
Submitted:
02 November 2023
Posted:
03 November 2023
You are already at the latest version
Abstract
Vulvovaginal candidiasis (VVC) is a prevalent condition affecting women worldwide. This study aimed to develop a rapid qPCR assay for accurate identification of VVC etiological agents and reduced azole susceptibility. One hundred and twenty nine vaginal samples from an outpatient clinic (Bilbao-Spain) were analyzed using culture-based methods and multiplex qPCR targeting fungal species, Candida albicans being the predominant species. Antifungal susceptibility tests revealed reduced azole susceptibility in three (3,48%) isolates. Molecular analysis identified several mutations in genes associated with azole resistance and novel mutations in TAC1 and MRR1 genes were also identified, which could contribute to drug resistance.
Keywords:
Vulvovaginal Candidiasis
; qPCR
; Candida species
; antifungal susceptibility
; resistance-related mutations
1. Introduction
Vulvovaginal candidiasis (VVC) is a common condition characterized by vulvovaginal inflammation in combination with the detection of one or more Candida or fungal species [1]. Approximately 75% of women will experience at least one episode of VVC in their lifetime [2], while some of them will develop recurrent VVC (RVVC), which is arbitrarily defined as four or more episodes every year; this fact translates into disease, affecting millions of women worldwide [3,4]. Even though it is not a life-threatening infection, for some women, VVC implies a severe loss of life-quality, mainly due to the wide variety of clinical symptoms, which include vulvovaginal pruritus, soreness or dyspareunia, and also to the fact that acute recurrences often lead to depression and anxiety [5]. Although it is widely recognized that misdiagnosis of vaginal complaints is extremely common, the lack of an accurate diagnosis is still widespread [6]. This fact and the availability of the topical over-the-counter (OTC) drugs, compel patients to self-diagnose and self-medicate, ultimately leading to a bias of epidemiological data and to an increase in less drug-susceptible isolates [4]. The latter may occur due to the appearance of non-albicans Candida (NAC) isolates, some of which are less susceptible or intrinsically resistant to azoles, particularly Nakaseomyces glabrata (formerly known as Candida glabrata), Candida tropicalis or Pichia kudriavcevii (formerly known as Candida krusei) [3]. In this line, some authors have described a trend shift towards NAC species in vaginal samples [7]. Another possibility is the increase of C. albicans resistant isolates, and recent data show a worrying high level of resistance to various azoles in vulvovaginal isolates [7,8]. The long-term treatment with fungistatic fluconazole may entail ideal conditions for C. albicans strains to evolve into a resistant phenotype [4]. The main trajectory for resistance development is up-regulation of CDR1, CDR2, or MDR1 genes related to efflux-pumps. In addition, over-expression or mutations in the ERG11 gene (encoding 14-α demethylase) and loss-of-function mutations in the ERG3 gene (encoding Δ5,6 desaturase), both involved in ergosterol synthesis, are frequently reported in C. albicans resistant isolates [9]. Furthermore, gain-of-function (GOF) mutations in TAC1, UPC2, MRR1 [10,11,12], and more recently MRR2 genes [13], transcription regulators of some of the genes mentioned above, lead to constitutive over-expression, resulting in increased resistance. Besides, genome aneuploidies and loss of heterozygosity are also involved in azole resistance [14]. Many C. albicans isolates exhibit a combination of resistance mechanisms resulting in high levels of drug resistance and clinical failure [15].
Based on isolates identified in VVC, here, we report a real-time PCR assay targeting the multi-copy ITS1 gene of six yeasts species namely C. albicans, C. parapsilosis, C. tropicalis, N. glabrata, P. kudriavcevii, and Pichia guilliermondii. We aimed to identify the etiologic agent as accurately and quickly as possible. The association of VVC/RVVC and some risk factors were also studied. Finally, we also characterized polymorphisms in genes related to azole resistance.
2. Patients and Methods
2.1. Vaginal Samples
One hundred and twenty nine vaginal samples were obtained from 115 women (aged >17 years) attending the outpatient clinic Bombero-Etxaniz (Bilbao, Spain) from February 2013 to July 2015. The research protocol was approved by the ethics committee of the Hospital of Basurto and the University of the Basque Country (UPV/EHU), and all subjects gave their informed consent to participate. Samples were obtained from the lateral vaginal wall with a sterile cotton-tip swab stored at 4°C and processed no later than 48 h. A case of VVC was defined as a patient with vulvar itching, vaginal discharge, and a positive culture.
2.2. Culture and Fungal Identification
Swab content was resuspended in 400 µL of sterile Phosphate Buffered Saline (PBS) and divided into two aliquots. One of the aliquots was cultured on chromogenic agar (Condalab, Spain) for 48h at 37°C, and the other one was stored at -20°C for later multiplex qPCR identification.
2.2.1. Isolates Identification by PCR or ID32C
Green colonies grown on chromogenic medium were further identified by PCR following the protocol described by Romeo and Criseo (2008), to allow discrimination between C. albicans, Candida dubliniensis and Candida africana [16]. Briefly, two colonies from fresh culture were added to 200 µL of PBS with glass-beads, vortexed and added directly to PCR mixture, which included 10 µL of DNA Polymerase Biomix buffer (Bioline, United Kingdom), and 0.8 µL of each Cr primer (25 µM). Not green colonies on chromogenic medium were identified using the ID32C system (bioMérieux, France). Isolates not identified by any of the above-described methods were derived to rDNA amplification (White et al, 1990) [17] and sequencing was performed at the SGIker for identification.
2.3. Molecular Identification of Fungal Species by Multiplex qPCR
2.3.1. Automated DNA Extraction
Vulvovaginal samples were thawed, 90 mg of glass-beads were added and vortexed for 5 minutes. Supernatants were transferred to a new collecting tube and 360 µL of MagNA Pure Bacteria Lysis Buffer (Roche, Germany) and 40 µL of Proteinase K (20 mg/mL) (Roche, Germany) were added. After incubating for 1 hour at 65°C and 10 min al 95°C, automated DNA extraction was performed by using 400 µL of the supernatant in the MagNA Pure Compact nucleic acid extraction instrument (Roche, Germany) using the protocol DNA_Bacteria_V3_2 and the extraction kit MagNA Pure Compact Nucleic Acid Isolation Kit I (Roche, Germany). The extracted DNA was eluted in 50 µL of buffer.
