Submitted:
02 November 2023
Posted:
03 November 2023
You are already at the latest version
Abstract
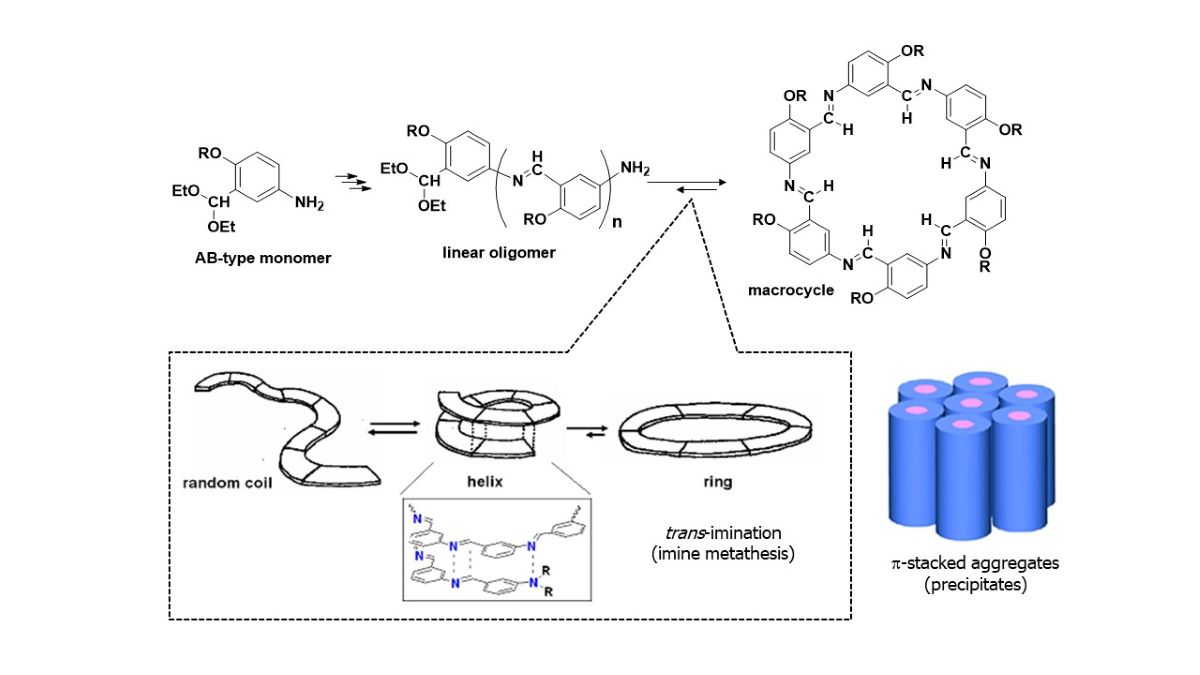
Hexakis(2-alkoxy-1,5-phenyleneimine) macrocycles were synthesized using a simple one−pot procedure through by precipitation−driven cyclization. The acetal−protected AB−type monomers, 2-alkoxy-5-aminobenzaldehyde diethyl acetals, underwent polycondensation in water or acid−containing tetrahydrofuran. The precipitation−driven cyclization based on imine dynamic covalent chemistry and π−stacked columnar aggregation played a decisive role in the one−pot synthesis. The progress of the reaction was analyzed by MALDI−TOF mass spectrometry. The macrocycles with alkoxy chains were soluble in specific organic solvents such as chloroform, allowing their structures to be analyzed by NMR. The shape−anisotropic, nearly planar, and persistent macrocycles aggregated into columnar assemblies in polymerization solvents, driven by aromatic π−stacking. The octyloxylated macrocycle OcO−Cm6 exhibited an enantiotropic columnar liquid crystal−like mesophase between 165°C and 197°C. In the SEM image of (S)-(−)-3,7-dimethyloctyloxylated macrocycle (−)BCO-Cm6, columnar substances with a diameter of 100−200 nm were observed. The polymerization solution for 2-(2-methoxyethoxy)ethoxy)ethoxylated macrocycle (TEGO−Cm6) gelled and showed thixotropic properties by forming a hydrogen bond network.
Keywords:
simple one−pot procedure
; imine−based macrocycles
; dynamic covalent polymer
; Schiff base
; azomethine
; precipitation−driven cyclization
; −stacked columnar aggregates
; columnar liquid crystal
; physical gel
; thixotropy
1. Introduction
Shape−persistent macrocycles are attractive in the fields of supramolecular chemistry and material science because of their unique structure and novel properties [1]. Tradition-ally, the widespread utilization of these macromolecules was hindered by tedious prepa-ration, often requiring dilute conditions, small scales, difficult separations, and low over-all yields. The efficient preparation of functionalized macrocycles has been a challenging task. In the past two decades, many efficient synthesis methods for shape−persistent macrocycles through cross−coupling [2,3] or dynamic covalent chemistry (DCC) [4,5,6,7,8,9] have been reported. A dynamic covalent bond is one of the ideal linkages for the construction of large and robust organic architectures. Better results have been obtained by a dynamic covalent approach involving reversible metathesis reactions that afford macrocycles in one step. Mechanistic studies demonstrate that macrocycle formation is thermodynamically controlled by the route [10]. Among macrocyclic compounds, imine macrocycles constitute an important and diverse class of compounds, which have applications in catalysis, recognition, separation, and medical diagnostics [11,12,13,14,15]. Imine−based macrocycles are generally prepared by AA−BB type polycondensations from dialdehydes and diamines. The [n+n] condensation may result in the formation of various macrocyclic products, such as [2+2], [3+3], etc., as well as linear oligomeric and polymeric imines. The selectivity and yields are dramatically enhanced in condensation reactions templated by transition metal ions such as zinc (II), copper (II), or nickel (II) [16,17,18,19,20,21,22,23]. Lisowski, J. reported that the [3+3] macrocycles were sometimes obtained in high yields by direct condensation without a metal template in the condensation of aromatic dialdehydes with chiral diamines such as 1,2-trans-diaminocyclohexane [24]. Lehn, J. −M. et al. investigated the self−assembly and self−sorting behavior of dynamic covalent organic architectures which makes possible the parallel generation of multiple discrete products in a simple one−pot procedure. They reported the self−assembly of covalent organic macrocycles and macrobicyclic cages from dialdehyde and polyamine components via multiple [2+2] and [3+2] polyimine condensations [25]. Imine chemistry is among the most well-established reversible reactions and has also been a main synthetic tool. Various shape−persistent macrocycles and covalent organic polyhedrons have been efficiently constructed in one step through dynamic imine chemistry. Zhang, W. and co−workers prepared imine−linked porous polymer networks, which exhibit permanent porosity with high specific surface areas. Their most recent contribution is the discovery of a recyclable polyimine material whose self−healing can be activated simply by heating or water treatment [26]. Zhang, D. and co−workers developed covalent polymeric networks composed of imine cross−linkages which exhibit malleability and self−healing characteristics [27].
Hughes, T. and co−workers reported on the efficient synthesis of an imine−based macrocycle from an AB−type monomer [28,29]. They synthesized a novel ortho-phenylene-para-phenyleneimine macrocycle through one−pot reduction and cyclooligomerization of 2-(4-nitrophenyl)benzaldehyde. Specifically, Fe(0) and aqueous HCl reduce the nitroaldehyde to the AB monomer aminoaldehyde which then undergoes spontaneous macrocyclization to give moderate yields of the macrocycle. This methodology is facile, requires no purification of the products, and is environmentally friendly. Recently, Mori, K. et al. developed a concise access to C3−symmetric imine−linked macrocycles from AB−type monomers with an aldehyde group and Boc−protected amine [30]. When the monomers were treated with an excess amount of concentrated HCl in 1,4-dioxane, the detachment of the Boc group followed by a trimerization reaction via imine formation proceeded smoothly to afford the macrocycles in good chemical yields.
Discotic liquid crystals have disc−shaped rigid cores and flexible side chains that ra-diate from the core, and are also called columnar liquid crystals because they form col-umns by stacking. Hexakis(m-phenyleneimine) macrocycle Cm6, which is a disc−shaped molecule and has the property of stacking in a columnar shape [31], is expected to show a liquid crystal phase by introducing a flexible long side chain into Cm6. Tew, G. and co−workers reported that shape−persistent macrocycles with branched alkoxy− and/or tri-ethylene glycol (TEG) side chains exhibited a columnar liquid crystal phase [32]. Tanaka, K. successfully obtained a giant macrocycle with long and branched side chains, which exhibited a rectangular columnar liquid crystal phase over a wide temperature range [33]. Macrocyclic compounds can form supramolecular gels, also known as physical gels. Granata, G. and co−workers reported a supramolecular nanohydrogel formed by a bio-friendly micellar self−assembling choline−calix [4]arene derivative in the presence of curcumin, a natural and multitarget pharmacologically relevant drug [34]. In these gels, small molecules (gelators) self−assemble through non−covalent interactions, usually into a network of fibers, to trap solvent. Many physical gels are responsive to stimuli and often these types of gels can be reversibly converted from gel to sol (thixotropic property). The properties make them ideal candidates for investigation into a range of potential applications, including biomedical, smart materials, sensors, and catalysts [35].
Our research focuses on the design and synthesis of discrete molecular architectures, such as macrocycles through a simple one−pot procedure and explores their unique properties. In the previous paper we reported a highly efficient one−pot synthesis of a shape-persistent macrocycle, hexakis(m-phenyleneimine) macrocycle Cm6, based on imine dynamic covalent chemistry [31]. The present article describes the synthesis of hexakis(m-phenyleneimine) macrocycles with long alkoxy side chains from the corresponding AB−type monomers through dynamic covalent chemistry, which includes their characterization and unique properties such as columnar liquid crystal and physical gel formation.
2. Materials and Methods
2.1. Materials and Instruments
2-Hydroxy-5-nitrobenzaldehyde, platinum (IV) oxide, 5% palladium on active carbon (Pd/C), sodium hydrogen carbonate, 95% sulfuric acid, trifluoroacetic acid, dichloromethane, diethyl ether, phosphorus tribromide, and magnesium sulfate were obtained from FUJIFILM Wako Pure Chemical Co. (Wako) (Tokyo, Japan). 1-Bromooctane, (S)-(−)-3,7-dimethyl-6-octen-1-ol, triethoxymethane, and dichloromethane were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Potassium carbonate was donated by Kanto Chemical Co., Inc. (Tokyo, Japan). Tetrahydrofuran, ethanol, N,N-dimethylformamide (DMF), and ethyl acetate were obtained from Wako and distilled over calcium hydride. Unless otherwise stated, all reagents were used as received without further purification.
The 1H− and 13C−NMR spectra were obtained using a JEOL JNM−LA500 spectrometer (JEOL Ltd., Tokyo, Japan). The proton signals in the 1H−NMR spectrum were assigned from the H,H−COSY and C,H−COSY spectra. FT−IR spectra were recorded using a JASCO VALOR−III Fourier transform spectrometer (JASCO Co., Tokyo, Japan). MALDI−TOF MS measurements were conducted with an Applied Biosystems Voyager-DETEPRO−T spectrometer (Applied Biosystems, Waltham, Massachusetts, USA). 2,5-Dihydroxybenzoic acid (DHB) was used as a matrix. Middle pressure liquid chromatography (MPLC) was performed on a YAMAZEN MPLC system (YAMAZEN Co., Osaka, Japan) consisting of a YAMZEN 540 pump, a YAMAZEN Prep UV−10V detector, and a silica gel column. TEM (JEM−2000EXII, JEOL Ltd., Tokyo, Japan) and SEM (JSM−5310, JEOL Ltd., Tokyo, Japan) were used to observe the morphology. WAXD (Rint-2000, Rigaku Co., Tokyo, Japan) was conducted using CuKα radiation of wavelength 1.54056 Å generated at a voltage of 40 kV and a current of 30 mA. WAXD data were acquired using a scintillation counter detector in a diffraction angle (2θ) range of 3−30° at a scanning speed of 2°/min. Thermal characterization of OcO−Cm6 and (−)BCO−Cm6 was performed by differential scanning calorimetry (DSC) using a DSC220U calorimeter from SII (SEIKO Instruments Inc., Chiba, Japan). The weight of the samples ranged from 3 to 5 mg, and the scan range was from 30 °C to 300 °C with a heating rate of 10 K/min for (−)BCO−Cm6 and 1 K/min for OcO−Cm6. Measurements using a polarizing optical microscope (POM) were carried out with an OLYMPUS DP70 polarizing microscope (OLYMPUS Co., Tokyo, Japan), equipped with a METTLER−TOLEDO FP82HT hot stage (METTLER−TOLEDO Co., Tokyo, Japan). Circular dichroism (CD) spectra were measured in chloroform at a concentration of 0.1 mmol/L. Spectra were recorded with a JASCO J820 spectropolarimeter (JASCO Co., Tokyo, Japan) in the 200–700 nm range (100 nm/min, 2 nm slit width) in a 10 mm path−length quartz cell. The baseline spectrum was recorded from pure chloroform.
