
Submitted:
01 November 2023
Posted:
02 November 2023
You are already at the latest version
Abstract
In eukaryotes, the Dph1•Dph2 dimer is a non-canonical radical SAM enzyme. Using iron-sulfur (FeS) clusters, it cleaves the cosubstrate S-adenosyl-methionine (SAM) to form a 3-amino-3-carboxy-propyl (ACP) radical for synthesis of diphthamide. The latter decorates a histidine residue on elongation factor 2 (EF2) conserved from archaea to yeast and humans and is important for accurate mRNA translation and protein synthesis. Guided by evidence from archaeal orthologues, we searched for a putative SAM binding pocket in Dph1•Dph2 from Saccharomyces cerevisiae. We predict a SAM binding pocket near the FeS cluster domain that is conserved across eukaryotes in Dph1 but not Dph2. Site-directed DPH1 mutagenesis and functional characterization by assays diagnostic for loss of diphthamide reveal the SAM pocket is essential for synthesis of the décor on EF2 in vivo. Further evidence from structural modeling suggests particularly critical residues close to the methionine moiety of SAM. Presumably, they facilitate a geometry specific for SAM cleavage and ACP radical formation that distinguishes Dph1•Dph2 from classical radical SAM enzymes, which generate canonical 5′-deoxyadenosyl (dAdo) radicals.
Keywords:
Saccharomyces cerevisiae
; SAM
; radical SAM enzymes
; EF2 diphthamide modification
; Dph1•Dph2
; diphtheria toxin
; ADP ribosylation
1. Introduction
In all domains of life, radical SAM (RS) enzymes use iron-sulfur (FeS) clusters together with S-adenosyl-methionine (SAM) as co-substrate for biocatalysis [1,2]. Members of the classical RS enzyme family possess a SAM motif (-CX3CX2C-) with each cysteine coordinating one iron of a cubic 4Fe-4S cluster [3]. The fourth, unique iron binds SAM to set the catalytic framework of a site-differentiated cluster for electron transfer and reductive SAM cleavage. As a result, 5′-deoxyadenosyl (dAdo) radicals are formed for use in biosynthetic reactions including, but not limited to chemical decorations of biological macromolecules, i.e., lipids, proteins and nucleic acids [4,5].
Considered ‘radically’ different from the classical RS family [6] are two related enzymes conserved among archaea (Dph2•Dph2) and eukaryotes including yeast, plants and humans (Dph1•Dph2) [7,8,9,10]. They incorporate FeS clusters and bind SAM [11,12] but rather than producing dAdo radicals, generate from SAM a non-canonical 3-amino-3-carboxy-propyl (ACP) radical in order to prime modification of translation elongation factor 2 (EF2) with diphthamide [13,14]. The décor can be attacked for ADP ribosylation and inactivation of EF2 by diphtheria toxin (DT), causative agent of the human diphtheria disease – hence its name [15,16,17]. Diphthamide is formed in four steps encoded by a complex gene (DPH1-DPH8) network [18,19,20] and supports EF2 accuracy during mRNA translation and protein synthesis. Hence, loss of diphthamide can trigger mistranslation and frame-shift errors with negative consequences onto proteostasis, cell proliferation and development [21,22,23,24].
The latter notion is particularly manifest by traits linked with DPH gene mutations in animal or human cells and collectively known as diphthamide deficiency syndrome (DDS) [10,25,26,27]. Prominent DDS phenotypes include developmental traits such as embryonic lethality in DPH gene-knockout mice [28,29] or craniofacial and neurodegenerative features observed in humans with DPH1, DPH2 or DPH5 gene mutations [25,26,27,28,29,30]. Molecularly, DDS underlies reduced activity of diphthamide synthesis enzymes including DPH1•DPH2, which upon mutation, can block ACP formation and thus fail to initiate the diphthamide pathway [8,10,11,14]. Taking the emerging diphthamide relevance for DDS into account, we further studied the ACP formation step using yeast Dph1•Dph2 as non-canonical RS enzyme model. Sequence alignments and structure modelling with CmnDph2•Dph2, an orthologue of the yeast dimer from the archaeon Candidatus methanoperedens nitroreducens [14], guided into site-directed yeast DPH1 gene mutagenesis. This way we identified candidate residues for a SAM pocket, which we show is essential for diphthamide synthesis on EF2 presumably by conferring SAM binding to the yeast RS enzyme Dph1•Dph2.
2. Materials and Methods
2.1. Strains, Media, Growth Conditions and Assays
S. cerevisiae strains used or generated throughout this study are listed (Table S1). BY4741-derived yeast strains carrying site-specific substitution or deletion mutations at the DPH1 chromosomal locus were generated using PCR-mediated protocols, oligonucleotides, gene specific primers (Table S2) and plasmid templates as previously described [10,19,31]. HA-epitope-tagging at the DPH1 wild-type and mutant loci for immunological Dph1 gene product detection involved previously described PCR-methods [32]. For genomic manipulations, DNA transformations utilized a standard lithium-acetate protocol [33]. Strains were grown in complete yeast peptone dextrose (YPD) or minimal synthetic defined (SD) media [34] at 30˚C unless otherwise stated. For antifungal response assays, ten-fold serial cell dilutions of S. cerevisiae tester strains (starting OD600: 1.5) were spotted onto YPD plates lacking or containing 5-15 μg/mL sordarin (Sigma-Aldrich). Incubation was for 2-4 days at 30˚C. Growth assays in response to diphtheria toxin (DT) fragment A involved transformation with single-copy vector pSU9 [19] for galactose-inducible expression of the cytotoxic ADP-ribosylase activity as previously described [10,21].
2.2. Sequence Alignments and Protein Modelling Based on Archaeal CmnDph2 Structure
The amino acid sequence of CmnDph2 (UniProt-ID A0A062UZ78) was aligned to Dph1 & Dph2 from S. cerevisiae (UniProt-IDs P40487 & P32461), A. thaliana (UniProt-IDs Q8RWW3 & A0A1I9LRW3), D. melanogaster (UniProt-IDs Q9VTM2 & Q9VFE9), M. musculus (UniProt-IDs Q5NCQ5 & Q9CR25) or H. sapiens (UniProt-IDs Q9BZG8 & Q9BQC3) with ClustalOmega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and illustrated with Jalview (https://www.jalview.org/development/archive/Version-2_11_2_7/). The structure of CmnDph2 in complex with SAM (PDB:6BXN) is solved [14] and guided into Dph1•Dph2 models using AlphaFold and ColabFold platforms [35,36] with yeast Dph1 and Dph2 protein sequences (for UniProt-ID details see above) as recently described (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) [10]. Visualization of structures used PyMOL (https://pymol.informer.com/1.3/); candidate SAM pocket residues were curated manually.
2.3. Assaying Diphthamide-Modified EF2 and ADP Ribosylation (ADPR) of EF2 by DT
Diagnosis of EF2 diphthamide modification states in vivo involved Western blots on total yeast cell extracts and antibodies that detect global EF2 pools irrespective of diphthamide modification (anti-EF2[pan]) or specifically recognize unmodified forms of EF2 (anti-EF2[no diphthamide]) [37]. Both antibodies were originally raised to detect human EF2 [37]. As diphthamide contexts in human (708-TLHADAIHRGGGQIIPT-724) and yeast (692-TLHADAIHRGGGQIIPT-708) cells are identical, anti-EF2(no diphthamide) is also suited to differentiate diphthamide modification states of EF2 from S. cerevisiae [21].
Total yeast cell extracts were generated as previously described [38] and protein concentrations determined by the Bradford assay [39]. Lämmli samples were run by SDS-PAGE (12% [w/v] polyacrylamide) and blotted onto PVDF membranes (Millipore). These were probed overnight at 4˚C with the anti-EF2(pan) and anti-EF2(no diphthamide) antibodies [21] and developed with anti-rabbit secondary antibody HRP-conjugate (Dianova; working concentration: 1:5000) and Lumi-Light Western blotting substrate (Roche) as described [21,37]. Protein loading was controlled in parallel Western blots with anti-Cdc19 antibodies, kindly donated by Dr Jeremy Thorner (University of California, USA) and recognizing yeast pyruvate kinase (i.e., Cdc19). Similarly, anti-HA Western blots (Invitrogen 2-2.2.14) were performed to verify expression of HA-tagged Dph1 gene products. Diphthamide-dependent ADPR acceptor activity of EF2 in presence of DT was tested in vitro [40] using total yeast extracts and biotinylated NAD+ as ADP-ribosyl donor for the DT reaction essentially as previously described with human and yeast EF2 resources [10,27,40].
