for the Fms-like tyrosine kinase 3 receptor (Flt-3R), and binding to Flt-3R triggers the differentiation of HPCs and monocytes, thereby stimulating HCMV reactivation.
3.3. Immune cell process caused by HCMV infection
Innate immunity serves as the first line of defense against HCMV. Upon initial contact with host cells, viral envelope proteins stimulate an innate immune response. The TLR-1/TLR-2 heterodimer acts as a functional receptor for HCMV, recognizing glycoproteins gB and gH. Recognition of HCMV by TLR2 activates the nuclear factor-kappaB (NF-κB) pathway, leading to the production of inflammatory cytokines. However, HCMV gene products interfere with TLR activity. For example, HCMV miR-UL112-3p modulates the TLR2/NF-κB signaling pathway and downregulates TLR2 expression. HCMV-encoded proteins, such as US7 and US8, also inhibit the TLR pathway, thereby antagonizing innate immunity. NK cells and IFNs play crucial roles in natural immune defense against HCMV. TLR9 recognition of natural interferon-producing cells and dendritic cells (DCs) leads to the secretion of cytokines like IL-12, which activates the antiviral activity of NK cells. Activated NK cells can control HCMV replication through cytolytic and non-cytolytic mechanisms. They utilize perforin to lyse infected cells and secrete lymphotoxin α and TNF, which induce IFN-β expression in target cells. IFN-γ produced by NK cells confers resistance to HCMV infection in other cells. The combined action of IFN-β and IFN-γ inhibits HCMV replication without causing cell lysis after IE gene expression. HCMV employs various strategies to evade NK cell elimination. The periplasmic protein pp65 inhibits NK cell cytotoxicity by interacting with the activation receptor NKp30. HCMV actively downregulates NKG2D ligands, including UL16 binding proteins (ULBP) 1/2/6 and MHC class I-associated chain B (MICB), mediated by the glycoprotein UL16. UL148A proteins downregulate MICA, while HCMV-miR-UL112 specifically suppresses MICB expression, resulting in reduced NKG2D binding and decreased NK cell killing.
Adaptive immunity provides long-lasting protection against primary HCMV infection and latent activation in healthy individuals. The role of antibodies in protecting against HCMV infection is still a topic of debate. While individuals with HCMV-specific T cells may not be completely protected from viral infection and disease episodes, this suggests that humoral immunity, particularly neutralizing antibodies, may play a role in resistance to HCMV infection. Studies have shown that HCMV immune serum is capable of neutralizing fibroblast and epithelial/endothelial cell infections. However, it is important to note that neutralizing antibodies are found at much higher levels in epithelial/endothelial cells compared to fibroblasts. In one study, the titers of neutralizing antibodies that effectively blocked HCMV infection were 128-fold higher in epithelial/endothelial cells than in fibroblasts. Additionally, immunoreactivity induced by HCMV immunoglobulin preparations was 50-fold higher in epithelial cells compared to fibroblasts. These findings suggest that neutralizing antibodies may be more effective in protecting against HCMV infection in epithelial and endothelial cells compared to fibroblasts. However, further research is needed to fully understand the role of antibodies in HCMV immunity and their effectiveness in different cell types.
CD8+ T cells play a crucial role in the immune response to HCMV by recognizing a wide range of structural, early, and late antigens, including HCMV-encodedomodulators. The most dominant antigens targeted by HCMV-specific CD8+ T cells are UL123 (IE-1), UL122 (IE-2), and UL83 (pp65). These cytotoxic CD8+ T cells recognize HCMV antigenic peptides presented on MHC class I molecules by antigen-presenting cells (APCs). They can inhibit intracellular viral replication by secreting IFN-γ or TNF-α, and they can also directly kill infected cells by releasing granzymes and perforins. Additionally, HCMV-specific CD8+ T cells undergo oligoclonal replication, leading to their accumulation with age. In fact, they can constitute more than 40% of the CD8+ T cell pool, a phenomenon known as "memory expansion," which is also observed in HCMV-specific CD4+ T cell responses. The clonal amplification and differentiation of cytotoxic CD8+ T cells during HCMV infection support the notion that HCMV may contribute to immune senescence. CD4+ T cells are also essential in controlling HCMV infection. They recognize MHC class II viral antigens presented by APCs and secrete INF-γ and IL-2, which inhibit viral replication. Furthermore, CD4+ T cells induce the proliferation of CD8+ T cells and macrophages. There are subpopulations of HCMV-specific effector CD4+ T cells that directly counteract viral infection, as described by Costa-García. Regulatory T (Treg) cells are involved in the immune response to HCMV as well. A study by Aandahl et al. demonstrated that depletion of CD25+ Treg cells from peripheral blood mononuclear cells enhances the immune response of CD8+ T cells to HCMV antigens.
HCMV has evolved various strategies to evade host immunity, with one of the main strategies being the targeting of MHC class I presentation of viral proteins to CD8+ T cells. The HCMV IE gene US3 encodes an endoplasmic reticulum (ER)-resident glycoprotein that retains protein-loaded MHC class I in the ER, preventing their transport to the cell surface. US6 interacts with the transporters associated with antigen processing in the ER, inhibiting the intracellular transport of MHC class I molecules. Additionally, US2 and US11 downregulate the expression of MHC class I heavy chain.
HCMV also hampers antigen presentation through the MHC class II pathway. US2 leads to the degradation of HLA-DR-α and HLA-DM-α, essential proteins in the MHC class II pathway. US3 can mediate the degradation of DR-α and DM-α and inhibit the assembly of MHC class II complexes. Normally, IFN-γ activates the MHC class II transactivator gene (CIITA) to promote MHC class II expression. However, HCMV inhibits MHC class II expression by blocking IFN-γ signal transduction through the Jak/Stat pathway. Moreover, HCMV regulates CIITA transcript levels in mature Langerhans-type dendritic cells and myeloid cell lines, reducing MHC class II biosynthesis.
Another evasion mechanism employed by HCMV is the production of multiple functional homologues. For instance, UL111A encodes a homologue of human IL-10 called cmvIL-10, which interacts with the human IL-10 receptor. IL-10 is an immunosuppressive cytokine that inhibits the ability of antigen-presenting cells (APCs) to present antigens to T cells, thereby suppressing the immune response[184].
4. Conclusion
The presence of herpesviruses, including HSV-1 and HCMV, has been detected in surgically removed tissues from patients with Meniere's disease, and viral infection is considered a leading hypothesis for the development of vestibular neuritis. However, the exact role of herpesviruses in the pathogenesis of vestibular neuritis is still not fully understood. In this paper, we aim to summarize the effects of herpesvirus infections, particularly HSV-1 and HCMV, on host cells and immune system processes. This information can be valuable in predicting neuronal cell damage, as well as the infiltration and killing of immune cells following viral infection. We hope that this review will stimulate further work and efforts to advance the prevention and treatment of diseases like vestibular neuritis that are potentially caused by viral infections.
