
Submitted:
03 November 2023
Posted:
03 November 2023
You are already at the latest version
Abstract
The skeletal muscle plays a critical role in regulating systemic blood glucose homeostasis. Impaired skeletal muscle glucose homeostasis associated with type 2 diabetes mellitus (T2DM) has been observed to significantly affect the whole-body glucose homeostasis, thereby resulting in other diabetic complications. T2DM does not only affect skeletal muscle glucose homeostasis, but it also affects skeletal muscle structure and functional capacity. Given that T2DM is a global health burden, there is an urgent need to develop therapeutic medical therapies that will aid in the management of T2DM. Prediabetes is a prominent risk factor of T2DM that usually goes unnoticed in many individuals as it is an asymptomatic condition. Hence, research on prediabetes is essential because establishing diabetic biomarkers during the prediabetic state would aid in preventing the development of T2DM, as prediabetes is a reversible condition if it is detected in the early stages. Literature predominantly documents the changes in skeletal muscle during T2DM, but the changes in skeletal muscle during prediabetes remain unknown. In this review, we seek to review the existing literature on prediabetic and T2DM associated changes in skeletal muscle function.
Keywords:
Type 2 diabetes mellitus
; Prediabetes
; skeletal muscle
; satellite cells
; myogenic regulatory factors
; insulin resistance
; muscle fibers
; inflammation
; oxidative stress
Introduction
The skeletal muscle is one of the most prominent insulin-sensitive tissue in the body and functions as the primary site for insulin-stimulated glucose uptake [1]. Skeletal muscle satellite cells are among the most paramount progenitor cells responsible for maintaining skeletal muscle health under physiological and pathophysiological conditions [2]. Satellite cells are proposed to play a critical role in muscle fiber maintenance, repair, and remodeling, ultimately maintaining skeletal muscle plasticity [3]. Alterations in skeletal muscle health can affect whole-body glucose homeostasis as it is the skeletal muscle that is chiefly involved in regulating glucose uptake and maintaining glucose homeostasis. Chronic metabolic diseases, such as diabetes mellitus have been observed to affect skeletal muscle health by negatively modulating satellite cell quantity or functionality [2].
Diabetes mellitus is a metabolic disorder characterized by chronically elevated blood glucose levels due to defective insulin release or function [2]. Approximately 422 million people have been diagnosed with diabetes mellitus globally, with the majority living in low and middle-income countries [4]. Type 2 diabetes mellitus (T2DM) is the most prevalent type, accounting for approximately 90% of global diabetes cases [2,5]. T2DM is anticipated to affect almost 8% of the worldwide population by 2030 [6]. T2DM is characterized by insulin resistance, where the body cells cannot effectively respond to insulin action, which leads to hyperglycemia. Unhealthy lifestyle behaviors, such as sedentary lifestyle combined with chronic consumption of high caloric diets, result in the onset of impaired glucose tolerance and insulin resistance seen in T2DM (7). Prediabetes is characterized by blood glucose levels higher than those in the homeostatic range, but below the threshold for diabetes mellitus for a diagnosis of T2DM [8]. It is observed that both fasting glucose levels and glucose tolerance are impaired during the prediabetic state [8]. T2DM has been observed to considerably compromise skeletal muscle health, a phenomenon known as diabetic myopathy [2]. Diabetic myopathy is associated with reduced physical capacity, strength, and muscle mass [9,10,11]. Diabetic myopathy is one of the understudied complications of diabetes mellitus, and it is proposed to be directly involved in the rate of comorbidity development [2].
However, the onset of T2DM is often preceded by an asymptomatic condition known as prediabetes [8]. There are several studies that have suggested that the onset of complications associated with T2DM begin during the prediabetic state. Literature predominantly documents the changes in skeletal muscle during T2DM, but the changes in skeletal muscle during prediabetes remain unknown. In this review, we seek to review the existing literature on prediabetic and T2DM associated changes in skeletal muscle function.
Skeletal Muscle Progenitor Cells and Muscle Strength
Skeletal muscle can adapt to various stimuli via the modulation of muscle size, fiber-type distribution, and metabolism. This phenomenon is due to the skeletal muscle progenitor cells, particularly satellite cells, playing a role in skeletal muscle maintenance and plasticity [2]. The skeletal muscle is regarded as the principal regulator of systemic glucose homeostasis in the body [12]. Thus, dysregulation in skeletal muscle glucose homeostasis can affect whole-body glucose homeostasis [2]. Approximately 80% of postprandial glucose is delivered to the skeletal muscle via translocation of insulin-dependent glucose transporters such as glucose transporter 4 (GLUT4), which contributes to the maintenance of the individual's physical and metabolic well-being [13]. The following section describes the role of skeletal muscle satellite cells in skeletal muscle health maintenance and skeletal muscle strength.
Role of Satellite Cells in Skeletal Muscle
Skeletal muscle satellite cells are vital for skeletal muscle fiber maintenance, repair, and remodeling [3]. The term "satellite cell" arose from the anatomical location of the satellite cells between the sarcolemma and basal lamina of their associated muscle fiber [3]. Satellite cells are generally latent in adult skeletal muscle and become only functional upon stimulation. Stimulation of satellite cells results in satellite cell activation, proliferation, and differentiation [3]. Myoblasts, the progeny of satellite cells, play a role in skeletal muscle growth and regeneration of satellite cells. Skeletal muscle growth mediated by myoblasts occurs by the combination of myoblasts to form new myofibers or combine with an existing muscle fiber and donate their nucleus during the fusion process. Satellite cell regeneration occurs when myoblasts return to a quiescent state which replenishes the resident pool of satellite cells [14].
Paired box transcription factor 7 (Pax7) and Myogenic regulatory factors (MRFs), such as MyoD, Myf5, MRF4, and myogenin, are observed to regulate the function of satellite cells during myogenesis [3]. Pax7 is mainly expressed in quiescent satellite cells, and plays a role in self-renewal and maintenance of basal satellite pool [15]. Studies illustrate that there is a functional overlap between the MRFs in establishing myogenesis. MyoD and Myf5 are proposed to induce myoblast activation and proliferation, whereas myogenin and MRF4 are proposed to induce terminal differentiation of satellite cells [16]. In a study of newborn mice lacking MyoD and Myf5, it was observed that the mice were devoid of myoblasts and myofibers [17]. Contrarily, mice with myogenin deficiency generated myoblasts but demonstrated insufficient skeletal muscle differentiation, with minimum and smaller myotubes [18,19]. Hence the coordinated action of MRFs is vital for establishing myogenic lineage and terminal myogenic phenotype [20]. MRFs consist of a basic helix-loop-helix (bHLH) domain that enables them to recognize and bind to the E box sequence (CANNTG), known as the muscle enhancer factor-1 (MEF-1) site [21] (Figure 1). Heterodimerization of MRFs with Jun D, a ubiquitously expressed E-protein family of bHLH proteins (i.e., E12), orchestrates the binding of MRFs to MEF-1 (Figure 1).
The presence of growth factors (GF) during skeletal muscle growth is observed to stimulate the proliferation of myoblasts, which lack the expression of differentiation markers such as myogenin. Growth factors are observed to induce the expression of inhibitor of differentiation (Id) protein which forms a dimerization with E12 and prevents the heterodimerization between MRF and Jun D, hence inhibiting MEF-1 DNA binding activity. Growth factors also lead to the activation of protein kinase C (PKC), which phosphorylates MRF and inhibits DNA-binding activity. Hence, inhibition of mitogenic factors decreases Id and promotes the formation of bHLH heterodimers, which bind to their DNA targets and induce muscle-specific gene transcription involved in myogenesis (Figure 1). Myoblasts can only differentiate when proliferating cells exhibit a low mitogen-containing environment [21].
Studies document that subpopulations of satellite cells can undergo asymmetric divisions to synthesize myogenic progenitors or symmetric divisions to increase satellite cell pool [22]. Moreover, satellite cells are observed to also commit to the myogenic lineage and proliferate to give rise to committed myogenic progenitors, which can asymmetrically divide or directly differentiate into myocytes that will fuse and form new myofibers [22] (Figure 2). The ability of satellite cells to be able to choose between performing asymmetric or symmetric divisions enables them to coordinate their activity with the needs of the regenerating muscle. The increased propensity of symmetric division during muscle regeneration would stimulate the expansion of the satellite cell pool [23]. In contrast, the asymmetric division would favor the generation of myogenic progenitors and maintenance of the stem cell pool (Figure 2). Thus, a dynamic balance must be established between the fluctuating symmetric and asymmetric divisions that occur during the different stages of muscle regeneration, as an imbalance would result in muscle regeneration impairment [22]. Hence MRFs can be used as a biomarker to assess satellite cell function in myogenesis.