3. Results and Discussion
3.1. In Search for a Potential SAM Pocket in the Yeast Dph1•Dph2 Heterodimer
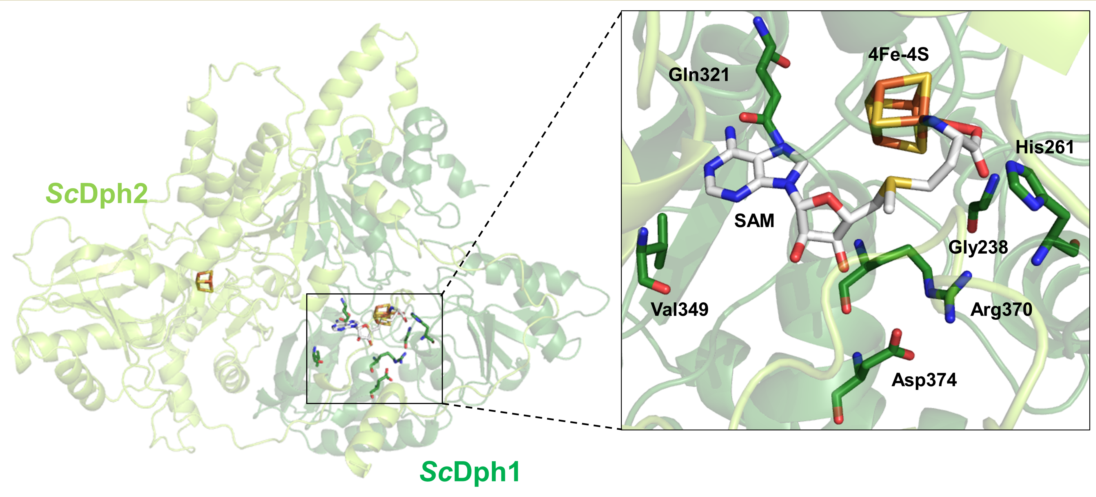
Structural studies with prokaryotic orthologues of the yeast Dph1•Dph2 heterodimer showed the presence of FeS clusters in each subunit of the Dph2•Dph2 homodimer from the archaea Pyrococcus horikoshii (Ph) [7,11], Candidatus methanoperedens nitroreducens (Cmn) [14] and Methanobrevibacter smithii (Ms) [41]. In addition, crystallization of the CmnDph2•Dph2 dimer in complex with SAM demonstrated [14] a subset of amino acid residues (Figure 1A) surrounding the FeS and SAM cofactors (i.e., Gly-158, His-180, Gln-237; Val-265, Arg-285, Asp-289, Asp-290). Although intramolecular contacts to SAM are suggestive for a potential role of these residues in SAM coordination (Figure 1A), there is hardly evidence in support of their catalytic relevance except for Arg-291 from MsDph2 (equivalent to Arg-289 in PhDph2 and Arg-285 in CmnDph2; Figure 1A), which proposedly guides the ACP radical to react with EF2 [41].
Therefore, we examined whether the residues of interest were invariant between archaeal and eukaryotic enzymes including the Dph1•Dph2 dimer from yeast. In search for a potential SAM binding pocket that is conserved among eukarya, the amino acid sequence of CmnDph2 was aligned to both Dph1 and Dph2 from Saccharomyces cerevisiae (Sc), Arabidopsis thaliana (At), Drosophila melanogaster (Dm), Mus musculus (Mm) and Homo sapiens (Hs) (Figure S1). Except for one residue (i.e., Asp-290) all others appeared to be invariant across all species in Dph1, but strikingly not in the eukaryal Dph2 subunit (Figures 1B and S2). CmnDph2 Val-265 is considered to be conserved since Dph1 sequences from the eukaryotes above contain either a valine residue (ScDph1 Val-349) or instead, a branched chain amino acid, e.g., leucine or isoleucine, at the position of interest, i.e., AtDph1 Leu-349, DmDph1 Ile-331, MmDph1 Ile-323 and HsDph1 Ile-328, respectively (Figure 1B). Of note, in contrast to the Dph1 subunits, no eukaryal Dph2 query sequences showed overall high degrees of conservation (Figure S2). Thus, only a few rare, randomly scattered residues are found, i.e., AtDph2 Asp-342 as well as DmDph2 Ile-309 & Asp-335 (Figure S2), to be similar between archaeal and eukaryal Dph2 sequences.
Using AlphaFold2-based models of the CmnDph2•Dph2 dimer we next aligned the conserved Dph1 residues, i.e., Gly-238, His-261, Gln-321, Val-349, Arg-370 and Asp-374 (Figure 1) to the SAM-bound structure of CmnDph2 [14]. In line with the SAM pocket in CmnDph2 near the 4Fe-4S cluster [14], all the conserved Dph1 residues were found to position around the two cofactors, too. As for the rare residues scattered among a few eukaryal Dph2 sequences excluding yeast (Figure S2) and similar to CmnDph2, our models would not suggest a putative SAM pocket in the Dph2 subunit of the heterodimer.
In conclusion, evidence from our sequence alignments supports the idea of a SAM pocket in the yeast Dph1•Dph2 heterodimer and possibly related to the archaeal Dph2•Dph2 homodimer (Figure 1). Moreover, this potential SAM binding pocket is exclusively conserved in Dph1 subunits from eukaryotes, not Dph2. This raises the intriguing question as to whether and if so, how such asymmetry in the ability to bind SAM between the two FeS cluster carrying subunits contributes to catalysis of the dimeric RS complex. In a previous report by Dong et al. 2019, the two FeS clusters were proposed to differ by catalytic versus accessory/regulatory functions [8].
3.2. Conserved Residues in Dph1 Qualify for a SAM Pocket Relevant for Diphthamide synthesis
To examine the relevance of the conserved Dph1 residues, site-directed substitutions were generated at the chromosomal DPH1 locus using PCR-based engineering that allowed for protein expression under native promoter control. In tandem with parallel HA epitope-tagging and by Western blots, we detected the mutant Dph1 proteins (G238A, H261A, Q321A, V349A, R370A & D374A) at expression levels comparable to a wild-type (DPH1) reference (Figure S3) and much less affected than C368S, a highly labile catalytic mutant [8] used as a negative control. Next, their diphthamide synthesis capacity was investigated in vivo with assays that monitor diphthamide-dependent inhibition of EF2 and cell growth by DT and the antifungal sordarin (Figure 2A).
First, the mutant collection was transformed with a plasmid (pSU9) allowing for cytotoxic expression of the catalytic DT subunit under GAL1 promoter control [19,21]. Upon cultivation under inducing conditions, i.e., with galactose added to the growth medium as sole carbon source (Figure 2B), a sensitive DT phenotype leading to cell death was seen with the diphthamide-proficient wild-type (DPH1) strain (Figure 2B). In contrast, significant DT resistance traits were typical of the diphthamide-deficient DPH1 deletion strain (dph1Δ) used as a negative control and in association with the G238A, H261A, R370A and D374A substitution mutants (Figure 2B). Intriguingly, both the Q321A and the V349A mutants appeared to express a DT sensitive phenotype (Figure 2B) similar to wild-type. To complement our DT data and further analyze the ability to produce diphthamide, the mutant collection was assayed with sordarin (Figure 2B), an antifungal that kills yeast by inhibiting EF2 in a fashion dependent on diphthamide but different from DT mode of action [42,43,44]. The sordarin phenotype of the D374A mutant and the diphthamide-deficient dph1∆ control compared to each other showing robust resistance against growth inhibition by the antifungal (Figure 2B). In addition, the substitution mutants G238A, H261A and R370A showed protection against toxic sordarin doses, too, yet to a lesser degree than either D374A or the dph1∆ control (Figure 2B). The two other mutants (Q321A & V349A) found to be DT sensitive above, also appeared unchanged from wild-type DPH1 cells with regards to their sordarin responses (Figure 2B).
As shown previously, resistance towards DT and sordarin are bona fide phenotypes diagnostic for failure to initiate or complete diphthamide synthesis on EF2 in yeast cells [10,19,21,43,44]. Thus, four out of the six DPH1 substitution tested, apparently cause defects that are characteristic for a diphthamide loss-of-function (dph1Δ) mutant. This confirms that the majority of candidate Dph1 residues targeted for mutagenesis are functionally relevant and play important roles in vivo for the Dph1•Dph2 enzyme to decorate EF2 with diphthamide in yeast.