5. Patents
Author Contributions
YS conceived and designed the manuscript. ZZ and LX wrote the manuscript. YZ and SX have drawn a schematic diagram. YS reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Innovative Research Groups of Hubei Province (No. 2023AFA038), the National Natural Science Foundation of China (No. 82071058), and the National Key Research and Development Program of China (No. 2021YFF0702303).
References
- Weidner-Glunde, M.; Kruminis-Kaszkiel, E.; Savanagouder, M., Herpesviral Latency-Common Themes. Pathogens 2020, 9, (2). [CrossRef]
- Theil, D.; Arbusow, V.; Derfuss, T.; Strupp, M.; Pfeiffer, M.; Mascolo, A.; Brandt, T., Prevalence of HSV-1 LAT in human trigeminal, geniculate, and vestibular ganglia and its implication for cranial nerve syndromes. Brain Pathol 2001, 11, (4), 408-13. [CrossRef]
- Arbusow, V.; Schulz, P.; Strupp, M.; Dieterich, M.; von Reinhardstoettner, A.; Rauch, E.; Brandt, T., Distribution of herpes simplex virus type 1 in human geniculate and vestibular ganglia: implications for vestibular neuritis. Ann Neurol 1999, 46, (3), 416-9. [CrossRef]
- Furuta, Y.; Takasu, T.; Fukuda, S.; Inuyama, Y.; Sato, K. C.; Nagashima, K., Latent herpes simplex virus type 1 in human vestibular ganglia. Acta Otolaryngol Suppl 1993, 503, 85-9. [CrossRef]
- Schulz, P.; Arbusow, V.; Strupp, M.; Dieterich, M.; Rauch, E.; Brandt, T., Highly variable distribution of HSV-1-specific DNA in human geniculate, vestibular and spiral ganglia. Neurosci Lett 1998, 252, (2), 139-42. [CrossRef]
- Morgenstein, K. M.; Seung, H. I., Vestibular neuronitis. Laryngoscope 1971, 81, (1), 131-9.
- Davis, L. E., Viruses and vestibular neuritis: review of human and animal studies. Acta Otolaryngol Suppl 1993, 503, 70-3. [CrossRef]
- Arbusow, V.; Strupp, M.; Wasicky, R.; Horn, A. K.; Schulz, P.; Brandt, T., Detection of herpes simplex virus type 1 in human vestibular nuclei. Neurology 2000, 55, (6), 880-2. [CrossRef]
- Suzuki, S., [Detection of latent herpes simplex virus in human vestibular ganglia]. Hokkaido Igaku Zasshi 1996, 71, (5), 561-71.
- Esaki, S.; Goshima, F.; Kimura, H.; Ikeda, S.; Katsumi, S.; Kabaya, K.; Watanabe, N.; Hashiba, M.; Nishiyama, Y.; Murakami, S., Auditory and vestibular defects induced by experimental labyrinthitis following herpes simplex virus in mice. Acta Otolaryngol 2011, 131, (7), 684-91. [CrossRef]
- Hirata, Y., [An experimental herpes simplex virus infection in the vestibular nerve]. Nihon Jibiinkoka Gakkai Kaiho 1994, 97, (7), 1191-9. [CrossRef]
- Karasneh, G. A.; Shukla, D., Herpes simplex virus infects most cell types in vitro: clues to its success. Virol J 2011, 8, 481. [CrossRef]
- AlMukdad, S.; Harfouche, M.; Farooqui, U. S.; Aldos, L.; Abu-Raddad, L. J., Epidemiology of herpes simplex virus type 1 and genital herpes in Australia and New Zealand: systematic review, meta-analyses and meta-regressions. Epidemiol Infect 2023, 151, e33. [CrossRef]
- Vallbracht, M.; Backovic, M.; Klupp, B. G.; Rey, F. A.; Mettenleiter, T. C., Common characteristics and unique features: A comparison of the fusion machinery of the alphaherpesviruses Pseudorabies virus and Herpes simplex virus. Adv Virus Res 2019, 104, 225-281.
- Möhl, B. S.; Chen, J.; Longnecker, R., Gammaherpesvirus entry and fusion: A tale how two human pathogenic viruses enter their host cells. Adv Virus Res 2019, 104, 313-343.
- Nishimura, M.; Mori, Y., Entry of betaherpesviruses. Adv Virus Res 2019, 104, 283-312.
- Connolly, S. A.; Jardetzky, T. S.; Longnecker, R., The structural basis of herpesvirus entry. Nat Rev Microbiol 2021, 19, (2), 110-121. [CrossRef]
- Atanasiu, D.; Saw, W. T.; Cohen, G. H.; Eisenberg, R. J., Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 2010, 84, (23), 12292-9. [CrossRef]
- Cooper, R. S.; Heldwein, E. E., Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses 2015, 7, (12), 6552-69. [CrossRef]
- Fontana, J.; Atanasiu, D.; Saw, W. T.; Gallagher, J. R.; Cox, R. G.; Whitbeck, J. C.; Brown, L. M.; Eisenberg, R. J.; Cohen, G. H., The Fusion Loops of the Initial Prefusion Conformation of Herpes Simplex Virus 1 Fusion Protein Point Toward the Membrane. mBio 2017, 8, (4). [CrossRef]
- Saksena, M. M.; Wakisaka, H.; Tijono, B.; Boadle, R. A.; Rixon, F.; Takahashi, H.; Cunningham, A. L., Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J Virol 2006, 80, (7), 3592-606. [CrossRef]
- Lee, G. E.; Murray, J. W.; Wolkoff, A. W.; Wilson, D. W., Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J Virol 2006, 80, (9), 4264-75. [CrossRef]
- Kharkwal, H.; Smith, C. G.; Wilson, D. W., Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J Virol 2014, 88, (24), 14467-78.
- Miranda-Saksena, M.; Armati, P.; Boadle, R. A.; Holland, D. J.; Cunningham, A. L., Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J Virol 2000, 74, (4), 1827-39. [CrossRef]
- Miranda-Saksena, M.; Denes, C. E.; Diefenbach, R. J.; Cunningham, A. L., Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, (2). [CrossRef]
- Maday, S.; Twelvetrees, A. E.; Moughamian, A. J.; Holzbaur, E. L., Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, (2), 292-309. [CrossRef]
- Liu, J. J., Regulation of dynein-dynactin-driven vesicular transport. Traffic 2017, 18, (6), 336-347.
