Submitted:
02 November 2023
Posted:
03 November 2023
You are already at the latest version
Abstract
Glycosaminoglycans (GAGs) with unique structures from marine animals show intriguing pharmacological activities and negligible biological risks, providing more options for us to explore safer agents. Swim bladder is a tonic food and folk medicine, and its GAGs show good anticoag-ulant activity. In this study, two GAGs SJG-1.0 and GMG-1.0 have been extracted and isolated from the swim bladder of Sphyraena jello and Gadus morhua. The physicochemical properties, pre-cise structural characteristics, and anticoagulant activities of these GAGs were determined. The analysis results of the SJG-1.0 and GMG-1.0 showed that they were chondroitin sulfate (CS)/dermatan sulfate (DS) hybrid chains with molecular weights of 109.32 kDa and 123.13 kDa, respectively. They were mainly composed of the repeating disaccharide unit of -{IdoA-α1,3-GalNAc4S-β1,4-}- (DS-A). The DS-B disaccharide unit of -{IdoA2S-α1,3-GalNAc4S-β1,4-}- also existed in both SJG-1.0 and GMG-1.0. SJG-1.0 had a higher proportion of CS-O disaccharide unit -{-GlcA-β1,3-GalNAc-β1,4-}-, while GMG-1.0 contained higher proportion of CS-E disaccha-ride unit -{-GlcA-β1,3-GalNAc4S6S-β1,4-}-. The disaccharide compositions of the GAGs varied in a species-specific manner. Anticoagulant activity assay revealed that both SJG-1.0 and GMG-1.0 had potent anticoagulant activity, which can significantly prolong activated partial thrombosis time. GMG-1.0 also can prolong the thrombin time. SJG-1.0 showed no intrinsic tenase inhibition ac-tivity, while GMG-1.0 can obviously inhibit intrinsic tenase with EC50 of 58 nM. Their significantly different anticoagulant activities may be due to their different disaccharide structural units and proportions. These findings suggested that the swim bladder CS/DS hybrid chains with well-defined structure may be safer anticoagulants with low bleeding risk.
Keywords:
Swim bladder
; glycosaminoglycan
; CS/DS hybrid chain
; structure
; anticoagulant activity
1. Introduction
Glycosaminoglycans (GAGs) are complex acidic polysaccharides composed of repeating disaccharide units formed by hexosamine and uronic acid (or galactose (Gal)). The common GAGs can be divided into chondroitin sulfate (CS), dermatan sulfate (DS), heparin (HP), heparan sulfate (HS), hyaluronic acid (HA), and keratan sulfate (KS) according to their monosaccharide compositions and sulfate substitution positions. GAGs are widely distributed in animal kingdom, and their structures are related to the animal tissue, organ and species [1,2]. The structural complexity of GAGs results in their diverse activities. Various GAGs have been isolated and found to possess anticoagulant, antithrombotic, nerve regeneration, and anti-inflammatory activities [3]. The most typical example is heparin, a very famous GAG, which has been widely used as an anticoagulant in clinical for over eighty years [4,5]. Another widely known GAG is CS, which is used in the treatment of joint diseases [6]. HA has also been used in skin regeneration, wound healing, and cosmetic fields [7]. The wide application of these GAGs encourages researchers to look for GAGs from different animal sources with unique structure and remarkable activity.
Numerous commercially available GAGs including HP are extracted from terrestrial mammalian tissues, such as bovine lung and porcine intestine. However, they have some inevitable problems, including religious concerns and potential risk of contamination by pathogens, such as prion virus and African swine fever virus [8]. Marine animal resources are abundant and most marine animals contain GAGs with novel structures, intriguing pharmacological functions, and negligible biological risks, which provide more options for us to explore safer agents [9].
Swim bladder is one of the byproducts of fish processing and has long been used as not only tonic food but also folk medicine in Asia, particularly in southern China [10]. The swim bladder weight accounts for ~1.3% of the final fish weight [11], and the scale of swim bladder production in Asia is quite large. The swim bladders from some fishes are dried and sold as fish maw, which have a great market demand for their high nutritional values and good pharmacological activities [12]. Collagen and peptide are the major components of swim bladder and have been studied extensively [13,14,15]. Some polysaccharides also have been isolated from swim bladder and found to possess preventive effect on the gastric injury, therapeutic effect on lupus nephritis, and anticancer activity [16,17,18]. However, the structural information of these polysaccharides is very limited. In recent years, GAGs from swim bladder were isolated and their basic structural characteristics were analyzed. For example, in 2018, the disaccharide compositions of swim bladder GAG were analyzed by compositional analysis of GAG disaccharides using heparin lyase I, II, III and chondroitin lyase ABC and by 1H NMR spectroscopy, indicating that the GAG from the commercial dried fish maw, “hudiejiao” consisted of CS (95%) and HS (5%) of the total GAG [19]. Subsequently, GAGs isolated from the swim bladders of Lateolabrax japonicus and Aristichthys nobili were identified to be CS-A mainly constituted by the repeating disaccharide unit -{GlcA-β1,3-GalNAc4S-β1,4-}- [20,21]. In addition, these GAGs have exhibited a wide range of activities, such as wound healing, anticoagulant, and anti-inflammatory activities and intervention effects against arsenic-induced damage [19,21,22].
Although several studies have shown that swim bladder is rich in GAGs, the detailed structure and anticoagulant activity of GAGs from various fish species have not been deeply studied. In this study, the GAG fractions were isolated from the swim bladder of Sphyraena jello (SJG-1.0) and Gadus morhua (GMG-1.0) and identified as CS/DS hybrid chains. Further analysis of structure and activity revealed that SJG-1.0 and GMG-1.0 are mainly composed of DS rather than CS units and exhibit potent anticoagulant activity. These findings suggest that SJG-1.0 and GMG-1.0 has potential in the development of anticoagulants, which will facilitate the high-value utilization of the swim bladder resources from fish processing.
2. Results and Discussion
2.1. Extraction and Purification of SJG-1.0 and GMG-1.0
The yields of crude GAGs extracted from Sphyraena jello and Gadus morhua were 0.24% and 0.36% by dry weight of swim bladders, respectively. The crude GAG was further purified by strong anion-exchange chromatography and resulted in two main fractions SJG-1.0 and GMG-1.0 with yields of 27.4% and 27.9%, respectively by dry weight of the crude GAGs.
2.2. Chemical compositions of SJG-1.0 and GMG-1.0
The protein content in SJG-1.0 was 0.91%, and no protein was detected in GMG-1.0 by the Bradford method indicating that proteins were almost removed by deproteinization and purification. The uronic acid contents in SJG-1.0 and GMG-1.0 were 35.04% and 23.04%, respectively, and their sulfate contents were 29.49% and 20.49%, respectively. The molar ratio of -OSO3−/-COO− of SJG-1.0 and GMG-1.0 were 1.16 and 1.56, respectively. The ultraviolet-visible absorption spectra of SJG-1.0 and GMG-1.0 showed that no absorption peak was observed at the wavelength of 280 and 260 nm, indicating no proteins and nucleic acids contained in these compounds (Figure S1).
2.3. Purity and molecular weight of SJG-1.0 and GMG-1.0
The purity and weight mean molecular mass (Mw) of the samples were determined by high-performance gel permeation chromatography (HPGPC), and the results are shown in Figure 1. Both SJG-1.0 and GMG-1.0 showed a single symmetrical peak, which indicated that they were homogeneous polysaccharides. The results of cellulose acetate electrophoresis further confirmed high purity of SJG-1.0 and GMG-1.0 because of only one band shown by these GAGs in the cellulose acetate strip ((Figure S2)). The Mws of SJG-1.0 and GMG-1.0 were calculated to be 109.32 kDa and 123.13 kDa, respectively, according to their standard curve. Their Mws were similar to the purified sulfate GAG from Aristichthys nobilis swim bladder [21], but smaller than the major GAG fraction from a swim bladder, whose species was not identified [19].
