
Submitted:
03 November 2023
Posted:
03 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
In addition to nucleic acids, various viral proteins are involved in the assembly of virion. If it is possible to create a biologically active agent that leads to the degradation of one of the key proteins for virus assembly and/or destroys the viral factory, then this agent will effectively cope with virus replication. One of these key proteins for the SARS-CoV-2 virus is the nucleocapsid protein (N-protein). As such a bioactive agent, we offer a modular nanotransporter (MNT) developed by us, which, in addition to an antibody mimetic to the N-protein, contains an amino acid sequence for the attraction of the Keap1 E3 ubiquitin ligase. This should lead to the subsequent degradation of the N-protein. We have shown that functional properties of modules within the MNT permit its internalization into target cells, endosome escape into the cytosol, and binding to the N-protein. Using flow cytometry and Western blot, it was demonstrated that significant degradation of N-protein is observed when A549 and A431 cells transformed with N-protein are incubated with the developed MNTs. The proposed MNTs open up a new approach for the treatment of viral diseases.
Keywords:
modular nanotransporters
; Keap1E3 ligase
; N-protein degradation
; SARS-CoV-2 virus
; flow cytometry
; Western blot
; intracellular concentrations
; thermophoresis
; cathepsin B
1. Introduction
The importance of tools selectively modulating target protein function has been reinforced by the SARS-CoV-2 pandemic, temporally moving other notorious indications to second place. Indeed, many proteins could be considered crucial elements of pathological processes, and abolishing their activity might have therapeutic effects. Despite several available groups of methods (chemical inhibitors, DNA-based, and RNA-based) known to contribute to the suppression of specific cellular targets, our capabilities to decrease target protein activity seem not to be excessive. Only a small fraction of the human proteome is amenable to inhibition via small molecules [1], mainly proteins with appropriate binding pockets such as ion channels or enzymes. Although gene knockouts and RNA interference are methods of choice for some applications, they exert an indirect effect on a protein level, which poses an impediment to targeting stable, long-lived proteins and raises questions about lack of temporal control and possible activation of compensatory mechanisms [2].
Recently emerged direct protein degradation technologies, recruiting cellular protein control mechanisms such as ubiquitin-proteasome and autophagy systems [3,4], are in many respects better suited for protein targeting. Compared to the aforementioned approaches, protein degradation strategy offers a number of strengths: direct elimination of proteins, all target protein-associated functions attenuation by one shot, substoichiometric activity due to the catalytic mechanism of action, and long-lasting inhibition after washout determined by the target protein turnover rate. The most developed technology in this domain is PROTACs (PROteolysis-TArgeting Chimeras), with several molecules having already reached clinical trials [5,6]. Nevertheless, the Achilles’ heel of the PROTACs is their heavy dependence on discovered small-molecule ligands on the shelf. This constraint hinders the progression toward a universal target protein degradation tool, limiting the druggable space of proteins that could be reached via such regulation. Antibodies and antibody-like proteins could be raised against different protein targets, including those without relevant small-molecule ligand. Switching from small-molecule ligands to miniproteins, such as antibody-like proteins, may alleviate the PROTACs drawback. The strategy of antibody-like protein employment as a binding domain for target protein degradation was explored in several articles [7,8,9,10,11], but the intracellular delivery method for possible therapeutic applications is still an issue, as it is for antibody delivery itself [12,13]. Earlier, we developed modular nanotransporter (MNT) technology, utilizing receptor-dependent endocytosis, active endosomal escape, and intracellular transport signals for targeted cargo delivery into the target cell compartment [14]. The applicability of this approach for intracellular delivery of antibody-like proteins was demonstrated using SARS-CoV-2 N-protein [15] as targets. In this study, we modified the SARS-CoV-2 N-protein targeting MNT to hijack cellular Keap1 E3-ligase for N-protein degradation. The functionality of this MNT, its cellular effects on cells expressing SARS-CoV-2 N-protein, and its ability to suppress the target were investigated.
2. Materials and Methods
2.1. Cell Lines
The adenocarcinoma human alveolar basal epithelial cells (A549) and human epidermoid carcinoma cells (A431) were maintained according to the specifications of the American Type Culture Collection (ATCC, Manassas, VA, USA).
2.2. SARS-CoV2 N-Protein Stable Cell Lines Generation
The plasmids from Addgene were used for lentiviral transduction. pMD2.G and psPAX2 were gifts from Didier Trono (Addgene plasmids # 12259 and # 12260, respectively). pHAGE N-mRuby3 (from SARS-CoV-2) IRES puro was a gift from Raphael Gaudin (Addgene plasmid # 170466). Recombinant lentiviral particles were produced by cotransfection of HEK293T cells in a T25 flask with 2.3 μg pMD2.G, 4.3 μg psPAX2, and 10.2 μg pHAGE N-mRuby3 (from SARS-CoV-2) IRES puro using Calcium Phosphate Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA). The virus suspension was collected at 72 h after transfection. The lentivirus was concentrated using the Lenti-X concentrator (Takara Bio, Otsu, Japan) according to the manufacturer’s protocol. Human cancer cell lines A549 and A431 were seeded in 24-well plates (1 × 104 cells/well) one day before viral infection. For lentivirus transduction, lentiviral particles were suspended in culture medium containing 10 μg/mL polybrene (Sigma-Aldrich, Burlington, MA, USA). Stable cell lines were selected with puromycin (1 μg/mL, Acros Organics, Geel, Belgium).
2.3. A549 Transient Transfection with SARS-CoV2 N-Protein
A549 cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum with 50 µg/mL gentamicin and maintained in a tissue culture incubator at 37°C with 5% CO2. After passaging in a T25 flask and reaching about 70–80% confluence, cells were seeded into POCmini chambers (Cell Cultivation System: Open Cultivation, PeCon, Erbach, Germany), 50,000 cells per chamber in 1 ml of DMEM medium with 10% bovine fetal serum, and cultivated at 37 °C under 5% CO2. The next day cells were transfected with pHAGE N-mRuby3 (from SARS-CoV-2) IRES puro (Addgene plasmid # 170466) using Lipofectamine 3000, according to the manufacturer’s instructions.
2.4. Recombinant Proteins Used in the Work
The main object of the study is a modular nanotransporter of the following composition (Figure 1): affibody(EGFR)-HisTag-DTox-HMP-FKFL-Keap1_E3BP-NC2, where affibody(EGFR) is an antibody mimetic that binds to the epidermal growth factor receptor (EGFR), HisTag is a peptide of six histidines, necessary for the isolation and purification of MNTs, DTox – endosomolytic module based on the translocation domain of diphtheria toxin, HMP – hemoglobin-like protein of E. coli, acting as a carrier module, FKFL – one of the most optimal cleavage sites by the endosomal protease cathepsin B [16], Keap1_E3BP is the amino acid sequence DPETGEYL, selected on the basis of the peptide PROTAC molecule proposed in the article [17] and binding the ubiquitin ligase Keap1 with nanomolar affinity, and NC2 – monobody NC2 to N-protein. In the following, this monobody is designated as MNT1. In addition, MNT0 will be designated as a transporter that lacks Keap1_E3BP. The control transporter, MNTcon, is MNT0 lacking also the FKFL site and NC2 monobody.
2.5. Protein Isolation and Purification
E. coli cells of the Ros(DE3)pLysS strain were transformed with the plasmid containing the resulting MNT1 construct. An overnight culture of these cells was seeded in 1 liter of Terrific modified medium (Dia-M, Moscow, Russian Federation), with the addition of kanamycin (30 μg/mL), chloramphenicol (40 μg/mL), 2.9 g/l glucose, 7.6 g/l lactose, and 11.2 g/l glycerol. These cells were grown at 37°C with constant stirring until an optical density of 0.8 units was reached at a wavelength of 600 nm. Autoinduction of protein expression was carried out by incubation for 48 hours at a temperature of +18°C and constant stirring. After this, the cells were separated from the medium by centrifugation for 30 min at 9000 rpm (JA-10 rotor, Beckman, Brea, CA, USA). Cell lysis was carried out for 1.5 hours at 4°C in a buffer of the following composition: 50 mM Na2HPO4, 500 mM NaCl, 5 mg/mL lysozyme, 1 cOmplete™ tablet (EDTA-free Protease Inhibitor Cocktail (Roche)), 2.5 units /mL benzonase (purity >90%, Novagen), 0.5% Triton X-100, and 10% glycerol, pH 8. The lysate was then centrifuged for 30 min at 15,000 rpm (JA-20 rotor, Beckman, Brea, CA, USA), the supernatant was collected and imidazole was added to it to a final concentration of 20 mM and NaCl to 500 mM, after which it was applied to a metal affinity chromatography column Ni HisTrap FF, 5 mL (Cytiva). The column was washed with a buffer of the following composition: 50 mM Na2HPO4, 500 mM NaCl, 25 mM imidazole, 0.5% Triton X-100, and 5% glycerol, pH 7.8. The MNT1 was eluted using a BioLogic LP low-pressure chromatography device (Bio-Rad, Hercules, CA, USA) in a buffer consisting of 50 mM Na2HPO4, 500 mM NaCl, and 700 mM imidazole at pH 8 (elution buffer). Fractions containing protein were dialyzed three times against a buffer of 10 mM Na2HPO4, 150 mM NaCl, pH 8.0 (PBS). The purity of the isolated MNT1 as well as the purity of other proteins were assessed in the Image Lab software (BioRad, Hercules, CA, USA) based on the results of denaturing polyacrylamide gel electrophoresis (PAGE), according to Laemmli. The purity of the resulting MNT1 was 86.6%.
E. coli strain Ros(DE3)pLysS cells were transformed with the plasmid containing the resulting MNT0 construct. Cells were seeded in LB Broth Miller nutrient medium (Luria-Bertani) and grown at a temperature of 37°C with constant stirring until an optical density of 0.8 units was reached at a wavelength of 600 nm. Induction of protein expression was carried out by adding isopropyl-β-D-1-thiogalactopyranoside (IPTG) to the nutrient medium to a concentration of 500 μM and incubating for 18 hours at a temperature of +17°C and constant stirring. After this, the cells were separated from the nutrient medium by centrifugation. Lysis, purification and elution were carried out in the same way as for MNT1. The purity of the resulting MNT0 was 96%.
E. coli strain BL21(DE3) cells were transformed with the plasmid containing the resulting MNTcon construct. Production, lysis and purification were carried out the same way as for MNT1. The MNTcon was sequentially eluted from the column with buffers containing 500 mM NaCl, 50 mM Na2HPO4, and 70 mM imidazole, pH 8; 500 mM NaCl, 50 mM Na2HPO4, and 100 mM imidazole, pH 8; 500 mM NaCl, 50 mM Na2HPO4, and 700 mM imidazole, pH 8. Fractions containing pure MNTcon were dialyzed 3 times against PBS buffer (10 mM Na2HPO4, 150 mM NaCl, pH 8.0). The purity of the resulting MNTcon was 93%.