- Hirokawa, N.; Niwa, S.; Tanaka, Y., Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, (4), 610-38. [CrossRef]
- Antinone, S. E.; Zaichick, S. V.; Smith, G. A., Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J Virol 2010, 84, (24), 13019-30. [CrossRef]
- Smith, G. A.; Gross, S. P.; Enquist, L. W., Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 2001, 98, (6), 3466-70. [CrossRef]
- Villanueva-Valencia, J. R.; Tsimtsirakis, E.; Evilevitch, A., Role of HSV-1 Capsid Vertex-Specific Component (CVSC) and Viral Terminal DNA in Capsid Docking at the Nuclear Pore. Viruses 2021, 13, (12). [CrossRef]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R. A., DNA virus replication compartments. J Virol 2014, 88, (3), 1404-20. [CrossRef]
- Charman, M.; Weitzman, M. D., Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses 2020, 12, (2). [CrossRef]
- Hidalgo, P.; Gonzalez, R. A., Formation of adenovirus DNA replication compartments. FEBS Lett 2019, 593, (24), 3518-3530. [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T. S.; Mitrea, D. M.; Zhu, L.; Richardson, T. M.; Kriwacki, R. W.; Pappu, R. V.; Brangwynne, C. P., Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, (7), 1686-1697. [CrossRef]
- Brangwynne, C. P.; Eckmann, C. R.; Courson, D. S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A. A., Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, (5935), 1729-32. [CrossRef]
- Hyman, A. A.; Weber, C. A.; Jülicher, F., Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 2014, 30, 39-58. [CrossRef]
- Kobiler, O.; Weitzman, M. D., Herpes simplex virus replication compartments: From naked release to recombining together. PLoS Pathog 2019, 15, (6), e1007714. [CrossRef]
- Seyffert, M.; Georgi, F.; Tobler, K.; Bourqui, L.; Anfossi, M.; Michaelsen, K.; Vogt, B.; Greber, U. F.; Fraefel, C., The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments. Int J Mol Sci 2021, 22, (9). [CrossRef]
- Steiner, I.; Kennedy, P. G.; Pachner, A. R., The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 2007, 6, (11), 1015-28. [CrossRef]
- Kramer, T.; Enquist, L. W., Directional spread of alphaherpesviruses in the nervous system. Viruses 2013, 5, (2), 678-707.
- Chayavichitsilp, P.; Buckwalter, J. V.; Krakowski, A. C.; Friedlander, S. F., Herpes simplex. Pediatr Rev 2009, 30, (4), 119-29; quiz 130.
- Roizman, B., Polykaryocytosis induced by viruses. Proc Natl Acad Sci U S A 1962, 48, (2), 228-34. [CrossRef]
- Avitabile, E.; Di Gaeta, S.; Torrisi, M. R.; Ward, P. L.; Roizman, B.; Campadelli-Fiume, G., Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J Virol 1995, 69, (12), 7472-82. [CrossRef]
- Heeg, U.; Dienes, H. P.; Müller, S.; Falke, D., Involvement of actin-containing microfilaments in HSV-induced cytopathology and the influence of inhibitors of glycosylation. Arch Virol 1986, 91, (3-4), 257-70. [CrossRef]
- Hampar, B.; Ellison, S. A., Chromosomal aberrations induced by an animal virus. Nature 1961, 192, 145-7. [CrossRef]
- Roizman, B., The programming of herpes virus multiplication in doubly-infected and in puromycin-treated cells. Proc Natl Acad Sci U S A 1963, 49, (2), 165-71. [CrossRef]
- Roizman, B.; Roane, P. R., Jr., THE MULTIPLICATION OF HERPES SIMPLEX VIRUS. II. THE RELATION BETWEEN PROTEIN SYNTHESIS AND THE DUPLICATION OF VIRAL DNA IN INFECTED HEP-2 CELLS. Virology 1964, 22, 262-9.
- Aubert, M.; Blaho, J. A., The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J Virol 1999, 73, (4), 2803-13. [CrossRef]
- Finlay, D.; Cantrell, D. A., Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol 2011, 11, (2), 109-17. [CrossRef]
- Dufour, F.; Sasseville, A. M.; Chabaud, S.; Massie, B.; Siegel, R. M.; Langelier, Y., The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 2011, 16, (3), 256-71.
- Dufour, F.; Bertrand, L.; Pearson, A.; Grandvaux, N.; Langelier, Y., The ribonucleotide reductase R1 subunits of herpes simplex virus 1 and 2 protect cells against poly(I · C)-induced apoptosis. J Virol 2011, 85, (17), 8689-701. [CrossRef]
- Finnen, R. L.; Johnston, S. M.; Neron, C. E.; Banfield, B. W., Nucleocytoplasmic shuttling of the HSV-2 serine/threonine kinase Us3. Virology 2011, 417, (1), 229-37. [CrossRef]
- Mori, I., Herpes simplex virus US3 protein kinase regulates host responses and determines neurovirulence. Microbiol Immunol 2012, 56, (6), 351-5. [CrossRef]
- Benetti, L.; Roizman, B., Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 2004, 101, (25), 9411-6.
- Lussignol, M.; Queval, C.; Bernet-Camard, M. F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A., The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 2013, 87, (2), 859-71. [CrossRef]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; Cao, Y., ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J Biol Chem 2011, 286, (28), 24785-92. [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D. A.; Levine, B., HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, (1), 23-35. [CrossRef]
- Zhu, S.; Viejo-Borbolla, A., Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, (1), 2670-2702. [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O., Pathogen recognition and innate immunity. Cell 2006, 124, (4), 783-801.
- Paludan, S. R.; Bowie, A. G.; Horan, K. A.; Fitzgerald, K. A., Recognition of herpesviruses by the innate immune system. Nat Rev Immunol 2011, 11, (2), 143-54.
- Takeda, K.; Akira, S., TLR signaling pathways. Semin Immunol 2004, 16, (1), 3-9.
- Vandevenne, P.; Sadzot-Delvaux, C.; Piette, J., Innate immune response and viral interference strategies developed by human herpesviruses. Biochem Pharmacol 2010, 80, (12), 1955-72. [CrossRef]
- Gianni, T.; Leoni, V.; Campadelli-Fiume, G., Type I interferon and NF-κB activation elicited by herpes simplex virus gH/gL via αvβ3 integrin in epithelial and neuronal cell lines. J Virol 2013, 87, (24), 13911-6.