3.3. Unmodified EF2 from SAM Pocket Mutants Escapes ADP Ribosylation by DT
We next examined directly whether EF2 from the candidate SAM pocket mutants can be hijacked in a diphtamide-dependent fashion by DT for ADP ribosylation. To do so, we subjected total protein extracts from the various substitution mutants to assays that can monitor ADP ribosylation (ADPR) of EF2 by DT in vitro, a reaction known to be strictly dependent on the diphthamide modification. The protocol used DT, biotinylated NAD+ as ADP-ribosyl donor, EF2 extracts as ADPR acceptors and an HRP-streptavidin conjugate to detect biotin in the ADPR reaction product [10,27,40]. EF2 from wild-type, V349A and Q321A extracts were ADP ribosylated by DT with significantly weak signals seen in the Q321A material (Figure 3). These are read-outs diagnostic for wild-type like diphthamide amounts present on EF2 from V349A cells and reduced (but not entirely abolished) diphtamide-modified EF2 amounts in the Q321A mutant [10,27,40]. In contrast, EF2 from the other SAM pocket mutants (G238A, H261A, R370A and D374) and dph1Δ cells, lacked detectable ADPR acceptor activity indicating loss of diphthamide on EF2 and evasion of the DT attack in these backgrounds (Figure 3).
The fact that the Q321A mutant produced weak, yet detectable ADPR signals on EF2 in presence of DT (Figure 3) suggests residual diphthamide modification and may be in part accountable for DT and sordarin sensitive phenotypes observed in vivo (Figure 2B). In an effort to further characterize the seemingly less important but conserved residues (Gln-321 & Val-349; Figure 1) and understand their contribution, if any, to catalysis by Dph1•Dph2, we probed for potential genetic interaction between the two SAM pocket sites. On comparing each single mutant (Q321A or V349A) alone with the double carrying both mutations (Q321A V349A), we found the two together were phenotypically additive and enhanced sordarin and DT resistance traits (Figure 4A). Since these phenotypes were otherwise not elicited in each single mutant alone, the read-out implies that in Dph1, residues Gln-321 and Val-349 may back-up each other in function so that their combined loss is negative adding up to the phenotype of each single mutant alone (Figure 4A).
Next, we examined the capacity of the single and double mutants to initiate step one of the diphthamide pathway, i.e., formation of ACP-modified EF2 (Figure 2A). To do so, we subjected total protein extracts from control strains (DPH1, dph1Δ) and the mutants (Q321A, V349A, Q321A V349A) to Western blots using antibodies, i.e., anti-EF2(no diphthamide) and anti-EF2(pan) (Figure 4B), previously shown to distinguish unmodified EF2 from global EF2 amounts [10,21,37]. Except for the single V349A mutant, which accumulated diphthamide-modified EF2 pools similar to wild-type (DPH1), the single Q321A and double Q321A V349A mutants produced immune-responsive signals diagnostic for unmodified EF2 and a defect in the diphthamide pathway (Figure 4B). As with the ADPR assays above (Figure 3), the EF2 modification defect of the single Q321A mutant was weaker than the robust signal of the dph1Δ null-mutant (Figure 4B). In contrast, the double mutant Q321A V349A displayed a pronouced EF2 modification defect indistinguishable from the dph1Δ control (Figure 4B).
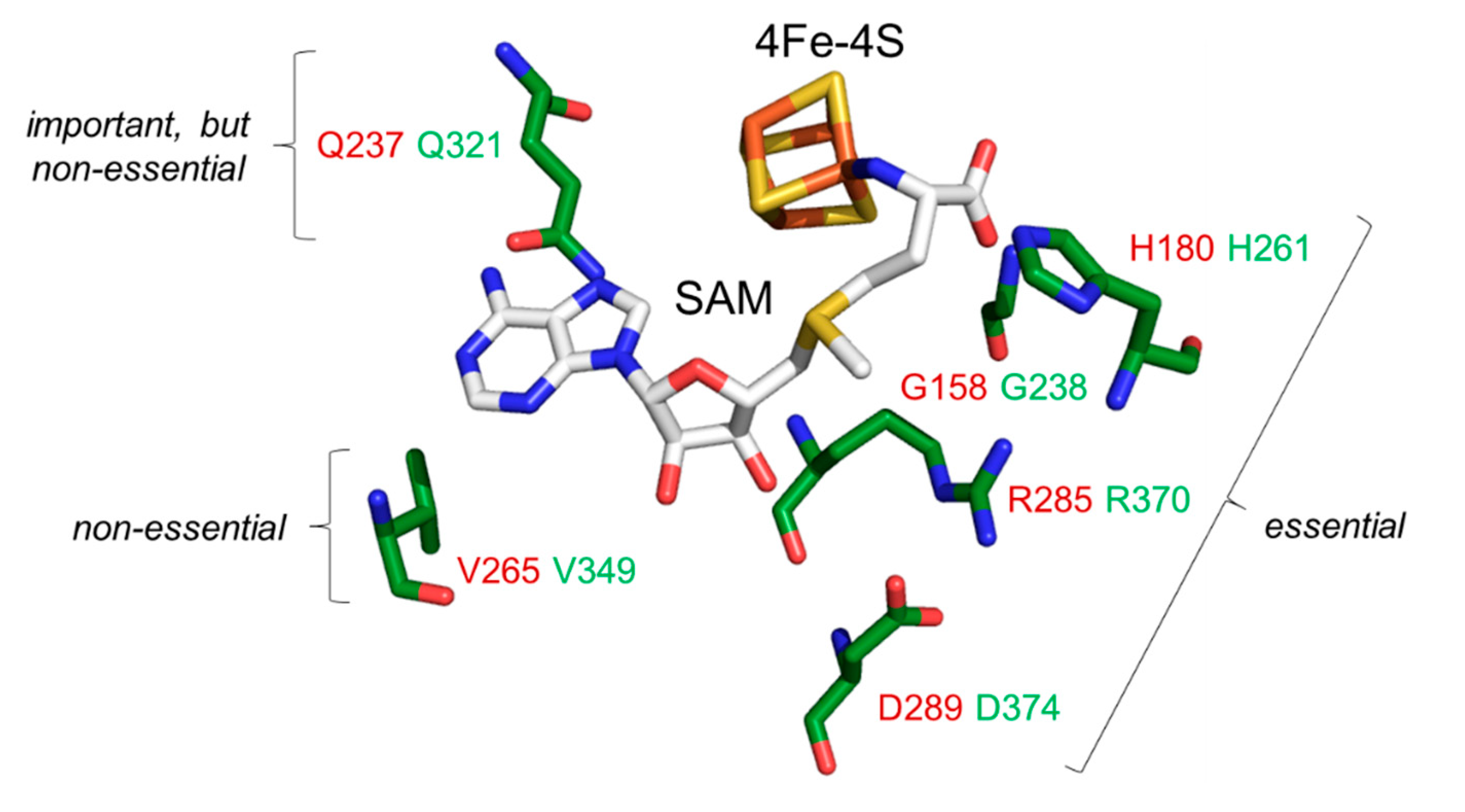
These findings together with data from the genetic and phenotypic interaction assays above (Figure 4A), strongly suggest that interaction between residues Gln-321 & Val-349, which map side-by-side in the SAM pocket (Figure 5), is required for full catalytic capacity of the Dph1•Dph2 enzyme. In line with this notion, which implies that SAM pocket residues in Dph1 can be heterogenous and classified according to their functionality (Figure 5), we found that mutation of the other conserved residues, i.e., Gly-238, His-261, Arg-370 and Asp-374 abolished diphthamide synthesis on EF2. Thus, in correspondence to their DT and sordarin resistance phenotypes in vivo (Figure 2B and Figure 4A) and their lack of EF2-based ADPR acceptor activity in vitro (Figure 3), the G238A, R370A, H261A and D374A mutants produced robust anti-EF2(no diphthamide) Western signals (Figure S4). These are defects typical bona fide dph1Δ (or dph2Δ) diphthamide mutants qualifyng the Dph1 residues Gly-238, His-261, Arg-370 and Asp-374 as essential for Dph1•Dph2 enzyme activity in vivo (Figure 5).
In sum, our work demonstrates that EF2 produced from the single and double substitution mutants is not or cannot be properly modified by diphthamide. This leads us to conclude that the integrity of residues Gly-238, His-261, Gln-321, Arg-370, Asp-374 and possibly Val-349 (in-tandem with Gln-321) is catalytically vital (Figure 5). It is highly likely that the Dph1 residues identified contribute to SAM binding and cleavage by Dph1•Dph2 in order to initiate the modification pathway (Figure 2A).
4. Conclusions and Perspectives
Collectively, our work reveals critical residues in Dph1 that likely contact SAM and are functionally relevant for diphtamide synthesis in the model eukaryote S. cerevisiae (Figure 5). In addition, our work provides a molecular explanation for pathological effects of clinically relevant mutations in human DPH1 recently shown to associate with DDS [10,26]. The essential contact residue His-261 in yeast Dph1 (Figure 5) corresponds to His-240 in human DPH1 (Figure 1B; Figure S2), for which the H240R allele has been shown to compromize diphthamide synthesis activity [10]. Moreover, human L350H variant is a DDS candidate allele [10] and corresponds to Leu-371 in yeast Dph1, a direct neighbour of Arg-370, which we show is an essential SAM pocket residue (Figure 5). A similar rationale may apply to human DDS variant P348S (Pro-369 in yeast) [10] that flanks the FeS cluster ligand Cys-347 (Cys-368 in yeast) [8]. Thus, pathogenic L350H and P348S variants likely affect side chain positioning of Arg-349, the SAM pocket residue in human DPH1 conserved to yeast Arg-370 (Figure 5).
