
Submitted:
03 November 2023
Posted:
06 November 2023
You are already at the latest version
Abstract
Colorectal cancer is a high-incidence tumor that has a high mortality rate due to its frequent metastasis to the liver. The difference in genes, proteins, and immune microenvironment between the primary and metastatic sites causes them to show different responses to treatment. Colorectal cancer liver metastasis patients also tend to show poorer treatment response and prognosis. Therefore, in this paper, we summarize the heterogeneity exhibited after colorectal cancer liver metastasis from five aspects (gene, transcriptome, protein, metabolism, and immunity), and we found that except for the genetic heterogeneity, the other four aspects exhibite significant heterogeneity, which might serve as a new therapeutic direction and a prognostic marker for patients with liver metastasis. Finally, the therapeutic modalities regarding tumors are rapidly evolving, and we have also summarize the new clinical therapeutic modalities currently proposed based on these heterogeneities, aiming to provide new therapeutic ideas for the clinical treatment of patients with colorectal cancer liver metastases.
Keywords:
colorectal cancer liver metastasis
; heterogeneity
; gene
; transcriptome
; protein
; metabolism
; immune
; therapy
1. Introduction
Colorectal cancer (CRC) is a tumor with a high incidence and mortality rate. The number of new CRC cases worldwide reached 1.93 million in 2020, third only to breast cancer and lung cancer, and the number of CRC deaths reached 940,000, second only to lung cancer, making it the second most deadly tumor worldwide [1]. In China, according to the 2016 national cancer statistics published by the National Cancer Center, a total of 4.06 million tumor patients were diagnosed in 2016, and there were approximately 408,000 CRC patients, accounting for 10.00% of the total, second only to lung cancer, while the total number of cancer deaths in 2016 was approximately 2.41 million, and approximately 196,000 CRC patients died, accounting for 8.10% of the total.
The high metastasis rate of CRC is one of the reasons why the mortality rate is so high. CRC frequently metastasizes, especially to the liver, with approximately 20% of patients having liver metastasis by the time CRC is diagnosed. The extremely high rate of colorectal liver metastasis (CRLM) reduces the effectiveness of treatment for CRC patients [2,3]. For one, these patients are often diagnosed with advanced tumors, and their disease is poorly controlled. For another, the current treatment modalities for CRLM patients are relatively limited. Surgical resection is the main treatment for CRLM patients, but only a small proportion of patients can be cured by resection of liver metastasis, and the prognosis of other patients is poor [4,5]. Immunotherapy is particularly effective for dMMR and MSI-H CRC patients [6,7,8], but it is not as effective in CRLM patients, possibly due to the immunosuppressive microenvironment of the liver [9].
It is well known that tumor heterogeneity has a large impact on the treatment outcome of tumor patients [10]. Although tumor heterogeneity is very complex, the study of tumor heterogeneity remains a hot topic. Tumor heterogeneity can be divided into intratumor heterogeneity and intertumor heterogeneity, as well as temporal heterogeneity and spatial heterogeneity. Temporal heterogeneity means that the nature of tumors changes over time, while spatial heterogeneity can indicate that the nature of different cell subpopulations within a tumor at the same site are different and that the nature of the primary tumor lesion and its corresponding metastasis are also different.
CRC is a highly heterogeneous disease, especially after CRLM, and the unique microenvironment of the liver makes CRC exhibit stronger spatial heterogeneity in all aspects, including gene expression, tumor microenvironment, and biological behavior [11].
Therefore, the strong heterogeneity in CRLM is a major reason for its poor response to treatment. To improve the treatment effect of CRLM patients, it is important to examine the heterogeneity between CRC and CRLM. In this article, we summarize the heterogeneity between CRC and CRLM at the genetic, transcriptional, protein, metabolic, and immune levels and discuss the prognostic value of this heterogeneity and its impact on clinical decision-making.
2. Genetic heterogeneity
The adenoma-carcinoma sequence underlies the development of CRC and involves many changes, including tumor suppressor gene inactivation (APC, TP53), oncogene activation (BRAF, PI3KA, and RAS), chromosomal instability (CIN), CpG island methylation phenotype (CIMP), and microsatellite instability (MSI) pathways [12,13]. These alterations result in a highly unstable genome in CRC. Yet when we explore the sources of genetic heterogeneity in CRLM, we are surprised to find little genetic heterogeneity between primary tumors and CRLM.
2.1. Key driver genes
Five key driver genes, APC, TP53, RAS, BRAF, and PIK3CA, play a critical role in the adenoma-carcinoma sequence of CRC, and the mutational status of these genes can influence the clinical outcome of CRC patients. Patients with KRAS wild-type tumors have significantly better clinical outcomes than patients with KRAS mutation tumors [14,15]. Patients with BRAF mutation and PI3KA mutation tumors also show poorer clinical outcomes [16,17]. Therefore, we summarize here the heterogeneity of these five genes among CRLM patients. Unexpectedly, these genes do not show heterogeneity between the primary tumor and corresponding liver metastasis [18,19,20,21,22]. Stephan et al. examined mutation sites in KRAS, BRAF, and PI3KA in 20 CRLM patients and found the same mutation status in 18 patients with primary tumor and liver metastasis [18]. Jiayun Hou et al. investigated the heterogeneity of the KRAS pathway in CRC and found that the frequency of KRAS mutations was significantly higher in the lung (62.0%) and brain (56.5%) than in the liver (32.5%), KRAS mutation could be an independent predictor of lung metastasis but played a less significant role in CRLM [23]. Another study reached a similar conclusion [24].
2.2. Chromosomal instability (CIN)
CIN is a common feature of solid tumors, including CRC, and it causes genomic instability in approximately 70% of CRC patients [12,25]. CIN includes instability of chromosome number (numerical CIN) and instability of chromosome structure (structural CIN), numerical CIN refers to the increase or decrease of chromosome copy number, and structural CIN includes deletions, translocations, and derivative chromosome, among other [26].
Previous studies have found that chromosome instability is significantly higher in metastatic breast cancer cells than in the primary, and it is also a driver for metastasis [27]. In CRC, some studies have reached the same conclusion [28,29,30,31]. Soulafa et al. found by whole-genome sequencing that the CNVs of the MMP9 and CDX2 genes were significantly increased in CRLM [31]. MMP9 belongs to the matrix metalloproteinase family, which can degrade various protein components in the extracellular matrix (ECM) and disrupt the histological barrier that prevents tumor cell invasion, and therefore plays a key role in tumor invasion and metastasis [32]. CDX2 is involved in the proliferation and differentiation of intestinal epithelial cells [33].
