
Submitted:
01 November 2023
Posted:
02 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
In its prefusion state, the SARS-CoV-2 Spike protein (similarly to other class I viral fusion proteins) is metastable, which is considered to be an important feature for optimizing or regulating their functions. After the binding process of its S1 subunit (S1) with the ACE2, the Spike protein (S) suffers a dramatic conformational change where S1 splits from the S2 subunit, which then penetrates the membrane of the host cell, promoting the fusion of the viral and cell membranes. This results in the infection of the host cell. In a previous work, we showed -using large scale molecular dynamics simulations- that the application of external electric fields (EF) induces drastic changes and damage in the receptor-binding domain (RBD) of the wild type Spike protein, as well of the Alpha, Beta and Gamma variants, leaving a structure which cannot be recognized any more by ACE2. In this work, we first extend the study to the Delta and Omicron variants and confirm the high sensitivity and extreme vulnerability of S to moderate EF (as weak as 104 V m-1), but, more importantly, we also show that, in contrast, the postfusion state of the spike protein does not suffer structural damage even if electric field intensities four orders of magnitude higher are applied. These results provide a solid scientific basis to confirm the prefusion state metastability roots of the SARS-CoV-2 Spike protein susceptibility to be damaged by EF. After the virus docks to the ACE2 receptor, the stable and robust postfusion conformation develops, which exhibits a similar resistance to EF (damage threshold higher than 108 V m-1) like most globular proteins.
Keywords:
SARS-CoV-2
; Spike protein
; structural stability
; Molecular dynamics simulations
; Electric fields
1. Introduction
The SARS-CoV-2 virus continues to be a significant global health concern. Since its emergence in 2019, SARS-CoV-2 has mutated and spread widely among the human population, leading to the emergence of several variants of concern (VOCs) [1]. Among the many monitored variants (Alpha, Beta, Gamma, Epsilon, Lota, Kappa, Mu, Zeta), three have had a significant worldwide impact: Alpha, Delta, and Omicron [2,3]. The latter is particularly noteworthy due to its distinctive feature of having approximately 30 or more mutations in its surface glycoprotein, in sharp contrast to the Delta variant, which has only a few mutations [3]. While the developed vaccines offer protection, some VOCs can evade immunity, highlighting the need for ongoing transmission control measures.
During the process of viral entry, enveloped viruses, such as SARS-CoV-2 depend on the fusion of their lipid-enveloped structure with the cell membranes of host organisms. In the case of coronaviruses, this crucial fusion event is driven by the spike (S) protein, a class I viral transmembrane fusion protein that exhibits distinctive features [4,5]. The S protein consists of trimeric polypeptide chains with glycosylated residues on the surface. Each monomeric unit contains both S1 and S2 subunits, which remain noncovalently bonded until viral fusion is initiated [6]. Within the S1 subunit, there is an N-terminal domain along with a receptor-binding domain (RBD) responsible for recognizing and binding to the host receptor, angiotensin-converting enzyme 2 [7]. The S2 subunit mediates viral cell membrane fusion and mainly comprises two regions known as heptad repeats (HR1 and HR2). These heptad repeats consist of repetitive heptapeptides characterized by α-helical structures and various hydrophobic residues, which are involved in the transition from the prefusion state to the postfusion conformation [8].
The free energy required for viral membrane fusion to overcome kinetic barriers is derived from the energy released during a substantial conformational shift in the viral envelope S protein [9]. Similar to other class I viral fusion proteins, the S protein typically resides in a prefusion conformation, trapped in a high-energy, metastable state [10,11,12,13]. Upon interaction with the host cell, a remarkable structural rearrangement of the S protein towards a lower energy, stable postfusion state occurs. This process involves the sequential folding of HR2 onto HR1, forming a structure called a 6-helix bundle or 6HB in an antiparallel configuration at the fusion core [14]. As a result, the viral membrane is drawn toward the host cell membrane and firmly adheres to it, ultimately allowing the fusion of the two membranes. The energy barrier restraining the prefusion state has been observed to be particularly low in the case of coronaviruses S protein [15,16].
Both theoretical predictions and experimental evidence have indicated that the application of electric fields (EFs) can induce significant conformational changes in proteins [17,18,19,20]. This phenomenon mainly arises from the balance between conformational and electrostatic energies, along with entropic contributions [21,22]. In a previous study, employing molecular dynamics (MD) simulations, we demonstrated that low to moderate electric fields with intensities as low as 105 V m−1 can affect the wild-type (WT), the Alpha, the Beta and the Gamma RBD’s of the S protein in such a way that they can overcome a non-thermal energy barrier, and therefore shifting it to a state exhibiting a conformation between the prefusion and postfusion states [23]. The purpose of this study is twofold. On the one side we extend our analysis to the Delta and Omicron variants of the RBD of the SARS-CoV-2 S protein and show that, remarkably, EF intensities as low as 104 V m-1 are enough to destabilize the RBD structure of these two variants and to induce irreversible changes on a sub-microsecond timescale. On the other hand, and as a central result of this paper, we assess the impact of external EFs on the conformational stability of the postfusion form, taking into account its dynamic structural changes. Interestingly, we observed no significant structural alterations, even when applying high-intensity EFs (108 V m-1), which is a threshold for significant conformational changes reported for many globular proteins.
2. Materials and Methods
2.1. Simulation Setup
The initial coordinates of the protein chains were obtained from the Protein Data Bank with IDs: 6VSB, 7V8B, 7WBP and 7COT, which were captured at 3.46, 3.20, 3.00 and 2.16 Å resolution, respectively [24]. The simulation cell consisting of the protein chains, waters, and ions was set up with the CHARMM-GUI [25,26,27] . The protein chains were centered in cubic cell boxes of sizes 198 nm for postfusion and 106 nm for delta/omicron, and then solvated with TIP3P waters [28] and Na+ and Cl- ions at 150 mM concentration.
The dynamical propagation of the Newton equations was achieved with the GROMACS simulation package solver (v. 2021) [29,30,31]. A cutoff distance of 12 Å was employed for solving the short-range interactions in both the electrostatic, with the particle mesh Ewald (PME) method [32], and in the van der Waals terms. In the former a fourth order of cubic interpolation method and a grid size of 1.2 Å was used. Hydrogen atoms were constrained with the LINCS algorithm [33].
The protocol for minimization and equilibration of the initial structure was similar to the one in our previous published work [23].
2.2. Principal-Component-Analysis:
Principal Components Analysis (PCA) was performed on the trajectories for both EF-on and EF-off states to characterize and visualize these trajectories and their respective states. In order to accomplish this, each trajectory was projected onto a two-dimensional space obtained through dimensionality reduction. Generalized coordinates, specifically dihedral angles, were employed to define the structural states, effectively separating the internal protein motion from its overall motion [34]. Furthermore, each dihedral angle was partitioned into two metric coordinates, signifying its sine and cosine components, respectively. This transition from dihedral space to a linear metric space introduced a well-defined Euclidean distance, ensuring a unique representation while circumventing artifacts resulting from the periodic nature of angles [35].
Subsequently, Principal Components Analysis was applied to the aforementioned metric coordinates to derive a reduced space. The first two components, corresponding to the highest eigenvalues, were selected to define the reduced two-dimensional space. PCA, as a technique, was employed to identify correlated motion patterns by diagonalizing the covariance matrix, where the eigenvectors delineated the directions of collective motion, and the eigenvalues, ranked in descending order, quantified their respective amplitudes.