2.4. Monosaccharide compositions of SJG-1.0 and GMG-1.0
Monosaccharide compositions of SJG-1.0 and GMG-1.0 are shown in Figure 1B. Both SJG-1.0 and GMG-1.0 were composed of IdoA, GalNAc, GlcA and GlcNAc with different molar ratios. The molar ratio of IdoA, GalNAc, GlcA and GlcNAc was 19.69: 22.76: 12.48: 1.00 for SJG-1.0 and 36.43: 26.28: 3.95: 1.00 for GMG-1.0. In addition, we also observed that there was an unknown peak (labeled x) appeared at approximately 25 min in the HPLC profiles, which did not match with any standard monosaccharide. It was reported that there were acidolysis-resistant diasaccharides when GAGs were not sufficiently hydrolyzed [23,24]. The unknown peak may be the diasaccharide units derived from SJG-1.0 or GMG-1.0. To prove our hypothesis, the PMP-derivatized sample was further analyzed by the UPLC-MS, resulting in three pseudo-molecular ions from the unknown peak with mass-to-charge values of 175.0871, 510.2349, and 686.2661, which were consistent with [PMP + H]+, [GalNAc-2PMP + H]+, and [GlcA/IdoA-GalNAc-PMP + H]+, respectively (Figure 1C). The IdoA and GlcA in SJG-1.0 or GMG-1.0 cannot be differentiated by monosaccharide analysis but can be identified by the 1D/2D NMR spectroscopy. Based on the results of monosaccharide composition and cellulose acetate electrophoresis, it can be speculated that both SJG-1.0. and GMG-1.0 may be CS/DS hybrid chains.
2.5. Disaccharide compositions of SJG-1.0 and GMG-1.0
Disaccharide compositions of SJG-1.0 and GMG-1.0 were identified by treatment with chondroitin ABC lyase and analysis of the released unsaturated disaccharides. As shown in Figure 1D, the disaccharide compositions with molar percentages of SJG-1.0 was ΔDi4S (78.61%), ΔDi6S (8.96%), ΔDi0S (7.56%), ΔDi2,4S (3.21%), ΔDi2,6S (0.64%), and ΔDi4,6S (0.24%). GMG-1.0 also mainly contained the disaccharide ΔDi4S (87.25%) and a small amount of ΔDi4,6S (5.18%), ΔDi2,4S (5.05%), ΔDi0S (1.54%), ΔUA-2S (0.37%), ΔDi2,4,6S (0.37%), and ΔDi2,6S (0.24%). Taken together, these data indicated that both SJG-1.0 and GMG-1.0 were mainly composed of ΔDi4S unit, a DS-A disaccharide unit, confirmed by the following NMR analysis. SJG-1.0 had a higher proportion of ΔDi0S unit, while GMG-1.0 contained higher proportion of ΔDi4,6S disaccharide unit. In a previous study, a purified GAG from a commercial swim bladder was determined to have 59.7% of ΔDi4S (CS-A/DS-A) but contain more ΔDi4,6S (36.5%) than SJG-1.0 and GMG-1.0 [19]. Another study reported that a heparin-like GAG from Aristichthys nobilis swim bladder was composed almost exclusively of ΔDi4S (CS-A) [21]. Therefore, the disaccharide compositions of GAG from swim bladders varied in a species-specific manner.
2.6. IR spectrum analysis of of SJG-1.0 and GMG-1.0
The FT-IR spectra of SJG-1.0 and GMG-1.0 are shown in Figure S3. The broad intense characteristic peaks around 3421 cm−1 in SJG-1.0 and 3435 cm−1 in GMG-1.0 were due to O-H stretching vibration. The bands around 2926/2943 cm−1 were attributed to C-H stretching vibration [25]. The bands around 1630 cm−1 and 1418 cm−1 were attributed to the stretching vibration of C=O and C-O, respectively, suggesting the existence of uronic acid. Absorptions at approximately 1260 cm−1 and 820-860 cm−1 were derived from the S=O asymmetric stretching vibration and C-O-S stretching vibration, respectively, indicating the presence of sulfate groups in both GAGs [26]. In addition, the absorption peaks of C-O-S stretching vibration at around 855 cm−1 in SJG-1.0 and 843 cm−1 in GMG-1.0 indicated that C-4 position of GalNAc or GlcNAc residues was sulfated [27].
2.7. Structural analysis of SJG-1.0 and GMG-1.0 by NMR spectroscopy
The detailed structural features of SJG-1.0 and GMG-1.0 were further elucidated by 1D/2D NMR analyses. First, some structural information obtained from above physicochemical analyses can be further confirmed by the 1H and 13C NMR spectra (Figure 2 and Figure 3). According to the literature [28], the relatively downfield chemical shifts (>4.8 ppm) of the anomeric signals suggested the α configuration of residues A–C. Relatively upfield chemical shifts (<4.7 ppm) of the anomeric proton signals indicated β configuration of residues D–I. The signals (5.27, 5.18, and 4.86 ppm) in the anomeric region may be from α-d-IdoA residues, while the anomeric singal of 4.47 ppm may be due to β-d-GlcA residues according to literature [29]. In addition, the anomeric protons at δ 4.67, 4.62, 4.61, 4.56, and 4.53 ppm may be attributed to the β-d-GalNAc residues. The intense signals with upfield resonance appeared at around 2.03-2.07 ppm may be due to the acetyl methyl groups in the amino sugars, such as GlcNAc and GalNAc residues in these GAGs. The amino sugar residues D–H were almost acetylated according to their peak area integration of the anomeric proton and the methyl from the acetyl groups. In the 13C spectra, the most downfield resonance, δH at 176–178 ppm, can be ascribed to two carbonyl groups in IdoA, GlcA, GlcNAc and GalNAc residues. The anomeric carbon signals were at 103–107 ppm. The relative upfield signals appearing at approximately 55 ppm can be arbitrarily assigned as C-2 resonance of GlcNAc and GalNAc residues because of the presence of the amino group at this position. The most upfield signals at approximately 25 ppm may be attributed to the acetyl methyl groups in the GlcNAc and GalNAc residues. Subsequently, the 2D NMR spectra (1H–1H COSY, TOCSY, ROESY, 1H–13C HSQC, HSQC-TOCSY, and HMBC) (Figure 4 and Figures S4–S6) were applied to assign all the chemical shifts of various residues compared with the data available in the literature [30,31]. The assignment results are shown in Table 1.
SJG-1.0 showed nine intra-residue spin coupling systems in 1H–1H COSY, TOCSY, and ROESY spectra (Figure 4A–C), indicating that it contained nine kinds of sugar residues linked to various sugar residues. The obvious signals at 5.27/103.6, 5.18/103.9, 4.86/106.3, 4.67/105.1, 4.62/105.0, 4.61/105.0, 4.56/103.8, 4.53/105.6, and 4.47/106.7 ppm in the HSQC spectrum were assigned to the anomeric signals of various sugar residues designated as A, B, C, D, E, F, G, H, and I, respectively (Figure 4E). The cross-signals at 4.05/54.3, 4.08/54.7, 4.10/54.3, 4.02/54.5, and 3.86/55.1 ppm in the HSQC spectrum can be readily assigned to H-2/C-2 of the D–H residues, which indicated that they were β-d-GalNAc or β-d-GalN residues. Chemical shifts of H-2 of A–I can be readily obtained from the COSY spectrum, and their C-2 chemical shifts can be assigned by the HSQC spectrum. The residues A, B, C, and I were then identified to the α-d-IdoA or β-d-GlcA residues because they had a relatively large C-2 signal compared with the amino sugar. The proton signals of the nine systems from H-3 to H-6 can be also assigned carefully using the 1H–1H COSY, TOCSY and ROESY spectra although some signals in these spectra were weak. The downfield chemical shift of H-5 of residues A–C (δH > 4.7 ppm) further confirmed that they were d-IdoA residues for their C-5 epimerization [30]. Then, the residue I was confirmed to be the d-GlcA residue. The detailed carbon signals of various sugar residues from C-3 to C-6 were assigned based on the assignment of the protons, using the 1H–13C HSQC and HSQC-TOCSY spectra (Figure 4E and Figure S4). Therefore, all signals from the 1D/2D NMR spectra can be clearly assigned as shown in Table 1.
The sequence of sugar residues in SJG-1.0 was confirmed by the 1H–1H ROESY and 1H–13C HMBC NMR spectra (Figure 4C and D). For example, the cross signal (4.86, 4.03 ppm) in the ROESY spectrum showed that the residue C had a strong inter-residue ROE connected to H-3 of residue D, confirming that residue C was linked to the C-3 position of residue D. The cross signal (4.67,4.08 ppm) indicated that residue D was linked to the C-4 position of residue C. Similarly, the linkages (I-β1,3-F/G/H), (E/G/H-β1,4-I), (A-α1,3-E), (B-α1,3-G), (F-β1,4-A/B) were confirmed by the cross peaks at (4.47, 4.00 ppm), (4.62/4.56/4.53, 3.78 ppm), (5.27,4.14 ppm), (5.17,3.96 ppm), and (4.61, 4.10/4.13 ppm) in the ROESY spectrum. The β1,3-linkages between residue I and residues F/G/H were further confirmed by the correlation signals (4.47, 78.6 ppm) in the HMBC spectrum. The cross signals (3.78, 103.8 ppm) further confirmed the β1,4-linkages between residue G and residue I.