2.3.2. Primer and Taqman Probe Design for qPCR Assays
Sequences of the 18S-ITS1-5.8S region were aligned using the MUSCLE software [18]. All the sequences were obtained from the NCBI Entrez Nucleotide Database (https://www.ncbi.nlm.nih.gov/nucleotide/). Homologous areas of the 18S-ITS1-5.8S region were used to design Diamol-F and Diamol-R primers with the Primer Express software (Applied Biosystems). Furthermore, 12 specific Taqman probes that were designed hybridized into the ITS1 region. Six of them were labelled with 5'-FAM, four with 5'-HEX and two with 5'-JOE; all of them were labelled with 3'-BHQ1. On the other hand, an internal control (IC) of amplification and a probe (ICP) hybridizing this IC were designed to detect inhibition of qPCR. The 5' end of the IC included the same sequence of the primer Diamol-F, and the 3' end the reverse-complementary sequence of the primer Diamol-R. The ICP sequence was located four bases downstream the Diamol-F primer sequence, the rest was a chimeric sequence designed not to hybridize with any or the target species, neither with cohabitating species of the vulvovaginal niches nor with human DNA. ICP was labelled with 5'-ROX and 3'-BHQ2. Table 1 lists the sequences of primers and probes designed.
Probes were marked with different fluorescent reporters attached at the 5' end (FAM, JOE, HEX, and ROX) and Black Hole Quencher (BHQ) at the 3' end. These were supplied by Metabion International AG, Germany, IDT, and/or Sigma-Aldrich. Internal Control (IC) 5' end includes the same sequence (italic letters) of the primer Diamol-F, and the 3' end the reverse-complementary sequence of the primer Diamol-R; Probe-ICP sequence is highlighted in bold. Locked Nucleic Acids are shown in square brackets [].
2.3.3. Multiplex qPCR
All PCR protocols followed the recommendations of Khot and Fredricks (2009) to avoid both DNA contamination and PCR inhibition [19]. Briefly, DNA extraction, PCR reaction mixes, amplification, and PCR product analysis were performed in different rooms. In addition, PCR reaction mixtures were performed in a laminar-flow biosafety cabinet, whose surfaces were cleaned with Termi-DNA-tor (Biotools, Spain).
Before proceeding with multiplex assays, singleplex assays were carried out in a 7300 Real Time Cycler (Applied Biosystem, USA). Amplification mixtures with a final volume of 25 µL contained 10 µL of Premix Ex TaqTM (Takara, Japan), 0.65 µM of each primer, 0.3 µM of each probe, and 1 pmol of the IC. Twenty nanograms of DNA extracted from microorganisms cultures or 2 µL of DNA extracted from vaginal samples were added as template. To determine the analytical sensitivity and reproducibility of the qPCR, decreasing amounts of DNA, from 20 ng to 20 fg, extracted from cultures of 6 fungal species were tested. All samples were assayed in triplicate and on 3 different days. Afterwards, probes were combined in pairs, considering the fluorophore label.
2.4. Antifungal Susceptibility Testing
Fungal isolates were tested for in vitro susceptibility to fluconazole and clotrimazole (Sigma, USA). Fluconazole testing was performed as described in the document M27-4th ed. from the Clinical Laboratory Standards Institute (CLSI) [20], and result interpretations were performed according to M27M44S document [21]. On the other hand, since there is no standardized procedure nor MIC limit ranges for microdilution test for clotrimazole, we followed the protocol proposed by Pelletier et al. (2000), which is based on CLSI M27-A3 document with some modifications [22].
The isolates showing reduced susceptibility to fluconazole or clotrimazole were also tested by Sensititre YeastOne10® (Trek Diagnostic System, UK) colorimetric method.
2.5. Gene Expression Analysis by RT-qPCR
The expression levels of CDR1, CDR2 and MDR1 genes were measured in three resistant (Be-113, Be-114 and Be-129), and three susceptible yeast strain (SC5314, Be-47 and Be-90); for normalization purposes, expression of two reference genes, ACT1 (actin) and PMA1 (H+-ATPase) was also measured [12,23] (Table S1 upper panel). Total RNA was extracted from mid-log cultures in YPD (1% Yeast Extract, 2% Peptone, 2% Glucose) using the MasterPure™ Yeast RNA Purification Kit (Epicentre) following manufacturer’s instructions with some modifications; proteinase K treatment was carried out at 50°C for 20 minutes and DNase treatment was extended up to 30 minutes. In every mutant obtained by CRISPR-Cas9 RNA was extracted from three independent cultures and it was quantified with NanoDrop 1000 (ThermoFisher Scientific). After checking the quality and integrity of RNA the cDNA was synthesized with PrimeScript™ RT reagent Kit (Perfect Real Time; Takara) following manufacturer’s instructions and negative controls (RT-minus) of the reverse-transcription were performed. RNA and cDNA samples stored at - 80°C until use. RT-qPCR was performed in an Applied Biosystems 7300 Real-Time PCR System with SYBR® Premix ExTaq™ (Tli RNaseH Plus; Takara). Technical triplicates of each sample were performed and for each gene a non-template control was included. Stability of the reference genes used for normalization was assessed with the web-based RefFinder tool (https://www.heartcure.com.au/reffinder/). The fold-change was calculated with the 2-ΔΔCt method [24]. RNA transcript levels of the resistant clinical isolates were compared to the average expression of the susceptible ones, which was set to one, and each gene was considered to be overexpressed when fold-change in expression was ≥ 2.
2.6. Sequencing Analysis of ERG11, TAC1, UPC2, MRR1 and MRR2 Genes
Those C. albicans isolates that showed reduced or dose dependent azole susceptibility were selected for sequencing of resistance related genes. DNA was extracted using the DNeasy® Plant Mini Kit (QIAGEN) following manufacturer’s instructions with modifications. Briefly, 4-5 yeast colonies grown in Sabouraud agar O/N at 37°C were suspended in 400 µL of sterile PBS, 90 mg of sterile glass beads (SIGMA) were added and vortexed for 5 minutes. The lysate was transferred to another tube, and 300 µL of Tris buffer (50 mM Tris-HCl pH8, 25 mM Na-EDTA, 75 mM NaCl) and 15 U of lyticase were added. After an incubation at 30°C for 20 minutes, 4 µL of RNAse (QIAGEN) was added and incubated at 37°C for 20 minutes. Then 100 µL of 10% SDS and 40 µL of Proteinase K (ROCHE) were added and incubated at 55°C for 20 minutes. At this point, the manufacturer’s kit protocol was followed from the fourth step. The DNA sample was quantified in NanoDrop 1000 (ThermoFisher Scientific) and stored at -20°C until use.