Nonetheless, some studies have come to the opposite conclusion. Leonie used a high-resolution array of comparative genomic hybridization to study 62 primary colorectal cancers and 68 matched metastatic lesions (22 liver, 11 lung, 12 ovary, 12 omentum, and 11 distant lymph nodes). They found that patterns of DNA copy number aberrations were highly similar between all primary and metastatic lesions [34]. By allelic copy number analysis of 33 CRC samples, Shogo identified several chromosomal aberrations common in CRC patients, with gains on 20p13-p12.1 and 20q11.21-q13.33 and LOH on 6q14.1-q25.1 more common in CRLM patients. By genetic analysis of metastatic lesions, they found that allelic imbalances in CRLM were very similar to those in CRC and that these aberrations on chromosomes 20p, 20q, and 6q were also present in CRLM, suggesting that they may promote CRLM [35]. Previous studies have also shown that only a few mutations are needed to transform highly aggressive tumor cells into metastatic ones [36]. These results indicate that CRC cells maintain relative chromosome stability during metastasis.
2.3. Microsatellite instability (MSI) status
Approximately 15% of CRC patients are affected by MSI pathways [37]. MSI is caused by functional defects in genes such as DNA mismatch repair genes (hMSH2, hMLH1, hMSH3, hMSH6, hPMSH1, and hPMSH2). There are two main methods currently used to detect MSI status: 1) immunohistochemistry (IHC), which detects the expression of four mismatch repair proteins (MLH1, PMS2, MSH2, and MSH6) in the nucleus to detect the presence of mismatch function defects; and 2) molecular testing, which detects the length of microsatellite sequences in tumor tissue to determine whether MSI is present at that site. Through IHC and molecular assays, the current studies found a very high similarity of MSI status between primary CRC and CRLM [38,39,40,41]. Among them, Wen-Zhuo He and Jiyoon Jung's study found partial differences, but the differences were concentrated in peritoneal and ovarian metastasis, and no differences were found in CRLM [38,40].
We summarize the genetic heterogeneity of CRLM in the table below (Table 1). In summary, the genetic heterogeneity of CRLM was not significant in terms of the above 3 aspects, a result that suggests that these genes play similar roles in CRC and CRLM, whereas the heterogeneity of CRC is more focused on other aspects.
3. Transcriptomic heterogeneity
3.1. MicroRNAs (miRNAs)
MicroRNAs (miRNAs), the most studied class of noncoding RNAs, are a class of short RNA molecules ranging in size from 19 to 25 nucleotides that are primarily responsible for regulating posttranscriptional gene expression [42,43]. MiRNAs have been linked to many diseases, including CRC. They are involved in colorectal carcinogenesis and can be used as a marker for CRC metastasis [44,45].
After CRC metastasizes to the liver, different types of miRNAs enable cancer cells to adapt to the new environment of the liver by regulating the expression of their respective target genes. Using genome-wide expression profiling, Petra et al. identified that miR-143, miR-10b and miR-28-5p were downregulated, while miR-122, miR-122*, and miR-885-5p were upregulated in the liver metastasis compared to their primary tumor [46]. Keun Hur and Tao Zhang reached the same conclusion [47,48]. MiR-122 is a liver-specific miRNA and a recognized suppressor of liver cancer. It exerts its effects by regulating the expression of important miRNAs in the liver, and it has been shown to have a strong relationship with the prognosis of patients with liver cancer [49,50]. Another upregulated miRNA, miR-885-5P, promotes the proliferation and migration of CRC cells by stimulating the EMT pathway. Epithelial-mesenchymal transition (EMT) is a crucial first step in the process of tumor metastasis, and tumor cells have the opportunity to metastasize to distant organs only after losing their epithelioid characteristics through EMT. Therefore, CRC patients with high miR-885-5p expression tend to have a worse prognosis [51,52]. However, research on miR-10b is still controversial. Several studies have confirmed that it is an oncogenic miRNA because its high expression is associated with worse outcomes [46,47,53]. Interestingly, J.-J. Song et al. found that miR-10b inhibits the growth of CRC by regulating EMT in animal experiments [54]. There may be two reasons for the two opposing conclusions. First, the organ characteristics of mice may be quite different from those of humans, and second, the miRNA may be responsible for regulating multiple mRNAs, which may have opposite effects. Therefore, the true role of miR-10b in CRC has not yet been determined.
In addition, Keun Hur et al. also found that the expression of miR-203 and miR-200c in CRLM was much higher than CRC [55,56]. MiR-200c promotes EMT mainly by suppressing the overexpression of target genes (ZEB1, ETS1, and FI1, three EMT-related genes) and therefore promotes the growth and metastasis of CRC. In addition to detecting miR203 expression in tissues, it is significantly more expressed in liver metastasis than in primary sites. Hur also found that miR203 is a secreted miRNA and that metastatic lesions of CRC secrete miR203 into circulation resulting in high serum miR203, thus high serum miR203 is usually associated with distant metastasis. However, miR-203 is a potent tumor suppressor miRNA in many other tumors [57,58]. Sofía Torres et al. found significant upregulation of miR-424-3p, miR-503, and miR-1292 expression in CRLM, and all these miRNAs might promote CRC metastasis [59].
3.2. circRNAs
Circular RNAs (circRNAs) are single-stranded, covalently closed RNA molecules [60]. Initially, circRNAs were considered to be "junk" with little function [61]. However, with the development of technologies such as immunohistochemistry (IHC) and high-throughput RNA sequencing (RNA-seq), it has been shown that circRNAs are involved in the development of many diseases [62,63]. In CRC, some circRNAs have also played a great role; for example, circ001971 and circ3823 can both promote tumor metastasis and angiogenesis [64,65,66]. Similar to miRNAs, the expression of some circRNAs changes in CRLM to promote tumor progression. Hanchen Xu et al. analyzed three cases by RNA sequencing and found that 92 circRNAs were upregulated in CRLM compared to CRC, and 21 circRNAs were downregulated in CRLM [67]. Among them, circRNA_0001178 and circRNA_0000826 were most significantly upregulated in CRLM and were considered promising markers of CRLM. In addition, Ri-Xin Chen et al. and Chenjing Zhang et al. identified circNSUN2 and hsa_circ_0006401 were also upregulated in CRLM, and promoted tumor progression [68,69]. CircNSUN2 was an m6A-modified circRNA. It forms a CircNSUN2/IGF2BP2/HMGA2 complex with insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) and high mobility group AT-hook 2 (HMGA2). This complex could improve the stability of HMGA2 RNA, thereby increasing the expression of HMGA2 protein. As reported by Yang Li et al, HMGA2 induces EMT and promotes CRC progression [70]. Ri-Xin Chen also analyzed the changes in EMT-related proteins after circNSUN2 overexpression and found that the expression of the epithelial marker E-cadherin was decreased and the expression of the mesenchymal marker Vimentin was increased. This further suggests that circNSUN2 can promote EMT in CRC cells through the HMGA2 pathway.
3.3. LncRNAs
Long noncoding RNAs (lncRNAs) are the third class of noncoding RNAs in addition to miRNAs and circRNAs. Currently, great progress has been made in the study of the role of lncRNAs in CRC. Nevertheless, only a few studies have examined the heterogeneity of lncRNAs in CRLM —— two lncRNAs associated with glucose metabolism (lncRNA GAL and lncRNA MIR17HG) have been found to be upregulated in CRLM [71,72]. Interestingly, the MIR17HG/miR-138-5p/hexokinase (HK1/2) pathway enhances glycolysis, and the increased lactate (a metabolite of glycolysis) activates the p38/ELK1 pathway, which promotes the expression of MIR17HG, thus forming a positive feedback loop for promoting tumor invasion and metastasis.