Skeletal Muscle Strength
Skeletal muscle strength is determined by the number and size of muscle fibers present in the skeletal muscle tissue [21]. Skeletal muscle fibers are categorized into two fiber types, i.e., "slow-twitch" (type I) and "fast-twitch" (type II) muscle fibers. Skeletal muscle fiber types are observed to play a role in skeletal muscle strength [24]. Skeletal muscle fibers contain four major myosin heavy chain (MHC) isoforms: the "slow" MHC and three "fast" type (IIa, IIx, and IIb) MHCs; and three major myosin light-chain (MLC) isoforms, the "slow" MLC1s and the two "fast" MLC1f and MLC3f [25]. MHC IIb, MHC IIa, and MCH IIx account for 90% of MHC in adult muscles [26]. "Slow-twitch" muscle fibers are associated with contraction endurance with lesser strength and rely on oxidative metabolism for energy production. "Fast-twitch" muscle fibers supply short-lived bursts of energy to the muscle and depend on glycolytic metabolism for energy production [24]. Thus, "fast-twitch" muscle fibers are observed to have considerable muscle strength and contraction speed, but only for short bursts of anaerobic activity before the muscle fatigue [27]. Type IIa/IIx fibers comprise heterogeneous characteristics of type I and type IIb fibers. They have intermediate numbers of mitochondria and oxidative potential, promoting moderate strength and improved resistance to fatigue [27].
Gene transcription of light and heavy myosin chain is regulated by MRFs, particularly MyoD, and MEF-1 DNA binding activity (26). Studies illustrate that MyoD is required for muscle fiber maintenance as it is observed to promote the development of slow and fast muscle fibers [28]. The myosin creatine kinase (MCK) gene is the most abundant nonmitochondrial mRNA expressed in all skeletal muscle fibers, which becomes activated when myoblasts commit to terminal differentiation into myocytes [29]. Thus, the MCK enzyme plays a pivotal role in differentiated skeletal muscle function (26). Myosin creatine kinase catalyzes the transfer of high-energy phosphate from ATP to creatine to promote energy storage in the form of phosphocreatine, thereby maintaining ATP homeostasis for the differentiated myocytes, ensuring optimum myocyte function [30]. The MEF-1 site has been observed in the enhancer region of MCK, which is considered to be prominent for MCK transcription [31]. The expression of MCK protein, as well as its enzymatic product, creatine phosphate, are observed to be considerably higher in fast-twitch muscles than in slow-twitch muscles [32]. The following section will outline the effect of T2D on satellite cell function and muscular strength.
Type 2 Diabetes Mellitus
Type 2 diabetes is one of the leading types of DM, accounting for approximately 90% of global DM cases [2]. T2DM is associated with peripheral insulin resistance, impaired regulation of hepatic glucose production, and decreased pancreatic β-cell function, eventually leading to β-cell failure [33]. An oral glucose tolerance test (OGTT), a fasting glucose test, a postprandial glucose test, or glycated haemoglobin (HbA1c) test can be used to diagnose T2DM [33]. Type 2 diabetes is established when fasting blood glucose (FBG) levels are ≥ 7mmol/L, postprandial glucose concentration ≥11.1mmol, and glycated haemoglobin concentrations ≥6,5% [34]. Other organs implicated in T2DM development, other than the pancreas, include the liver, skeletal muscle, kidneys, brain, small intestine, and adipose tissue [35]. T2DM results in micro and macrovascular complications such as nephropathy, neuropathy, cardiovascular diseases, and diabetic myopathy [2]. Diabetic myopathy is associated with reduced physical capacity, strength, and muscle mass [11]. Diabetic myopathy is proposed to be directly involved in the rate of comorbidity development; however, it is a relatively understudied diabetic complication [2].
Effects of T2DM on Skeletal Muscle Progenitor Cells and Muscle Strength
Type 2 diabetes mellitus is observed to affect skeletal muscle metabolism, structure, and function [2]. The skeletal muscle alterations observed in T2D include muscle atrophy [36,37], fiber-type transition [38], impaired glucose uptake [39], glycogen synthesis [40,41], and defective myokine secretion [42,43], which eventually result to muscle weakness and compromised muscle functional capacity [44]. Diminished appendicular lean mass and decreased skeletal muscle strength are usually observed in T2D patients regardless of gender and ethnicity, and the incidence increases with aging [45,46]. T2D is proposed to affect skeletal muscle progenitor cells, particularly the satellite cells, by altering progenitor cell quantity and function, thereby affecting overall skeletal muscle health [2]. This section will outline the effect of T2DM on skeletal muscle satellite cells and muscle strength.
Effects of T2DM on Skeletal Muscle Satellite Cells
T2DM has been observed to alter the function of satellite cells involved in muscle growth and regeneration [2]. Satellite cell function has been proposed to be considerably affected by hyperglycemic and lipotoxic conditions associated with type 2 diabetic states. For instance, a study discovered that three weeks of high-fat feeding (HFF) affected satellite cell content and functionality. The latter was characterized as the quantity of regenerating fibers present following injury [47]. Another study documented reduced muscle regeneration following eight months of HFF, which was proposed to be induced by delayed myofiber maturation [48]. A study conducted in the Obese Zuker Rat model for metabolic syndrome documented reduced satellite cell proliferative capacity; however, quiescent species remained unaltered [49]. A similar outcome was reported in another T2D study, whereby SC cell proliferation and activation were compromised, thereby affecting muscle regeneration [50].
Oxidative stress associated with T2DM is proposed to impair myogenesis [2]. Myogenesis is regulated by an integrated interaction between myogenic regulatory factors (MRFs), such as Myo, Jun D, and myogenin. These MRFs specifically bind to the muscle enhancer factor (MEF)-1 site, which regulates gene transcription of light and heavy chains of myosin and myosin creatine kinase (MCK) [51,52,53]. Disruption in the coordinated interaction between MRFs and the MEF-1 site can affect muscle protein synthesis and subsequent compromised skeletal muscle health [26]. Studies illustrate reduced myogenic factors (MyoD, myogenin, and Jun D) in STZ-diabetic and Zucker diabetic rodents. MEF-1 DNA binding activity is also observed to be altered in T2D rats [26]. Myosin creatine kinase and myosin expression are also observed to be impaired due to reduced MEF-1 DNA binding activity associated with T2DM [54]. The binding of the homo and heterodimers of MFRs to the MEF-1 site tightly regulates the development and differentiation of skeletal muscle progenitor cells into multinucleated myotubes [55].
Effect of T2DM on Skeletal Muscle Strength
Studies document that T2D individuals have reduced type I muscle fibers and elevated type IIx muscle fiber proportions, potentially accounting for the reduced functional capacity observed in T2D individuals (56, 57). Type I levels directly correlate with insulin sensitivity hence the insulin resistance associated with T2D considerably affects the expression of Type I fibers (58). In another study, biopsies from T2DM patients illustrated a reduced oxidative metabolism program, along with increased type 2x fibers (59). MCK synthesis is observed to be reduced in T2D rats, which is proposed to possibly result from the loss of MEF-1 binding activity observed in T2D conditions (26). Furthermore, MHC IIb synthesis is reduced in the gastrocnemius muscle of T2D rats. MLC1 and MLC3 isoforms are also observed to be decreased in T2D rats (26). The reduction in skeletal muscle fiber MHC and MLC isoforms results in diminished muscle fiber number and size, thereby affecting skeletal mass and muscle strength (21).
