
Submitted:
02 November 2023
Posted:
02 November 2023
You are already at the latest version
Abstract
Akebia trifoliata is an economically important self-incompatible fruit tree in the family Lardiza-balaceae. Asexual propagation is the main strategy used to maintain excellent agronomic traits. However, the generation of adventitious roots during asexual propagation is very difficult. To study the important role of the WUSCHEL-related homeobox (WOX) transcription factor in fruit and adventitious root growth and development, we characterized this transcription factor family in the whole genome of A. trifoliata. A total of 10 AktWOXs were identified, with the fol-lowing characteristics: length (657~11328 bp), exon number (2~5), isoelectric point (5.65~9.03), amino acid number (176~361 AA), and molecular weight (20.500~40.173 kDa). 10 AktWOXs were classified into modern (6), intermediate (2), and ancient clades (2) and that all AktWOXs had undergone strong purifying selection during evolution. The expression profile of AktWOXs dur-ing A. trifoliata fruit development and adventitious root formation indicated that AktWOXs are widely involved in the development of the three fruit tissues, flesh, seeds and rind, and play an important role in the regulation of adventitious root development. Overall, this is the first study to identify and characterize the WOX family in A. trifoliata and will be helpful for further re-search on A. trifoliata fruit development and adventitious root formation.
Keywords:
Akebia trifoliata
; WUSCHEL-related homeobox
; transcription factor
; adventitious roots.
1. Introduction
Akebia trifoliata (Thunb.) Koidz. (2n=2x=32) belongs to the flowering plant family Lardizabalaceae [1]. As the third generation of emerging fruit, the flesh of A. trifoliata, which is deeply loved by people, not only has a delicate texture and sweet taste but also contains many free essential amino acids[2]. Therefore, the artificial cultivation of A. trifoliata has been rapidly increasing in Southwest China and the middle and lower reaches of the Yangtze River in recent years. However, A. trifoliata breeding techniques are not yet able to meet the needs of farmers because A. trifoliata is a cross-pollinated plant and good maternal traits can only be maintained through asexual reproduction [3].
To date, research on asexual breeding methods for A. trifoliata has mainly focused on tissue culture breeding and cutting breeding. In the exploration of tissue culture, Wu et al. (2015) established and optimized an efficient callus culture system using leaves as explants and established a method for the rapid propagation of stems with leaf buds, with induction and rooting rates of more than 80% in the optimized medium [4]. This method can shorten the seedling cycle of seedlings. However, since the formation of endogenous toxins and adventitious roots of A. trifoliata is hard to achieve, it is still difficult to establish a complete tissue culture system of A. trifoliata[5]. The method of culturing cuttings of A. trifoliata has also attracted the attention of researchers. Studies have shown that fine river sand + nutrient soil is the preferred medium for cuttings of A. trifoliata[6]. In addition, plant growth regulators are also used to promote the growth of A. trifoliata roots. There is a report showing that ABT2 rooting- powder treatment can effectively promote the rooting of A. trifoliata cuttings [7]. In China, the A. trifoliata cutting system has slowly begun to mature. The unstable roots produced by cuttings and tissue culture are liable to fall off and do not easily survive field transplantation. The root system plays a crucial role in the growth and development of the whole plant. Therefore, the study of the formation and development process of adventitious roots helps us to obtain high-quality saplings of A. trifoliata.
The formation of adventitious roots is one of the key steps of plant asexual propagation [8], and the WUSCHEL-related homeobox (WOX) transcription factors widely present in plant genomes have been shown to be involved in the regulation of adventitious root formation [9]. For example, in Arabidopsis thaliana, WOX11 and WOX12 respond to auxin induction and then activate the expression of WOX5 and WOX7 to change the cell fate from root invasive cells to root primordium cells and achieve adventitious root regeneration [9,10]; in Oryza sativa, OsWOX3A leads to an increase in plant lateral root number, indicating that OsWOX3A may be involved in the regulation of GA-IAA crosstalk in rice root development 11; in the gymnosperms Picea-Abies and Populus nigra, PsWOX3 is expressed in a few cells on the peripheral surface of the shoot apical meristem, and PaWOX3 is highly expressed in the root tip [12,13]. Overexpression of MdWOX11 promotes adventive root primordium formation in apple, while interference of MdWOX11 inhibits adventive root primordium production 1414. Therefore, some members of the WOX transcription factor family play important roles in the growth and development of adventitious roots.
At present, genome-wide identification of the WOX transcription factor family has been completed in many plants. The WOX family is a group of plant-specific transcription factors and belongs to the homeobox (HB) transcription factor family. The typical homeodomain (HD) of the HB superfamily has 60-66 amino acid residues that fold into a “helix−loop−helix−turn−helix” spatial structure, where a combination of the second and third helices forms a “helix−turn−helix” that can bind to specific DNA sequences 15. WUSCHEL (WUS) is the most primitive gene of the WOX transcription factor family. In 2004, Haecker et al. identified 14 other members with similar structures by using homology search methods for A. thaliana WUS genes 16. According to phylogenetic tree analysis, it can be divided into three clades: the first clade is the modern/WUS clade (WUS, AtWOX1-AtWOX7), which exists in higher plants; the second clade is the intermediate clade (AtWOX8, AtWOX9, AtWOX11 and AtWOX12), which originates from tracheophytes; and the third clade is the ancient clade (AtWOX10, AtWOX13 and AtWOX14), which originated from phycophyta [17,18]. The ancient origin of the WOX transcription factor and other evolutionary branches derived from plant evolution suggest that this gene family is essential for plant survival.
In the present study, we comprehensively identified the WOX genes from the A. trifoliata genome. We first determined the AktWOX gene structures, motif compositions, and chromosomal distributions. Furthermore, we analyzed the phylogenetic relationships and evolutionary patterns of the AktWOXs. In addition, the expression patterns of AktWOXs in different A. trifoliata tissues and under different AR formation conditions were determined. Our results provide insights for further understanding WOX family genes in A. trifoliata, clarify their evolutionary history, and facilitate their application in gene transformation for improving plants.
2. Results
2.1. Systemic Characterization of the WOX Gene Family in A. trifoliata
A total of 10 WOX genes were identified from the A. trifoliata genome through HMM analysis. They were sequentially named AktWOX1–9 and AktWUS (chromosome 2) (Table 1) according to their positions on the chromosome 19. The 10 AktWOXs had a wide range in gene length (from 657 bp to 11328 bp) and exon number (from 2 to 5). In terms of protein properties, the 10 AktWOXs had obvious differences in amino acid length (from 176 to 361), molecular weight (from 20.500 to 40.173), and isoelectric point (from 5.65 to 9.03). Subcellular localization analysis showed that these proteins were spatially located in the nucleus but had no obvious signal peptide signature.
The secondary structure of 10 AktWOX proteins was predicted and analyzed (Table S1). The α-helical structure and β-folded structure are ordered structures of proteins that have high stability, and random curling is a disordered structure of proteins. The results showed that 10 AktWOX proteins were mainly randomly curled, accounting for 56.28% to 71.75% of the secondary structure, followed by α helices. This indicated that the protein secondary structure of the AktWOX family genes was unstable as a whole. The instability coefficient of the proteins in this family was greater than 40, and the hydrophilicity value was less than 0, indicating that they were poorly stable and hydrophilic proteins.
2.2. Phylogenetic Analysis of AktWOX
A phylogenetic tree of the WOX protein family was constructed based on the amino acid sequences of 39 WOX proteins from A. trifoliata (10), O. sativa (14) and A. thaliana (15). According to the evolutionary tree, the 39 WOX proteins were divided into three main branches, and the 10 AktWOXs of A. trifoliata were unevenly distributed into the three branches (Figure 1). Among them, six WOX members were assigned to the modern clade, including AktWUS, AktWOX9, AktWOX6, AktWOX3, AktWOX7, and AktWOX5. AktWOX1 and AktWOX8 were assigned to intermediate clades, and AktWOX2 and AktWOX4 were assigned to ancient clades.
2.3. Gene Structure and Conserved Motifs of AktWOXs
Domain analysis showed that 10 AktWOX protein sequences had conserved HD and WUS-box domains (Figure S1). Further motif analysis showed that the 10 AktWOX proteins contained 10 relatively conserved motifs (Table S2). Motif 1 and motif 2 were found in all 10 AktWOXs and contained a highly conserved "helix-ring-helix-corner-helix" HD domain (Figure S2). Motif 5 was the WUS-box motif (Figure S3) and existed in AktWUS, AktWOX9, AktWOX6, AktWOX3, AktWOX7, and AktWOX5 (modern evolution branch) (Figure S2). Motif 7 existed only in AktWOX1 and AktWOX8 (intermediate clades), while motif 4 and motif 8 existed only in AktWOX2 and AktWOX4 (ancient clades).
The analysis of its exon and intron structure revealed that the AktWOX gene contained 2-4 CDSs (coding DNA sequences); AktWOX3, AktWOX5, AktWOX6 and AktWOX9 contained 2 CDSs; AktWOX7 had 4 CDSs; and AktWUS, AktWOX3 and 4 members of the middle branch and ancient branch contained 3 CDSs (Figure 2c).