2.2. Preparation of Monomers
2.2.1. 2-Alkoxy-5-nitrobenzaldehydes
Three types of 2-alkoxy-5-nitrobenzaldehydes, including 2-octyloxy-, 2-(S)-(−)-(3,7-dimethyloctyloxy)- and 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde were prepared from 2-hydroxy-5-nitrobenzaldehyde and the corresponding 1-bromoalkanes according to the Williamson ether synthesis [36]. A representative procedure for 2-octyloxy-5-nitrobenzaldehyde is as follows: In a 300−mL pear−shaped flask with an Allihn condenser, 2-hydroxy-5-nitrobenzaldehyde (4.01g, 23.0 mmol), potassium carbonate (5.05 g, 36.0 mmol), and DMF (solvent, 100 mL) were placed. 1-Bromooctane (5.07 g, 26.0 mmol) was added dropwise to the solution and then it was refluxed at 100 oC for 60 h. The product was extracted with dichloromethane and the extract was dried over magnesium sulfate then concentrated by evaporation to give 2-octyloxy-5-nitrobenzaldehyde. The other 2-alkoxy-5-nitrobenzaldehydes were synthesized according to a similar procedure as mentioned above except that 1-bromooctane was replaced by (S)-(−)-1-bromo-3,7-dimethyloctane for 2-(S)-(−)-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde, and by 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane for 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde. (S)-(−)-1-Bromo-3,7-dimethyloctane and 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane were prepared by bromination of (S)-(−)-3,7-dimethyloctanol and triethylene glycol monomethyl ether with phosphorus tribromide, respectively. 2-(S)-(−)-(3,7-Dimethyloctyloxy)-5-nitrobenzaldehyde and 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde were purified by YAMAZEN Automated Flash Chromatograph (YAMAZEN Co., Osaka, Japan). Experimental procedures of (S)-(−)-1-bromo-3,7-dimethyloctane, 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane and the NMR spectra were described in Supplementary Materials (Experimental procedures and Figures S1−S6).
2-Octyloxy-5-nitrobenzaldehyde: Yield 87%. 1H−NMR(CDCl3, δ): 0.89(t,J=6.1Hz,3H,H−d), 1.30(m,8H), 1.51(quin,J=7.7Hz,2H,H−c), 1.91(quin,J=6.7Hz,2H,H−b), 4.22(t,J=6.4Hz,2H,H−a), 7.10(d,J=9.2Hz,1H,H−5), 8.41(quar,J=6.1Hz,1H,H−4), 8.69(d,J=2.8Hz,1H,H−2), 10.48(s,1H,CHO). 13C−NMR(CDCl3, δ): 14.1(C−b), 28.9, 29.16, 29.23, 31.8, 69.9(C−a), 112.9(C−5), 124.5(C−2), 124.7(C−4), 130.6(C−1), 141.5(C−3), 165.3(C−6), 187.6(CHO). (Figures S7, S8)
2-(S)-(−)-(3,7-Dimethyloctyloxy)-5-nitrobenzaldehyde: Yield 81%. 1H−NMR(CDCl3, δ): 0.87(d,J=6.7Hz,6H), 1.00(d,J=6.1Hz,3H), 1.14-1.39(m,6H), 1.49-1.57(m,1H), 1.93−1.99(m,2H), 4.25−4.32(m,2H), 7.15(d,J=9.1Hz,1H), 8.40(d,J=9.4Hz,1H), 8.64(d,J=2.7Hz,1H), 10.45(s,1H). 13C−NMR(CDCl3, δ): 19.6, 22.6, 22.7, 24.7, 28.0, 29.9, 35.7, 37.2, 39.2, 68.4, 113.1, 124.3, 124.6, 130.6, 141.4, 165.3, 187.5. (Figures S9−S15)
2-(2-(2-(2-Methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde: Yield 87%. 1H−NMR(CDCl3, δ): 3.37(s,3H,H−14), 3.53-3.55(m,2H,H−13), 3.63-3.69(m,4H,H−11,12), 3.74−3.77(m,2H,H−10), 3.98(t,J=4.5,2H,H−9), 4.42(t,J=4.5Hz,2H,H−8), 7.19(d,J=9.4Hz,1H,H−5), 8.41(d,J=6.4Hz,1H,H−4), 8.66(d,J=2.7Hz,1H,H−2), 10.48(s,1H,H−7). 13C-NMR(CDCl3, δ): 59.0(C−14), 69.2(C−8(9)), 69.4(C−9(8)), 70.60(C−10(11 or 12), 70.64(C−11(10 or 12), 71.0(C−12(10 or 11), 71.9(C−13), 113.5(C−5), 124.3(C−2), 124.8(C−1), 130.5(C−4), 141.7(C−3), 165.1(C−6), 187.6(C−7). (Figures S16−S21).
2.2.2. 2-Alkoxy-5-nitrobenzaldehyde Diethyl Acetals
2-Alkoxy-5-nitrobenzaldehyde diethyl acetals were synthesized by acid−catalyzed acetalation from the corresponding aldehydes. A representative procedure for 2-octyloxy-5-nitrobenzaldehyde diethyl acetal is as follows: In a 200−mL pear−shaped flask with an Allihn condenser, 2-octyloxy-5-nitrobenzaldehyde (5.86 g, 20 mmol), triethoxymethane (4.65 g, 31 mmol), EtOH (solvent, 100 mL), and H2SO4 (50 μL) were placed and refluxed for 60 h. The reaction mixture was neutralized with saturated NaHCO3 aq. solution and the product was extracted with dichloromethane and the extract was dried over magnesium sulfate then concentrated by evaporation. The other 2-alkoxy-5-nitrobenzaldehydes were synthesized according to a similar procedure as mentioned above except that 2-octyloxy-5-nitrobenzaldehyde was replaced by 2-(S)-(−)-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde for 2-(S)-(−)-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal, and by 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde for 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethyl acetal.
2-Octyloxy-5-nitrobenzaldehyde diethyl acetal: Yield 80%. 1H−NMR(CDCl3, δ): 0.89(t,J=6.7Hz,3H,H−e), 1.25(t,J=7.0Hz,6H,H−d), 1.29(m,8H), 1.49(quin,J=7.6Hz,2H), 1.85(quin,J=7.0Hz,2H), 3.63(m,4H,H−c), 4.10(t,J=6.4Hz,2H,H-b), 5.72(s,1H,−CH(acetal)), 6.91(d,J=8.9Hz,1H,H−5), 8.18(quar,J=6.1Hz,1H,H−4), 8.47(d,J=2.8Hz,1H,H−2). 13C−NMR(CDCl3, δ): 14.1(C−e), 15.2(C−d), 29.0, 29.23, 29.27, 31.8, 62.6(C−c), 69.1(C−b), 96.7(C−a), 111.1(C−5), 123.6(C−2), 125.8(C−4), 128.7(C−1), 141.2(C−3), 161.5(C−6). (Figures S22−S23).
2-(S)-(−)-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal: Yield 92%. 1H−NMR(CDCl3, δ): 0.87(d,J=6.7Hz,6H), 0.96(d,J=6.4Hz,3H), 1.14−1.36(m,6H), 1.24(t,J=7.0Hz,6H), 1.49−1.73(m,3H), 1.86−1.92(m,1H), 3.56−3.71(m,4H), 4.11−4.15(m,2H), 6.93(d,J=9.1Hz,1H), 8.19(d,J=9.1Hz,1H), 8.48(d,J=2.7Hz,1H). 13C−NMR(CDCl3, δ): 15.2, 19.6, 22.6, 22.7, 24.7, 28.0, 29.8, 36.0, 37.2, 39.2, 62.6, 67.5, 96.7, 111.0, 123.6, 125.8, 128.7, 141.2, 161.4. (Figures S24−S25).
2-(2-(2-(2-Methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethyl acetal: Yield 92%. 1H−NMR(CDCl3, δ): 1.21(t,J=7.0Hz,6H), 3.37(s,3H), 3.53−3.55(m,2H), 3.59−3.71(m,8H), 3.73−3.74(m,2H), 3.91(t,J= 4.8Hz,2H), 4.28(t,J=4.5Hz,2H), 5.74(s,1H), 6.98(d,J=9.1Hz,1H), 8.10(d,J=6.1Hz,1H), 8.47(d,J= 3.0Hz,1H). 13C−NMR(CDCl3, δ): 15.2, 59.0, 62.7, 68.7, 69.4, 70.6, 70.7, 70.9, 72.0, 96.6, 111.6, 123.6, 125.6, 129.1, 141.5, 161.2. (Figures S26−S27).
2.2.3. 2-Alkoxy-5-aminobenzaldehyde Diethyl Acetals
2-Alkoxy-5-aminobenzaldehyde diethyl acetals were prepared by catalytic hydrogenation of the corresponding nitro compounds. A typical procedure for 2-octyloxy-5-aminobenzaldehyde diethyl acetal is as follows: 2-Octyloxy-5-nitrobenzaldehyde diethyl acetal (5.92 g, 16 mmol), platinum (IV) oxide (0.04 g, 0.18 mmol), and tetrahydrofuran (solvent, 30 mL) were placed in a 100−mL glass−autoclave TEM−V100 (TAIATSU TECHNO Corp., Tokyo, Japan) equipped with a mechanical stirrer and a thermocouple. Hydrogen gas was supplied to the apparatus for 23 h at room temperature while maintaining a constant pressure of 1.0 MPa. The catalyst was filtered off through Celite® and the filtrate was dried with MgSO4, then condensed to dryness by evaporation. The resulting amino−acetal compound was used as a monomer for macrocycle synthesis without further purification. 2-(S)-(−)-(3,7-Dimethyloctyloxy)-5-aminobenzaldehyde diethyl acetal and 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-aminobenzaldehyde diethyl acetal were synthesized according to a similar procedure as mentioned above except that 2-octyloxy-5-nitrobenzaldehyde diethyl acetal was replaced by 2-(S)-(−)-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal for 2-(S)-(−)-(3,7-dimethyloctyloxy)-5-aminobenzaldehyde diethyl acetal, and by 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethyl acetal for 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-aminobenzaldehyde diethyl acetal.
2.3. Synthesis of Hexakis(2-Alkoxy-1,5-Phenyleneimine) Macrocycles
2.3.1. Hexakis(2-octyloxy-1,5-phenyleneimine) Macrocycle (OcO−Cm6)
(A) Water Method: A 100−mL sample bottle was filled with 2-octyloxy-5-aminobenzaldehyde diethylacetal (5.19 g, 16 mmol), tetrahydrofuran (50 mL), and H2O (1 mL). The mixture was then stirred magnetically at room temperature until precipitates appeared. Yield: 1.73 g (44%). FT−IR(KBr, cm-1): 2925(νC−H,CH3), 2852(νC−H,CH2), 1618(νCH=N), 1494, 1253, 1021. (Figure S28). 1H−NMR(CDCl3, δ): 0.83(t,J=7.0Hz,3H,H−15), 1.24(m,8H,H−11,12,13,14), 1.48(quin,J=10.4Hz,2H,H−10), 1.81(quin,J=8.0Hz,2H,H−9), 4.02(t,J=6.7Hz,2H,H−8), 6.90(d,J=8.9Hz,1H,H−3), 7.30(quar,J=2.7Hz,1H,H−4), 8.35(d,J=2.7Hz,1H,H−6), 8.98(s,1H,H−7).
(B) AcOH Method: 1 M acetic acid was used instead of water. A 100−mL sample bottle was filled with 2-octyloxy-5-aminobenzaldehyde diethylacetal (0.89 g, 2.7 mmol), tetrahydrofuran (10 mL), and 1 M acetic acid (1 mL). The mixture was then stirred magnetically at room temperature until precipitates appeared. Yield: 0.20 g (44%).
2.3.2. Hexakis(2-((S)-(−)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) Macrocycle ((−)BCO−Cm6)
A 100−mL glass autoclave container equipped with a mechanical stirrer and a thermocouple was filled with 2-((S)-(−)-3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal (3.20 g, 83 mmol), tetrahydrofuran (50 mL), and 5% Pd/C (1.50 g, 0.71 mmol as Pd). Hydrogen gas was introduced into the container at a pressure of 1.0 MPa and catalytic hydrogenation was performed for 20 hours. Pd/C was removed by Celite® filtration, and the filtrate was dried with MgSO4 and evaporated to give 2-((S)-(−)-3,7-dimethyloctyloxy)-5-aminobenzaldehyde diethyl acetal as a yellow liquid. The resulting amino−acetal compound was transferred to a 100−mL sample bottle and polymerization was initiated under conditions where tetrahydrofuran (20 mL) and H2O (1 mL) were added, followed by trifluoroacetic acid (0.5 mL) after one week. The polymerization suspension was poured into hexane and the solid was collected by suction filtration, then washed with tetrahydrofuran. Yield: 0.55g (18%). FT−IR(KBr, cm-1): 2925(νC−H,CH3), 2869(νC−H,CH2), 1623(νCH=N), 1494, 1383, 1364, 1265, 1016. 1H−NMR(CDCl3, δ): 0.82(d,J=6.7Hz,6H,H−16,17), 0.98(d,J=6.4Hz,3H,H−15), 1.08−1.39(m,6H,H−11,12,13), 1.45−1.72(m,2H,H−10,14), 1.87−1.93(m,2H,H−9), 4.08−4.14(m,2H,H−8), 6.98(d,J=8.8Hz,1H,H−3), 7.37(d,J=8.8Hz,1H,H−4), 8.09(d,J=2.7Hz,1H,H−6), 9.05(s,1H,H−7). 13C−NMR(CDCl3, δ): 19.8(C−15), 22.6(C−17), 22.7(C−16), 24.7(C−12), 27.9(C−14), 30.1(C−10), 36.2(C−9), 37.3(C−11), 39.2(C−13), 67.4(C−8), 112.7(C−6), 125.7(C−4), 145.8(C−3), 157.5(C−7).