3.4. Transcription factors
Transcription factors are a large class of proteins that specifically bind to target genes and are important parts of transcriptomic regulation [73]. Transcription factors are inextricably linked to tumors, and they can alter their activity in tumors and promote tumor proliferation and invasion through chromosomal mutations, gene amplifications or deletions, and point mutations [74]. For example, promyelocytic leukemia protein (PML)-retinoic acid receptor α (RARα) is a driver of leukemia, and overexpression of ETS translocation variant 1 (ETV1) is also associated with melanoma and gastrointestinal stromal tumors (GIST) [75,76]. In CRC, death domain-associated protein (DAXX) is a tumor suppressor that acts as a transcriptional repressor in the nucleus and affects the progression of CRC. Vertebrate zinc finger E-box binding homeobox (ZEB) proteins are a family of transcription factors. Yanliang Liu et al. found that the expression of DAXX was downregulated in CRLM compared with CRC [77]. As a transcriptional repressor, DAXX inhibited the expression of ZEB-mediated E-cadherin.
Signal transducers and activators of transcription proteins (STATs) are another large class of transcription factors that are key regulators of cell growth and differentiation. A variety of cytokines (such as interferons and interleukins) are known to be involved in tumorigenesis through the Janus kinase (JAK)/STAT signaling pathway. Using multiplex bead-based immunoassay technologies, Fee Klupp et al. analyzed the expression patterns of STAT1, STAT3, STAT4, and STAT5 in 104 patients [78]. The results showed that STAT1 and STAT3 were significantly upregulated in CRLM compared with CRC, while STAT4 and STAT5 were opposite. STAT1 is currently considered to be a tumor suppressor, and the growth rate of tumors is significantly reduced after knocking down STAT1 [79]. As with STAT1, STAT4 and STAT5 can suppress tumors. STAT3, on the other hand, is a pro-oncogenic factor that promotes EMT activity and works with NF-kappaB to regulate inflammatory mediators with oncogenic functions [80,81]. Finally, Fee Klupp also showed for the first time that an increased ratio of STAT3/STAT5 is an indicator of poor prognosis in CRC patients. MYC and hypoxia-inducible factor-1 (HIF1α) were found increased in liver metastasis compared to their primary tumors [82]. Due to the long-term hypoxia of tumor cells, HIF1α is also in a state of high expression. The high expression of HIF1α activates downstream effector genes, and the MYC gene is essential for HIF1α to promote cell proliferation [83].
To summarize, we elaborate on the transcriptomic heterogeneity of CRLM from the above four aspects (miRNAs, circRNAs, LncRNAs, and Transcription factors) and summarize them in the following table (Table 2). We can find that transcriptomic heterogeneity is more obvious compared to genetic heterogeneity. The functions of these differentially expressed RNAs vary, but it can be found that most of them are involved in the EMT process of CRC cells and contribute to their progression and metastasis.
4. Protein heterogeneity
4.1. EMT-related proteins
As discussed above, EMT is an essential process in CRLM progression. In this process, due to the action of the involved proteins through signaling pathways, epithelial cells lose connection and apical-basal polarity, which enables tumor cells to acquire greater motility, thus enabling metastasis. Among them, adhesion-related proteins (E-cadherin, N-cadherin, tight junction family proteins, etc.), α-SMA, Snail, and Twist proteins play a huge role in this process [84].
For migration-associated proteins, Xuefei Yin et al. found that cell migration-related protein vitronectin (VTN) and actin-related protein (ARP3) expression was higher in CRLM than in CRC by large-scale quantitative proteomic analysis [85]. Using a similar approach, X. Liu et al. also identified 311 proteins that were dysregulated in CRLM, including fibronectin 1 (FN1), tissue inhibitor of metalloproteinases 1 (TIMP1), Versican (VCAN), periostin (POSTN) and thrombospondin-1 (THBS1), which have been identified as the five most critical proteins that promote CRC metastasis [86]. For example, THBS1 promotes CRC metastasis by enhancing EMT; FN1, TIMP1, VCAN, and POSTN have also all been shown to play a role in the process of CRC metastasis [87,88,89,90,91]. In addition, insulin-like growth factor binding protein 7 (IGFBP7) has also been found to be downregulated in CRLM, which inhibits EMT to block CRC metastasis [92].
Decreased adhesion of cancer cells to each other and thus separation and detachment from the primary lesion is a key step in metastasis. Integrins are the major cell adhesion receptors and claudins are tight junction proteins [93,94], both of which maintain the adhesion between cells and thus prevent cancer cells from shedding. Xuefei Yin et al. found that integrin alpha5 (ITA5) expression is decreased in CRLM [85]. Kun Wang et al. and Rania Georges et al. also found that claudin-1, claudin-4, and claudin-7 were downregulated in CRLM [95,96]. Another protein, Rho GTPase-activating protein 5 (ARHGAP5), was significantly upregulated in CRLM. ARHGAP5 is a GAPs regulating the Rho family of small GTPases, and the researchers found that knocking down this protein, Down-regulation of E-Cadherin expression, up-regulation of N-Cadherin and Vimentin expression, and the metastasis of CRC were inhibited. It was demonstrated that it could affect the invasion and metastasis of CRC cells by regulating the activity of EMT [97]. The decreased expression of these proteins reduces the adhesion between cells so that cancer cells can take the first step of metastasis.
It is clear that both the Wnt/β-catenin signaling pathway and the MAPK signaling pathway are the two most important pathways in CRC progression, and both of these pathways can promote EMT in CRC cells and thus promote CRC cells invasion and metastasis [98,99]. Bo Tang et al. found that a protein involved in the MAPK signaling pathway, PEA15, was significantly more highly expressed in CRLM than in CRC [100]. Phosphoprotein enriched in astrocytes-15 kDa (PEA15) can promote EMT by activating the MAPK signaling pathway. Two other proteins (ATP6L and FILIP1L) involved in the Wnt signaling pathway have also been found to be heterogeneous. ATP6L, the C subunit of the V-ATPase V0 domain, has previously been shown to enhance the invasion and metastasis of breast cancer cells in vitro [101]. Jingyi Wang et al. demonstrated the role of ATP6L in CRC through in vivo experiments in mice [102]. ATP6L is required for the activation of the Wnt/β-catenin signaling pathway, and it is also responsible for regulating the acidic tumor microenvironment, which could induce cancer cells to secrete pro-angiogenesis factors, such as interleukin-8 and vascular endothelial growth factor and is therefore beneficial to tumor angiogenesis and growth. Another protein, filamin A-interacting protein 1-like (FILIP1L), differs from ATP6L in that overexpression of FILIP1L in CRC cells inhibits the WNT signaling pathway, thereby inhibiting EMT. Xin Ku et al. used mass spectrometry to compare the proteomic profiles of CRC patients (n=9) and found that in CRLM, FILIP1L expression was significantly lower than that in CRC [103]. This result also suggests stronger EMT activity in CRLM.