The primers used for sequencing TAC1, ERG11, UPC2, MRR1 and MRR2 genes were designed to amplify the regions where mutations related to resistance to azoles had been previously identified in the literature and are listed in Table S1 (lower panel) [25,26]. The sequences of the amplicons were provided by the Sequencing and Genotyping Service SGIker of the UPV/EHU. The analysis was done with Chromas 2.5.1 software (Technelysium) to identify heterozygous mutations and, in order to identify the homozygous ones, BioEdit Sequence Alignment Editor 7.2.5 was used to align the sequences obtained with reference sequences for TAC1 (GeneBank accession no. DQ393587), for UPC2 (GeneBank accession no. EU583451), for ERG11 (GeneBank accession no. X13296), for MRR1 (GeneBank accession no. XM_711520) from Morio and collaborators (2013) [15], while for MRR2 the sequence with the GeneBank accession no. XM_705846 was used.
2.7. Statistical Analysis
We used the 2x2 contingency table to calculate sensitivity, specificity, and predictive values of the multiplex qPCR, and X2 test for the association between risk factors and VVC. P values ≤ 0.05 were considered statistically significant.
3. Results
Of the 115 patients enrolled in this study, VVC was confirmed in 86 (66.67%) cases and, among them, 65 (75.58%) were classified as RVVC. The median age of the patients with VVC/RVVC was 30 years (range 17 to 51 years), slightly lower than the total population analyzed, 32 years (range 17 to 57 years). Among positive patients, 46 (53.49%) were pregnant, 17 (19.77%) had received antibiotic treatment in the last month, 13 (15.12%) had a history of allergic disorders, including allergic rhinitis, asthma, or sinusitis, 11 (12.8%) were taking oral contraceptives, 3 (3.49%) male partners of the patients with VVC had symptoms of Candida infection, and 1 (1.16%) was diabetic. Pregnancy was the only risk factor statistically associated to VVC/RVVC.
A total of 86 isolates were obtained from 129 vaginal specimens, including 81 (94.2%) C. albicans, 2 (2.32%) C. parapsilosis, 2 (2.32%) N. glabrata, and 1 (1.16%) C. tropicalis. Only one specimen registered a mixed infection with C. albicans and N. glabrata.
3.1. Yeast Identification by Multiplex qPCR
3.1.1. Collection of Microorganisms
The Diamol primers pair and the combinations of Calb-Cpar3, Cgla-Cgui4, and Ctro2-Ckru2 probes allowed the specific detection of C. albicans-C. parapsilosis, N. glabrata-M. guilliermondii and C. tropicalis-P. kudriavzevii, respectively (Table 2). Almost all the other species in our collection analyzed were negative for all the probes mentioned above, with the exception of the Cgla probe, which hybridized with Saccharomyces cerevisiae DNA. We decided to remove the IC, excluding the possibility of detecting qPCR inhibition, since probe-ICP had to be labelled with ROX; and this impeded the detection of any signal when three probe pairs were combined (data not shown).
On the other hand, Calb, Cpar3 and Cgla probes were tested in order to evaluate their specificity within the C. albicans, C. parapsilosis and N. glabrata complexes, respectively (Table S3). Cpar3 did not recognized C. metapsilosis nor C. orthopsilosis cryptic species, while Calb and Cgla probes hybridized with all species of their respective complexes. New Calb2 and Cgla2 new probes were designed, Calb2 did not hybridize with C. dubliniensis, while Cgla2 did not improve its specificity with respect to the Cgla probe.
3.1.2. Analytical Sensitivity or Limit of Detection (LOD) of the Multiplex qPCR
Three independent standard curves ranging 20 fg to 20 ng of DNA for C. albicans, N. glabrata, C. parapsilosis, M. guilliermondii, C. tropicalis, and P. kudriavzevii showed LOD of 20 fg for all the probes (Figure 1).
3.1.3. Validation of the Multiplex qPCR with Vaginal Swab Samples/Analytical Sensitivity and Specificity of Multiplex qPCR Compared with Conventional Culture
The multiplex qPCR developed was applied to 129 human vulvovaginal samples. Along with these samples, DNA extraction controls were performed, and multiplex qPCR was applied to these controls in order to monitor fungal/DNA contamination and detect false positives.
Of the 129 vaginal swabs, 123 (96.09%) were PCR-positive for C. albicans, 3 (2.325%) for N. glabrata, 2 (1.55%) for C parapsilosis and M. guilliermondii, 3 (2.325%) for C. tropicalis, and 4 (3.1%) P. kudriavzevii. Given such a high rate of positivity, probably because of the non-sterile origin of the samples, a Ct cut-off value of 30 cycles was stablished, so that samples with Ct values above 30 were considered negative. Notably, PCR contamination could be excluded, as evidenced by the lack of amplification in Non Template Controls (NTC) for the 6 probes used (data not shown). The majority of species identified in vaginal swabs by fungal culture and multiplex qPCR were C. albicans, therefore, it was only determined the analytical sensitivity of the qPCR for Calb probe. For that purpose, we built a contingency table (Table S4); the analytical sensitivity of the qPCR for Calb probe using fungal culture as the gold standard was 91.35%, the analytical specificity was 89.6%, the positive predictive value was 93.67%, and the negative predictive value 86%.
We were able to detect the two N. glabrata culture-positive samples with Cgla probe, but we obtained a discordant result negative-culture positive-PCR in one sample. Ctro2 probe detected the only sample culture positive for C. tropicalis, but we were not able to detect the two C. parapsilosis culture-positive samples with Cpar3 probe. Finally, Cgui4 and Ckru2 probes did not hybridized with any sample, so no false-positives were obtained for these probes.
3.2. Antifungal Susceptibility
A total of 78 isolates were tested for susceptibility to fluconazole and clotrimazole (8 isolates lost viability), MIC ranges, MIC50s and MIC90s of C. albicans isolates are summarized in Table S5. Most isolates were susceptible to both azoles. Three C. albicans isolates (Be-113, Be-114 and Be-129), showing high MICs of fluconazole or clotrimazole, were also tested with caspofungin, micafungin, anidulafungin, amphotericin B, 5-flucytosine, voriconazole, posaconazole, itraconazole using the Sensititre YeastOne method (Table S6). Be-113 was resistant to posaconazole and itraconazole, and susceptible-dose-dependent to fluconazole, voriconazole and clotrimazole; Be-114 was susceptible-dose-dependent to fluconazole and clotrimazole. Be-113 and Be-114 showed a MIC of 0.25 µg/ml for clotrimazole, a notably higher value than the rest of the vulvovaginal isolates, therefore they were considered susceptible-dose-dependent. Finally, Be-129 was resistant to fluconazole and voriconazole, and itraconazole susceptible-dose-dependent. Related to NAC strains, the two N. glabrata isolates showed dose dependent-susceptibility to fluconazole and clotrimazole, while C. parapsilosis and C. tropicalis strains were susceptible to both azoles.
