
Submitted:
01 November 2023
Posted:
02 November 2023
You are already at the latest version
Abstract
Leukodystrophies, also known as demyelinating diseases, mainly affect the CNS. Clinical presentation of different types of leukodystrophies can be nonspecific and thus, imaging techniques like MRI can be used for a more definitive diagnosis. These diseases are characterized as cerebral lesions with characteristic demyelinating patterns, which can be used as differentiating tools. In this review, we talk about these MRI imaging findings for each leukodystrophy, associated genetics, blood work that can help differentiate, emerging diagnostics, and follow up imaging strategy. The leukodystrophies discussed in this paper include X-linked adrenoleukodystrophy, Metachromatic leukodystrophy, Krabbe’s disease, Pelizaeus-Merzbacher disease, Alexander’s disease, Canavan disease, and Aicardi-Goutières Syndrome.
Keywords:
Leukodystrophies
; Demyelinating disorders
; Magnetic Resonance Imaging
; Myelin Imaging
; White matter disorders
1. Introduction
Magnetic resonance imaging (MRI) is the primary imaging technique to identify, localize, and characterize cerebral lesions in patients with leukodystrophy. Leukodystrophies, also known as demyelinating diseases, threaten the integrity of the brain and peripheral nerves [1,2]. Clinical presentation of leukodystrophies can be nonspecific, and imaging techniques like MRI are helpful in establishing a more definitive diagnosis [3]. Current research suggests that early detection of leukodystrophy allows for more optimal implementation of therapy treatments, highlighting the importance of early disease detection [3,4].
Quantitative MRI is a form of noninvasive imaging that provides information regarding myelin and axonal content, condition, and chemical composition of white matter. Having information about a patient’s white matter microstructural involvement not only helps in the diagnosis of leukodystrophy but in the progression of the disease as well [5]. In addition to MRI, genetic testing may also contribute help to a specific diagnosis of leukodystrophy. Leukodystrophies are monogenic disorders with distinct alterations in specific genes. A study in children with leukodystrophy showed that exome sequencing had a diagnostic yield of 82.9%, single-gene sequencing with 54%, targeted panels with 33.3%, and chromosomal microarray with 8% and that access to advanced sequencing accelerated speed of diagnosis [6].
In this review we will focus on the following leukodystrophies: X-linked adrenoleukodystrophy, Metachromatic leukodystrophy, Krabbe’s disease, Pelizaeus-Merzbacher disease, Alexander’s disease, Canavan disease, and Aicardi-Goutières Syndrome (Figure 1). Leukodystrophies often present with nonspecific symptoms including movement disturbance, vision problems, hearing impairment, imbalance, memory loss, behavioral changes, and attention deficits [7,8]. While similar clinical presentation may make distinguishing between these conditions more challenging, more distinctions seen on MRI may assist in coming to a more conclusive and specific diagnosis.
2. Types of Leukodystrophies
2.1. X-linked adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is an X-linked genetic condition impacting either the central or peripheral nervous system along with the adrenal cortex [9]. X-ALD primarily affects boys and presents clinically with adrenal insufficiency, dysarthria, dysgraphia, vision and hearing deficits, and neurocognitive and neurobehavioral issues [10]. This condition is characterized by a mutation in the ABCD1 gene on the X chromosome [11]. This gene then encodes the ALD protein, a transmembrane protein responsible for the transport of very long chain fatty acid (VLCFA)-CoA esters into the peroxisome [12]. Therefore, mutations in the ABCD1 gene result in diminished transport of VLCFAs and accumulation throughout the body [13]. Approximately almost all affected males develop adrenocortical insufficiency, with onset occurring around age 7.5 while the development of progressive myelopathy and peripheral neuropathy occurs in adulthood [9,11]. 60% of male patients will present with cerebral demyelination, which is often progressive and fatal [14]. The use of MRI is instrumental in the detection of presymptomatic demyelinating lesions, with early detection improving patient outcomes [15].
Newborn screening for X-ALD has been recommended for implementation into the uniform screening panel in the United States. To date, over half of the states have begun screening for X-ALD. Additionally, some states in the U.S. have also incorporated the use of molecular genetic testing and family screenings. However, to establish the diagnosis of X-ALD, detection of the ABCD1 pathogenic variant and detection of accumulation of VLCFAs is necessary [9].
Patients with cerebral ALD often present with large areas of white matter demyelination and inflammation [11]. These lesions are subdivided into 3 zones. The innermost zone is characterized by the loss of axons, oligodendrocytes, and myelin sheaths. The middle zone is characterized by loss of myelin sheaths and results in gadolinium enhancement. The outermost zone is characterized by axon preservation and the active involvement of macrophages responsible for myelin destruction [11,16,17]. These lesions can be visualized on MRI and often impact the dorsal columns and corticospinal tracts of the spinal cord [18,19]. Within the brain, 80% of cases result in hyperintense confluent lesions of the corpus callosum and parieto-occipital white matter [20]. In adulthood, MRI demonstrates white matter lesions of the frontopontine and corticospinal projection fibers, eventually impacting the entire cerebral white matter as the disease progresses [21]. Therefore, MRIs of the brain are repeated every 6 months until the age of 12, followed by yearly MRIs to monitor the progression of X-ALD lesions [22]. Unlike imaging of the brain, spinal cord imaging is not used in follow-up of X-ALD or for prognostic significance. However, spinal cord MRI demonstrates corticospinal tract and dorsal column degeneration resulting in the appearance of a flattened spinal cord and the reduction of anteroposterior diameters [23,24,25]. The pathophysiology of X-ALD is summarized in Figure 2.
Although MRI remains the gold standard for the detection of lesions in X-ALD, studies in other neurodegenerative disorders have demonstrated that Neurite Orientation Dispersion and Density Imaging (NODDI) shows increased orientation dispersion and detection of a higher surface area of neurodegeneration than structural MRI and diffusion tensor imaging [26,27]. Additionally, myelin water fraction (MWF) imaging detects the quantity of myelin through the use of specific water pools, opening the possibility of MWF being used to detect early lesions of X-ALD in the future [28].
2.2. Metachromatic leukodystrophy
Metachromatic leukodystrophy is an autosomal recessive lysosomal storage disease characterized by a deficiency of arylsulfatase A (ASA) due to a mutation in the arylsulfatase A gene on chromosome 22q13.3-qter [29,30]. ASA is involved in the degradation of sulphatide, a membrane lipid expressed in myelin, the distal tubules of the kidney, and bile duct epithelia [29]. Therefore, a deficiency of ASA leads to the accumulation of sulphatide as degradation is prevented [29]. Accumulation of sulphatide functionally impairs the nervous system with minimal impact on the kidneys or gallbladder. This presents clinically as progressive demyelination leading to ataxia, optic atrophy, dementia, and decerebrate posturing [31].
Metachromatic leukodystrophy is often suspected in children who are unable to meet developmental milestones along with a decline in gross and fine motor skills [30]. Diagnosis of metachromatic leukodystrophy is made by laboratory studies evaluating the levels of ASA [10]. The range of values that meet the criteria for diagnosis ranges from undetectable to less than 10% of the normal value. However, metachromatic leukodystrophy needs to be differentiated from arylsulfatase A pseudodeficiency, which is present in around 1% of the general population and presents with decreased ASA values without any evidence of clinical or radiographic manifestations of the disease. Therefore, to differentiate the two conditions, urine sulfatide levels, radiolabeled sulfatide fibroblast loading, and DNA analysis can be used.
Additionally, as a demyelinating condition, brain demyelination will be present on MRI along with abnormalities of nerve conduction [32,33]. The central and periventricular white matter are often impacted first with the subcortical structures being affected as the disease progresses. In extreme cases, projection fibers are also impacted resulting in a “tigroid pattern” and “leopard skin” pattern (Figure 3) [34,35]. One study found that nearly all patients with metachromatic leukodystrophy displayed splenial corpus callosum demyelination [36]. Additionally, MRI demonstrates T2-weighted FLAIR symmetric, confluent hyperintense areas of the periventricular white matter. Hyperintense areas are visible on T1-weighted images as metachromatic leukodystrophy is a demyelinating disorder [37].