2.4. Chromosomal Location and Evolutionary Analyses of AktWOXs
Chromosomal location analysis showed that 10 AktWOXs were distributed on 7 chromosomes of A. trifoliata (Figure 3), and two AktWOXs were located on chromosome 15. The remaining five AktWOXs are found on chromosomes 1, 3, 6, 8, and 9.
In terms of evolution, intraspecies collinearity analysis showed that dispersed and segmental or whole-genome duplication (WGD) events were the main sources of AktWOX expansion (Figure 3), but the majority (8; 80%) AktWOXs were derived by dispersed replication, and the minority (2; 20%) AktWOXs were derived by WGD events.
To further understand the gene duplication mechanism of the WOX gene family in A. trifoliata, a comparative map was generated with the dicotyledonous plants A. thaliana, Liriodendron tulipifera, Populus x canescens, Solanum lycopersicum, Glycine max, Solanum tuberosum and Amborella trichopoda and the monocotyledonous plants O. sativa, Zea mays, and Andropogon gerardi. They were analyzed with the Chlorophyta plant Chlamydomonas reinhardtii (Figure 4). The number of homologs between A. trifoliata and A. thaliana was 8, L. tulipifera was 12, Populus x canescens was 18, S. lycopersicum was 7, G. max was 19, S. tuberosum and A. trichopoda were 6 and 6, the monocotyledonous plant O. sativa was 3, Z. mays was 5, A. gerardi was 14, and the chlorophyta plant C. reinhardtii did not contain any homologs, indicating a strong direct homology between the A. trifoliata WOXs and the dicotyledons members, which showed a high degree of evolutionary divergence compared with the monocotyledons.
Determining the Ka/Ks ratio can effectively improve the understanding of the evolutionary constraints of the WOX gene family. The Ka/Ks values of all 45 homologous AktWOX pairs were much lower than 1 and varied from 0.01 to 0.33 (Table S3), indicating that the AktWOXs could have experienced strong purifying selection during their evolutionary history.
2.5. Identification of Cis-Acting Elements of the AktWOX Gene Family
The cis-element analysis results of the upstream sequence of AktWOXs are shown in Figure 5. The types of AktWOX cis-elements included hormone-responsive elements and environment-responsive elements, and each element had 5 and 7 subtypes, respectively.
There were three cis-acting elements related to stress resistance: defense and stress response elements (13), low-temperature induction response elements (8), light responsiveness elements (131), anaerobic induction elements (22), zein metabolism regulation elements (9), elements involved in endosperm expression (9) and elements involved in flavonoid biosynthesis genes (2). Cis-acting elements related to hormone regulation mainly included auxin (7), gibberellin (11), abscisic acid (33), MeJA responsiveness elements (42) and salicylic acid response elements (8). Cis-acting elements related to substance synthesis: elements involved in metabolic regulation of zein, elements involved in endosperm expression, and MYB-binding site elements involved in flavonoid biosynthesis genes.
Both the type and the number of cis-acting elements also widely varied among members of the AktWOXs (Table S5). We found that every AktWOX had a light-responsive element with numbers ranging from 6 to 19, and AktWOX5 and AktWUS had the most (51) and least (17) cis-acting elements, respectively. The number of cis-acting element subtypes varied from 6 (AktWUS, AktWOX8 and AktWOX9) to 9 (AktWOX4, AktWOX6 and AktWOX7), and the AktWOX3 genes contained seven cis-acting element subtypes, and the AktWOX1, AktWOX2 and AktWOX5 genes contained eight cis-acting element subtypes.
2.6. GO Enrichment Analysis of AktWOX Genes
The 10 AktWOX genes were divided into three categories, molecular functions (MFs), cellular components (CCs), and biological processes (BPs), by GO enrichment analysis, with 9, 12, and 183 subcategories (Table S7), respectively (Figure 6). Eight AktWOX genes were involved in MFs, such as transcriptional regulatory activity and DNA-binding transcription factor activity; four were involved in CCs; and seven were involved in BPs, such as RNA biosynthesis, the regulation of cell metabolism, the regulation of biosynthesis, nucleic acid metabolism, and transcription regulation.
2.7. AktWOXs Expression in Fruit and Root of A. trifoliata
The 10 AktWOXs were grouped into four groups based on their relative expression levels: (A) they were expressed at different developmental stages and in different tissues, and their expression levels were higher in the overall expression level (AktWOX2, AktWOX4); (B) they were tissue-specific, expressed almost exclusively at the seed growth stage (AktWUS, AktWOX8), at the third flesh growth stage (AktWOX7), and in the rind and flesh of the fruit (AktWOX3); and (C) they showed low or no expression throughout the developmental stages (AktWOX5, AktWOX6, AktWOX9, AktWOX1). It has been speculated that genes in group A may positively regulate fruit growth and development in A. trifoliata, while genes in group C may play a minor role in the growth and development of the flesh, seed, and rind and may even have a negative regulatory effect.
Sequence homology alignment revealed that six AktWOX genes (AktWOX1, AktWOX2, AktWOX3, AktWOX4, AktWOX8, and AktWOX9) were homologous to AtWOX4, AtWOX5, AtWOX7, AtWOX9, AtWOX11, AtWOX12, AtWOX13, and AtWOX14 in Arabidopsis 20. We further examined the expression of these six genes during the growth of adventitious roots of A. trifoliata. These genes are reportedly related to root growth and development.
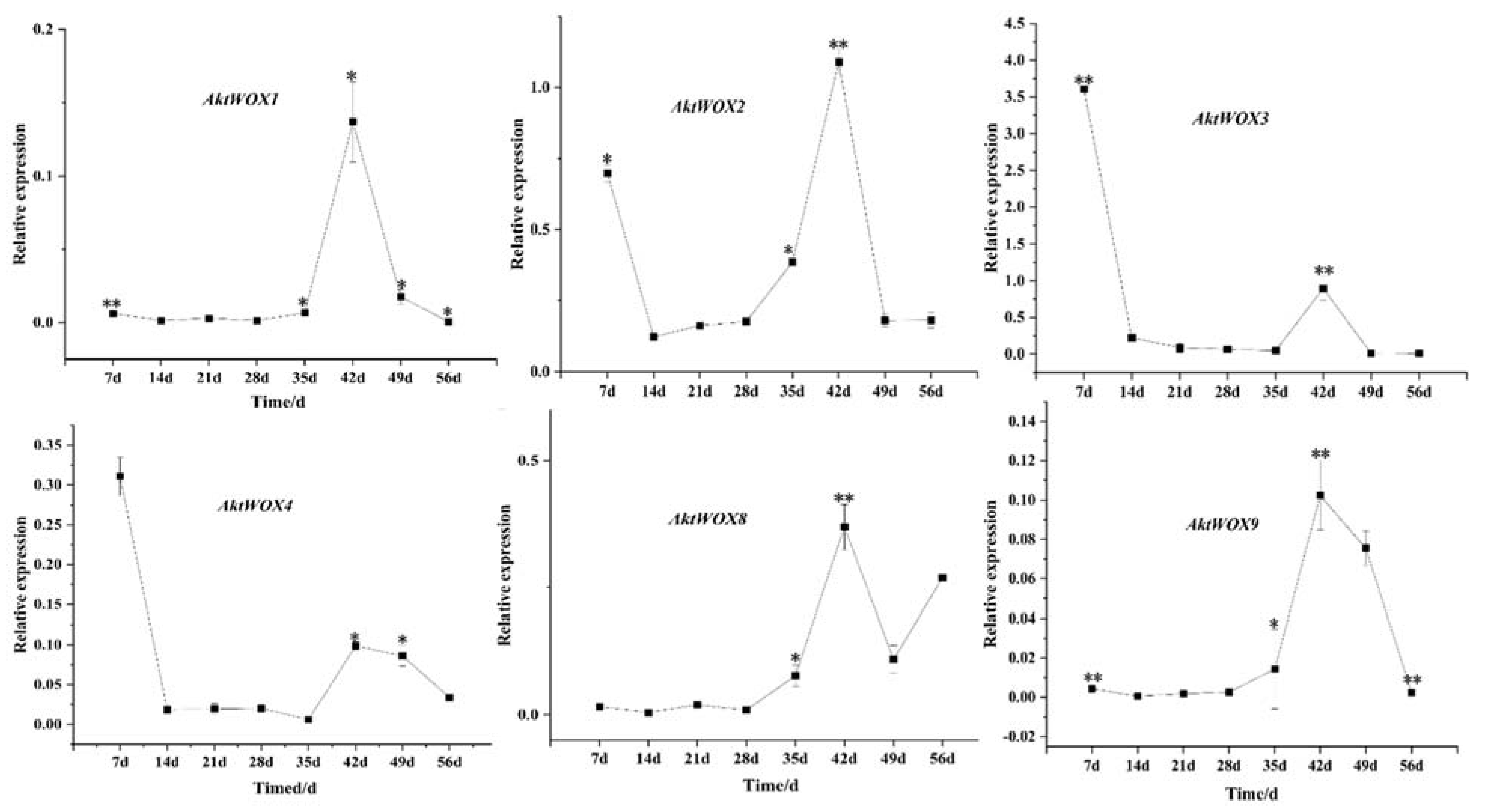
The RT‒qPCR results showed that the expression of the WOX genes during adventitious root formation of AktWOX1, AktWOX2, AktWOX8 and AktWOX9 increased to the highest values at 42 d of development, and then their expression gradually decreased to below the initial levels. Their expression increased at 28 d and decreased slightly with increasing development time but was still above the initial level. AktWOX2, AktWOX3 and AktWOX4 expression patterns were similar throughout adventitious root formation, with their expression decreasing at the beginning of development, being rapidly upregulated at 35 d and continuing until 42 d, after which their expression again decreased.