3.3. Expression of CDR1, CDR2 and MDR1 Genes of Azole Resistant Isolates
We measured the expression levels of these genes, prior to sequencing, in order to identify which isolates could harbor GOF mutations in Tac1, Mrr1 and Mrr2 transcription factors. Be-113 overexpressed CDR1 and CDR2 (expression level>x2), while Be-114 reached an overexpression close to 2 and Be-129 did not overexpress any of the genes analyzed.
3.4. Amino Acid Substitutions in Erg11, Tac1, Upc2, Mrr1 and Mrr2
Be-113 and Be-129 isolates harbored mutations leading to amino acid substitutions in Erg11 (A114S, Y132H, Y257H and G450E) and Upc2 (G648S, in heterozigosis) that have been previously described and related to azole resistance (Table 3) [12,15,27,28,29,30]. Moreover, Be-113 displayed a homozygous genetic alteration in Tac1, S758F, which is described for the first time in this study. Finally, Be-114 isolate did not harbor any of the known resistance-related mutations that could be related to reduced azole susceptibility, but it had a new mutation, A311V.
4. Discussion
We have developed a multiplex-qPCR assay to identify several Candida and fungal species simultaneously from vaginal swabs. Assay sensitivity and specificity for the C. albicans probe (Calb) and cut-off Ct value of 30 were 91.35% and 89.6%, respectively. Our results improve those obtained by Giraldo and colleagues (S=29.4%) [31], Weissenbacher and colleagues (S=42%) [32] or Mårdh and colleagues (E∽50%) [33] and are similar to the excellent of Cartwright and colleagues [34] or, more recently, of Tardif and Schlaberg or Gaydos and colleagues [1,35]. Non-albicans Candida isolates were scarce so we were unable to validate the qPCR assay for the other probes, but Cgla and Ctro2 probes served to detect two infections caused by N. glabrata and one by C. tropicalis, respectively. The Cpar3 probe could not detect two samples of C. parapsilosis, probably because of the low fungal load of these samples (1-2 CFU). Cgui4 and Ckru2 probes, designed to detect P. guilliermondii and P. kudriavzevii, did not cross-react with any of the clinical samples, highlighting the specificity of these two probes.
Differentiation of Candida/fungal species is clinically relevant, not only for epidemiological reasons but also for differences in virulence and antifungal susceptibility among species [36]. Accurate diagnosis is, therefore, of utmost importance to provide optimal treatment. We were able to detect cryptic species of the Candida albicans and Candida parapsilosis complexes since Calb2 and Cpar3 probes only hybridized with their respective C. albicans and C. parapsilosis strains from our collection. We could not distinguish C. albicans and C. africana using the Calb probe, but both species are highly similar and several studies have also failed to achieve this milestone [36]. Furthermore, some authors do not even consider them different species [37]. Due to differences in virulence and antifungal susceptibility patterns among species within the same complex, the differentiation of Candida cryptic species has also become clinically relevant [36]. Finally, regarding the specificity of the multiplex qPCR assay, none of our probes hybridized with any of the tested microorganisms that can be present in human microbiota, not even with human DNA. An exception came from Cgla probe, which cross-reacted with DNA from S. cerevisiae, a species very much closer to N. glabrata than other species of Candida are [38]. This mismatch implies that this probe must be optimized, and designing a new probe with locked nucleic acid (LNA) technology could be a good option.
Compared with culture, the gold standard for VVC [39], this multiplex qPCR assay might imply an improvement in diagnosing of VVC, especially including accuracy, reliability, and time needed to finish the tests. In this sense, our assay provided an answer for clinical swabs in 5 hours compared to 24-96 hours required for culture detection [6]. Given this delay, many women do not seek medical diagnosis resulting in underdiagnoses of VVC, making it impossible to perform epidemiologic studies of this infection [5]. In this study, 15/115 (13,04%) of patients suspecting fungal infection that attended the outpatient clinic were finally diagnosed with Gardnerella vaginalis, a bacteria responsible for bacterial vaginosis. The later and VVC are common infections that frequently are misdiagnosed [40]. Additionally, over-the-counter (OTC) availability of antifungal agents is convenient for a rapid symptom relief, but they have also worsened the misdiagnosis of candidiasis. Furthermore, azole overuse and overexpose might be associated with a high resistance rate in C. albicans isolates and several authors have reported a trend towards less susceptible vulvovaginal Candida spp. isolates [41,42,43]. Regarding antifungal susceptibility we found three (3,48%) susceptible dose-dependent or azole-resistant C. albicans isolates. All of them were isolated from patients suffering from RVVC for a long time. Two of these isolates harbored mutations related to azole resistance: A114S and Y257H in Erg11p for Be-113 isolate; and Y132H and G450E in Erg11p and G648S in Upc2p for Be-129 [12,15,27,28,29,30]. The aforementioned mutations could explain the high MIC values of Be-113 and Be-129 isolates and the treatment failure for these patients. On the contrary, the Be-114 isolate did not harbor any of the already known resistance-related mutations; firstly classified as clotrimazole SDD, it was reinterpreted with new CLSI cut-off values [21] and categorized as an azole susceptible isolate. Nevertheless, since it was obtained from a patient who did not respond to azole treatment, we would expect Be-114 to be a resistant strain. Bearing in mind that CLSI M27-A4 antifungal drug susceptibility test (AST) does not consider vulvovaginal pH and drug pharmacokinetics in this anatomical niche, Sobel and Akins compared MIC values for 125 C. albicans vaginal isolates at pH 7.0 and pH 4.5 and observed that the lower the pH, the higher the MIC values, as a result of reduction in drug activity Consequently, they proposed that AST for vulvovaginal isolates should be performed at pH 4.5 [8]. This could explain why Be-114 isolate was reported by the lab as sensible, although it behaved as clinically resistant isolate in situ.