2.3.3. Hexakis(2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy-1,5- phenyleneimine) Macrocycle (TEGO−Cm6)
A 100−mL glass autoclave container was filled with 2-(2-(2-(2-Methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethyl acetal (5.25 g, 0.013 mol), tetrahydrofuran (50 mL), and 5% Pd/C (1.53 g, 0.72 mmol as Pd). Catalytic hydrogen reduction was performed for 20 hours. The 5% Pd/C was removed by Celite® filtration, and after dehydration with MgSO4, the filtrate was evaporated to obtain a reddish-brown liquid. The synthesized product was transferred to a 100−mL sample bottle and polymerization was initiated under conditions where tetrahydrofuran (20 mL) and H2O (10 mL) were added. After 3 weeks, the solution gelled.
3. Results
3.1. Preparation of Monomers
The synthetic pathway of monomers (2-alkoxy-5-aminobenzaldehyde diethyl acetals), consisting of three steps, is illustrated in Scheme 1. 2-Alkoxy-5-nitrobenzaldehydes, including 2-octyloxy-, 2-((S)-(−)-3,7-dimethyloctyloxy)-, and 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehydes were prepared from 2-hydroxy-5-nitrobenzaldehyde and the corresponding 1-bromoalkanes or its analogue according to Williamson ether synthesis. (S)-(−)-1-Bromo-3,7-dimethyloctane and 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane were synthesized from (S)-(−)-3,7-dimethyl-6-octen-1-ol and triethylene glycol monomethyl ether, respectively. The synthetic routes involving catalytic hydrogenation and bromination reactions to the bromoalkanes are shown in Scheme 2. The chemical structures of the monomers and their precursors were determined by NMR spectra, and the signals were assigned using DEPT and 2D NMR (H,H−cosy and C,H−cosy) techniques. The spectra are provided in Supplementary Materials Section as Experimental Procedures and Figures S1−S6.
3.2. Synthesis of Hexakis(2-Alkoxy-1,5-phenyleneimine) Macrocycles
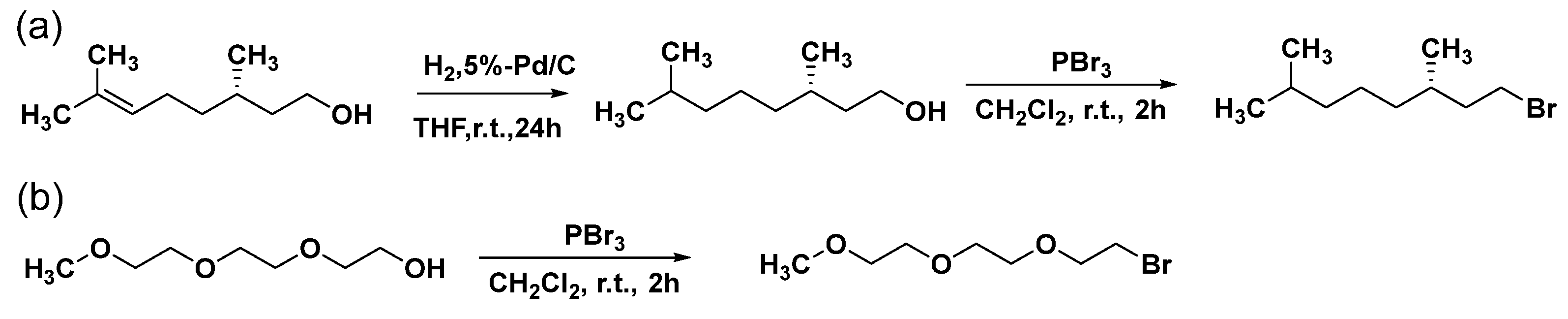
Hexakis(2-alkoxy-1,5-phenyleneimine) macrocycles were synthesized by a one−pot procedure where the corresponding acetal−protected AB−type monomers, 2-alkoxy-5-aminobenzaldehyde diethyl acetals were polycondensed in tetrahydrofuran containing water or acid at room temperature. The general synthetic route of the macrocycle is illustrated in Scheme 3.
The acetal−protected AB−type monomer, 2-alkoxy-5-aminobenzaldehyde diethyl acetal is slowly deprotected with water or acid, which supplies a condensable aldehyde−amino monomer in the polymerization system at a low concentration. The monomers undergo polycondensation to yield linear imine oligomers in an equilibrium state, as shown in Scheme 3. A random−coil oligomer forms a helical foldamer with a strong intramolecular association of hexameric macrocycles having the same backbone structure. A trans−imination (imine metathesis) occurs between two imine linkages or between an amino group and a neighboring imine linkage to yield a macrocycle [27]. Shape−anisotropic, nearly planar, and persistent macrocycles are known to aggregate into columnar assemblies in polar solvents driven by aromatic π−stacking. The macrocycle is stabilized due to the free energy gained from the intermolecular and noncovalent interactions upon aggregation, and therefore the aggregates become thermodynamically the most stable species and precipitate [37].
3.2.1. Hexakis(2-octyloxy-1,5-phenyleneimine) Macrocycle (OcO−Cm6)
The macrocycle OcO−Cm6 was synthesized by polycondensation of 2-octyloxy-5-aminobenzaldehyde diethyl acetal at room temperature in a mixed solvent of tetrahydrofuran and water (THF/H2O=50/1(v/v)). The MALDI−TOF MASS spectra of the reaction mixture are shown in Figure 1. In the spectrum of the reaction solution, two weeks after the reaction started (Figure 1a), the peaks of the unreacted monomer (A1), the dimer (A2), and the macrocycle OcO−Cm6 are observed. After an additional six weeks (totally two months), the peak of OcO−Cm6 increases and becomes predominant (Figure 1b).
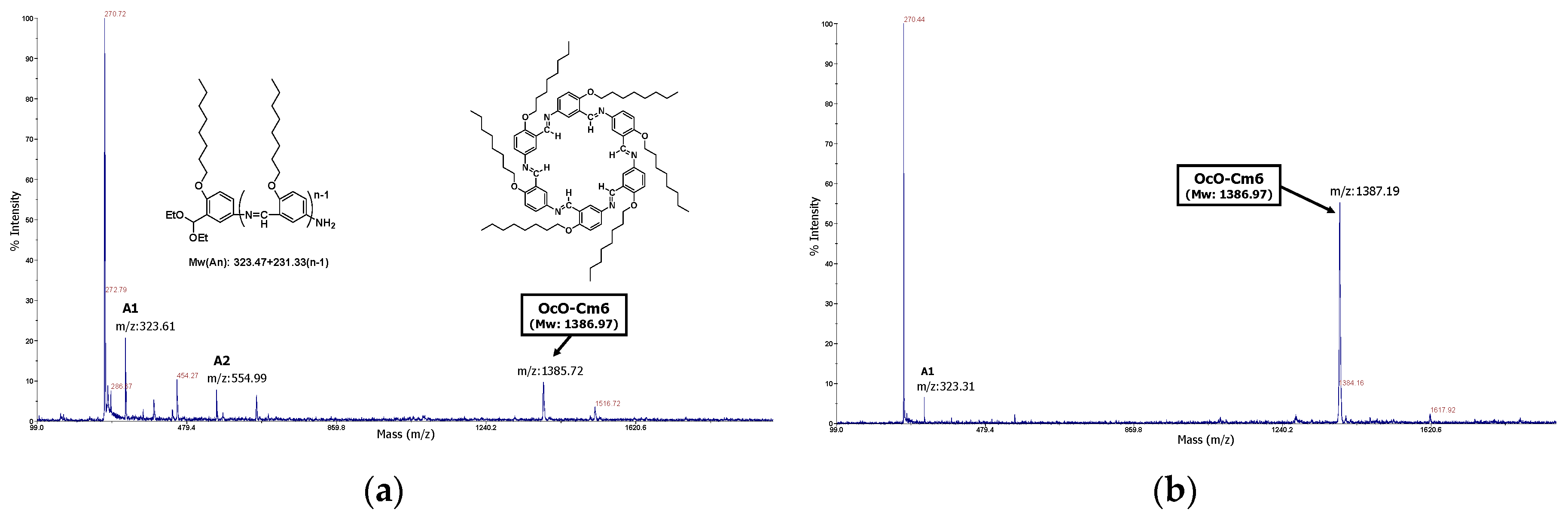
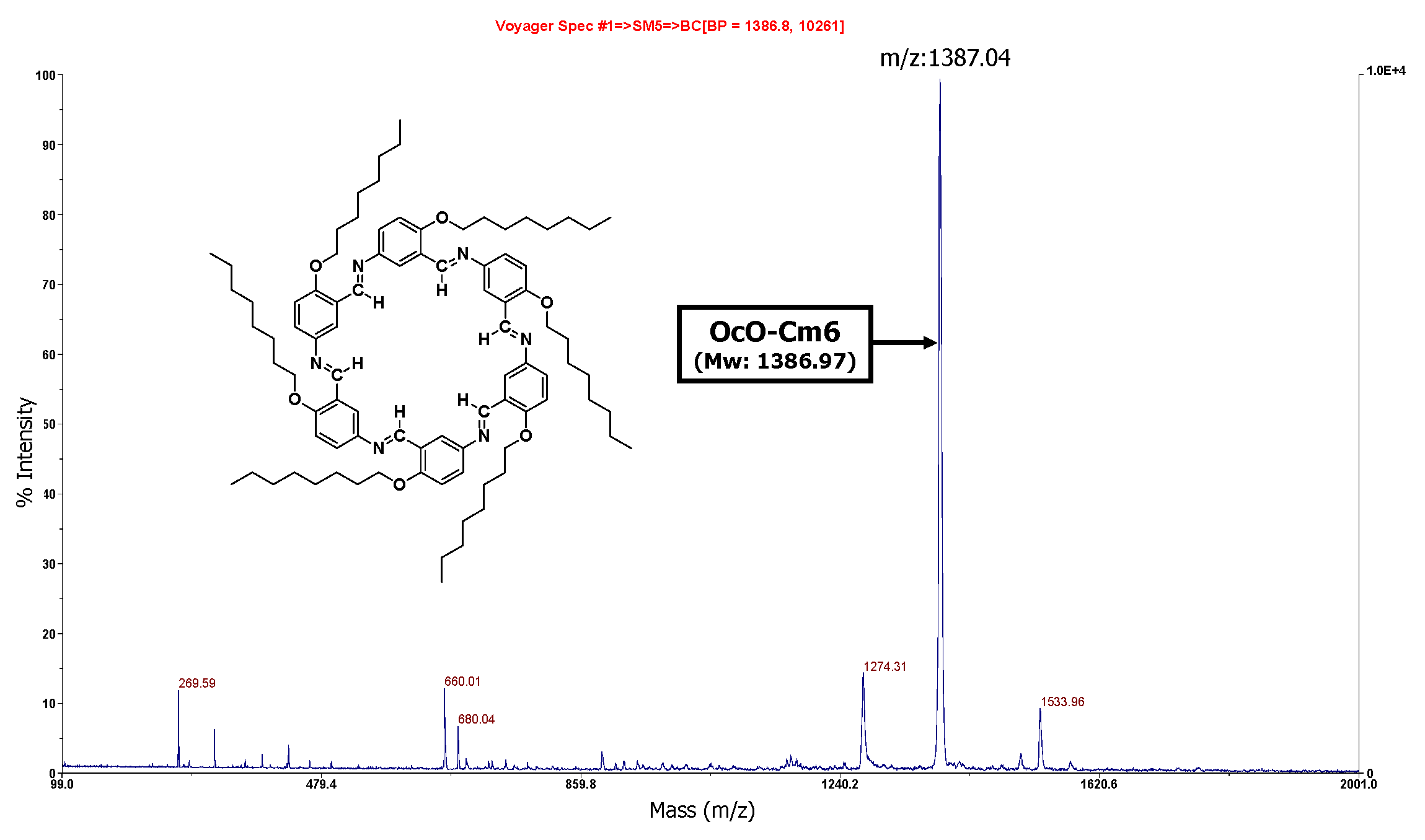
Figure 1.
MALDI−TOF MS spectra of reaction mixture: 0.31 M, THF/H2O =50/1(v/v), r.t., in air, magnetically stirring. Sampling: (a) two days later, homogeneous solution, (b) two months later, suspension containing precipitates.
Figure 1.
MALDI−TOF MS spectra of reaction mixture: 0.31 M, THF/H2O =50/1(v/v), r.t., in air, magnetically stirring. Sampling: (a) two days later, homogeneous solution, (b) two months later, suspension containing precipitates.
Figure 2.
MALDI−TOF MS spectrum of reaction mixture: 0.245 M, THF/AcOH =50/1(v/v), r.t., in air, magnetically stirring. Sampling: one month later, suspension containing precipitates.
Figure 2.