4.2. Other proteins
In addition to FILIP1L, Xin Ku et al. also identified the remaining 46 differentially expressed proteins, by further ANOVA (Tukey test), they identified plasminogen (PLG), a protein that was most up-regulated [103]. Plasmin is required by CRC cells to hydrolyze the extracellular matrix, and PLG is involved in the plasminogen activation system (PAS), meaning that the upregulation of PLG expression allows CRC cells to undergo distant metastasis through the extracellular matrix. Eun-Kyung Kim et al. performed mass spectrometry on five CRLM patients and validated the results by western blotting (WB) [104]. Out of 164 proteins, they observed reduced expression of 51 proteins and increased expression of 7 proteins. The reduced proteins were mainly in the mitochondrial matrix, the mitochondrial intermembrane space, the proteasome complex, and the actin cytoskeleton and play a role in protein and ATP synthesis and actin dynamics. Thus, actin dynamics, protein degradation, and ATP synthesis are reduced in CRLM compared to CRC. In contrast, the seven proteins with increased expression were mainly serpin family A member 1 (SERPINA1), apolipoprotein A1 (APOA1A), carbonic anhydrase 1 (CA1), and succinate dehydrogenase complex flavoprotein subunit A (SDHA). Serpin A1 is a protease inhibitor that is regulated by the Snail protein and can promote tumor cell invasion and metastasis. It has been found in many studies to be elevated in serum in patients with a variety of tumors, including ovarian, gastric, and cervical cancers [105,106,107]. SDHA is considered to be a tumor suppressor, and its loss of function is associated with the development of kidney cancer and breast cancer [108,109], but its upregulation in CRLM is intriguing, and perhaps it plays an opposite role in CRC.
Only after EMT can tumor cells undergo distant metastasis, and during the process of EMT, a large number of proteins change to support the transformation and metastasis of tumor cells, and thus these cells also show significant heterogeneity after metastasis. In the following table (Table 3), we summarize the protein heterogeneity of CRLM, most of which promote CRLM by affecting EMT, including increased expression of migration-related proteins, decreased expression of adhesion-related proteins, or acting by affecting EMT-related pathways.
5. Metabolic heterogeneity
Metabolic reprogramming is one of the hallmarks of cancer [110]. Compared with normal tissues, tumor cells often require more energy to maintain their growth. Due to the different microenvironments of metastatic organs, tumor cells still need to undergo metabolic reprogramming to obtain energy for growth in different metastatic organs.
Tumor cells are often in a state of aerobic glycolysis, the so-called "Warburg effect"——Even with sufficient oxygen, cells prioritize glycolysis to quickly generate energy rather than through the tricarboxylic acid cycle (TCA cycle). After CRLM, some specific growth factors and enzymes in the liver make this effect more obvious in the metastatic lesions. The expression of glucose transporter 3 (GLUT3) and pyruvate kinase muscle isozyme 2 (PKM2) is also significantly higher in CRLM [111]. Increased glucose uptake mediated by GLUT3 can promote the occurrence of various tumors, including liver cancer, breast cancer, and lung cancer [112,113,114]. The overexpression of GLUT3 activates Yes-associated protein (YAP), which in turn promotes the expression of GLUT3 and glycolytic genes; conversely, the expression of GLUT3 and glycolytic genes is decreased after YAP is knocked down. Meanwhile, YAP also interacts with PKM2 through the WW domain and together enhance the expression of GLUT3. GLUT3 and YAP/PKM2 constitute a positive feedback pathway that enhances glycolysis in CRLM [111]. Some studies have also reported the mechanism of GLUT3 upregulation in CRLM. High mobility group proteins (HMGs) are a class of structural transcription factors that do not have transcriptional activity, but they can regulate the transcription of target genes by binding with their structures. Meijing Yang et al. found that HMGA1 can promote the expression of GLUT3 in CRLM, thereby enhancing the GLUT3-YAP signaling pathway [115].
Next, the expression of phosphorylated PKM2 is higher in CRLM than in CRC, and it can act as a transcriptional cofactor for hypoxia-inducing factor 1 (HIF-1), thus promoting the expression of glycolytic genes, including LDHA, PDK1, and SLC2A1 (GLUT1) [116]. In addition to the two proteins PKM2 and GLUT3, Fengliu Deng et al. also identified another differentially expressed protein, dickkopf-associated protein 2 (DKK2), which promotes aerobic glycolysis in CRC cells [117]. By comparing the proteomes of CRC and CRLM from seven patients, Fahrner et al. also found that most of the proteins upregulated in CRLM were involved in glucose metabolism, including pyruvate carboxylase, fructose-bisphosphate aldolase B, and fructose-1,6-bisphosphatase 1 [118]. Finally, according to Bu et al., the expression of aldolase B (ALDOB), an enzyme involved in fructose metabolism, is increased in CRLM, and overexpressed ALDOB enhances fructose metabolism, thereby generating more propanose phosphate [119]. The production of large amounts of propyl phosphate also promotes glycolysis in CRC cells.
In addition, enhanced cholesterol synthesis and upregulated expression of some fatty acids, acylcarnitines, and polyamines have also been found in CRLM. As described above, SREBP2 is a key transcription factor for lipid synthesis. Kai-Li Zhang et al. found that the expression of SREBP2 and its downstream target genes LDLR and SRB1 were significantly upregulated in CRLM [120]. These authors subsequently knocked down SREBP2 and found that total cholesterol levels in tumor cells were significantly reduced and tumor cell growth was restricted. After screening several liver-rich growth factors, they finally found that hepatocyte growth factor (HGF) in the liver promotes the PI3K/AKT/mTOR pathway, which stimulates SREBP2 and thus stimulates cholesterol synthesis in CRLM [120]. Finally, Williams et al. also found that several phosphatidylcholines, carnitine, bile acids, nucleotides, oxidative compounds (glutathione), and polyamines (putrescine) were significantly more highly expressed in CRLM than in CRC [121]. Glutathione (GSH) protects cells against oxidative stress and polyamines are important growth factors required for cell growth.
Taken together, we summarize in the following table (Table 4) the changes in metabolic reprogramming of CRLM that allow CRC cells to adapt more quickly to the metabolic state of the liver, thereby promoting their growth in the liver. From the table, we can find that glycolysis-related heterogeneity is the most obvious, probably because glycolysis can generate a large amount of energy, and it provides energy in the process of CRC cell metastasis and colonization in the liver. Of course, the rest of the metabolites also upregulate and promote the growth of CRC cells. From our conclusion, we can see that CRLM possesses a more active metabolic state to maintain cell growth compared to CRC.
6. Immune heterogeneity
The immune microenvironment of tumors is a complex system, and immune cells in the microenvironment have been shown to influence tumor progression and response to immunotherapy [122,123]. Current research on the heterogeneity of the immune microenvironment is still mainly focused on immune cells. During tumor metastasis, immune cells are dynamically heterogeneous, which means that cell types, numbers, and sizes change [124]. It is thought that CRLM contains more immunosuppressive cells than CRC.