2.3. Krabbe's disease
Another type of leukodystrophy that can prove to be a diagnostic dilemma is Krabbe disease (KD). KD is an inherited lysosomal storage disease that was first discovered by Knud Krabbe in 1916 and has been extensively studied ever since [41]. It is characterized by a defect in the GALC gene, which encodes for the galactocerebrosidase protein; furthermore, the galactocerebrosidase protein is necessary for the catabolic processes responsible for the breakdown of myelin. It leads to an accumulation of toxic myelin products in cells [42]. KD is also known as Globoid Cell Leukodystrophy, as it is characterized by globoid cells with multiple nuclei.
KD classically affects infants under the age of six but can also occur in adolescents or adults [42]. Common symptoms of KD include muscle weakness with spasticity and hypertonia, myoclonic seizures, sensory deficits such as blindness or deafness, and fever without an underlying infectious etiology [42]. As mentioned, many of these symptoms are nonspecific and have a significant overlap with other neurodegenerative disorders, such as Alexander Disease, metachromatic leukodystrophy, or GM2 gangliosidosis [43]. Currently, no treatment options are available for KD. In infants, the disease is fatal before age 2, but it has a milder onset and progression when it has a later onset. At the presymptomatic stage, stem cell transplant can improve patient prognosis; however, without a family history of KD, the disorder is often not identified until the symptomatic stage. Once symptomatic, the patient becomes ineligible for transplantation [44]. For this reason, the goal of care is to screen infants for KD and identify it before symptoms appear.
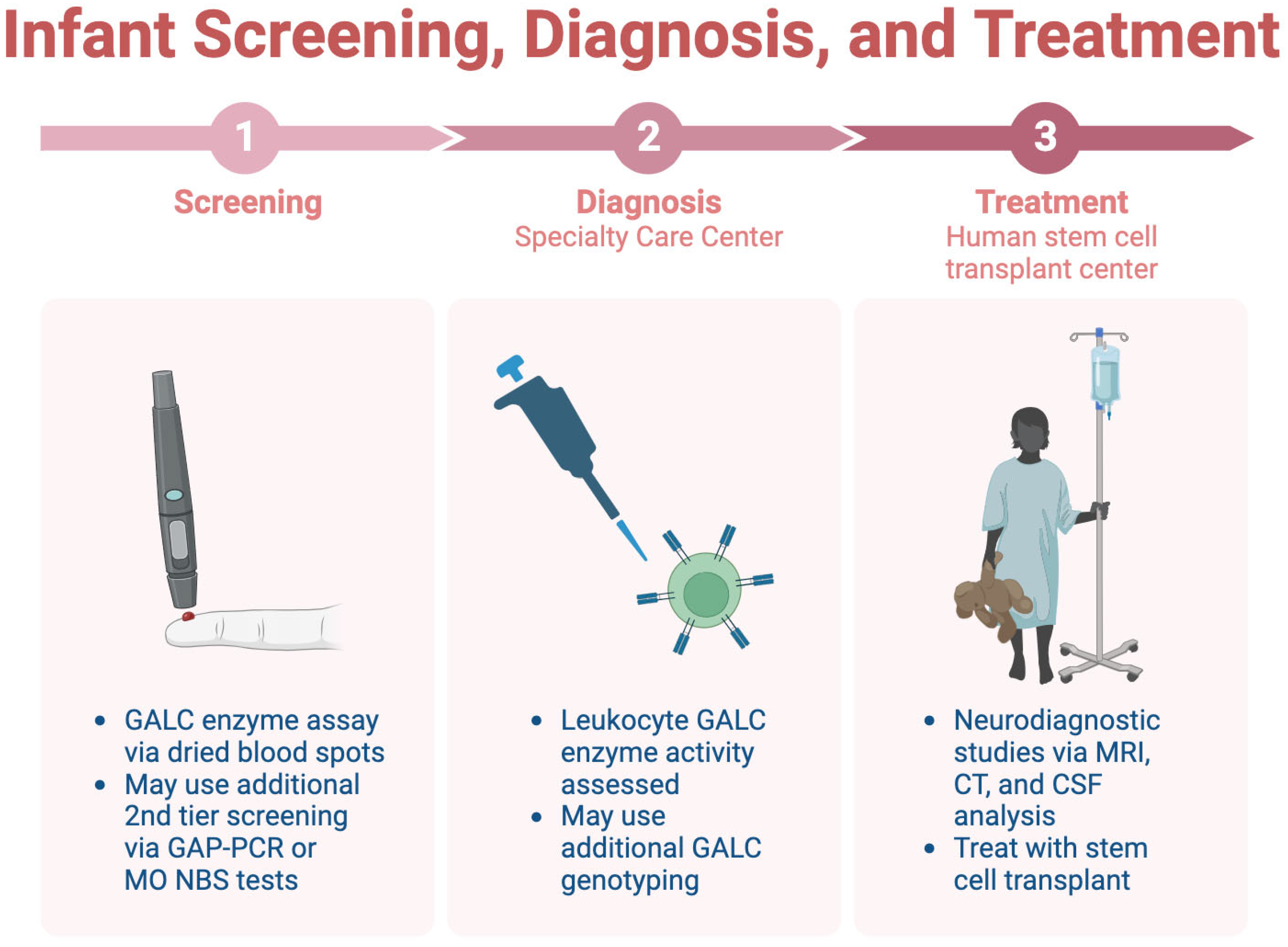
Only a few states across the U.S. screen infants for KD, one of which is New York [45]. Consensus guidelines for KD outline a three-step process for screening, diagnosis, and treatment [44]. In the first step, dried blood spots are utilized for a GALC enzyme assay and can be analyzed via tandem mass spectrometry-based assays or fluorometry. Other tiers of screening may be added to the GALC enzyme assay, as this assay has low specificity. After that, the patient is referred to a specialty care center (SCC), which conducts diagnostic tests. Leukocyte GALC enzyme activity is the preferred diagnostic test. GALC genotyping may also be used, but its ability to detect gene deletion is limited, restricting its diagnostic capabilities. Once the diagnosis is confirmed, the patient is referred to a human stem cell transplantation center (HSCT), where neurodiagnostic studies are completed, including MRI and CSF analysis. These neurodiagnostic tests should not be conducted at other stages where the diagnosis is not yet confirmed; they will be performed in the HSCT [44]. This process is summarized in Figure 4.
MRI findings of KD include deep cerebral and cerebellar white matter changes with the involvement of the dentate hilum [46]. One study involving 38 MRI scans from 27 patients revealed distinct differences in findings across different subtypes of KD [46]. For instance, the infantile form of KD presented with optic nerve and cervical cord enlargement [46,47]. In the adult form of KD, T2-hyperintense changes were observed in the corticospinal tracts, the posterior limb of the internal capsule, and the pyramidal tracts in the brainstem [3,46]. The MRI scan in the adult form of KD could also be normal [3]. The juvenile form of KD displayed a mix of both findings.
Another study assessed CT findings in 3 patients with KD and found hyperdense regions in the thalami, cerebellar cortex, and corona radiata [48]. The CT scans also revealed a plaque-like high signal intensity in the periventricular region and cerebellum [48].
Psychosine, also known as galactosyl sphingosine, also accumulates in the CSF and can be used as a diagnostic tool for KD, as well as a potential target for gene therapy [44,49]. Another study involving nine children with KD found that most cases presented with elevated protein in CSF and peripheral neuropathy on nerve conduction studies [50]. This study also revealed that GALC levels in peripheral blood leukocytes were low or absent in all of these cases [50]. Hence, the authors concluded that imaging holds diagnostic significance for KD in the absence of the availability of enzyme diagnosis [50].
Overall, KD is a severe neurodegenerative disease that is fatal and can be difficult to diagnose. Stem cell transplant is a treatment option for infants with KD, but it must be screened and diagnosed in the asymptomatic stage in order for a patient to be eligible. Neurodiagnostic studies aid in the assessment of KD, and GALC levels are the most promising option diagnostic test available for patients.