MALDI−TOF MS spectrum of reaction mixture: 0.245 M, THF/AcOH =50/1(v/v), r.t., in air, magnetically stirring. Sampling: one month later, suspension containing precipitates.
To accelerate the macrocycle formation, 1 M acetic acid (1.0 mL) was used instead of water. The MALDI−TOF MS spectrum of the reaction mixture is illustrated in Figure 2. Despite the short reaction time of one month, a strong peak at 1387.04 m/z corresponding to the macromolecule OcO−Cm6 appears. It can be seen that AcOH acceleratesd the deprotection reaction of the acetal group and facilitated the formation of OcO−Cm6. In the FT−IR spectrum of the isolated product, the characteristic peaks were observed at 2925 cm-1 (νC−H, CH3), 2852 cm-1 (νC−H, CH2), and 1618 cm-1 (νC=N) (Figure S28). The macrocycle with long alkoxy chains was soluble in specific organic solvents such as chloroform, whereas Cm6 without side chains was practically insoluble, allowing their structures to be analyzed by NMR. The 1H−NMR spectrum of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO−Cm6) is illustrated in Figure 3. The imine proton signal (H−7) is observed at 8.98 ppm and three phenylene−ring proton signals appear in a range from 6.90 to 8.35 ppm. The proton signal of methylene attached to the oxygen atom in the alkyl side chain (H−8) is observed at 4.02 ppm, and the other protons of the alkyl side chain appear in a range from 0.83 to 1.81 ppm. All the 1H−NMR signals appear at reasonable positions without excess or deficiency.
The WAXD patterns of finely ground powdered OcO−Cm6 and non−alcoxylated Cm6 are illustrated in Figure 4 and WAXD data (2θ and d value) of OcO−Cm6 are listed in Table 1. The macrocycles π−stack with a distance of 5.5 Å for OcO−Cm6 and 5.4 Å for Cm6, and both of them form a columnar packing with a cylindrical channel. The outer diameters of OcO−Cm6 and Cm6 are estimated to be 26.3 Å and 16.8 Å, respectively. In the solid state, these columns aggregate with a hexagonally closest−packed structure.
The transmission electron microscopic (TEM) images of OcO−Cm6 are displayed in Figure 5. The two crystals from different locations (Figure 5a and 5b) are both hexagonal plate−shaped with one side of about 10 μm, and OcO−Cm6 is expected to have hexagonal close−packed nanocolumns that are π−stacked like Cm6, which is consistent with the results of powder X-ray diffraction. However, the crystals were too thick in the (001) direction to measure the electron diffraction (ED).
The DSC profile of OcO−Cm6 is depicted in Figure 6. Upon heating, small endothermic peaks are observed at 177°C and 194°C, along with a large endothermic peak at 200°C. During cooling, a significant exothermic peak is observed at 159°C. The DSC measurement results indicate that the melting point (Tm) of OcO−Cm6 is approximately 200°C and the crystallization temperature (Tc) is around 160°C. Furthermore, the presence of small peaks besides Tm and Tc suggests that OcO−Cm6 may be a liquid crystal molecule. The phase transition of OcO−Cm6 was examined using a polarizing microscope with a hot−stage. Upon heating, a phase resembling a mesophase was observed at 190−197°C, transitioning to an isotropic phase above 197°C. During cooling, a phase appearing to be a mesophase was observed at 165−175°C, followed by crystallization at 159°C. Based on these findings, it is proposed that OcO−Cm6 may be an enantiotropic columnar liquid crystal molecule. However, further detailed investigation is required

Figure 5.
Transmission electron microscopic images of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO-Cm6) crystals at different locations; (a): hexagonal plate-shaped crystal at a location, (b): that at another location.
Figure 5.
Transmission electron microscopic images of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO-Cm6) crystals at different locations; (a): hexagonal plate-shaped crystal at a location, (b): that at another location.
3.2.2. Hexakis(2-((S)-(−)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) Macrocycle ((−)BCO-Cm6)
2-(S)-(−)-(3,7-Dimethyloctyloxy)-5-aminobenzaldehyde diethyl acetal (8.3 mmol) was polymerized in tetrahydrofuran (20 mL) containing water (1.0 mL) at room temperature with magnetic stirring. Water serves to deprotect the acetal group, regenerating the aldehyde group. The polycondensation between the regenerated aldehyde and an amino group of another monomer occurs, meaning that water acts as an initiator of the polymerization. The reaction process was analyzed by MALDI-TOF MS spectroscopy.
In the MALDI-TOF MS spectrum of the reaction solution one day after the reaction started (Figure 7a), the peaks of the unreacted monomer and the dimer are predominantly observed, indicating that most of the acetal groups were not deprotected with water. Consequently, trifluoroacetic acid (0.5 mL) was added to the reaction mixture and allowed to react for an additional two weeks. The spectrum is shown in Figure 7b, where the peak corresponding to macrocycle (−)BCO-Cm6 is observed at 1556.06(m/z). The suspension was poured into hexane, and the precipitates were collected then washed with tetrahydrofuran (Yield 18%). The spectrum of the isolated product is shown in Figure 7c, in which the intense peak corresponding to (-)BCO-Cm6 appears clearly.
In the FT−IR spectrum, characteristic peaks were observed at 2925 cm-1(νC−H,CH3), 2869 cm-1(νC−H,CH2), and 1623 cm-1(νC=N) (Figure 7d).
Figure 8.
NMR spectra of hexakis(2-((S)-(−)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) (−)BCO−Cm6. (a) 1H−NMR(CDCl3, δ), (b) NOESY(CDCl3, δ): cross-peaks of H6−H7 and H3−H8.
Figure 8.
NMR spectra of hexakis(2-((S)-(−)-3,7-dimethyloctyloxy)-1,5-phenyleneimine) (−)BCO−Cm6. (a) 1H−NMR(CDCl3, δ), (b) NOESY(CDCl3, δ): cross-peaks of H6−H7 and H3−H8.
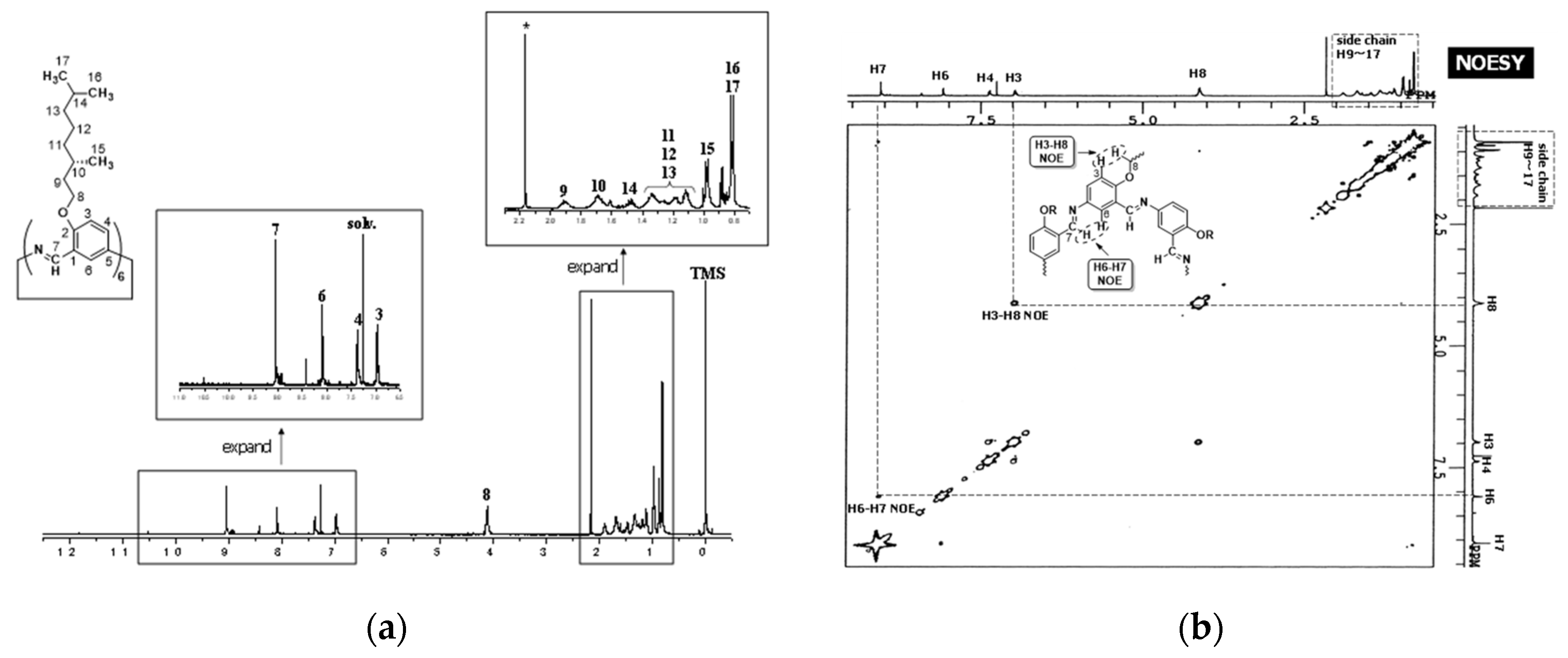
The 1H−NMR and NOESY spectra are illustrated in Figure 8. The imine proton signal is observed at 9.05 ppm (Figure 8a). Benzene−ring proton signals appear in a range from 6.98 to 8.09 ppm. The proton signal of the methylene attached to the oxygen atom in the alkyl side chain (H−8) is observed around 4.1 ppm, and the other protons of the alkyl side chain appear in a range from 0.82 to 1.93 ppm. These chemical shifts and integral values support the chemical structure. All the proton signals were assigned from the H,H−COSY spectrum (Figures S12, S13). In the NOESY spectrum (Figure 8b), the cross-peak of H6 and H7 protons is observed, indicating that these two hydrogen atoms are close to each other in three−dimensional space. This suggests that the imine C−H bonds are oriented toward the inside of the macrocycle ring and that six nitrogen atoms align on the outer rim of the ring. The cross−peak of H3−H8 implies that these two hydrogen atoms are spatially close to each other because the six atoms (C2−O−C8−H8−H3−C3) containing these hydrogens form a pseudo-hexagonal shape. The Circular Dichroism (CD) spectra of macrocycle (−)BCO−Cm6 and the monomer precursor, 2-((S)-(−)-3,7-dimethyloctyloxy)-5-nitrobenzaldehyde are illustrated in Figure 9. In both spectra, negative peaks are observed in a wavelength range from 250 nm to 350 nm, and their CD profiles remain almost unchanged. This indicates that the absolute configuration of the C−10 carbon in the side chain is maintained as S-(−) throughout all synthetic steps.
The DSC profile is illustrated in Figure 10. The as−synthesized (−)BCO−Cm6 retains various characteristics of the synthesis process, such as the encapsulation of the reaction solvent. The first heating curve exhibits several peaks that represent these characteristics. Consequently, the second run curve is typically utilized to assess the thermal properties of the material itself using DSC. During the second heating process of (−)BCO−Cm6, the crystallization temperature (Tc), glass transition temperature (Tg), and melting point (Tm) are observed at 108.1°C, 150.8°C, and 165.0°C, respectively. The morphology of (−)BCO−Cm6 was examined using scanning electron microscopy (SEM). Figure 11 displays the SEM images obtained at magnifications of (a) 1,000 and (b) 10,000. Columnar−like structures with a diameter of 100−200 nm are observed. Given that Cm6 molecules exhibit the property of self−assembling through π−stacking [31], it is inferred that (−)BCO−Cm6 aggregates in a similar fashion.
3.2.3. Hexakis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy-1,5-phenyleneimine) Macrocycle (TEGO−Cm6)
2-(2-(2-Methoxyethoxy)ethoxy)ethoxy-5-aminobenzaldehyde diethyl acetal (13 mmol) was polymerized in tetrahydrofuran (20 mL) containing water (10 mL) at room temperature with magnetic stirring. The polymerization solution gelled after three weeks. The reaction process was analyzed by MALDI−TOF MS spectroscopy. In the spectrum of the reaction solution one week after the reaction initiation (Figure 12a), the peaks of the linear oligomers terminated with an acetal group and an amino group are observed. The peak corresponding to macrocycle TEGO−Cm6 appears at 1592.42 m/z in the spectrum of the reaction mixture obtained two weeks later (Figure 12b). After 3 weeks, the solution gelled.
The gel that forms transitions to a liquid state when shaken or subjected to mechanical stress, and reverts to a gel when left undisturbed. In other words, the TEGO−Cm6 hydrogel exhibits thixotropic properties (Figure 13a). Aida, T. et al. reported that a hexa-peri-hexabenzocoronene (HBC) amphiphile bearing lipophilic dodecyl chains and hydrophilic triethylene glycol chains can self−assemble in THF to form well−defined nanotubes [38]. Similarly, TEGO−Cm6 also forms nanocolumns (nanotubes) in solution like the aforementioned amphiphilic HBC, and it is suggested that it exhibits thixotropic properties by forming a hydrogen bond network and extending into a fibrous shape (Figure 13b). We hypothesized that the thixotropic phenomenon of the TEGO−Cm6 polymerization solution was influenced by the concentration condition, and examined the relationship between concentration and the gelation phenomenon. Table 2 displays the polymerization conditions and the time when the polymer solution gelled. Entry 1 gelled on the 20th day after the start of polymerization, but it was not possible to determine whether it exhibited thixotropy due to the small volume of solution. Entries 2 and 3 gelled on the 20th and 24th day after the start of polymerization, respectively, and demonstrated thixotropy. Entry 4 did not gel even after more than 120 days. From these results, it was determined that the thixotropy of this TEGO−Cm6 polymer solution depended on the concentration, and no gelation occurred at low concentration conditions (0.09 mol/L). This is thought to be because TEGO−Cm6 could not form a network based on hydrogen bonds or nanocolumns could not develop into fibrous shapes when the concentration was low.