The down-field chemical shifts of protons and carbons caused by the sulfation can identify the sulfated positions on residues A–I in comparison with the corresponding unsubstituted monosaccharide. Compared with the H/C-4 chemical shifts (4.21/70.3, 4.17/70.3 ppm) of residues G and H, the downfield H/C-4 chemical shifts (4.66/79.2, 4.63/79.1, 4.74/79.4 ppm) of residues D, E, and F indicated that these positions were sulfated. Furthermore, the downfield chemical shift of H/C-6 (4.17/4.25, 70.0 ppm) confirmed that residue F was sulfated at the C-6 position. Similarly, the downfield H/C-2 chemical shifts (3.83/79.1, 3.79/79.2 ppm) of residues A and B were obviously higher than the corresponding chemical shift (3.53/72.4 ppm) of residue C, indicating that C-2 positions of residues A and B were sulfated.
Based on the above analysis, the proposed polysaccharide sequence of SJG-1.0 was -{-C-α1,3-D-β1,4-}m-{-A-α1,3-E-β1,4-}n-{-I-β1,3-H-β1,4-}o-{-I-β1,3-F-β1,4-}p-{-B-α1,3-G-β1,4-}q-{-I-β1,3-G-β1,4-}r. According to the integral area of the anomeric proton of amino sugars in the 1H spectrum (Figure 2A), the proportion of various disaccharide units in the SJG-1.0 can be calculated to be m:n:o:p:q:r = 26:6:2:2:1:9.
Based on the above results of NMR analysis of SJG-1.0, the detailed chemical structure of GMG-1.0 was also further confirmed by its 1D/2D NMR analysis (Figure 4F, Figures S5 and S6). The 1H and 13C NMR spectra and the chemical shifts from identified 2D NMR spectra are shown in Figure 2B, 3B and Table S1. It was observed that most signals from the same residues of GMG-1.0 were similar to those of SJG-1.0. GMP-1.0 was proposed to have a similar polysaccharide sequence to SJG-1.0, except that the GMP-1.0 had a obviously different disaccharide proportion of m:n:o:p:q:r = 69.5:6:3:3.5:1:1 and contained higher content of disaccharide unit -{-GlcA-β1,3-GalN4S6S-β1,4-}-. These results indicated that both SJP-1.0 and GMG-1.0 were rich in DS domain, i.e., they were mainly composed of repeating disaccharide units of -{IdoA-α1,3-GalNAc4S-β1,4-}-. However, the proportions of various disaccharide units in these two GAGs were obviously different, which may be due to the different species.
Based on the results of monosaccharide, disaccharide composition, and NMR analyses, SJG-1.0 and GMG-1.0 were confirmed to be CS/DS hybrid chains with high amounts of DS-A disaccharide unit. Since some disaccharide units are extremely low in these CS/DS chains, especially in the GMG-1.0, having low resolution of the ROESY and HMBC spectra, their precise structure of these GAGs may be elucidated by analyzing their oligosaccharide fragments as carried out in our previous studies [9,32].
DS often occurs in co-polymeric form with CS, thus forming a CS/DS hybrid chain. To date, many CS/DS hybrid chains with varying proportions of CS and DS disaccharide units have been isolated from marine animals, such as shark skin and brittlestars [33,34]. These hybrid chains from different species of marine animals displayed enormous structural diversity mainly due to the variability of sulfate substitution, and showed multiple biological activities, such as neuritogenic activity, wound healing, and anticoagulant activity, which had potential as therapeutic agents [33,34,35]. In 2017, GAGs from fish swim bladder were determined to contain 95% CS of the total GAG [21]. Considering that the CS may contain CS-B (DS) unit, it was probably a CS/DS hybrid chain. The integration of the peak area for 1H–IdoA and 1H–GlcA in the 1H NMR spectrum suggested that the ratio of DS disaccharide unit to CS-A disaccharide unit was 1:1.4. In our present studies, the CS/DS hybrid chains SJG-1.0 and GMG-1.0 are mainly composed of DS-A disaccharide unit and a small amount of DS-B and CS disaccharide units. Therefore, the structures of SJG-1.0 and GMG-1.0 are obviously different from those of the CS/DS hybrid chains in previous reports [19,21].
2.11. Anticoagulant activity
The anticoagulant activities of SJG-1.0 and GMG-1.0 were investigated by the classical coagulation assays, such as the activated partial thromboplastin time (APTT), thrombin time (TT), and prothrombin time (PT) assays, and the results are shown in Table 2. Both SJG-1.0 and GMG-1.0 showed significant anticoagulant activity by prolonging APTT. The concentration 0.226 μM of GMG-1.0 was required to double the APTT, indicating that it had strong intrinsic anticoagulant activity that was stronger than that of low-molecular weight heparin (LMWH). The concentration of GMG-1.0 required to double the TT was 12.035 μM, indicating that it can obviously inhibit common pathway of the coagulation cascade. SJG-1.0 did not exhibit TT and PT prolonging activity, indicating it had higher selective to inhibit the intrinsic coagulation pathway than the GMG-1.0. Both SJG-1.0 and GMG-1.0 showed no PT prolonging activity, indicating that they had no effect on the extrinsic coagulation pathways. The obviously different anticoagulant activity of these two GAGs may be due to their different structural units and proportions.
Considering that SJG-1.0 and GMG-1.0 can obviously prolong APTT, they may have potential to inhibit the coagulation factors, such as factor XIIa, factor XIa, factor IXa and intrinsic tenase associated with the intrinsic coagulation pathway. The activity assay revealed that GMG-1.0 potently inhibited the intrinsic tenase with EC50 value of 58 nM (Table 2). The intrinsic tenase is the rate-limiting enzyme in the intrinsic pathway, and inhibitors of this enzyme complex, such as depolymerized product and nonasaccharide from fucosylated glycosaminoglycans, exhibit strong anticoagulant and antithrombotic activities while avoiding adverse effects [32,36]. The reason is that intrinsic tenase inhibition has no effect on extrinsic coagulation pathway and preserves the hemostatic function [37]. Therefore, the intrinsic tenase has been recognized as a potential target for developing anticoagulant inhibitors. The application value of GMG-1.0 as a potent and safe intrinsic tenase inhibitor to prevent thrombus formation deserves further investigation.
3. Materials and Methods
3.1. Materials and reagents
Two commercial dried swim bladders were purchased from local sea food market in Nanning city and identified as Sphyraena jello and Gadus morhua by Professor Jing Wen in Lingnan Normal University. d-GlcA, d-GalA, d-glucose (Glc), d-Gal, d-GalNAc, d-GlcNAc, and Chondroitinase ABC from Proteus vulgaris (EC 4.2.2.4) were obtained from Sigma–Aldrich (St. Louis, MO, USA). d-IdoA was purchased from Shanghai ZZBIO Co., Ltd. (Shanghai, China). Alcian blue 8GX, d-mannose (Man), and l-fucose (Fuc) were obtained from Aladdin Chemical Reagent Co., Ltd. (Shanghai, China). Standard pullulans were obtained from Sepax Technologies, Inc. (Delaware, USA). Deuterium oxide (D2O) with 99.9 % atom D and D2O containing 0.05 wt% 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP) sodium salt were obtained from Sigma-Aldrich (China). CS and DS disaccharide standard mix (CD Mix), including ΔDi0S (ΔUA-1,3-GalNAc), ΔDi6S (ΔUA-1,3-GalNAc6S), ΔDi4S (ΔUA-1,3-GalNAc4S), ΔDi2S (ΔUA2S-1,3-GalNAc), ΔDi2,6diS (ΔUA2S-1,3-GalNAc6S), ΔDi4,6diS (ΔUA-1,3-GalNAc4S6S), ΔDi2,4diS (ΔUA2S-1,3-GalNAc4S), and ΔDi2,4,6triS (ΔUA2S-1,3-GalNAc4S6S) were obtained from Iduron (Manchester, England). Enoxaparin (Mw: 4500 Da, 0.4 mL × 4000 AXaIU) was purchased from Sanofi-Aventis (France). Tris-HCl (>99.5 %) was purchased from Amresco (USA). Coagulation control plasma, 0.05 M CaCl2 solution, and APTT, PT, and TT assay kits were obtained from TICO GmbH (Germany). Human FVIII was obtained from Shanghai RAAS Blood Products Co., Ltd. (China). Biophen FVIII: C kit was from Hyphen Biomed (France). All other chemicals and reagents used were of analytical grade.