2.4. Pelizaeus-Merzbacher disease
Pelizaeus-Merzbacher disease (PMD) is a rare leukodystrophy that is also a demyelinating disease of the CNS. The disease was first discovered by Friederich Pelizaeus and Ludwig Merzbacher in 1885 when a German family presented with this clinical syndrome, including the symptoms of nystagmus, spastic paralysis, and ataxia [51]. PMD is characterized by deteriorating coordination, motor abilities, and cognitive function [52]. The disease has an X-linked recessive inheritance, and it is caused by mutations of the proteolipid protein 1 (PLP1) gene, which is located on the long arm of the X chromosome [51]. Various mutations, including deletions, missense, and deletions, lead to different levels of decreased myelin production. Hence, PMD is classified into Type I, II, and III based on the mutation, with Type I being the most severe [51].
The diagnosis of PMD is again challenging and complex, because of many overlapping symptoms with other leukodystrophies. Most other leukodystrophies should be ruled out before diagnosing a patient with PMD [51]. The neonatal onset of PMD is more severe and associated with a worse prognosis than the adult form, which is nearly benign [53]. Various studies have attempted to create feasible screening and diagnostic tests for PMD.
One study by Inoue et al. created an interphase fluorescence in situ hybridization (FISH) assay to diagnose PMD duplications [54]. As an efficient screening test for PMD, 13 patients were successfully diagnosed with PMD, and female carriers of the disease were also detected [54]. The size and location of the gene duplications, which strongly impact patient prognosis, can further be identified through molecular analysis [54]. These authors then further investigated the applicability of the FISH assay in detecting neonatal gene duplications—in two families with a history of PMD, the FISH assay was able to detect PMD duplications from amniotic fluid [55]. Postnatal blood samples and haplotype analysis confirmed both diagnoses after birth [55].
In terms of imaging, generally, MRIs display gliosis around areas of demyelination [51]. T1-weighted sections will display hypointensity, representing the absence of white matter [53]. T2-weighted images display hyperintensity in places that should have lower intensity, conveying demyelination [53]. Magnetic resonance spectroscopy (MRS) has inconsistent findings across cases of PMD, but more research is required to assess its utility in diagnosing PMD [53,56]. A study by Sumida et al. retrospectively assessed the MRI scans of 19 patients with a confirmed PMD diagnosis to correlate the images with the severity of the clinical presentation of the disease [57]. The authors found that the severity of the disease was increased when the entire brainstem or corticospinal tract displayed a high intensity on T2-weighted images [57]. Additionally, CSF analysis of patients with PMD revealed a low level of N-acetyl aspartate (NAA), which correlated to axonal damage and severity of disease [58].
Although the FISH assay, MRI, and CSF analysis contribute to the identification and diagnosis of PMD, overlapping findings lead to a reliance on diagnosis by exclusion of other leukodystrophies. The treatment for PMD is currently supportive and focused on reducing symptoms. Overall, more research is needed to diagnose and treat patients with PMD to improve their quality of life.
2.5. Alexander Disease
First diagnosed in 1949, Alexander disease (AxD) describes a form of leukodystrophy that affects the white matter of the CNS characterized by the degeneration of the myelin sheath via a defect in the Glial Fibrillary Acidic Protein (GFAP) gene. [59,60]. While mainly associated with infants, clinical presentation can occur at neonatal, infantile, juvenile, and adult ages [61]. The major hallmark of AxD diagnostically is the appearance of Rosenthal fibers and eosinophilic granular bodies with light microscopy which is divided into two subtypes: Type I and Type II [62].
Furthermore, the only known defect that implicates AxD is from mutations in the GFAP gene on chromosome 17q21.31 encoding a protein expressed in astrocytes. This defect causes GFAP aggregates to form called Rosenthal fibers, leading to astrocyte degeneration and demyelination [63]. GFAP levels can be collected and quantified via CSF and are significantly elevated in patients with AxD, although levels are only marginally elevated if blood is collected [64].
In terms of imaging, MRI findings typically consist of abnormal signals from the frontal white matter, the periventricular rim, or various structures such as the caudate head, thalamus, and brain stem [65]. As demonstrated in Figure 4, MRI findings indicate increased T2-weighted signal intensity, and decreased T1-weighted signal intensity [66]. Van der Knaap et al defined five MR imaging criteria of which four should be met for an imaging-based diagnosis: extensive cerebral frontal white matter abnormalities, periventricular rim with decreased T2 signals and increased T1 signals, basal ganglia and thalami abnormalities, brain stem abnormalities, and contrast enhancement of one or more of the following: ventricular lining, periventricular rim of tissue, the white matter of the frontal lobes, optic chiasm, fornix, basal ganglia, thalamus, dentate nucleus, and brain stem structures [65]. Hindbrain structural abnormalities, such as the brainstem are characteristic of later onset presentation, showing signs of atrophy of the medulla and cervical spinal cord [60].
Figure 5.
MRIs characteristic of Alexander Disease; The right image demonstrates an increased signal of T2, the middle image demonstrates a decreased signal of T1, and the right image is a T2 midline sagittal section demonstrating atrophy [66].
Figure 5.
MRIs characteristic of Alexander Disease; The right image demonstrates an increased signal of T2, the middle image demonstrates a decreased signal of T1, and the right image is a T2 midline sagittal section demonstrating atrophy [66].
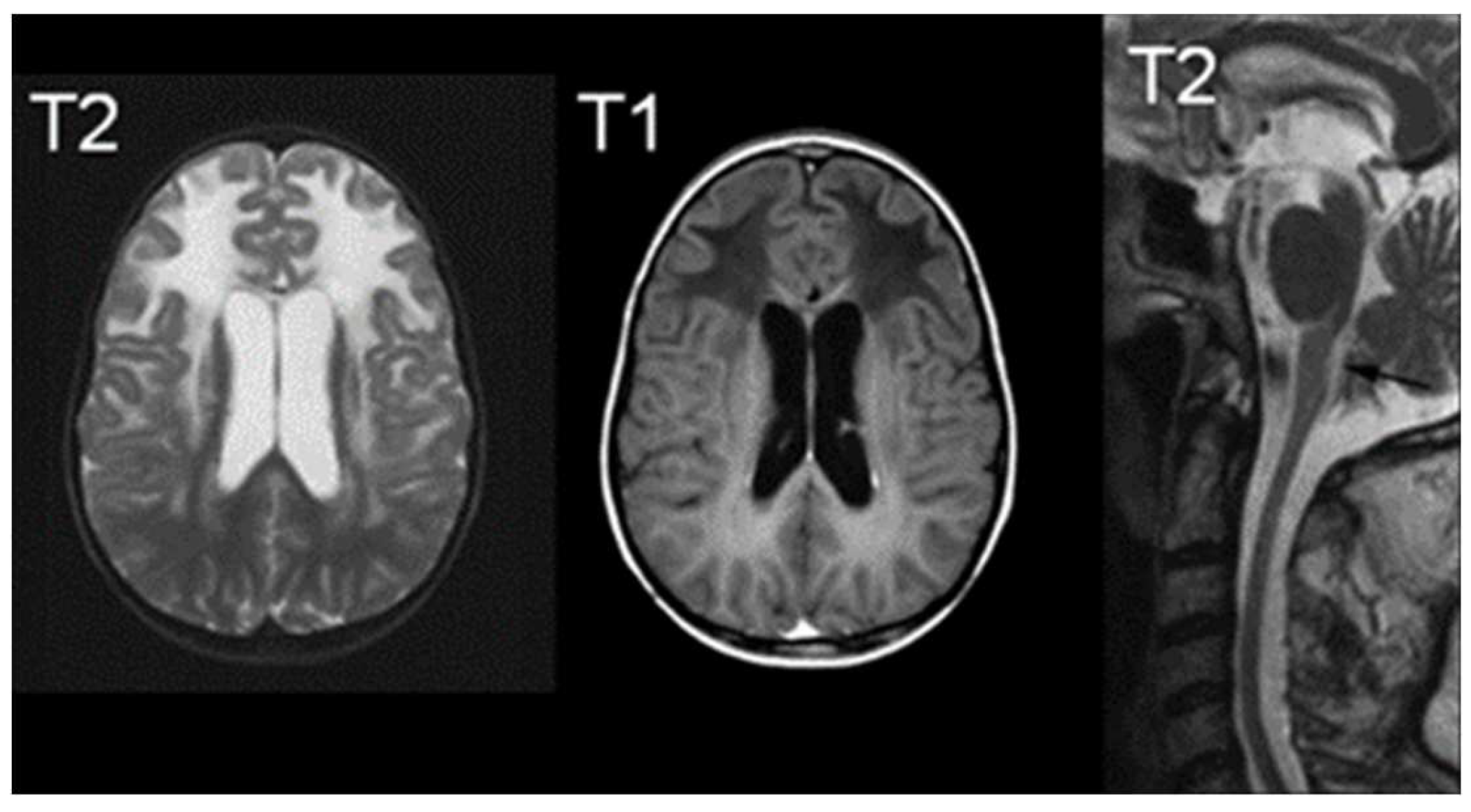
Lastly, while autopsies are the only definitive diagnostic tool for Rosenthal fiber analysis, current diagnosis techniques revolve around histological examination via brain biopsy, MRI diagnosis, and gene analysis for the GFAP gene [67].