PCA was executed using the scikit-learn Python library [36]. Following this, each trajectory was projected onto the reduced PCA space. It is important to note that while we employed components associated with trajectories under an electric field (EF) intensity of 107 V m−1 for projection, similar plots could be generated using principal components corresponding to runs with different EF intensities. Therefore, the conclusions drawn from this analysis are not dependent on the specific choice of EF intensity.
2.3. Free-Energy Landscape Estimation
The estimation of free energy was carried out along by previously recording the root mean square displacements (RMSD) of the system. For this purpose we utilized a path-sampling method [37,38,39] to approximate the potential of mean force (PMF) for each condition (no-EF, EF-on, and EF-off) and for each strength of the electric field. Considering the RMSD as the x-axis to characterize the states of the protein, we determined the free energy profile by the equation:
where represents the segmented RMSD value of the j position along the trajectory, kB stands for the Boltzmann constant, T is the temperature, and δ (... ) denotes the Dirac delta function. Each trajectory was discretized into 20 windows along the RMSD coordinate.
2.4. MD Analysis
The trajectory files were processed and analyzed using GROMACS tools or the MDAnalysis Python library [40]. The MD trajectories were visualized, and molecular representations were drawn with the assistance of the VMD software [41].
Customized Python scripts and the MDAnalysis library were employed to calculate various residues and atomic distances, including those related to the crucial amino acids. To analyze interactions by determining the distance between individual residues, the mean distance between all the atoms of each residue was computed.
The STRIDE algorithm, integrated into the VMD software package version 1.9.4a38 (2019), was employed to estimate changes in the secondary structure of the RBD over time under the conditions of no-EF, EF-on, and EF-off. The STRIDE algorithm relies on hydrogen bond energy and statistically derived backbone torsion angle data to characterize secondary structures within trajectories previously generated by GROMACS.
2.5. Electrostatic Potential Surface Calculations
The calculation of all potential maps on the PDB 7V8B and 7WBP structural data and final frames from the MD trajectories was carried out using the Adaptive Poisson-Boltzmann Solver (APBS) algorithm [42]. The PDB formats were initially prepared by the PDB2PQR web server and converted to PQR format using the CHARMM force field, with PROPKA set at pH = 7.0 [43]. Subsequently, the APBS analysis was conducted via the Linearized Poisson-Boltzmann Equation in the VMD software, with settings parameters including a solvent dielectric constant of 78.5, a solvent radius of 1.4 Å, a solute dielectric constant of 2.0, a system temperature of 300 K, a surface density of 10.0 points/Å, and the use of harmonic average smoothing for surface definition.
3. Results
3.1. Global Structural Changes in the S1 and S2 Subunits of the Spike Protein under Electric Fields
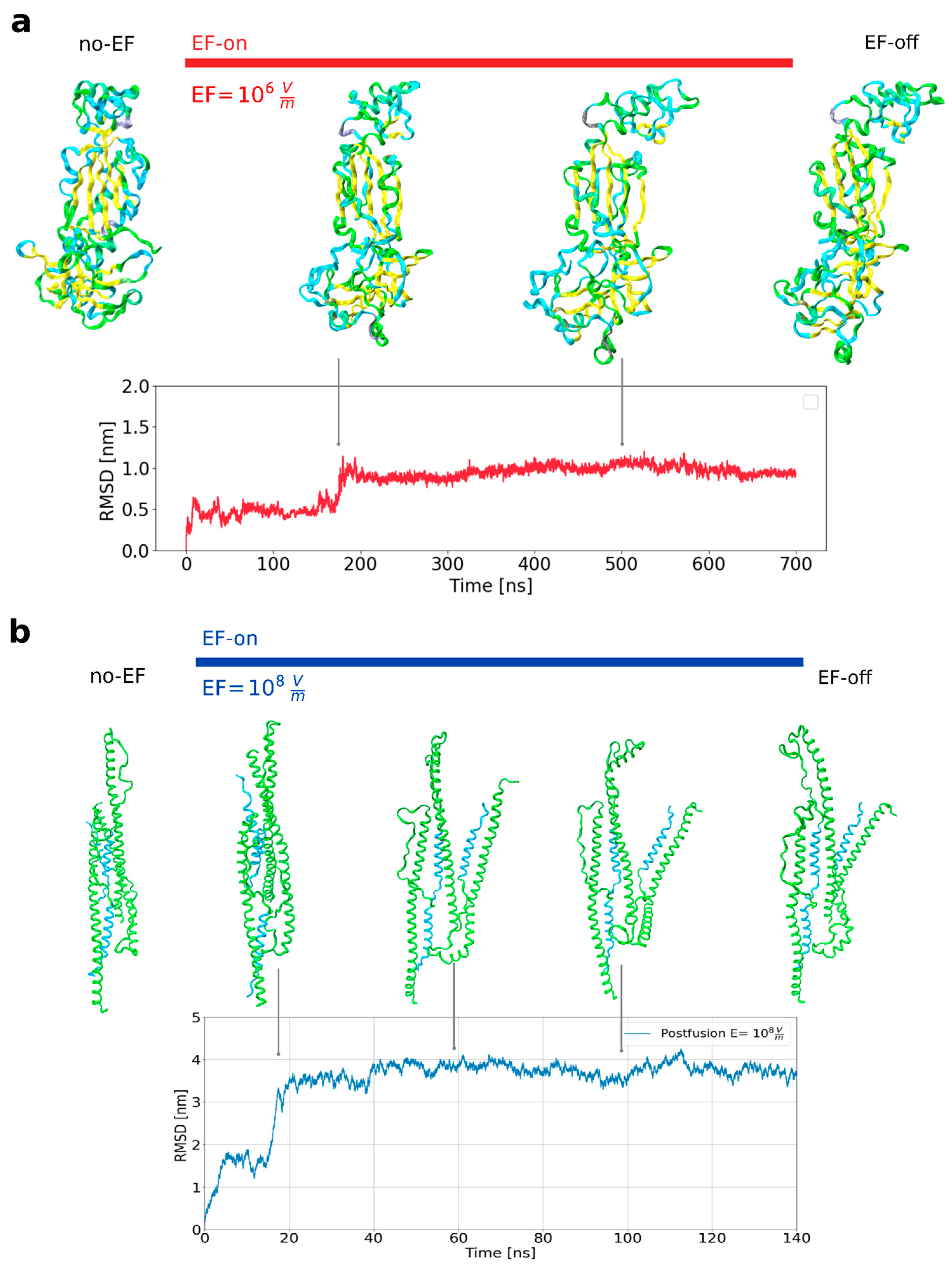
The impact of external electric fields (EFs) on the overall structure of the S1 and S2 subunits of the S protein was investigated through molecular dynamics simulations. We studied the S1 subunit with a selected segment of the S protein in an "up" conformation, spanning from residue 319 to residue 686 in the prefusion conformation. This segment encompasses the entire Receptor-Binding Domain (RBD), subdomains SD1 and SD2, as well as the interface connecting S1 and S2 (as illustrated in the upper panel of Figure 5). In the simulations without applied EFs, this segment displayed the local structure, dynamic properties and biochemical attributes consistent with those exhibited in the complete protein, therefore supporting the choice of a segment for the numerical modeling [44]. In this manner, simulations were conducted within a confined spatial domain without compromising the generalizability of the findings (also refer to the Methods section). Regarding the S2 subunit, the primary focus was placed on the postfusion core directly using the asymmetric unit as deposited in the Protein Data Bank without applying symmetry operations to obtain the 6HB [45]. The simulations were initiated using structures from the PDB (PDB ID 6VSB for the S1 subunit and 7COT for the S2 subunit), and missing residues were added (see Methods). The first production run aimed to attain thermal equilibrium for the system in the absence of an EF (referred to as the "no-EF" run), ensuring that the protein reached thermodynamic equilibrium at 30 °C. The conformation obtained from the experimentally derived segment closely resembled a stable equilibrium folding state, and the thermalization run primarily served to relax any residual structural tension. Subsequently, using the thermally equilibrated structure as the initial state, simulations were conducted on each subunit of the S protein in the presence of an electric field for a duration of 700 nanoseconds ( "EF-on" runs). For the study of the prefusion subunit, low field intensities were applied (V m−1), based on our previous results [23]. For the stable postfusion S2 subunit, we performed simulations assuming an EF intensity of 108 V m−1, which is just below the damage threshold for globular proteins. In the case of the prefusion subunit, protein elongation was observed in the trajectories due to the alignment of permanent local dipoles and the displacement of charges parallel to the electric field (as observed in Figure 1a), also including a loss of tertiary protein structures within a few nanoseconds. The conformational shifts of the prefusion S1 subunit protein under the influence of an electric field are clearly visible in the time-dependent changes in the root-mean-square displacements (RMSD) of the protein backbone relative to the initial structures (Figure 1a). The transition from the initial conformation to a new stable structure occurs within the first 200 nanoseconds.