E. coli cells of strain BL21(DE3), transformed with a plasmid encoding the N-protein of SARS-CoV-2, were seeded in LB medium and grown at a temperature of 37ºC and constant stirring until optical density 0.8 units at a wavelength of 600 nm. Induction of N-protein expression was carried out by adding IPTG to the nutrient medium to a concentration of 500 μM and incubating for 3 hours at a temperature of 37°C and constant stirring. After this, the cells were separated from the nutrient medium by centrifugation. Cell lysis was carried out for 3 hours at a temperature of +4°C in a buffer of the following composition: 500 mM NaCl, 50 mM Na2HPO4, 10 mg/ml lysozyme, 0.5% Triton X-100, 1 mM PMSF, and 50 KIU/ml aprotinin. The inclusion body fraction was separated from the lysate by centrifugation. Inclusion bodies were washed sequentially twice with a TE50/20 solution (50 mM Tris, 20 mM EDTA, and 1 mM PMSF, pH 8.0) with the addition of NaCl and Triton X-100 to a final concentration of 0.5 M and 2%, respectively, then once with a Tris-NaCl solution (20 mM Tris, 1 M NaCl, and 1 mM PMSF, pH 8.0) with the addition of Triton X-100 to a final concentration of 2%, after which the inclusion bodies were washed with a Tris-NaCl solution (20 mM Tris, 1 M NaCl, and 1 mM PMSF, pH 8.0). The inclusion body pellet was resuspended for two hours at room temperature in a buffer containing 500 mM NaCl, 50 mM Na2HPO4, and 8 M urea, pH 7, and then centrifuged. The supernatant was applied to an affinity chromatographic column Protino® Ni-TED Resin, after which the column was washed with buffers of the following composition: 500 mM NaCl, 50 mM Na2HPO4, and 1 M urea, pH 7, as well as 500 mM NaCl, 50 mM Na2HPO4, 1 M urea, and 0.06% SDS, pH 7. The N-protein was eluted from the column with a buffer containing 500 mM NaCl, 50 mM Na2HPO4, 1 M urea, and 700 mM imidazole, pH 7. The protein was dialyzed 3 times against HBS buffer (10 mM HEPES, 150 mM NaCl pH 8.0). The purity of the resulting N-protein was 91%.
After purification, all protein solutions were centrifuged and sterilized by filtration. The concentrations of the purified MNTs were measured by the Bradford method.
2.6. Flow Cytometry
Flow cytometry was used to study the ability of MNT1 to bind to EGFR receptors on the surface of A431 cells. The MNT1 was labeled with the AF488 fluorescent dye. To do this, an 8-fold molar excess of the activated AF488-N-hydroxysuccinimide ester (Lumiprobe, Moscow, Russia) was added to the MNT1 in 65 mM carbonate buffer (pH 8.5), and the mixture was incubated for 1h at room temperature with constant stirring. The MNT1 with the attached AF488 was purified from the unreacted dye using a PD10 gel filtration column. As a result, an average of two AF488 molecules were attached to one MNT1 molecule. A431 cells seeded in a 24-well plate were incubated with 500 nM MNT1 for a given time, then washed twice with Versene solution, then detached with an 0.25% trypsin solution in Versene, and finally resuspened in Hanks solution. The amount of MNT1-AF488 bound to cells was determined using a CytoFLEX S flow cytometer (Beckman Coulter, Inc., Brea, CA, USA) in the fluorescence channel of 500-550 nm. Fluorescence was excited by a laser with a wavelength of 488 nm. The average fluorescence value per cell was determined from 7–13 replicates at different incubation times with MNT1-AF488. Large cell aggregates were excluded from consideration.
To study N-protein degradation, A549 and A431cell lines, stably transfected with N-protein fused to the fluorescent protein mRuby3, were used. A431 cells or A549 cells seeded in a 24-well or a 48-well plate were incubated with MNT1 or MNT0 for a given time, then washed twice with Versene solution, then removed with a 0.25% trypsin solution in Versene, and then transferred to Hanks solution. The average fluorescence value per cell was determined from 5-19 replicates at different incubation times with 500 nM MNT1 or 500 nM MNT0 using a CytoFLEX S flow cytometer in the fluorescence channel of 564–606 nm. Fluorescence was excited by a laser with a wavelength of 561 nm. The average fluorescence value per cell was determined from 7–13 replicates at different incubation times with MNT1-AF488. Large cell aggregates and cellular debris were excluded from consideration.
2.7. Thermophoresis
The interaction affinity between MNT1 or cleaved MNT1 and N-protein was measured with a Monolith NT.115 instrument (NanoTemper Technologies, München, Germany) in phosphate buffer (25 mM NaH2PO4, 150 mM NaCl, pH 8.0). The N-protein was labeled with the AF488 fluorescent dye. To do this, a 4-fold molar excess of the activated AF488-N-hydroxysuccinimide ester was added to the N-protein in 65 mM carbonate buffer (pH 8.5), and the mixture was incubated for 1 h at room temperature with constant stirring. The N-protein with attached AF488 was purified from the free unreacted dye via gel filtration on PD10 chromatographic column. As a result, an average of 2.2 AF488 molecules per one N-protein molecule modification was obtained. At a fixed concentration of N-protein-AF488 (5 nM), thermophoresis curves were obtained. Four such curves were obtained for each experiment, and the whole experiment was repeated three or four times. For each curve, the dissociation constant of the N-protein complex with MNT1 or cleaved MNT1 was determined by Monolith NT.115 Instruments software, then the constant was averaged over all 14–17 curves, and the relative measurement error was determined. Cleaved MNT1 was prepared by incubating 4 μM MNT1 with 4 μg/ml activated native human cathepsin B (ab90387, Abcam) for 20 hours at 37°C. Activation of cathepsin B was carried out as described by Kern et al. [18].
2.8. Western Blot
N-protein degradation was studied in A549 and A431 cells stably transfected with the fluorescent protein mRuby3 using Western blots with antibodies to the N-protein. Cells were harvested via trypsin treatment and centrifugation (200 × g, 5 min). Then cells were suspended in a buffer (pH 8,0) containing 25 mM NaH2PO4, 150 mM NaCl, cOmplete, (EDTA free protease inhibitor cocktail, 11873580001, www.sigmaaldrich.com) and 10 mM EDTA. The number of cells was quantified using a CytoFLEX S flow cytometer. The cells were lysed by four freezing-thawing cycles. Freezing was performed in liquid nitrogen. Then, the cell lysates were centrifuged (10000× g, 5 min). The supernatants of the cell lysates were applied to denaturing electrophoresis, followed by a Western blot stained by anti-N-protein mAb (SARS-CoV-2 Nucleocapsid Polyclonal Antibody, PA5-116894, Invitrogen, Waltham, MA, USA) and secondary antibodies goat-antirabbit+Peroxidase (G21234, Thermo Fisher Scientific, Waltham, MA, USA). Sample electrophoresis was performed using standard 10% SDS-PAGE. Given the frequent inhomogeneous coloration of samples obtained using Western blot, all samples were loaded on the gel in either duplicate or triplicate. This enables us to exclude random outliers of individual bands from the analysis and obtain more reliable averaged data. Samples were transferred from the gel to a supported nitrocellulose membrane (0.22 μm) using the Trans-Blot Turbo Transfer System (Bio-RAD, Hercules, CA, USA). For each sample, the band intensity in the selected area and the background area were measured, and they were subtracted from each other. The obtained band intensity was averaged for two or three identical samples and normalized to cell concentration and to band intensity at the initial time point. For each cell line and staining with the anti-N-protein antibodies, Western blots were performed in n = 6–19 replicates. In A431 cells, the effect of the proteasomal degradation inhibitor MG-132 (S2619, Selleckchem.com) and the autophagy inhibitor Bafilomycin A1 (54645S, Cell Signaling Technology) on N-protein degradation was also studied. To do this, MNT1 was incubated for 24 hours with 5 μM MG-132 or 100 nM Bafilomycin A1. Next, the relative amount of N-protein in the cells was determined using Western blot in n = 8–15 replicates.
2.9. Confocal Microscopy
Images of A549 cells transiently transformed with N-protein fused to the fluorescent protein mRuby3 were obtained on a confocal microscope STELLARIS 5 (Leica, Germany), using a 63× objective with a NA = 1.3. Excitation of mRuby3 fluorescence was carried out with a laser with a wavelength of 561 nm, and fluorescence registration was carried out in the range of 580–640 nm. Nuclei were stained with the fluorescent dye Hoechst 33342. This dye was excited by a laser with a wavelength of 405 nm, and its fluorescence was recorded in the range of 415–420 nm. For the mRuby3 fluorescence channel, the cell cytoplasm region was selected in the ImageJ Fiji software, and then the cell-average standard deviation of fluorescence intensity was calculated. Next, such standard deviations were averaged over all cells, and the standard deviation of noise signal, which was calculated in areas where there are no cells, was subtracted from the resulting value.
3. Results
First of all, it was necessary to study the retention of the functional activities of all modules within the MNT1. The ability of the MNT1 labeled with the fluorescent dye AF488 to bind to its receptors was studied on the A431 cells. Figure 2 shows the average fluorescence values per cell for various incubation times of 200 nM MNT1-AF488 with A431 cells. The average cell fluorescence after 15 minutes of incubation was significantly (p < 0.05, ANOVA multiple comparisons, nonparametric test) higher than the control, to which MNT1-AF488 was not added. To verify that internalization of MNT1, containing an affibody to the epidermal growth factor receptor (EGFR), occurs due to binding to this receptor, a control experiment was performed in which A431 cells were first incubated for 1 hour with 1 μM epidermal growth factor, EGF, and then these cells were incubated with 200 nM MNT1-AF488, to which EGF was added at a concentration of 2 μM. In this case, the average fluorescence was significantly (p < 0.05, ANOVA multiple comparisons, nonparametric test) lower than when MNT1-AF488 was incubated for the same time without EGF (Figure 2). Thus, the internalization of MNT1 into A431 cells is mediated mainly by interaction with EGFR, i.e., the affibody in MNT1 retains its functional activity.
The ability of the MNT1 to make membrane pores via the endosomolytic module DTox was tested using leakage of calcein-loaded liposomes at various pHs. The new MNT1 did not cause liposomal damage at pH 7.5 (Figure 3). The liposome leakage due to the MNT1 was detected in acidic medium including the range characteristic for endosomal pH 5–6 (Figure 3). Thus, the endosomolytic module within MNT1 retains its functional activity.
The ability of the NC2 monobody in the MNT1 molecule to bind to the N-protein was tested by thermophoresis. Based on the obtained dependence of relative fluorescence on MNT1 concentrations at a constant concentration of N-protein (Figure 4a), the equilibrium dissociation constant of the complexes of MNT1 with N-protein was 47.2 ± 2.5 nM (± SE). Thus, the NC2 monobody retains its functional activity as part of MNT1.
MNT1 includes the FKFL site, which allows cathepsin B to cleave the NC2 monobody, and the sequence for binding the Keap1 E3 ubiquitin ligase. Figure 5a shows the kinetics of this cleavage upon the addition of activated cathepsin B. From the kinetic curve of cleavage obtained from analysis of the intensity of the band corresponding to cleaved MNT1, it can be seen that after 4 hours of incubation of MNT1 with cathepsin B, the proportion of cleaved MNT1 reaches a plateau (Figure 5b). Interpolating this curve (Figure 5b) with the dependence y = a×(1-exp(-k×t)), where a is the maximum proportion of cleaved MNT1, and k is the cleavage rate constant, k was 0.89 ± 0.07 hr-1. Thus, in MNT1 the FKFL site is accessible for cleavage by the endosomal protease cathepsin B.