2.6. Canavan Disease
First described in 1931, Canavan Disease is an autosomal recessive leukodystrophy that primarily affects the white matter of the brain [68]. The affected gene on chromosome 17p13.2 is the ASPA gene that encodes for aspartoacylase; a deficiency in this gene leads to an accumulation of N-acetyl-L-aspartic acid (NAA), which is linked to oligodendrocyte dysfunction and degradation of myelin [68]. Prevalent in infants, the disease typically affects Ashkenazi Jews but has been described in other populations [69]. It is thought that NAA is a component in a series of pathways that help produce cerebroside, a component of myelin; excess amounts may lead to abnormal myelination and spongy degeneration of tissue [69,70]. As of present, there are no specific therapies that can cure this disease, although there is continuing research on introducing the ASPA gene into functional neural progenitor cells, and has shown relative success in mouse models [71,72].
The modality for confirming the diagnosis of Canavan disease is to culture skin fibroblasts with NAA and determine the level of NAA through gas chromatography Mass Spectrometry after incubation; elevated levels indicate an aspartoacylase deficiency [73]. This is in contrast with other leukodystrophies, like Alexander’s disease, which demonstrate normal NAA levels. Another technique is to determine aspartoacylase expression via chorionic villus biopsy for prenatal diagnoses [74].
Furthermore, like other white matter diseases, Canavan disease is characteristic of hyperintense T2-weighted images in subcortical U fibers [75]. T1 is characterized by hypointense signals with diffuse signals throughout the white matter and brainstem [76]. MRI images show diffuse cerebral white matter degeneration; other structures such as the periventricular rim are preserved. Follow-up MRI scans for infants can demonstrate progressive ventriculomegaly as well as atrophy [77,78]. Other modalities, like CT, could show the hypodensity of white matter [78].
2.7. Aicardi-Goutières Syndrome
A Aicardi-Goutières Syndrome (AGS) is a rare genetically inherited neuroinflammatory disorder that impacts the brain, immune system, and skin. AGS commonly presents progressively with clinical presentation including dystonia/spasticity, hepatosplenomegaly, elevated liver enzymes, thrombocytopenia, chilblain-like skin lesions, and neurological abnormalities including microcephaly, CSF lymphocytosis, developmental delays, and other neurological impairments [79,80,81]. These symptoms mimic TORCH congenital infections despite the absence of an active viral infection [80,82,83]. Considering AGS is not of infectious origins, it is commonly referred to as Pseudo-TORCH syndrome. Commonly, AGS presents with an early-onset encephalopathy at birth and is considered the more severe form of the disease, but it can also present as a late-onset within a few months after birth. Both are associated with significant intellectual and physical disability [82]. The diagnosis of AGS is usually multifaceted and involves a comprehensive understanding of its distinctive clinical presentation, specific imaging findings, intricate genetic underpinnings, discerning cerebrospinal fluid (CSF), and bloodwork biomarkers, facilitating more accurate diagnosis [84]. Furthermore, effective management and prognosis evaluation often necessitate vigilant follow-up imaging procedures to monitor disease progression and assess treatment efficacy.
MRI Imaging findings play a pivotal role in identifying characteristic brain abnormalities associated with AGS. These findings commonly include loss of white matter, particularly in the periventricular and deep white matter regions as well as calcifications in the basal ganglia and dentate nuclei [79,81,85]. Additionally, the presence of ventriculomegaly and thinning of the corpus callosum are frequently observed, contributing to the distinctive radiological profile that aids in distinguishing AGS from other neurological conditions [80].
AGS is inherited in an autosomal recessive pattern and is closely linked to several previously described genetic mutations, particularly involving genes associated with the intracellular metabolism of nucleic acids [80,83]. Some of these genetic mutations include mutations in genes like TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1 [79,82,86,87,88,89,90]. These mutations lead to increased calcium deposits in the brain, which in turn are believed to result in an overactive immune system [91]. underscoring the significance of genetic testing for accurate diagnosis and genetic counseling. Also, a strong association between AGS and a predisposition to other autoimmune conditions like systemic lupus erythematosus has been revealed in recent research [92,93].
Additionally, specific abnormalities in the CSF, such as elevated levels of interferon-alpha, neopterin, and other proinflammatory cytokines, along with the presence of autoantibodies, can aid in differentiating AGS from other related conditions with overlapping clinical manifestations [79,85,86].
The evolving landscape of diagnostic techniques for AGS has witnessed the integration of advanced genomic sequencing methods, enabling the identification of novel genetic variants and the elucidation of intricate disease mechanisms. Next-generation sequencing (NGS) and whole-exome sequencing (WES) have significantly augmented diagnostic accuracy and facilitated the identification of atypical AGS cases, thereby enabling more precise prognostication and personalized therapeutic interventions [79].
Follow-up imaging, including serial MRI examinations, is crucial for monitoring disease progression and treatment response in individuals with AGS. Longitudinal MRI assessments help track the evolution of white matter abnormalities, calcifications, and cerebral atrophy, providing essential insights into disease progression and response to therapeutic interventions. Moreover, timely and comprehensive follow-up imaging serves as a valuable tool for prognostic evaluation and the optimization of individualized management strategies, emphasizing the significance of a multidisciplinary approach in the care of individuals affected by AGS.
3. Conclusions
Leukodystrophies are a wide group of disorders with heterogeneous genetic and acquired causes. Even though they primarily affect the CNS, there’s a wide array of clinical presentation that affects patients of any age. At times, these clinical presentations can be similar, and thus, imaging can be used as a differentiator. As mentioned, MRI is the current diagnostic foundation for these diseases, and in combination with certain specific features, the diseases can be systemized efficiently with minimal testing. The demyelinating pattern seen on MRI can be used to distinguish between different types of leukodystrophies. Following an abnormal MRI sequence, the patients can be further worked up to other genetic and biochemical testing. Despite these advances, certain patients can present with ambiguous MRI findings, and for them, other diagnostic techniques and genetic sequencing are currently being explored.