Figure 8.
qRT−PCR analysis of the expression of some AktWOXs. Transcript levels of AktWOXs were calculated using the 2−∆∆Ct method. * represents the significance between peak expression (**, p < 0.01).
Figure 8.
qRT−PCR analysis of the expression of some AktWOXs. Transcript levels of AktWOXs were calculated using the 2−∆∆Ct method. * represents the significance between peak expression (**, p < 0.01).
3. Discussion
3.1. The AktWOX gene structure is extremely conserved during evolution
In plants, the WOX family is an extremely important gene family, and the proteins it encodes are involved in the growth and development of almost every organ of angiosperms 21. As an increasing number of plant genomes are sequenced and released, many WOX genes in angiosperms have been systematically identified and studied. Enriching the number of reported WOX genes in basal dicots will further improve our understanding of the evolution of the WOX gene family 20. In this study, we identified 10 WOX genes from the genome of the basal dicot A. trifoliata. We found that although the AktWOX genes have wide differences at the DNA sequence level, mainly reflected in the number of introns and sequence length, the physical and chemical properties of the proteins they encode are extremely similar, including similar protein lengths and molecular weights and the instability and hydrophilicity of the protein structure (Table 1). Moreover, these characteristics of WOX genes in A. trifoliata are very similar to those of monocots, including wheat 15 and four Euphorbiaceae plants 22, as well as core dicots, including A. thaliana 16 and G. max 23. This indicates that WOX genes are highly conserved during evolution, and the gain and loss of introns/exons are the driving forces for the evolution of this gene family.
From an evolutionary perspective, genes duplicated by different mechanisms, such as WGDs, and tandem and dispersed duplications, are primary raw materials for new gene origins and evolution and ultimately result in functional novelty and specialization Error! Reference source not found.. Some studies have shown that following WGD events, genes encoding TFs are preferentially retained 24. Two WGD events occurred in A. trifoliata approximately 85 and 140 million years ago (ɵ event), respectively. The former is a specific WGD event in A. trifoliata, and the latter occurred during the early stages of dicotyledonous plant differentiation (ɵ event) 26. In this study, 8 (80%) of 10 identified AktWOXs were found to be derived from dispersed duplication, 2 AktWOXs were found to be derived from WGD (Figure 3), which suggested that dispersal was the major force of AktWOX origin, and the AktWOX gene family was involved in only one WGD event. We reconstructed the evolution of twelve species over time (Figure S4) and showed that the AktWOX gene family was involved in a specific genome-wide duplication event in A. trifoliata. In addition, the fact that all Ka/Ks values of the homologous AktWOX pairs were much lower than 1 (Table S4) further suggested that all AktWOXs experienced strong purifying selection during their evolutionary history. The Ka/Ks value of two combinations between AktWUS and both AkWOX2 and AktWOX4 was very close to 0.004, while the combination (AktWUS and AktWOX8) with the largest Ka/Ks value was also related to AktWUS (Table S3), which indicated that AktWUS could be an ancestral gene of the AktWOX family. This evolutionary evidence further demonstrates that AktWOXs are highly conserved.
3.2. AktWOX Gene Family Members May Have Greatly Diverged Functions
Many reports have confirmed that WOX transcription factors play important roles in regulating plant growth and development, including embryonic development, maintenance of meristematic stem cells, seed formation, regeneration of isolated tissues and organs, and response to abiotic stress. For instance, WOX genes play different roles in the development of O. sativa roots, stems, and leaves 27. The OsWOX6 gene plays a major role in the regulation of seed development, especially for the growth and development of seeds under water-deficient conditions 28. WOX genes are widely involved in the growth and development of different plant organs as well as physiological and biochemical processes, but their protein structure and gene number are very conserved, which indicates that this gene family has extensive functional differentiation. In this study, sequence analysis of the AktWOX promoter results showed that AktWOXs not only play important roles in the response to light signals and resistance to stress but also play a role in endosperm or seed development and meristem formation. GO enrichment analysis indicated seven AktWOXs widely involved in various growth development and tissue metabolism processes in biological processes of A. trifoliata. The results indicate that the AktWOX gene family is functionally diverse.
In addition, our transcriptomic data analysis revealed that some AktWOXs in the whole fruit, such as AktWOX1, AktWOX5, AktWOX6 and AktWOX9, exhibited very low expression, while some AktWOXs, such as AktWOX2 and AktWOX4, exhibited high expression. In addition, AktWUS, AktWOX2, AktWOX4, and AktWOX8 had seed-specific expression patterns, while AktWUS, AktWOX7, and AktWOX8 had developmental stage-specific patterns. These results indicate that AktWOXs may be functionally diverse. The HD and WUS-box domains are two conserved domains of the WOX family 16. At present, research on the WUS-box domain is mainly based on the WUS gene. In the process of WUS participating in maintaining the characteristics of stem cells in the plant stem meristem, the WUS-box mainly exerts inhibitory activity and maintains the dynamic balance of stem cell proliferation regulation 29. Studies have shown that the WUS box plays an important role in the maintenance of stem cell characteristics 29. Therefore, the modern branch of AktWOXs may be involved in regulating the development of stem cells. This is further evidence that members of the AktWOX gene family may have wide functional differences.
3.3. The AktWOX Gene may be Involved in adventitious root Regulation
The WOX gene family is widely involved in the formation of adventitious roots. In A. thaliana, AtWOX4, AtWOX5, AtWOX7, AtWOX9, AtWOX11, AtWOX12, AtWOX13, and AtWOX14 are associated with root growth and development 20, while there are no homologous genes for AtWOX7, AtWOX12, and AtWOX14 in AktWOX. De novo root organogenesis from tissue explants requires consecutive cell fate transition steps to finally form an adventitious root. The first step of cell fate transition is priming, which results in the formation of adventitious root founder cells. The second step of cell fate transition is initiation, which results in the formation of the dome-shaped root primordium via cell division. The expression levels of AtWOX11/12 decrease and those of AtWOX5/7 increase as the root founder cells transition into the root primordium 9. In the formation of adventive roots of A. trifoliata, AktWOX2 and AktWOX8 were highly expressed in the late stage, and AktWOX3 and AktWOX4 were highly expressed in the early stage. This result, which is similar to that of Hu et al. (2016), may indicate that AktWOX3 and AktWOX4 are related to the initiation of adventitia root cells in A. trifoliata, while AktWOX2 and AktWOX8 may be related to the initiation of adventitious root cells.
4. Materials and Methods
4.1. Identification and Physicochemical Characterization of AktWOX Sequences
To identify WOX genes in the A. trifoliata genome, hidden Markov model (HMM) ID PF00046 files downloaded from the Pfam database (http://smart.embl.de/smart/batch.pl, accessed on 28 December 2021) were used to filter protein sequences of A. trifoliata with the 10e-5 e-value parameter 30. After obtaining the A. trifoliata WOX genes, the AktWOX protein sequence was submitted to the conserved domain database (CDD: https: //www.ncbi.nlm.nih.gov/Structure/bwrpsb/BWRPSB. Cgi) for structural domain filtering to determine the final implant AktWOX transcription factor family members 31. Gene position on chromosomes and collinearity mapping using TBtools software 30.
The ExPASy's ProtParam online tool (http://www.ExPASy.org/tools/protparam.html/) was used to predict the physical and chemical properties of the AktWOX transcription factors 32. We used SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/) 33 to predict the secondary structure of the WOX gene in A. trifoliata. DataProtComp9.0 (http://linux1.softberry.com/berry.phtml?topic=protcomppl&group=Programs&subgroup=proloc) and SignalIP5.0 (http://www.cbs.dtu.dk/services/SignalP/) were used for subcellular localization and signal peptide prediction 34.
4.2. Sequence Characteristic Analysis, Phylogenetic Analyses, GO Enrichment Analysis and Collinearity of AktWOXs
Multiple alignments of the full-length protein sequences were executed by using ClustalW (https://www.genome.jp/tools-bin/clustalw, accessed on 20 May 2023). A phylogenetic tree was constructed using MEGA 11 software (version 11.0.10) via the maximum likelihood (ML) method with 1000 bootstrap replicates [35,36,37,38]. The GFF3 file of the A. trifoliata genomic annotation was used to analyze the gene sequence characteristics. The Gene Structure Display Server (http://gsds.gao-lab.org/, accessed on 20 May 2023) was used to count the number and location of exons/introns of the AktWOXs. The conserved motifs of the A. trifoliata proteins were analyzed by MEME Suite (https://meme-suite.org/meme/tools/meme, accessed on 20 May 2023) 39, where the maximum motif number was set to 10 and the other settings were set to their default values. The above results were subsequently visualized by TBtools 30 software (version 1.0876). To display the evolutionary selection pressure between collinear gene pairs 40, the Ka/Ks ratio was calculated by TBtools 30 software (version 1.0876). The reference genome sequences of A. thaliana, L. tulipifera, Populus x canescens, S. lycopersicum, G. max, S. tuberosum and A. trichopoda, the monocotyledonous plants O. sativa, Z. mays, and A. gerardi, and the Chlorophyta plant C. reinhardtii (accession on 9 August 2023). We downloaded data from the NCBI database and used them to perform a collinearity analysis with the sequence of A. trifoliata. The PlantCARE online website (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 26 May 2022) was then used to analyze the cis-acting elements in the 2000 bp promoter region upstream of A. trifoliata 42. Timetree5 (http://timetree.org/) was used to reconstruct the evolution of twelve species over time 43. The Metascape (Metascape.org) web-based portal was used for comprehensive gene annotation and analysis resources 44. A bubble chart was plotted using the Bioinformatics (www.bioinformatics.com.cn) free online platform for bioinformatics-related data analysis 44.