MNT1 (4 μM), which was incubated for 20 hours with cathepsin B (4 μg/ml), was considered as the most cleaved. At a fixed concentration of N-protein-AF488 (5 nM), the thermophoresis was used to obtain the dependence of the relative fluorescence on the concentration of cleaved MNT1 (Figure 4b). The dissociation constant of the NC2 monobody complex with N-protein-AF488 determined from this dependence was 17.2 ± 4.4 nM, compared with 47.2 ± 2.5 nM for the full-sized MNT1 (Figure 4). Thus, if the NC2 monobody with Keap1_E3PB is cleaved from MNT1 in endosomes, then its affinity for the N-protein increases.
We have previously shown that MNT0 can interact with N-protein in A431 cells transiently transfected with N-protein fused to the fluorescent protein mRuby3 [15]. It is known that in cells, N-protein causes liquid-liquid phase separation with the formation of so-called biocondensates [19,20]. The destruction of these biocondensates significantly affects both the stability of the N-protein and the formation of the viral capsid, ultimately leading to the suppression of virus replication [19,20]. Biocondensate-like structures are observed in A431 cells transiently transfected with an N-protein fusion to mRuby3 (Figure 6). Moreover, incubation of these cells with MNT0 leads to their destruction and the appearance of more uniform fluorescence (Figure 6a and Figure 6b). In contrast, incubation with the control MNT, MNTcon, does not lead to the destruction of these structures (Figure 6c and Figure 6d). To quantitatively describe this process, the standard deviation of fluorescence intensity in the cytoplasm can be used [21]. Indeed, when cells are incubated with MNT0, this standard deviation decreases, while when cells are incubated with MNTcon, it does not change (Figure 6e). Thus, the interaction of the studied MNTs with the N-protein in the cell leads to the destruction of biocondensates formed by the N-protein.
The N-protein degradation in A549 and A431 cells stably transfected with N-protein fused with the red fluorescent protein mRuby3 can be observed by a decrease in the fluorescence of this protein. To do this, cells were incubated with MNT1 and MNT0 for different time intervals, washed, and the average mRuby3 fluorescence per cell was measured using flow cytometry. Figure 7 shows the obtained dependences of cell fluorescence on the time of cell incubation with these MNTs. Starting from 15 h incubation with MNTs cells incubated with MNT1 exhibit significantly (p < 0.05, Mann-Whitney test) diminished fluorescence compared to those incubated with MNT0 (Figure 7). Thus, the presence of a sequence that recruits the Keap1 E3 ligase in MNT1 leads to a noticeable decrease in the fluorescence of the mRuby3 fused to the N-protein, which may indicate degradation of the N-protein.
Since a decrease in fluorescence does not directly indicate degradation of the N-protein, the relative amount of N-protein in the cell during incubation with MNT1 was also measured using Western blot (Figure 8a and Figure 8b). The intensity of the corresponding bands was normalized to the cell concentration and to the intensity of the band for cells to which MNT1 was not added. Figure 8c and Figure 8d show a comparison of fluorescence data with Western blot data for A549 and A431 cells, respectively. It can be seen that the relative amount of N-protein indeed decreases greatly, especially for the A431 cells (Figure 8d), and noticeably faster than the fluorescence of the cells. Thus, MNT1 causes degradation of the N-protein in A549 and A431 cells stably transformed with an N-protein fused to mRuby3. Cell fluorescence also decreases, but it does not reflect the entire proportion of N-protein that has undergone degradation.
To determine the pathway through which the N-protein fused with mRuby3 is degraded, we used the proteasomal degradation inhibitor MG132 and the autophagy inhibitor Bafilomycin A1. Figure 9 shows that for A431 cells, the addition of MG132 together with MNT1 does not lead to significant inhibition of N-protein degradation (p > 0.05, Mann Whitney test). In contrast, the addition of Bafilomycin A1 together with MNT1 causes significant inhibition (p < 0.05, Mann Whitney test) of degradation of the N-mRuby3 fusion protein (Figure 9). Thus, the degradation of the N-mRuby3 protein occurs not through the proteasomal pathway, but through the autophagy pathway.
4. Discussion
For targeted delivery of drugs and bioactive molecules, we have developed modular nanotransporters (MNTs) [14]. Their peculiarity is that they can bind to an internalizable receptor on the surface of target cells using a ligand module. Then they are internalized and enter endosomes, from which they exit due to the endosomolytic module. Delivery is then carried out to a selected compartment in the cell and/or to a selected target protein using an intracellular targeting and/or effector module. All modules are combined together by a carrier module, to which the delivered substance can also be attached. In this work, the ligand module was an antibody-like molecule, affibody, to the human epidermal growth factor receptor (EGFR) [22,23]. The E. coli hemoglobin-like protein acted as a carrier module, and the translocation domain of diphtheria toxin acted as an endosomolytic module. The target protein was the nucleocapsid protein (N-protein) of the SARS-CoV-2 virus. The binding of the developed MNTs to this protein was carried out by another antibody mimetic, monobody NC2 [24]. According to the literature data, the equilibrium dissociation constant of the NC2 complex with the N-protein is the smallest of this type of monobody and is 6.7 nM [24]. The proposed MNTs can not only bind to the N-protein, but also provide its degradation due to the presence of an amino acid sequence that attracts the Keap1 E3 ligase. Thus, the binding of these MNTs to the N-protein should lead to ubiquitination of the N-protein and its further degradation. To increase the efficiency of the N-protein targeting part of MNT, consisting of the NC2 monobody fused with a site that attracts E3 ligase, the possibility of its endosomal cleavage via endosomal protease cathepsin B was introduced. For this, we previously selected the most optimal cleavage site for this protease [16]. The kinetics of cleavage of short peptide sequences were studied, which potentially, based on the literature data, can be cleaved by cathepsin B [16]. It was shown that the FKFL and FRRG sequences are cleaved most quickly at endosomal pH (pH 6.0), with FKFL cleaved three times faster than FRRG [16]. Moreover, the FKFL sequence is cleaved an eightfold less efficient at pH 7.5 (outside the cell) than at pH 6.0 (in endosomes) [16], so it is assumed that the cleavage of the resulting MNT will occur mainly in endosomes. Presumably, this cleavage should improve the release from endosomes of the N-protein targeting part of MNT and increase its affinity for the N-protein. To simplify reading the MNT where its N-protein targeting part consists of NC2 monobody fused with a site that attracts E3 ligase was designated MNT1, and similar MNT, but lacking a site for E3 ligase was designated MNT0.
First of all, it was necessary to establish whether all modules within MNT1 retain their functional properties. The ability of the ligand module, the anti-EGFR affibody, to bind to its receptor was tested using flow cytometry for MNT1 labeled with a fluorescent dye. A significant increase in cell fluorescence was shown after 15 minutes of incubation of A431 cells with this transporter. To test the proportion of nonspecific binding, A431 cells were preincubated with epidermal growth factor, EGF, for 1 hour. This, on the one hand, should block the EGF receptors and, on the other hand, strongly reduce their number on cell surface due to their massive internalization. As a result this allows us to determine nonspecific interactions of MNT1 with A431 cells, after subsequent incubation of MNT1 in excess EGF with EGF-pretreated cells. We observed that in this case, cell fluorescence drops almost to the control level of cells incubated without MNT1 (Figure 2). Thus, the increase in the average fluorescence of A431 cells upon the addition of MNT1 labeled with a fluorescent dye is mainly associated with receptor-specific binding of MNT1 to EGFR and the further internalization of their complex. In other words, the affibody, as part of MNT1, retains its functional activity.
The preservation of the ability of the endosomolytic module to cause defects in the lipid membrane was tested at different pH values by the release of the fluorescent dye calcein from liposomes loaded with it to the self-quenching concentration. As we showed earlier, two peaks should be observed on the curve of this release, one corresponding to the membranolytic activity of HMP (pH less than 5), and another to the membranolytic activity of the endosomolytic module DTox (pH 5.5–6) [14]. For MNT1, a second peak at pH 5.5–6 is also observed (Figure 3), which means that endosomolytic module DTox retains its functional activity within this transporter.
The functional activity of monobody NC2 within MNT1 was tested using thermophoresis. The equilibrium dissociation constant of the MNT1 complex with N-protein was 47.2 ± 2.5 nM. Thus, the NC2 monobody within MNT1 retains its ability to bind to the N-protein of the SARS-CoV-2 virus.
To test whether the FKFL site is accessible for binding and subsequent cleavage by the endosomal protease cathepsin B, the kinetics of MNT1 cleavage by the activated protease cathepsin B was studied in solution at pH 5.5 (Figure 5). It turned out that cleavage kinetics is quite fast, with a cleavage rate constant equal to 0.89 ± 0.07 hr-1. Thus, the FKFL site within MNT1 is accessible to the action of the cathepsin B protease, and in endosomes, the NC2 monobody with a site that attracts the E3 ligase Keap1 can be cleaved from MNT1.
The NC2 monobody within MNT1, and the free monobody possess different affinities for the N-protein. Indeed, according to the literature, the equilibrium dissociation constant of the NC2 monobody complex with the N-protein of the SARS-CoV-2 virus is 6.7 nM [24], whereas within the MNT1, its constant is 47.2 ± 2.5 nM. If MNT1 is subjected to cleavage by cathepsin B, the resulting dissociation constant hits 17.2 ± 4.4 nM. A similar post-cleavage increase in affinity was observed for for MNT0, lacking a site for E3 ligase [15]. The equilibrium dissociation constants of the MNT0 or cleaved MNT0 complex with the N-protein of the SARS-CoV-2 virus were 116 ± 20 and 10 ± 3 nM, respectively [15]. Based on this, we can suppose that when the monobody NC2 is released from MNT in endosomes its affinity to the N-protein increases noticeably.
Our MNTs can interact with N-protein not only in vitro but also in cells. For example, for A431 cells transiently transfected with an N-protein fused to the red fluorescent protein mRuby3, it was shown using a cellular thermal shift assay that MNT0 interacts with the N-protein [15]. For certain types of proteins, in particular the nucleocapsid protein of SARS-CoV-2, so-called liquid-liquid phase separation is observed in cells [19,20]. In this case, drop-shaped structures called biocondensates are formed, in which the concentration of the proteins responsible for their formation is significantly higher than in the rest of the solution [25]. For viruses, these biocondensates play an important role acting as viral factories, preserving viral proteins from degradation and, on the other hand, serving as scaffolds for the construction of the viral capsid [19,20]. Previously, it was shown that simply destroying such structures by some molecule interacting with a given protein can lead to blocking the production of the virus in the cell [19]. In the present work, we observed biocondensate-like structures in A431 cells transiently transfected with an N-protein fused to mRuby3 (Figure 6). These structures behave differently depending on whether these cells are incubated with MNT0 or control MNTcon (Figure 6). Indeed, upon MNTcon addition, the biocondensates do not change; on the contrary, upon MNT0 addition, they start to disappear. To quantitatively describe this process, we used the standard deviation of fluorescence intensity in the cytoplasm. This standard deviation did not change in cells incubated with MNTcon, but decreased markedly in cells incubated with MNT0 (Figure 6e). Thus, even the modular transporters lacking the site for the E3 ligase, MNT0, can cause the destruction of biocondensates formed by the N-protein, thereby preventing the formation of the viral capsid and, probably, increasing the rate of N-protein degradation.