4. Conclusions
A simple one−pot synthesis of three types of hexakis(m-phenyleneimine) macrocycles, each with a long alkoxy side chain, has been successfully accomplished from acetal-protected AB−type monomers. These monomers were polymerized in tetrahydrofuran containing either water or acid. The presence of acid expedited the deprotection reaction of the acetal group, facilitating the formation of macrocycles. The alkoxylated macrocycles formed nanocolumns through π−stacked self−assembly, and the columns of octyloxylated macrocycle OcO−Cm6 aggregated into a hexagonally closest−packed structure like Cm6 without the side chain. Oct−Cm6 exhibited an enantiotropic columnar liquid crystal−like mesophase between 165°C and 197°C. In the SEM image of (−)BCO−Cm6, columnar substances with diameters ranging from 100 to 200 nm were observed. The polymerization solution for the 2-(2-methoxyethoxy)ethoxy)ethoxylated macrocycle (TEGO−Cm6) gelled and displayed thixotropic properties by forming a hydrogen bond network and extending in a fibrous shape. No gelation occurred under low monomer−concentration conditions. These thixotropic properties make them ideal candidates for investigation into a range of potential applications, including biomedical smart materials and sensors.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Experimental procedures; Scheme S1: Synthesis of 3,7-dimethyloctan-1-ol; Scheme S2: Synthesis of 1-bromo-3,7-dimethyloctane; Scheme S3: Synthesis of 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane; Figure S1: 1H−NMR spectrum of 3,7-dimethyloctan-1-ol; Figure S2: 13C-NMR spectrum of 3,7-dimethyloctan-1-ol; Figure S3: 1H−NMR spectrum of 1-bromo-3,7-dimethyloctane; Figure S4: 13C−NMR spectrum of 1-bromo-3,7-dimethyloctane; Figure S5: 1H−NMR spectrum of 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane; Figure S6: 13C−NMR spectrum of 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane; Figure S7: 1H−NMR spectrum of 2-octyloxy-5-nitrobenzaldehyde; Figure S8: 13C−NMR spectrum of 2-octyloxy-5-nitrobenzaldehyde; Figure S9: 1H−NMR spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde; Figure S10: 13C−NMR spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde; Figure S11: 13C−NMR(DEPT-135) spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde; Figure S12: H,H−cosy spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde; Figure S13: H,H−cosy spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde(expanded); Figure S14: C,H−cosy spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde; Figure S15: C,H−cosy spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde(expanded); Figure S16: 1H−NMR spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehydel; Figure S17: 13C−NMR spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehydel; Figure S18: H,H−cosy spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde; Figure S19: H,H−cosy spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde(expanded); Figure S20: C,H−cosy spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde; Figure S21: C,H−cosy spectrum of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde(expanded); Figure S22: 1H−NMR spectrum of 2-octyloxy-5-nitrobenzaldehyde diethyl acetal; Figure S23: 13C−NMR spectrum of 2-octyloxy-5-nitrobenzaldehyde diethyl acetal; Figure S24: 1H−NMR spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal; Figure S25: 13C−NMR spectrum of 2-(3,7-dimethyloctyloxy)-5-nitrobenzaldehyde diethyl acetal; Figure S26. 1H−NMR of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethylacetal; Figure S27: 13C−NMR of 2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-5-nitrobenzaldehyde diethylacetal; Figure S28: FT−IR spectrum of hexakis(2-octyloxy-1,5-phenyleneimine) OcO-Cm6.
Funding
This work was supported financially by the Ministry of Education, Culture, Sports, Science and Technology (Japan) Grant−in−Aid for Challenging Exploratory Research (22655037).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the author.
Acknowledgments
The author thanks Tetsuya Uchida, Okayama University, for TEM−ED measurement. He is also thankful to Kozo Ishida and Seitaro Oishi of Tokyo Polytechnic University for their technical assistance. This paper has been supported by the Ministry of Education, Culture, Sports, Science and Technology’s project for the promotion of advanced private universities ‘Academic Frontier Project’ (Japan).
Conflicts of Interest
The author declares no conflict of interest.
References
- Zhang, W.; Moore, J.S. Shape-persistent macrocycles: structures and synthetic approaches from arylene and ethynylene building blocks. Angew. Chem., Int. Ed. 2006, 45, 4416-4439. [CrossRef]
- Jiang, B.; Zhao M., Li, S.; Xu, Y.; Loh, T. Macrolide synthesis through intramolecular oxidative cross-coupling of alkenes. Angew. Chem. 2018, 130, 564-568. [CrossRef]
- Kotha, S.; Chavan, S.A.; Shaikh, M. Diversity-oriented approach to macrocyclic cyclophane derivatives by Suzuki–Miyaura cross-coupling and olefin metathesis as key steps. J. Org. Chem. 2012, 77, 482–489. [CrossRef]
- Zhang, W.; Moore J.S. Arylene ethynylene macrocycles prepared by precipitation-driven alkyne metathesis. J. Am. Chem. Soc. 2004, 126, 12796. [CrossRef]
- Kotha, S.; Meshram, M.; Dommaraju Y. Design and synthesis of polycycles, heterocycles, and macrocycles via strategic utilization of ring-closing metathesis. The Chemical Record, 2018, 18, 1613-1632. [CrossRef]
- Huang, S.; Lei, Z.;Jin, Y.; Zhang, W. By-design molecular architectures via alkyne metathesis. Chem. Sci. 2021, 12, 9591-9606. [CrossRef]
- Jin, Y.; Zhang, A.; Huanga Y.; Zhang, W. Shape-persistent arylenevinylene macrocycles (AVMs) prepared viaacyclic dienem-etathesis macrocyclization (ADMAC). Chem. Commun. 2010, 46, 8258-8260. [CrossRef]
- MacLachlan, M.J. Conjugated shape−persistent macrocycles via Schiff−base condensation: New motifs forsupramolecular chem-istry. Pure Appl. Chem. 2006, 78, 873–888. [CrossRef]
- Lisowski, J. Imine− and amine−type macrocycles derived from chiral diamines and aromatic dianhydrides. Molecules 2022, 27, 4097. [CrossRef]
- Zhang, W.; Moore, J. S. Shape-Persistent Macrocycles: Structures and Synthetic Approaches from Arylene and Ethynylene Building Blocks. Angew. Chem., Int. Ed. 2006, 45, 4416-4439. [CrossRef]
- He, Z; Ye, G.; Jiang, W. Imine macrocycle with a deep cavity: guest-selected formation of syn/anti configuration and guest-controlled reconfiguration. Chemistry A European Journal 2015, 21, 3005-3012. [CrossRef]
- Zhao, D.; Moore, J. S. Synthesis and self-association of an imine-containing m-phenylene ethynylene macrocycle. J. Org. Chem. 2002, 67, 3548–3554. [CrossRef]
- Yang, Z.; Esteve, F.; Antheaume, C.; Lehn, J.−M. Dynamic covalent self−assembly and self−sorting processes in the formation of imine−based macrocycles and macrobicyclic cages. Chem. Sci. 2023, 14, 6631–6642. [CrossRef]
- Lisowski, J., Imine- and amine-type macrocycles derived from chiral diamines and aromatic dianhydrides. Molecules 2022, 27, 4097. [CrossRef]
- MacLachlan, M.J. Conjugated shape-persistent macrocycles via Schiff-base condensation: New motifs for supramolecular chem-istry. Pure Appl. Chem. 2006, 78, 873–888. [CrossRef]
- Chinnaraja, E.; Arunachalam, R.; Pillai, R.S.; Peuronen, A.; Rissanen, K.; Subramanian, P.S. One-pot synthesis of [2 + 2]-helicate-like macrocycle and 2 + 4- μ4-oxo tetranuclear open frame complexes: Chiroptical properties and asymmetric oxidative coupling of 2-naphthols. Appl. Organomet. Chem. 2020, 34, e5666. [CrossRef]
- Chinnaraja, E.; Arunachalam, R.; Samanta, J.; Natarajan, R.; Subramanian, P.S. Desymmetrization of meso diols using enantiopure zinc (II) dimers: Synthesis and chiroptical properties. Appl. Organomet. Chem. 2019, 33, e4827. [CrossRef]
- Janczak, J.; Prochowicz, D.; Lewiński, J.; Fairen-Jimenez, D.; Bereta, T.; Lisowski, J. Trinuclear cage-like Zn(II) macrocyclic com-plexes: Enantiomeric recognition and gas adsorption properties. Chem. Eur. J. 2016, 22, 598–609. [CrossRef]
- Sarnicka, A.; Starynowicz, P.; Lisowski, J. Controlling the macrocycle size by the stoichiometry of the applied template ion. Chem. Commun. 2012, 48, 2237–2239. [CrossRef]
- Byun, J.C.; Lee, N.H.; Mun, D.H.; Park, K.M. Synthesis and characterization of dinuclear copper(II) complexes, [Cu2( [20] -DCHDC) (La)2 (La = N3−, NCS− or S2O32−) with tetraazadiphenol macrocyclic ligand having cyclohexane rings. Inorg. Chem. Commun. 2010, 13, 1156–1159. [CrossRef]
- Gao, J.; Liu, Y.-G.; Zhou, Y.; Boxer, L.M.; Woolley, F.R.; Zingaro, R.A. Artificial Zinc(II) complexes regulate cell cycle and apop-tosis-related genes in tumor cell lines. ChemBioChem 2007, 8, 332–340. [CrossRef]
- Gao, J.; Woolley, F.R.; Zingaro, R.A. Catalytic asymmetric cyclopropanation at a chiral platform. Org. Biomol. Chem. 2005, 3, 2126–2128. [CrossRef]
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally defined catalysts for enantioselective oxidative coupling reactions. Angew. Chem. 2003, 115, 6190–6194. [CrossRef]
- Lisowski, J. Imine- and amine-type macrocycles derived from chiral diamines and aromatic dialdehydes. Molecules 2022, 27, 4097. [CrossRef]
- Yang, Z.; Esteve, F.; Antheaume, C.; Lehn, J.-M. Dynamic covalent self-assembly and self-sorting processes in the formation of imine-based macrocycles and macrobicyclic cages. Chem. Sci. 2023, 14, 6631–6642. [CrossRef]
- Jin, Y.; Wang, Q.; Taynton, P.; Zhang, W. Dynamic covalent chemistry approaches toward macrocycles, molecular cages, and polymers. Acc. Chem. Res. 2014, 47, 1575–1586. [CrossRef]
- Chao, A.; Negulescu, I.; Zhang, D. Dynamic covalent polymer networks based on degenerative imine bond exchange: Tuning the malleability and self-healing properties by solvent. Macromolecules 2016, 17, 6277–6284. [CrossRef]
- Korich A. L; Hughes, T. S. Arylene imine macrocycles of C3h and C3 symmetry from reductive imination of nitroformylarenes. Org. Lett. 2008, 10, 5405–5408. [CrossRef]
- Hughes, T.S.; Korich, A.L. Efficient synthesis of an ortho-phenylene-para-phenylene-imine macrocycle; Abstracts of Papers. In Proceedings of the 230th ACS National Meeting, Washington, DC, USA, 28 August–1 September 2005.
- Moriya, Y.; Yamanaka, M.; Mori, K. Synthesis of C3−symmetric macrocyclic triimines from monomers having Boc−protected amine and formyl group. Chem. Lett. 2022, 51, 217–220. [CrossRef]
- Matsumoto, T. Highly efficient one-pot synthesis of hexakis(m-phenyleneimine) macrocyle Cm6 and the thermostimulated self-healing property through dynamic covalent chemistry. Polymers 2023, 15, 3542. [CrossRef]
- Seo, S. H,; Jones, T. V.; Seyler, H.; Peters, J. O.; Kim, T. H.; Chang, J. Y.; Tew, G. N. Liquid crystalline order from ortho-phenylene ethynylene macrocycles J. Am. Chem. Soc. 2006, 128, 9264–9265. [CrossRef]
- Kawano, S.; Kato, M.; Soumiya, S.; Nakaya, M.; Onoe, J.; Tanaka, K. Columnar liquid crystals from a giant macrocycle mesogen, Angew. Chem., Int. Ed. 2017, 57, 167-171. [CrossRef]
- Granata, G.; Petralia, S.; Forte, G.; Conoci S., Consoli, G. M. L. Injectable supramolecular nanohydrogel from a micellar self-assembling calix[4]arene derivative and curcumin for a sustained drug release. Materials Science and Engineering: C 2020, 111, 110842. [CrossRef]
- Coh, C. Y,; Mocerino, M.; Ogden M. I. Macrocyclic gelators. Supramolecular Chemistry 2013, 25, 555-566. [CrossRef]
- Williamson, A. W. Theory of etherification J. Chem. Soc., 1852, 4, 106-112. [CrossRef]
- Zhao, D.; Moore, J.S. Folding-driven reversible polymerization of oligo(m-phenylene ethynylene) imines: Solvent and starter sequence studies. Macromolecules 2003, 36, 2712–2720. [CrossRef]
- Jin, W.; Fukushima, T.; Niki, M.; Aida, T. Self-assembled graphitic nanotubes with one-handed helical arrays of a chiral am-phiphilic molecular graphene. PNAS 2005, 102, 10801-10806. [CrossRef]
Scheme 1.