3.2. Extraction and Isolation of GAGs from swim bladder
The dried swim bladders were ground to powder using a homogenizer. GAGs were extracted according to a procedure described by Vieira et al. with slight modifications [38]. Briefly, the swim bladder powder was suspended in distilled water (1 g/10 mL) and treated with 1% papain solution at 55 °C for 16 h. The mixture was digested by NaOH solution at the final concentration of 0.5 M at 60 °C for 2 h. After cooling to room temperature, the pH of the mixture was adjusted to 2–3 by addition of 6 M HCl solution. The supernatant was obtained by centrifugation, and the pH was adjusted to 7.0. Ethanol was then added to the final concentration of 75% (v/v), standing overnight at 4 °C. The precipitate was collected after centrifugation at 4816 ×g for 15 min and lyophilization.
The crude GAGs were dissolved in distilled water, applied to a column packed with Amberlite FPA98Cl anion-exchange resin, and eluted with increasing concentrations of NaCl solution (0, 0.5, 1.0, 1.5, 2.0 M). The main acidic fractions eluted by 1.0 M NaCl were collected, precipitated by ethanol, desalted by a dialysis bag with molecular weight cut-off of 3.5 kDa, and lyophilized to obtain white powders named SJG-1.0 and GMG-1.0.
3.3. Physicochemical analysis
Protein contents in the SJG-1.0 and GMG-1.0 samples were determined by the method described by Bradford [39] using bovine serum albumin as a standard. The uronic acid content was determined using the Blumenkrantz and Asboe-Hansen procedure [40] using GalA as a standard. The sulfate group content was measured by the turbidimetric method [41]. The sulfate/carboxyl ratio was determined by a conductimetric method as described in our previous study [42].
The purity and Mw of GAG were estimated by HPGPC as described in our previous study [25]. The samples were injected into the Shodex OHpak SB-804 HQ column (7 µm, 8×300 mm) and eluted with 0.1 M NaCl at a flow rate of 0.5 mL/min. For the Mw calculation, a standard curve was made using standard pullulans with Mw of 1.08, 5.9, 9.6, 21.1, 47.1, 107.0, and 200.0 kDa.
Cellulose acetate electrophoresis was performed as reported previously [43,44] with minor modifications. Briefly, the samples and standard GAGs, such as HP, CS, and DS, were prepared at a concentration of 5 mg/mL. The cellulose acetate strips were stated in 50% methanol overnight, and then soaked in electrophoretic buffer (0.1 M barium acetate buffer, pH 5.0) for 30 min. The samples and GAG standards were placed at the origin of the cellulose acetate strip, and run in the electrophoretic buffer for 1 h 55 min. After migration, the strip was stained by alcian blue for 15 min, and the excess stain was then removed by soaking in 2% acetate buffer for 10 min.
PMP pre-column derivatization combined with HPLC was used to analyze the monosaccharide composition [45]. Briefly, samples were hydrolyzed with 4 M TFA. The released monosaccharides were then derivatized by PMP and further analyzed by HPLC equipped with an Agilent ZORBAX Eclipse Plus C18 column (4.6 × 250 mm, 5 μm). The unknown peak in the HPLC profiles of derivatives was further analyzed by ESI-MS in a positive-ion model. The identification was performed on a ACQUITY UPLC BEH C18 column (2.1 × 100 mm; 1.7 μm) using an ACQUITY UPLC I-Class & Xevo G2-XS QTOF HRMS spectrometer (Waters, USA).
FT-IR spectra were determined by the methods as described previously [46]. The spectra were scanned from 4000 to 400 cm-1 with an iS50 infrared spectrometer (Thermo Fisher Scientific, USA).
3.4. Enzymatic treatment and disaccharide composition analysis
The disaccharide composition analysis was carried out as reported previously [47]. Briefly, the sample was incubated with chondroitin ABC lyase at 37 °C for 48 h. After heating in boiled water for 5 min, the mixture was centrifugated to obtain the supernatant and analyzed by the SAX-HPLC (Welch Ultimate XB-SAX, 4.6 × 250 mm). The mobile phase was a mixture of 2 mM Na2HPO4 (pH 3.0, solvent A) and 2 mM Na2HPO4 containing 1.2 M NaClO4 (pH 3.0, solvent B). The gradient was programmed as 97% A at the beginning, the mobile phase B linearly increased from 3% to 35% during 120 min. The flow rate was 0.6 mL/min, and the detection wavelength was 232 nm.
3.5. NMR spectroscopy
The dried samples (10–20 mg) were dissolved in 0.5 mL of D2O without the internal standard TSP and freeze-dried thrice to replace the exchangeable protons with deuterium. The samples were then redissolved in 0.5 mL of D2O containing TSP for NMR analysis. The NMR spectra were recorded on a Bruker AVANCE NEO 600 M spectrometer at 298 K. The 13C spectra were recorded with numbers of scans of 16384.
3.6. Assay of anticoagulant activity
The anticoagulant activity of the SJG-1.0 and GMG-1.0 samples was investigated by the APTT, TT, and PT assays as described in our previous study [46]. The APTT assay was carried out by mixing 5 μL of samples at various concentrations and 45 μL of standard human plasma and incubating at 37 °C for 2 min. Fifty microliters of APTT reagent was then added, and the mixture was kept at 37 °C for 3 min. The clotting time was immediately recorded after addition of 50 μL of 0.02 M CaCl2 solution. For PT assay, 5 μL of samples at various concentrations were mixed with 45 μL of standard human plasma at 37 °C and incubated for 2 min. The clotting time was obtained after adding the PT reagent (100 μL). The TT assay was carried out by mixing 10 μL of samples at various concentrations and 90 μL of standard human plasma and incubating at 37 °C for 2 min. The clotting time was recorded after addition of 50 μL of TT reagent. Tris-HCl buffer and LMWH were used as the blank and positive control, respectively.
The activity of intrinsic tenase inhibition was determined by a colorimetric method using a 96-well plate kinetic assay as previously described [32]. Briefly, equal volumes of sample solution, factor VIII, and factor IXa were mixed and incubated at 37 °C for 2 min. The R1 solution was then added to the mixture and incubated at 37 °C for 1 min. Finally, R3 sloution was added and mixed well, and the absorbance was recorded at 405 nm.
3.7. Statistical analysis
The data were analyzed by the IBM SPSS software (Chicago, USA) and expressed as the mean ± standard deviation (SD). One-way analysis of variance (One-Way ANOVA) and Duncan's new multiple range test were used for the statistical analysis. P < 0.05 was statistically significant.
4. Conclusions
In this study, two crude GAGs were extracted from the swim bladders of Sphyraena jello and Gadus morhua by enzymatic and alkaline hydrolysis, and the purified GAGs SJG-1.0 and GMG-1.0 were obtained after isolation and purification using the anion-exchange chromatography. The detailed structures of these purified GAGs were elucidated by the physicochemical analyses, such as chemical composition, monosaccharide and disaccharide composition analyses, and 1D/2D NMR spectroscopy. The polysaccharide sequences of SJG-1.0 and GMG-1.0 were confirmed to be the CS/DS hybrid chains mainly composed of the repeating disaccharide unit DS-A and a small quantity of DS-B and CS disaccharide units. SJG-1.0 had a higher proportion of CS-O disaccharide unit, while GMG-1.0 contained higher proportion of CS-E disaccharide unit. The disaccharide compositions and proportions of GAGs from swim bladders varied in a species-specific manner. Both SJG-1.0 and GMG-1.0 showed potent anticoagulant activity mainly by inhibiting the intrinsic coagulation pathway. Further studies on the anticoagulant mechanism indicated that GMG-1.0 with strong inhibition activity of intrinsic tenase had a potential to be developed as an intrinsic tenase inhibitor. The significant difference in the anticoagulant activity between the SJG-1.0 and GMG-1.0 may be attributed to their differences in their disaccharide compositions and proportions. More effort is deserved to investigate the detailed structure-activity relationships and the action mechanisms of anticoagulant activity of these CS/DS hybrid chains.