6.1. Macrophages
Tumor-associated macrophages (TAMs) are closely associated with tumor progression and angiogenesis [125]. SPP1+ TAMs are immunosuppressive cells that have been previously reported to be highly expressed in CRC compared to normal tissues, which can promote CRC progression and metastasis and are associated with the prognosis and response to immunotherapy in CRC patients [126,127]. Yedan Liu et al. found that SPP1+ TAMs are malignancy-associated and are linked to CRLM [128]. They also compared the angiogenesis and phagocytosis properties of three types of TAMs, MKI67+ TAMs, SPP1+ TAMs, and C1QC+ TAMs in the context of CRC. The results revealed that SPP1+ TAMs possessed the strongest angiogenic function, which confirmed their immunosuppressive and protumorigenic functions. Yingcheng Wu et al. used single-cell RNA sequencing and spatial transcriptomics to determine 97 CRC paired samples and derived a single-cell spatial map of CRLM [3]. The results demonstrated that MRC1+ CCL18+ TAMs, SPP1+ TAMs, and neutrophils were significantly increased in CRLM compared to matched CRC. Neutrophils have been reported to be potential tumor-promoting cells [129]. Yingcheng Wu et al. focused their research on MRC1+ CCL18+ TAMs. They suggested that MRC1+ CCL18+ TAMs might originate from Kupffer cells in the liver and found that M2 polarization-related genes (APOE, MARCO) were significantly upregulated in MRC1+ CCL18+ TAMs of CRLM, while MRC1+ CCL18+ TAMs of CRC showed higher expression of inflammatory cytokines (TNF, IL1B, CCL3, and CCL4). In addition, they found that the MRC1+ CCL18+ TAMs of CRLM possessed strong metabolic activity, mainly in terms of phenylalanine metabolism, whereas the MRC1+ CCL18+ TAMs of CRC were dominated by oxidative phosphorylation. Moreover, both SPP1+ TAMs and MRC1+ CCL18+ TAMs showed enhanced antigen processing and presentation and complex activity. Wei Tu et al. also found more TAM enrichment in CRLM and dominance of M2 TAMs [130]. They found that this phenomenon was associated with elevated expression of TCF4 in CRLM. TCF4, a transcription factor involved in the WNT/TCF signaling pathway, recruits TAMs and promotes TAMs M2 polarization mainly by promoting the expression of two monocyte chemokines, CCL2 and CCR2.
In addition, earlier studies have shown that macrophages are morphologically heterogeneous. For example, M1-like macrophages are often round or flat, whereas M2-like macrophages are elongated [131,132], and macrophages acquire different geometries in different tissues [133,134], so it is conceivable that during CRC metastasis to the liver, macrophages change not only in type and gene expression but also in morphology. Matteo Donadon et al. investigated this phenomenon and found a significant increase in the area and circumference of macrophages in CRLM, which they termed large (L-TAMs) macrophages [135]. These L-TAMs have a strong lipid metabolizing capacity, while inflammation-related pathways (leukocyte extravasation, acute phase response, and NF-κB signaling) are downregulated. Finally, both complement-related pathways and their genes were highly expressed in these L-TAMs, a result that is consistent with the findings of Wu [3].
6.2. T cells
T cells play an important role in tumor progression and metastasis. Cytotoxic T cells can secrete granzyme and perforin to kill tumor cells, while regulatory T cells (Tregs) can suppress the immune response and promote tumor cell development [136]. During tumor progression, large numbers of CD4+ T cells and CD8+ T cells are usually depleted, and studies have demonstrated that during the course of CRLM the number of these two cells is significantly reduced, and more CD4+FOXP3+ Tregs are found instead in liver metastasis [137,138]. In addition, two other Tregs (Treg-IL10 and Treg-CTLA4) are also enriched in CRLM. Treg-IL10 show high expression of IL10, IL23, and IL1R1. The role of IL10 in tumors is controversial, as on the one hand, it can inhibit the function of antigen-presenting cells and block T-cell killing function against tumors, while on the other hand, it can inhibit angiogenic factors and activate CD8+ T cells [139,140]. The chronic inflammation-associated cytokine IL23 can promote tumor progression [141]. Treg-CTLA4 is highly expressed in Treg activation-related factors, including LAYN, CCR8, and TIGIT.
6.3. Dendritic cells
Dendritic cells (DCs) can be divided into plasmacytoid DCS (pDCs) and conventional DCS (cDCs) in both human and mouse [142]. cDCs can be further divided into two phenotypically and functionally distinct subsets. cDC1 expresses Toll-like receptors (TLR) and secretes pro-inflammatory cytokines, including IL-12p70 and IFN-α, to induce Th1 responses. cDC2 mainly acts as an antigen presenting cell and activates effector T cells, including Th2 and Th17 [142,143]. At present, there are few studies on the heterogeneity of dendritic cells in CRLM. Yedan Liu et al. identified 10 DC subsets in CRLM patients, and they found great heterogeneity in two types of cDC2 (cDC2-C1QC and cDC2-TIMP1) [128]. Because cDC2-C1QC was highly expressed in C1QA, CD68, CD163, and CD14, similar to the recently identified DC3 population [144,145], it was identified as DC3s. cDC2-C1QC showed a higher proinflammatory profile. cDC2-TIMP1 revealed a high expression of maturation markers (such as CCR7) and angiogenesis-related genes (EREG, CREM, and VEGFA). cDC2-TIMP1 expressed more anti-inflammatory genes compared to DC3s. The results revealed that cDC2-C1QC was enriched in CRC, in contrast to cDC2-TIMP1, which was more abundant in CRLM.
Immune cells play a huge role in the immune response to tumors, and different types of immune cells either kill tumor cells or promote their immune escape. In the following table (Table 5), we summarize the types of cells with the most pronounced heterogeneity in CRLM, in which we can find immunosuppressive cells occupying the majority. Compared to colorectal, the accumulation of these immunosuppressive cells in the liver makes the probability of immune escape from the tumor greater and reduces the response of CRLM to immunotherapy.
7. Discussion
Due to the complex chain reaction in vivo, tumor progression and metastasis must involve changes in many biological processes and their corresponding factors. CRC has a high incidence rate and often metastasizes to the liver, so it is particularly important to clarify the heterogeneity between primary tumors and tumor metastasis. In this review, we summarize the heterogeneity between CRC and CRLM at the genetic, transcriptomic, protein, metabolic, and immune levels.
As shown in Table 1, Table 2, Table 3, Table 4 and Table 5, heterogeneity is evident at all 4 levels except for genetic heterogeneity. Although the differentially expressed noncoding RNAs, transcription factors, and proteins are diverse and each performs different functions, the vast majority of them promote CRC progression and metastasis by promoting EMT and angiogenesis(Figure 1). Enhanced glycolysis, fatty acid synthesis, and other processes also provide liver metastatic cells with more energy to sustain growth. In addition, a decrease in the number of cytotoxic cells, such as CD4+ T cells and CD8+ T cells, can also be found in the liver, which is replaced by more immunosuppressive cells, such as Tregs and TAMs(Figure 2).