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G., Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-κB. J Virol 2012, 86, (12), 6555-62. [CrossRef]
- Kurt-Jones, E. A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M. M.; Knipe, D. M.; Finberg, R. W., Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 2004, 101, (5), 1315-20. [CrossRef]
- Marino, A.; Pergolizzi, S.; Cimino, F.; Lauriano, E. R.; Speciale, A.; D'Angelo, V.; Sicurella, M.; Argnani, R.; Manservigi, R.; Marconi, P., Role of Herpes Simplex Envelope Glycoprotein B and Toll-Like Receptor 2 in Ocular Inflammation: An ex vivo Organotypic Rabbit Corneal Model. Viruses 2019, 11, (9). [CrossRef]
- Gantt, S.; Muller, W. J., The immunologic basis for severe neonatal herpes disease and potential strategies for therapeutic intervention. Clin Dev Immunol 2013, 2013, 369172. [CrossRef]
- Casanova, J. L., Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A 2015, 112, (51), E7128-37. [CrossRef]
- Reinert, L. S.; Harder, L.; Holm, C. K.; Iversen, M. B.; Horan, K. A.; Dagnæs-Hansen, F.; Ulhøi, B. P.; Holm, T. H.; Mogensen, T. H.; Owens, T.; Nyengaard, J. R.; Thomsen, A. R.; Paludan, S. R., TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 2012, 122, (4), 1368-76. [CrossRef]
- Leib, D. A., Herpes simplex virus encephalitis: toll-free access to the brain. Cell Host Microbe 2012, 12, (6), 731-2. [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A., Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 2003, 198, (3), 513-20. [CrossRef]
- Krug, A.; Luker, G. D.; Barchet, W.; Leib, D. A.; Akira, S.; Colonna, M., Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, (4), 1433-7. [CrossRef]
- Verjans, G. M.; Hintzen, R. Q.; van Dun, J. M.; Poot, A.; Milikan, J. C.; Laman, J. D.; Langerak, A. W.; Kinchington, P. R.; Osterhaus, A. D., Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A 2007, 104, (9), 3496-501. [CrossRef]
- van Velzen, M.; Jing, L.; Osterhaus, A. D.; Sette, A.; Koelle, D. M.; Verjans, G. M., Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog 2013, 9, (8), e1003547. [CrossRef]
- Zhu, J.; Koelle, D. M.; Cao, J.; Vazquez, J.; Huang, M. L.; Hladik, F.; Wald, A.; Corey, L., Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med 2007, 204, (3), 595-603. [CrossRef]
- Zhu, J.; Peng, T.; Johnston, C.; Phasouk, K.; Kask, A. S.; Klock, A.; Jin, L.; Diem, K.; Koelle, D. M.; Wald, A.; Robins, H.; Corey, L., Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature 2013, 497, (7450), 494-7.
- Posavad, C. M.; Zhao, L.; Dong, L.; Jin, L.; Stevens, C. E.; Magaret, A. S.; Johnston, C.; Wald, A.; Zhu, J.; Corey, L.; Koelle, D. M., Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal Immunol 2017, 10, (5), 1259-1269. [CrossRef]
- Jing, L.; Haas, J.; Chong, T. M.; Bruckner, J. J.; Dann, G. C.; Dong, L.; Marshak, J. O.; McClurkan, C. L.; Yamamoto, T. N.; Bailer, S. M.; Laing, K. J.; Wald, A.; Verjans, G. M.; Koelle, D. M., Cross-presentation and genome-wide screening reveal candidate T cells antigens for a herpes simplex virus type 1 vaccine. J Clin Invest 2012, 122, (2), 654-73.
- Moss, N. J.; Magaret, A.; Laing, K. J.; Kask, A. S.; Wang, M.; Mark, K. E.; Schiffer, J. T.; Wald, A.; Koelle, D. M., Peripheral blood CD4 T-cell and plasmacytoid dendritic cell (pDC) reactivity to herpes simplex virus 2 and pDC number do not correlate with the clinical or virologic severity of recurrent genital herpes. J Virol 2012, 86, (18), 9952-63. [CrossRef]
- Laing, K. J.; Magaret, A. S.; Mueller, D. E.; Zhao, L.; Johnston, C.; De Rosa, S. C.; Koelle, D. M.; Wald, A.; Corey, L., Diversity in CD8(+) T cell function and epitope breadth among persons with genital herpes. J Clin Immunol 2010, 30, (5), 703-22. [CrossRef]
- Koelle, D. M.; Corey, L.; Burke, R. L.; Eisenberg, R. J.; Cohen, G. H.; Pichyangkura, R.; Triezenberg, S. J., Antigenic specificities of human CD4+ T-cell clones recovered from recurrent genital herpes simplex virus type 2 lesions. J Virol 1994, 68, (5), 2803-10. [CrossRef]
- Valyi-Nagy, T.; Deshmane, S. L.; Raengsakulrach, B.; Nicosia, M.; Gesser, R. M.; Wysocka, M.; Dillner, A.; Fraser, N. W., Herpes simplex virus type 1 mutant strain in1814 establishes a unique, slowly progressing infection in SCID mice. J Virol 1992, 66, (12), 7336-45. [CrossRef]
- Wakim, L. M.; Jones, C. M.; Gebhardt, T.; Preston, C. M.; Carbone, F. R., CD8(+) T-cell attenuation of cutaneous herpes simplex virus infection reduces the average viral copy number of the ensuing latent infection. Immunol Cell Biol 2008, 86, (8), 666-75. [CrossRef]
- Hoshino, Y.; Pesnicak, L.; Cohen, J. I.; Straus, S. E., Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J Virol 2007, 81, (15), 8157-64. [CrossRef]
- Derfuss, T.; Segerer, S.; Herberger, S.; Sinicina, I.; Hüfner, K.; Ebelt, K.; Knaus, H. G.; Steiner, I.; Meinl, E.; Dornmair, K.; Arbusow, V.; Strupp, M.; Brandt, T.; Theil, D., Presence of HSV-1 immediate early genes and clonally expanded T-cells with a memory effector phenotype in human trigeminal ganglia. Brain Pathol 2007, 17, (4), 389-98. [CrossRef]
- Geiger, K. D.; Nash, T. C.; Sawyer, S.; Krahl, T.; Patstone, G.; Reed, J. C.; Krajewski, S.; Dalton, D.; Buchmeier, M. J.; Sarvetnick, N., Interferon-gamma protects against herpes simplex virus type 1-mediated neuronal death. Virology 1997, 238, (2), 189-97.