To facilitate a direct comparison of stability between the prefusion and postfusion states, a 140 nanosecond simulation was conducted while applying a high electric field to the postfusion structure (PDB: 7COT). The postfusion state of the S protein, under a field strength of 108 V m−1, exhibits significantly greater stability in its secondary structure compared to the metastable prefusion state. The overall three-dimensional structure underwent changes during the initial 20 nanoseconds of the dynamics, while maintaining the interaction between the two Heptad Repeat (HR) regions in each protomer. The simulation starts from an asymmetric unit within which the three HR1-linker-HR2 chains are related by a three-fold axis forming a homotrimeric complex. A few seconds after applying the EF, as a result of conformational rearrangements, the single chains move away from each other and each molecule acts as a protomer. As can be seen in the RMSD (Figure 1b), this conformation is maintained throughout the entire simulation without significant additional change. This global dynamics has its explanation in the presence of flexible loops that connect HR1 and CH in the prefusion state, which are preserved in the asymmetric 7COT unit along with the flexible linker that connect HR1 and HR2. These results could have functional implications in terms of avoiding the formation of the HR1-HR2 six-helix bundle, critical for viral entry mediated by class I fusion proteins [46].
Figure 1.
Large amplitude conformational changes are observable in the studied segments of the SARS-CoV-2 spike protein. (A) Snapshots of the prefusion conformation (S1 subunit) as it evolves with EF application (upper row). Deviations from the initial structure are quantified with the Root Mean Square Deviation (RMSD) (lower row). (B) Snapshots of the postfusion conformation (S2 subunit, PDB 7COT) under EF (upper row) and deviations from initial structure as RMSD (lower row).
Figure 1.
Large amplitude conformational changes are observable in the studied segments of the SARS-CoV-2 spike protein. (A) Snapshots of the prefusion conformation (S1 subunit) as it evolves with EF application (upper row). Deviations from the initial structure are quantified with the Root Mean Square Deviation (RMSD) (lower row). (B) Snapshots of the postfusion conformation (S2 subunit, PDB 7COT) under EF (upper row) and deviations from initial structure as RMSD (lower row).
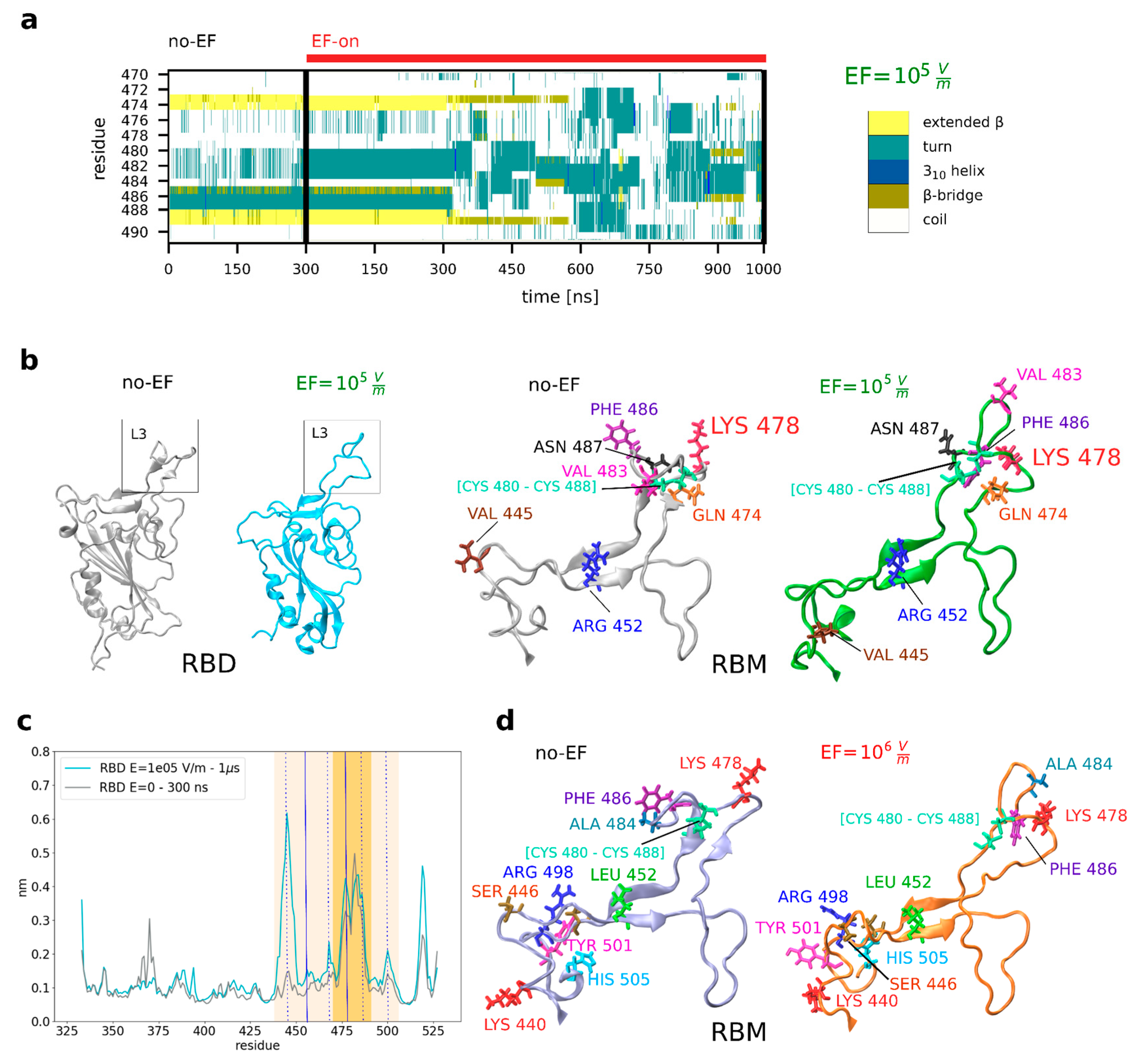
3.2. Effects of Moderate Electric Fields on the Secondary Structure of the Receptor Binding Domain
The RBD has a unique three-dimensional structure that contains critical amino acid residues relevant to the virus specificity and binding affinity to the ACE2 receptor. The receptor binding motif (RBM) is the primary functional component within the RBD which forms the interface responsible for the interaction between the spike protein and the ACE2. Mutations located distally from the binding region have been identified as factors affecting the structural stability of the prefusion spike protein and its affinity to ACE2, highlighting the important role played by the precise spatial arrangement of RBM residues involved in receptor binding. Loop 3 (L3) encompasses amino acids from Tyr470 to Pro491 and is one of the four loops constituting the RBM. L3 is crucial in S protein-ACE2 interaction, due to the presence of critical β-strands (Cys488-Tyr489 and Tyr473-Gln474) that significantly enhance WT SARS-CoV-2 affinity for ACE2. This enhanced affinity, estimated to be approximately 15-20 times greater than that of SARS-CoV-1, results from the structured β-strands within L3, which stand in contrast to the unstructured L3 in SARS-CoV-1 [47]. In our earlier work, the influence of EF on the stability of the RBD and its essential residues involved in the local interaction with ACE2 was examined for the wild-type RBD and several VOCs. The current objective is centered on the extension and generalization of results to the impact of EFs on the ability of S to dock into the ACE receptors. To achieve this, a series of simulations were conducted to determine whether the Delta and Omicron VOCs are also susceptible to damage caused by EFs of low to moderate strength. MD simulations were conducted running over 700 ns for every variant, using a model based on the structure of the unbound RBD, which was derived from experimental data acquired from the PDB structure of the RBD-ACE2 complex (PDB 7V8B and 7WBP).