To summarize, molecular characterization of resistance-related polymorphisms is plausible for rapid diagnosis of resistance in C. albicans isolates. We used Sanger sequencing to identify mutations associated with resistance, which is laborious and does not permit the analysis of complete genes. However, probe-based real-time PCR assays are easier to perform, and when Next Generation Sequencing (NGS) platforms become cheaper, such broad analyses could replace or encompass culture-based diagnosis [44]. Here we described two new mutations, S758F in Tac1p and A311V in Mrr2p, in two isolates with reduced susceptibility and/or treatment failure, whose putative involvement in fluconazole resistance must be empirically tested, and, to this end, CRISPR-Cas9 seems a promising technology. In addition, vulvovaginal C. albicans isolates may be considered a model to detect new mutations and/or mechanisms related to azole resistance or tolerance, since they are faced to repeated short courses of low antifungal concentration, favoring their adaptation to azole pressure [45].
5. Conclusions
In conclusion, our multiplex-qPCR assay provides a rapid method to accurately diagnose yeast vulvovaginal infections, a condition usually underdiagnosed. Furthermore, the existence of resistance-related mutations emphasizes the need for precise resistance diagnosis, and molecular characterization holds promise for rapid reporting.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Primers for gene expression analysis by RT-qPCR and primers for sequencing of C. albicans resistance-associated genes; Table S2: Collections of microorganisms tested by multiplex qPCR; Table S3: Amplifications results of the probes designed for cryptic species of C. albicans, N. glabrata and C. parapsilosis. Table S4: Comparison of multiplex qPCR assay and culture. Table S5: In vitro antifungal susceptibility of vulvovaginal isolates to several azoles; Table S6: Sensititre YeastOne microdilution method results of 3 resistant C. albicans isolates.
Author Contributions
conceptualization, I.A.A., P.M.M. and M.D.M.; methodology, in vitro assays, writing-original draft preparation I.A.A. and P.M.M.; data collection, data recording I.A.A., G.C. and A.D.; supervision M.D.M.; project administration, funding acquisition I.F.L. and L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Basque Government project, number IT 913-16_Basque University System Research Groups (to M.D.M.), University of the Basque Country project, GIU21/07_Research Groups of the University of the Basque Country UPV/EHU (to I.F.L.), and a Collaborative Research Project among UPV/EHU Groups (to M.D.M., I.F.L and I.A.) P.M.M received a predoctoral grant from the Department of Education, Universities, and Research of the Basque Government (Programa de Formación de Personal Investigador No Doctor).
Institutional Review Board Statement
This study was approved by the ethics committee of the University Hospital of Basurto and the University of the Basque Country (UPV/EHU).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank nurses M.J. Zabala and A. del Val from the outpatient clinic Bombero-Etxaniz (Bilbao-Spain) and Dr. María Soledad Cuétara from the University Hospital Severo-Ochoa (Madrid-Spain) for her unevaluable assistance in providing clinical samples and clinical isolates.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tardif, K.D.; Schlaberg, R. Development of a real-time PCR assay for the direct detection of Candida species causing Vulvovaginal candidiasis. Diagn. Microbiol. Infect. Dis. 2017, 88, 39-40. Available online: https://www.sciencedirect.com/science/article/pii/S0732889317300317.
- Sobel, J.D. Vulvovaginal candidosis. The Lancet 2007, 369, 1961-1971. Available online: https://www.sciencedirect.com/science/article/pii/S0140673607609179.
- Denning, D.W.; Kneale, M.; Sobel, J.D.; Rautemaa-Richardson, R. Global burden of recurrent vulvovaginal candidiasis: a systematic review. Lancet Infect. Dis. 2018, 18, e339-e347. Available online: https://www.sciencedirect.com/science/article/pii/S1473309918301038.
- Marchaim, D.; Lemanek, L.; Bheemreddy, S.; Kaye, K.S.; Sobel, J.D. Fluconazole-Resistant Candida albicans Vulvovaginitis. Obstet. Gynecol. 2012, 120, 1407-1414. Available online: https://journals.lww.com/greenjournal/fulltext/2012/12000/fluconazole_resistant_candida_albicans.22.aspx.
- Sobel, J.D. Recurrent vulvovaginal candidiasis. Am. J. Obstet. Gynecol. 2016, 214, 15-21. Available online: https://www.sciencedirect.com/science/article/pii/S0002937815007164.
- Sobel, J.D.; Akins, R.A. The Role of PCR in the Diagnosis of Candida Vulvovaginitis—a New Gold Standard? Curr. Infect. Dis. Rep. 2015, 17, 33. [CrossRef]
- Arechavala, A.; Negroni, R.; Santiso, G.; Depardo, R.; Bonvehí, P. Chronic recurrent vulvovaginitis is not only due to Candida. Rev. Iberoam. Micol. 2021, 38, 132-137. Available online: https://www.sciencedirect.com/science/article/pii/S1130140621000243.
- Sobel, J.D.; Akins, R. Determining Susceptibility in Candida Vaginal Isolates. Antimicrob. Agents Chemother. 2022, 66, e0236621. [CrossRef]
- Berkow, E.L.; Lockhart, S.R. Fluconazole resistance in Candida species: a current perspective. Infect. Drug Resist. 2017, 237-245. [CrossRef]
- Coste, A.T.; Karababa, M.; Ischer, F.; Bille, J.; Sanglard, D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 2004, 3, 1639-1652. [CrossRef]
- Dunkel, N.; Blaß, J.; Rogers, P.D.; Morschhäuser, J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol. Microbiol. 2008, 69, 827-840. [CrossRef]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289-1299. [CrossRef]
- Nishimoto, A.T.; Zhang, Q.; Hazlett, B.; Morschhäuser, J.; Rogers, P.D. Contribution of clinically derived mutations in the gene encoding the zinc cluster transcription factor Mrr2 to fluconazole antifungal resistance and CDR1 expression in Candida albicans. Antimicrob. Agents Chemother. 2019, 63, e00078-19. [CrossRef]
- Nishimoto, A.T.; Sharma, C.; Rogers, P.D. Molecular and genetic basis of azole antifungal resistance in the opportunistic pathogenic fungus Candida albicans. J. Antimicrob. Chemother. 2019, 75, 257-270. [CrossRef]
- Morio, F.; Pagniez, F.; Besse, M.; Gay-andrieu, F.; Miegeville, M.; Le Pape, P. Deciphering azole resistance mechanisms with a focus on transcription factor-encoding genes TAC1, MRR1 and UPC2 in a set of fluconazole-resistant clinical isolates of Candida albicans. Int. J. Antimicrob. Agents 2013, 42, 410-415. Available online: https://www.sciencedirect.com/science/article/pii/S0924857913002720.
- Romeo, O.; Criseo, G. First molecular method for discriminating between Candida africana, Candida albicans, and Candida dubliniensis by using hwp1 gene. Diagn. Microbiol. Infect. Dis. 2008, 62, 230-233. Available online: https://www.sciencedirect.com/science/article/pii/S0732889308002757.