It is now certain that intratumor heterogeneity is a major challenge in the clinical treatment of oncology patients. Thus, as shown in Table 1, Table 2, Table 3, Table 4 and Table 5, the heterogeneity reflected in these five levels may serve as new molecular biological markers and new targets for the treatment of CRC patients, providing new therapeutic directions for the clinical treatment of CRLM patients.
First, targeting the EMT process in CRC is also a hot topic in current clinical research, and targeting EMT-related genes, RNAs and proteins can be a way to inhibit EMT. Nan Zhang et al. have detailed a summary of current drugs that target EMT, such as fresolimumab, a monoclonal antibody targeting TGF-β, and regorafenib, which targets factors such as BRAF and VEGF [146]. These drugs can improve the prognosis of CRLM patients when used as adjuvant therapy or chemopreventive agents.
Targeted metabolic approaches have also been shown to be highly effective in cancer treatment, including targeting aerobic glycolysis to inhibit glucose uptake by tumor cells and targeting fatty acid synthesis and amino acid metabolism [147]. Bu et al. also provided a new direction for targeting metabolism by targeting fructose metabolism [119]. Similarly, it is clear that increased GSH in CRLM is associated with tumor progression and drug resistance. Increased GSH can lead to drug resistance in CRC cells by binding to drugs, interacting with reactive oxygen species, preventing protein or DNA damage, or participating in DNA repair processes [148,149]. Therefore, GSH is a potential therapeutic target for CRC patients. Studies have demonstrated that GSH depletion therapy combined with reactive oxygen species-based therapy (photodynamic therapy (PDT), sonodynamic therapy (SDT), and chemodynamic therapy (CDT)) may improve the therapeutic effect for CRLM patients [150].
Finally, the liver has a unique immunosuppressive environment [151]. Table 5 shows that CRLM contains more immunosuppressive macrophages, and the infiltration of CD8+ T cells and CD4+ T cells is also significantly reduced, which indicates that liver metastasis aggravate the immunosuppressive microenvironment of the liver and reduce the antitumor immune response. Thus, the application effect of immune checkpoint inhibitors (such as PD-1/PD-L1 inhibitors) in patients with CRLM is weakened. A recent study also found that this immunosuppressive effect of the liver is achieved by Tregs, and CTLA-4 inhibitors can inhibit the effect of Tregs, so the combination of CTLA-4 inhibitors and PD-1 inhibitors has a significantly better antitumor effect than PD-1 inhibitors alone [152]. This conclusion is supported by the increase in Treg-CTLA4 shown in Table 5. Therefore, the addition of CTLA-4 inhibitors is a good choice for the treatment of CRLM patients. In addition, Wu et al. found that neoadjuvant chemotherapy (NAC) can inhibit the activity of MRC1+ CCL18+ TAMs and SPP1+ TAMs, thereby enhancing the response of CRLM to immunotherapy [3]. These authors also proposed new clinical combination regimens, such as combining NAC, metabolism checkpoint inhibitors, and immunotherapy. Targeting these immunosuppressive cells in combination with immunotherapy may become the primary treatment modality for patients with CRLM in the future.
In conclusion, in this review we have summarized the intratumor heterogeneity between CRC and CRLM. The clinical effects of these heterogeneities need further investigation and may hold promise as new targets for the treatment of CRLM patients.
Author Contributions
Conceptualization, H.C. and J.S. ; writing—original draft preparation, H.C. and C.Z. ; writing—review and editing, J.S. and W.H. ; visualization, X.X. ; supervision, H.W. . All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y. , Brocks, D. & Gerhauser, C. Intratumor heterogeneity in epigenetic patterns. Semin Cancer Biol 2018, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov 2022, 12, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Yu, X. et al. Emerging Role of Immunotherapy for Colorectal Cancer with Liver Metastasis. Onco Targets Ther 2020, 13, 11645–11658. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. et al. Chinese guidelines for the diagnosis and comprehensive treatment of colorectal liver metastases (version 2018). J Cancer Res Clin Oncol 2019, 145, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K. et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, F. , Barbolini, M., Spallanzani, A., Pugliese, G. & Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat Rev 2016, 51, 19–26. [Google Scholar] [CrossRef]
- Willis, J. A. , Reyes-Uribe, L., Chang, K., Lipkin, S. M. & Vilar, E. Immune Activation in Mismatch Repair-Deficient Carcinogenesis: More Than Just Mutational Rate. Clinical Cancer Research : an Official Journal of the American Association For Cancer Research 2020, 26, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, M. et al. Metastatic site as a predictor of nivolumab efficacy in patients with advanced non-small cell lung cancer: A retrospective multicenter trial. PLoS One 2018, 13, e0192227. [Google Scholar] [CrossRef]
- Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Hou, Y. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016, 26, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S. D. & Bertagnolli, M. M. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Sagaert, X. , Vanstapel, A. & Verbeek, S. Tumor Heterogeneity in Colorectal Cancer: What Do We Know So Far? Pathobiology 2018, 85, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Koulouridi, A. et al. Prognostic Value of Mutations in Colorectal Cancer Patients. Cancers (Basel) 2022, 14, 3320. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A. et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Santos, C. et al. Phase II study of high-sensitivity genotyping of KRAS, NRAS, BRAF and PIK3CA to ultra-select metastatic colorectal cancer patients for panitumumab plus FOLFIRI: the ULTRA trial. Ann Oncol 2019, 30, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F. et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Brunsell, T. H. et al. High Concordance and Negative Prognostic Impact of RAS/BRAF/PIK3CA Mutations in Multiple Resected Colorectal Liver Metastases. Clin Colorectal Cancer 2020, 19, e26–e47. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S. E. et al. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 2010, 16, 790–799. [Google Scholar] [CrossRef]
- Knijn, N. et al. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer 2011, 104, 1020–1026. [Google Scholar] [CrossRef]
- Brannon, A. R. et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E. et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Hou, J. , Zhang, Y. & Zhu, Z. Gene heterogeneity in metastasis of colorectal cancer to the lung. Semin Cell Dev Biol 2017, 64, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tie, J. et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res 2011, 17, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L. , Vanhaesebroeck, B. & Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nature Reviews. Clinical Oncology 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondón, N. , Villegas, V. E. & Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers (Basel) 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A. et al. Intra-patient Inter-metastatic Genetic Heterogeneity in Colorectal Cancer as a Key Determinant of Survival after Curative Liver Resection. PLoS Genet 2016, 12, e1006225. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, K. et al. Copy number and transcriptome alterations associated with metastatic lesion response to treatment in colorectal cancer. Clin Transl Med 2021, 11, e401. [Google Scholar] [CrossRef]