T2D skeletal muscles portray diminished contractile force in humans and mice [60]. Studies document that the hands are a target for several diabetes-induced complications. Hence, low handgrip strength is observed to be related to hand disability in T2DM [61]. Previous studies on the relationship between handgrip strength and T2DM have been conflicting [62]. Some studies document a significant inverse association between handgrip strength and T2DM [63,64,65,66], and some studies observed no significant association between handgrip strength and T2DM [67,68]. Studies illustrate that handgrip strength differs between ethnic groups, possibly accounting for the conflicting findings [66]. Low grip strength associated with T2DM is observed to be substantially higher in the South Asian population than in the Western population [66]. The inflammation associated with T2DM is also proposed to be related to low muscular strength [69]. In another study, high tumor necrosis factor-alpha (TNF-α) levels induced a decline in muscle strength [70]. The loss of skeletal muscle mass and strength caused by T2DM results in decreased surface area for glucose transport, further exacerbating insulin resistance [71]. Studies propose that fat accumulation in skeletal muscle observed under T2D conditions, combined with low mitochondrial oxidative capacity, is associated with low muscle strength [72].
Prediabetes
Prediabetes is an asymptomatic condition that usually precedes the onset of type 2 diabetes [8]. A large proportion of the global population is predisposed to prediabetes. The global prevalence rate of prediabetes in 2017 was estimated to be 352,1 million (7.3%) of the adult population, and it is anticipated to increase to 587 million (8.3%) individuals by 2045 [73]. Prediabetes is characterized by higher than normal blood glucose levels but not high enough to establish a T2DM diagnosis and presents as a risk for the onset of T2DM [8]. According to the World Health Organisation (WHO), the prediabetes diagnostic criteria include individuals presenting with one or both of impaired fasting glucose (IFG) or impaired glucose tolerance (IGT). IFG is characterized by fasting plasma glucose (FPG) concentration ≥6.1 mmol/L and <7 mmol/L and IGT is characterized by FPG concentration ˂6.1 mmol/L and a 2-hour post-load plasma glucose concentration between ≥7.8 mmol/L and <11.1 mmol/L measured during the oral glucose tolerance test (OGTT) [74]. The glycated hemoglobin A1c (HbA1c) levels between 5.7 and 6.4% is also used for prediabetes diagnosis [75]. Several factors such as genetic predisposition, insulin resistance, glucotoxicity, lipotoxicity, and β-cell dysfunction result in prediabetes development [8].
Studies document prediabetes to be related to early forms of micro and macrovascular diabetic complications such as nephropathy, chronic kidney disease, small fiber neuropathy, diabetic retinopathy, and heightened risk of macrovascular disease [76]. Studies have observed an increased risk of coronary disease during the prediabetic state [77,78]. Considering that the onset of T2DM complications occurs during the prediabetic state, prediabetic conditions need to be well elucidated as this would aid in preventing some of the overlapping prediabetic and T2D complications.
Effects of Prediabetes on Skeletal Muscle Glucose Homeostasis
The insulin resistance associated with prediabetes is proposed to contribute to endothelial dysfunction [79] (Figure 3). Insulin is vital for endothelial function and glucose metabolism [80]. Studies document that insulin induces vasodilation in resistant arterioles, increases compliance of large arteries, promotes capillary recruitment, and maintains capillary permeability to support nutrient delivery [81,82]. The effects of insulin require coordinated downstream events to keep the vascular tone in the basal state while the vasodilatory response to insulin is elevated in the postprandial state. The skeletal muscle microvasculature, therefore, links insulin's vascular and metabolic functions by increasing the surface area for tissue perfusion. Considering that the skeletal muscle is the principal tissue for insulin-stimulated glucose disposal, these insulin actions represent the role of the endothelium in regulating glucose homeostasis [80].
Increased FFAs induce insulin resistance and endothelial dysfunction in obese patients with prediabetes [79]. A high-fat diet triggers endothelial dysfunction in mice [85], and eating a meal high in fat reduces brachial artery reactivity in humans [86]. FFAs decrease tyrosine phosphorylation of IRS-1/2 and inhibit the PI3K/Akt pathway, resulting in reduced glucose transport and reduced phosphorylation of eNOS [87,88,89] (Figure 4). FFAs activate NADPH oxidase via protein kinase C (PKC) to generate reactive oxygen species (ROS) [90]. Activated PKC contributes to endothelial permeability [91] and extracellular matrix (ECM) expansion [92].
Elevated oxidative stress is associated with the activation of several serine/threonine kinases and the activation of transcription factors NF-kB and activator protein (AP-1), which result in insulin resistance [93]. The activation of serine/threonine kinases c-Jun NH2-terminal kinase (JNK), PKCs, and IkB kinase complex β (IKKβ) leads to serine phosphorylation of IRS-1, which disrupts its ability to bind and activate PI 3-kinase. Thus, there is reduced activation of downstream kinases Akt and PKC-ζ, which reduces the translocation of GLUT4 and glucose transport [94,95,96] (Figure 4). Current literature mainly focuses on microvascular and macrovascular prediabetic complications such as endothelial dysfunction and cardiovascular disease. The skeletal muscle plays a substantial role in glucose homeostasis [93]. Hence, more studies must be conducted to contribute to the current understanding of prediabetic complications, within the context of skeletal muscle.
Effects of Prediabetes on Skeletal Muscle Structure
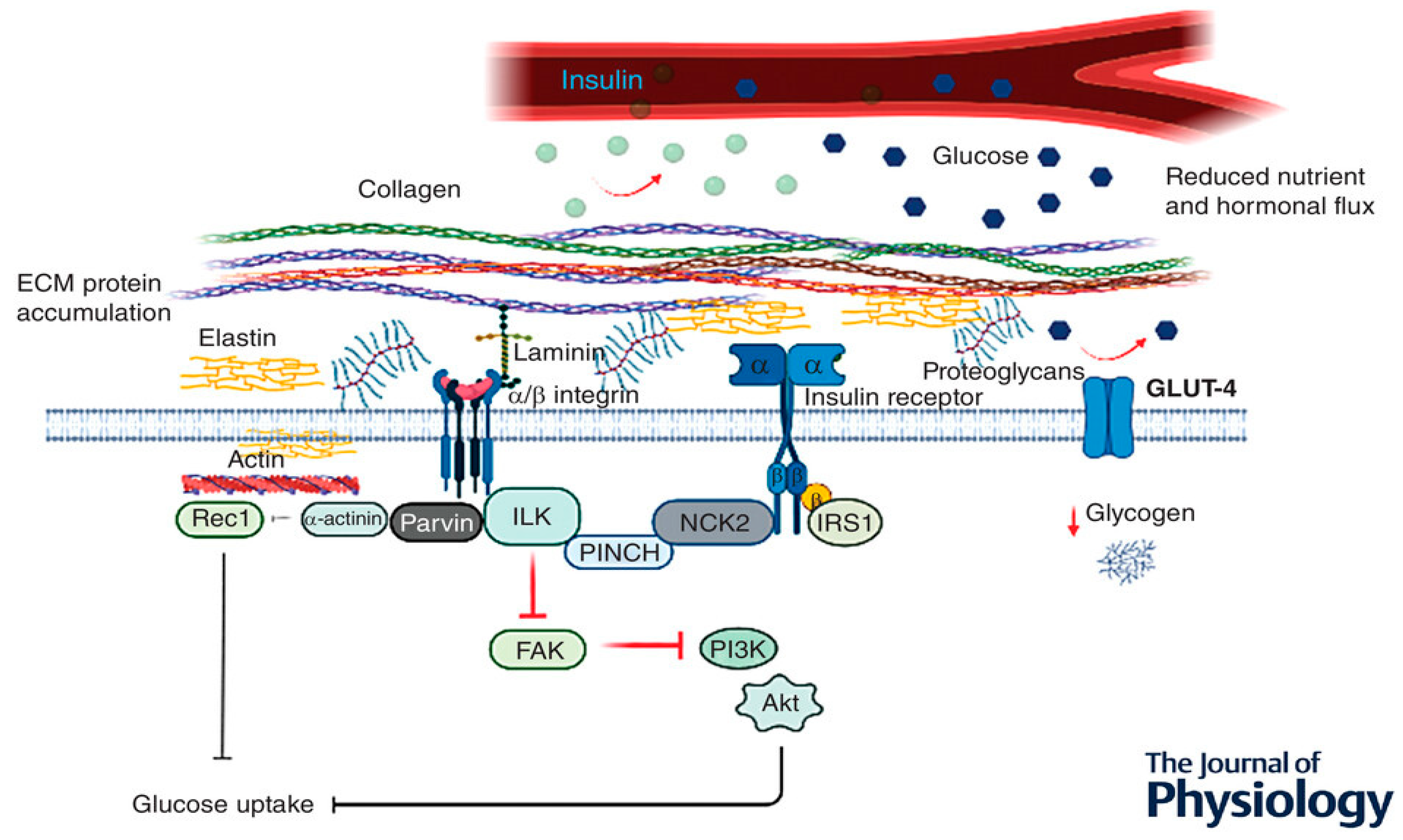
The proinflammatory prediabetic state promotes increased skeletal muscle collagens and other ECM proteins [97] , including fibronectin, proteoglycans, and connective tissue growth factors, and ECM remodelling [98]. The glycosaminoglycans hyaluronan is elevated in tissues of prediabetic animals [99,100]. Hyaluronan is a prominent component of the glycocalyx of capillary lumens, which may affect insulin access to tissues [79]. Reduced hyaluronan using PEGylated hyaluronidase in high-fat-fed mice ameliorates insulin action [99]. Expansion of the muscle ECM and decreased muscle capillary are proposed to contribute to muscle insulin resistance [101].