- 17. White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; pp 315-322.
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792-1797. [CrossRef]
- Khot, P.D.; Fredricks, D.N. PCR-based diagnosis of human fungal infections. Expert Rev. Anti-Infe. 2009, 7, 1201-1221. [CrossRef]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Fourth Edition M27; CLSI: Wayne, PA, USA, 2017.
- Clinical and Laboratory Standards Institute. Performance Standards for Antifungal Susceptibility Testing of Yeasts: Third Edition; CLSI Supplement M27M44S: CLSI, 2022.
- Pelletier René; Joanne, P.; Cynthia, A.; Corina, G.; Lauren, W.; Walsh Thomas, J. Emergence of Resistance of Candida albicans to Clotrimazole in Human Immunodeficiency Virus-Infected Children: In Vitro and Clinical Correlations. J. Clin. Microbiol. 2000, 38, 1563-1568. [CrossRef]
- Morio, F.; Pagniez, F.; Lacroix, C.; Miegeville, M.; Le Pape, P. Amino acid substitutions in the Candida albicans sterol Δ5,6-desaturase (Erg3p) confer azole resistance: characterization of two novel mutants with impaired virulence. J. Antimicrob. Chemother. 2012, 67, 2131-2138. [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402-408. Available online: https://www.sciencedirect.com/science/article/pii/S1046202301912629.
- Coste, A.; T.; Crittin, J.; Bauser, C.; Rohde, B.; Sanglard, D. Functional Analysis of cis- and trans-Acting Elements of the Candida albicans CDR2 Promoter with a Novel Promoter Reporter System. Eukaryot. Cell. 2009, 8, 1250-1267. [CrossRef]
- Wang, Y.; Liu, J.; Shi, C.; Li, W.; Zhao, Y.; Yan, L.; Xiang, M. Mutations in transcription factor Mrr2p contribute to fluconazole resistance in clinical isolates of Candida albicans. Int. J. Antimicrob. Agents 2015, 46, 552-559. Available online: https://www.sciencedirect.com/science/article/pii/S0924857915002897.
- Xiang, M.; Liu, J.; Ni, P.; Wang, S.; Shi, C.; Wei, B.; Ni, Y.; Ge, H. Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res. 2013, 13, 386-393. [CrossRef]
- Hiroshi, K.; Yoshitsugu, M.; Haruko, M.; Katherine, N.; Brian, G.; Bennett John, E. Genetic Analysis of Azole Resistance in the Darlington Strain of Candida albicans. Antimicrob. Agents Chemother. 2000, 44, 2985-2990. [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E. Y132H substitution in Candida albicans sterol 14α-demethylase confers fluconazole resistance by preventing binding to haem. FEMS Microbiol. Lett. 1999, 180, 171-175. [CrossRef]
- Sanglard, D.; Ischer, F.; Koymans, L.; Bille, J. Amino Acid Substitutions in the Cytochrome P-450 Lanosterol 14α-Demethylase (CYP51A1) from Azole-Resistant Candida albicans Clinical Isolates Contribute to Resistance to Azole Antifungal Agents. Antimicrob. Agents Chemother. 1998, 42, 241-253. [CrossRef]
- Giraldo, P.; von Nowaskonski, A.; Gomes, F.A.M.; Linhares, I.; Neves, N.A.; Witkin, S.S. Vaginal colonization by Candida in asymptomatic women with and without a history of recurrent vulvovaginal candidiasis. Obstet. Gynecol. 2000, 95, 413-416. Available online: https://www.sciencedirect.com/science/article/pii/S0029784499005773.
- Weissenbacher, T.; Witkin, S.S.; Ledger, W.J.; Tolbert, V.; Gingelmaier, A.; Scholz, C.; Weissenbacher, E.R.; Friese, K.; Mylonas, I. Relationship between clinical diagnosis of recurrent vulvovaginal candidiasis and detection of Candida species by culture and polymerase chain reaction. Arch. Gynecol. Obstet. 2009, 279, 125-129. [CrossRef]
- Mårdh, P.A.; Novikova, N.; Witkin, S.S.; Korneeva, I.; Rodriques, A.R. Detection of Candida by polymerase chain reaction vs microscopy and culture in women diagnosed as recurrent vulvovaginal cases. Int. J. STD AIDS 2003, 14, 753-756. [CrossRef]
- Cartwright Charles, P.; Lembke Bryndon, D.; Kalpana, R.; Body Barbara, A.; Nye Melinda, B.; Rivers Charles, A.; Schwebke Jane, R. Comparison of Nucleic Acid Amplification Assays with BD Affirm VPIII for Diagnosis of Vaginitis in Symptomatic Women. J. Clin. Microbiol. 2020, 51, 3694-3699. [CrossRef]
- Gaydos, C.A.; Beqaj, S.; Schwebke, J.R.; Lebed, J.; Smith, B.; Davis, T.E.; Fife, K.H.; Nyirjesy, P.; Spurrell, T.; Furgerson, D.; Coleman, J.; Paradis, S.; Cooper, C.K. Clinical Validation of a Test for the Diagnosis of Vaginitis. Obstet. Gynecol. 2017, 130, 181-189. Available online: https://journals.lww.com/greenjournal/fulltext/2017/07000/clinical_validation_of_a_test_for_the_diagnosis_of.25.aspx.
- Arastehfar, A.; Fang, W.; Pan, W.; Liao, W.; Yan, L.; Boekhout, T. Identification of nine cryptic species of Candida albicans, C. glabrata, and C. parapsilosis complexes using one-step multiplex PCR. BMC Infect. Dis. 2018, 18, 480, 10.1186/s12879-018-3381-5. [CrossRef]
- Theill, L.; Dudiuk, C.; Morano, S.; Gamarra, S.; Nardin, M.E.; Méndez, E.; Garcia-Effron, G. Prevalence and antifungal susceptibility of Candida albicans and its related species Candida dubliniensis and Candida africana isolated from vulvovaginal samples in a hospital of Argentina. Rev. Argent. Microbiol. 2016, 48, 43-49. Available online: https://www.sciencedirect.com/science/article/pii/S0325754115001492.