The degradation of the N-protein can be conveniently monitored by the fluorescence of the mRuby3 protein fused to it. For this purpose, we obtained A549 and A431 cells stably transfected with this N-protein. Using flow cytometry, the fluorescence of A549 and A431 cells was studied when they were incubated for various times with MNT1 and MNT0 (Figure 7). Both of these proteins are able to interact with the N-protein. As mentioned above, this interaction itself can cause the destruction of biocondensates formed by the N-protein, which, in turn, can lead to an increase in the degradation of this protein. Indeed, for all MNTs, a decrease in cell fluorescence is observed. However, after 15 hours of incubation, this decrease for MNT1 is significantly greater than for MNT0 (Figure 7). This difference is most prominent in the A431 cells (Figure 7b). Thus, the MNT that is able to bind to the N-protein and contains a site for E3 ligase appears to cause more prominent degradation of the target protein N-mRuby3, compared to the MNT that does not contain this site.
To prove that we observe actual N-protein degradation and not just a decrease in mRuby3 fluorescence, a Western blot with N-protein antibodies was used (Figure 8a and Figure 8b). We showed that N-protein degradation is indeed observed, and at late incubation times, the proportion of degraded protein according to Western blot data is significantly greater than according to flow cytometry (Figure 8). In other words, fluorescence does not reflect the entire proportion of degraded N-protein. To understand why this might be related, we used inhibitors of proteasomal and autophagy degradation. For this purpose, a common inhibitor of proteasomal degradation, MG132 [26], and an autophagy inhibitor, Bafilomycin A1 [27], were selected. It turned out that Bafilomycin A1, in contrast to MG132, leads to inhibition of N-mRuby3 protein degradation (Figure 9). Based on these data, it can be assumed that N protein degradation occurs predominantly through the autophagy pathway rather than the proteasomal pathway. It is known that ubiquitinylation of proteins can lead not only to their proteasomal degradation but also to autophagy [28,29,30]. Moreover, for fluorescent proteins, it is known that, as a result of autophagy, they are not completely degraded [31,32]. If such fragment remains after mRuby3 degradation and fluoresces, then this could explain the difference we observed between cell fluorescence and Western blot data (Figure 8c and Figure 8d).
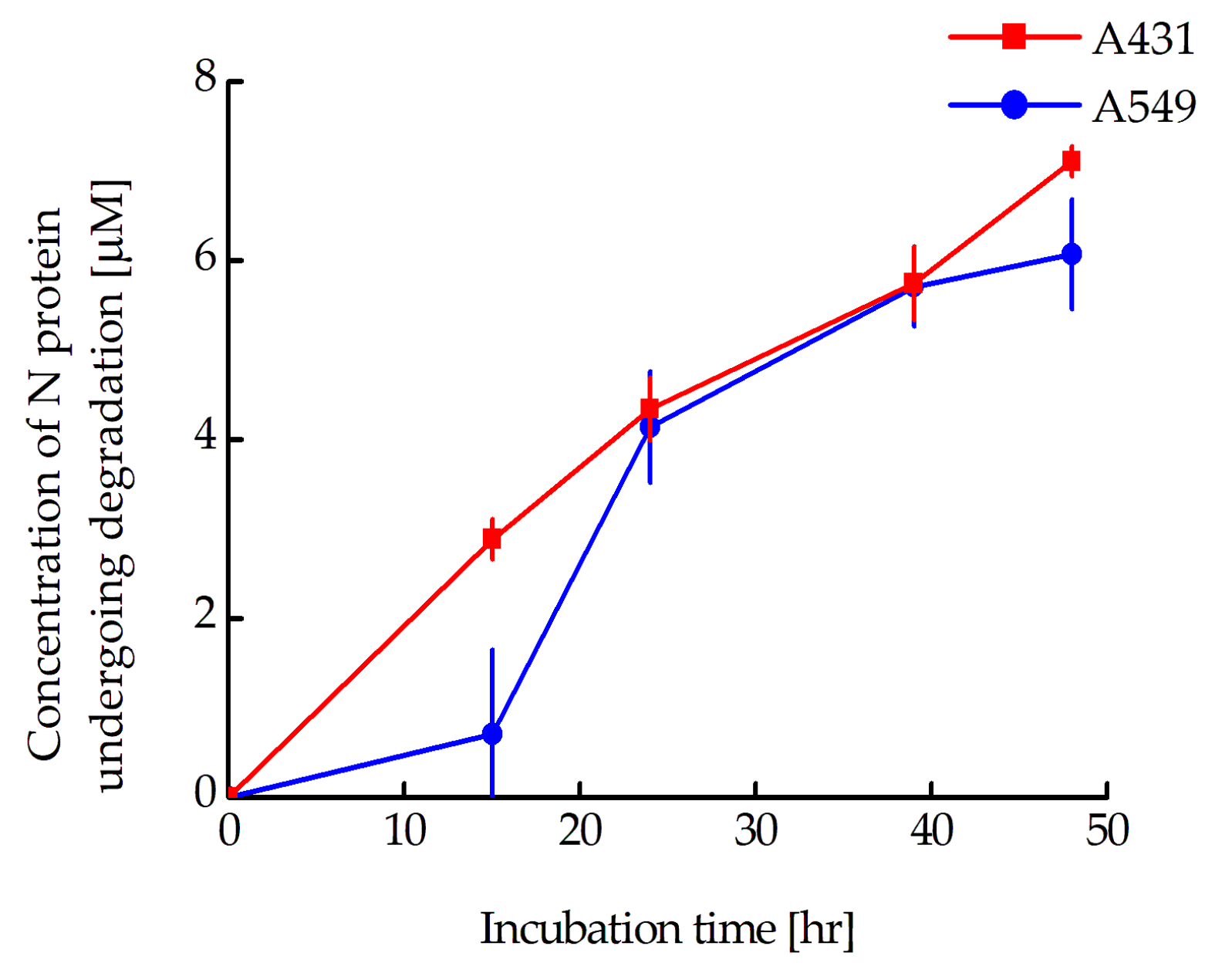
According to Western blot data, there is a significant difference in the proportion of N-protein that has undergone degradation for the A549 and A431 cells. In a paper being prepared for publication, we assessed the concentrations of the N-mRuby3 protein in A431 and A549 cells stably transfected with this protein. They were 9.0 ± 1.8 and 19.0 ± 1.3 µM for A431 and A549 cells, respectively. In other words, if we plot the change in the concentration of N-protein undergoing degradation from the time of incubation with MNT1 for A431 and A549 cells, they will coincide well with each other (Figure 10). Thus, the rate of N-protein degradation for these two cell lines is the same and is about 130 nM per hour. It should be noted that, as we previously estimated, the concentration of Keap1 does not depend on the cell type and is approximately 270 nM [33]. Moreover, the concentration of Keap1, which is capable of interacting with MNT1, will be significantly lower. Considering that the rate of N-protein degradation for A431 and A549 cells is the same, we can assume that the extent of its degradation is limited not by the concentration of MNT1, but by the concentration of Keap1, which is capable of interacting with this MNT1.
It is known that the N-proteins of coronaviruses have a degree of homology of more than 90% [34]. As we have shown previously, three of the seven monobodies capable of interacting with the N-protein of the SARS-CoV virus are also capable of interacting with the N-protein of the SARS-CoV-2 virus [35]. Thus, this developed modular nanotransporter may also be effective in the treatment of SARS-CoV and MERS coronaviruses. Moreover, our proposed approach can be extended to treat other viral diseases. It can be done, for example, by obtaining an antibody-like molecule capable of interacting with a viral protein that is a key for virus assembly/replication and subsequent inclusion of this antibody-like molecule into the modular nanotransporter we have developed.
5. Conclusions
This work proposes a modular nanotransporter containing an antibody mimetic molecule, affibody, to the epidermal growth factor receptor, an endosomolytic module, a sequence that attracts Keap1 E3-ligase, and another antibody mimetic molecule, monobody NC2, to the SARS-CoV-2N-protein. All of these modules retain their functional activity as part of the proposed nanotransporter. This nanotransporter contains a cleavage site for the endosomal protease cathepsin B, which can lead to cleavage off the monobody in endosomes and a noticeable increase in its affinity for the selected viral protein, as was shown in the work for the NC2 monobody against the N-protein of the SARS-CoV-2 virus. Together with the antibody mimetic, the amino acid site that attracts the E3 ligase Keap1 is also cleaved off. The interaction of a monobody containing this site with the viral protein should lead to ubiquitinylation of the protein and its subsequent degradation. The ability of the proposed modular nanotransporter to cause degradation of the N-protein was studied in the A549 and A431 cell lines stably transfected with the N-protein fused with the fluorescent protein mRuby3. Already after one day of incubation of this nanotransporter with cells, a significant decrease in both the fluorescence of the mRuby3 protein and the amount of N-protein in the cells determined by Western blot was observed. The inhibitory analysis performed suggests that the degradation of the N-protein fused to mRuby3 occurs via the autophagy pathway. Thus, we have proposed modular nanotransporters that are capable of causing degradation of a selected viral protein, thereby allowing similar transporters to be used in the future to treat a wide range of viral diseases.
Supplementary Materials
no.