The chronic systematic inflammation associated with a high-fat diet [102,103] is suggested to heighten ECM protein synthesis and decrease ECM degradation, leading to increased deposition and ECM remodelling [104,105] (Figure 5). The increased protein expression within the ECM is hypothesised to induce a physical barrier, impeding normal insulin action and glucose diffusion across the sarcolemma [99,106] (Figure 5).
The increased protein expression is suggested to be associated with collagen, fibronectin and proteoglycan proteins, which accumulate in the interstial space, resulting in increased diffusion distance and prevention of substrate and hormonal delivery [98,106]. In support of this hypothesis, Kang et al. [99] illustrated that hyaluronan (a significant ECM component) in skeletal muscle was remarkably increased in insulin-resistant diet-induced obesity (DIO) mice when compared to normal chow-fed mice. Interestingly, the same authors also demonstrated that treatment with long-acting pegylated human recombinant PH20 hyaluronidase (PEGPH20) induced a dose-dependent decrease in muscle hyaluronan content and improved skeletal muscle insulin resistance in DIO mice [99]. These results suggest that depletion of ECM polysaccharide promotes muscle insulin sensitivity in obese mice, and contrarily, ECM protein accumulation seems to aggravate muscle insulin resistance [99].
Another hypothesised factor that is suggested to be linked to the underlying mechanism of ECM-associated insulin resistance in DIO, is that the muscle ECM may expand to disrupt vascular function and neovascular growth, provided the close contact between the ECM and endothelium [106]. Nutrient delivery to the contracting muscle requires functional blood flow to establish sufficient glucose (during exercise) and insulin (post-exercise) availability to facilitate glucose uptake and glycogen resynthesis, respectively [105]. Hence, ECM vascular dysfunction and capillary rarefaction have been associated with insulin resistance and T2D [106,107]. Studies show that 40% of insulin-stimulated glucose uptake is attributed to augmented muscle perfusion; however, in the insulin-resistant state, this hemodynamic response is observed to be absent [108,109,110]. Insulin-resistant models and humans are documented to present with capillary rarefaction, thereby highlighting the importance of sufficient muscle capillarization for insulin-mediated disposal [108,111].
Knockout mice lacking vascular endothelial growth factor (mvegf-/-) are proposed to have reduced insulin-mediated glucose disposal [108]. Importantly, Bonner et al. discovered that the reduction in skeletal muscle insulin-mediated glucose uptake was not associated with a dysregulation in intracellular insulin signalling (IRS-1, p85 and phosphorylated total (p/t) Akt), proposing that reduced insulin-stimulated muscle glucose uptake was caused by inadequate muscle perfusion [108]. Consequently, it has been suggested that it is challenging to elucidate insulin signalling in skeletal muscle as it is possible that integrin-associated signalling could have been implicated in mvegf-/- rodent model [108]. The studies above suggest that ECM remodelling contributes to skeletal muscle insulin resistance via endothelial dysfunction and capillary rarefaction [106].
Conclusions
The research discussed in this review suggests that the skeletal muscle plays a substantial role in glucose homeostasis and illustrates the mechanisms involved in the onset of insulin resistance in the prediabetic and T2D states. Prediabetes is suggested to be related to early forms of T2D complications such as diabetic myopathy. A plethora of factors influence skeletal muscle function, among which are the satellite cells of skeletal muscles. Skeletal muscle satellite cells are proposed to maintain skeletal muscle health in physiological and pathophysiological conditions. Hence, the mechanisms involved in regulating skeletal muscle satellite cells' effective functioning need to be well elucidated in the prediabetic state, as literature mainly documents that satellite cell function is potentially impacted during the T2D state. Understanding the modifications that occur in the skeletal muscle during the prediabetic state can allow us to be able to target and prevent the processes that contribute toward the development of diabetic myopathy in the prediabetic state.
Author Contributions
Conceptualization, M.D. and A.K.; writing—original draft preparation, M.D.; writing—review and editing, A.K.; visualization, M.D. and A.K.; supervision, A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef] [PubMed]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.C.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef]
- WHO. Diabetes: World Health Organization. Available online: Diabetes (who.int) (Accessed on 24 April 2022).
- Forouhi NG, Wareham NJ. Epidemiology of diabetes. Medicine (Abingdon). 2014, 42, 698–702.
- Shaw, J.; Sicree, R.; Zimmet, P. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pr. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Diagnosis and classification of diabetes mellitus. . 2006;29 Suppl 1:S43-8. Diabetes Care 2006, 29 Suppl 1, S43–S48.
- Bergman, M. Pathophysiology of prediabetes and treatment implications for the prevention of type 2 diabetes mellitus. Endocrine 2012, 43, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Gadeberg, P.C.; Brock, B.; Jakobsen, J. Muscular atrophy in diabetic neuropathy: a stereological magnetic resonance imaging study. Diabetologia 1997, 40, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Andersen H, Gjerstad MD, Jakobsen J. Atrophy of foot muscles: a measure of diabetic neuropathy. Diabetes Care. 2004, 27, 2382–2385.
- Andersen, H.; Schmitz, O.; Nielsen, S. Decreased isometric muscle strength after acute hyperglycaemia in Type 1 diabetic patients. Diabet. Med. 2005, 22, 1401–1407. [Google Scholar] [CrossRef]
- Otto Buczkowska E, Dworzecki T. [The role of skeletal muscle in the regulation of glucose homeostasis]. Endokrynol Diabetol Chor Przemiany Materii Wieku Rozw. 2003, 9, 93–97.
- Reaven, GM. Insulin resistance in noninsulin-dependent diabetes mellitus: does it exist and can it be measured? The American journal of medicine. 1983, 74, 3–17. [Google Scholar] [CrossRef]
- Schmalbruch, H. The morphology of regeneration of skeletal muscles in the rat. Tissue Cell 1976, 8, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Olguin, H.C.; Yang, Z.; Tapscott, S.J.; Olwin, B.B. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J. Cell Biol. 2007, 177, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; Jaenisch, R. The MyoD family of transcription factors and skeletal myogenesis. BioEssays 1995, 17, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, M.A.; Schnegelsberg, P.N.; Stead, R.H.; Braun, T.; Arnold, H.-H.; Jaenisch, R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esuml, E.; Li, S.; Nonaka, I.; Nabeshima, Y.-I. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Maltin, C.; Delday, M.; Sinclair, K.; Steven, J.; Sneddon, A. Impact of manipulations of myogenesis in utero on the performance of adult skeletal muscle. Reproduction 2001, 122, 359–374. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Muscle stem cells at a glance. J. Cell Sci. 2014, 127, 4543–4548. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C.; Stuart, C.A.; Lee, M.L.; South, M.A.; Howell, M.E.A.; Stone, M.H.; Velten, B.P.; Welch, J.K.C.; Reho, J.J.; et al. Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. 1994, 77, 493–501. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R.; Catalano, M.G.; Brignardello, E.; Danni, O.; Boccuzzi, G. Oxidative Stress Impairs Skeletal Muscle Repair in Diabetic Rats. Diabetes 2004, 53, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur-S, D.K.; et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. USA 2014, 112, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.M.; Koishi, K.; Rudnicki, M.; Maggs, A.M. MyoD protein is differentially accumulated in fast and slow skeletal muscle fibres and required for normal fibre type balance in rodents. Mech. Dev. 1997, 61, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.A.; Robinson, A.; Hartigan-O’Connor, D.; Williams-Gregory, D.; Buskin, J.N.; Apone, S.; Kirk, C.J.; Hardy, S.; Hauschka, S.D.; Chamberlain, J.S. Analysis of Muscle Creatine Kinase Regulatory Elements in Recombinant Adenoviral Vectors. Mol. Ther. 2000, 2, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Hettling H, Van Beek JH. Analyzing the Functional Properties of the Creatine Kinase System with Multiscale’ Sloppy’ Modeling. PLoS Computational Biology. 2011, 7, e1002130.