- Mogensen, M. B. et al. Genomic alterations accompanying tumour evolution in colorectal cancer: tracking the differences between primary tumours and synchronous liver metastases by whole-exome sequencing. BMC Cancer 2018, 18, 752. [Google Scholar] [CrossRef]
- Mamlouk, S. et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat Commun 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M. & Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002, 2, 161–174. [Google Scholar] [PubMed]
- Saad, R. S. , Ghorab, Z., Khalifa, M. A. & Xu, M. CDX2 as a marker for intestinal differentiation: Its utility and limitations. World J Gastrointest Surg 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Mekenkamp, L. J. M. et al. Chromosomal copy number aberrations in colorectal metastases resemble their primary counterparts and differences are typically non-recurrent. PLoS One 2014, 9, e86833. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S. et al. Identification of chromosomal aberrations of metastatic potential in colorectal carcinoma. Genes Chromosomes Cancer 2010, 49, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Jones, S. et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [PubMed]
- Jass, J. R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef] [PubMed]
- He, W.-Z. et al. Comparison of Mismatch Repair Status Between Primary and Matched Metastatic Sites in Patients With Colorectal Cancer. J Natl Compr Canc Netw 2019, 17, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Evrard, C. et al. Heterogeneity of Mismatch Repair Status and Microsatellite Instability between Primary Tumour and Metastasis and Its Implications for Immunotherapy in Colorectal Cancers. International Journal of Molecular Sciences 2022, 23, 4427. [Google Scholar] [CrossRef]
- Jung, J. et al. Comparison of the Mismatch Repair System between Primary and Metastatic Colorectal Cancers Using Immunohistochemistry. J Pathol Transl Med 2017, 51, 129–136. [Google Scholar] [CrossRef]
- Haraldsdottir, S. et al. Mismatch repair deficiency concordance between primary colorectal cancer and corresponding metastasis. Fam Cancer 2016, 15, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lim, L. P. et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. X. & Rothenberg, M. E. MicroRNA. J Allergy Clin Immunol 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, H. et al. Circulating microRNA-1290 as a novel diagnostic and prognostic biomarker in human colorectal cancer. Ann Oncol 2016, 27, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Nassar, F. J. et al. Circulating miRNA as Biomarkers for Colorectal Cancer Diagnosis and Liver Metastasis. Diagnostics (Basel) 2021, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Vychytilova-Faltejskova, P. et al. Genome-wide microRNA Expression Profiling in Primary Tumors and Matched Liver Metastasis of Patients with Colorectal Cancer. Cancer Genomics Proteomics 2016, 13, 311–316. [Google Scholar] [PubMed]
- Hur, K. et al. Identification of a metastasis-specific MicroRNA signature in human colorectal cancer. J Natl Cancer Inst 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T. et al. Identifying the key genes and microRNAs in colorectal cancer liver metastasis by bioinformatics analysis and in vitro experiments. Oncol Rep 2019, 41, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-C. et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology 2009, 49, 1571–1582. [Google Scholar] [CrossRef]
- Thakral, S. & Ghoshal, K. miR-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr Gene Ther 2015, 15, 142–150. [Google Scholar]
- Lam, C. S.-C. et al. Identification of microRNA 885-5p as a novel regulator of tumor metastasis by targeting CPEB2 in colorectal cancer. Oncotarget 2017, 8, 26858–26870. [Google Scholar] [CrossRef] [PubMed]
- Su, M. , Qin, B., Liu, F., Chen, Y. & Zhang, R. miR-885-5p upregulation promotes colorectal cancer cell proliferation and migration by targeting suppressor of cytokine signaling. Oncol Lett 2018, 16, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Pizzini, S. et al. Impact of microRNAs on regulatory networks and pathways in human colorectal carcinogenesis and development of metastasis. BMC Genomics 2013, 14, 589. [Google Scholar] [CrossRef] [PubMed]
- Song, J. J. & Li, W. MiR-10b suppresses the growth and metastasis of colorectal cancer cell by targeting FGF13. Eur Rev Med Pharmacol Sci 2019, 23, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Hur, K. et al. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut 2017, 66, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Hur, K. et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2013, 62, 1315–1326. [Google Scholar] [CrossRef]
- Furuta, M. et al. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y. et al. miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J Biol Chem 2014, 289, 529–539. [Google Scholar] [CrossRef]
- Torres, S. et al. Combined miRNA profiling and proteomics demonstrates that different miRNAs target a common set of proteins to promote colorectal cancer metastasis. J Pathol 2017, 242, 39–51. [Google Scholar] [CrossRef]
- Vo, J. N. et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176. [Google Scholar] [CrossRef]
- Capel, B. et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Greene, J. et al. Circular RNAs: Biogenesis, Function and Role in Human Diseases. Front Mol Biosci 2017, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Guo, Y. et al. Circ3823 contributes to growth, metastasis and angiogenesis of colorectal cancer: involvement of miR-30c-5p/TCF7 axis. Mol Cancer 2021, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. et al. The circular RNA 001971/miR-29c-3p axis modulates colorectal cancer growth, metastasis, and angiogenesis through VEGFA. J Exp Clin Cancer Res 2020, 39, 91. [Google Scholar] [CrossRef] [PubMed]
- Long, F. et al. Comprehensive landscape and future perspectives of circular RNAs in colorectal cancer. Mol Cancer 2021, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Xu, H. , Wang, C., Song, H., Xu, Y. & Ji, G. RNA-Seq profiling of circular RNAs in human colorectal Cancer liver metastasis and the potential biomarkers. Mol Cancer 2019, 18, 8. [Google Scholar] [CrossRef]
- Chen, R.-X. et al. N-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun 2019, 10, 4695. [Google Scholar] [CrossRef]
- Zhang, C. et al. Circular RNA hsa_circ_0006401 promotes proliferation and metastasis in colorectal carcinoma. Cell Death Dis 2021, 12, 443. [Google Scholar] [CrossRef]
- Li, Y. et al. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett 2014, 355, 130–140. [Google Scholar] [CrossRef]
- Zhao, S. et al. LncRNA MIR17HG promotes colorectal cancer liver metastasis by mediating a glycolysis-associated positive feedback circuit. Oncogene 2021, 40, 4709–4724. [Google Scholar] [CrossRef]
- Li, B. et al. LncRNA GAL promotes colorectal cancer liver metastasis through stabilizing GLUT1. Oncogene 2022, 41, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S. A. et al. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J. H. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Chi, P. et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 2010, 467, 849–853. [Google Scholar] [CrossRef]
- Jané-Valbuena, J. et al. An oncogenic role for ETV1 in melanoma. Cancer Res 2010, 70, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. et al. Death Domain-Associated Protein Promotes Colon Cancer Metastasis through Direct Interaction with ZEB1. J Cancer 2020, 11, 750–758. [Google Scholar] [CrossRef]
- Klupp, F. et al. Expressional STAT3/STAT5 Ratio is an Independent Prognostic Marker in Colon Carcinoma. Ann Surg Oncol, 2015, 22 Suppl 3, S1548-S1555. [CrossRef]
- Malilas, W. et al. Cancer upregulated gene 2, a novel oncogene, enhances migration and drug resistance of colon cancer cells via STAT1 activation. Int J Oncol 2013, 43, 1111–1116. [Google Scholar] [CrossRef]
- Colomiere, M. et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef]