Initially, for both Delta and Omicron variants of the RBD, thermalization simulations without electric field (EF = 0 V m-1, “no-EF”) were conducted, and it was observed that the secondary and tertiary structure were preserved in comparison to the original crystal structure. Throughout the thermalization simulations, the mentioned β-strands in the L3 loop remained intact, in agreement with prior studies that emphasize the rigidity of β-sheets in WT SARS-CoV-2, contributing to the stability of L3. Concerning the Delta and Omicron variants, they present mutated residues located in the RBM region. While the Delta variant has two mutated residues, Leu452Arg and Thr478Lys, Omicron exhibits 15 substitutions throughout the RBD, which are expected to cause a change in the spatial organization of the RBD residues as well as the RBD-ACE2 interactions [48,49]. Nevertheless, previous research has emphasized the importance of L3 stability in the ACE2-interacting interface of the RBD [48]. Furthermore, the overall secondary structure of the RBD was closely monitored and found to exhibit remarkable stability throughout the thermalization run (Figures 2 a, b, d).
To investigate the impact of EFs, simulations were carried out at different field intensities: EF = 104, 105, 106, and 107 V m−1. Figure 2a shows the time evolution analysis of L3 secondary structure (for the Delta variant, EF = 105 V m−1) obtained using the STRIDE algorithm implemented in VMD [41] on the simulation files. A gradual reduction in the β-sheets was observed during the initial 300 ns of the EF-on simulation, resulting in their eventual deconstruction into turns or random coils before 1 μs. A representative snapshot of the no-EF and EF-on runs (an example for EF = 105 V m−1) for the Delta variant shows that the secondary structure of the RBD experienced disruptions at multiple segments, with a notable impact on L3 (Figure 2b). As a result, L3 goes through a transition from its compact structure with the two β-sheets to an open and entirely unstructured coil, similar to the conformation of L3 in SARS-CoV-1 [47,50]. In addition to the previous analysis, the impact of EF on the flexibility of the RBD was also investigated, considering the distinct flexibility properties of coil or loop structures compared to highly ordered secondary structures like β-sheets and α-helices in proteins. To quantify these changes, root-mean-square fluctuations (RMSF) analysis of the RBD, which describes the flexibility of the residues, was employed. In Figure 2c, it is demonstrated that the flexibility of the RBD is modified by the EF in a non-uniform manner, with a more impact on the L3 loop and the RBM region.
The same type of simulations and evaluation to assess the impact of EFs was performed on the Omicron variant, where it was observed that the disruption of beta sheets is an irreversible process, and L3 turned into an open and unstructured state (Figure 2d). These findings agree with our prior research on the wild-type and other S protein variants [23]. Taken together, these observations strongly imply that the initial conformation of the RBD is destabilized by the application of an EF. As a result, the secondary structure of crucial RBM segments involved in the docking to ACE2 undergoes changes, leading to the disruption of the spatial atomic organization of backbone and side chain residues in key positions. These rearrangements have important implications for RBD-ACE2 interactions, which are weakened.
Figure 2.
EFs of different moderate intensities induce changes in the relative positions and orientations of residues in the RBD spike protein of the Variant of Concern Delta and Omicron of SARS-CoV-2. (A) Evolution of the secondary structure in the residues corresponding to the loop L3 of the RBM of the prefusion structure of the Delta variant under EF (PDB 7V8B). The beta-sheets (yellow) disappear completely after 600ns. (B) The relative position and structure of the residues in the loop L3 (Delta variant) are changed after applying 105 V/m, underlying the removal of the beta-sheet structure (left panel) and the repositioning of residues (right panel). (C) Root Mean Square Fluctuations of the RBD reveal flexibilization of the backbone structure under application of EF. (D) Close-up view showing the key residues of the RBD that participate in the binding with ACE2 for the Omicron variant, both before and after EF application.
Figure 2.
EFs of different moderate intensities induce changes in the relative positions and orientations of residues in the RBD spike protein of the Variant of Concern Delta and Omicron of SARS-CoV-2. (A) Evolution of the secondary structure in the residues corresponding to the loop L3 of the RBM of the prefusion structure of the Delta variant under EF (PDB 7V8B). The beta-sheets (yellow) disappear completely after 600ns. (B) The relative position and structure of the residues in the loop L3 (Delta variant) are changed after applying 105 V/m, underlying the removal of the beta-sheet structure (left panel) and the repositioning of residues (right panel). (C) Root Mean Square Fluctuations of the RBD reveal flexibilization of the backbone structure under application of EF. (D) Close-up view showing the key residues of the RBD that participate in the binding with ACE2 for the Omicron variant, both before and after EF application.