- Buskin, J.N.; Hauschka, S.D. Identification of a Myocyte Nuclear Factor That Binds to the Muscle-Specific Enhancer of the Mouse Muscle Creatine Kinase Gene. Mol. Cell. Biol. 1989, 9. [Google Scholar] [CrossRef]
- Yamashita, K.; Yoshioka, T. Profiles of creatine kinase isoenzyme compositions in single muscle fibres of different types. J. Muscle Res. Cell Motil. 1991, 12, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Mahler RJ, Adler ML. Type 2 Diabetes Mellitus: Update on Diagnosis, Pathophysiology, and Treatment. The Journal of Clinical Endocrinology & Metabolism. 1999, 84, 1165–1171.
- Authors/Task Force Members; Ryden, L. ; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.P.; et al. ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The task force on diabetes, pre-diabetes, and cardiovascular diseases of the european society of cardiology (ESC) and developed in collaboration with the european association for the study of diabetes (EASD). Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar]
- Defronzo, RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009, 58, 773–795. [Google Scholar]
- Andersen, H.; Gadeberg, P.C.; Brock, B.; Jakobsen, J. Muscular atrophy in diabetic neuropathy: a stereological magnetic resonance imaging study. Diabetologia 1997, 40, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Sishi, B.; Loos, B.; Ellis, B.; Smith, W.; du Toit, E.F.; Engelbrecht, A.-M. Diet-induced obesity alters signalling pathways and induces atrophy and apoptosis in skeletal muscle in a prediabetic rat model. Exp. Physiol. 2011, 96, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schön, M.R.; Blüher, M.; Punkt, K. Altered Fiber Distribution and Fiber-Specific Glycolytic and Oxidative Enzyme Activity in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Abrams, L.; Nikoulina, S.; Mudaliar, S.; Henry, R.R. Glucose transport in cultured human skeletal muscle cells. Regulation by insulin and glucose in nondiabetic and non-insulin-dependent diabetes mellitus subjects. J. Clin. Investig. 1995, 96, 2820–2827. [Google Scholar] [CrossRef] [PubMed]
- Damsbo, P.; Vaag, A.; Hother-Nielsen, O.; Beck-Nielsen, H. Reduced glycogen synthase activity in skeletal muscle from obese patients with and without Type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1991, 34, 239–245. [Google Scholar] [CrossRef]
- Nikoulina, S.E.; Ciaraldi, T.P.; Carter, L.; Mudaliar, S.; Park, K.S.; Henry, R.R. Impaired Muscle Glycogen Synthase in Type 2 Diabetes Is Associated with Diminished Phosphatidylinositol 3-Kinase Activation. J. Clin. Endocrinol. Metab. 2001, 86, 4307–4314. [Google Scholar] [CrossRef]
- Macdonald, I.A. A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur. J. Nutr. 2016, 55, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Ryan, A.J.; Mudaliar, S.R.; Henry, R.R. Altered Myokine Secretion Is an Intrinsic Property of Skeletal Muscle in Type 2 Diabetes. PLOS ONE 2016, 11, e0158209. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Huang, P. The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function. Stem Cell Res. Ther. 2019, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Bruunsgaard, H.; Weis, N.; Hendel, H.W.; Andreassen, B.U.; Eldrup, E.; Dela, F.; Pedersen, B.K. Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 2003, 124, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Lee, J.S.; Kuller, L.H.; Boudreau, R.; de Rekeneire, N.; Harris, T.B.; Kritchevsky, S.; Tylavsky, F.A.; Nevitt, M.; et al. Excessive Loss of Skeletal Muscle Mass in Older Adults With Type 2 Diabetes. Diabetes Care 2009, 32, 1993–1997. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.; Isganaitis, E.; Cerletti, M.; Fitzpatrick, C.; Wagers, A.J.; Jimenez-Chillaron, J.; Patti, M.E. Early Life Nutrition Modulates Muscle Stem Cell Number: Implications for Muscle Mass and Repair. Stem Cells Dev. 2011, 20, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, H.; Lee, I.H.; Modi, S.; Wang, X.; Du, J.; Mitch, W.E. PTEN Inhibition Improves Muscle Regeneration in Mice Fed a High-Fat Diet. Diabetes 2010, 59, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Bryner, R.W.; Alway, S.E.; Acevedo, L.M.; Raya, A.I.; Ríos, R.; Aguilera-Tejero, E.; Rivero, J.-L.L.; Bennett, B.T.; Wilson, J.C.; et al. Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. Am. J. Physiol. Physiol. 2008, 295, C521–C528. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-H.; Cheng, M.; Koh, T.J. Impaired Muscle Regeneration in Ob/ob and Db/db Mice. Sci. World J. 2011, 11, 1525–1535. [Google Scholar] [CrossRef]
- Amacher, S.L.; Buskin, J.N.; Hauschka, S.D. Multiple Regulatory Elements Contribute Differentially to Muscle Creatine Kinase Enhancer Activity in Skeletal and Cardiac Muscle. Mol. Cell. Biol. 1993, 13. [Google Scholar] [CrossRef]
- Wheeler, M.T.; Snyder, E.C.; Patterson, M.N.; Swoap, S.J.; Miura, S.; Kai, Y.; Tadaishi, M.; Tokutake, Y.; Sakamoto, K.; Bruce, C.R.; et al. An E-box within the MHC IIB gene is bound by MyoD and is required for gene expression in fast muscle. Am. J. Physiol. Physiol. 1999, 276, C1069–C1078. [Google Scholar] [CrossRef] [PubMed]
- Wentworth BM, Donoghue M, Engert JC, Berglund EB, Rosenthal N. Paired MyoD-binding sites regulate myosin light chain gene expression. Proc Natl Acad Sci U S A. 1991, 88, 1242–1246.
- Vettor, R.; Fabris, R.; Serra, R.; Lombardi, A.; Tonello, C.; Granzotto, M.; Marzolo, M.; Carruba, M.; Ricquier, D.; Federspil, G.; et al. Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int. J. Obes. 2002, 26, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Cheng, P.F.; Weintraub, H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc. Natl. Acad. Sci. USA 1993, 90, 8028–8032. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.S.; Carey, J.O.; Azevedo, J.L.; Houmard, J.A.; Pories, W.J.; Israel, R.G.; Dohm, G.L. Skeletal muscle fiber composition is related to adiposity and in vitro glucose transport rate in humans. Am. J. Physiol. Metab. 1995, 268, E453–E457. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.J.; Barakat, H.A.; Dohm, G.L.; Pories, W.J.; MacDonald, K.G.; Cunningham, P.R.G.; Swanson, M.S.; Houmard, J.A. Muscle fiber type is associated with obesity and weight loss. Am. J. Physiol. Metab. 2002, 282, E1191–E1196. [Google Scholar] [CrossRef]
- Stuart, C.A.; McCurry, M.P.; Marino, A.; South, M.A.; Howell, M.E.A.; Layne, A.S.; Ramsey, M.W.; Stone, M.H. Slow-Twitch Fiber Proportion in Skeletal Muscle Correlates With Insulin Responsiveness. J. Clin. Endocrinol. Metab. 2013, 98, 2027–2036. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Hanaoka, T.; Imagita, H.; Yasui, T.; Takeshita, D.; Abe, M.; Kawata, S.; Yamakami, T.; Okada, K.; Washio, H.; et al. Long-term wheel-running prevents reduction of grip strength in type 2 diabetic rats. Physiol. Rep. 2021, 9, e15046. [Google Scholar] [CrossRef]
- Redmond, C.L.; Bain, G.I.; Laslett, L.L.; McNEIL, J.D. Hand Syndromes Associated with Diabetes: Impairments and Obesity Predict Disability. J. Rheumatol. 2009, 36, 2766–2771. [Google Scholar] [CrossRef]
- Lee MR, Jung SM, Bang H, Kim HS, Kim YB. Association between muscle strength and type 2 diabetes mellitus in adults in Korea: Data from the Korea national health and nutrition examination survey (KNHANES) VI. Medicine (Baltimore). 2018, 97, e10984.