Author Contributions
Conceptualization, Y.V.K., A.S.S., A.A.R. and T.A.S.; Methodology, Y.V.K., A.V.U. and A.A.R.; Validation, Y.V.K. and T.N.L.; Formal Analysis, Y.V.K.; Investigation, Y.V.K., A.A.R., A.V.U., T.N.L., T.A.S. and E.S.B.; Writing—Original Draft Preparation, Y.V.K., A.V.U.; Writing—Review and Editing, A.A.R. and A.S.S.; Visualization, Y.V.K. and T.N.L.; Supervision, A.S.S. and G.P.G.; Project Administration, A.S.S. and G.P.G.; Funding Acquisition, G.P.G. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by the grant 21-14-00130 of the Russian Science Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to Alexander S. Saburov for assistance in isolating MNT. Experiments were carried out with the use of equipment from the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, IGB RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Clift, D.; McEwan, W.A.; Labzin, L. I.; Konieczny, V.; Mogessie, B.; James, L.C.; Schuh, M. A method for the acute and rapid degradation of endogenous proteins. Cell 2017, 171, 1692–1706. [Google Scholar] [CrossRef]
- Verma, R.; Mohl, D.; Deshaies, R.J. Harnessing the power of proteolysis for targeted protein inactivation. Molecular cell 2020, 77, 446–460. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduction Targeted Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discovery 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Song, J.; Hu, M.; Zhou, J.; Xie, S.; Li, T.; Li, Y. Targeted protein degradation in drug development: Recent advances and future challenges. Eur. J. Med. Chem. 2023, 115839. [Google Scholar] [CrossRef]
- Fulcher, L.J.; Hutchinson, L.D.; Macartney, T.J.; Turnbull, C.; Sapkota, G.P. Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open biology 2017, 7, 170066. [Google Scholar] [CrossRef]
- Ibrahim, A.F.; Shen, L.; Tatham, M.H.; Dickerson, D.; Prescott, A.R.; Abidi, N.; Xirodimas, D.P.; Hay, R.T. Antibody RING-mediated destruction of endogenous proteins. Molecular cell 2020, 79, 155–166. [Google Scholar] [CrossRef]
- Ju Shin, Y.; Kyun Park, S.; Jung Jung, Y.; Na Kim, Y.; Sung Kim, K.; Kyu Park, O.; Kwon, S.H.; Ho Jeon, S.; Trinh, L.A.; Fraser, S.E. Nanobody-targeted E3-ubiquitin ligase complex degrades nuclear proteins. Sci. Rep. 2015, 5, 14269. [Google Scholar] [CrossRef]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). PNAS 2020, 117, 5791–5800. [Google Scholar] [CrossRef]
- Portnoff, A.D.; Stephens, E.A.; Varner, J.D.; DeLisa, M.P. Ubiquibodies, synthetic E3 ubiquitin ligases endowed with unnatural substrate specificity for targeted protein silencing. J. Biol. Chem. 2014, 289, 7844–7855. [Google Scholar] [CrossRef]
- Niamsuphap, S.; Fercher, C.; Kumble, S.; Huda, P.; Mahler, S.M.; Howard, C.B. Targeting the undruggable: emerging technologies in antibody delivery against intracellular targets. Expert Opin. Drug Delivery 2020, 17, 1189–1211. [Google Scholar] [CrossRef]
- Tolmachev, V.M.; Chernov, V.I.; Deyev, S.M. Targeted nuclear medicine. Seek and destroy. Russ. Chem. Rev. 2022, 91, RCR5034. [Google Scholar] [CrossRef]
- Gilyazova, D.G.; Rosenkranz, A.A.; Gulak, P.V.; Lunin, V.G.; Sergienko, O.V.; Khramtsov, Y.V.; Timofeyev, K.N.; Grin, M.A.; Mironov, A.F.; Rubin, A.B. Targeting cancer cells by novel engineered modular transporters. Cancer research 2006, 66, 10534–10540. [Google Scholar] [CrossRef]
- Khramtsov, Y.V.; Ulasov, A.V.; Lupanova, T.N.; Georgiev, G.P.; Sobolev, A.S. Modular Nanotransporters Capable of Binding to SARS-CoV-2 Virus Nucleocapsid Protein in Target Cells. Dokl. Biochem. Biophys. 2023, 510, 87–90. [Google Scholar] [CrossRef]
- Khramtsov, Y.V.; Georgiev, G.P.; Sobolev, A.S. Selection of an Amino Acid Site with One of the Fastest Cleavage Kinetics by the Endosomal Protease Cathepsin B for Potential Use in Drug Delivery Systems. Dokl. Biochem. Biophys. 2023, 509, 78–80. [Google Scholar] [CrossRef]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef]
- Kern, H.B.; Srinivasan, S.; Convertine, A.J.; Hockenbery, D.; Press, O.W.; Stayton, P.S. Enzyme-cleavable polymeric micelles for the intracellular delivery of proapoptotic peptides. Mol. Pharmaceutics 2017, 14, 1450–1459. [Google Scholar] [CrossRef]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, C. Phase Separation: The Robust Modulator of Innate Antiviral Signaling and SARS-CoV-2 Infection. Pathogens 2023, 12, 243. [Google Scholar] [CrossRef]
- Puthenveedu, M. A.; Bachert, C.; Puri, S.; Lanni, F.; Linstedt, A. D. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 2006, 8, 238–248. [Google Scholar] [CrossRef]
- Kim, D.; Yan, Y.; Valencia, C.A.; Liu, R. Heptameric targeting ligands against EGFR and HER2 with high stability and avidity. 2012.
- Stеhl, S.; Graslund, T.; Karlstram, A.E.; Frejd, F.Y.; Nygren, P.Е.; Lofblom, J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, T.H.; Meng, X.; Shi, Y.; Hu, M.; Yuan, S.; La, C. Y.; Li, S.X.; Liu, S.; Li, J. Development of high affinity monobodies recognizing SARS-CoV-2 antigen. 2020.
- Semerdzhiev, S.A.; Fakhree, M.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M. Interactions between SARS-CoV-2 N-protein and α-synuclein accelerate amyloid formation. ACS Chem. Neurosci. 2021, 13, 143–150. [Google Scholar] [CrossRef]
- Guo, N.; Peng, Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia-Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef]
- Li, L.Q.; Xie, W.J.; Pan, D.; Chen, H.; Zhang, L. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumor Biology 2016, 37, 653–659. [Google Scholar] [CrossRef]
- Jee, S. C.; Cheong, H. Autophagy/Mitophagy Regulated by Ubiquitination: A Promising Pathway in Cancer Therapeutics. Cancers 2023, 15, 1112. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C. H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Takahashi, D.; Arimoto, H. Selective autophagy as the basis of autophagy-based degraders. Cell Chem. Biol. 2021, 28, 1061–1071. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Matveenko, A.G.; Ryzhkova, V.E.; Zaytseva, N.A.; Danilov, L.G.; Mikhailichenko, A.S.; Barbitoff, Y.A.; Zhouravleva, G.A. Processing of Fluorescent Proteins May Prevent Detection of Prion Particles in [PSI+] Cells. Biology 2022, 11, 1688. [Google Scholar] [CrossRef]
- Khramtsov, Y.V.; Ulasov, A.V.; Rosenkranz, A.A.; Slastnikova, T.A.; Lupanova, T.N.; Georgiev, G.P.; Sobolev, A.S. An Approach to Evaluate the Effective Cytoplasmic Concentration of Bioactive Agents Interacting with a Selected Intracellular Target Protein. Pharmaceutics 2023, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Tilocca, B.; Soggiu, A.; Musella, V.; Britti, D.; Sanguinetti, M.; Urbani, A.; Roncada, P. Molecular basis of COVID-19 relationships in different species: a one health perspective. Microbes and Infection 2020, 22, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, Y.V.; Ulasov, A.V.; Lupanova, T.N.; Georgiev, G.P.; Sobolev, A.S. Among Antibody-Like Molecules, Monobodies, Able to Interact with Nucleocapsid Protein of SARS-CoV Virus, There Are Monobodies with High Affinity to Nucleocapsid Protein of SARS-CoV-2 Virus. Dokl. Biochem. Biophys. 2022, 503, 90–92. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme of the MNT1. A description of the modules is given in the text.

Figure 2.
Interaction of MNT1 fluorescently labeled with AF488 with A431 cells possessing EGFR. A control group without MNT1-AF488 addition, groups in which 200 nM MNT1-AF488 were incubated with cells for 15, 30, 45, and 60 minutes, and a group in which 200 nM MNT1-AF488 was incubated with 2 μM EGF are shown. The fluorescence of the cells incubated with MNT1-AF488 for 60 minutes was taken as 100%. Mean values are given ± 95% confidence intervals (n = 7–13).
Figure 2.
Interaction of MNT1 fluorescently labeled with AF488 with A431 cells possessing EGFR. A control group without MNT1-AF488 addition, groups in which 200 nM MNT1-AF488 were incubated with cells for 15, 30, 45, and 60 minutes, and a group in which 200 nM MNT1-AF488 was incubated with 2 μM EGF are shown. The fluorescence of the cells incubated with MNT1-AF488 for 60 minutes was taken as 100%. Mean values are given ± 95% confidence intervals (n = 7–13).
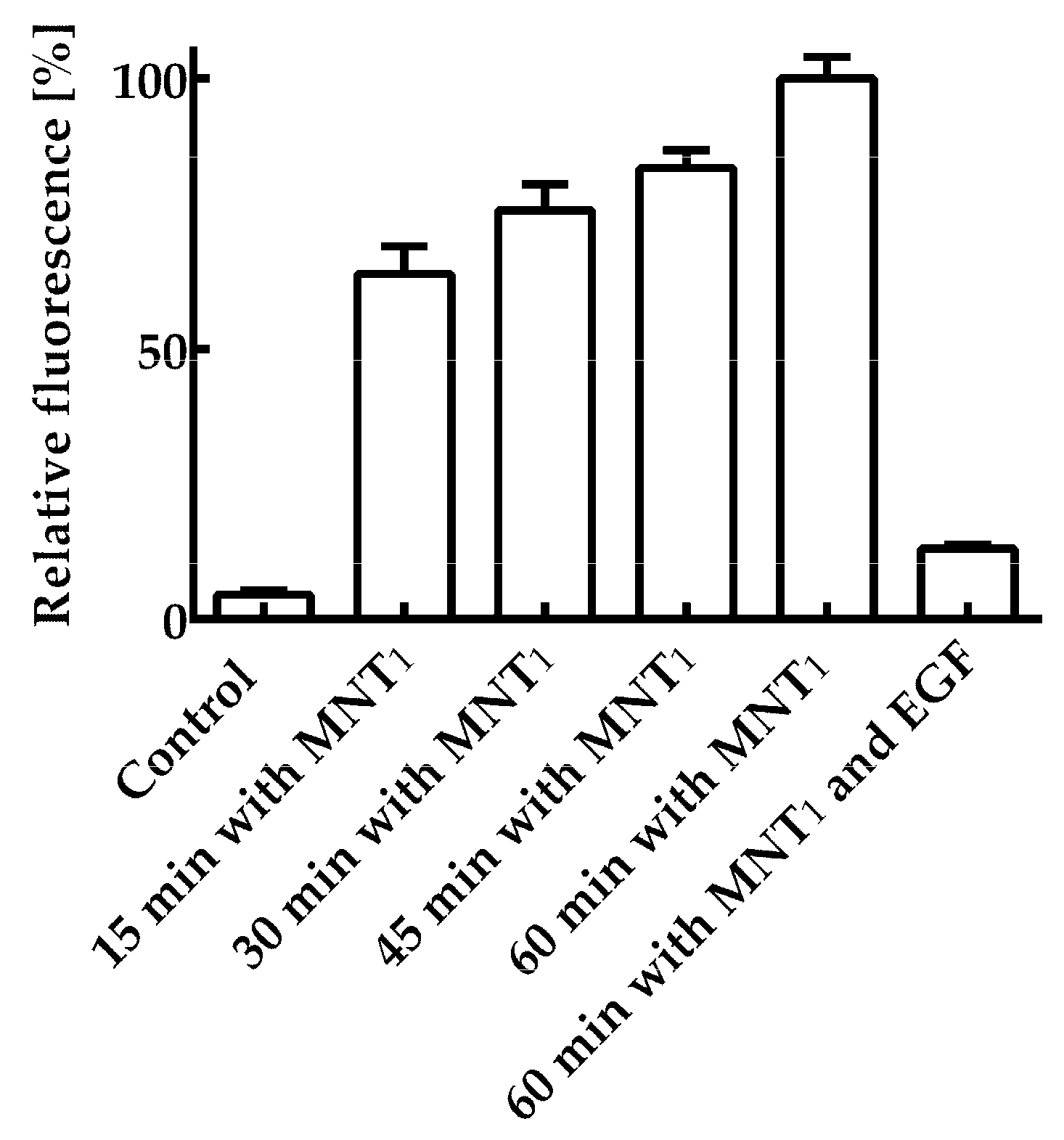
Figure 3.