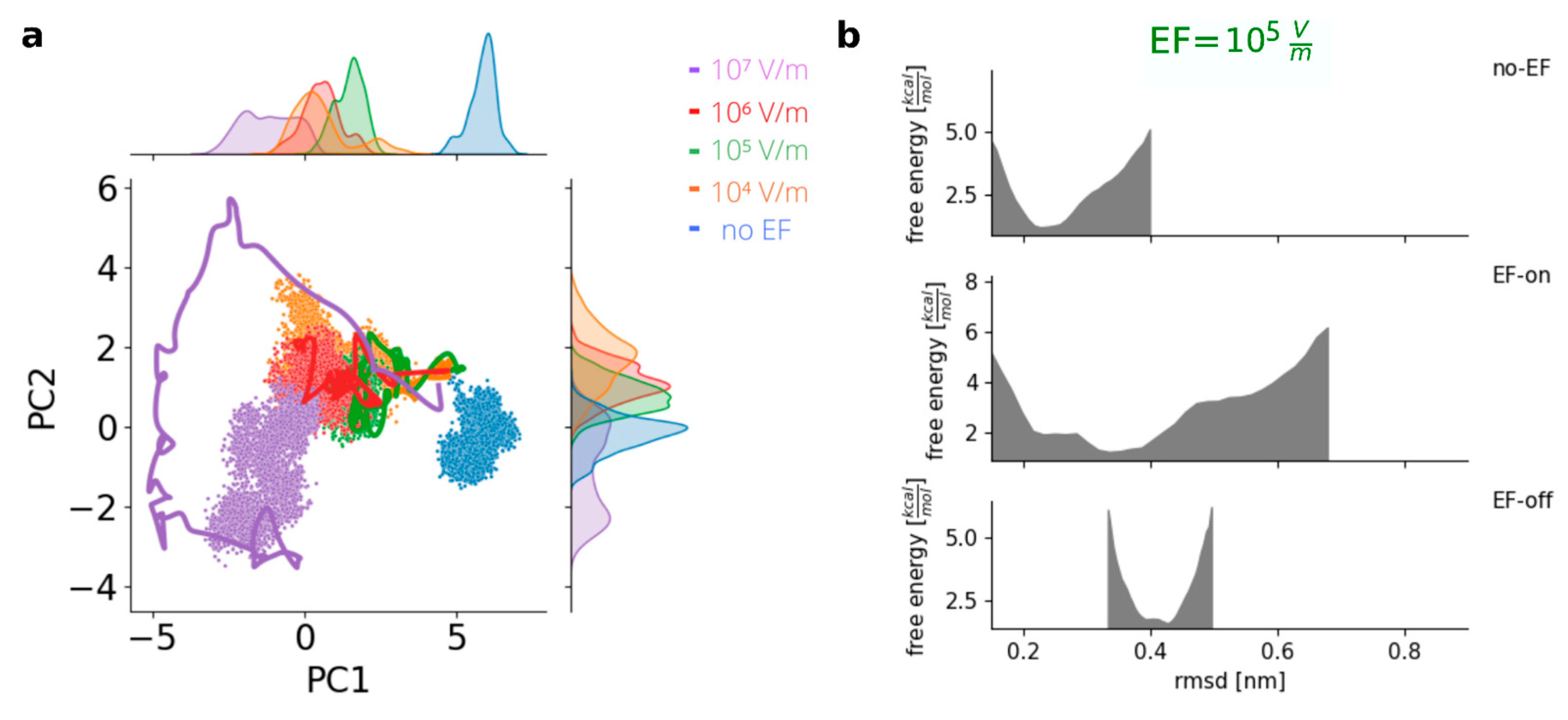
3.3. Stability of the Field-Induced Final Conformational States in the RDB Spike Protein
To gain insights into the conformational changes, a Principal Component Analysis (PCA) was performed on the EF-on trajectories to represent and visualize the protein states in a two-dimensional space obtained through dimensionality reduction. By considering a subspace defined by the two most relevant principal components for the run at EF = 107 V m−1, the trajectories for all EF-off runs were projected onto that plane (Figure 3a, Delta variant). It was observed that under the influence of EFs with different intensities, different paths in the phase space were explored by the protein. The final conformation at the end of each EF-on run was found to depend on the specific intensity of the applied field, suggesting field-dependent directions of movement in the high-dimensional space of internal coordinates. Once the external field was deactivated, during the EF-off runs, the protein structure remained in the vicinity of the EF-induced new conformations, and there was no return to the initial structures. Figure 3a, shows the points corresponding to the trajectories clustered around the final states, with almost no overlapping regions in the reduced phase space, in agreement with previous results [23].
To investigate the stability of the new, EF-induced conformations of the S protein, the EF was deactivated, and the simulations were continued in the absence of fields (“EF-off” run). On average, no more than 200 ns were required for each EF-off run, as the protein exhibited limited motion around the structure formed after the EF application for all EF intensities. The existence of a new stable minimum for all studied EF intensities was confirmed by the estimated free energy plots, in line with previously published, and the presence of an energy barrier preventing a return to the original conformation was also revealed. In Figure 3b, the middle panel shows both the initial state and the damaged state of the system, both under the influence of the EF. The free energy profile connecting both states in the absence of fields was also determined, clearly showing a barrier separating the two states. An observation worth noting at this point is that the minimum state achieved after applying the external field is not a transient or rapidly decaying metastate. Instead, it proves to be remarkably stable and long-lasting.
Figure 3.
Irreversible states are achieved in the Variant of Concern Delta after application of EF. (A) Principal Component Analysis shows distinct stable states after application of EF (lines: smoothed trajectory during EF, dots: states visited with EF-off). (B) Energy profile estimation of the prefusion conformation (PDB 7V8B) before application of EF (no-EF), during application of EF (EF-on) and after the application (EF-off), example for EF = 105 V m−1.
Figure 3.
Irreversible states are achieved in the Variant of Concern Delta after application of EF. (A) Principal Component Analysis shows distinct stable states after application of EF (lines: smoothed trajectory during EF, dots: states visited with EF-off). (B) Energy profile estimation of the prefusion conformation (PDB 7V8B) before application of EF (no-EF), during application of EF (EF-on) and after the application (EF-off), example for EF = 105 V m−1.
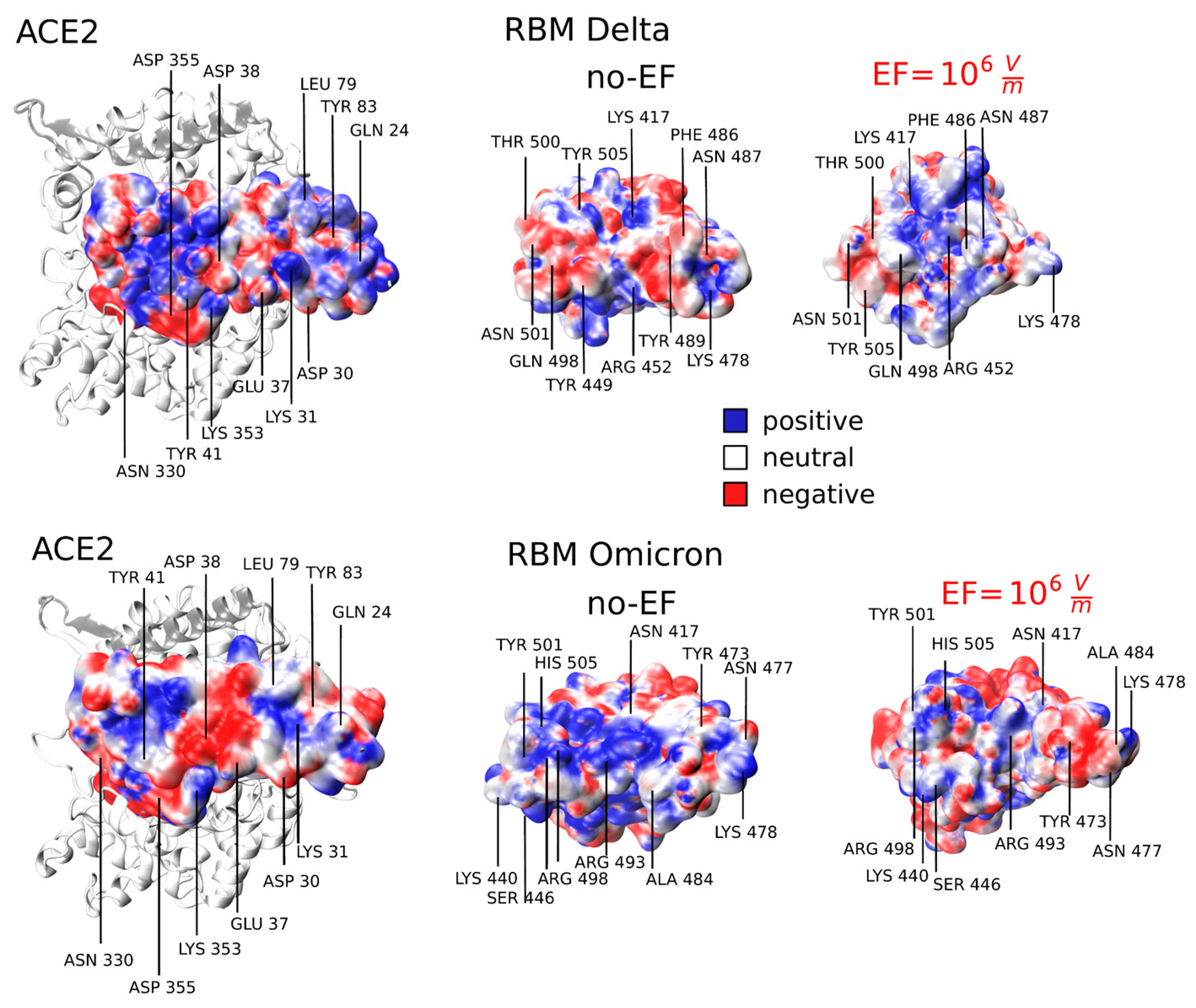
3.4. Disruption of the Charge Complementarity between RBD and ACE2 upon the Application of an Electric Field
Electrostatic complementarity in protein-protein interactions plays a crucial role in molecular recognition and binding processes, as more than 20% of all amino acids in proteins become ionized under physiological conditions, and polar groups are present in sidechains [51]. The results presented in the previous sections demonstrate that the changes in the secondary structure and atomic rearrangement in the S protein, induced by the EF, are likely to weaken its interaction with ACE2.