- Li, J.J.; Wittert, G.A.; Vincent, A.; Atlantis, E.; Shi, Z.; Appleton, S.L.; Hill, C.L.; Jenkins, A.J.; Januszewski, A.S.; Adams, R.J. Muscle grip strength predicts incident type 2 diabetes: Population-based cohort study. Metabolism 2016, 65, 883–892. [Google Scholar] [CrossRef] [PubMed]
- McGrath, R.; Vincent, B.M.; Al Snih, S.; Markides, K.S.; Peterson, M.D. The Association Between Muscle Weakness and Incident Diabetes in Older Mexican Americans. J. Am. Med Dir. Assoc. 2017, 18, 452–e7. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.D.; Zhang, P.; Choksi, P.; Markides, K.S.; Al Snih, S. Muscle Weakness Thresholds for Prediction of Diabetes in Adults. Sports Med. 2016, 46, 619–628. [Google Scholar] [CrossRef] [PubMed]
- van der Kooi, A.-L.L.F.; Snijder, M.B.; Peters, R.J.G.; van Valkengoed, I.G.M. The Association of Handgrip Strength and Type 2 Diabetes Mellitus in Six Ethnic Groups: An Analysis of the HELIUS Study. PLOS ONE 2015, 10, e0137739. [Google Scholar] [CrossRef] [PubMed]
- Leong DP, Teo KK, Rangarajan S, Lopez-Jaramillo P, Avezum Jr A, Orlandini A, et al. Prognostic value of grip strength: findings from the Prospective Urban Rural Epidemiology (PURE) study. The Lancet. 2015, 386, 266–273.
- Xu, L.; Hao, Y.T. Effect of handgrip on coronary artery disease and myocardial infarction: a Mendelian randomization study. Sci. Rep. 2017, 7, 954. [Google Scholar] [CrossRef]
- Agostinis-Sobrinho, C.A.; Moreira, C.; Abreu, S.; Lopes, L.; Sardinha, L.B.; Oliveira-Santos, J.; Oliveira, A.; Mota, J.; Santos, R. Muscular fitness and metabolic and inflammatory biomarkers in adolescents: Results from LabMed Physical Activity Study. Scand. J. Med. Sci. Sports 2016, 27, 1873–1880. [Google Scholar] [CrossRef]
- Schaap, L.A.; Pluijm, S.M.F.; Deeg, D.J.H.; Harris, T.B.; Kritchevsky, S.B.; Newman, A.B.; Colbert, L.H.; Pahor, M.; Rubin, S.M.; Tylavsky, F.A.; et al. Higher Inflammatory Marker Levels in Older Persons: Associations With 5-Year Change in Muscle Mass and Muscle Strength. Journals Gerontol. Ser. A Biomedical Sciences and Medical Sciences 2009, 64A, 1183–1189. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: the effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef]
- Phielix, E.; Meex, R.; Ouwens, D.M.; Sparks, L.; Hoeks, J.; Schaart, G.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. High Oxidative Capacity Due to Chronic Exercise Training Attenuates Lipid-Induced Insulin Resistance. Diabetes 2012, 61, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Hostalek, U. Global epidemiology of prediabetes—present and future perspectives. Clin. Diabetes Endocrinol. 2019, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997, 20, 1183–1197. [CrossRef] [PubMed]
- Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003, 26 Suppl 1, S5–S20. [CrossRef] [PubMed]
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: a high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010, 375, 2215–2222.
- Collaboration ERF. Diabetes mellitus, fasting glucose, and risk of cause-specific death. New England Journal of Medicine. 2011, 364, 829–841.
- Wasserman, D.H.; Wang, T.J.; Brown, N.J. The Vasculature in Prediabetes. Circ. Res. 2018, 122, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Meza CA, La Favor JD, Kim D-H, Hickner RC. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? International Journal of Molecular Sciences. 2019, 20, 3775.
- Zheng, C.; Liu, Z. Vascular function, insulin action, and exercise: an intricate interplay. Trends Endocrinol. Metab. 2015, 26, 297–304. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Breslin, J.W.; Perrin, R.; Gaudreault, N.; Guo, M.; Kargozaran, H.; Wu, M.H. Microvascular Permeability in Diabetes and Insulin Resistance. Microcirculation 2007, 14, 363–373. [Google Scholar] [CrossRef]
- Baron AD, Clark MG. Role of blood flow in the regulation of muscle glucose uptake. Annual review of nutrition. 1997, 17, 487–499.
- Kim J-a, Koh KK, Quon MJ. The union of vascular and metabolic actions of insulin in sickness and in health. Am Heart Assoc 2005, 889–891.
- Keogh, J.B.; Grieger, J.A.; Noakes, M.; Clifton, P.M. Flow-Mediated Dilatation Is Impaired by a High–Saturated Fat Diet but Not by a High-Carbohydrate Diet. Arter. Thromb. Vasc. Biol. 2005, 25, 1274–1279. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a Single High-Fat Meal on Endothelial Function in Healthy Subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef]
- Wang, X.L.; Zhang, L.; Youker, K.; Zhang, M.-X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Shen, Y.H. Free Fatty Acids Inhibit Insulin Signaling–Stimulated Endothelial Nitric Oxide Synthase Activation Through Upregulating PTEN or Inhibiting Akt Kinase. Diabetes 2006, 55, 2301–2310. [Google Scholar] [CrossRef]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Lynch, J.J.; Ferro, T.J.; A Blumenstock, F.; Brockenauer, A.M.; Malik, A.B. Increased endothelial albumin permeability mediated by protein kinase C activation. J. Clin. Investig. 1990, 85, 1991–1998. [Google Scholar] [CrossRef]
- Koya, D.; Jirousek, M.R.; Lin, Y.W.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J. Clin. Investig. 1997, 100, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Kim J-a, Montagnani M, Koh KK, Quon MJ. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation. 2006, 113, 1888–1904.
- Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. The Journal of clinical investigation. 2017, 114, 1752–1761.
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine Phosphorylation of Insulin Receptor Substrate 1 by Inhibitor κB Kinase Complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-Induced Muscle Insulin Resistance Is Associated With Extracellular Matrix Remodeling and Interaction With Integrin α2β1 in Mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; De Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol. Metab. 2006, 290, E560–E565. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan Accumulates With High-Fat Feeding and Contributes to Insulin Resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Chajara, A.; Raoudi, M.; Delpech, B.; Leroy, M.; Basuyau, J.P.; Levesque, H. Increased Hyaluronan and Hyaluronidase Production and Hyaluronan Degradation in Injured Aorta of Insulin-Resistant Rats. Arter. Thromb. Vasc. Biol. 2000, 20, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Mayes, W.H.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice. Diabetologia 2014, 57, 603–613. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E. Effect of High-Fat Diets on Oxidative Stress, Cellular Inflammatory Response and Cognitive Function. Nutrients 2019, 11, 2579. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [PubMed]
- Draicchio, F.; Behrends, V.; Tillin, N.A.; Hurren, N.M.; Sylow, L.; Mackenzie, R. Involvement of the extracellular matrix and integrin signalling proteins in skeletal muscle glucose uptake. J. Physiol. 2022, 600, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P. Endothelial dysfunction in insulin resistance and type 2 diabetes. J. Intern. Med. 2007, 262, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.S.; Lantier, L.; Hasenour, C.M.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Muscle-Specific Vascular Endothelial Growth Factor Deletion Induces Muscle Capillary Rarefaction Creating Muscle Insulin Resistance. Diabetes 2013, 62, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Maloney, E.; Rizzo, N.O.; Morton, G.J.; Wisse, B.E.; Kirk, E.A.; Chait, A.; Schwartz, M.W.; B, D.; et al. Vascular Inflammation, Insulin Resistance, and Reduced Nitric Oxide Production Precede the Onset of Peripheral Insulin Resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell metabolism. 2011, 13, 294–307.