Synthesis pathway of 2-alkoxy-5-aminobenzaldehyde diethyl acetals. OcO: octyloxy, (−)BCO: (S)-(−)-3,7-dimethyloctyloxy, TEGO: 2-(2-(2-methoxyethoxy)ethoxy)ethoxy.
Scheme 1.
Synthesis pathway of 2-alkoxy-5-aminobenzaldehyde diethyl acetals. OcO: octyloxy, (−)BCO: (S)-(−)-3,7-dimethyloctyloxy, TEGO: 2-(2-(2-methoxyethoxy)ethoxy)ethoxy.

Scheme 2.
Synthesis of (a) (S)-(−)-1-bromo-3,7-dimethyloctane and (b) 1-bromo-2-(2-methoxyethoxy)ethoxy)ethane.
Scheme 2.
Synthesis of (a) (S)-(−)-1-bromo-3,7-dimethyloctane and (b) 1-bromo-2-(2-methoxyethoxy)ethoxy)ethane.
Scheme 3.
Synthetic route to the macrocycles from acetal−protected 2-alkoxy-5-aminobenzaldehyde diethyl acetals.
Scheme 3.
Synthetic route to the macrocycles from acetal−protected 2-alkoxy-5-aminobenzaldehyde diethyl acetals.
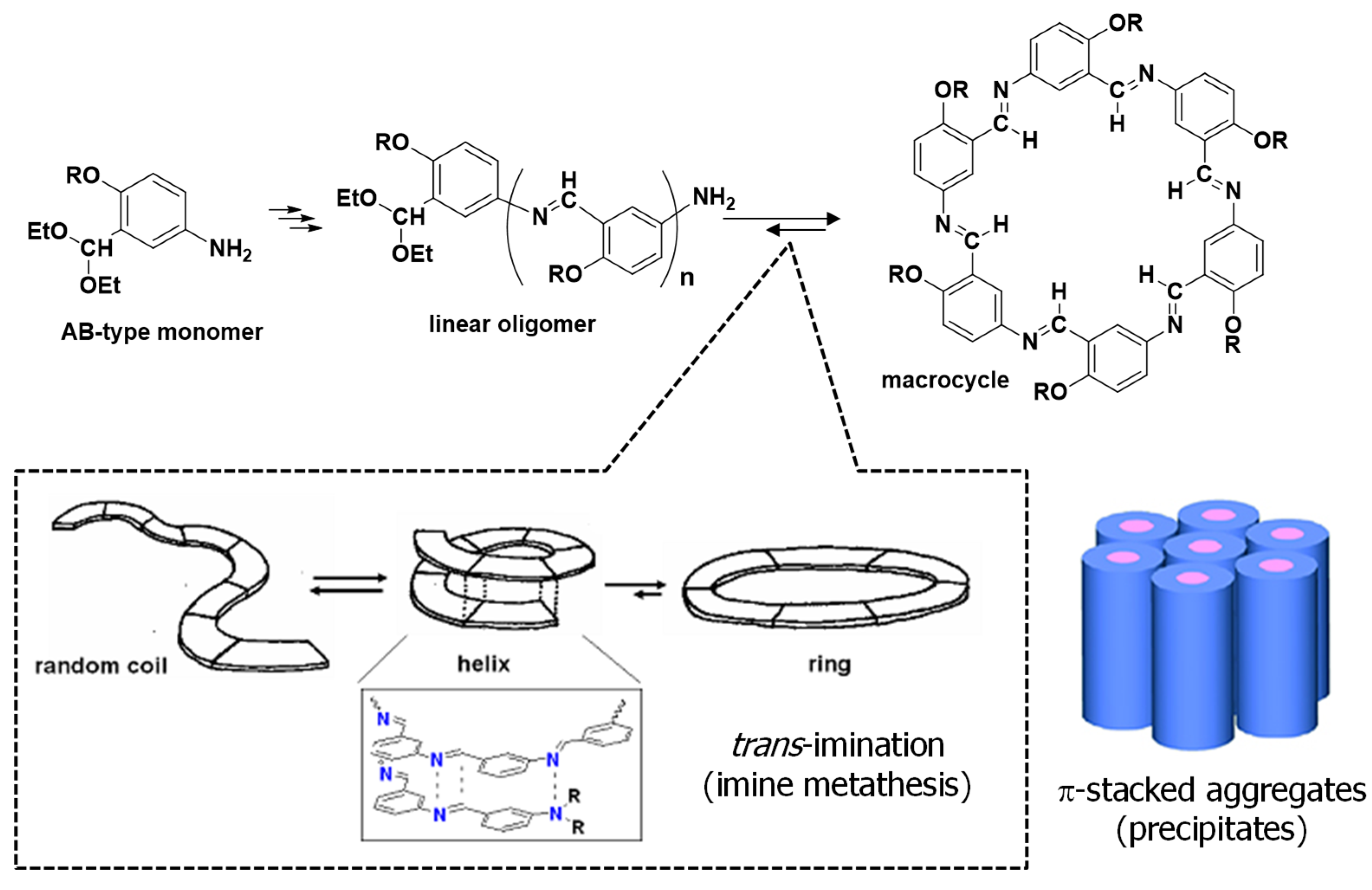
Figure 3.
1H-NMR spectrum of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO−Cm6) in CDCl3.
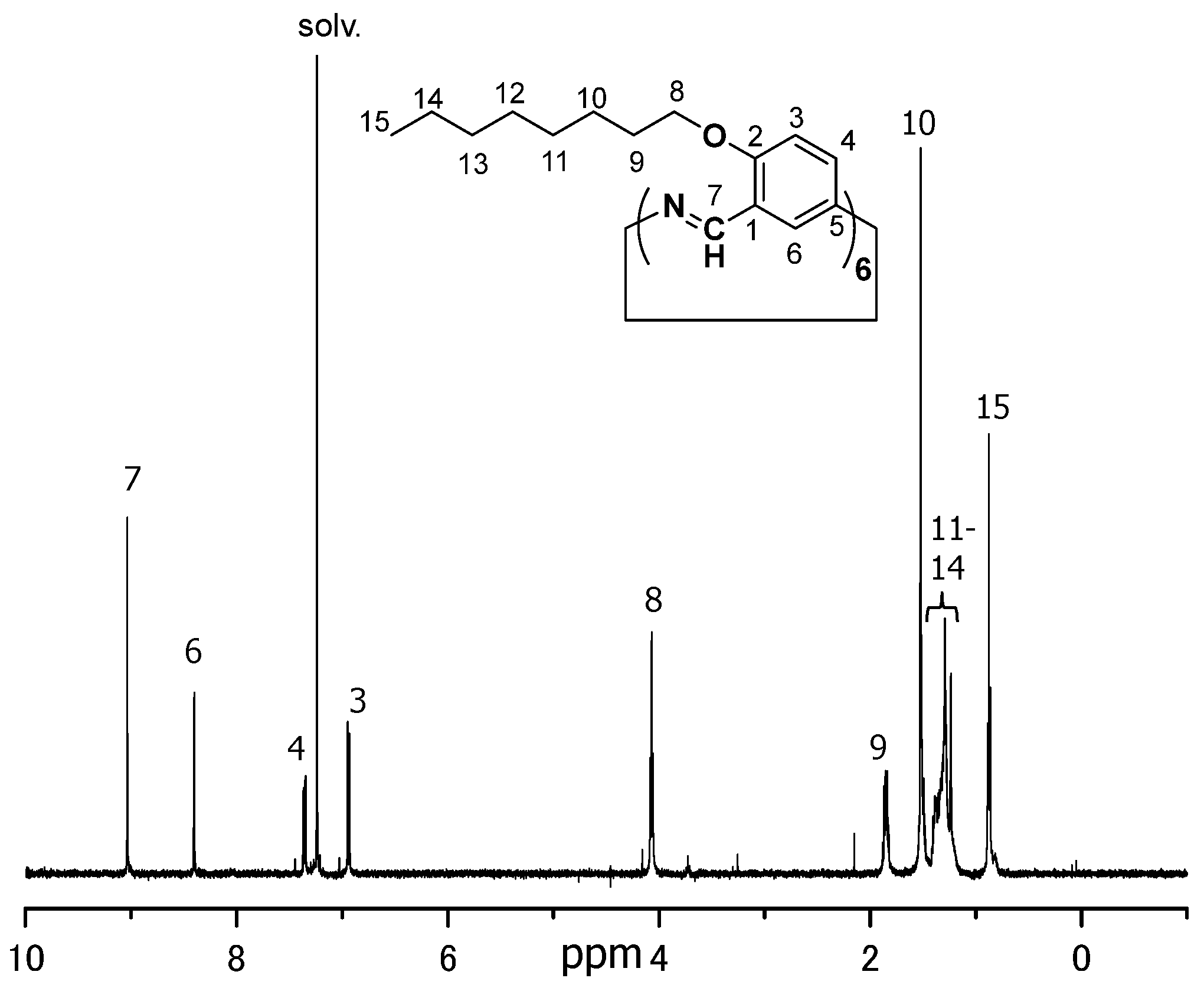
Figure 4.
WAXD petters of of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO−Cm6) and hexakis(1,5-phenyleneimine) macrocycle (Cm6). Line color scheme; red: OcO-Cm6, blue: Cm6.
Figure 4.
WAXD petters of of hexakis(2-octyloxy-1,5-phenyleneimine) macrocycle (OcO−Cm6) and hexakis(1,5-phenyleneimine) macrocycle (Cm6). Line color scheme; red: OcO-Cm6, blue: Cm6.
Figure 6.
The DSC profile of OcO−Cm6. a heating program from 30 °C to 250 °C at a heating rate of 1 K/min. The figures within the figure show polarized microscope photographs.
Figure 6.
The DSC profile of OcO−Cm6. a heating program from 30 °C to 250 °C at a heating rate of 1 K/min. The figures within the figure show polarized microscope photographs.
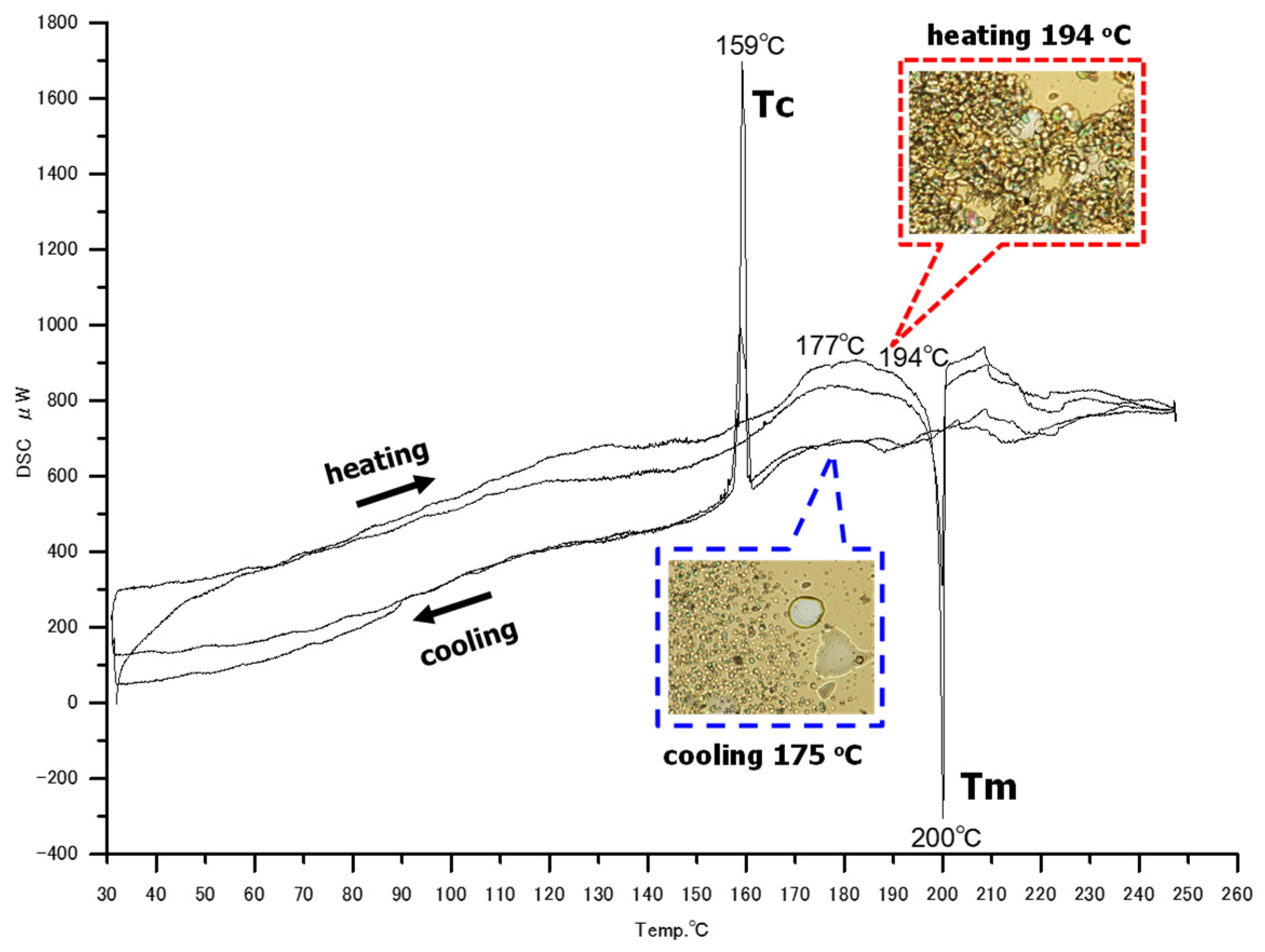
Figure 7.