Author Contributions
Conceptualization, B.L.-W.; Writing—original draft preparation, R.N.T., I.P.K., D.P., R.A., P.R. and D.T.F.; Writing—reviewing & editing, R.N.T., I.P.K., D.P., R.A., P.R. and D.T.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were collected or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cheon, J.E.; Kim, I.O.; Hwang, Y.S.; Kim, K.J.; Wang, K.C.; Cho, B.K.; Chi, J.G.; Kim, C.J.; Kim, W.S.; Yeon, K.M. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics : a review publication of the Radiological Society of North America, Inc 2002, 22, 461–476. [Google Scholar] [CrossRef] [PubMed]
- van de Stadt, S.I. W.; Huffnagel, I.C.; Turk, B.R.; van der Knaap, M.S.; Engelen, M. Imaging in X-Linked Adrenoleukodystrophy. Neuropediatrics 2021, 52, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Resende, L.L.; de Paiva, A.R. B.; Kok, F.; da Costa Leite, C.; Lucato, L.T. Resende, L.L.; de Paiva, A.R. B.; Kok, F.; da Costa Leite, C.; Lucato, L.T. Adult Leukodystrophies: A Step-by-Step Diagnostic Approach. Radiographics : a review publication of the Radiological Society of North America, Inc. 2019; 39, 153–168. [Google Scholar] [CrossRef]
- Waldman, A.T. (2018). Leukodystrophies. Continuum (Minneapolis, Minn.), 24(1, Child Neurology), 130–149. [CrossRef]
- Stellingwerff, M.D.; Pouwels, P.J.W.; Roosendaal, S.D.; Barkhof, F.; van der Knaap, M.S. Quantitative MRI in leukodystrophies. NeuroImage. Clinical 2023, 38, 103427. [Google Scholar] [CrossRef] [PubMed]
- Zerem, A.; Libzon, S.; Ben Sira, L.; Meirson, H.; Hausman-Kedem, M.; Haviv, N.; Yosovich, K.; Mory, A.; Baris Feldman, H.; Lev, D.; Lerman-Sagie, T.; Fattal-Valevski, A.; Hacohen, Y.; Marom, D. Utility of genetic testing in children with leukodystrophy. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society 2023, 45, 29–35. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Bugiani, M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta neuropathologica 2017, 134, 351–382. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Bernard, G.; Leventer, R.J.; van der Knaap, M.S.; van Hove, J.; Pizzino, A.; McNeill, N.H.; Helman, G.; Simons, C.; Schmidt, J.L.; Rizzo, W.B.; Patterson, M.C.; Taft, R.J.; Vanderver, A. GLIA Consortium A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Molecular genetics and metabolism 2015, 114, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Moser, A.B.; Fatemi, A. (1999). X-Linked Adrenoleukodystrophy. In M. P. Adam (Eds.) et. al.; GeneReviews®. University of Washington, Seattle.
- Hong, X.; Kumar, A.B.; Daiker, J.; Yi, F.; Sadilek, M.; De Mattia, F.; Fumagalli, F.; Calbi, V.; Damiano, R.; Della Bona, M.; la Marca, G.; Vanderver, A.L.; Waldman, A.T.; Adang, L.; Sherbini, O.; Woidill, S.; Suhr, T.; Kurtzberg, J.; Beltran-Quintero, M.L.; Escolar, M.; … Gelb, M.H. Leukocyte and Dried Blood Spot Arylsulfatase A Assay by Tandem Mass Spectrometry. Analytical chemistry 2020, 92, 6341–6348. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; Poll-The, B.T. X-linked adrenoleukodystrophy: pathogenesis and treatment. Current neurology and neuroscience reports 2014, 14, 486. [Google Scholar] [CrossRef]
- Singh, I.; Moser, A.E.; Moser, H.W.; Kishimoto, Y. Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatric research 1984, 18, 286–290. [Google Scholar] [CrossRef]
- Berger, J.; Forss-Petter, S.; Eichler, F.S. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 2014, 98, 135–142. [Google Scholar] [CrossRef]
- de Beer, M.; Engelen, M.; van Geel, B.M. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology 2014, 83, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, E.I.; Eisengart, J.B.; Shanley, R.; Nascene, D.; Raymond, G.V.; Shapiro, E.G.; Ziegler, R.S.; Orchard, P.J.; Miller, W.P. Neurocognitive Trajectory of Boys Who Received a Hematopoietic Stem Cell Transplant at an Early Stage of Childhood Cerebral Adrenoleukodystrophy. JAMA neurology 2017, 74, 710–717. [Google Scholar] [CrossRef] [PubMed]
- van der Voorn, J.P.; Pouwels, P.J.; Powers, J.M.; Kamphorst, W.; Martin, J.J.; Troost, D.; Spreeuwenberg, M.D.; Barkhof, F.; van der Knaap, M.S. Correlating quantitative MR imaging with histopathology in X-linked adrenoleukodystrophy. AJNR. American journal of neuroradiology 2011, 32, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Melhem, E.R.; Breiter, S.N.; Ulug, A.M.; Raymond, G.V.; Moser, H.W. Improved tissue characterization in adrenoleukodystrophy using magnetization transfer imaging. AJR. American journal of roentgenology 1996, 166, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; DeCiero, D.P.; Cox, C.; Richfield, E.K.; Ito, M.; Moser, A.B.; Moser, H.W. The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. Journal of neuropathology and experimental neurology 2001, 60, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Liu, Y.; Moser, A.B.; Moser, H.W. The inflammatory myelinopathy of adreno-leukodystrophy: cells, effector molecules, and pathogenetic implications. Journal of neuropathology and experimental neurology 1992, 51, 630–643. [Google Scholar] [CrossRef]
- Melhem, E.R.; Loes, D.J.; Georgiades, C.S.; Raymond, G.V.; Moser, H.W. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression. AJNR. American journal of neuroradiology 2000, 21, 839–844. [Google Scholar] [PubMed]
- van der Knaap, M.S.; Valk, J.; de Neeling, N.; Nauta, J.J. Pattern recognition in magnetic resonance imaging of white matter disorders in children and young adults. Neuroradiology 1991, 33, 478–493. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet journal of rare diseases 2012, 7, 51. [Google Scholar] [CrossRef]
- Kumar, A.J.; Köhler, W.; Kruse, B.; Naidu, S.; Bergin, A.; Edwin, D.; Moser, H.W. MR findings in adult-onset adrenoleukodystrophy. AJNR. American journal of neuroradiology 1995, 16, 1227–1237. [Google Scholar]
- Israel, H.; Ostendorf, F.; Stiepani, H.; Ploner, C.J. Spinal cord atrophy in adrenomyeloneuropathy. Archives of neurology 2005, 62, 1157. [Google Scholar] [CrossRef] [PubMed]
- Castellano, A.; Papinutto, N.; Cadioli, M.; Brugnara, G.; Iadanza, A.; Scigliuolo, G.; Pareyson, D.; Uziel, G.; Köhler, W.; Aubourg, P.; Falini, A.; Henry, R.G.; Politi, L.S.; Salsano, E. Quantitative MRI of the spinal cord and brain in adrenomyeloneuropathy: in vivo assessment of structural changes. Brain : a journal of neurology, 2016; 139, (Pt 6), 1735–1746. [Google Scholar] [CrossRef]
- Broad, R.J.; Gabel, M.C.; Dowell, N.G.; Schwartzman, D.J.; Seth, A.K.; Zhang, H.; Alexander, D.C.; Cercignani, M.; Leigh, P.N. Neurite orientation and dispersion density imaging (NODDI) detects cortical and corticospinal tract degeneration in ALS. Journal of neurology, neurosurgery, and psychiatry 2019, 90, 404–411. [Google Scholar] [CrossRef] [PubMed]
- By, S.; Xu, J.; Box, B.A.; Bagnato, F.R.; Smith, S.A. Application and evaluation of NODDI in the cervical spinal cord of multiple sclerosis patients. NeuroImage. Clinical 2017, 15, 333–342. [Google Scholar] [CrossRef] [PubMed]
- MacKay, A.L.; Laule, C. Magnetic Resonance of Myelin Water: An in vivo Marker for Myelin. Brain plasticity (Amsterdam, Netherlands) 2016, 2, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Gieselmann, V. Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options. Acta paediatrica (Oslo, Norway : 1992) 2008, 97, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, A.; Rocha Cabrero, F. (2023). Metachromatic Leukodystrophy. In StatPearls. StatPearls Publishing.