Supplementary Materials
The following supporting information can be downloaded at: , Figure S1: UV spectrum of SJG-1.0 and GMG-1.0; Figure S2: Electrophoretogram of SJG-1.0 (Track 2) and GMG-1.0 (Track 3); Figure S3: FT-IR spectra of SJG-1.0 and GMG-1.0; Figure S4. 1H-13C HSQC-TOCSY spectrum of SJG-1.0; Figure S5: 1H-1H COSY spectrum of GMG-1.0; Figure S6: 1H-1H TOCSY spectrum of GMG-1.0; Table S1: Assignment of 1H and 13C NMR signals of GMG-1.0.
Author Contributions
Conceptualization, methodology, Q.Y. and L.Z.; investigation, Q.Y., Y.Y., H.M., H.T, Z.L., and J.H.; formal analysis, Y.Y., H.M., H.T, Z.L., and J.H.; data curation, Q.Y. and L.Z.; writing—original draft preparation, Q.Y.; writing—review and editing, Q.Y. and L.Z.; supervision, Q.Y., L.Z., and X.Y.; project administration, Q.Y. and L.Z.; funding acquisition, Q.Y. and L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Guangxi Zhuang Autonomous Region (No. 2020GXNSFBA159022 and 2020GXNSFFA297005), the National Natural Science Foundation of China (No. 32160220 and 82373788), the Development Program of High-level Talent Team under the Qihuang Project of Guangxi University of Chinese Medicine (No. 2021004), the Guangxi University of Chinese Medicine “GuiPai Traditional Chinese Medicine Inheritance and Innovation Team” Project (No. 2022A007), and Guangxi First-class Discipline: Chinese Materia Medica (Scientific Research of Guangxi Education Department [2022] No. 1).
Data Availability Statement
The original data presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Acknowledgments
The authors thank Dr. Xiaohuo Shi from the Instrumentation and Service Center for Molecular Sciences, Westlake University for performing the NMR experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arima, K.; Fujita, H.; Toita, R.; Imazu-Okada, A.; Tsutsumishita-Nakai, N.; Takeda, N.; Nakao, Y.; Wang, H.; Kawano, M.; Matsushita, K.; et al. Amounts and compositional analysis of glycosaminoglycans in the tissue of fish. Carbohydr. Res. 2013, 366, 25–32. [Google Scholar] [CrossRef]
- Maccari, F.; Galeotti, F.; Volpi, N. Isolation and structural characterization of chondroitin sulfate from bony fishes. Carbohydr. Polym. 2015, 129, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel, J.; Novoa-Carballal, R.; Pérez-Martín, R.; Reis, R.L.; Vázquez, J.A. Glycosaminoglycans from marine sources as therapeutic agents. Biotechnol. Adv. 2017, 35, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Messmore, H.L.; Wehrmacher, W.H.; Coyne, E.; Fareed, J. Heparin to pentasaccharide and beyond: The end is not in sight. Semin. Thromb. Hemost. 2004, 30, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. Heparin/heparan sulphate-based drugs. Drug Discov. Today 2010, 15, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Uebelhart, D. Clinical review of chondroitin sulfate in osteoarthritis. Osteoarthr. Cartil. 2008, 16, S19–S21. [Google Scholar] [CrossRef] [PubMed]
- Price, R.D.; Berry, M.G.; Navsaria, H.A. Hyaluronic acid: the scientific and clinical evidence. J. Plast. Reconstr. Aesthetic Surg. 2007, 60, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Caputo, H.E.; Straub, J.E.; Grinstaff, M.W. Design, synthesis, and biomedical applications of synthetic sulphated polysaccharides. Chem. Soc. Rev. 2019, 48, 2338–2365. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, Q.; Lv, K.; Ma, H.; Gao, C.; Liu, Y.; Zhang, S.; Zhao, L. Low-molecular-weight fucosylated glycosaminoglycan and its oligosaccharides from sea cucumber as novel anticoagulants: A review. Carbohydr. Polym. 2021, 251, 117034. [Google Scholar] [CrossRef]
- Sadovy de Mitcheson, Y.; To, A.W.L.; Wong, N.W.; Kwan, H.Y.; Bud, W.S. Emerging from the murk: threats, challenges and opportunities for the global swim bladder trade. Rev. Fish Biol. Fish. 2019, 29, 809–835. [Google Scholar] [CrossRef]
- González-Félix, M.L.; Perez-Velazquez, M.; Castellanos-Rico, M.; Sachs, A.M.; Gray, L.D.; Gaines, S.D.; Goto, G.M. First report on the swim bladder index, proximate composition, and fatty acid analysis of swim bladder from cultured Totoaba macdonaldi fed compound aquafeeds. Aquac. Reports 2021, 21, 100901. [Google Scholar] [CrossRef]
- Clarke, S. Understanding pressures on fishery resources through trade statistics: A pilot study of four products in the Chinese dried seafood market. Fish Fish. 2004, 5, 53–74. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, W.Y.; Wu, M.F.; Wang, Y.M.; Zhu, W.Y.; Chi, C.F.; Wang, B. Eighteen novel bioactive peptides from monkfish (Lophius litulon) swim bladders: Production, identification, antioxidant activity, and stability. Mar. Drugs 2023, 21, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kaewdang, O.; Benjakul, S.; Kaewmanee, T.; Kishimura, H. Characteristics of collagens from the swim bladders of yellowfin tuna (Thunnus albacares). Food Chem. 2014, 155, 264–270. [Google Scholar] [CrossRef]
- Cruz-López, H.; Rodríguez-Morales, S.; Enríquez-Paredes, L.M.; Villarreal-Gómez, L.J.; True, C.; Olivera-Castillo, L.; Fernández-Velasco, D.A.; López, L.M. Swim bladder of farmed Totoaba macdonaldi: A source of value-added collagen. Mar. Drugs 2023, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qian, Y.; Li, G.J.; Tan, J. Preventive effects of the polysaccharide of Larimichthys crocea swim bladder on carbon tetrachloride (CCl4) -induced hepatic damage. Chin. J. Nat. Med. 2015, 13, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.H.; Zhao, X.; Luo, H.L.; Zhu, K. Therapeutic effect of polysaccharide of large yellow croaker swim bladder on lupus nephritis of mice. Nutrients 2014, 6, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Suo, H.Y.; Song, J.L.; Zhou, Y.L.; Liu, Z.H.; Yi, R.K.; Zhu, K.; Xie, J.; Zhao, X. Induction of apoptosis in HCT-116 colon cancer cells by polysaccharide of Larimichthys crocea swim bladder. Oncol. Lett. 2015, 9, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.X.; Wang, P.P.; Zhang, F.M.; Yu, Y. .; Zhang, X.; Lin, L.; Linhardt, R.J. Glycosaminoglycans from fish swim bladder: isolation, structural characterization and bioactive potential. Glycoconj. J. 2018, 35, 87–94. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Zhong, S.Y.; Su, W.M.; Du, Z.M.; Chen, J.P.; Hong, P.Z.; Zhang, C.H. Isolation, purification and structural identification of heparinoids from fish swim bladder. Food Sci. 2019, 40, 84–91. [Google Scholar]