- Chung, A.W.; Hsiang, Y.N.; Matzke, L.A.; McManus, B.M.; van Breemen, C.; Okon, E.B.; A, C.; H, Y.; J, K.; M, S.; et al. Reduced Expression of Vascular Endothelial Growth Factor Paralleled With the Increased Angiostatin Expression Resulting From the Upregulated Activities of Matrix Metalloproteinase-2 and -9 in Human Type 2 Diabetic Arterial Vasculature. Circ. Res. 2006, 99, 140–148. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of myogenic regulatory factor (MRF) regulation of muscle-specific gene expression (MGE).
Figure 1.
Schematic representation of myogenic regulatory factor (MRF) regulation of muscle-specific gene expression (MGE).
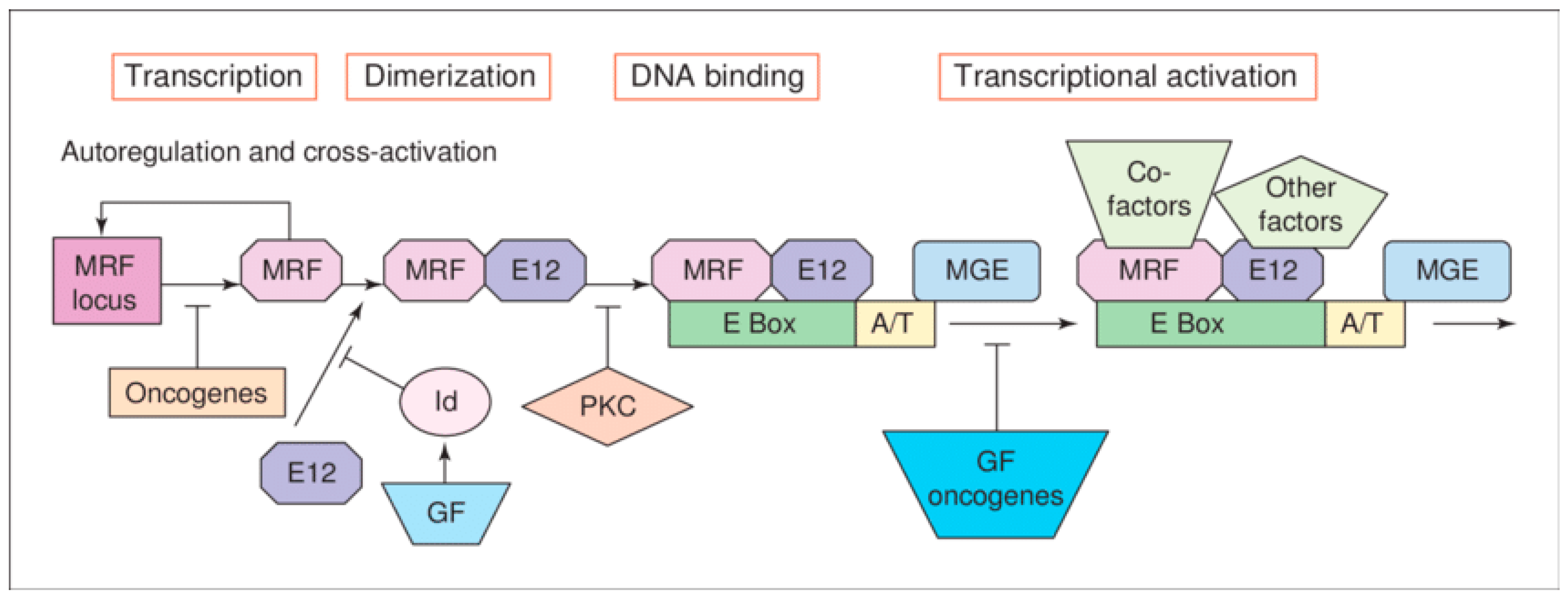
Figure 2.
Schematic representation of symmetric and asymmetric satellite cell divisions.
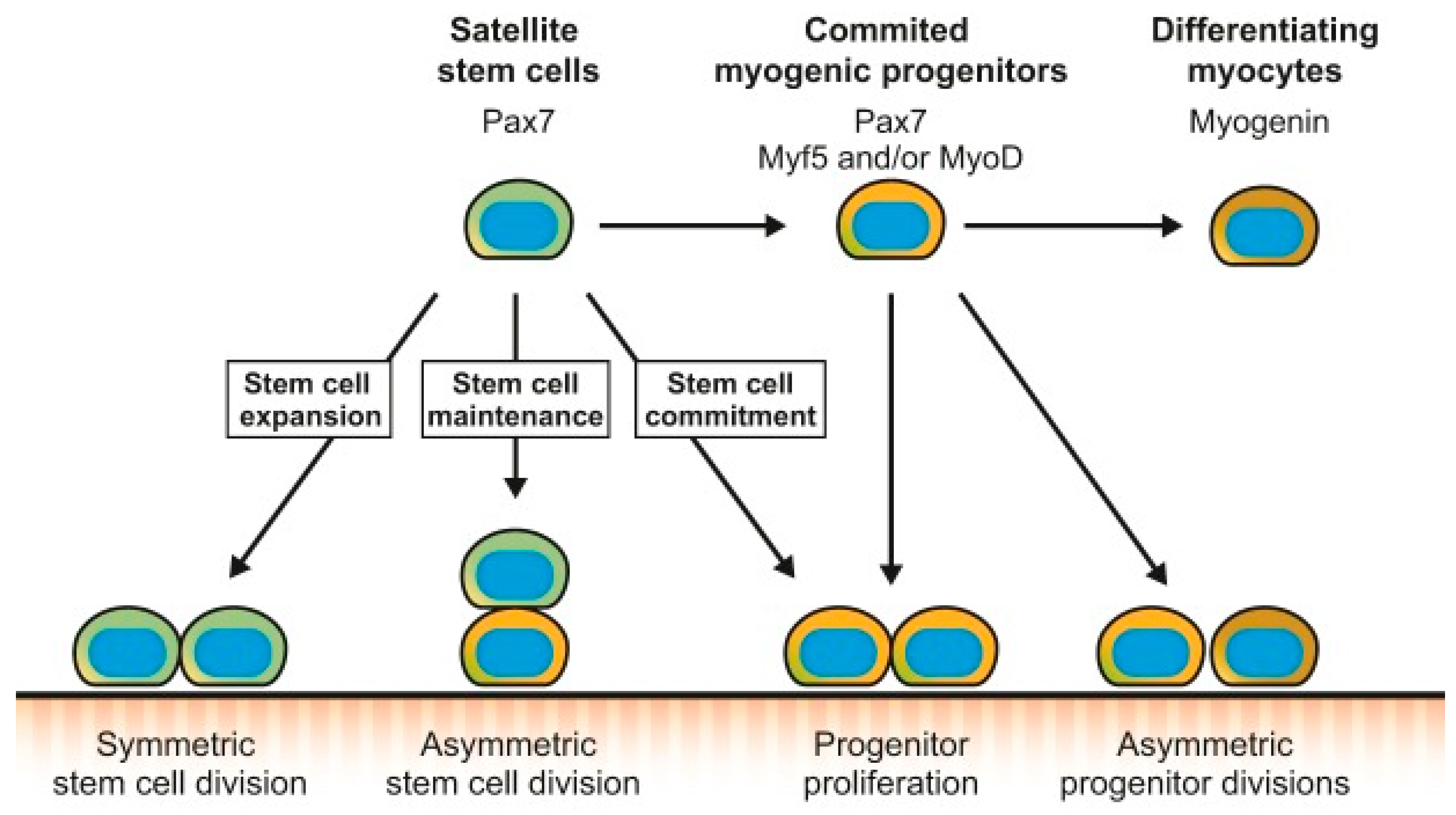
Figure 3.
Endothelial insulin resistance, hyperglycemia, and increased free fatty acids (FFAs) give rise to oxidative stress, inflammation, endothelial dysfunction, and fibrinolytic dysfunction in prediabetes. PAI-1 indicates plasminogen activator inhibitor-1; RAAS renin–angiotensin–aldosterone system; and VAT, visceral adipose tissue.
Figure 3.