MNT1-induced leakage from egg yolk phosphatidylcholine liposomes loaded with the fluorescent dye calcein at the fluorescence self-quenching concentration. The appearance of fluorescence indicates liposome leakage. Error bars are SEM (n = 3).
Figure 3.
MNT1-induced leakage from egg yolk phosphatidylcholine liposomes loaded with the fluorescent dye calcein at the fluorescence self-quenching concentration. The appearance of fluorescence indicates liposome leakage. Error bars are SEM (n = 3).
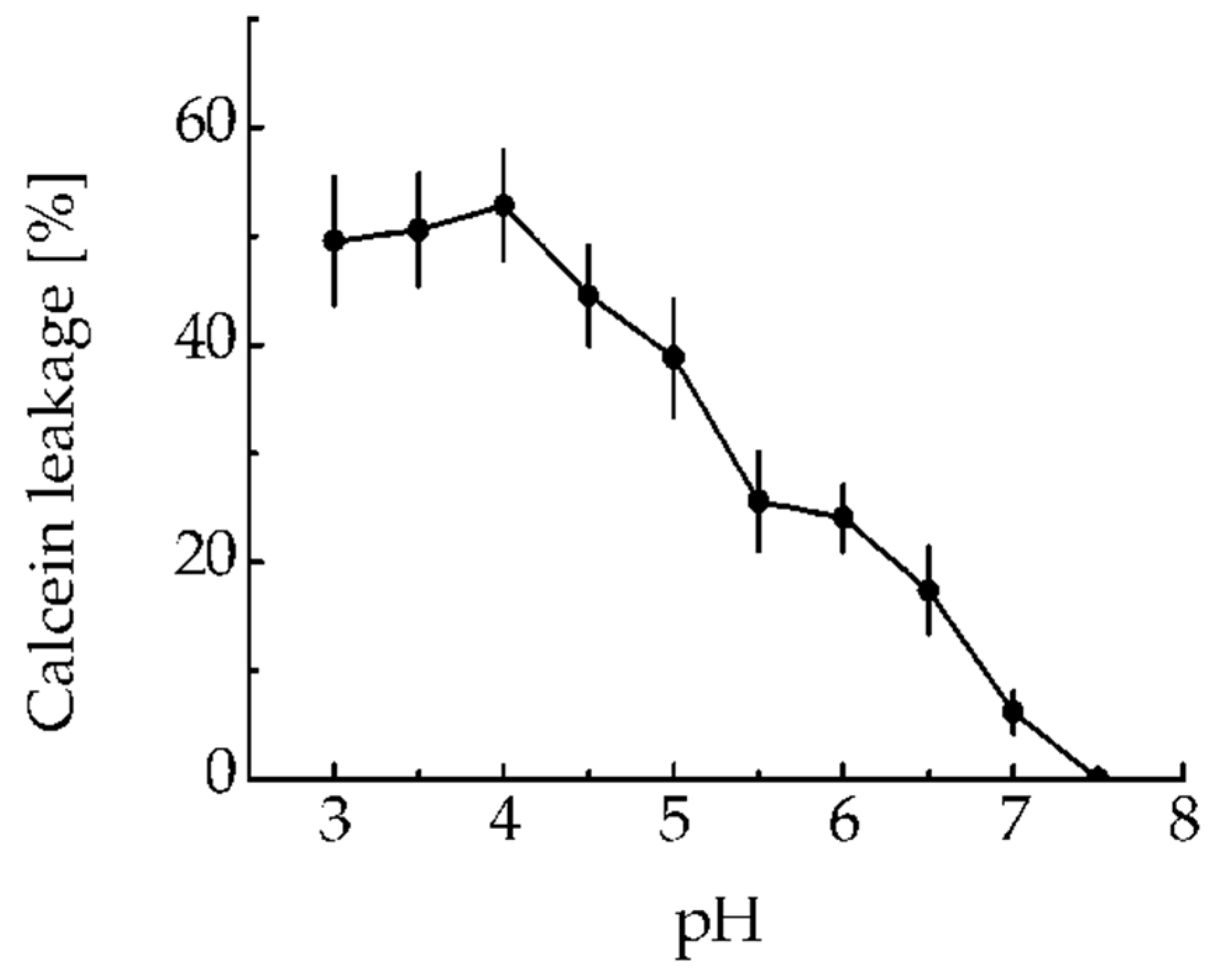
Figure 4.
The interaction of MNT1 (a) and cleaved MNT1 (b) with N-protein-AF488 shown with thermophoresis. Dependences of relative fluorescence intensities (fluorescence intensity before the start of thermophoresis is taken as 100%) at 20 s after the start of thermophoresis on the concentration of the MNT1 (a) or cleaved MNT1 (b) at a constant concentration of the N-protein-AF488 (5 nM). Standard errors (SE) of relative fluorescence intensities are shown (n = 14–17). The equilibrium dissociation constant of the MNT1 or cleaved MNT1 complex with the N-protein, Kd, is indicated.
Figure 4.
The interaction of MNT1 (a) and cleaved MNT1 (b) with N-protein-AF488 shown with thermophoresis. Dependences of relative fluorescence intensities (fluorescence intensity before the start of thermophoresis is taken as 100%) at 20 s after the start of thermophoresis on the concentration of the MNT1 (a) or cleaved MNT1 (b) at a constant concentration of the N-protein-AF488 (5 nM). Standard errors (SE) of relative fluorescence intensities are shown (n = 14–17). The equilibrium dissociation constant of the MNT1 or cleaved MNT1 complex with the N-protein, Kd, is indicated.
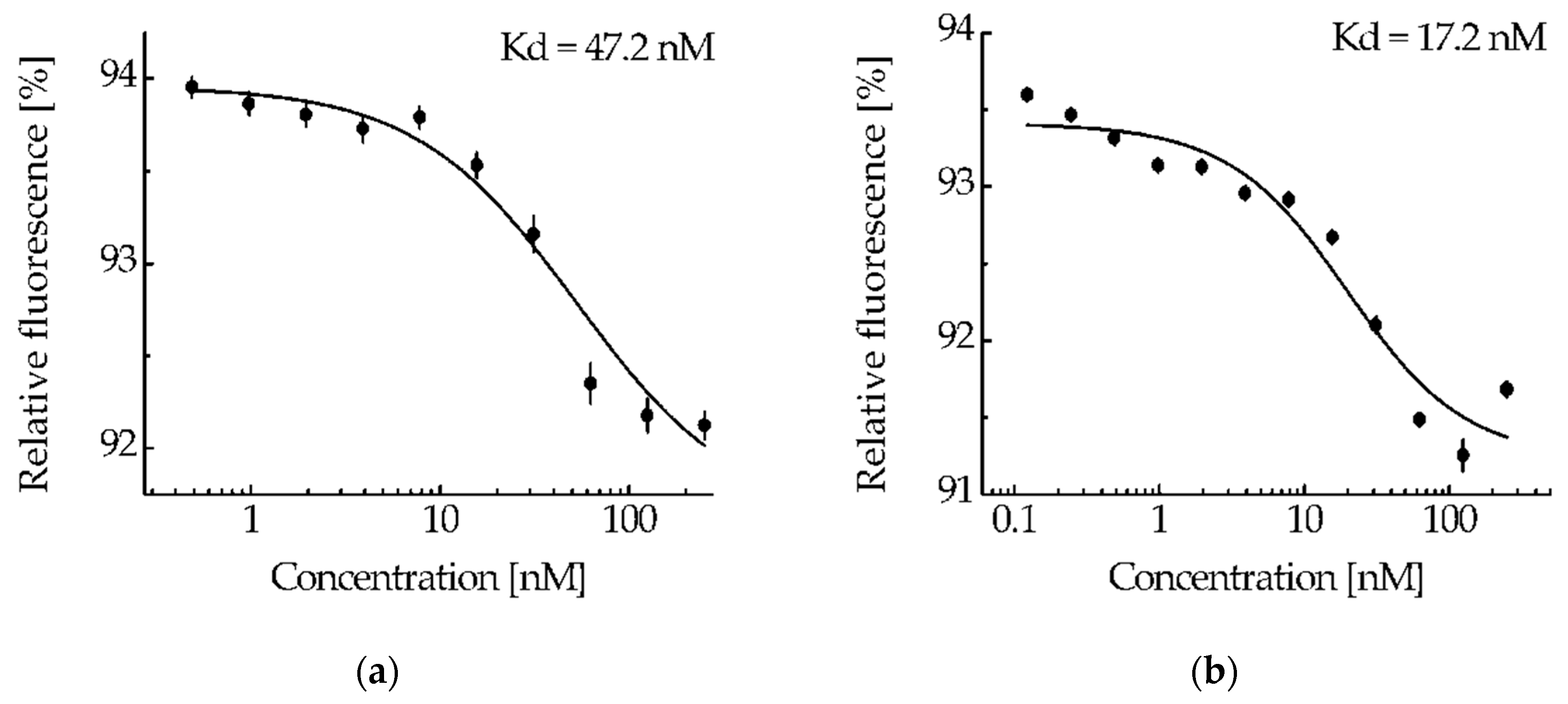
Figure 5.
(a) Availability of the cathepsin B cleavage site in MNT1. Cleavage of MNT1 (4 μM) with cathepsin B (4 μg/ml); polyacrylamide gel electrophoresis. Samples 1 – MNT1 without cathepsin B, 2 – 1 hour of incubation of MNT1 with cathepsin B, 3 – 2 hours of incubation of MNT1 with cathepsin B, 4 – 3 hours of incubation of MNT1 with cathepsin B, 5 – 4 hours of incubation of MNT1 with cathepsin B, 6 – 20 hours of incubation of MNT1 with cathepsin B. M – protein standards. MNT1 – original MNT1, MNTcl – cleaved MNT1. (b) Kinetics of cleavage of MNT1 (4 µM) by the endosomal protease cathepsin B (4 µg/ml) at pH 5.5. Shown means ± SE (n = 4–5).
Figure 5.
(a) Availability of the cathepsin B cleavage site in MNT1. Cleavage of MNT1 (4 μM) with cathepsin B (4 μg/ml); polyacrylamide gel electrophoresis. Samples 1 – MNT1 without cathepsin B, 2 – 1 hour of incubation of MNT1 with cathepsin B, 3 – 2 hours of incubation of MNT1 with cathepsin B, 4 – 3 hours of incubation of MNT1 with cathepsin B, 5 – 4 hours of incubation of MNT1 with cathepsin B, 6 – 20 hours of incubation of MNT1 with cathepsin B. M – protein standards. MNT1 – original MNT1, MNTcl – cleaved MNT1. (b) Kinetics of cleavage of MNT1 (4 µM) by the endosomal protease cathepsin B (4 µg/ml) at pH 5.5. Shown means ± SE (n = 4–5).