- Chen, J.; Zhou, S. y.; Wang, Z.; Liu, S.C.; Li, R.; Jia, J.; Chen, J.P.; Liu, X.F.; Song, B.B.; Zhong, S.Y. Anticoagulant and anti-inflammatory effects of a degraded sulfate glycosaminoglycan from swimming bladder. Food Res. Int. 2022, 157, 111444. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.Y.; Wang, Z.; Huang, H.P.; Chen, J.; Liu, X.F.; Jia, X.J.; Song, B.B.; Cheong, K.L.; Gao, Y.; Zhong, S.Y. Intervention effects of sulfate glycosaminoglycan from swim bladder against arsenic-induced damage in IEC-6 cells. Int. J. Biol. Macromol. 2023, 252, 126460. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, X.; Zhang, X.; Liu, L.L.; Cong, D.P.; Zhao, X.; Yu, G.L. Acidolysis-based component mapping of glycosaminoglycans by reversed-phase high-performance liquid chromatography with off-line electrospray ionization-tandem mass spectrometry: Evidence and tags to distinguish different glycosaminoglycans. Anal. Biochem. 2014, 465, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.X.; Li, H.; Wang, Q.; Sun, S.J.; Fang, Z.Y.; Tang, H.; Shi, X.H.; Wen, J.; Huang, L.H.; Bai, M.; et al. Deaminative-cleaved S. monotuberculatus fucosylated glycosaminoglycan: Structural elucidation and anticoagulant activity. Carbohydr. Polym. 2022, 298, 120072. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.L.; Yuan, Q.X.; Li, H.; Li, T.T.; Ma, H.Q.; Gao, C.H.; Zhang, S.Y.; Liu, Y.H.; Zhao, L.Y. Chlorella pyrenoidosa polysaccharides as a prebiotic to modulate gut microbiota: Physicochemical properties and fermentation characteristics in vitro. Foods 2022, 11, 725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Mi, S.; Li, J.; Gao, J.; Wang, X.H.; Sang, Y.X. Purification, characterisation and antioxidant activities of chondroitin sulphate extracted from Raja porosa cartilage. Carbohydr. Polym. 2020, 241, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.R.; Tsai, M.F.; Shieh, C.J.; Arakawa, O.; Dong, C.D.; Huang, C.Y.; Kuo, C.H. Ultrasonic-assisted extraction and structural characterization of chondroitin sulfate derived from jumbo squid cartilage. Foods 2021, 10, 2363. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, X.; Ma, M.; Long, T.; Xiao, C.; Zhang, J.; Liu, J.; Zhao, L. Immunoenhancing glucuronoxylomannan from Tremella aurantialba Bandoni et Zang and its low-molecular-weight fractions by radical depolymerization: Properties, structures and effects on macrophages. Carbohydr. Polym. 2020, 238, 116184. [Google Scholar] [CrossRef]
- Panagos, C.G.; Thomson, D.; Moss, C.; Bavington, C.D.; Ólafsson, H.G.; Uhrín, D. Characterisation of hyaluronic acid and chondroitin/dermatan sulfate from the lumpsucker fish, C. lumpus. Carbohydr. Polym. 2014, 106, 25–33. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; He, Z.; Gao, N.; Shang, F.; Xu, J.; Li, Z.; Yang, Z.; Wu, M.; Zhao, J. Structural analysis and biological activity of a highly regular glycosaminoglycan from Achatina fulica. Carbohydr. Polym. 2018, 181, 433–441. [Google Scholar] [CrossRef]
- Volpi, N.; Maccari, F. Structural characterization and antithrombin activity of dermatan sulfate purified from marine clam Scapharca inaequivalvis. Glycobiology 2009, 19, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wu, M.; Xiao, C.; Yang, L.; Zhou, L.; Gao, N.; Li, Z.; Chen, J.; Chen, J.; Liu, J.; et al. Discovery of an intrinsic tenase complex inhibitor: Pure nonasaccharide from fucosylated glycosaminoglycan. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, R.; Namburi, R.B.; Ortega-Martinez, O.; Shi, X.; Zaia, J.; Dupont, S.T.; Thorndyke, M.C.; Lindahl, U.; Spillmann, D. Brittlestars contain highly sulfated chondroitin sulfates/dermatan sulfates that promote fibroblast growth factor 2-induced cell signaling. Glycobiology 2014, 24, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Nandini, C.D.; Itoh, N.; Sugahara, K. Novel 70-kDa chondroitin sulfate/dermatan sulfate hybrid chains with a unique heterogenous sulfation pattern from shark skin, which exhibit neuritogenic activity and binding activities for growth factors and neurotrophic factors. J. Biol. Chem. 2005, 280, 4058–4069. [Google Scholar] [CrossRef] [PubMed]
- Bougatef, H.; Ghlissi, Z.; Kallel, R.; Amor, I. Ben; Boudawara, T.; Gargouri, J.; Sahnoun, Z.; Volpi, N.; Sila, A.; Bougatef, A. Chondroitin/dermatan sulfate purified from corb (Sciaena umbra) skin and bone: In vivo assessment of anticoagulant activity. Int. J. Biol. Macromol. 2020, 164, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.D.; Lock, J.B.; Van’t Veer, C.; Gaffney, D.P.; Mann, K.G. Blood clotting in minimally altered whole blood. Blood 1996, 88, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhao, L.; Gao, N.; Yin, R.; Li, S.; Sun, H.; Zhou, L.; Zhao, G.; Purcell, S.W.; Zhao, J. From multi-target anticoagulants to DOACs, and intrinsic coagulation factor inhibitors. Blood Rev. 2020, 39, 100615. [Google Scholar] [CrossRef]
- Vieira, R.P.; Mulloy, B.; Mouriio, P.A.S. Structure of a fucose-branched chondroitin sulfate from sea cucumber. J. Biol. Chem. 1991, 266, 13530–13536. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Blumenkrantz, N.; Asboe-hansen, G. New method for quantitative determination of uronic acids. Anal. Biochem. 1973, 54, 484–489. [Google Scholar] [CrossRef]
- Dodgson, K.S.; Price, R.G. A note on the determination of the ester sulphate content of sulphated polysaccharides. Biochem. J. 1962, 84, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lai, S.; Huang, R.; Wu, M.; Gao, N.; Xu, L.; Qin, H.; Peng, W.; Zhao, J. Structure and anticoagulant activity of fucosylated glycosaminoglycan degraded by deaminative cleavage. Carbohydr. Polym. 2013, 98, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.R.; Lyon, M.; Gallagher, J.T.; Johnson, E.A.; Pepys, M.B. Isolation and characterization of the integral glycosaminoglycan constituents of human amyloid A and monoclonal light-chain amyloid fibrils. Biochem. J. 1991, 275, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bougatef, H.; Ghlissi, Z.; Kallel, R.; Amor, I.B.; Boudawara, T.; Gargouri, J.; Sahnoun, Z.; Volpi, N.; Sila, A.; Bougatef, A. Chondroitin/dermatan sulfate purified from corb (Sciaena umbra) skin and bone: In vivo assessment of anticoagulant activity. Int. J. Biol. Macromol. 2020, 164, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.X.; Xie, Y.F.; Wang, W.; Yan, Y.H.; Ye, H.; Jabbar, S.; Zeng, X.X. Extraction optimization, characterization and antioxidant activity in vitro of polysaccharides from mulberry (Morus alba L.) leaves. Carbohydr. Polym. 2015, 128, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.Q.; Yuan, Q.X.; Tang, H.; Tan, H.J.; Li, T.T.; Wei, S.Y.; Huang, J.W.; Yao, Y.; Hu, Y.P.; Zhong, S.P.; et al. Structural elucidation of a glucan from Trichaster palmiferus by its degraded products and preparation of its sulfated derivative as an anticoagulant. Mar. Drugs 2023, 21, 148. [Google Scholar] [CrossRef]
- da Silva, H.A.M.; de Queiroz, I.N.L.; Francisco, J.S.; Pomin, V.H.; Pavão, M.S.G.; de Brito-Gitirana, L. Chondroitin sulfate isolated from the secretion of the venom-producing parotoid gland of Brazilian bufonid. Int. J. Biol. Macromol. 2019, 124, 548–556. [Google Scholar] [CrossRef]
Figure 1.
HPGPC profiles of SJG-1.0 and GMG-1,0 (A), chromatograms of PMP derivatives of mixed monosaccharide standards, SJG-1.0 and GMG-1,0 (B), positive-ion ESI-TOF-MS spectrum (C) of PMP-labeled acidolysis-resistant disaccharide (labeled x in B), and HPLC profiles of disaccharide composition analysis of SJG-1.0 and GMG-1,0 (D). The disaccharide standards (D) include 1. ΔDi0S (ΔUA-1,3-GalNAc), 2. ΔDi6S (ΔUA-1,3-GalNAc6S), 3. ΔDi4S (ΔUA-1,3-GalNAc4S), 4. ΔDi2S (ΔUA2S-1,3-GalNAc), 5. ΔDi2,6diS (ΔUA2S-1,3-GalNAc6S), 6. ΔDi4,6diS (ΔUA-1,3-GalNAc4S6S), 7. ΔDi2,4diS (ΔUA2S-1,3-GalNAc4S), and 8. ΔDi2,4,6triS (ΔUA2S-1,3-GalNAc4S6S).
Figure 1.