Guided by the archaeal sructure in complex with the SAM cosubstate, our study thus pinpoints to a potential SAM binding pocket in eukaryal Dph1•Dph2 heterodimers that is structurally similar to archaeal Dph2•Dph2 homodimers but exclusively restricted to one of the two subunits in the yeast RS enzyme: Dph1 (Figure 5). In contrast to archaeal homodimers (Dph2•Dph2), the eukaryal enzyme is made of different subunits (Dph1•Dph2). We show in here that this asymmetry not only reflects subunit composition but also the exclusive presence of a potential SAM binding pocket in subunit Dph1 (Figure 5), not Dph2 ! Thus, in terms of evolution, the archaeal prototype Dph2 is much more conserved to Dph1 than to Dph2 from any of the model eukaryotes used in our query. Whether this suggests the yeast or human enzymes arose from an ancient DPH2 gene duplication and diversification event that may explain today’s peculiar heterodimeric structure, remains to be elucidated, for example using gene shuffle and complementation studies in budding yeast or other eukaryotic models of interest.
Nonetheless, the uniqueness of a SAM pocket in Dph1 that we show is essential for the initiation step of diphthamide synthesis urges for further studies into the individual roles played by the FeS clusters in either subunit of the Dph1•Dph2 enzyme [8,46]. While the FeS cluster in Dph1 operates catalytically in reductive SAM cleavage and ACP radical formation [11,14,41], the role for the metallic cofactor in Dph2 is far from clear. Potentially, it acts in regulation of the heterodimer rather than direct catalysis. Such option has been spurred by observations that proper electron flow to Dph1•Dph2 from donor proteins (Cbr1, Dph3•Dph8/Kti11•Kti13) may help control physiological radical reactions and avoid harmful ones [20,45,46,47,48].
With archaeal (Dph2•Dph2) and eukaryotic (Dph1•Dph2) enzymes being able to form ACP radicals, dimer asymmetry obviously is not a prerequisite for the underlying SAM cleavage [11,14,41]. In our hands, it rather implies an FeS cluster geometry in the SAM binding pocket that facilitates regio-selective SAM cleavage to specifically liberate the ACP (rather than a classical dAdo) radical. Results obtained in here propose that pocket residues in proximity to the methionine moiety of SAM are essential, i.e., Gly-238, His-261, Arg-370 and Asp-374 (Figure 5). Whether these may determine, which of the three carbon bonds with the sulfonium in SAM to break and set ACP radicals free [14,41], is important to study since it may clarify the conceptual differences between classical members of the RS enzyme family and non-canonical ones of the diphthamide pathway, i.e., yeast Dph1•Dph2 or human DPH1•DPH2 [49,50].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Alignment between archaeal CmnDph2 and eukaryal Dph1 and Dph2 sequences; Figure S2: The SAM pocket from archaeal CmnDph2 is conserved in eukaryal Dph1, not Dph2; Figure S3: Dph1 protein expression analysis from wild-type and various mutant strain backgrounds; Figure S4: Detection of unmodified EF2 pools using Western blot analysis; Table S1: Yeast strains used and generated in this study; Table S2: Primers used for PCR-based gene engineering and genomic verification.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. Conceptualization, K.Ü. and R.S.; investigation, K.Ü., S.S. and K.M.; methodology, K.Ü., S.S. and K.M.; supervison, U.B. and R.S.; writing—original draft preparation, K.Ü., R.K. and R.S.; writing—review and editing, K.Ü., R.K., U.B and R.S.; funding acquisition, R.S.
Funding
This research work was supported by Deutsche Forschungsgemeinschaft (DFG Bonn, Germany) Priority Program 1927 Iron-Sulfur for Life to R.S. (SCHA750/21-1) and by a Diphthamide Pilotgrant to R.S. (#2887) from Zentraler Forschungsfonds (ZFF, Universität Kassel, Germany). Potential OAP funding statement …
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data can be found in the manuscript and the Supplement.
Acknowledgments
We would like to thank Universität Kassel, Germany for support of active exchange of team members between the Schaffrath and Brinkmann laboratories and greatly acknowledge Prof. Jeremy Thorner (University of California, Berkeley, USA) for kindly donating anti-Cdc19 antibodies we used in Western blots on total yeast cell extracts.
Conflicts of Interest
KM and UB are employed by and members of Roche Pharma Research & Early Development (pRED), and are co-inventors on patent applications that cover assays to detect presence or absence of diphthamide. Roche is interested in targeted therapies and diagnostics. All other authors declare no conflict of interest.
References
- Frey, P.A.; Hegeman, A.D.; Ruzicka, F.J. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63–88. [Google Scholar] [CrossRef]
- Landgraf, B.J.; McCarthy, E.L.; Booker, S.J. Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 2016, 85, 485–514. [Google Scholar] [CrossRef]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef]
- Yokoyama, K.; Lilla, E.A. C-C bond forming radical SAM enzymes involved in the construction of carbon skeletons of cofactors and natural products. Nat. Prod. Rep. 2018, 35, 660–694. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Broderick, W.E.; Broderick, J.B. Mechanism of radical initiation in the radical SAM enzyme superfamily. Annu. Rev. Biochem. 2023, 92, 333–349. [Google Scholar] [CrossRef]
- Broderick, J.B. Biochemistry: a radically different enzyme. Nature. 2010, 465, 877–878. [Google Scholar] [CrossRef]
- Zhu, X.; Dzikovski, B.; Su, X.; Torelli, A.T.; Zhang, Y.; Ealick, S.E.; Freed, J.H.; Lin, H. Mechanistic understanding of Pyrococcus horikoshii Dph2, a [4Fe-4S] enzyme required for diphthamide biosynthesis. Mol. Biosyst. 2011, 7, 74–81. [Google Scholar] [CrossRef]
- Dong, M.; Dando, E.E.; Kotliar, I.; Su, X.; Dzikovski, B.; Freed, J.H.; Lin, H. The asymmetric function of Dph1-Dph2 heterodimer in diphthamide biosynthesis. J. Biol. Inorg. Chem. 2019, 24, 777–782. [Google Scholar] [CrossRef]
- Zhang H, Quintana J, Ütkür K, Adrian L, Hawer H, Mayer K, Gong X, Castanedo L, Schulten A, Janina N, Peters M, Wirtz M, Brinkmann U, Schaffrath R, and Krämer U. Translational fidelity and growth of Arabidopsis require stress-sensitive diphthamide biosynthesis. Nat. Comm. 2022, 13, 4009. [CrossRef]
- Ütkür, K.; Mayer, K.; Khan, M.; Manivannan, T.; Schaffrath, R.; Brinkmann, U. DPH1 and DPH2 variants that confer susceptibility to diphthamide deficiency syndrome in human cells and yeast models. Dis. Model. Mech. 2023, 16, dmm050207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Torelli, A.T.; Lee, M.; Dzikovski, B.; Koralewski, R.M.; Wang, E.; Freed, J.; Krebs, C.; Ealick, S.E. , Lin, H. Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 2010, 465, 891–896. [CrossRef]
- Zhu, X.; Kim, J.; Su, X.; Lin, H. Reconstitution of diphthine synthase activity in vitro. Biochemistry 2010, 49, 9649–9657. [Google Scholar] [CrossRef] [PubMed]
- Lin, H. S-Adenosylmethionine-dependent alkylation reactions: when are radical reactions used? Bioorg. Chem. 2011, 39, 161–170. [Google Scholar] [CrossRef]
- Dong, M.; Kathiresan, V.; Fenwick, M.K.; Torelli, A.T.; Zhang, Y.; Caranto, J.D.; Dzikovski, B.; Sharma, A.; Lancaster, K.M.; Freed, J.H.; Ealick, S.E.; Hoffman, B.M.; Lin, H. Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis. Science 2018, 359, 1247–1250. [Google Scholar] [CrossRef]
- Van Ness, B.G. , Howard, J.B., and Bodley, J.W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J. Biol. Chem. 1980, 255, 10717–10720.