- Beaudet, A.L.; Scriver, C.R.; William, S.; Valle, D. The Metabolic and Molecular Bases of Inherited Disease (Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B.; eds.; 8th ed.; McGraw-Hill, New-York, 2001, 7012 p.; $550.00). Biochemistry (Moscow) 2004, 67, 611–612. [Google Scholar]
- Liaw, H.R.; Lee, H.F.; Chi, C.S.; Tsai, C.R. Late infantile metachromatic leukodystrophy: Clinical manifestations of five Taiwanese patients and Genetic features in Asia. Orphanet journal of rare diseases 2015, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Groeschel, S.; Kehrer, C.; Engel, C.; I Dali, C.; Bley, A.; Steinfeld, R.; Grodd, W.; Krägeloh-Mann, I. Metachromatic leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. Journal of inherited metabolic disease 2011, 34, 1095–1102. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic Leukodystrophy: Diagnosis, Modeling, and Treatment Approaches. Frontiers in medicine 2020, 7, 576221. [Google Scholar] [CrossRef]
- Nandhagopal, R.; Krishnamoorthy, S.G. Neurological picture. Tigroid and leopard skin pattern of dysmyelination in metachromatic leucodystrophy. Journal of neurology, neurosurgery, and psychiatry 2006, 77, 344. [Google Scholar] [CrossRef]
- Schoenmakers, D.H.; Beerepoot, S.; Krägeloh-Mann, I.; Elgün, S.; Bender, B.; van der Knaap, M.S.; Wolf, N.I.; Groeschel, S. Recognizing early MRI signs (or their absence) is crucial in diagnosing metachromatic leukodystrophy. Annals of clinical and translational neurology 2022, 9, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; van der Knaap, M.S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009, 72, 750–759. [Google Scholar] [CrossRef] [PubMed]
- van Rappard, D.F.; Klauser, A.; Steenweg, M.E.; Boelens, J.J.; Bugiani, M.; van der Knaap, M.S.; Wolf, N.I.; Pouwels, P.J. W. Quantitative MR spectroscopic imaging in metachromatic leukodystrophy: value for prognosis and treatment. Journal of neurology, neurosurgery, and psychiatry 2018, 89, 105–111. [Google Scholar] [CrossRef] [PubMed]
- van Rappard, D.F.; Königs, M.; Steenweg, M.E.; Boelens, J.J.; Oosterlaan, J.; van der Knaap, M.S.; Wolf, N.I.; Pouwels, P.J. W. Diffusion tensor imaging in metachromatic leukodystrophy. Journal of neurology 2018, 265, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Sevin, C.; Lazarus, C.; Bellesme, C.; Aubourg, P.; Adamsbaum, C. Toward a better understanding of brain lesions during metachromatic leukodystrophy evolution. AJNR. American journal of neuroradiology 2012, 33, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Compston, A. A new familial infantile form of diffuse brain-sclerosis. Brain : a journal of neurology 2013, 136 (Pt 9), 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- “Krabbe Disease.” National Institute of Neurological Disorders and Stroke, www.ninds.nih.
- Jain, M.; De Jesus, O. (2023). Krabbe Disease. In StatPearls. StatPearls Publishing.
- Kwon, J.M.; Matern, D.; Kurtzberg, J.; Wrabetz, L.; Gelb, M.H.; Wenger, D.A.; Ficicioglu, C.; Waldman, A.T.; Burton, B.K.; Hopkins, P.V.; Orsini, J.J. Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet journal of rare diseases 2018, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Lantos, J.D. Dangerous and expensive screening and treatment for rare childhood diseases: the case of Krabbe disease. Developmental disabilities research reviews 2011, 17, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, K.; Sudhakar, S.V.; Thomas, M.; Yoganathan, S.; Christudass, C.S.; Chandran, M.; Panwala, H.; Gibikote, S. Revisiting magnetic resonance imaging pattern of Krabbe disease - Lessons from an Indian cohort. Journal of clinical imaging science 2019, 9, 25. [Google Scholar] [CrossRef]
- Nagar, V.A.; Ursekar, M.A.; Krishnan, P.; Jankharia, B.G. Krabbe disease: unusual MRI findings. Pediatric radiology 2006, 36, 61–64. [Google Scholar] [CrossRef]
- Sasaki, M.; Sakuragawa, N.; Takashima, S.; Hanaoka, S.; Arima, M. MRI and CT findings in Krabbe disease. Pediatric neurology 1991, 7, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Jeffrey, B.A.; Jian, J.; Choudhury, G.R.; Michalson, K.; Mitchell, T.W.; Buza, E.L.; Chichester, J.; Dyer, C.; Bagel, J.; Vite, C.H.; Bradbury, A.M.; Wilson, J.M. Efficacy and Safety of a Krabbe Disease Gene Therapy. Human gene therapy 2022, 33, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Tullu, M.S.; Muranjan, M.N.; Kondurkar, P.P.; Bharucha, B.A. Krabbe disease--clinical profile. Indian pediatrics 2000, 37, 939–946. [Google Scholar] [PubMed]
- Singh, R.; Samanta, D. (2023). Pelizaeus-Merzbacher Disease. In StatPearls. StatPearls Publishing.
- “Pelizaeus-Merzbacher Disease | National Institute of Neurological Disorders and Stroke.” Www.ninds.nih.gov, www.ninds.nih.gov/health-information/disorders/pelizaeus-merzbacher-disease.
- Koeppen, A.H.; Robitaille, Y. Pelizaeus-Merzbacher disease. Journal of neuropathology and experimental neurology 2002, 61, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Osaka, H.; Imaizumi, K.; Nezu, A.; Takanashi, J.; Arii, J.; Murayama, K.; Ono, J.; Kikawa, Y.; Mito, T.; Shaffer, L.G.; Lupski, J.R. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Annals of neurology 1999, 45, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kanai, M.; Tanabe, Y.; Kubota, T.; Kashork, C.D.; Wakui, K.; Fukushima, Y.; Lupski, J.R.; Shaffer, L.G. Prenatal interphase FISH diagnosis of PLP1 duplication associated with Pelizaeus-Merzbacher disease. Prenatal diagnosis 2001, 21, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Nezu, A.; Kimura, S.; Takeshita, S.; Osaka, H.; Kimura, K.; Inoue, K. An MRI and MRS study of Pelizaeus-Merzbacher disease. Pediatric neurology 1998, 18, 334–337. [Google Scholar] [CrossRef]
- Sumida, K.; Inoue, K.; Takanashi, J.; Sasaki, M.; Watanabe, K.; Suzuki, M.; Kurahashi, H.; Omata, T.; Tanaka, M.; Yokochi, K.; Iio, J.; Iyoda, K.; Kurokawa, T.; Matsuo, M.; Sato, T.; Iwaki, A.; Osaka, H.; Kurosawa, K.; Yamamoto, T.; Matsumoto, N.; … Sato, N. The magnetic resonance imaging spectrum of Pelizaeus-Merzbacher disease: A multicenter study of 19 patients. Brain & development 2016, 38, 571–580. [Google Scholar] [CrossRef]
- Bonavita, S.; Schiffmann, R.; Moore, D.F.; Frei, K.; Choi, B.; Patronas MD, N.; Virta, A.; Boespflüg-Tanguy, O.; Tedeschi, G. Evidence for neuroaxonal injury in patients with proteolipid protein gene mutations. Neurology 2001, 56, 785–788. [Google Scholar] [CrossRef]
- Kuhn, J.; Cascella, M. (2023). Alexander Disease. In StatPearls. StatPearls Publishing.
- Messing, A. Refining the concept of GFAP toxicity in Alexander disease. Journal of neurodevelopmental disorders 2019, 11, 27. [Google Scholar] [CrossRef]
- Srivastava, S.; Waldman, A.; Naidu, S. (2002). Alexander Disease. In M. P. Adam (Eds.) et. al.; GeneReviews®. University of Washington, Seattle.
- Hagemann, T.L. Alexander disease: models, mechanisms, and medicine. Current opinion in neurobiology 2022, 72, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Ciammola, A.; Sangalli, D.; Sassone, J.; Poletti, B.; Carelli, L.; Banfi, P.; Pappacoda, G.; Ceccherini, I.; Grossi, A.; Maderna, L.; Pingue, M.; Girotti, F.; Silani, V. A Novel Mutation of GFAP Causing Adult-Onset Alexander Disease. Frontiers in neurology 2019, 10, 1124. [Google Scholar] [CrossRef]
- Jany, P.L.; Agosta, G.E.; Benko, W.S.; Eickhoff, J.C.; Keller, S.R.; Köehler, W.; Koeller, D.; Mar, S.; Naidu, S.; Marie Ness, J.; Pareyson, D.; Renaud, D.L.; Salsano, E.; Schiffmann, R.; Simon, J.; Vanderver, A.; Eichler, F.; van der Knaap, M.S.; Messing, A. CSF and Blood Levels of GFAP in Alexander Disease. eNeuro 2015, 2, ENEURO.0080–152015. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Naidu, S.; Breiter, S.N.; Blaser, S.; Stroink, H.; Springer, S.; Begeer, J.C.; van Coster, R.; Barth, P.G.; Thomas, N.H.; Valk, J.; Powers, J.M. Alexander disease: diagnosis with MR imaging. AJNR. American journal of neuroradiology 2001, 22, 541–552. [Google Scholar] [PubMed]
- Messing, A.; Brenner, M.; Feany, M.B.; Nedergaard, M.; Goldman, J.E. Alexander disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2012, 32, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- “Alexander Disease - NORD (National Organization for Rare Disorders).” NORD (National Organization for Rare Disorders), NORD, 2015, rarediseases.org/rare-diseases/alexander-disease/.