MALDI-TOF MS spectra of the reaction mixture; (a): one day after the reaction start(homogeneous), (b): two weeks after adding trifluoroacetic acid(inhomogeneous), (c): precipitates obtained by reprecipitation in hexane. (d): FT−IR spectrum of (−)BCO−Cm6 powder (KBr, cm-1): 2925(νC−H,CH3), 2869(νC−H,CH2), 1623(νCH=N), 1383, 1364, 1494, 1265, 1016.
Figure 7.
MALDI-TOF MS spectra of the reaction mixture; (a): one day after the reaction start(homogeneous), (b): two weeks after adding trifluoroacetic acid(inhomogeneous), (c): precipitates obtained by reprecipitation in hexane. (d): FT−IR spectrum of (−)BCO−Cm6 powder (KBr, cm-1): 2925(νC−H,CH3), 2869(νC−H,CH2), 1623(νCH=N), 1383, 1364, 1494, 1265, 1016.
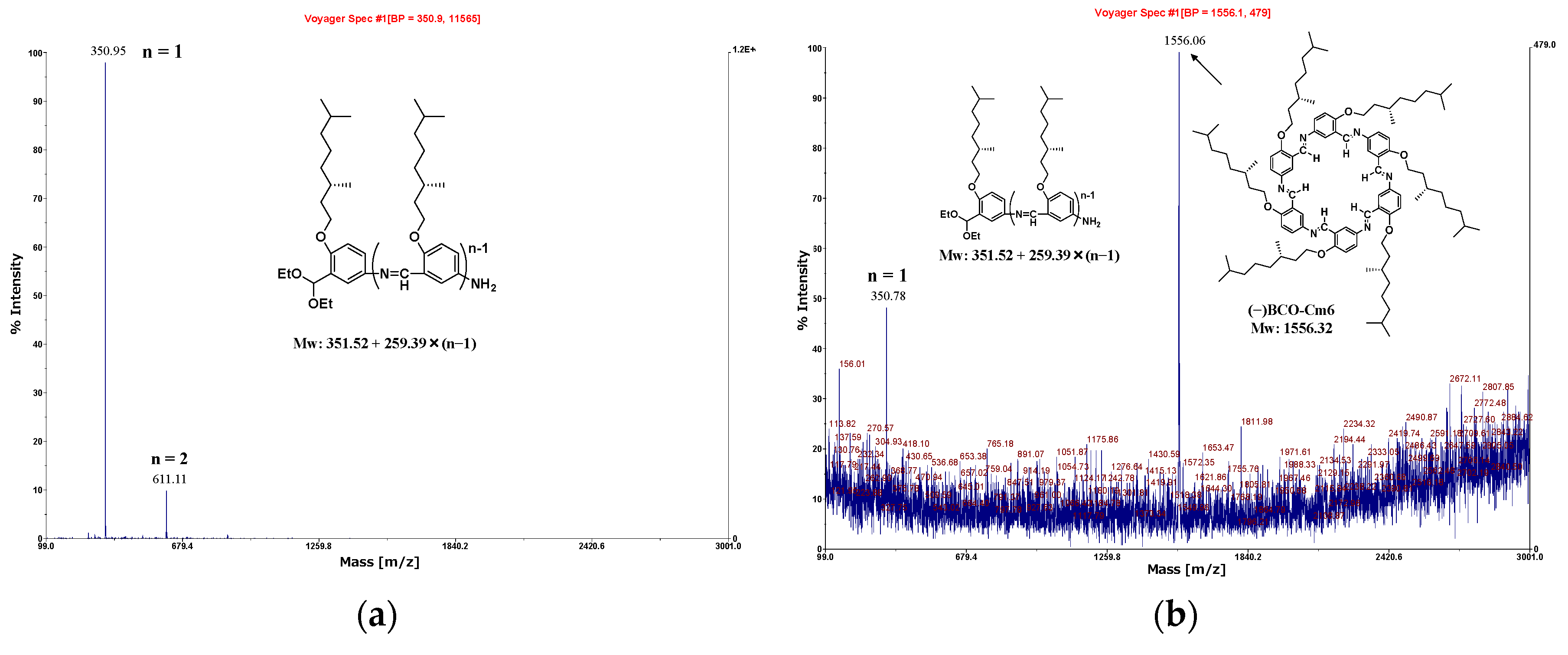
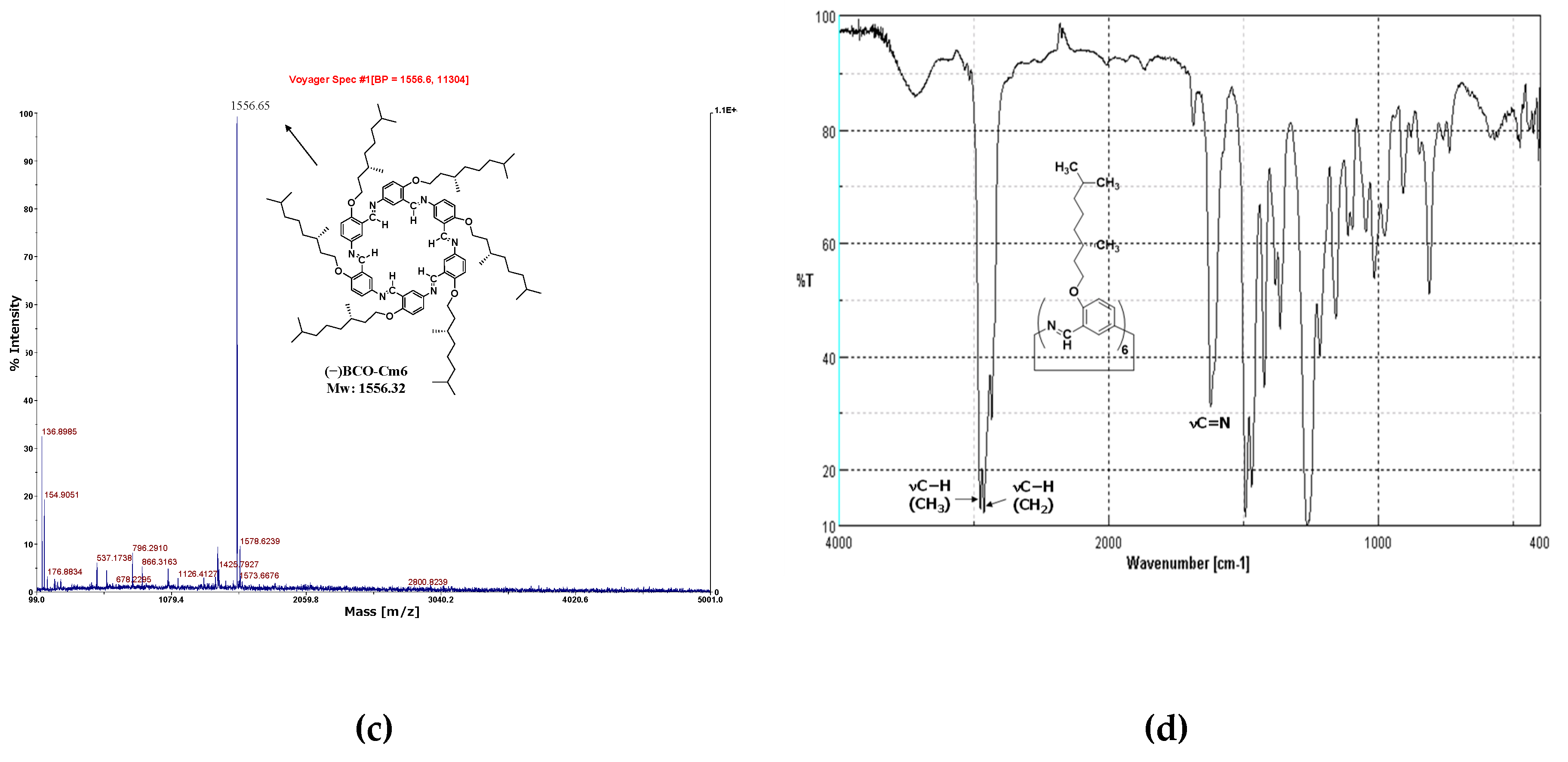
Figure 9.
The CD(Circular Dichroism) spectra of macrocycle (−)BCO−Cm6 and the monomer precursor. 2 mM in chloroform, 0.1 mm cell length. (a): (−)BCO−Cm6, (b): 2-((S)-(−)-3,7-dimethyloctyloxy)-5-nitrobenzaldehyde.
Figure 9.
The CD(Circular Dichroism) spectra of macrocycle (−)BCO−Cm6 and the monomer precursor. 2 mM in chloroform, 0.1 mm cell length. (a): (−)BCO−Cm6, (b): 2-((S)-(−)-3,7-dimethyloctyloxy)-5-nitrobenzaldehyde.
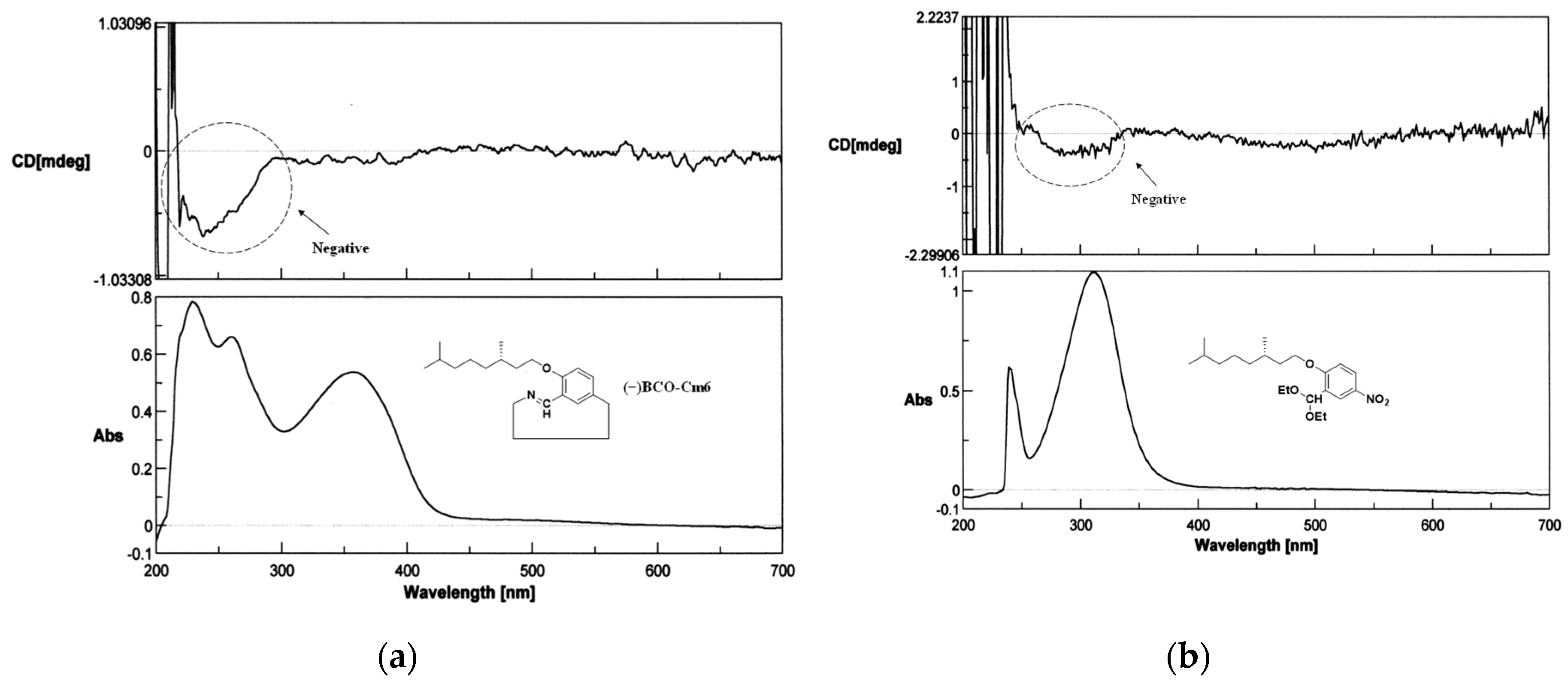
Figure 10.