Endothelial insulin resistance, hyperglycemia, and increased free fatty acids (FFAs) give rise to oxidative stress, inflammation, endothelial dysfunction, and fibrinolytic dysfunction in prediabetes. PAI-1 indicates plasminogen activator inhibitor-1; RAAS renin–angiotensin–aldosterone system; and VAT, visceral adipose tissue.
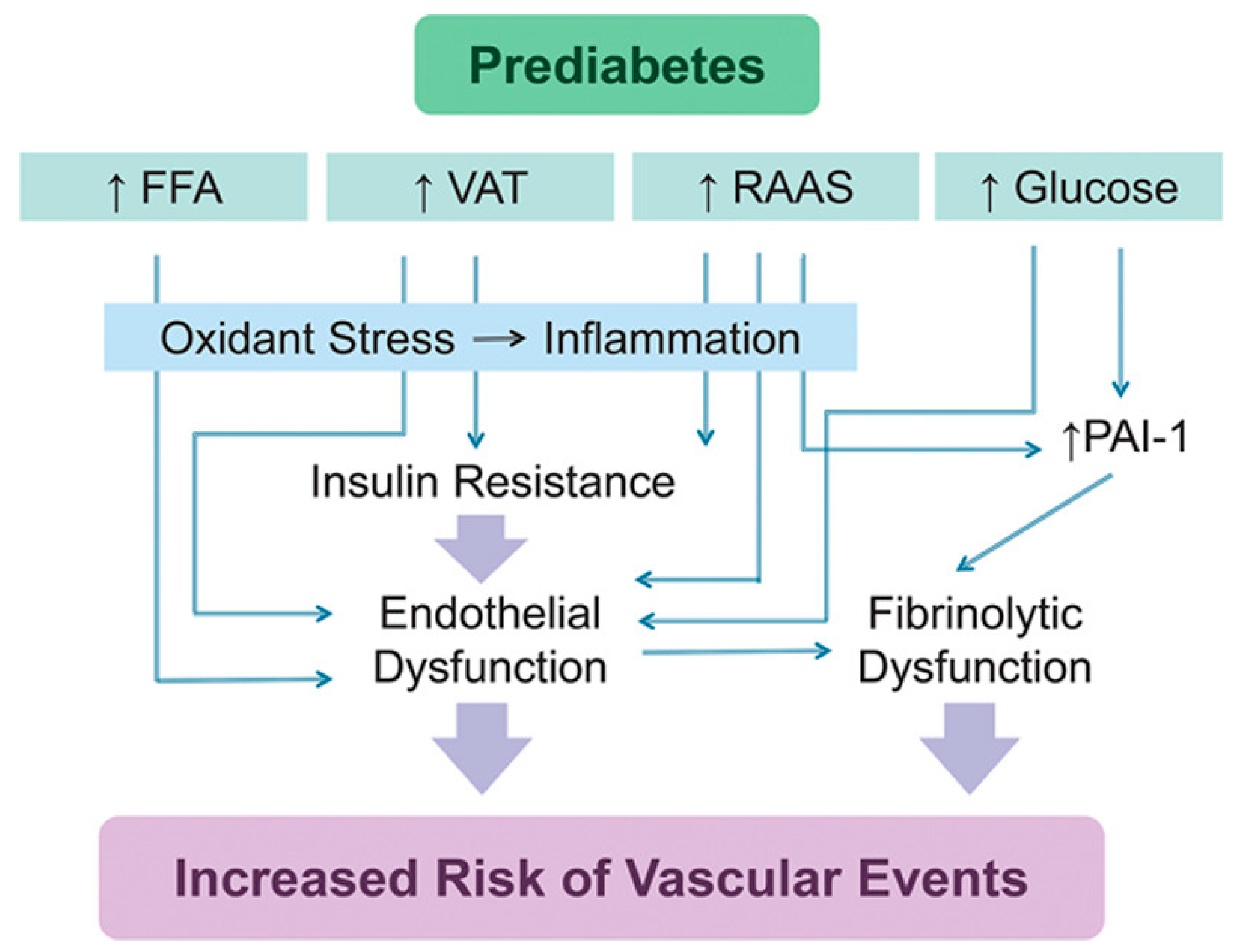
Figure 4.
The physiology and pathophysiology of the vascular and metabolic actions of insulin.
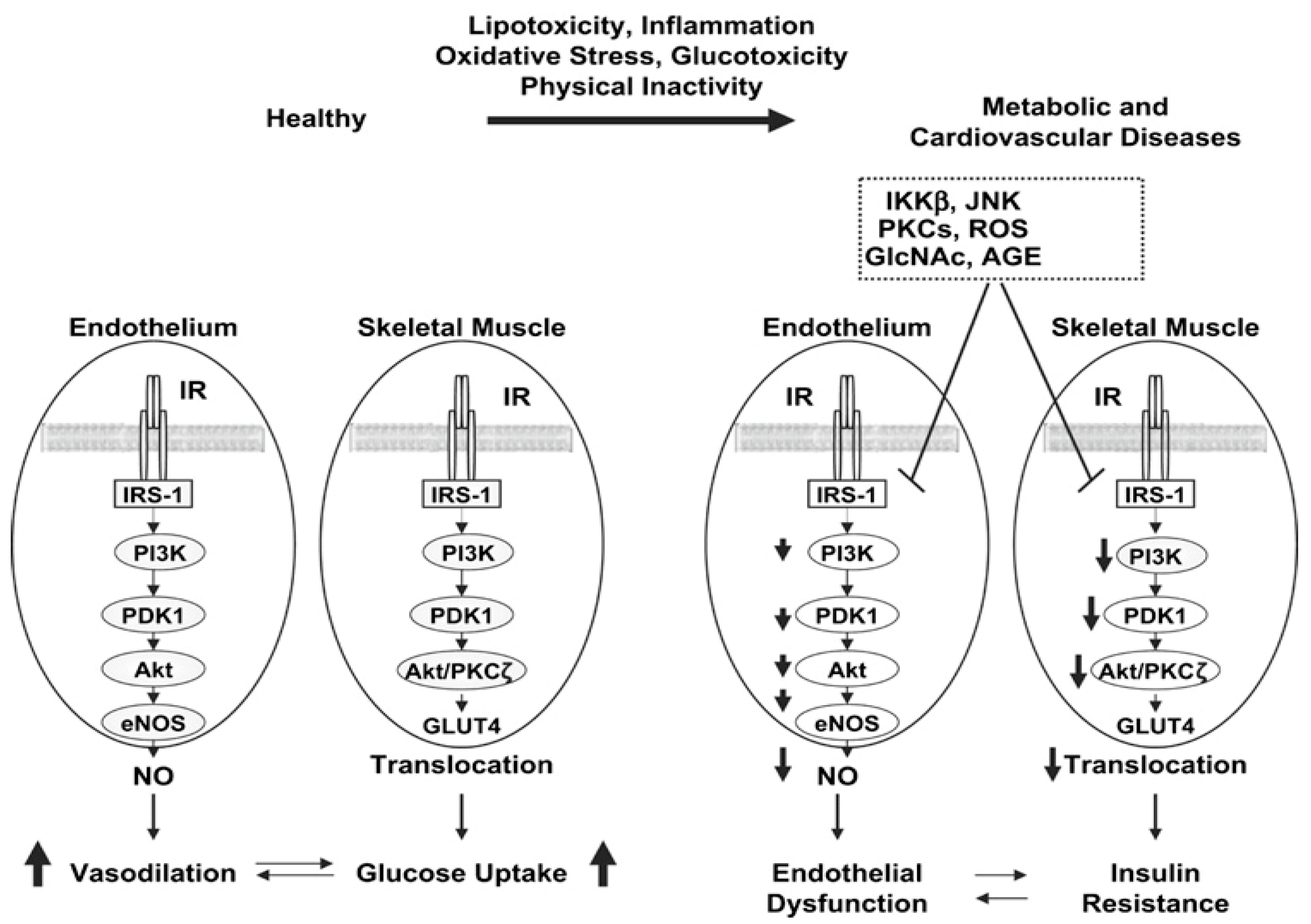
Figure 5.
Potential pathway linking integrins and their associated proteins in the regulation of glucose metabolism in skeletal muscle.
Figure 5.
Potential pathway linking integrins and their associated proteins in the regulation of glucose metabolism in skeletal muscle.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