- Uthman, S.; Liu, S.; Giorgini, F.; Stark, M.J.R.; Costanzo, M.; Schaffrath, R. Diphtheria disease and genes involved in formation of diphthamide, key effector of the diphtheria toxin. In: Insight and Control of Infectious Disease in Global Scenario, Kumar, R. (ed), INTECH OAP, 2012, pp. 333–356. [CrossRef]
- Jørgensen, R.; Merrill, A.R.; Andersen, G.R. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 2006, 34, 1–6. [Google Scholar] [CrossRef]
- Liu, S.; Milne, G.T.; Kuremsky, J.G.; Fink, G.R.; Leppla, S.H. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol. Cell. Biol. 2004, 24, 9487–9497. [Google Scholar] [CrossRef]
- Uthman, S.; Bär, C.; Scheidt, V.; Liu, S.; ten Have, S.; Giorgini, F.; Stark, M.J.R.; Schaffrath, R. The amidation step of diphthamide biosynthesis in yeast requires DPH6, a gene identified through mining the DPH1-DPH5 interaction network. PLoS Genet. 2013, 9, e1003334. [Google Scholar] [CrossRef]
- Arend, M.; Ütkür, K.; Hawer, H.; Mayer, K.; Ranjan, N.; Adrian, L.; Brinkmann, U.; Schaffrath, R. Yeast gene KTI13 (alias DPH8) operates in the initiation step of diphthamide synthesis on elongation factor 2. Microb. Cell 2023, 10, 195–203. [Google Scholar] [CrossRef]
- Hawer, H.; Ütkür, K.; Arend, M.; Mayer, K.; Adrian, L.; Brinkmann, U.; Schaffrath, R. Importance of diphthamide modified EF2 for translational accuracy and competitive cell growth in yeast. PLoS ONE 2018, 13, e0205870. [Google Scholar] [CrossRef]
- Pellegrino, S.; Demeshkina, N.; Mancera-Martinez, E.; Melnikov, S.; Simonetti, A.; Myasnikov, A.; Yusupov, M.; Yusupova, G.; Hashem, Y. Structural insights into the role of diphthamide on elongation factor 2 in mRNA reading-frame maintenance. J. Mol. Biol. 2018, 430, 2677–2687. [Google Scholar] [CrossRef]
- Djumagulov, M.; Demeshkina, N.; Jenner, L.; Rozov, A.; Yusupov, M.; Yusupova, G. Accuracy mechanism of eukaryotic ribosome translocation. Nature 2021, 600, 543–546. [Google Scholar] [CrossRef]
- Shin, B.-S.; Ivanov, I.P.; Kim, J.-R.; Cao, C.; Kinzy, T.G.; Dever, T.E. eEF2 diphthamide modification restrains spurious frameshifting to maintain translational fidelity. Nucleic Acids Res. 2023, 51, 6899–6913. [Google Scholar] [CrossRef]
- Loucks, C.M.; Parboosingh, J.S.; Shaheen, R.; Bernier, F.P.; Mcleod, D.R.; Seidahmed, M.Z.; Puffenberger, E.G.; Ober, C.; Hegele, R.A.; Boycott, K.M. ; Alkuraya FS, Innes AM. Matching two independent cohorts validates DPH1 as a gene responsible for autosomal recessive intellectual disability with short stature, craniofacial, and ectodermalanomalies. Hum. Mutat. 2015, 36, 1015–1019. [CrossRef]
- Hawer H, Mendelsohn BA, Mayer K, Kung A, Malhotra A, Tuupanen S, Schleit J, Brinkmann U, and Schaffrath R. Diphthamide-deficiency syndrome: a novel human developmental disorder and ribosomopathy. Eur. J. Hum. Genet. 2020, 28, 1497–1508. [CrossRef]
- Shankar, S.P.; Grimsrud, K.; Lanoue, L.; Egense, A.; Willis, B.; Hörberg, J.; AlAbdi, L.; Mayer, K.; Ütkür, K.; Monaghan, K.G.; Krier, J.; Stoler, J.; Alnemer, M.; Shankar, P.R.; Schaffrath, R.; Alkuraya, F.S.; Brinkmann, U.; Eriksson, L.A.; Lloyd, K.; Rauen, K.A. ; Undiagnosed Diseases Network. A novel DPH5-related diphthamide-deficiency syndrome causing embryonic lethality or profound neurodevelopmental disorder. Genet. Med. 2022, 24, 1567–1582. [CrossRef]
- Liu, S. , Wiggins, J.F., Sreenath, T., Kulkarni, A.B., Ward, J.M., and Leppla, S.H. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol. Cell. Biol. 2006, 26, 3835–3841. [CrossRef]
- Webb, T.R.; Cross, S.H.; McKie, L.; Edgar, R.; Vizor, L.; Harrison, J.; Peters, J.; Jackson, I.J. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J. Cell Sci. 2008, 121, 3140–3145. [Google Scholar] [CrossRef]
- Liu, S.; Bachran, C.; Gupta, P.; Miller-Randolph, S.; Wang, H.; Crown, D.; Zhang, Y.; Wein, A.N.; Singh, R.; Fattah, R.; Leppla, S.H. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc. Natl. Acad. Sci. USA 2012, 109, 13817–13822. [Google Scholar] [CrossRef]
- Toulmay, A.; Schneiter, R. A two-step method for the introduction of single or multiple defined point mutations into the genome of Saccharomyces cerevisiae. Yeast 2006, 23, 825–831. [Google Scholar] [CrossRef]
- Janke, C.; Magiera, M.M.; Rathfelder, N.; Taxis, C.; Reber, S.; Maekawa, H.; Moreno-Borchart, A.; Doenges, G.; Schwob, E.; Schiebel, E.; Knop, M. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004, 21, 947–962. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S.A.A.; Ballard, A.J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A.W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Stahl, S.; da Silva Mateus Seidl, A.R.; Ducret, A.; Kux van Geijtenbeek, S.; Michel, S.; Racek, T.; Birzele, F.; Haas, A.K.; Rueger, R.; Gerg, M.; Niederfellner, G.; Pastan, I.; Brinkmann, U. Loss of diphthamide pre-activates NF-kappaB and death receptor pathways and renders MCF7 cells hypersensitive to tumor necrosis factor. Proc. Natl. Acad. Sci. USA 2015, 112, 10732–10737. [Google Scholar] [CrossRef]
- Zachariae, W.; Shin, T.H.; Galova, M.; Obermaier, B.; Nasmyth, K. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science 1996, 274, 201–1204. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Chem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.; Schröder, A.; Schnitger, J.; Stahl, S.; Brinkmann, U. Influence of DPH1 and DPH5 protein variants on the synthesis of diphthamide, the target of ADPRibosylating toxins. Toxins 2017, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, M.K.; Dong, M.; Lin, H.; Ealick, S.E. The crystal structure of Dph2 in complex with elongation factor 2 reveals the structural basis for the first step of diphthamide biosynthesis. Biochemistry 2019, 58, 4343–4351. [Google Scholar] [CrossRef]
- Shao, Y.; Molestak, E.; Su, W.; Stankevič, M.; Tchórzewski, M. Sordarin – an anti-fungal antibiotic with a unique modus operandi. Br. J. Pharmacol. 2022, 179, 1125–1145. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Zabel, R.; Liu, S.; Stark, M.J.; Schaffrath, R. A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3. Mol. Microbiol. 2008, 69, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Botet, J.; Rodriguez-Mateos, M.; Ballesta, J.P.; Revuelta, J.L.; Remacha, M. A chemical genomic screen in Saccharomyces cerevisiae reveals a role for diphthamidation of translation elongation factor 2 in inhibition of protein synthesis by sordarin. Antimicrob. Agents Chemother. 2008, 52, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Dong, M.; Zhang, Y.; Lee, E.A.; Lin, H. Cbr1 is a Dph3 reductase required for the tRNA wobble uridine modification. Nat. Chem. Biol. 2016, 12, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Su, X.; Dzikovski, B.; Dando, E.E.; Zhu, X.; Du, J.; Freed, J.H.; Lin, H. Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. J. Am. Chem. Soc. 2014, 136, 1754–1757. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, D.; Dzikovski, B.; Majer, S.H.; Coleman, R.; Chandrasekaran, S.; Fenwick, M.K.; Crane, B.R.; Lancaster, K.M.; Freed, J.H.; Lin, H. Dph3 enables aerobic diphthamide biosynthesis by donating one iron atom to transform a [3Fe-4S] to a [4Fe-4S] cluster in Dph1-Dph2. J. Am. Chem. Soc. 2021, 143, 9314–9319. [Google Scholar] [CrossRef]
- Glatt, S.; Zabel, R.; Vonkova, I.; Kumar, A.; Netz, D.J.; Pierik, A.J.; Rybin, V.; Lill, R.; Gavin, A.C.; Balbach, J.; Breuing, K.D.; Müller, C.W. Structure of the Kti11/Kti13 heterodimer and its double role in modification of tRNA and eukaryotic elongation factor 2. Structure 2015, 23, 149–160. [Google Scholar] [CrossRef]
- Schaffrath, R.; Abdel-Fattah, W.; Klassen, R.; Stark, M.J. The diphthamide modification pathway from Saccharomyces cerevisiae – Revisited. Mol. Microbiol. 2014, 94, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Tsuda-Sakurai, K; Miura, M. The hidden nature of protein translational control by diphthamide: the secrets under the leather. J. Biochem. 2019, 165, 1–8. [CrossRef]
Figure 1.