- Knickelbein, J. E.; Khanna, K. M.; Yee, M. B.; Baty, C. J.; Kinchington, P. R.; Hendricks, R. L., Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, (5899), 268-71. [CrossRef]
- Frank, G. M.; Lepisto, A. J.; Freeman, M. L.; Sheridan, B. S.; Cherpes, T. L.; Hendricks, R. L., Early CD4(+) T cell help prevents partial CD8(+) T cell exhaustion and promotes maintenance of Herpes Simplex Virus 1 latency. J Immunol 2010, 184, (1), 277-86. [CrossRef]
- Singh, N.; Tscharke, D. C., Herpes Simplex Virus Latency Is Noisier the Closer We Look. J Virol 2020, 94, (4). [CrossRef]
- Liu, T.; Khanna, K. M.; Chen, X.; Fink, D. J.; Hendricks, R. L., CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med 2000, 191, (9), 1459-66. [CrossRef]
- van Velzen, M.; Laman, J. D.; Kleinjan, A.; Poot, A.; Osterhaus, A. D.; Verjans, G. M., Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. J Immunol 2009, 183, (4), 2456-61. [CrossRef]
- Gobeil, P. A.; Leib, D. A., Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 2012, 3, (5), e00267-12. [CrossRef]
- Lubinski, J. M.; Jiang, M.; Hook, L.; Chang, Y.; Sarver, C.; Mastellos, D.; Lambris, J. D.; Cohen, G. H.; Eisenberg, R. J.; Friedman, H. M., Herpes simplex virus type 1 evades the effects of antibody and complement in vivo. J Virol 2002, 76, (18), 9232-41. [CrossRef]
- Weisblum, Y.; Panet, A.; Haimov-Kochman, R.; Wolf, D. G., Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin Immunopathol 2014, 36, (6), 615-25. [CrossRef]
- Navti, O. B.; Al-Belushi, M.; Konje, J. C., Cytomegalovirus infection in pregnancy - An update. Eur J Obstet Gynecol Reprod Biol 2021, 258, 216-222. [CrossRef]
- Chandler, S. H.; Holmes, K. K.; Wentworth, B. B.; Gutman, L. T.; Wiesner, P. J.; Alexander, E. R.; Handsfield, H. H., The epidemiology of cytomegaloviral infection in women attending a sexually transmitted disease clinic. J Infect Dis 1985, 152, (3), 597-605. [CrossRef]
- Gerna, G.; Baldanti, F.; Revello, M. G., Pathogenesis of human cytomegalovirus infection and cellular targets. Hum Immunol 2004, 65, (5), 381-6. [CrossRef]
- Gretch, D. R.; Kari, B.; Rasmussen, L.; Gehrz, R. C.; Stinski, M. F., Identification and characterization of three distinct families of glycoprotein complexes in the envelopes of human cytomegalovirus. J Virol 1988, 62, (3), 875-81. [CrossRef]
- Mach, M.; Kropff, B.; Dal Monte, P.; Britt, W., Complex formation by human cytomegalovirus glycoproteins M (gpUL100) and N (gpUL73). J Virol 2000, 74, (24), 11881-92. [CrossRef]
- Mach, M.; Kropff, B.; Kryzaniak, M.; Britt, W., Complex formation by glycoproteins M and N of human cytomegalovirus: structural and functional aspects. J Virol 2005, 79, (4), 2160-70. [CrossRef]
- Kari, B.; Gehrz, R., A human cytomegalovirus glycoprotein complex designated gC-II is a major heparin-binding component of the envelope. J Virol 1992, 66, (3), 1761-4.
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P. A.; Cianfrocco, M. A.; Gerrein, R.; Feire, A. L.; Barnett, S. W.; Lilja, A. E.; Rappuoli, R.; Norais, N.; Settembre, E. C.; Carfi, A., Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 2015, 112, (6), 1767-72. [CrossRef]
- Wang, D.; Shenk, T., Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 2005, 79, (16), 10330-8. [CrossRef]
- Wang, D.; Shenk, T., Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 2005, 102, (50), 18153-8. [CrossRef]
- Lin, E.; Spear, P. G., Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci U S A 2007, 104, (32), 13140-5. [CrossRef]
- Reimer, J. J.; Backovic, M.; Deshpande, C. G.; Jardetzky, T.; Longnecker, R., Analysis of Epstein-Barr virus glycoprotein B functional domains via linker insertion mutagenesis. J Virol 2009, 83, (2), 734-47. [CrossRef]
- Maurer, U. E.; Zeev-Ben-Mordehai, T.; Pandurangan, A. P.; Cairns, T. M.; Hannah, B. P.; Whitbeck, J. C.; Eisenberg, R. J.; Cohen, G. H.; Topf, M.; Huiskonen, J. T.; Grünewald, K., The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 2013, 21, (8), 1396-405.
- Soroceanu, L.; Akhavan, A.; Cobbs, C. S., Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, (7211), 391-5. [CrossRef]
- Wang, X.; Huong, S. M.; Chiu, M. L.; Raab-Traub, N.; Huang, E. S., Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, (6947), 456-61. [CrossRef]
- Feire, A. L.; Roy, R. M.; Manley, K.; Compton, T., The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J Virol 2010, 84, (19), 10026-37. [CrossRef]
- Feire, A. L.; Koss, H.; Compton, T., Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A 2004, 101, (43), 15470-5. [CrossRef]
- Ryckman, B. J.; Jarvis, M. A.; Drummond, D. D.; Nelson, J. A.; Johnson, D. C., Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 2006, 80, (2), 710-22.
- Martinez-Martin, N.; Marcandalli, J.; Huang, C. S.; Arthur, C. P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A. M.; Shriver, S.; Payandeh, J.; Leitner, A.; Lanzavecchia, A.; Perez, L.; Ciferri, C., An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, (5), 1158-1171.e19. [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B. M.; Druz, A.; Zhang, B.; Geiger, R.; Pagani, M.; Sallusto, F.; Kwong, P. D.; Corti, D.; Lanzavecchia, A.; Perez, L., Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat Microbiol 2016, 1, (8), 16082.
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B., Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-α as a key for entry. PLoS Pathog 2017, 13, (4), e1006281.
- Compton, T.; Nepomuceno, R. R.; Nowlin, D. M., Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 1992, 191, (1), 387-95. [CrossRef]
- Scherer, M.; Stamminger, T., Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J Virol 2016, 90, (13), 5850-5854. [CrossRef]
- Caragliano, E.; Bonazza, S.; Frascaroli, G.; Tang, J.; Soh, T. K.; Grünewald, K.; Bosse, J. B.; Brune, W., Human cytomegalovirus forms phase-separated compartments at viral genomes to facilitate viral replication. Cell Rep 2022, 38, (10), 110469. [CrossRef]
- Sanchez, V.; Greis, K. D.; Sztul, E.; Britt, W. J., Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 2000, 74, (2), 975-86. [CrossRef]
- Stagno, S.; Reynolds, D. W.; Tsiantos, A.; Fuccillo, D. A.; Long, W.; Alford, C. A., Comparative serial virologic and serologic studies of symptomatic and subclinical congenitally and natally acquired cytomegalovirus infections. J Infect Dis 1975, 132, (5), 568-77. [CrossRef]
- Griffiths, P.; Baraniak, I.; Reeves, M., The pathogenesis of human cytomegalovirus. J Pathol 2015, 235, (2), 288-97. [CrossRef]
- Cope, A. V.; Sweny, P.; Sabin, C.; Rees, L.; Griffiths, P. D.; Emery, V. C., Quantity of cytomegalovirus viruria is a major risk factor for cytomegalovirus disease after renal transplantation. J Med Virol 1997, 52, (2), 200-5.
- Cope, A. V.; Sabin, C.; Burroughs, A.; Rolles, K.; Griffiths, P. D.; Emery, V. C., Interrelationships among quantity of human cytomegalovirus (HCMV) DNA in blood, donor-recipient serostatus, and administration of methylprednisolone as risk factors for HCMV disease following liver transplantation. J Infect Dis 1997, 176, (6), 1484-90. [CrossRef]
- Emery, V. C.; Cope, A. V.; Bowen, E. F.; Gor, D.; Griffiths, P. D., The dynamics of human cytomegalovirus replication in vivo. J Exp Med 1999, 190, (2), 177-82. [CrossRef]
- Reeves, M.; Sinclair, J., Aspects of human cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 2008, 325, 297-313.