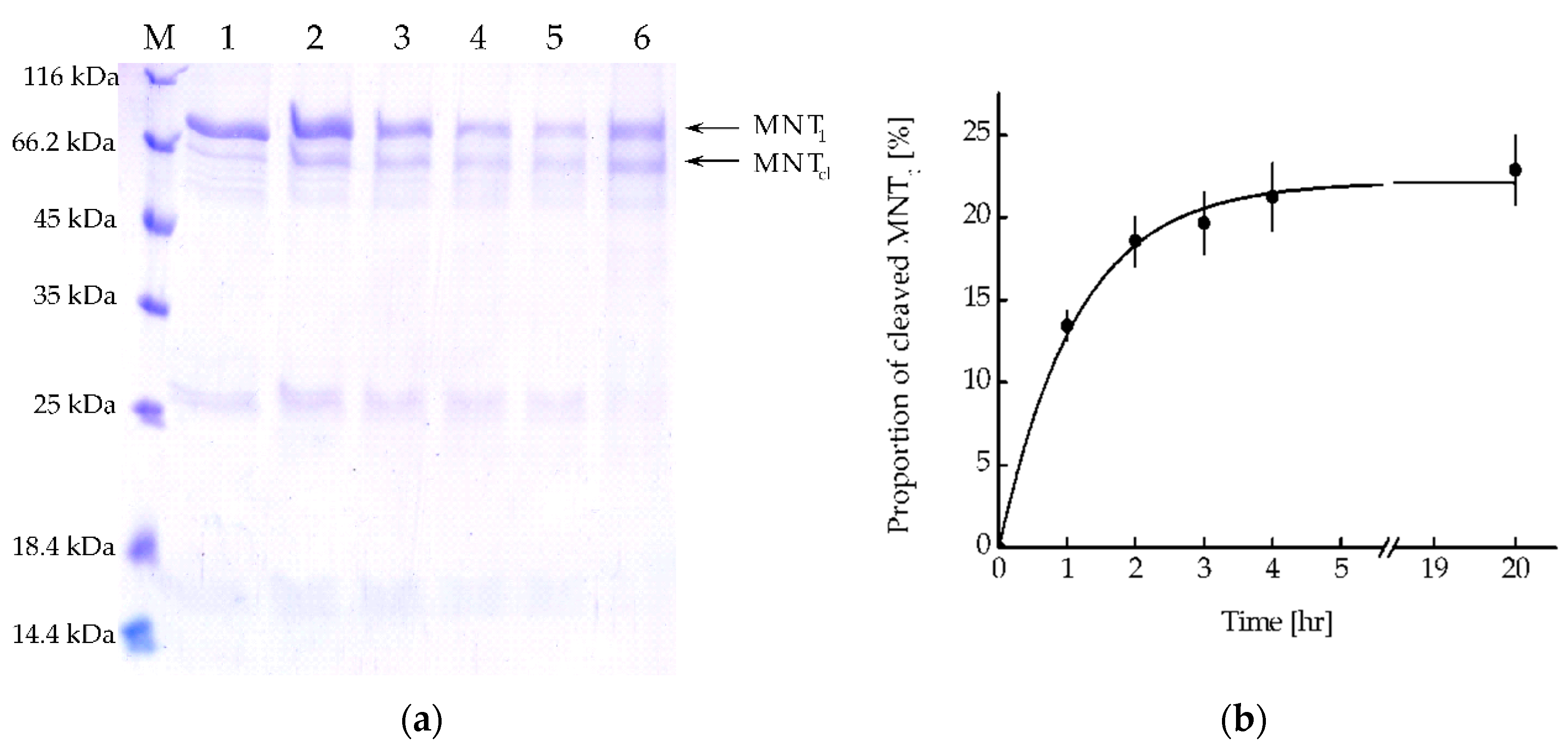
Figure 6.
Confocal microscopy images of A549 cells transiently transfected with N protein fused to the red fluorescent protein mRuby3 and incubated with 500 nM MNT0 for 0 min (a) and 80 min (b) or with 500 nM MNTcon for 0 min (c) and 80 min (d). Blue shows the staining of nuclei with Hoechst 33342 dye. Bar is 10 μm. (e) Dependence of the relative standard deviation of fluorescence intensity in the cytoplasm (the standard deviation of fluorescence intensity at the initial time point is taken as 100%), SD, on the time of incubation of A431 cells with 500 nM MNT0 or with 500 nM MNTcon. The background SD was subtracted. Shown means ± SE (n = 3–5).
Figure 6.
Confocal microscopy images of A549 cells transiently transfected with N protein fused to the red fluorescent protein mRuby3 and incubated with 500 nM MNT0 for 0 min (a) and 80 min (b) or with 500 nM MNTcon for 0 min (c) and 80 min (d). Blue shows the staining of nuclei with Hoechst 33342 dye. Bar is 10 μm. (e) Dependence of the relative standard deviation of fluorescence intensity in the cytoplasm (the standard deviation of fluorescence intensity at the initial time point is taken as 100%), SD, on the time of incubation of A431 cells with 500 nM MNT0 or with 500 nM MNTcon. The background SD was subtracted. Shown means ± SE (n = 3–5).
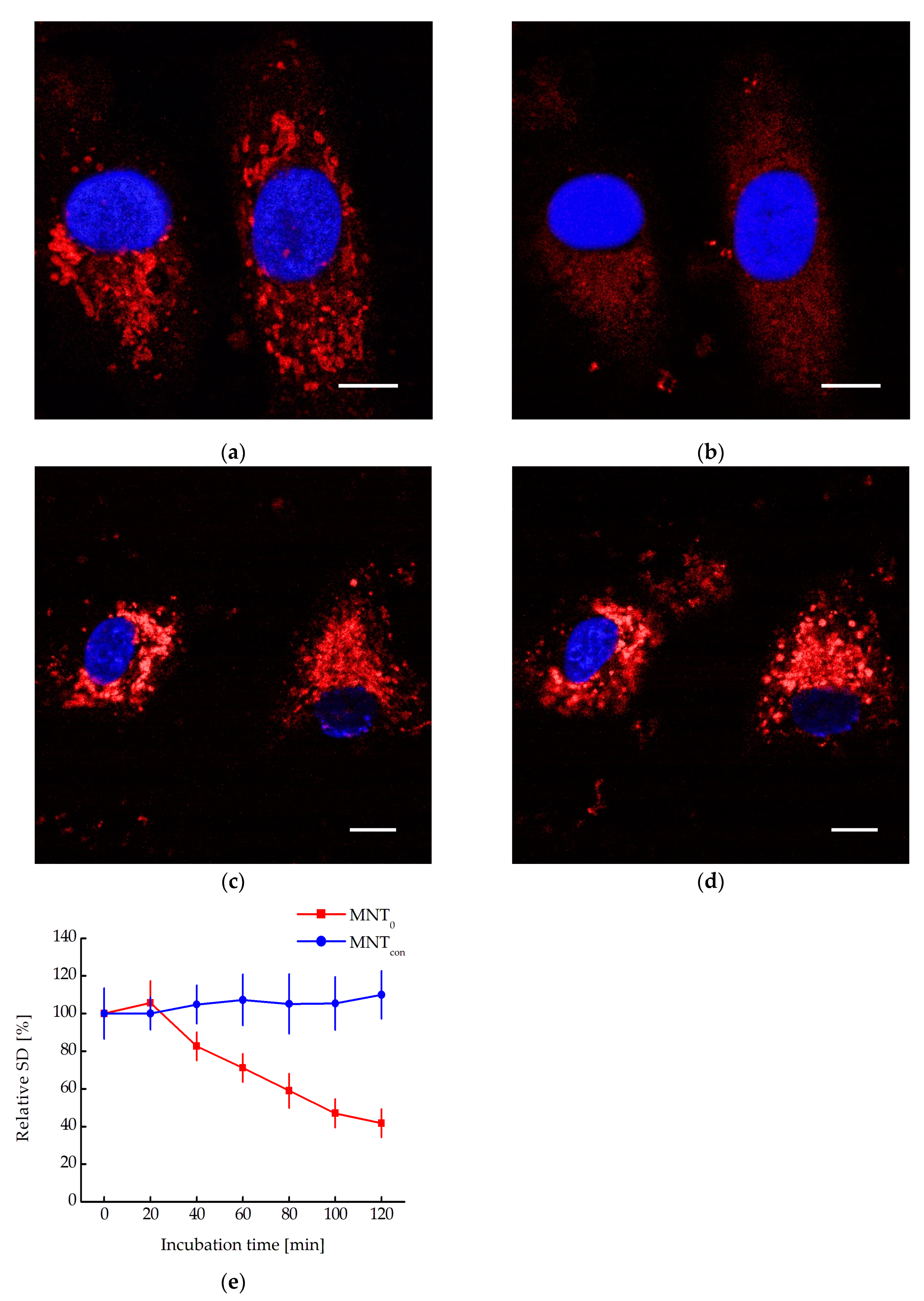
Figure 7.
Relative fluorescence of A549 (a) and A431 (b) cells (the fluorescence of cells incubated without MNT was taken as 100%) when they were incubated for different times with either 500 nM MNT1 or 500 nM MNT0. Shown means ± SE (n = 5–19).
Figure 7.
Relative fluorescence of A549 (a) and A431 (b) cells (the fluorescence of cells incubated without MNT was taken as 100%) when they were incubated for different times with either 500 nM MNT1 or 500 nM MNT0. Shown means ± SE (n = 5–19).
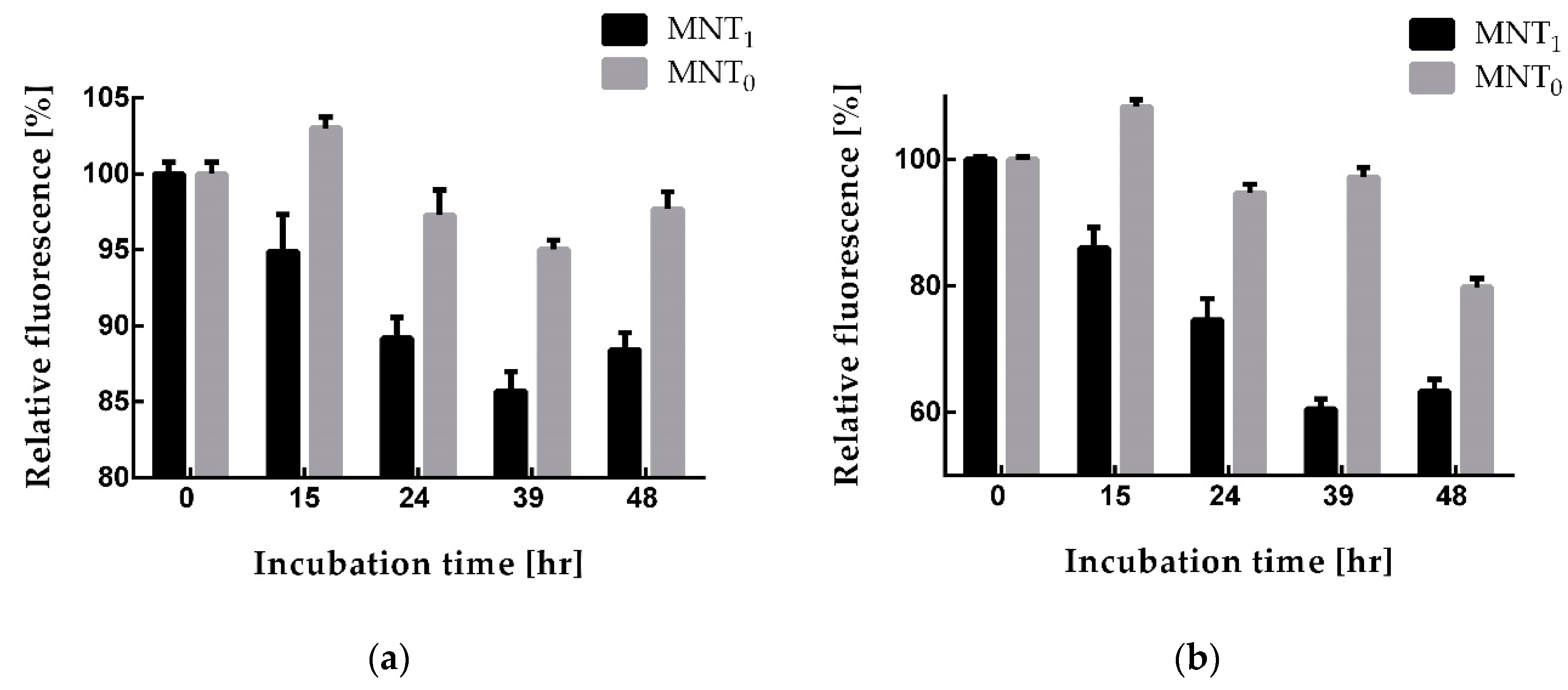
Figure 8.