The SAM binding pocket in archaeal Dph2 subunits of ACP synthase dimers from Candidatus methanoperedens nitroreducens (CmnDph2•Dph2) is conserved in the Dph1 subunit of eukaryal counterparts (Dph1•Dph2). (A) Crystal structure of CmnDph2•Dph2 homodimer in complex with SAM (PDB: 6BXN) revealed specific SAM interacting residues (orange dotted lines represent polar contacts): Gly-158, His-180, Gln-237, Val-265, Arg-285, Asp2-89 and Asp-290 [14]. (B) Excerpt of the amino acid sequence alignment between CmnDph2 (in red) and Dph1 subunits from model eukaryotes (in green): S. cerevisiae (Sc), A. thaliana (At), D. melanogaster (Dm), M. musculus (Mm) and H. sapiens (Hs). The table sums up eukaryal Dph1 residues that are conserved or similar to the SAM pocket from CmnDph2. Residues identical between CmnDph2 and ScDph1 are labelled in bold with the arrow highlighting the amino acid positions chosen for alanine specific substitution mutagenesis of yeast gene DPH1. For full alignment details, see Figures S1 and S2.
Figure 1.
The SAM binding pocket in archaeal Dph2 subunits of ACP synthase dimers from Candidatus methanoperedens nitroreducens (CmnDph2•Dph2) is conserved in the Dph1 subunit of eukaryal counterparts (Dph1•Dph2). (A) Crystal structure of CmnDph2•Dph2 homodimer in complex with SAM (PDB: 6BXN) revealed specific SAM interacting residues (orange dotted lines represent polar contacts): Gly-158, His-180, Gln-237, Val-265, Arg-285, Asp2-89 and Asp-290 [14]. (B) Excerpt of the amino acid sequence alignment between CmnDph2 (in red) and Dph1 subunits from model eukaryotes (in green): S. cerevisiae (Sc), A. thaliana (At), D. melanogaster (Dm), M. musculus (Mm) and H. sapiens (Hs). The table sums up eukaryal Dph1 residues that are conserved or similar to the SAM pocket from CmnDph2. Residues identical between CmnDph2 and ScDph1 are labelled in bold with the arrow highlighting the amino acid positions chosen for alanine specific substitution mutagenesis of yeast gene DPH1. For full alignment details, see Figures S1 and S2.
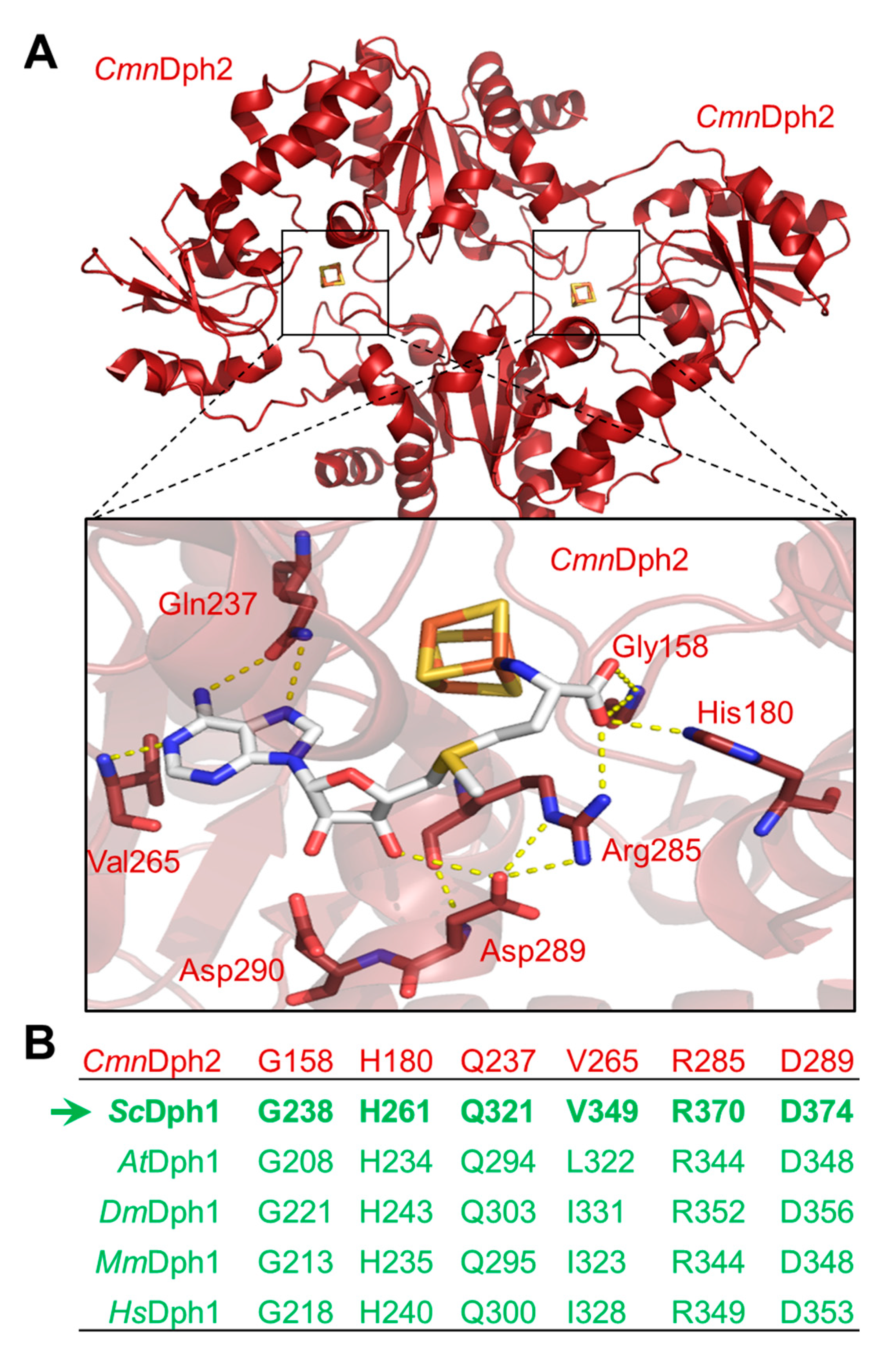
Figure 2.
Site-directed DPH1 mutagenesis uncovers a candidate SAM pocket in yeast Dph1•Dph2 relevant for dipthamide synthesis on EF2. (A) Simplified scheme showing diphthamide synthesis is initiated by the DPH1 and DPH2 gene products and other factors (et al.) to modify His-699 on yeast EF2 with ACP. Subsequent enzymatic steps that complete diphthamide are not shown in detail for simplicity. Diphthamide is pathologically relevant; it can be hijacked by diphtheria toxin (DT) for inhibitory ADP ribosylation of EF2 and induction of cell death (skull-crossbones) or complexed by antifungal sordarin (sor) to irreversibly stall elongating ribosomes. (B) Cell growth assays in response to DT and sordarin to phenotypically diagnose diphthamide synthesis on EF2. As indicated, yeast tester strains comprised wild-type (DPH1) and null-mutant (dph1∆) controls as well as the candidate SAM pocket mutants (G238A, H261A, Q321A V349A, R370A, D374A). Ten-fold serial cell dilutions were cultivated for 2-3 days at 30°C without DT or sordarin (left panel: control), under conditions of endogenous DT fragment A production from a galactose-inducible expression plasmid (middle panel: + DT) [19] or in presence of 12.5 µg/mL antifungal (right panel: + sordarin) sufficient to inhibit the wild-type (DPH1) control. Green arrows indicate DT and sordarin resistance.
Figure 2.
Site-directed DPH1 mutagenesis uncovers a candidate SAM pocket in yeast Dph1•Dph2 relevant for dipthamide synthesis on EF2. (A) Simplified scheme showing diphthamide synthesis is initiated by the DPH1 and DPH2 gene products and other factors (et al.) to modify His-699 on yeast EF2 with ACP. Subsequent enzymatic steps that complete diphthamide are not shown in detail for simplicity. Diphthamide is pathologically relevant; it can be hijacked by diphtheria toxin (DT) for inhibitory ADP ribosylation of EF2 and induction of cell death (skull-crossbones) or complexed by antifungal sordarin (sor) to irreversibly stall elongating ribosomes. (B) Cell growth assays in response to DT and sordarin to phenotypically diagnose diphthamide synthesis on EF2. As indicated, yeast tester strains comprised wild-type (DPH1) and null-mutant (dph1∆) controls as well as the candidate SAM pocket mutants (G238A, H261A, Q321A V349A, R370A, D374A). Ten-fold serial cell dilutions were cultivated for 2-3 days at 30°C without DT or sordarin (left panel: control), under conditions of endogenous DT fragment A production from a galactose-inducible expression plasmid (middle panel: + DT) [19] or in presence of 12.5 µg/mL antifungal (right panel: + sordarin) sufficient to inhibit the wild-type (DPH1) control. Green arrows indicate DT and sordarin resistance.