- Mocarski, E. S.; Kemble, G. W.; Lyle, J. M.; Greaves, R. F., A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc Natl Acad Sci U S A 1996, 93, (21), 11321-6. [CrossRef]
- Marchini, A.; Liu, H.; Zhu, H., Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 2001, 75, (4), 1870-8. [CrossRef]
- Martínez, F. P.; Cruz, R.; Lu, F.; Plasschaert, R.; Deng, Z.; Rivera-Molina, Y. A.; Bartolomei, M. S.; Lieberman, P. M.; Tang, Q., CTCF binding to the first intron of the major immediate early (MIE) gene of human cytomegalovirus (HCMV) negatively regulates MIE gene expression and HCMV replication. J Virol 2014, 88, (13), 7389-401. [CrossRef]
- Lau, B.; Poole, E.; Krishna, B.; Sellart, I.; Wills, M. R.; Murphy, E.; Sinclair, J., The Expression of Human Cytomegalovirus MicroRNA MiR-UL148D during Latent Infection in Primary Myeloid Cells Inhibits Activin A-triggered Secretion of IL-6. Sci Rep 2016, 6, 31205.
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F., Opposing Regulation of the EGF Receptor: A Molecular Switch Controlling Cytomegalovirus Latency and Replication. PLoS Pathog 2016, 12, (5), e1005655.
- Diggins, N. L.; Skalsky, R. L.; Hancock, M. H., Regulation of Latency and Reactivation by Human Cytomegalovirus miRNAs. Pathogens 2021, 10, (2). [CrossRef]
- Pan, C.; Zhu, D.; Wang, Y.; Li, L.; Li, D.; Liu, F.; Zhang, C. Y.; Zen, K., Human Cytomegalovirus miR-UL148D Facilitates Latent Viral Infection by Targeting Host Cell Immediate Early Response Gene 5. PLoS Pathog 2016, 12, (11), e1006007.
- Diggins, N. L.; Crawford, L. B.; Hancock, M. H.; Mitchell, J.; Nelson, J. A., Human Cytomegalovirus miR-US25-1 Targets the GTPase RhoA To Inhibit CD34(+) Hematopoietic Progenitor Cell Proliferation To Maintain the Latent Viral Genome. mBio 2021, 12, (2).
- Hancock, M. H.; Crawford, L. B.; Perez, W.; Struthers, H. M.; Mitchell, J.; Caposio, P., Human Cytomegalovirus UL7, miR-US5-1, and miR-UL112-3p Inactivation of FOXO3a Protects CD34(+) Hematopoietic Progenitor Cells from Apoptosis. mSphere 2021, 6, (1).
- Anderson, D.; DeFor, T.; Burns, L.; McGlave, P.; Miller, J.; Wagner, J.; Weisdorf, D., A comparison of related donor peripheral blood and bone marrow transplants: importance of late-onset chronic graft-versus-host disease and infections. Biol Blood Marrow Transplant 2003, 9, (1), 52-9. [CrossRef]
- Hancock, M. H.; Crawford, L. B.; Pham, A. H.; Mitchell, J.; Struthers, H. M.; Yurochko, A. D.; Caposio, P.; Nelson, J. A., Human Cytomegalovirus miRNAs Regulate TGF-β to Mediate Myelosuppression while Maintaining Viral Latency in CD34(+) Hematopoietic Progenitor Cells. Cell Host Microbe 2020, 27, (1), 104-114.e4. [CrossRef]
- Hancock, M. H.; Mitchell, J.; Goodrum, F. D.; Nelson, J. A., Human Cytomegalovirus miR-US5-2 Downregulation of GAB1 Regulates Cellular Proliferation and UL138 Expression through Modulation of Epidermal Growth Factor Receptor Signaling Pathways. mSphere 2020, 5, (4).
- Crawford, L. B.; Kim, J. H.; Collins-McMillen, D.; Lee, B. J.; Landais, I.; Held, C.; Nelson, J. A.; Yurochko, A. D.; Caposio, P., Human Cytomegalovirus Encodes a Novel FLT3 Receptor Ligand Necessary for Hematopoietic Cell Differentiation and Viral Reactivation. mBio 2018, 9, (2). [CrossRef]
- Boehme, K. W.; Guerrero, M.; Compton, T., Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol 2006, 177, (10), 7094-102. [CrossRef]
- Compton, T.; Kurt-Jones, E. A.; Boehme, K. W.; Belko, J.; Latz, E.; Golenbock, D. T.; Finberg, R. W., Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol 2003, 77, (8), 4588-96. [CrossRef]
- Landais, I.; Pelton, C.; Streblow, D.; DeFilippis, V.; McWeeney, S.; Nelson, J. A., Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway. PLoS Pathog 2015, 11, (5), e1004881.
- Park, A.; Ra, E. A.; Lee, T. A.; Choi, H. J.; Lee, E.; Kang, S.; Seo, J. Y.; Lee, S.; Park, B., HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat Commun 2019, 10, (1), 4670. [CrossRef]
- Krug, A.; French, A. R.; Barchet, W.; Fischer, J. A.; Dzionek, A.; Pingel, J. T.; Orihuela, M. M.; Akira, S.; Yokoyama, W. M.; Colonna, M., TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, (1), 107-19. [CrossRef]
- Loh, J.; Chu, D. T.; O'Guin, A. K.; Yokoyama, W. M.; Virgin, H. W. t., Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol 2005, 79, (1), 661-7. [CrossRef]
- Iversen, A. C.; Norris, P. S.; Ware, C. F.; Benedict, C. A., Human NK cells inhibit cytomegalovirus replication through a noncytolytic mechanism involving lymphotoxin-dependent induction of IFN-beta. J Immunol 2005, 175, (11), 7568-74.
- Wu, Z.; Sinzger, C.; Reichel, J. J.; Just, M.; Mertens, T., Natural killer cells can inhibit the transmission of human cytomegalovirus in cell culture by using mechanisms from innate and adaptive immune responses. J Virol 2015, 89, (5), 2906-17. [CrossRef]
- Arnon, T. I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; Porgador, A.; Honigman, A.; Plachter, B.; Mevorach, D.; Wolf, D. G.; Mandelboim, O., Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 2005, 6, (5), 515-23. [CrossRef]
- Rölle, A.; Mousavi-Jazi, M.; Eriksson, M.; Odeberg, J.; Söderberg-Nauclér, C.; Cosman, D.; Kärre, K.; Cerboni, C., Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J Immunol 2003, 171, (2), 902-8. [CrossRef]
- Eagle, R. A.; Traherne, J. A.; Hair, J. R.; Jafferji, I.; Trowsdale, J., ULBP6/RAET1L is an additional human NKG2D ligand. Eur J Immunol 2009, 39, (11), 3207-16.