4.3. Expression analysis of AktWOXs in fruit development
The transcriptomic data of A. trifoliata were downloaded from the NCBI database under BioProject ID PRJNA671772 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA671772; accessed on 25 April 2023). The A. trifoliata transcriptomic data contained data on three tissue types (fruit flesh, seeds, and rind) at four different stages (young, enlargement, coloring, and mature stages), and there were data for three biological replicates (young stage, SAMN16551934-36, enlargement stage; SAMN16551937-39, coloring stage; SAMN16551940-42, mature stage). FPKM values calculated by HISAT2 and DESeq2 were used to estimate gene expression levels 45. TBtools (version 1.0876) software was used to construct a heatmap of AktWOX expression 30.
4.4. AktWOX expression during adventitious root formation
The cuttings used for the experimental treatment were obtained from the same tree cuttings and were exposed to the same cultivation conditions. The cuttings were transplanted in the germplasm nursery of the Sichuan Agricultural University Chongzhou Research Station (30◦430 N, 103◦650 E); the RNA of 2 cm stem base and root mixed samples at 7, 14, 21, 28, 35, 42, 49 and 56 d during the cutting period of AktWOX Shusen 1 was extracted. Total RNA was extracted with an M5 Plant RNeasy Complex Mini Kit (Polysaccharides and Polyphenolics-rich) (JUHEMAI, Beijing, China). The integrity and purity of the RNA were assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Austin, TX, USA), respectively. Then, the RNA of the samples was reverse transcribed into cDNA using an EasyScript One-Step gDNA Removal and cDNA Synthesis Supermix Kit (TransGen Biotech, Beijing, China).
The primer pairs for the AktWOXs and GAPDH genes were designed using Primer 3.0, and the primer sequences and related details are listed in Table S3. The amount of cDNA was 1 µmol as the amplification substrate, and the reaction was carried out as follows: 92°C for 30 s, followed by 45 cycles of 5 s at 92°C and 30 s at 53°C. To determine the expression patterns of the AktWOXs, RT‒qPCR was conducted on a Thermal Cycler CFX96 Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA) together with PerfectStart Green qPCR SuperMix (TransGen Biotech, Beijing, China). Each sample included three technical replicates. The 2−∆∆Ct method was used to calculate the expression level of genes. Statistical analysis was performed with SPSS (version 20.0.0) and Origin 2018 software (version 9.5.1).
5. Conclusions
In this study, we identified 10 candidate AktWOXs that were unevenly distributed on 7 high-quality assembled chromosomes of the A. trifoliata genome. All 10 AktWOXs were classified into 3 groups, and in terms of evolution, they were mainly produced by dispersal events and underwent strong purifying selection. Many AktWOXs exhibited tissue and developmental stage-specific expression patterns. We further identified four genes, namely, AktWOX2, AktWOX3, AktWOX4, and AktWOX8, that could be involved in the response to adventitious root formation conditions. In addition, this study provides important information concerning the WOX genes of A. trifoliata and provides a theoretical reference for their functions in adventitious root formation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.P (Shengpeng).C. and P.L.; methodology, S.P (Shengpeng).C., and S.Z.; software, SP (Shengpeng).C., and H (huai).Y. ; validation, C.C., F.T., and T.R.; formal analysis, SP(Shengpeng).C. ; resources, P.L.; data curation, SP(Shengpeng).C. and Y.L.; writing—original draft preparation, S.P. (Shengpeng). ; writing—review and editing, S.P (Shengpeng).C., P.L.; visualization, H (huai).Y. ; supervision, Y.L., T.R. and F.T.; project administration, P.L.; funding acquisition, P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Science and Technology Department of Sichuan Province, grant numbers 2022ZHXC0002, 2022ZHXC0028 and 2022ZHXC0044.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data analyzed during this study are included in the manuscript and supplementary information files. Genome sequence files of A. trifoliata were downloaded from the National Genomics Data Center database under BioProject PRJCA003847. Transcriptome data for A. trifoliata were downloaded from the NCBI database under accession numbers PRJNA671772, SAMN16551931–33, SAMN16551934–36, SAMN16551937–39 SAMN16551940–42 and the National Genomics Data Center database under BioProject PRJCA014987.
Acknowledgments
We are grateful to the Science and Technology Department of Sichuan Province and the Bureau of Science and Technology of Chengdu for supporting this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Huang P, Zang F, Li C, et al. The Akebia Genus as a Novel Forest Crop: A Review of Its Genetic Resources, Nutritional Components, Biosynthesis, and Biological Studies. Front Plant Sci. 2022; 13: 936571. Published 2022 Jul 26. [CrossRef]
- Zou, S., Yao, X., Zhong, C., Zhao, T., Huang, H., Efectiveness of recurrent selection in Akebia trifoliata (Lardizabalaceae) breeding. Sci Hortic. 2019, 246: 79–85.
- Bai, Jie., Zhou, Tao., Ma, Jiang., Liu, Wen., Jiang, Zhiguo., Chen, Faju. Sporogenesis and Gametogenesis of Akebia trifoliata[J]. Bulletin of Botanical Research. 2022, 42(6): 946-955. [in Chinese].
- Wu, L. L., Ke, K. E. B. F., Gong, C., Ma, Y., Lei, X. L., and Li, J. A. Tissue culture and rapid propagation of Akebia trifoliate var. australis. Plant Physiol. J. 2015, 51, 903–908.
- Zou S, Yao X, Zhong C, et al. Recurrent somatic embryogenesis and development of somatic embryos in Akebia trifoliata (Thunb.) Koidz (Lardizabalaceae)[J]. Plant Cell, Tissue and Organ Culture (PCTOC), 2019, 139(3): 493-504. [CrossRef]
- Wu Xi-an, Tang Hui, Zhong Jian-hua, Wang Yue, Niu Juan, Xiao Jin-jun, Luan Ming-bao. Effects of Different Factors on the Rooting of Akebia trifoliata Cuttings Based on Orthogonal Experiment[J]. Hunan Agricultural Sciences, 2022, (05): 33-36. [in Chinese].
- Tian Dingke, Yu Zhimin. Analysis on the Cultivation Factors Affect the Success of Cuttage of Akebia trifoliata Koidz[J]. Hunan Agricultural Sciences, 2009, (7): 129-131. [in Chinese].
- Steffens, B., & Rasmussen, A. The Physiology of Adventitious Roots. Plant physiology, 2016, 170(2), 603–617.
- Hu X, Xu L. Transcription Factors WOX11/12 Directly Activate WOX5/7 to Promote Root Primordia Initiation and Organogenesis. Plant Physiol. 2016; 172(4): 2363-2373. [CrossRef]
- Liu J, Sheng L, Xu Y, et al. WOX11 and 12 are involved in the first-step cell fate transition during de novo root organogenesis in Arabidopsis. Plant Cell. 2014; 26(3): 1081-1093. [CrossRef]
- Cho SH, Kang K, Lee SH, Lee IJ, Paek NC. OsWOX3A is involved in negative feedback regulation of the gibberellic acid biosynthetic pathway in rice (Oryza sativa). J Exp Bot. 2016; 67(6): 1677-1687. [CrossRef]
- Nardmann J, Werr W. Symplesiomorphies in the WUSCHEL clade suggest that the last common ancestor of seed plants contained at least four independent stem cell niches. New Phytol. 2013; 199(4): 1081-1092. [CrossRef]
- Hedman, H., Zhu, T., von Arnold, S., & Sohlberg, J. J. Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in the conifer picea abies reveals extensive conservation as well as dynamic patterns. BMC plant biology, 2013, 13, 89.