To investigate whether the EF-induced atomic reorganization affects the electrostatic potential landscape of the RBD, potentially leading to a reduced interaction with the ACE2 receptor, we conducted an in-depth analysis of the electrostatic potential (𝜙) within the Receptor Binding Motif (RBM). To calculate 𝜙, we utilized the Adaptive Poisson-Boltzmann Solver (APBS) algorithm, which allowed us to solve the Poisson-Boltzmann equations for continuum electrostatics. Before the analysis, we prepared the PDB formats using the PDB2PQR web server, converting them to PQR format with the CHARMM force field, and adjusted PROPKA to pH = 7.057. Subsequently, we performed the APBS analysis using the Linearized Poisson-Boltzmann Equation within the VMD software (see Methods).
Figure 4 shows a significant distortion in the spatial distribution of 𝜙 on the RBM when an EF of intensity 106 V m-1 is applied. This distortion is present in both the Delta and Omicron variants. Since mutations in SARS-CoV-2 variants lead to different interactions at the RBD:ACE2 interface, it is important to take into account the specific ACE2 binding surface for each variant. The mutations at the RBD interface induce significant perturbations in the van der Waals and electrostatic interactions for both the Delta and Omicron variants [52,53]. In the S protein of the Delta variant, the ACE2 binding surface exhibits two prominent positive patches attributed to residues Lys 31 and Lys 353, which precisely match with the corresponding negative regions on the RBD (Tyr 489, Gln 498, and Thr 500) [49]. The negative areas are characterized by polar and acidic residues (Gln 24, Tyr 83, Asp 355, and Gln 493), establishing a stronger electrostatic complementarity at the binding interface with Lys 417, Asn 487, and Lys 478 [52] (see Figure 4). In particular, the Thr 478 to Lys and Leu 452 to Arg mutations alter the electrostatic surface of the RBM replacing uncharged amino acids with positively charged ones [52]. During the thermalization process (no-EF run), no significant changes were observed in this region. However, upon the rearrangement of L3 resulting from the EF application, a shift in the negative region within the RBD is observed. Specifically, positively charged residues, primarily Arg 452, shift and match the positive part of ACE2, resulting in the generation of a repulsive force. Simultaneously, the region of the RBD opposite to Lys 31 of ACE2 exposes non-polar residues (Figure 4).
In the case of the Omicron variant, and as shown in Figure 4, mutations entirely alter its interaction profile. The considerable number mutations, 15 in the RBD, leads to an increase of the positive electrostatic potential in the RBM when compared to the WT and other variants, enhancing the affinity for interactions with the large negative (or neutral) regions of the ACE2 receptor [53]. The size of the positively charged regions in Omicron is significantly larger compared to Delta and WT, explaining why the electrostatic attractive forces are stronger in the Omicron RBD-ACE2 interaction compared to WT and other VOCs [53]. Upon application of an EF, we can observe a disruption in the central positive patch which results in a repulsive contact with the ACE2 surface. These findings on the electrostatic properties indicate that the surface charge distribution in the RBM is significantly altered by the EF, disrupting the electrostatic complementarity between the S protein and ACE2. Consequently, we can argue that EF leads to a potential disruption in the bonding between the RBD and ACE2, resulting in a reduction of their electrostatic affinity.
Figure 4.
Electrostatic potential at ACE2 and the Receptor Binding Motif (RBM) interface under EF conditions (EF = 0 and EF = 106 V m−1). Red, white, and blue colors represent negative, neutral, and positive charges, respectively (0.3 eV/-0.4 eV). It visualizes the disruption of charge complementarity between RBD and ACE2 upon EF application.
Figure 4.
Electrostatic potential at ACE2 and the Receptor Binding Motif (RBM) interface under EF conditions (EF = 0 and EF = 106 V m−1). Red, white, and blue colors represent negative, neutral, and positive charges, respectively (0.3 eV/-0.4 eV). It visualizes the disruption of charge complementarity between RBD and ACE2 upon EF application.
3.5. Influence of Electric Fields on the Fusion Core Region of the S2 Subunit of the Spike Protein
In the postfusion state, numerous strong interactions exist between the HR1 and HR2 domains within the helical section identified as the fusion core. The HR1 domains form a trimeric coiled-coil core that runs in parallel, surrounded by three HR2 domains in an antiparallel fashion [54]. We demonstrated above that EF application induces conformational changes in the tertiary structure of the postfusion S2 subunit, possibly destabilizing the 6HB structure. To better understand the structural effects of EF on the secondary structure in the fusion core, we analyzed HR1-HR2 interactions represented in single-chain mode. As shown in Figure 5, relevant interacting residues of HR1 that contribute to binding with HR2 through the formation of hydrogen bonds and salt bridges were conserved [46].
These results suggest that under EF, the secondary structure is not affected in the postfusion S2 subunit. However, the conservation of interactions between the HR1 and HR2 domains does not necessarily imply further stabilization of the 6-HB structure, which may lead to increased virus infectivity [55].
Figure 5.
Schematic representation of the S1 and S2 subunits within the SARS-CoV-2 Spike protein, along with a sequence alignment of the HR1 and HR2 domains. The segments used in this study have been distinctly highlighted using colors (upper panel). The core of the S2 subunit in the postfusion state (PDB 6LXT) and structural details of the HR1 and HR2 region interactions are presented in cartoon form, with HR1 highlighted in purple and HR2 in cyan (PDB 7COT). Important residues are indicated and labeled. The fusion core regions for each protomer remain conserved after the application of EF.
Figure 5.
Schematic representation of the S1 and S2 subunits within the SARS-CoV-2 Spike protein, along with a sequence alignment of the HR1 and HR2 domains. The segments used in this study have been distinctly highlighted using colors (upper panel). The core of the S2 subunit in the postfusion state (PDB 6LXT) and structural details of the HR1 and HR2 region interactions are presented in cartoon form, with HR1 highlighted in purple and HR2 in cyan (PDB 7COT). Important residues are indicated and labeled. The fusion core regions for each protomer remain conserved after the application of EF.