HPGPC profiles of SJG-1.0 and GMG-1,0 (A), chromatograms of PMP derivatives of mixed monosaccharide standards, SJG-1.0 and GMG-1,0 (B), positive-ion ESI-TOF-MS spectrum (C) of PMP-labeled acidolysis-resistant disaccharide (labeled x in B), and HPLC profiles of disaccharide composition analysis of SJG-1.0 and GMG-1,0 (D). The disaccharide standards (D) include 1. ΔDi0S (ΔUA-1,3-GalNAc), 2. ΔDi6S (ΔUA-1,3-GalNAc6S), 3. ΔDi4S (ΔUA-1,3-GalNAc4S), 4. ΔDi2S (ΔUA2S-1,3-GalNAc), 5. ΔDi2,6diS (ΔUA2S-1,3-GalNAc6S), 6. ΔDi4,6diS (ΔUA-1,3-GalNAc4S6S), 7. ΔDi2,4diS (ΔUA2S-1,3-GalNAc4S), and 8. ΔDi2,4,6triS (ΔUA2S-1,3-GalNAc4S6S).
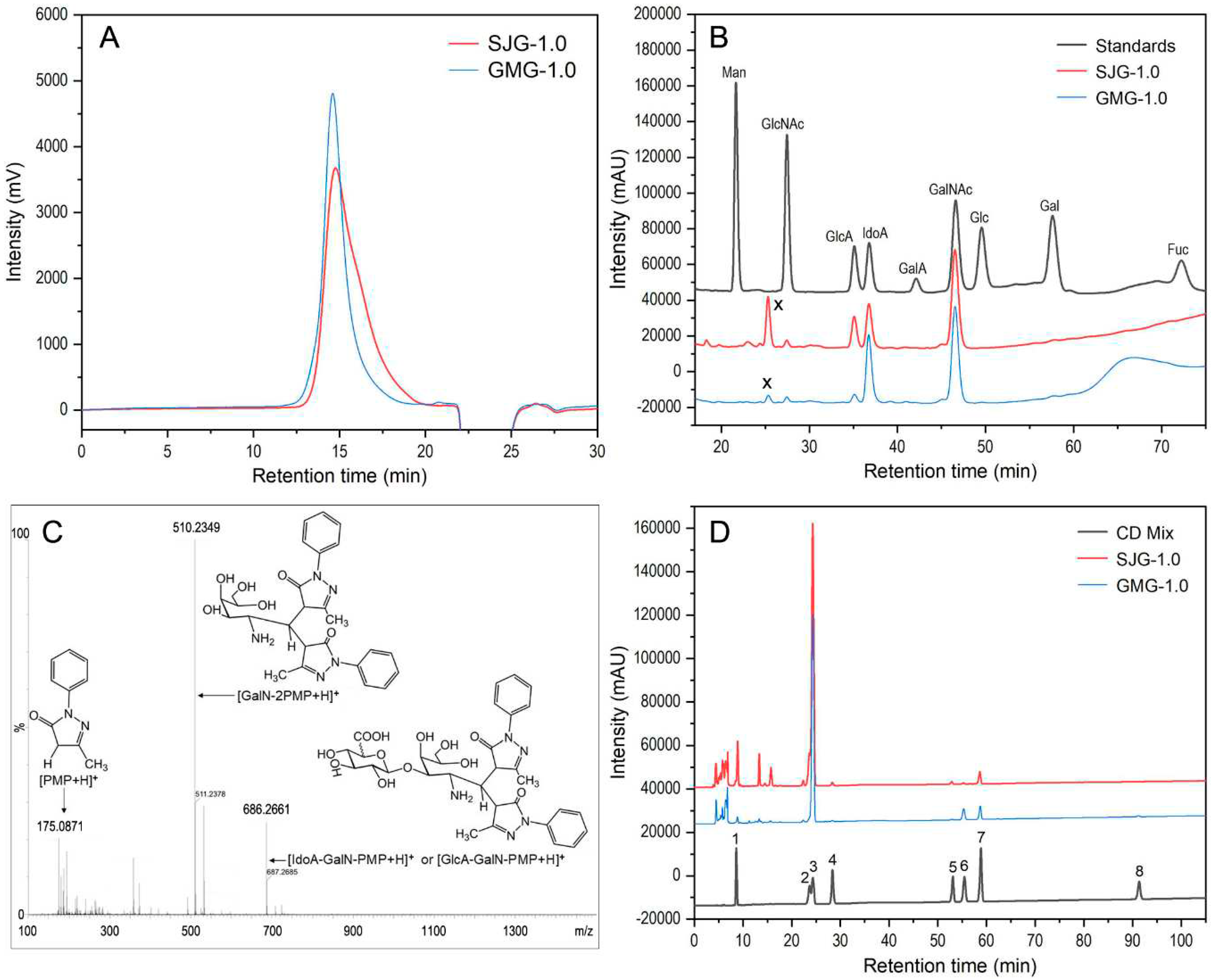
Figure 2.
1H NMR spectra of SJG-1.0 (A) and GMG-1.0 (B).

Figure 3.
13C and DEPT-135 NMR spectra of SJG-1.0 (A) and GMG-1.0 (B).
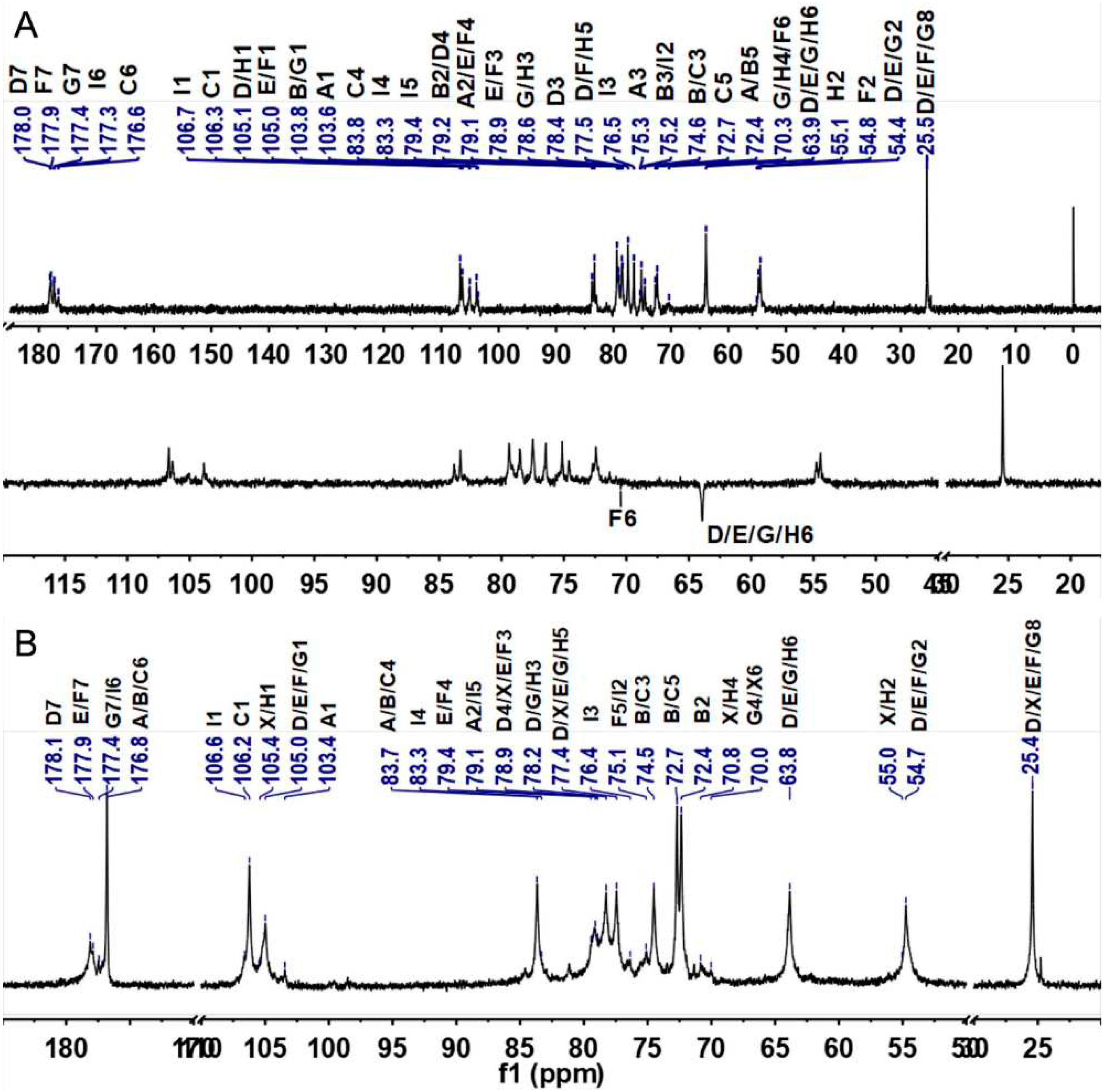
Figure 4.