DSC profile of (−)BCO−Cm6 measured in N2. 1st heating: 30−300 oC, 5 oC/min; 1st cooling: 300−30 oC, 20 oC/min; 2nd heating: 30−300 oC, 5 oC/min; 2nd cooling: 300−30 oC, 20 oC/min.
Figure 10.
DSC profile of (−)BCO−Cm6 measured in N2. 1st heating: 30−300 oC, 5 oC/min; 1st cooling: 300−30 oC, 20 oC/min; 2nd heating: 30−300 oC, 5 oC/min; 2nd cooling: 300−30 oC, 20 oC/min.
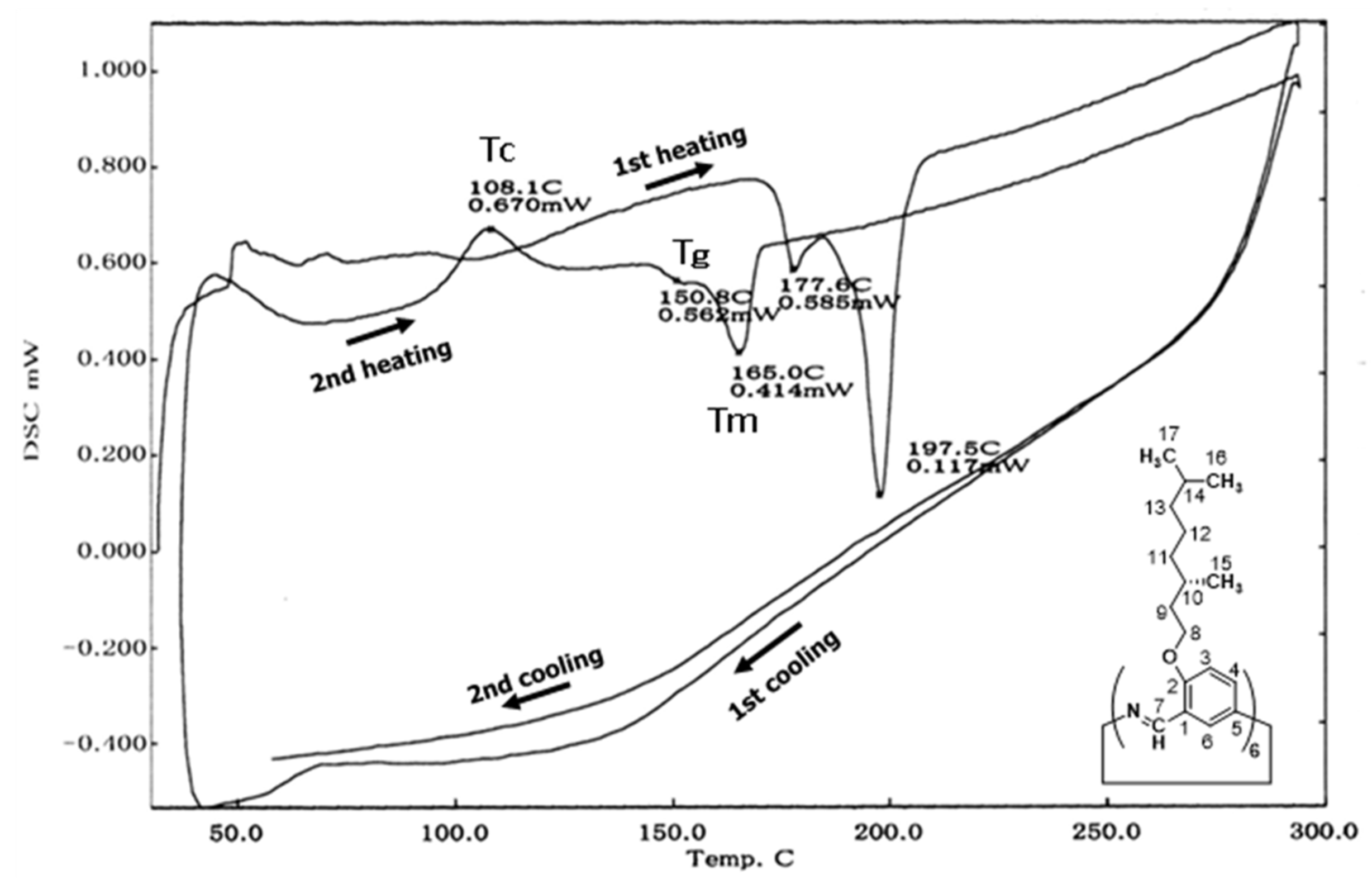
Figure 11.
SEM images of (−)BCO−Cm6 powders obtained at magnifications of (a) 1,000 and (b) 10,000.

Figure 12.
MALDI-TOF MS spectra of reaction mixture: 0.43 M, THF/H2O =20/10(v/v), r.t., in air, magnetically stirring. Sampling: (a) one week later, homogeneous solution; (b) two week later, homogeneous solution.
Figure 12.
MALDI-TOF MS spectra of reaction mixture: 0.43 M, THF/H2O =20/10(v/v), r.t., in air, magnetically stirring. Sampling: (a) one week later, homogeneous solution; (b) two week later, homogeneous solution.

Figure 13.
(a): Thixotropic property of the TEGO−Cm6 gel preprepared by polycondensation of 2-(2-(2-methoxyethoxy)ethoxy)ethoxy-5-aminobenzaldehyde diethyl acetal(0.43 M) in THF/H2O at room temperature for three weeks; (b): Imaginary rendering for hydrogel formation by self−assemble of the TEGO−Cm6 yielding nanofibers.
Figure 13.
(a): Thixotropic property of the TEGO−Cm6 gel preprepared by polycondensation of 2-(2-(2-methoxyethoxy)ethoxy)ethoxy-5-aminobenzaldehyde diethyl acetal(0.43 M) in THF/H2O at room temperature for three weeks; (b): Imaginary rendering for hydrogel formation by self−assemble of the TEGO−Cm6 yielding nanofibers.
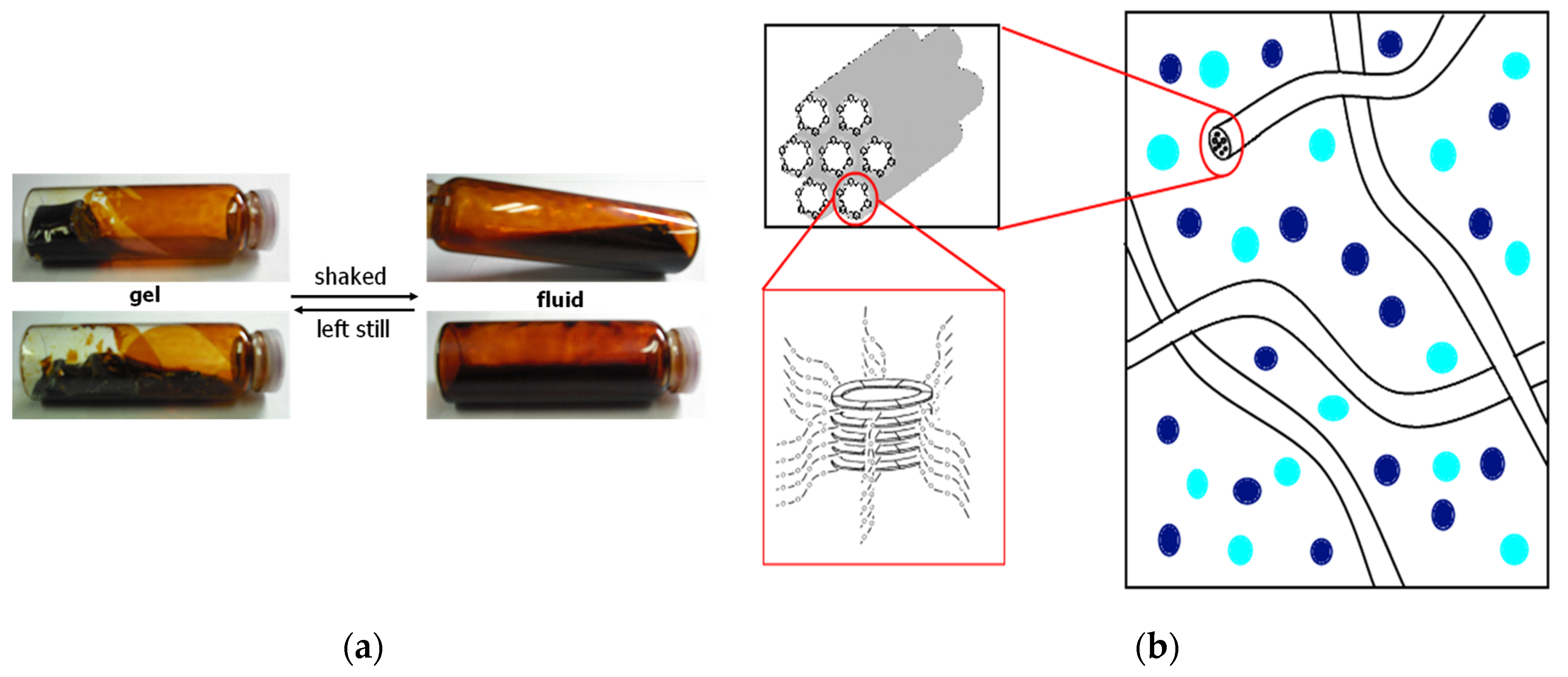

Table 1.
WAXD peak data (2θ and d value) of OcOCm6.
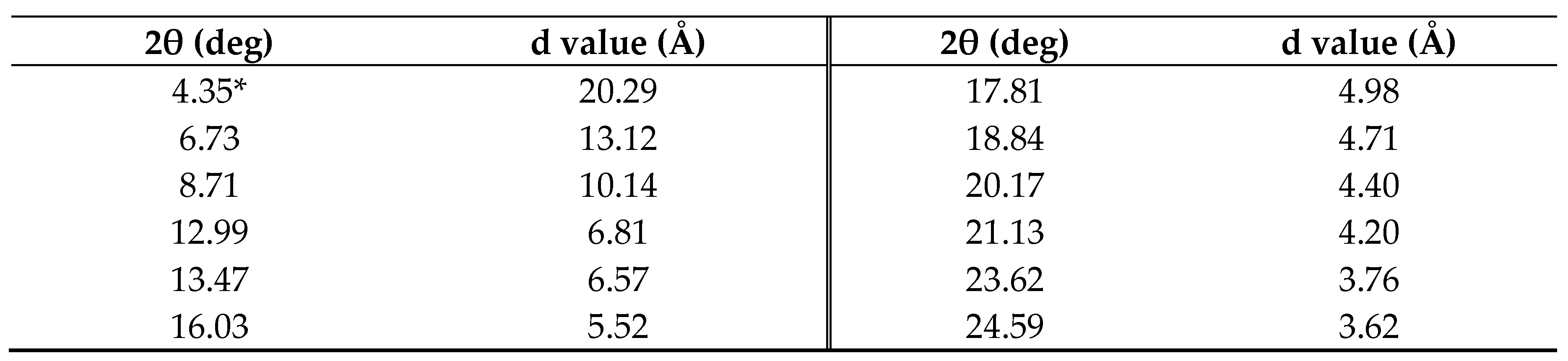
*Very weak.
Table 2.
The relationship between monomer concentration and gelation time 1.
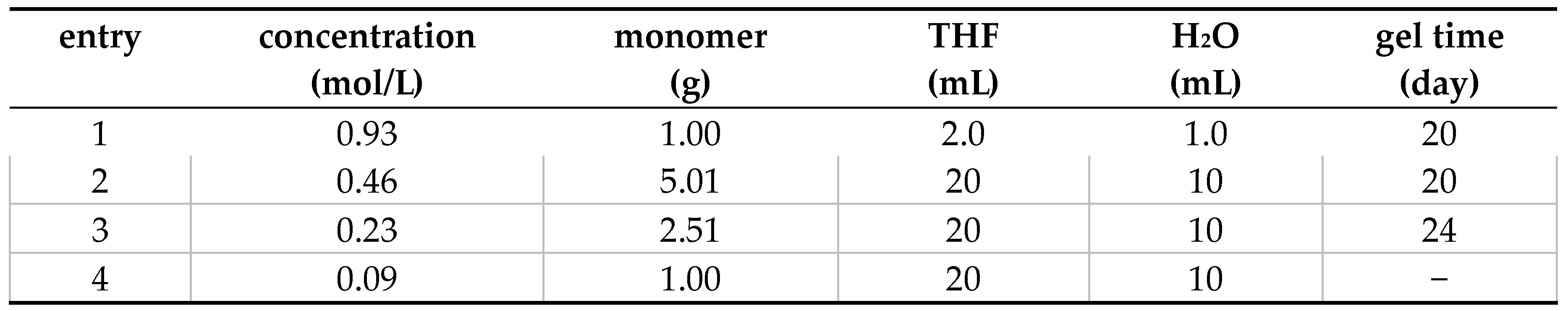
1 Monomer: 2-(2-(2-methoxyethoxy)ethoxy)ethoxy-5-aminobenzaldehyde diethyl acetal, magnetically stirring, room temperature.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