- Kantor, B.; McCown, T.; Leone, P.; Gray, S.J. Clinical applications involving CNS gene transfer. Advances in genetics 2014, 87, 71–124. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, M.R.; Samanta, D.; Bokhari, S.R. A. Canavan Disease. In StatPearls. StatPearls Publishing.
- Leone, P.; Shera, D.; McPhee, S.W.; Francis, J.S.; Kolodny, E.H.; Bilaniuk, L.T.; Wang, D.J.; Assadi, M.; Goldfarb, O.; Goldman, H.W.; Freese, A.; Young, D.; During, M.J.; Samulski, R.J.; Janson, C.G. (2012). Long-term follow-up after gene therapy for canavan disease. Science translational medicine 2023, 4, 165ra163. [Google Scholar] [CrossRef]
- Feng, L.; Chao, J.; Tian, E.; Li, L.; Ye, P.; Zhang, M.; Chen, X.; Cui, Q.; Sun, G.; Zhou, T.; Felix, G.; Qin, Y.; Li, W.; Meza, E.D.; Klein, J.; Ghoda, L.; Hu, W.; Luo, Y.; Dang, W.; Hsu, D.; … Shi, Y. Cell-Based Therapy for Canavan Disease Using Human iPSC-Derived NPCs and OPCs. Advanced science (Weinheim, Baden-Wurttemberg, Germany) 2020, 7, 2002155. [Google Scholar] [CrossRef]
- Fröhlich, D.; Kalotay, E.; von Jonquieres, G.; Bongers, A.; Lee, B.; Suchowerska, A.K.; Housley, G.D.; Klugmann, M. Dual-function AAV gene therapy reverses late-stage Canavan disease pathology in mice. Frontiers in molecular neuroscience 2022, 15, 1061257. [Google Scholar] [CrossRef]
- Matalon, R.; Michals, K.; Sebesta, D.; Deanching, M.; Gashkoff, P.; Casanova, J. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. American journal of medical genetics 1988, 29, 463–471. [Google Scholar] [CrossRef]
- Bennett, M.J.; Gibson, K.M.; Sherwood, W.G.; Divry, P.; Rolland, M.O.; Elpeleg, O.N.; Rinaldo, P.; Jakobs, C. Reliable prenatal diagnosis of Canavan disease (aspartoacylase deficiency): comparison of enzymatic and metabolite analysis. Journal of inherited metabolic disease 1993, 16, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Gowda, V.K.; Bharathi, N.K.; Bettaiah, J.; Bhat, M.; Shivappa, S.K. Canavan Disease: Clinical and Laboratory Profile from Southern Part of India. Annals of Indian Academy of Neurology 2021, 24, 347–350. [Google Scholar] [CrossRef]
- Perlman, S.J.; Mar, S. Leukodystrophies. Advances in experimental medicine and biology. 2012, 724, 154–171. [Google Scholar] [CrossRef] [PubMed]
- McAdams, H.P.; Geyer, C.A.; Done, S.L.; Deigh, D.; Mitchell, M.; Ghaed, V.N. CT and MR imaging of Canavan disease. AJNR. American journal of neuroradiology 1990, 11, 397–399. [Google Scholar]
- Hoshino, H.; Kubota, M. C anavan disease: Clinical features and recent advances in research. Pediatrics International 2014, 56, 477–483. [Google Scholar] [CrossRef]
- Crow, Y.J. (2005). Aicardi-Goutières Syndrome. In M. P. Adam (Eds.) et. al.; GeneReviews®. University of Washington, Seattle.
- Misk, R.A.; Qawasme, L.; Abunejma, F.M.; Abu Rahma, B.I.; Abuawwad, E.M.; Abu Iram, R.I.; Karaki, A.H.; Alzughayyar, T.Z.; Zalloum, J.S. A Case Report and Literature Review of Pseudo-TORCH Syndrome Type 2 (PTORCH2). Case reports in pediatrics 2022, 2022, 3555532. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, G.; Fazzi, E.; D'Arrigo, S. Aicardi-Goutières syndrome: a description of 21 new cases and a comparison with the literature. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society, 2002; 6 Suppl A, A9–A86. [Google Scholar] [CrossRef]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; Baxter, P.; Benko, W.S.; Bergmann, C.; Bertini, E.; Biancheri, R.; Blair, E.M.; Blau, N.; Bonthron, D.T.; Briggs, T.; Brueton, L.A.; … Crow, Y.J. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. American journal of human genetics 2007, 81, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Wolf, C.; Günther, C. Aicardi-Goutières syndrome: a model disease for systemic autoimmunity. Clinical and experimental immunology 2014, 175, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Tolmie, J.L.; Shillito, P.; Hughes-Benzie, R.; Stephenson, J.B. The Aicardi-Goutières syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). Journal of medical genetics 1995, 32, 881–884. [Google Scholar] [CrossRef]
- Goutières, F.; Aicardi, J.; Barth, P.G.; Lebon, P. Aicardi-Goutières syndrome: an update and results of interferon-alpha studies. Annals of neurology 1998, 44, 900–907. [Google Scholar] [CrossRef]
- Rice, G.I.; Forte, G.M.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; Barnerias, C.; Bernard, G.; Bodemer, C.; Botella, M.P.; Cereda, C.; Chandler, K.E.; Dabydeen, L.; Dale, R.C.; De Laet, C.; De Goede, C.G.; … Crow, Y.J. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. The Lancet. Neurology 2013, 12, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; Abdel-Hamid, M.S.; Abdel-Salam, G.M.; Ackroyd, S.; Aeby, A.; Agosta, G.; Albin, C.; Allon-Shalev, S.; Arellano, M.; Ariaudo, G.; Aswani, V.; … Rice, G.I. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. American journal of medical genetics. Part A 2015, 167A, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Shiromoto, Y.; Minakuchi, M.; Nishikura, K. The role of RNA editing enzyme ADAR1 in human disease. Wiley interdisciplinary reviews. RNA 2022, 13, e1665. [Google Scholar] [CrossRef] [PubMed]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 Functions and Human Diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef] [PubMed]
- Rigby, R.E.; Leitch, A.; Jackson, A.P. Nucleic acid-mediated inflammatory diseases. BioEssays : news and reviews in molecular, cellular and developmental biology 2008, 30, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.J.; Beal, P.A. Effects of Aicardi-Goutières syndrome mutations predicted from ADAR-RNA structures. RNA biology 2017, 14, 164–170. [Google Scholar] [CrossRef]
- Chahwan, C.; Chahwan, R. Aicardi-Goutieres syndrome: from patients to genes and beyond. Clinical genetics 2012, 81, 413–420. [Google Scholar] [CrossRef]
- Crow, Y.J.; Rehwinkel, J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Human molecular genetics 2009, 18, R130–R136. [Google Scholar] [CrossRef]
Figure 1.
Major MRI findings, CSF/ blood markers and other key features in leukodystrophies.

Figure 2.
Pathophysiology of X-Linked Adrenoleukodystrophy: Mutations in the ABCD1 gene on the X chromosome result in the prevention of very long chain fatty acids (VLCFAs) into the peroxisome, resulting in the clinical phenotype due to VLCFA accumulation throughout the body.
Figure 2.
Pathophysiology of X-Linked Adrenoleukodystrophy: Mutations in the ABCD1 gene on the X chromosome result in the prevention of very long chain fatty acids (VLCFAs) into the peroxisome, resulting in the clinical phenotype due to VLCFA accumulation throughout the body.