- Kurtzman, C.P.; Robnett, C.J. Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 5' end of the large-subunit (26S) ribosomal DNA gene. J. Clin. Microbiol. 1997, 35, 1216-1223. [CrossRef]
- Nyirjesy, P.; Brookhart, C.; Lazenby, G.; Schwebke, J.; Sobel, J.D. Vulvovaginal Candidiasis: A Review of the Evidence for the 2021 Centers for Disease Control and Prevention of Sexually Transmitted Infections Treatment Guidelines. Clin. Infect. Dis. 2022, 74, S162-S168. [CrossRef]
- Nyirjesy, P. Vulvovaginal Candidiasis and Bacterial Vaginosis. Infect. Dis. Clin. North Am. 2008, 22, 637-652. Available online: https://www.sciencedirect.com/science/article/pii/S0891552008000524.
- Sobel, J.D.; Sobel, R. Current treatment options for vulvovaginal candidiasis caused by azole-resistant Candida species. Expert Opin. Pharmacother. 2018, 19, 971-977. [CrossRef]
- Ying, C.; Zhang, H.; Tang, Z.; Chen, H.; Gao, J.; Yue, C. Antifungal susceptibility and molecular typing of 115 Candida albicans isolates obtained from vulvovaginal candidiasis patients in 3 Shanghai maternity hospitals. Med. Mycol. 2015, 54, 394-399. [CrossRef]
- Bulik, C.C.; Sobel, J.D.; Nailor, M.D. Susceptibility profile of vaginal isolates of Candida albicans prior to and following fluconazole introduction – impact of two decades. Mycoses 2011, 54, 34-38. [CrossRef]
- Morio, F.; Jensen, R.H.; Le Pape, P.; Arendrup, M.C. Molecular basis of antifungal drug resistance in yeasts. Int. J. Antimicrob. Agents 2017, 50, 599-606. Available online: https://www.sciencedirect.com/science/article/pii/S0924857917302054.
- Sobel, J.D. Resistance to Fluconazole of Candida albicans in Vaginal Isolates: a 10-Year Study in a Clinical Referral Center. Antimicrob. Agents Chemother. 2023, 67, 181, e00181-23. [CrossRef]
Figure 1.
Results of the representative standard curves with de singleplex qPCR assay: Logarithmic representation of fluorescence detected with (A) Calb, (C) Cgla, (E) Cpar3, (G) Cgui4, (I) Ctro2, and (K) Ckru2 probes with the C. albicans, N. glabrata, C. parapsilosis, M. guilliermondii, C. tropicalis, and P. kudriavzevii (respectively) DNA serial dilutions from 20 fg to 20 ng. (B, D, F, H, J, and L) Plot of the Ct values versus the logarithmic of the genomic DNA starting concentration. Numbers next to replicates indicate the detection threshold cycles mean values obtained for each DNA concentration ± SD.
Figure 1.
Results of the representative standard curves with de singleplex qPCR assay: Logarithmic representation of fluorescence detected with (A) Calb, (C) Cgla, (E) Cpar3, (G) Cgui4, (I) Ctro2, and (K) Ckru2 probes with the C. albicans, N. glabrata, C. parapsilosis, M. guilliermondii, C. tropicalis, and P. kudriavzevii (respectively) DNA serial dilutions from 20 fg to 20 ng. (B, D, F, H, J, and L) Plot of the Ct values versus the logarithmic of the genomic DNA starting concentration. Numbers next to replicates indicate the detection threshold cycles mean values obtained for each DNA concentration ± SD.
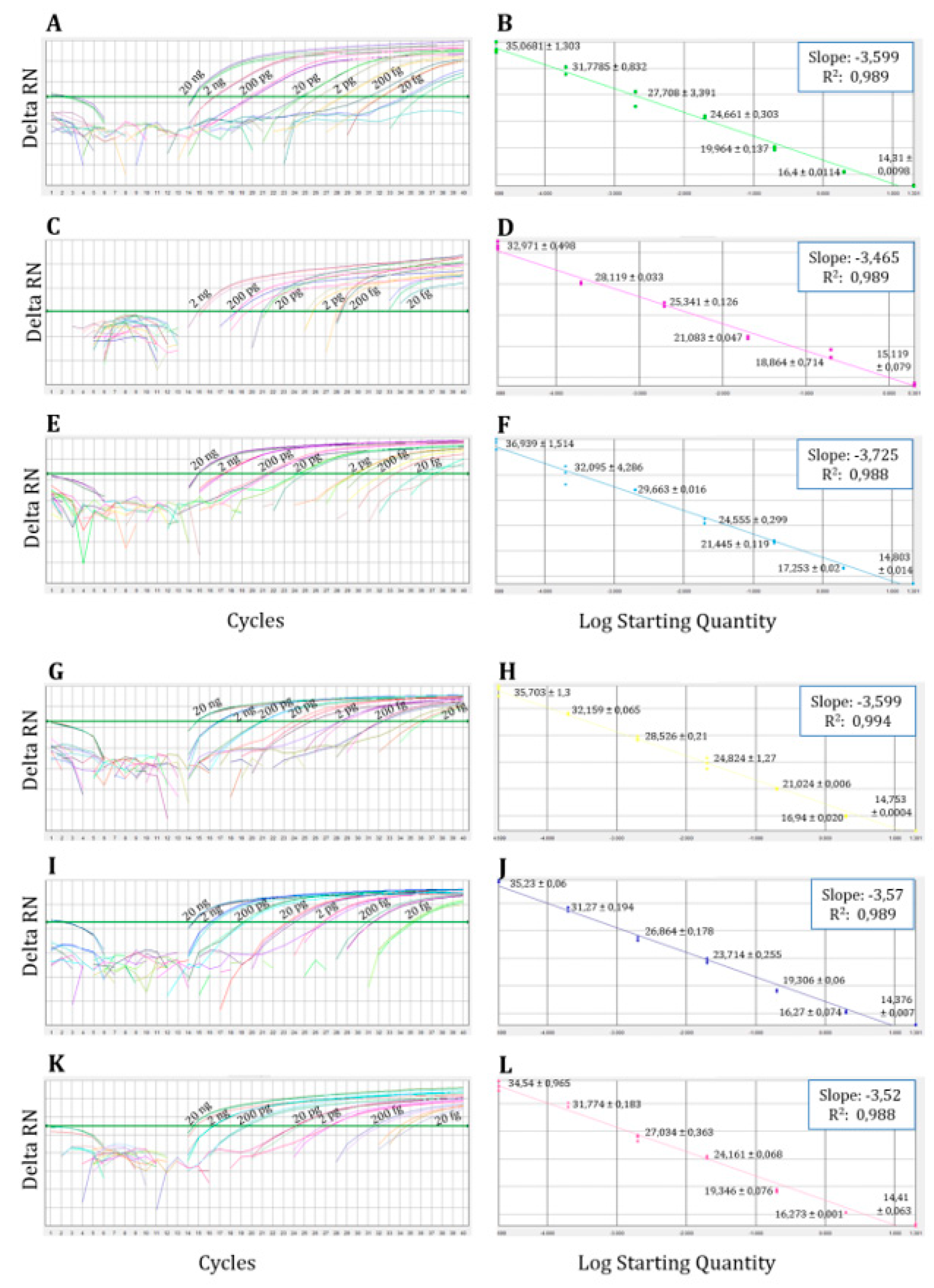

Table 1.