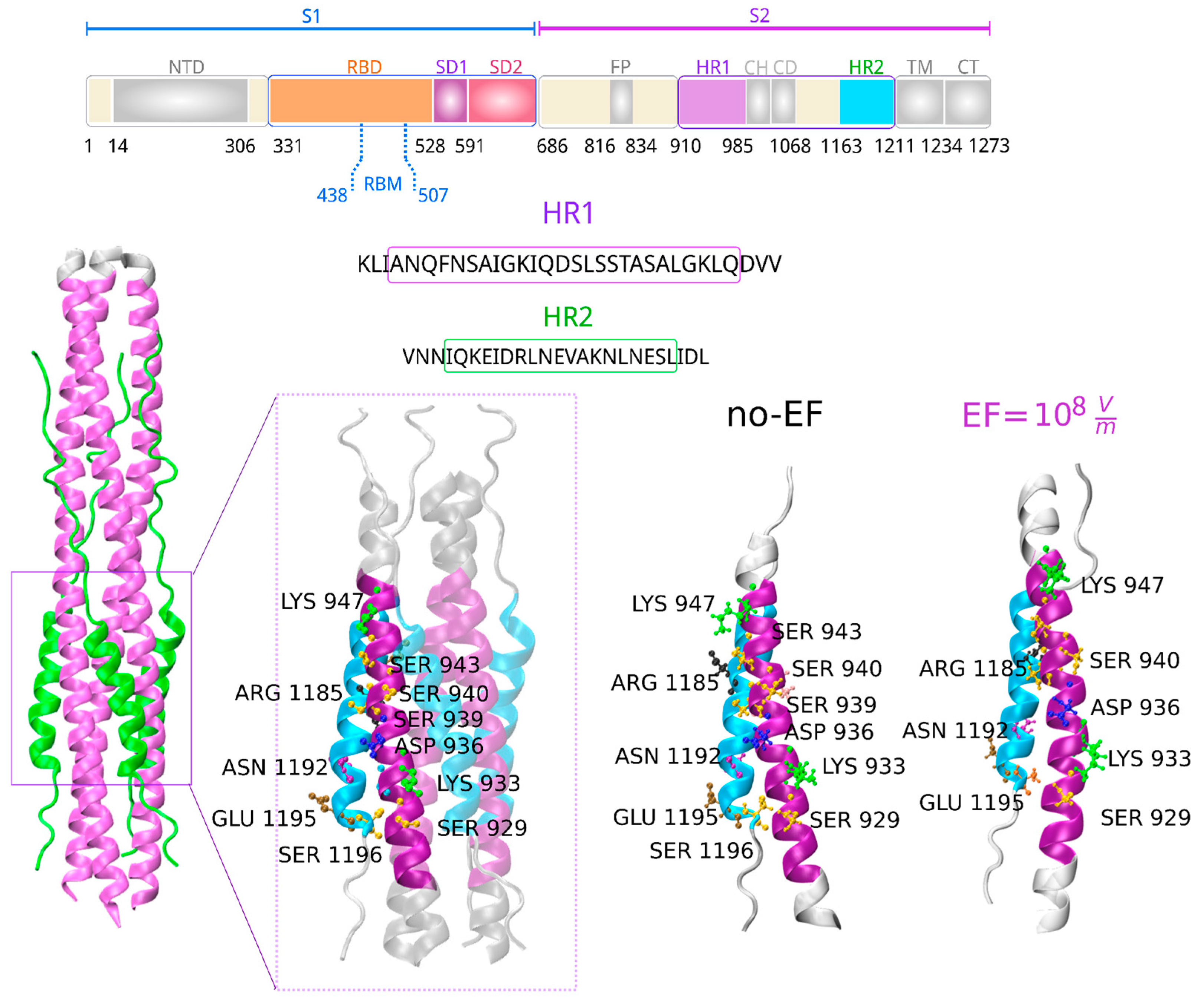
The fact that, according to our simulations, the postfusion conformation does not suffer irreversible damage in its secondary structure upon application of electric fields as high as 108 V m−1 is consistent with the widely accepted damage threshold between 5x108 V m−1 and 109 V m−1 for globular proteins, which was determined by different theoretical and experimental works. Notice also, that the alpha helices are much more robust and resistant to the application of electric fields, as has been analyzed in detail in Ref. [18]. It is therefore not surprising that the postfusion conformation is mainly composed of alpha helices, since this structure must withstand the electric field through the cell membrane, which typically has an intensity around 109 V m−1 .
4. Discussion
In this work, we studied the effects of external electric fields on the conformation of the SARS-CoV-2 spike protein using molecular dynamics simulations. We focused on the prefusion and postfusion conformations in order to characterize the differences in the stability and the vulnerability of both structures. Our study shows that the receptor binding domain of the prefusion conformation of the spike protein of the Delta and Omicron variants undergoes significant changes in the tertiary as well as the secondary structure under the application of EFs. In contrast, the postfusion conformation displays changes only in the tertiary structure, which can be attributed to its slender shape that contributes to its flexibility.
We characterized the changes in the structures by combining detailed analysis of the position and orientation of individual residues, and changes in the secondary structure of segments that participate in interaction with ACE2, together with global descriptors of the trajectories that are derived from the calculation of the free energy landscapes, dimensionality reduction and computation of the electrostatic potential in the surface of the protein.
A detailed analysis of the RMSF data revealed an increase in flexibility for L3 (residues 470 to 491) even in absence of fields for Delta variant (Figure 2c), similar findings were also obtained in previous works for other variants when compared to WT [48]. Under EF application, residues of the RBD that considerably increase their flexibility, particularly Val445, Phe456, Ile468, Phe486, and Thr500 (Figure 2c, dashed lines), along with a moderate increase in flexibility for the mutated residues Arg452 and Lys478 (Figure 2c, continuous lines), are critical and situated at the interface region to the ACE2 receptor. In the Delta variant, some of these residues have significant contributions to ACE2 binding through the formation of hydrogen bonds or salt bridges [56]. It is worth mentioning that, although Arg452 and Lys478 may not directly interact through bonds with ACE2, previous studies have highlighted their influence on the structural microenvironment of crucial interface residues, such as Gln493 and L3 [49,57,58]. A similar scenario occurs in the Omicron variant, where some of the mutations are not directly participating in the docking to ACE2, but may affect the microenvironment of interacting residues.
The analysis of the trajectories in the conformational space displayed by the proteins, which we performed via PCA and the calculation of the free energy landscapes along the visited states, reveal that the final states resulting from the application of EFs to the prefusion conformation are stable and clearly distinct from the initial structures. Follow-up work should address the comparison of the dynamics under EFs of the full Spike protein trimer in the prefusion and postfusion conformations, in order to identify the complete set of conformational changes and, in consequence, characterize the spatial- and residue-dependence of the susceptibility to EFs.
Overall, this work provides evidence that the reported vulnerability of the SARS-CoV-2 spike protein resides in its metastable characteristic, which is present in the prefusion but not in the postfusion conformation. Other class I viral fusion proteins are also metastable in their prefusion conformation, a fact that is described to participate in membrane fusion processes. This suggests the possibility that the reported vulnerability to EFs, which is demonstrated here for multiple variants of the SARS-CoV-2 spike protein, extends as a general feature in class I fusion proteins of other viruses.
Author Contributions
Conceptualization: M.E.G and C.A.; methodology: P.O.-M. and P.R; simulations: A.L. and C.A.; evaluation and analysis: A.L, C.A and P.R.; writing—original draft preparation: all authors; visualization, A.L., C.A. and P.R.; supervision, M.E.G, C.A. and P.O.-M..; project administration: M.E.G.; funding acquisition: M.E.G. All authors have read and agreed to the published version of the manuscript.