Western blot with N-protein antibodies for lysates of A549 (a) and A431 (b) cells stably transfected with N-protein fused with mRuby3. The times of incubation of cells with 500 nM MNT1 are indicated. Relative intensity of fluorescence or Western blot band for N-protein for A549 (c) and A431 (d) cells (the intensity of fluorescence or Western blot band for cells to which MNT was not added was taken as 100%) when they were incubated for different times with 500 nM MNT1. Shown means ± SE (n = 6–19).
Figure 8.
Western blot with N-protein antibodies for lysates of A549 (a) and A431 (b) cells stably transfected with N-protein fused with mRuby3. The times of incubation of cells with 500 nM MNT1 are indicated. Relative intensity of fluorescence or Western blot band for N-protein for A549 (c) and A431 (d) cells (the intensity of fluorescence or Western blot band for cells to which MNT was not added was taken as 100%) when they were incubated for different times with 500 nM MNT1. Shown means ± SE (n = 6–19).

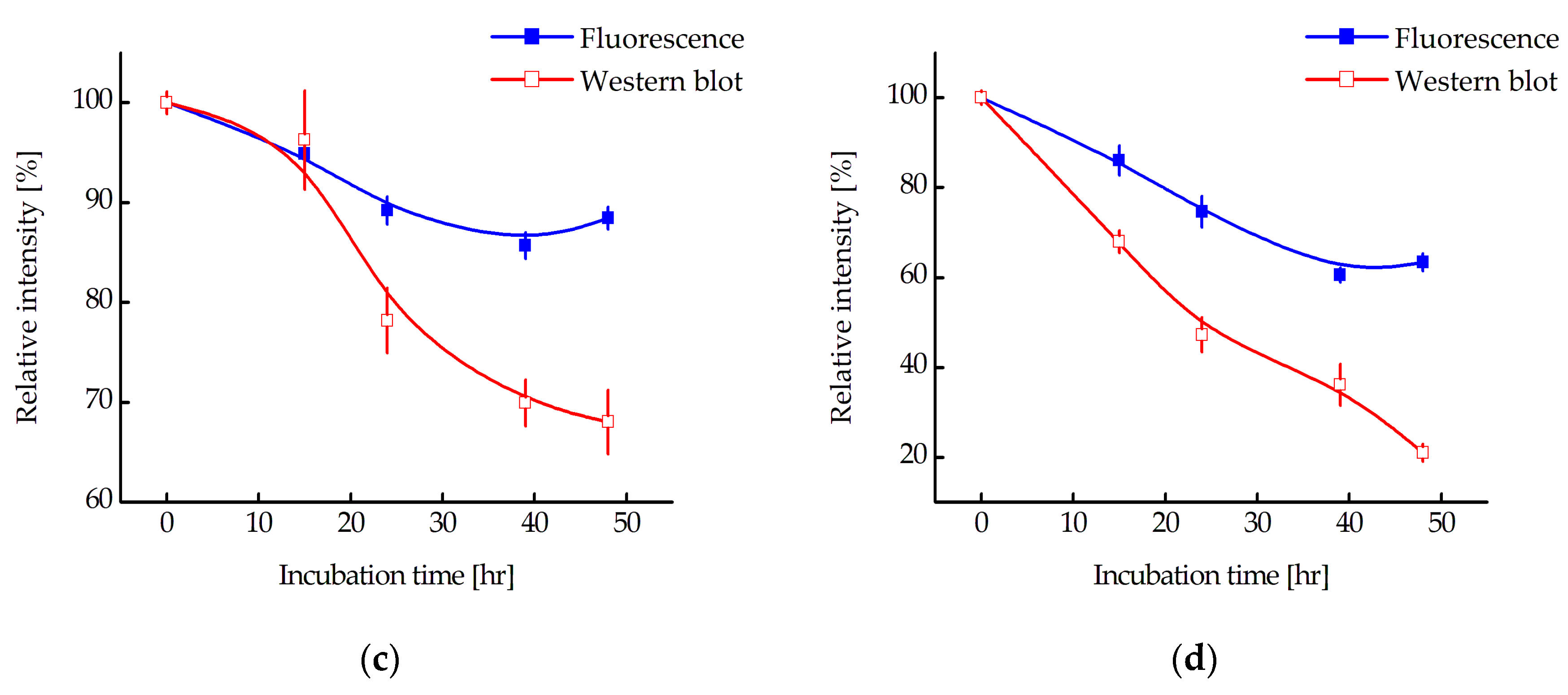
Figure 9.
The influence of MG132 and Bafilomycin A1 on MNT1 caused degradation of N-protein. Relative intensity of Western blot band for the N-protein for A431 cells (the intensity of Western blot band for cells to which MNT was not added was taken as 100%) when they were incubated for 24 hr with MNT1 (500 nM) and MNT1 (500 nM) with MG132 (5 μM) or Bafilomycin A1 (100 nM). Shown means ± SE (n = 8–15).
Figure 9.
The influence of MG132 and Bafilomycin A1 on MNT1 caused degradation of N-protein. Relative intensity of Western blot band for the N-protein for A431 cells (the intensity of Western blot band for cells to which MNT was not added was taken as 100%) when they were incubated for 24 hr with MNT1 (500 nM) and MNT1 (500 nM) with MG132 (5 μM) or Bafilomycin A1 (100 nM). Shown means ± SE (n = 8–15).
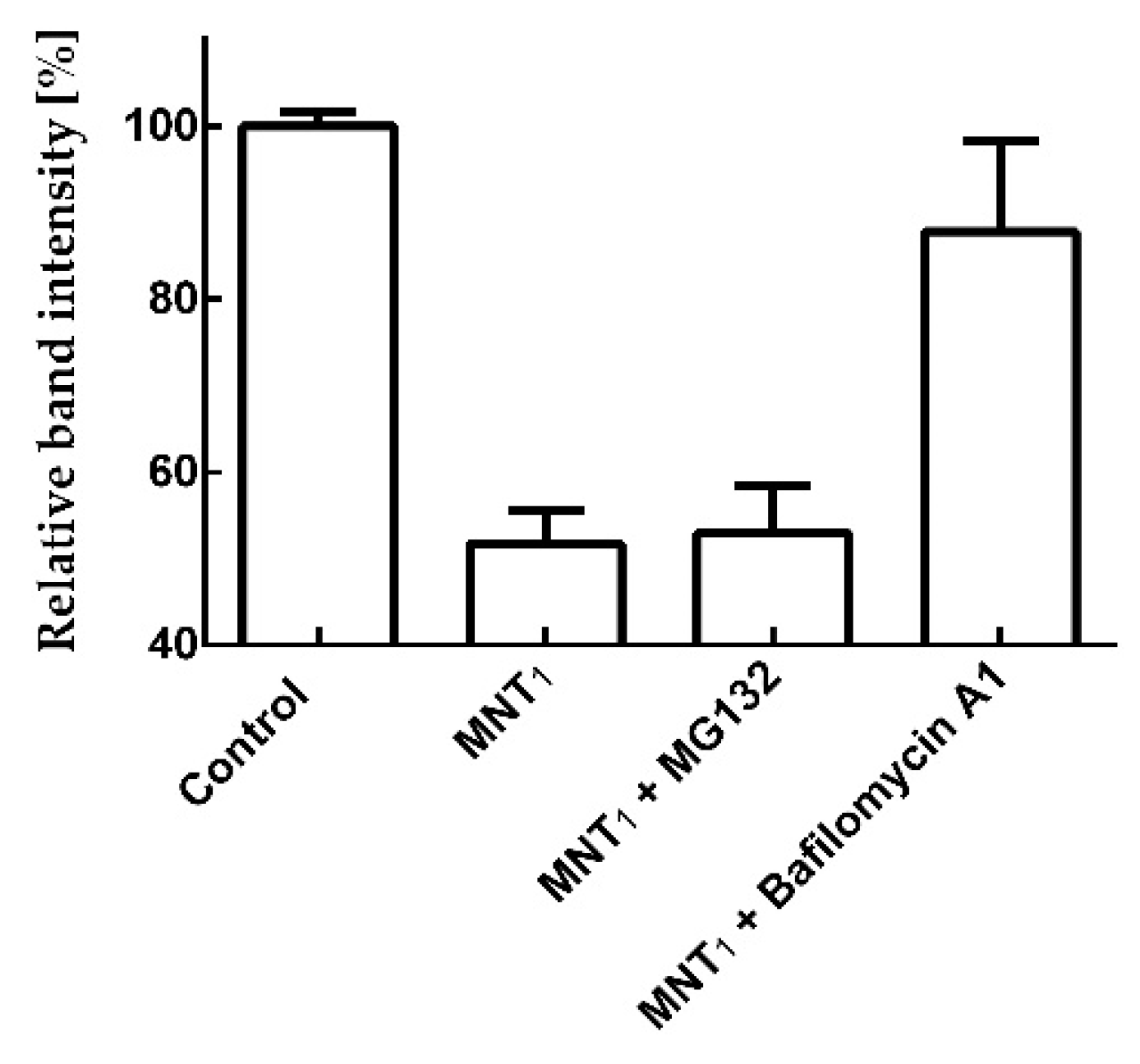
Figure 10.
Relationships between the concentration of degraded N-protein and incubation time in A549 and A431 cells stably transfected with mRuby3-fused N-protein. Shown means ± SE (n = 6–19).
Figure 10.
Relationships between the concentration of degraded N-protein and incubation time in A549 and A431 cells stably transfected with mRuby3-fused N-protein. Shown means ± SE (n = 6–19).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