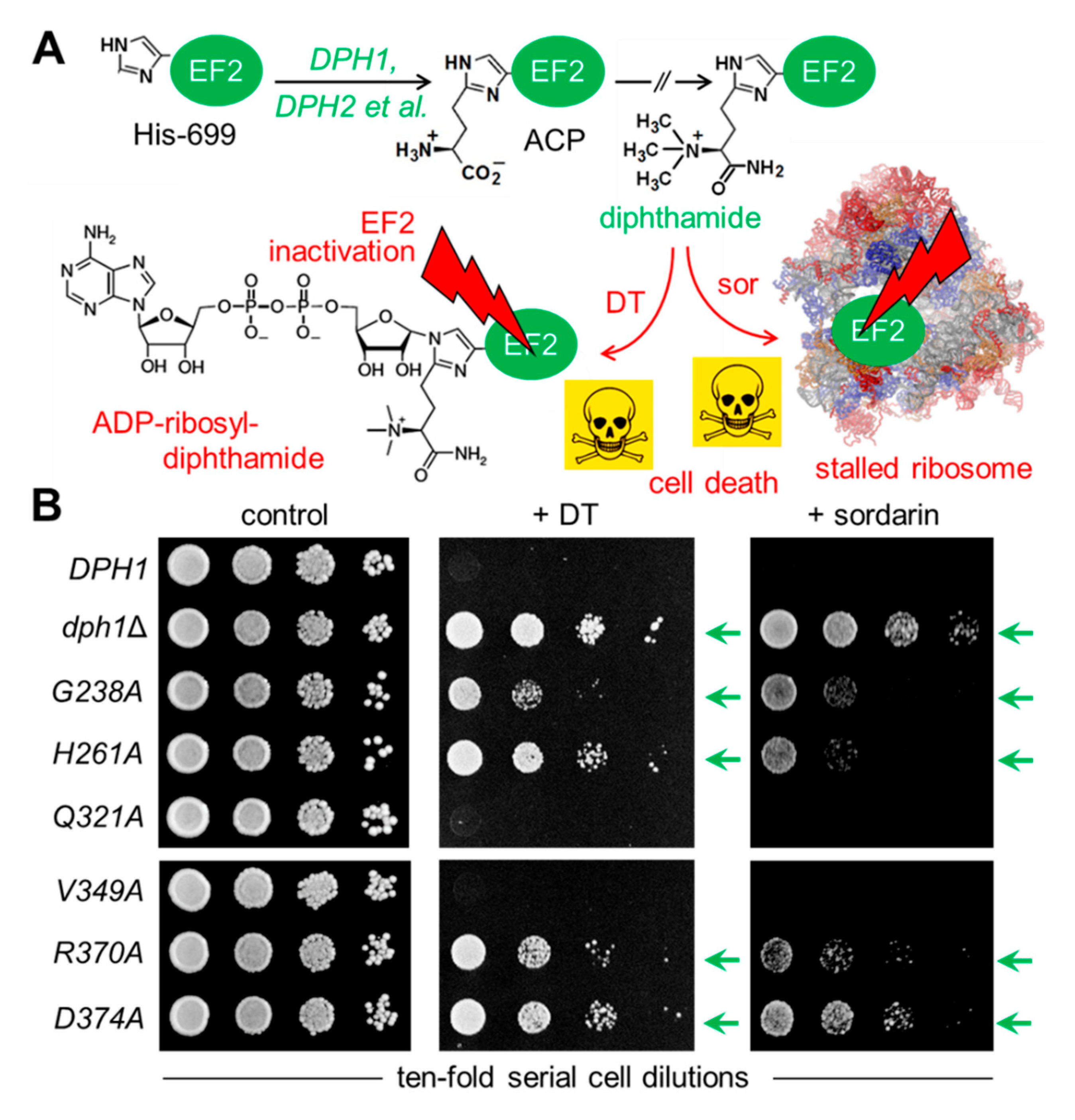
Figure 3.
Diphthamide-dependent ADP ribosylation of EF2 by DT in vitro. Total protein extracts obtained from yeast strains with the indicated genetic backgrounds were treated with (+) or without (-) DT (200 ng) in the presence of 5 µM biotin-NAD at 25°C for 1 h. Detection of the biotin moiety transferred with ADP-ribose to EF2 by DT involved Western blots with a streptavidin peroxidase conjugate. The reaction product (EF2-ADPR-biotin) with a molecular weight of ~100 kDa is denoted by dotted arrows. Unspecific bands marked with an asterisk likely represent endogenously biotinylated yeast proteins. Note that V349A produces wild-type like EF2-ADPR-biotin signals diagnostic for EF2 diphthamide modification, while significantly weaker signals from Q321A suggest reduced (but not entirely abolished) Dph1 activity.
Figure 3.
Diphthamide-dependent ADP ribosylation of EF2 by DT in vitro. Total protein extracts obtained from yeast strains with the indicated genetic backgrounds were treated with (+) or without (-) DT (200 ng) in the presence of 5 µM biotin-NAD at 25°C for 1 h. Detection of the biotin moiety transferred with ADP-ribose to EF2 by DT involved Western blots with a streptavidin peroxidase conjugate. The reaction product (EF2-ADPR-biotin) with a molecular weight of ~100 kDa is denoted by dotted arrows. Unspecific bands marked with an asterisk likely represent endogenously biotinylated yeast proteins. Note that V349A produces wild-type like EF2-ADPR-biotin signals diagnostic for EF2 diphthamide modification, while significantly weaker signals from Q321A suggest reduced (but not entirely abolished) Dph1 activity.
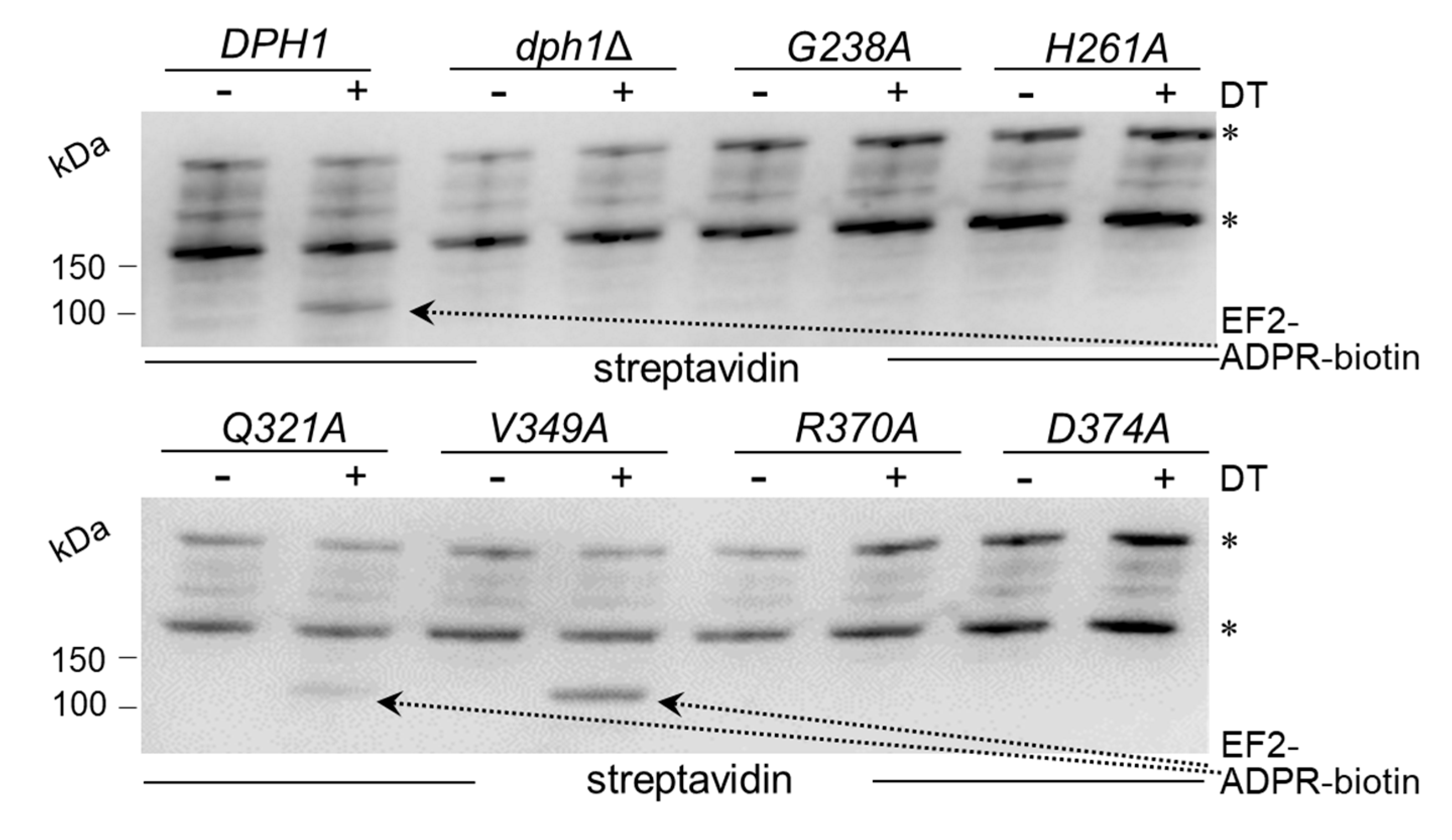
Figure 4.