- Wu, J.; Chalupny, N. J.; Manley, T. J.; Riddell, S. R.; Cosman, D.; Spies, T., Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J Immunol 2003, 170, (8), 4196-200. [CrossRef]
- Dassa, L.; Seidel, E.; Oiknine-Djian, E.; Yamin, R.; Wolf, D. G.; Le-Trilling, V. T. K.; Mandelboim, O., The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA To Avoid NK Cell Attack. J Virol 2018, 92, (17). [CrossRef]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D. G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; Goldman-Wohl, D.; Greenfield, C.; Yagel, S.; Hengel, H.; Altuvia, Y.; Margalit, H.; Mandelboim, O., Host immune system gene targeting by a viral miRNA. Science 2007, 317, (5836), 376-81. [CrossRef]
- Mena-Romo, J. D.; Pérez Romero, P.; Martín-Gandul, C.; Gentil, M.; Suárez-Artacho, G.; Lage, E.; Sánchez, M.; Cordero, E., CMV-specific T-cell immunity in solid organ transplant recipients at low risk of CMV infection. Chronology and applicability in preemptive therapy. J Infect 2017, 75, (4), 336-345. [CrossRef]
- Díaz, J.; Henao, J.; Rodelo, J.; García, A.; Arbeláez, M.; Jaimes, F., Incidence and risk factors for cytomegalovirus disease in a Colombian cohort of kidney transplant recipients. Transplant Proc 2014, 46, (1), 160-6. [CrossRef]
- Lu, L. L.; Suscovich, T. J.; Fortune, S. M.; Alter, G., Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol 2018, 18, (1), 46-61. [CrossRef]
- Lilleri, D.; Kabanova, A.; Lanzavecchia, A.; Gerna, G., Antibodies against neutralization epitopes of human cytomegalovirus gH/gL/pUL128-130-131 complex and virus spreading may correlate with virus control in vivo. J Clin Immunol 2012, 32, (6), 1324-31. [CrossRef]
- Gerna, G.; Percivalle, E.; Perez, L.; Lanzavecchia, A.; Lilleri, D., Monoclonal Antibodies to Different Components of the Human Cytomegalovirus (HCMV) Pentamer gH/gL/pUL128L and Trimer gH/gL/gO as well as Antibodies Elicited during Primary HCMV Infection Prevent Epithelial Cell Syncytium Formation. J Virol 2016, 90, (14), 6216-6223. [CrossRef]
- Gerna, G.; Sarasini, A.; Patrone, M.; Percivalle, E.; Fiorina, L.; Campanini, G.; Gallina, A.; Baldanti, F.; Revello, M. G., Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J Gen Virol 2008, 89, (Pt 4), 853-865. [CrossRef]
- Cui, X.; Cao, Z.; Wang, S.; Lee, R. B.; Wang, X.; Murata, H.; Adler, S. P.; McVoy, M. A.; Snapper, C. M., Novel trimeric human cytomegalovirus glycoprotein B elicits a high-titer neutralizing antibody response. Vaccine 2018, 36, (37), 5580-5590. [CrossRef]
- Elkington, R.; Walker, S.; Crough, T.; Menzies, M.; Tellam, J.; Bharadwaj, M.; Khanna, R., Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multispecific reactivities in healthy virus carriers. J Virol 2003, 77, (9), 5226-40. [CrossRef]
- Crough, T.; Khanna, R., Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 2009, 22, (1), 76-98, Table of Contents. [CrossRef]
- van Lier, R. A.; ten Berge, I. J.; Gamadia, L. E., Human CD8(+) T-cell differentiation in response to viruses. Nat Rev Immunol 2003, 3, (12), 931-9. [CrossRef]
- Day, E. K.; Carmichael, A. J.; ten Berge, I. J.; Waller, E. C.; Sissons, J. G.; Wills, M. R., Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol 2007, 179, (5), 3203-13. [CrossRef]
- Ouyang, Q.; Wagner, W. M.; Wikby, A.; Walter, S.; Aubert, G.; Dodi, A. I.; Travers, P.; Pawelec, G., Large numbers of dysfunctional CD8+ T lymphocytes bearing receptors for a single dominant CMV epitope in the very old. J Clin Immunol 2003, 23, (4), 247-57. [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J. A.; Sinclair, A. J.; Nayak, L.; Moss, P. A., Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol 2002, 169, (4), 1984-92. [CrossRef]
- Pourgheysari, B.; Khan, N.; Best, D.; Bruton, R.; Nayak, L.; Moss, P. A., The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J Virol 2007, 81, (14), 7759-65. [CrossRef]
- Pawelec, G.; Akbar, A.; Caruso, C.; Solana, R.; Grubeck-Loebenstein, B.; Wikby, A., Human immunosenescence: is it infectious? Immunol Rev 2005, 205, 257-68.
- Chiu, Y. L.; Lin, C. H.; Sung, B. Y.; Chuang, Y. F.; Schneck, J. P.; Kern, F.; Pawelec, G.; Wang, G. C., Cytotoxic polyfunctionality maturation of cytomegalovirus-pp65-specific CD4 + and CD8 + T-cell responses in older adults positively correlates with response size. Sci Rep 2016, 6, 19227. [CrossRef]
- Costa-García, M.; Ataya, M.; Moraru, M.; Vilches, C.; López-Botet, M.; Muntasell, A., Human Cytomegalovirus Antigen Presentation by HLA-DR+ NKG2C+ Adaptive NK Cells Specifically Activates Polyfunctional Effector Memory CD4+ T Lymphocytes. Front Immunol 2019, 10, 687. [CrossRef]
- Aandahl, E. M.; Michaëlsson, J.; Moretto, W. J.; Hecht, F. M.; Nixon, D. F., Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J Virol 2004, 78, (5), 2454-9. [CrossRef]
- Basta, S.; Bennink, J. R., A survival game of hide and seek: cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol 2003, 16, (3), 231-42. [CrossRef]
- Ahn, K.; Angulo, A.; Ghazal, P.; Peterson, P. A.; Yang, Y.; Früh, K., Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc Natl Acad Sci U S A 1996, 93, (20), 10990-5. [CrossRef]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T. R.; Wiertz, E. J.; Ploegh, H. L.; Peterson, P. A.; Yang, Y.; Früh, K., The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6, (5), 613-21. [CrossRef]
- Jones, T. R.; Sun, L., Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J Virol 1997, 71, (4), 2970-9. [CrossRef]
- Wiertz, E. J.; Jones, T. R.; Sun, L.; Bogyo, M.; Geuze, H. J.; Ploegh, H. L., The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, (5), 769-79. [CrossRef]
- Tomazin, R.; Boname, J.; Hegde, N. R.; Lewinsohn, D. M.; Altschuler, Y.; Jones, T. R.; Cresswell, P.; Nelson, J. A.; Riddell, S. R.; Johnson, D. C., Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 1999, 5, (9), 1039-43. [CrossRef]
- Hegde, N. R.; Tomazin, R. A.; Wisner, T. W.; Dunn, C.; Boname, J. M.; Lewinsohn, D. M.; Johnson, D. C., Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J Virol 2002, 76, (21), 10929-41. [CrossRef]
- Lim, E. Y.; Jackson, S. E.; Wills, M. R., The CD4+ T Cell Response to Human Cytomegalovirus in Healthy and Immunocompromised People. Front Cell Infect Microbiol 2020, 10, 202.