- Mao J, Niu C, Li K, et al. Cytokinin-responsive MdTCP17 interacts with MdWOX11 to repress adventitious root primordium formation in apple rootstocks. Plant Cell. 2023; 35(4): 1202-1221. [CrossRef]
- Shi L, Wang K, Du L, Song Y, Li H, Ye X. Genome-Wide Identification and Expression Profiling Analysis of WOX Family Protein-Encoded Genes in Triticeae Species. Int J Mol Sci. 2021; 22(17): 9325. Published 2021 Aug 28. [CrossRef]
- Haecker A, Gross-Hardt R, Geiges B, et al. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development. 2004;131(3): 657-668. [CrossRef]
- Lian G, Ding Z, Wang Q, Zhang D, Xu J. Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. Scientific World Journal. 2014; 2014: 534140. Published 2014 Jan 6. [CrossRef]
- Li M, Wang R, Liu Z, Wu X, Wang J. Genome-wide identification and analysis of the WUSCHEL-related homeobox (WOX) gene family in allotetraploid Brassica napus reveals changes in WOX genes during polyploidization. BMC Genomics. 2019; 20(1): 317. Published 2019 Apr 25. [CrossRef]
- Zhang Z, Yuan L, Liu X, Chen X, Wang X. Evolution analysis of Dof transcription factor family and their expression in response to multiple abiotic stresses in Malus domestica. Gene. 2018; 639: 137-148. [CrossRef]
- Liu W, Xu L. Recruitment of IC-WOX Genes in Root Evolution. Trends Plant Sci. 2018;23(6):490-496. [CrossRef]
- He P, Zhang Y, Liu H, et al. Comprehensive analysis of WOX genes uncovers that WOX13 is involved in phytohormone-mediated fiber development in cotton. BMC Plant Biol. 2019;19(1):312. Published 2019 Jul 15. [CrossRef]
- Wang Z, Cai Q, Xia H, et al. Genome-Wide Identification and Comparative Analysis of WOX Genes in Four Euphorbiaceae Species and Their Expression Patterns in Jatropha curcas. Front Genet. 2022; 13: 878554. Published 2022 Jun 30. [CrossRef]
- Hao Q, Zhang L, Yang Y, Shan Z, Zhou XA. Genome-Wide Analysis of the WOX Gene Family and Function Exploration of GmWOX18 in Soybean. Plants (Basel). 2019;8(7):215. Published 2019 Jul 11. [CrossRef]
- Magadum S, Banerjee U, Murugan P, Gangapur D, Ravikesavan R. Gene duplication as a major force in evolution. J Genet. 2013; 92(1): 155-161. [CrossRef]
- Feng C, Wang J, Wu L, et al. The genome of a cave plant, Primulina huaijiensis, provides insights into adaptation to limestone karst habitats. New Phytol. 2020; 227(4): 1249-1263. [CrossRef]
- Yang H, Chen W, Fu P, et al. Developmental stages of Akebia trifoliata fruit based on volume[J]. Horticultural science and technology, 2021: 39(6): 823-831.
- Han, W., Li, G., Feng, L., Yan, X., Bai, Y., Li, M., et al. (2019). Genome-Wide Characterization Analysis of WOX Transcription Factors and Response to Abiotic Stresses in Ricinus communis L. J. Agric. Sci. 33 (10), 1921–1927. [CrossRef]
- Cheng S, Huang Y, Zhu N, Zhao Y. The rice WUSCHEL-related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene. 2014; 549(2): 266-274. [CrossRef]
- Dolzblasz A, Nardmann J, Clerici E, et al. Stem Cell Regulation by Arabidopsis WOX Genes. Mol Plant. 2016; 9(7): 1028-1039. [CrossRef]
- Chen C, Chen H, Zhang Y, et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant. 2020; 13(8): 1194-1202. [CrossRef]
- Marchler-Bauer A, Bo Y, Han L, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017; 45(D1): D200-D203. [CrossRef]
- Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(Web Server issue): W597-W603. [CrossRef]
- Sawal HA, Nighat S, Safdar T, Anees L. Comparative In Silico Analysis and Functional Characterization of TANK-Binding Kinase 1-Binding Protein 1. Bioinform Biol Insights. 2023; 17: 11779322231164828. Published 2023 Apr 2. [CrossRef]
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420-423. [CrossRef]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4(4): 406-425. [CrossRef]
- Felsenstein J. CONFIDENCE LIMITS ON PHYLOGENIES: AN APPROACH USING THE BOOTSTRAP. Evolution. 1985; 39(4): 783-791. [CrossRef]
- Kaur G, Iyer LM, Subramanian S, Aravind L. Evolutionary convergence and divergence in archaeal chromosomal proteins and Chromo-like domains from bacteria and eukaryotes. Sci Rep. 2018;8(1):6196. Published 2018 Apr 18. [CrossRef]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35(6):1547-1549. [CrossRef]
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue): W202-W208. [CrossRef]
- Navarro A, Barton NH. Chromosomal speciation and molecular divergence--accelerated evolution in rearranged chromosomes. Science. 2003; 300(5617): 321-324. [CrossRef]
- Wang Y, Tang H, Debarry JD, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012; 40(7): e49. [CrossRef]
- Sazegari S, Niazi A, Ahmadi FS. A study on the regulatory network with promoter analysis for Arabidopsis DREB-genes. Bioinformation. 2015;11(2):101-106. Published 2015 Feb 28. [CrossRef]
- Kumar S, Suleski M, Craig JM, et al. TimeTree 5: An Expanded Resource for Species Divergence Times [published online ahead of print, 2022 Aug 6]. Mol Biol Evol. 2022; 39(8): msac174. [CrossRef]
- Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019; 10(1): 1523. Published 2019 Apr 3. [CrossRef]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [CrossRef]
Figure 1.
Phylogenetic tree analysis of WOX genes in A. trifolium and other species. At: A. thaliana (green); Os: O. sativa (yellow); Akt: A. trifoliata (purple).
Figure 1.
Phylogenetic tree analysis of WOX genes in A. trifolium and other species. At: A. thaliana (green); Os: O. sativa (yellow); Akt: A. trifoliata (purple).
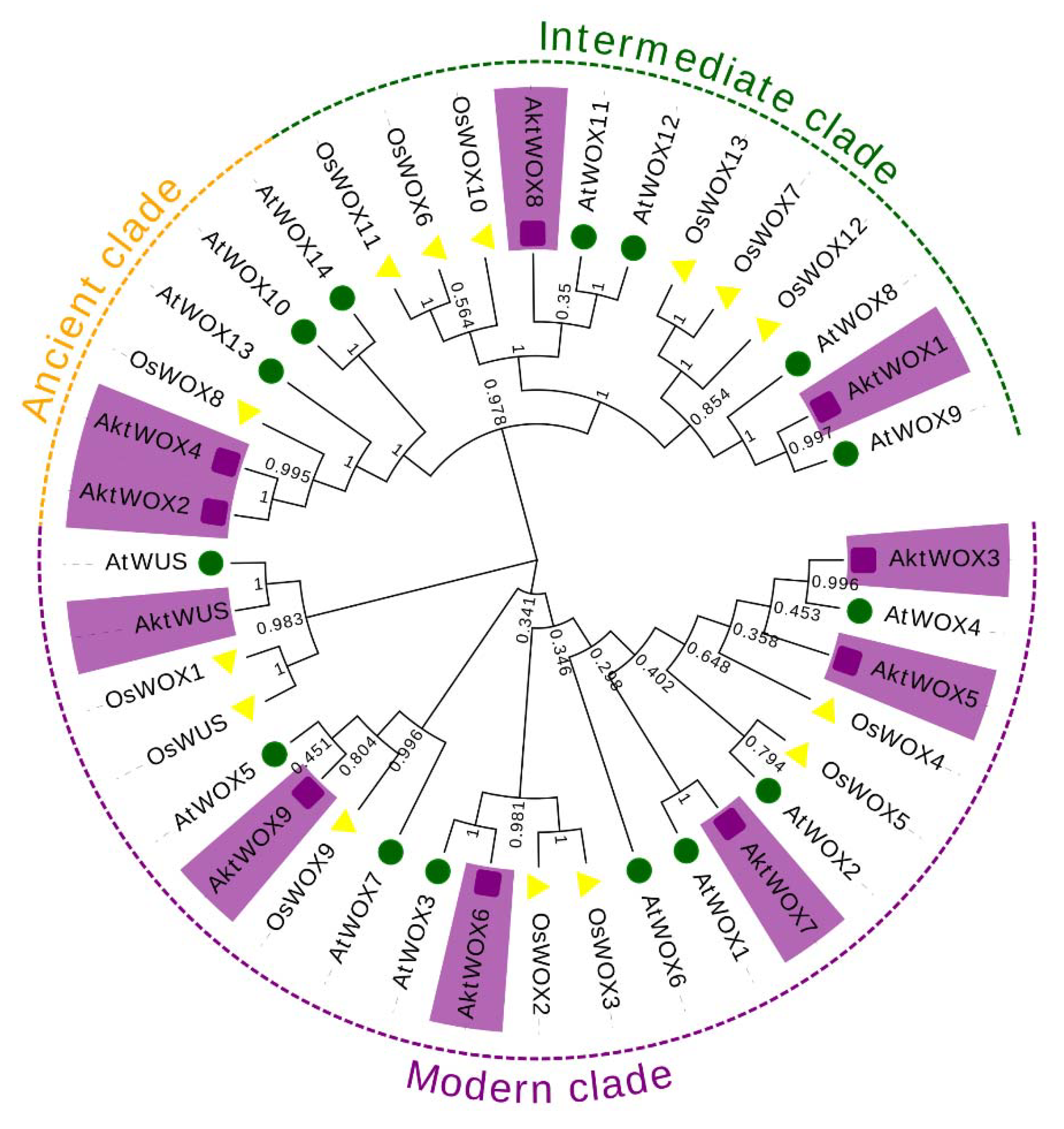
Figure 2.
Gene and protein structure analyses of the AktWOX family. (a) Phylogenetic tree of AktWOXs. (b) Motifs of AktWOX proteins. (c) Exon‒intron structures of AktWOXs.