1H–1H ROESY, 1H–13C HMBC, and HSQC NMR spectra of SJG-1.0 (A), and 1H–13C HSQC NMR spectrum of GMG-1,0 (B).
Figure 4.
1H–1H ROESY, 1H–13C HMBC, and HSQC NMR spectra of SJG-1.0 (A), and 1H–13C HSQC NMR spectrum of GMG-1,0 (B).
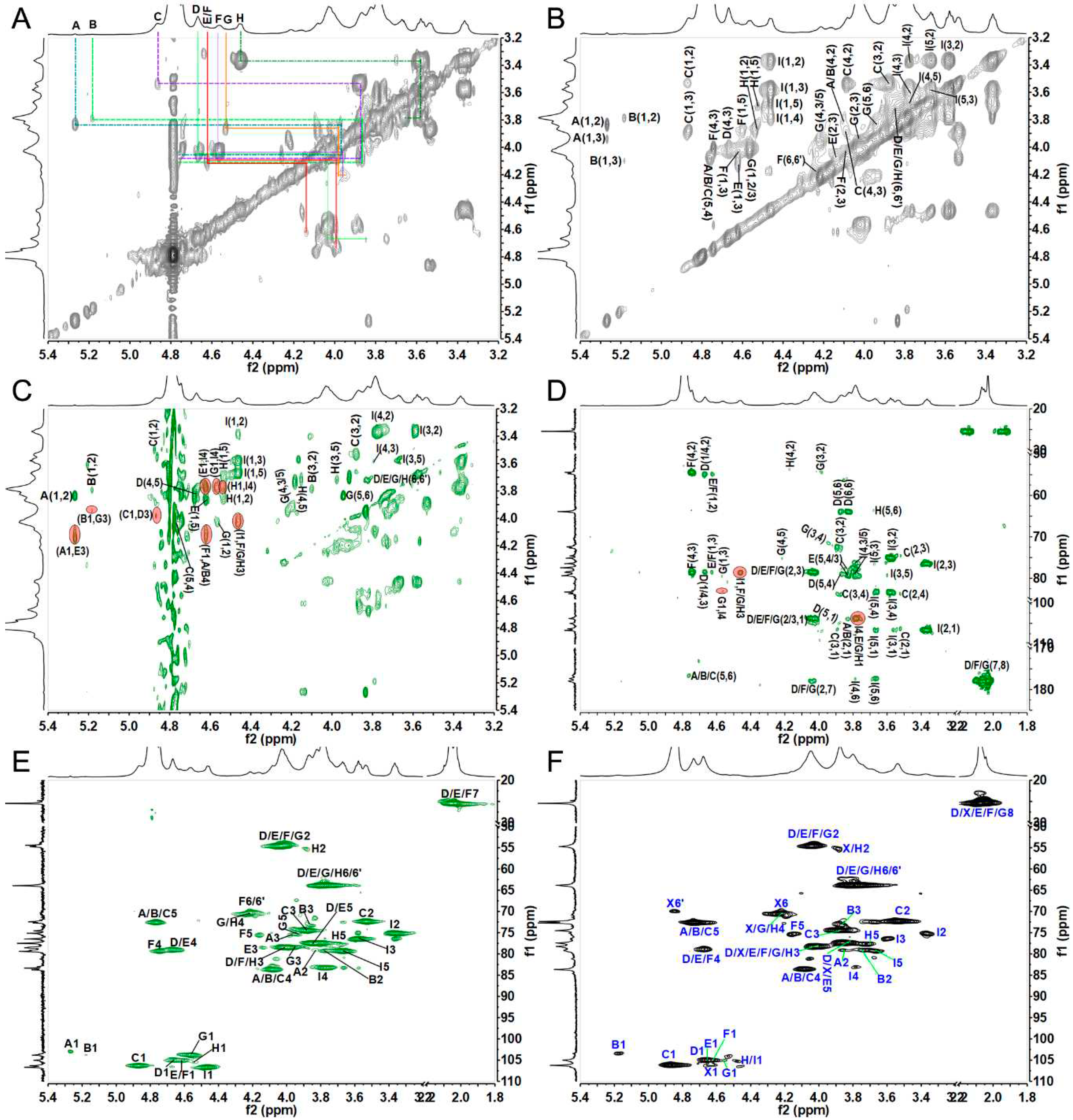

Table 1.
Assignment of 1H and 13C signals of SJG-1.0.
| Residues | H/C | Chemical shifts (δ,ppm) a |
Connection patterns Cross signals (ppm) |
|||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| A | H | 5.27 | 3.84 b | 3.95 | 4.10 c | 4.76 | A-E | |||
| α-D-IdoA2S | C | 103.6 | 79.1 | 75.5 | 83.0 | 72.4 | 176.8 | (5.27,4.14) | ||
| B | H | 5.18 | 3.79 | 4.10 | 4.13 | 4.78 | B-G | |||
| α-D-IdoA2S | C | 103.9 | 79.2 | 75.1 | 83.5 | 72.4 | 176.8 | (5.18,3.96) | ||
| C | H | 4.86 | 3.53 | 3.88 | 4.08 | 4.76 | C-D | |||
| α-D-IdoA | C | 106.3 | 72.4 | 74.6 | 83.8 | 72.7 | 176.6 | (4.86,4.03) | ||
| D | H | 4.67 | 4.05 | 4.03 | 4.66 | 3.87 | 3.73/3.84 | 2.03 | D-C | |
| β-D-GalNAc4S | C | 105.1 | 54.3 | 78.4 | 79.2 | 77.5 | 63.9 | 178.0 | 25.5 | (4.67,4.08) |
| E | H | 4.62 | 4.08 | 4.14 | 4.63 | 3.83 | 3.73/3.84 | 2.05 | E-I | |
| β-D-GalNAc4S | C | 105.0 | 54.7 | 78.6 | 79.1 | 77.7 | 63.9 | 177.9 | 25.5 | (4.62,3.78) |
| F | H | 4.61 | 4.10 | 4.04 | 4.74 | 4.15 | 4.25/4.17 | 2.04 | F-A/B | |
| β-D-GalNAc4S6S | C | 105.0 | 54.3 | 78.9 | 79.4 | 75.7 | 70.0 | 177.9 | 25.5 | (4.61,4.10/4.13) |
| G | H | 4.56 | 4.02 | 3.96 | 4.21 | 3.93 | 3.72/3.84 | 2.07 | G-I | |
| β-D-GalNAc | C | 103.8 | 54.5 | 78.6 | 70.3 | 75.7 | 63.9 | 177.4 | 25.5 | (4.56,3.78) |
| H | H | 4.53 | 3.86 | 4.00 | 4.17 | 3.70 | 3.74/3.82 | H-I | ||
| β-D-GalN2S | C | 105.6 | 55.1 | 78.6 | 70.3 | 77.3 | 63.9 | (4.53,3.78) | ||
| I | H | 4.47 | 3.37 | 3.58 | 3.78 | 3.67 | I-F/G/H | |||
| β-D-GlcA | C | 106.7 | 75.2 | 76.5 | 83.3 | 79.4 | 177.3 | (4.47,4.04/3.96/4.00) | ||
a The 600 MHz NMR spectra were recorded at 298 K. All chemical shifts are relative to TSP at 0 ppm; b, c Values with underline and in boldface indicate sulfated and glycosylated positions, respectively.
Table 2.
Effects of SJG-1.0 and GMG-1.0 on APTT, PT, TT, and intrinsic tenase (n = 2).
| Sample | APTT 1, μM | TT 1, μM | PT 1, μM | anti-tenase 2, μM |
| LMWH | 0.571 ± 0.003 | 0.658 ± 0.043 | / 3 | 0.010 ± 0.001 |
| SJG-1.0 | 0.586 ± 0.001 | / | / | / |
| GMG-1.0 | 0.226 ± 0.016 | 12.035 ± 0.404 | / | 0.058 ± 0.015 |
1 The activity of samples to prolong APTT, PT, or TT is expressed by each drug concentration (μM) required to double the APTT, PT, or TT; 2 EC50 value, the concentration of each sample required to inhibit 50% of tenase activity; 3 not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