Sequences of primers and probes used in the multiplex qPCR assays.
| Primers and probes | Species | Gene | Sequences (5’→ 3’) |
|---|---|---|---|
| Primers | |||
| Diamol-F | - | 18S | TAGGTGAACCTGCGGAAGGA |
| Diamol-R | - | 5.8S | TCGCTGCGTTCTTCATCGAT |
| Probes | |||
| Calb | C. albicans | ITS1 | FAM-CGGTGGGCCCAGCCTGCC-BHQ1 |
| Calb2 | C. albicans | FAM-ATCAA[C]TTGTCACA[C][C]AGA-ZNA4-BHQ1 | |
| Cpar2 | C. parapsilosis | JOE-AGGCC[C]CATA[T]AGAAGG[C]CTA-BHQ1 | |
| Cpar3 | C. parapsilosis | HEX-TGGCAGGCCCCATATAGAAGGCCTAC-BHQ1 | |
| Cgla | N. glabrata | FAM-ATTTCTCCTGCCTGCGCTTAAGTGCG-BHQ1 | |
| Cgla2 | N. glabrata | FAM-TTAAGTGCGCGG[T][T]GGTGG-ZNA4-BHQ1 | |
| Cgui3 | M. guilliermondii | JOE-AA[C]CTA[T]CT[C]TA[G]GC[C]AAA-BHQ1 | |
| Cgui4 | M. guilliermondii | HEX-CAGCGTTTAACTGCGCGGCGA-BHQ1 | |
| Ctro | C. tropicalis | HEX-CGGTAGGATTGCTCCCGCCA-BHQ1 | |
| Ctro2 | C. tropicalis | HEX-CGGTAGGATTGCTCCCGCCACC-BHQ1 | |
| Ckru | P. kudriavzevii | FAM-TTTAGGTGTTGTTGTTTTCGTTCCGCTC-BHQ1 | |
| Ckru2 | P. kudriavzevii | FAM-CTACACTGCGTGAGCGGAACGAAAAC-BHQ1 | |
| Control | |||
| Probe-ICP | - | - | ROX-AACGTGCGACGTTCCGAGCA-BHQ2 |
| IC | - | - | TAGGTGAACCTGCGGAAGGATCGAAACGTGCGACGCTTCCGAGCATGATCACTATGTCCTAATCCCATATATTATTCACTGTGTACTAGCCCTTCTTGGTTCTCGCATCGATGAAGAACGCAGCGA |
Table 2.
Results obtained by multiplex qPCR with some species from our microorganism collection including Ct mean values ± SD.
Table 2.
Results obtained by multiplex qPCR with some species from our microorganism collection including Ct mean values ± SD.
| Probes (Ct ± SD)b | ||||||||
|---|---|---|---|---|---|---|---|---|
| Calb | Cgla | Cpar3 | Cgui4 | Ctro2 | Ckru2 | |||
| Species | Straina | |||||||
| C. albicans | NCPF 3153 | 14.26 ± 0.056 | - | - | - | - | - | |
| N. glabrata | NCPF 3203 | - | 13.34 ± 0.056 | - | - | - | - | |
| C. parapsilosis | NCPF 3104 | - | - | 14.4 ± 0.106 | - | - | - | |
| M. guilliermondii | NCPF 3099 | - | - | - | 14.33 ± 0.891 | - | - | |
| C. tropicalis | NCPF 3111 | - | - | - | - | 14.31 ± 0.993 | - | |
| P. kudriavzevii | ATCC 6258 | - | - | - | - | - | 13.64 ± 0.007 | |
| Other yeasts | ||||||||
| Saccharomyces cerevisiae | CECT 1678 | - | 18.37 | - | - | - | - | |
| Saprochaete capitata | IHEM 5666 | - | - | - | - | - | - | |
| Yarrowia lipolytica | UPV 12-097 | - | - | - | - | - | - | |
| Rhodotorula mucilaginosa | CECT 11016 | - | - | - | - | - | - | |
| Filamentous fungi | ||||||||
| Aspergillus fumigatus | Af-293 | - | - | - | - | - | - | |
| Lomentospora prolificans | ATCC 64913 | - | - | - | - | - | - | |
| Cryptococcus neoformans | ATCC 90113 | - | - | - | - | - | - | |
| Bacteria | ||||||||
| Staphylococcus aureus | CECT 435 | - | - | - | - | - | - | |
| Streptococcus pyogenes | CECT 985 | - | - | - | - | - | - | |
| Streptococcus viridans | CECT 804 | - | - | - | - | - | - | |
| Streptococcus pneumoniae | CECT 993 | - | - | - | - | - | - | |
| Escherichia coli | CECT 434 | - | - | - | - | - | - | |
| Klebsiella pneumoniae | CECT 144 | - | - | - | - | - | - | |
| Pseudomonas aeruginosa | CECT 108 | - | - | - | - | - | - | |
| Proteus mirabilis | CECT 4168 | - | - | - | - | - | - | |
| Gardnerella vaginalis | ATCC 14018 | - | - | - | - | - | - | |
| Human DNA | ||||||||
| Human Genomic DNA (Promega, Spain) | G304A* | - | - | - | - | - | - | |
Table 3.
Amino acid substitutions in the transcription factors Tac1p, Upc2p, Mrr1p and Mrr2p and in Erg11p.
Table 3.
Amino acid substitutions in the transcription factors Tac1p, Upc2p, Mrr1p and Mrr2p and in Erg11p.
| Isolate | Erg11pa | Tac1pa | Upc2pa | Mrr1a | Mrr2a |
| Be-113 | A114S; Y257H | N396Sh; S758F | - | V341Eh; L592Fh; E1020Qh | - |
| Be-114 | - | A337Vh; N396S; N772K; D776N; E829Qh; S941Ph | - | V341E; E1020Q | A311V; A451A; V582L |
| Be-129 | Y132H; G450E | N396Sh; N772Kh; D776Nh; E829Qh;S935Lh; S941Ph | G648Sh | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