- Miller, D. M.; Rahill, B. M.; Boss, J. M.; Lairmore, M. D.; Durbin, J. E.; Waldman, J. W.; Sedmak, D. D., Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp Med 1998, 187, (5), 675-83. [CrossRef]
- Lee, A. W.; Wang, N.; Hornell, T. M.; Harding, J. J.; Deshpande, C.; Hertel, L.; Lacaille, V.; Pashine, A.; Macaubas, C.; Mocarski, E. S.; Mellins, E. D., Human cytomegalovirus decreases constitutive transcription of MHC class II genes in mature Langerhans cells by reducing CIITA transcript levels. Mol Immunol 2011, 48, (9-10), 1160-7. [CrossRef]
- Sandhu, P. K.; Buchkovich, N. J., Human Cytomegalovirus Decreases Major Histocompatibility Complex Class II by Regulating Class II Transactivator Transcript Levels in a Myeloid Cell Line. J Virol 2020, 94, (7). [CrossRef]
- Poole, E.; Neves, T. C.; Oliveira, M. T.; Sinclair, J.; da Silva, M. C. C., Human Cytomegalovirus Interleukin 10 Homologs: Facing the Immune System. Front Cell Infect Microbiol 2020, 10, 245. [CrossRef]
- Mittal, S. K.; Roche, P. A., Suppression of antigen presentation by IL-10. Curr Opin Immunol 2015, 34, 22-7. [CrossRef]
Figure 1.
A: Schematic diagram of the structure of herpesviruses. Herpesviruses are composed of double-stranded DNA, nucleocapsid, tegument, envelope proteins, and a lipid envelope. B: Schematic diagram of herpesvirus entry into cells via envelope proteins. While the types and composition of envelope proteins may vary among different viruses in the sporozoan virus family, the fusion of the viral envelope with the host cell membrane and the entry of the nucleocapsid rely on the binding of multiple envelope proteins to receptors on the host cell membrane and conformational changes.
Figure 1.
A: Schematic diagram of the structure of herpesviruses. Herpesviruses are composed of double-stranded DNA, nucleocapsid, tegument, envelope proteins, and a lipid envelope. B: Schematic diagram of herpesvirus entry into cells via envelope proteins. While the types and composition of envelope proteins may vary among different viruses in the sporozoan virus family, the fusion of the viral envelope with the host cell membrane and the entry of the nucleocapsid rely on the binding of multiple envelope proteins to receptors on the host cell membrane and conformational changes.
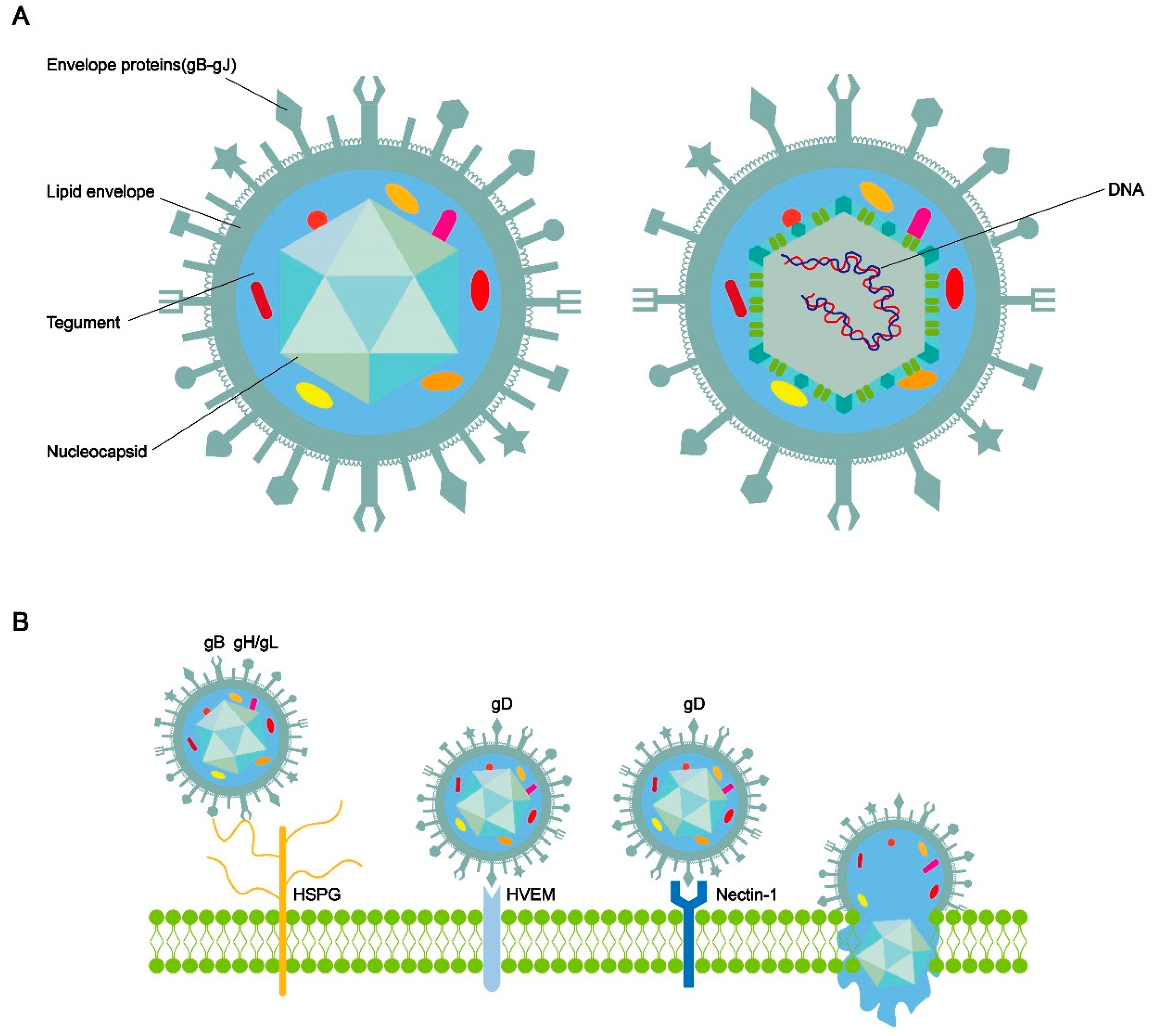

Table 1.
Viruses associated with VN.
| Viruses associated with VN | Family of viruses |
|---|---|
| Herpes simplex virus | Alphaherpesvirinae |
| Varicella-zoster virus | |
| Human Cytomegalovirus | Betaherpesvirinae |
| Epstein-Barr virus | Gammaherpesvirinae |
| Influenza virus A | Non-herpesvirus family |
| Influenza virus B | |
| Adenoviruses | |
| Rubella virus | |
| Parainfluenza virus |
|
R
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
|
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