Figure 2.
Gene and protein structure analyses of the AktWOX family. (a) Phylogenetic tree of AktWOXs. (b) Motifs of AktWOX proteins. (c) Exon‒intron structures of AktWOXs.
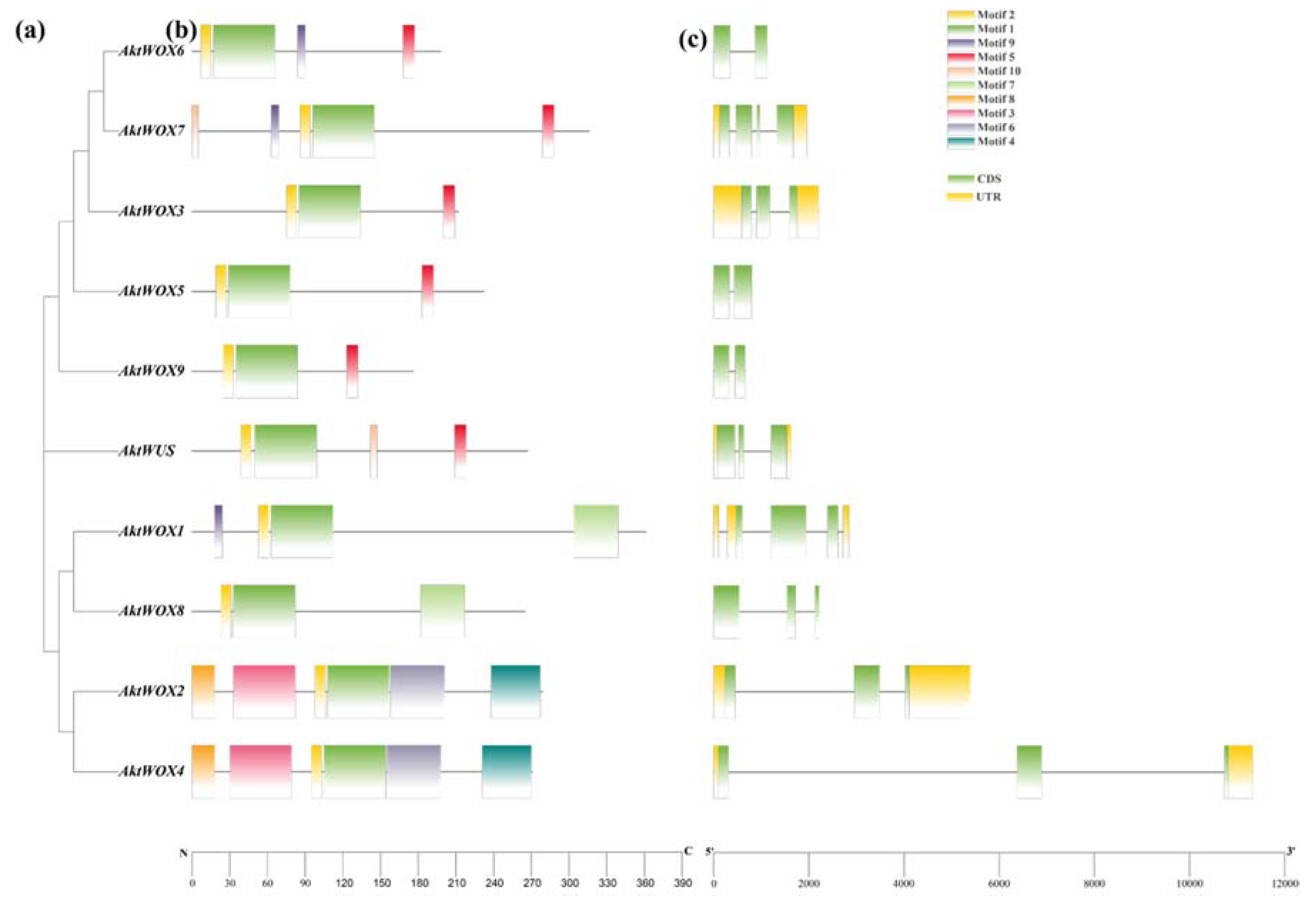
Figure 3.
Collinearity and gene duplication events and gene clusters of AktWOXs. The red line indicates the AktWOX collinear gene pair; the two gene duplication types (dispersed, WGD or segmental) are represented in blue and black, respectively.
Figure 3.
Collinearity and gene duplication events and gene clusters of AktWOXs. The red line indicates the AktWOX collinear gene pair; the two gene duplication types (dispersed, WGD or segmental) are represented in blue and black, respectively.
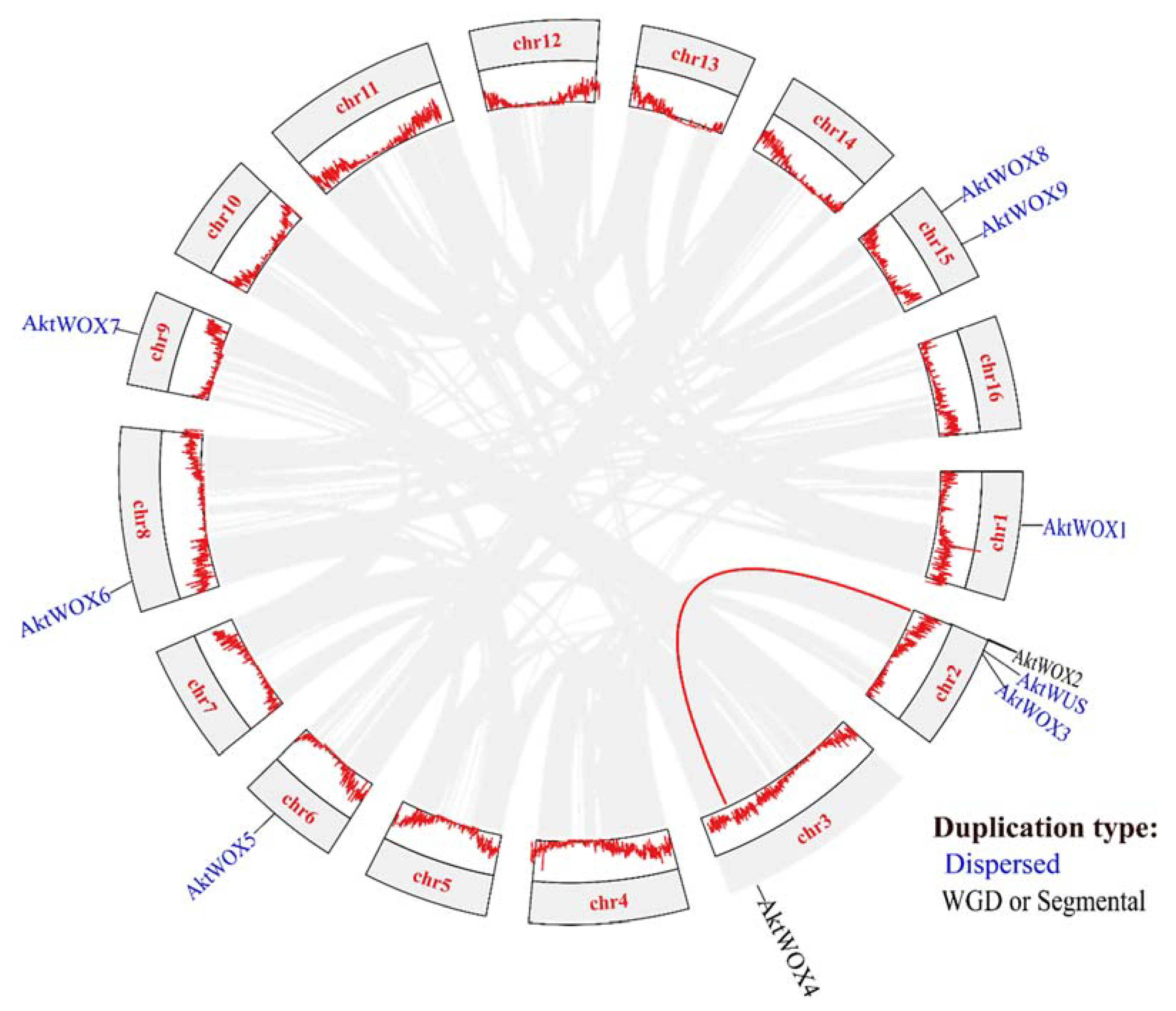
Figure 4.
Collinearity analysis between WOX genes of A. trifoliata and WOX genes of other species. Different species names and chromosomes are represented by different colors. The blue line indicates the homologous WOX gene pairs between other species and AktWOXs, and the number in parentheses after the species name indicates the number of collinear pairs between the WOX genes of the other species and AktWOXs.
Figure 4.
Collinearity analysis between WOX genes of A. trifoliata and WOX genes of other species. Different species names and chromosomes are represented by different colors. The blue line indicates the homologous WOX gene pairs between other species and AktWOXs, and the number in parentheses after the species name indicates the number of collinear pairs between the WOX genes of the other species and AktWOXs.