Characterization of single Q321A and V391A mutants alone and in-tandem. (A) Phenotypic spot assays diagnostic for diphthamide modification defects. As indicated, the tester strains comprised wild-type (DPH1) and null-mutant (dph1∆) controls as well as single (Q321A or V391A) and double (Q321A V391A) mutants. Ten-fold serial cell dilutions were cultivated and grown under conditions essentially described in Figure 2B legend. DT and sordarin resistance traits are indicated (green arrows). (B) Analysis of the EF2 modification state using anti-EF2(pan) antibodies for global EF2 recognition and anti-eEF2(no diphthamide) antibodies to specifically detect unmodified EF2 [37]. For Western blots, protein extracts were obtained from tester strains with the genetic backgrounds as indicated in (A). EF2 degradation products are marked with an asterisk. Detection of Cdc19 with anti-Cdc19 antibodies is shown in parallel Western blots. Note that the combination of the two single Q321A and V391A mutations in the double mutant is phenotypically additive (A) and causes diphthamide modification defects (B) comparable to the null-mutant (dph1∆) control.
Figure 4.
Characterization of single Q321A and V391A mutants alone and in-tandem. (A) Phenotypic spot assays diagnostic for diphthamide modification defects. As indicated, the tester strains comprised wild-type (DPH1) and null-mutant (dph1∆) controls as well as single (Q321A or V391A) and double (Q321A V391A) mutants. Ten-fold serial cell dilutions were cultivated and grown under conditions essentially described in Figure 2B legend. DT and sordarin resistance traits are indicated (green arrows). (B) Analysis of the EF2 modification state using anti-EF2(pan) antibodies for global EF2 recognition and anti-eEF2(no diphthamide) antibodies to specifically detect unmodified EF2 [37]. For Western blots, protein extracts were obtained from tester strains with the genetic backgrounds as indicated in (A). EF2 degradation products are marked with an asterisk. Detection of Cdc19 with anti-Cdc19 antibodies is shown in parallel Western blots. Note that the combination of the two single Q321A and V391A mutations in the double mutant is phenotypically additive (A) and causes diphthamide modification defects (B) comparable to the null-mutant (dph1∆) control.
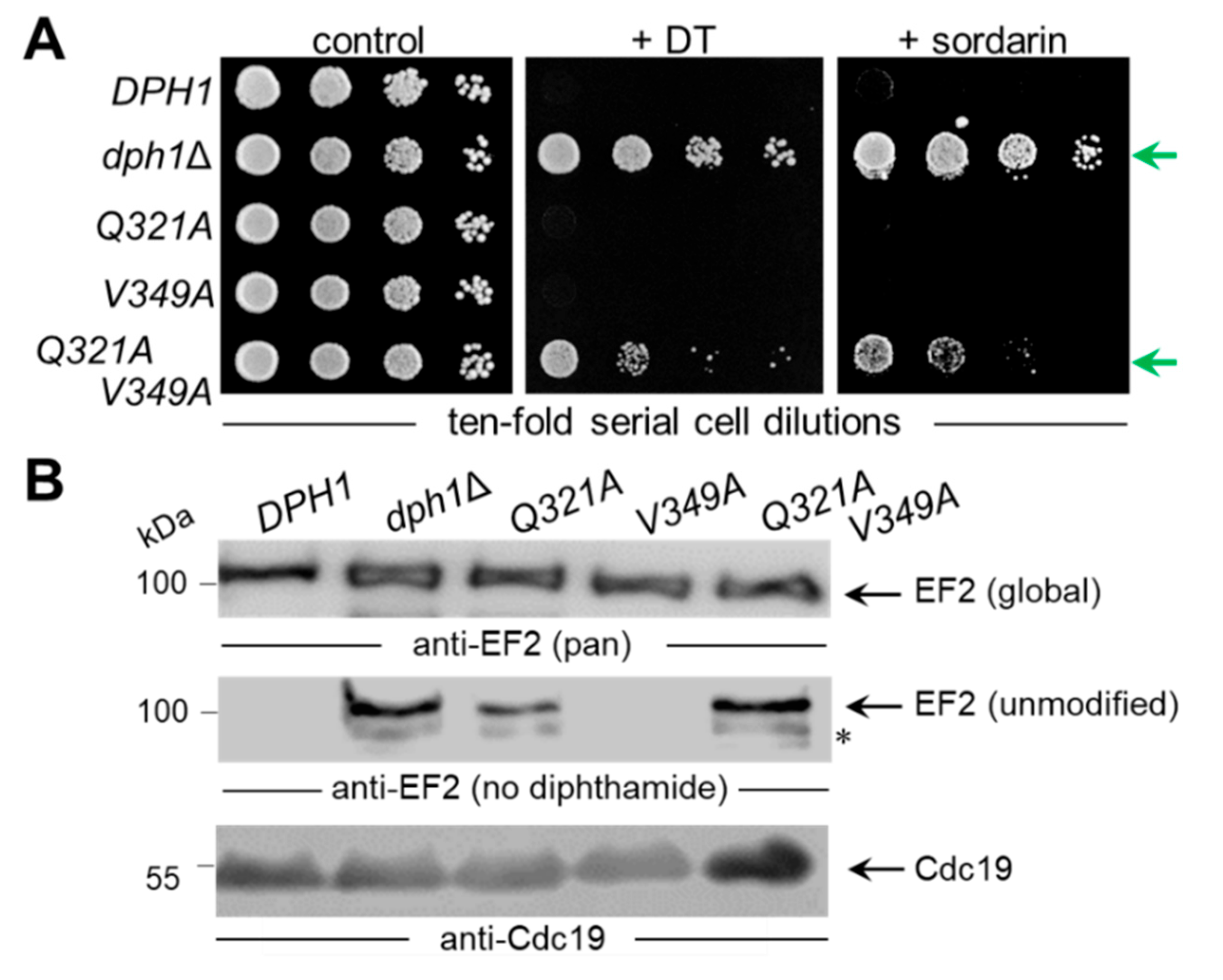
Figure 5.
Functional distinctions of individual SAM pocket residues identified in subunit Dph1 from the yeast Dph1•Dph2 dimer. An AlphaFold2-based structural model [35,36] of Dph1 was aligned to the structure of CmnDph2 in complex with SAM (PDB: 6BXN). Protein structures of CmnDph2 are hidden, while the 4Fe-4S and SAM cofactors remain shown as sticks. Dph1 amino acids (green) are part of a SAM pocket: Gly-238, His-261, Gln-321, Val-349, Arg-370 and Asp-374 conserved to CmnDph2 residues (red): Gly-158, His-180, Gln-237, Val-265, Arg-285, and Asp-289, respectively (see also Figure 1A). Residues in proximity to the SAM methionine moiety are essential (Gly-238, His-261, Arg-370, Asp-374), while amino acids close to the adenine are important but non-essential (Gln-321) or dispensable and non-essential (Val-349).
Figure 5.
Functional distinctions of individual SAM pocket residues identified in subunit Dph1 from the yeast Dph1•Dph2 dimer. An AlphaFold2-based structural model [35,36] of Dph1 was aligned to the structure of CmnDph2 in complex with SAM (PDB: 6BXN). Protein structures of CmnDph2 are hidden, while the 4Fe-4S and SAM cofactors remain shown as sticks. Dph1 amino acids (green) are part of a SAM pocket: Gly-238, His-261, Gln-321, Val-349, Arg-370 and Asp-374 conserved to CmnDph2 residues (red): Gly-158, His-180, Gln-237, Val-265, Arg-285, and Asp-289, respectively (see also Figure 1A). Residues in proximity to the SAM methionine moiety are essential (Gly-238, His-261, Arg-370, Asp-374), while amino acids close to the adenine are important but non-essential (Gln-321) or dispensable and non-essential (Val-349).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