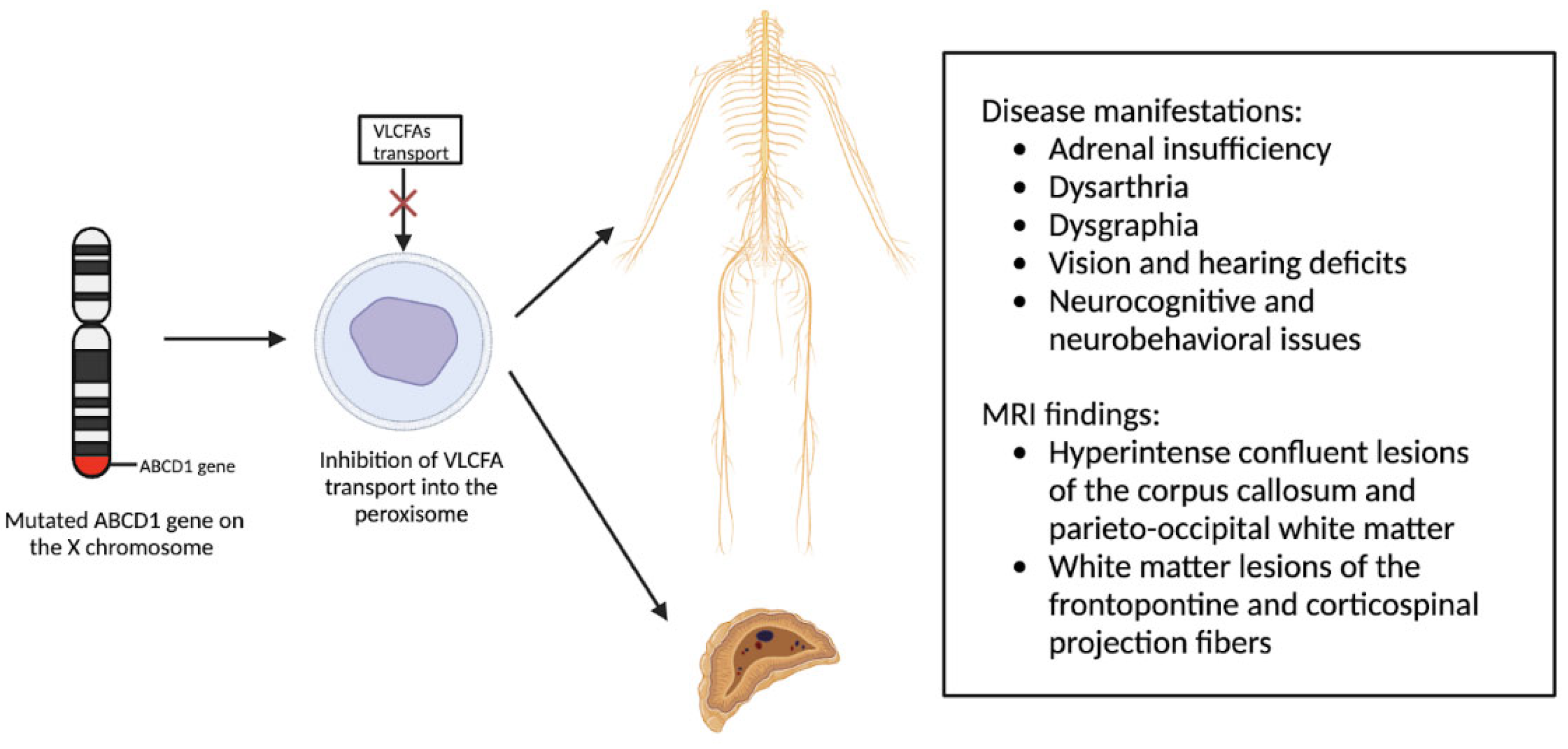
Figure 3.
MRI findings found in metachromatic leukodystrophy. (A) Hypointense radial stripes resembling tiger skin. (B) Hypointense dots resembling leopard skin [35].
Figure 3.
MRI findings found in metachromatic leukodystrophy. (A) Hypointense radial stripes resembling tiger skin. (B) Hypointense dots resembling leopard skin [35].
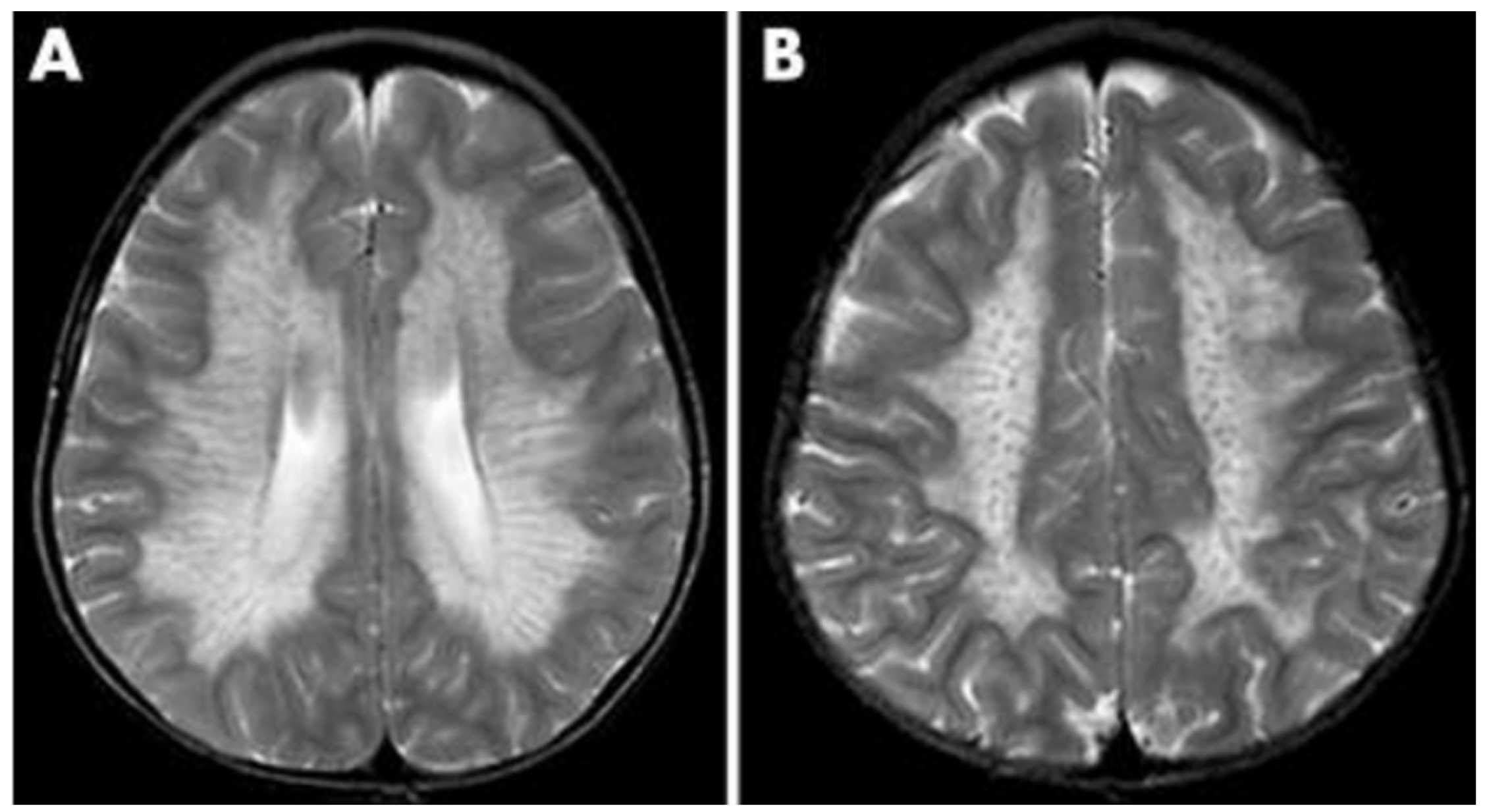
Figure 4.
The three-step process for infant screening for KD.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