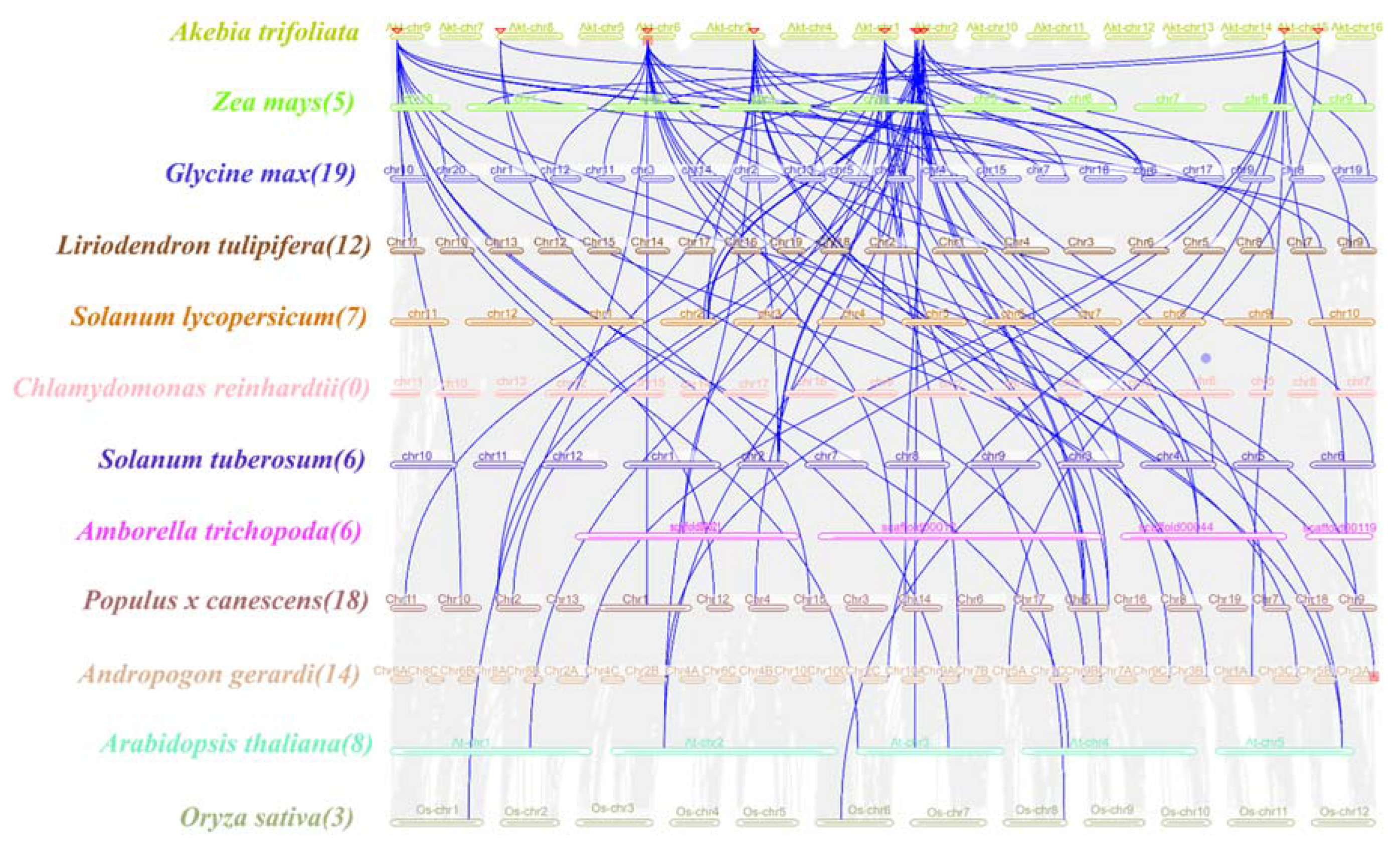
Figure 5.
Prediction of cis-elements in the WOX promoter of A. trifoliata. (A) The distribution of cis-acting elements in the 2000 bp region upstream of the transcription start site of AktWOXs; (B) the number of cis-acting elements of the two functional categories in AktWOXs, respectively indicated by different colors and numbers, cyan-white‒red represents the increasing number of cis-acting elements.
Figure 5.
Prediction of cis-elements in the WOX promoter of A. trifoliata. (A) The distribution of cis-acting elements in the 2000 bp region upstream of the transcription start site of AktWOXs; (B) the number of cis-acting elements of the two functional categories in AktWOXs, respectively indicated by different colors and numbers, cyan-white‒red represents the increasing number of cis-acting elements.
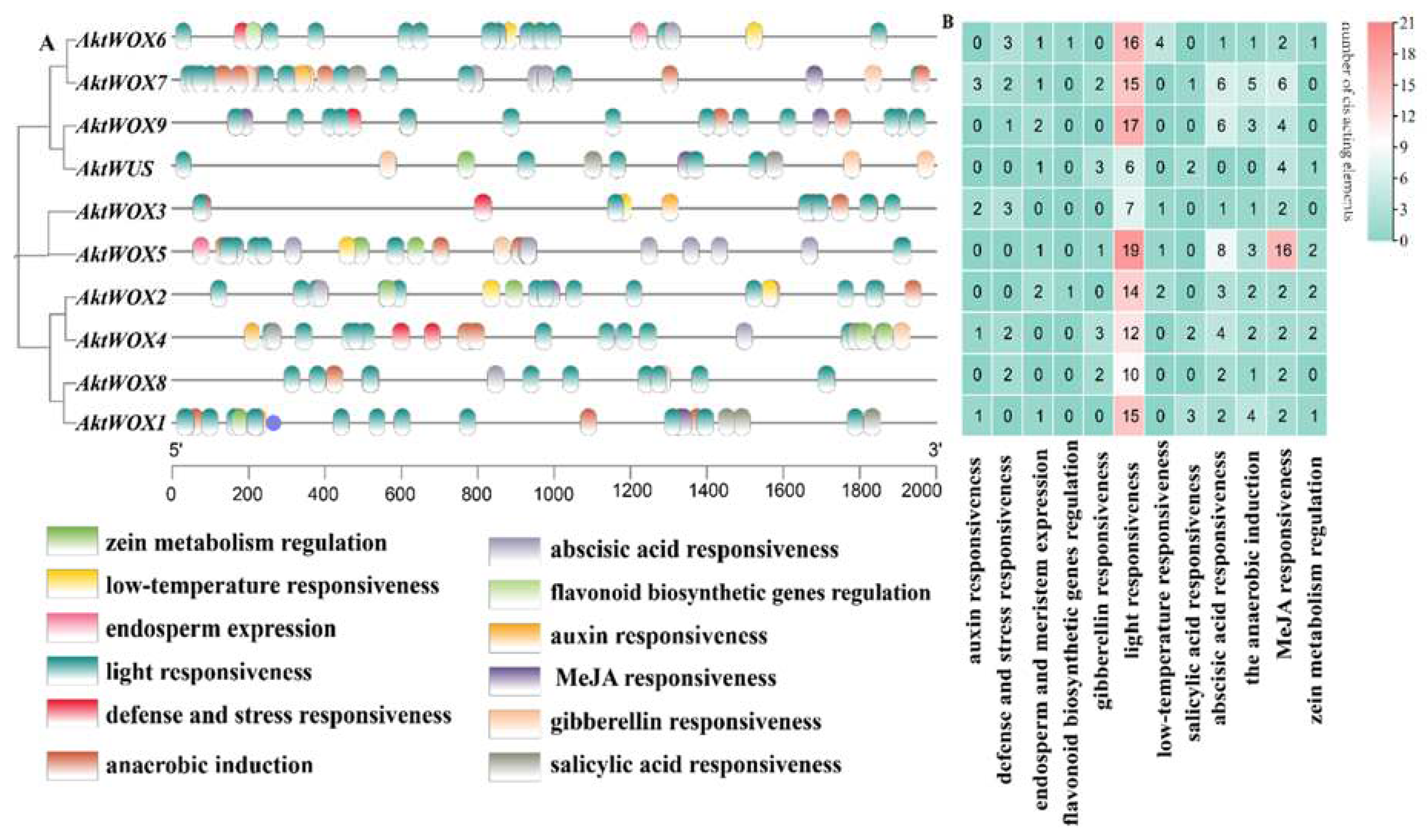
Figure 6.
GO function analysis histogram.