Funding
C.R.A. and M.E.G. acknowledge support by the PhosMOrg (P/1082 232) research unit of the University of Kassel. P.R. received support of the Joachim Herz Foundation through its Add-on Fellowship for Interdisciplinary Life Science. P.R. and M.E.G. received support from Zentrale Forschungsförderung (P2377) of the University of Kassel.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors thank Dr. Bernd Bauerhenne for his help with computing resources. Calculations for this research were performed on the Lichtenberg high-performance computer of the TU Darmstadt, the High Performance Computing Center North (HPC2N) at Umeå University, and at local dedicated workstations of our group.
Conflicts of Interest
The authors declare no conflict of interest.
References
- https://www.cdc.gov/coronavirus/2019-ncov/variants/.
- Tulimilli, S.V.; Dallavalasa, S.; Basavaraju, C.G.; Kumar Rao, V.; Chikkahonnaiah, P.; Madhunapantula, S.V.; Veeranna, R.P. Variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and vaccine effectiveness. Vaccines 2022, 10, 1751. [Google Scholar] [CrossRef]
- Pipitò, L.; Reynolds, C.A.; Mobarec, J.C.; Vickery, O.; Deganutti, G. A Pathway Model to Understand the Evolution of Spike Protein Binding to ACE2 in SARS-CoV-2 Variants. Biomolecules 2022, 12, 1607. [Google Scholar] [CrossRef]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nature Reviews Molecular Cell Biology 2003, 4, 309–319. [Google Scholar] [CrossRef]
- Benhaim, M.A.; Lee, K.K. New biophysical approaches reveal the dynamics and mechanics of type I viral fusion machinery and their interplay with membranes. Viruses 2020, 12, 413. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Current opinion in virology 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Ng, K.T.; Mohd-Ismail, N.K.; Tan, Y.J. Spike S2 subunit: the dark horse in the race for prophylactic and therapeutic interventions against SARS-CoV-2. Vaccines 2021, 9, 178. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Ranjan, A. The metastable states of proteins. Protein Science 2020, 29, 1559–1568. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef]
- Lee, C.; Park, S.H.; Lee, M.Y.; Yu, M.H. Regulation of protein function by native metastability. Proceedings of the National Academy of Sciences 2000, 97, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Agard, D.A. Kinetics versus thermodynamics in protein folding. Biochemistry 1994, 33, 7505–7509. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Lu, L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell research 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; McLellan, J.S. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S. ,... & Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Infez Med 2020, 28, 174–184. [Google Scholar] [PubMed]
- Hekstra, D.R.; White, K.I.; Socolich, M.A.; Henning, R.W.; Šrajer, V.; Ranganathan, R. Electric-field-stimulated protein mechanics. Nature 2016, 540, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-May, P.; Garcia, M.E. Electric field-driven disruption of a native β-sheet protein conformation and generation of a helix-structure. Biophysical journal 2010, 99, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shao, D.; Xu, P.; Jiang, Z. Effects of an electric field on the conformational transition of the protein: Pulsed and oscillating electric fields with different frequencies. Polymers 2021, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Urabe, G.; Katagiri, T.; Katsuki, S. Intense pulsed electric fields denature urease protein. Bioelectricity 2020, 2, 33–39. [Google Scholar] [CrossRef]
- Baumketner, A. Electric field as a disaggregating agent for amyloid fibrils. The Journal of Physical Chemistry B 2014, 118, 14578–14589. [Google Scholar] [CrossRef]
- De Biase, P.M.; Paggi, D.A.; Doctorovich, F.; Hildebrandt, P.; Estrin, D.A.; Murgida, D.H.; Marti, M.A. Molecular basis for the electric field modulation of cytochrome c structure and function. Journal of the American Chemical Society 2009, 131, 16248–16256. [Google Scholar] [CrossRef] [PubMed]
- Arbeitman, C.R.; Rojas, P.; Ojeda-May, P.; Garcia, M.E. The SARS-CoV-2 spike protein is vulnerable to moderate electric fields. Nature communications 2021, 12, 5407. [Google Scholar] [CrossRef] [PubMed]
- https://www.rcsb.org/.
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. Journal of computational chemistry 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell Jr, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. Journal of chemical theory and computation 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Mackerell Jr, A.D.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. Journal of computational chemistry 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of chemical theory and computation 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: fast, flexible, and free. Journal of computational chemistry 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R. ,... & Lindahl, E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. The Journal of chemical physics 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. Journal of chemical theory and computation 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins: Structure, Function, and Bioinformatics 2005, 58, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. The Journal of chemical physics 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Fabian, P. Scikit-learn: Machine learning in Python. Journal of machine learning research 2011, 12, 2825. [Google Scholar]
- Calvo, F. Sampling along reaction coordinates with the Wang-Landau method. Molecular Physics 2002, 100, 3421–3427. [Google Scholar] [CrossRef]
- Adjanor, G.; Athenes, M.; Calvo, F. Free energy landscape from path-sampling: application to the structural transition in LJ 38. The European Physical Journal B-Condensed Matter and Complex Systems 2006, 53, 47–60. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, Academic Press 2002, ISBN 0-12-267351-4.
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. Journal of computational chemistry 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. Journal of molecular graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic acids research 2004, 32 (suppl_2), W665–W667. [Google Scholar] [CrossRef]
- Spiga, O.; Bernini, A.; Ciutti, A.; Chiellini, S.; Menciassi, N.; Finetti, F.; Niccolai, N. Molecular modelling of S1 and S2 subunits of SARS coronavirus spike glycoprotein. Biochemical and biophysical research communications 2003, 310, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H. Crystallographic and biophysical analysis of the fusion core from SARS-CoV-2 spike protein. Journal of the Chinese Chemical Society 2023. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Lu, L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell research 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Saltalamacchia, A.; Magistrato, A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the hallmark for its enhanced infectivity? Insights from all-atom simulations. The journal of physical chemistry letters 2020, 11, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.; Bhattarai, N.; Hossen, M.L.; Stebliankin, V.; Gerstman, B.S.; Narasimhan, G.; Chapagain, P.P. Mutation-induced changes in the receptor-binding interface of the SARS-CoV-2 Delta variant B. 1.617. 2 and implications for immune evasion. Biochemical and biophysical research communications 2021, 574, 14–19. [Google Scholar] [CrossRef]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Hung, A.; Karagiannis, T. Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain. Computers in Biology and Medicine 2022, 149, 106035. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS pathogens 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Gilson, M.K. Theory of electrostatic interactions in macromolecules. Current opinion in structural biology 1995, 5, 216–223. [Google Scholar] [CrossRef]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular dynamics simulations of the delta and omicron SARS-CoV-2 spike–ACE2 complexes reveal distinct changes between both variants. Computational and structural biotechnology journal 2022, 20, 1168–1176. [Google Scholar] [CrossRef]
- Sang, P.; Chen, Y.Q.; Liu, M.T.; Wang, Y.T.; Yue, T.; Li, Y.; Yang, L.Q. Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. International Journal of Molecular Sciences 2022, 23, 14796. [Google Scholar] [CrossRef]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nature communications 2020, 11, 3618. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Jiang, S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. The Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Wang, P. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Zhao, X.; Luo, S.; Duan, L. Insights from computational analysis: how does the SARS-CoV-2 Delta (B. 1.617. 2) variant hijack ACE2 more effectively? Physical Chemistry Chemical Physics 2022, 24, 8683–8694. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The global epidemic of the SARS-CoV-2 delta variant, key spike mutations and immune escape. Frontiers in immunology 2021, 12, 751778. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