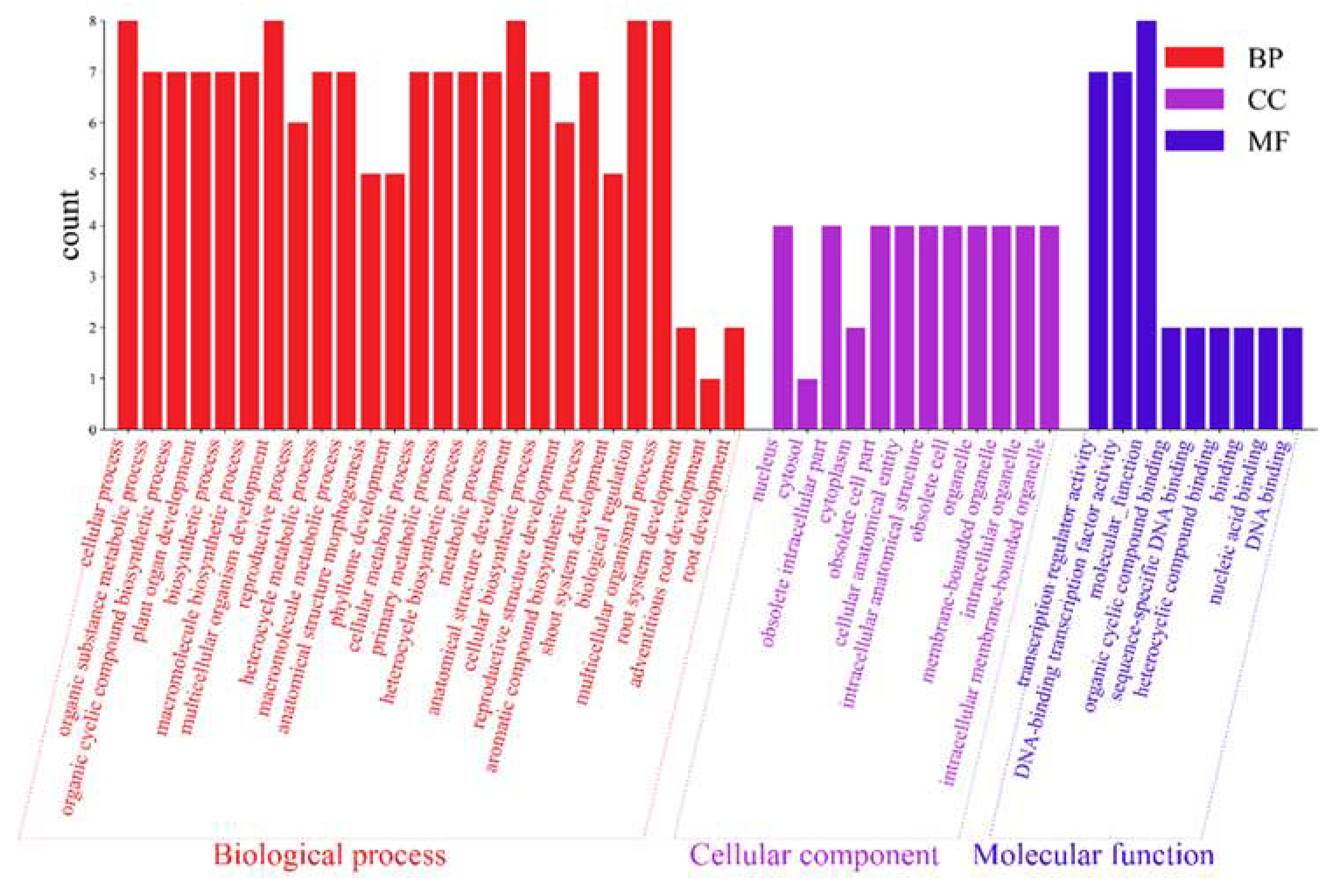
Figure 7.
Heatmap of the expression patterns of WOX genes in different tissues. FT: flesh, ST: seed, RT: rind.
Figure 7.
Heatmap of the expression patterns of WOX genes in different tissues. FT: flesh, ST: seed, RT: rind.
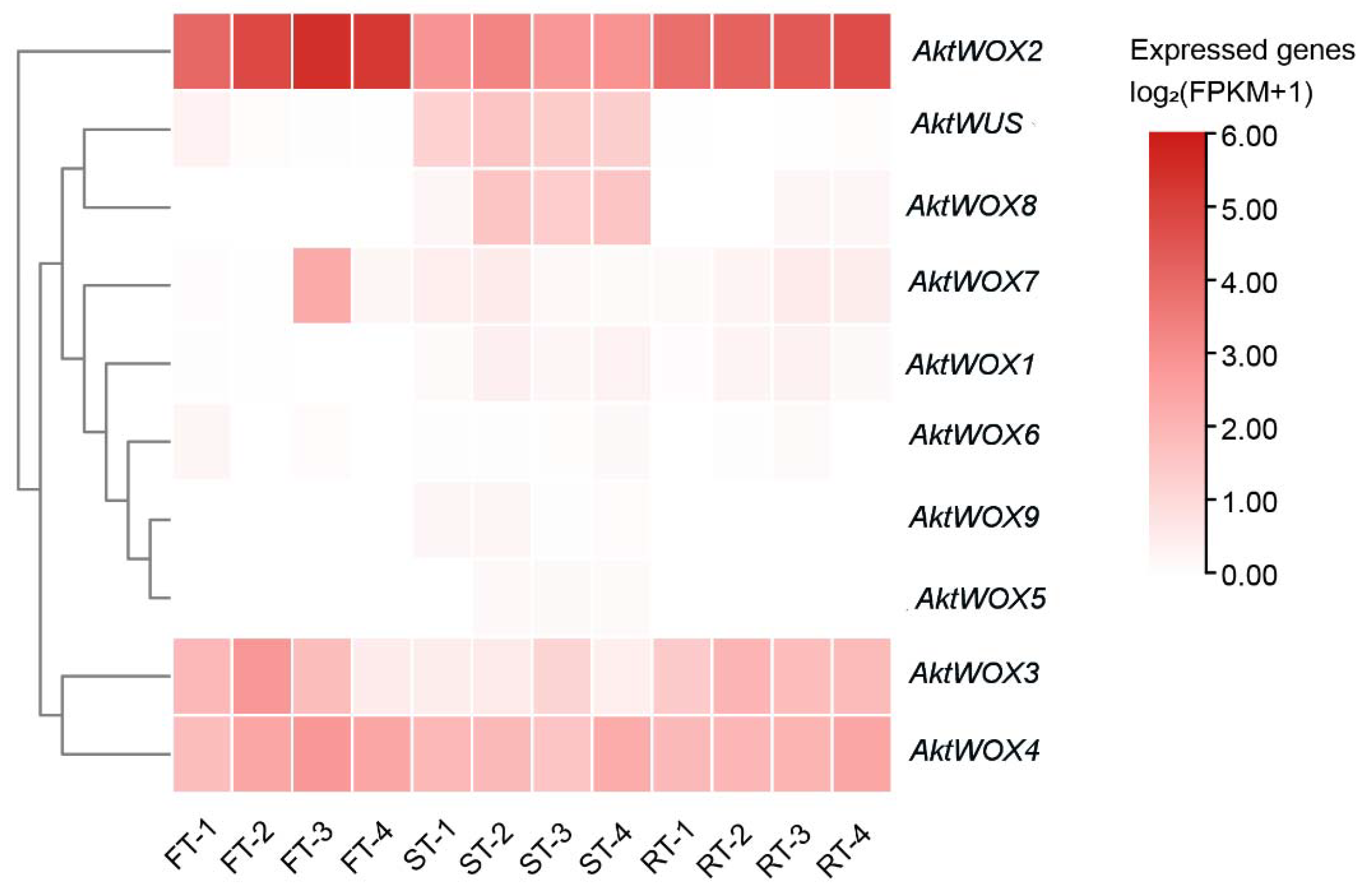

Table 1.
Characteristics of the identified WOX gene family members from the A. trifoliata genome.
| WOX genes | Gene length | Chromosome location | Exon | Cell location |
Putative protein | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Length AA | MW (kDa) | PI | Instability index | Hydrophilic | Signal peptide | |||||||
| AktWOX1 | 2840 | chr1 | 25213082 | 25215922 | 5 | Nucleus | 361 | 40.173 | 6.8 | 57.89 | -0.593 | 0.0009 |
| AktWOX2 | 5392 | chr2 | 896118 | 901510 | 3 | Nucleus | 279 | 31.869 | 6.02 | 59.79 | -0.946 | 0.0005 |
| AktWOX3 | 2204 | chr2 | 7184275 | 7186479 | 3 | Nucleus | 212 | 24.462 | 9.03 | 52.78 | -1.06 | 0.0023 |
| AktWOX4 | 11328 | chr3 | 50099349 | 50110677 | 3 | Nucleus | 307 | 35.238 | 5.65 | 65.16 | -0.927 | 0.0003 |
| AktWOX5 | 796 | chr6 | 6820221 | 6821017 | 2 | Nucleus | 232 | 26.506 | 8.42 | 51.72 | -0.919 | 0.0006 |
| AktWOX6 | 1120 | chr8 | 2341031 | 2342151 | 2 | Nucleus | 198 | 23.316 | 8.75 | 61.48 | -0.891 | 0.0003 |
| AktWOX7 | 1950 | chr9 | 5460894 | 5462844 | 4 | Nucleus | 316 | 36.673 | 8.44 | 75.25 | -0.949 | 0.015 |
| AktWOX8 | 2212 | chr15 | 429281 | 431493 | 3 | Nucleus | 265 | 29.411 | 5.85 | 60.72 | -0.317 | 0.0007 |
| AktWOX9 | 657 | chr15 | 27705270 | 27705927 | 2 | Nucleus | 176 | 20.500 | 8.85 | 53.15 | -0.903 | 0.0044 |
| AktWUS | 1622 | chr2 | 2976634 | 2978256 | 3 | Nucleus | 267 | 29.505 | 6.83 | 60.26 | -0.788 | 0.0012 |
AA, amino acids; PI, isoelectric point; MW, molecular weight. “Instability index” >40 means unstable; “hydrophilicity” <0 is hydrophilic, and >0 is hydrophobic.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