- He, G. & Karin, M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Kamal, Y. , Schmit, S. L., Hoehn, H. J., Amos, C. I. & Frost, H. R. Transcriptomic Differences between Primary Colorectal Adenocarcinomas and Distant Metastases Reveal Metastatic Colorectal Cancer Subtypes. Cancer Res 2019, 79, 4227–4241. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G. N. & Li, W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S. , Xu, J. & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Yin, X. et al. Large scale systematic proteomic quantification from non-metastatic to metastatic colorectal cancer. Sci Rep 2015, 5, 12120. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. et al. THBS1 facilitates colorectal liver metastasis through enhancing epithelial-mesenchymal transition. Clin Transl Oncol 2020, 22, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y. K. et al. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum Pathol 2013, 44, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Hope, C. et al. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J Immunol 2017, 199, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Song, G. et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J Exp Clin Cancer Res 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. et al. Periostin expression and its prognostic value for colorectal cancer. International Journal of Molecular Sciences 2015, 16, 12108–12118. [Google Scholar] [CrossRef]
- Yu, J. et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut 2015, 64, 636–645. [Google Scholar] [CrossRef]
- Li, Y. et al. Downregulated IGFBP7 facilitates liver metastasis by modulating epithelial-mesenchymal transition in colon cancer. Oncol Rep 2019, 42, 1935–1945. [Google Scholar] [CrossRef]
- Hamidi, H. & Ivaska, J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M. B. , Dhawan, P. & Baumert, T. F. Tight junction proteins in gastrointestinal and liver disease. Gut 2019, 68, 547–561. [Google Scholar] [CrossRef]
- Wang, K. et al. Claudin-7 downregulation induces metastasis and invasion in colorectal cancer via the promotion of epithelial-mesenchymal transition. Biochem Biophys Res Commun 2019, 508, 797–804. [Google Scholar] [CrossRef]
- Georges, R. et al. Sequential biphasic changes in claudin1 and claudin4 expression are correlated to colorectal cancer progression and liver metastasis. J Cell Mol Med 2012, 16, 260–272. [Google Scholar] [CrossRef]
- Tian, T. et al. Investigation of the role and mechanism of ARHGAP5-mediated colorectal cancer metastasis. Theranostics 2020, 10, 5998–6010. [Google Scholar] [CrossRef]
- Wang, B. et al. MYH9 Promotes Growth and Metastasis via Activation of MAPK/AKT Signaling in Colorectal Cancer. J Cancer 2019, 10, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. et al. RUNX1 promotes tumour metastasis by activating the Wnt/β-catenin signalling pathway and EMT in colorectal cancer. J Exp Clin Cancer Res 2019, 38, 334. [Google Scholar] [CrossRef] [PubMed]
- Tang, B. et al. PEA15 promotes liver metastasis of colorectal cancer by upregulating the ERK/MAPK signaling pathway. Oncol Rep 2019, 41, 43–56. [Google Scholar] [CrossRef]
- Cotter, K. et al. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J Biol Chem 2015, 290, 3680–3692. [Google Scholar] [CrossRef]
- Wang, J. et al. ATP6L promotes metastasis of colorectal cancer by inducing epithelial-mesenchymal transition. Cancer Sci 2020, 111, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Ku, X. et al. In-Depth Characterization of Mass Spectrometry-Based Proteomic Profiles Revealed Novel Signature Proteins Associated with Liver Metastatic Colorectal Cancers. Anal Cell Pathol (Amst) 2019, 2019, 7653230. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-K. , Song, M.-J., Jung, Y., Lee, W.-S. & Jang, H. H. Proteomic Analysis of Primary Colon Cancer and Synchronous Solitary Liver Metastasis. Cancer Genomics Proteomics 2019, 16, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C. H. et al. Serpin peptidase inhibitor clade A member 1 is a biomarker of poor prognosis in gastric cancer. Br J Cancer 2014, 111, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Normandin, K. et al. Protease inhibitor SERPINA1 expression in epithelial ovarian cancer. Clin Exp Metastasis 2010, 27, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, B. et al. The effect of combined therapy on activity of cathepsin D and alpha-1-antitrypsin in the blood serum of women with cervical cancer. Eur J Gynaecol Oncol 2008, 29, 617–619. [Google Scholar] [PubMed]
- Kim, S. , Kim, D. H., Jung, W.-H. & Koo, J. S. Succinate dehydrogenase expression in breast cancer. Springerplus 2013, 2, 299. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C. et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kuo, C.-C. et al. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics 2019, 9, 2526–2540. [Google Scholar] [CrossRef]
- Kuo, M.-H. et al. Glucose Transporter 3 is Essential for the Survival of Breast Cancer Cells in the Brain. Cells 2019, 8, 1568. [Google Scholar] [CrossRef] [PubMed]
- Gao, H. et al. Prognostic value of glucose transporter 3 expression in hepatocellular carcinoma. Oncol Lett 2020, 19, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Ali, A. et al. CAV1 - GLUT3 signaling is important for cellular energy and can be targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics 2019, 9, 6157–6174. [Google Scholar] [CrossRef] [PubMed]
- Yang, M. , Guo, Y., Liu, X. & Liu, N. HMGA1 Promotes Hepatic Metastasis of Colorectal Cancer by Inducing Expression of Glucose Transporter 3 (GLUT3). Med Sci Monit 2020, 26, e924975. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-J. et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism. Proc Natl Acad Sci U S A 2014, 111, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Deng, F. et al. Tumor-secreted dickkopf2 accelerates aerobic glycolysis and promotes angiogenesis in colorectal cancer. Theranostics 2019, 9, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M. , Bronsert, P., Fichtner-Feigl, S., Jud, A. & Schilling, O. Proteome biology of primary colorectal carcinoma and corresponding liver metastases. Neoplasia 2021, 23, 1240–1251. [Google Scholar] [CrossRef]
- Bu, P. et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-L. et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics 2021, 11, 6560–6572. [Google Scholar] [CrossRef]
- Williams, M. D. et al. Characterizing metabolic changes in human colorectal cancer. Anal Bioanal Chem 2015, 407, 4581–4595. [Google Scholar] [CrossRef]
- Halama, N. et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 2011, 71, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S. K. & Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I. , Shema, E., Loi, S. & Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H. et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP fibroblasts and SPP1 macrophages in colorectal cancer. Nat Commun 2022, 13, 1742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. et al. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Letters 2020, 470, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 2022, 40. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R. et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50. [Google Scholar] [CrossRef] [PubMed]
- Tu, W. , Gong, J., Zhou, Z., Tian, D. & Wang, Z. TCF4 enhances hepatic metastasis of colorectal cancer by regulating tumor-associated macrophage via CCL2/CCR2 signaling. Cell Death Dis 2021, 12, 882. [Google Scholar] [CrossRef]
- Ballotta, V. , Driessen-Mol, A., Bouten, C. V. C. & Baaijens, F. P. T. Strain-dependent modulation of macrophage polarization within scaffolds. Biomaterials 2014, 35, 4919–4928. [Google Scholar] [CrossRef]
- Waldo, S. W. et al. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol 2008, 172, 1112–1126. [Google Scholar] [CrossRef]
- Geissmann, F. , Gordon, S., Hume, D. A., Mowat, A. M. & Randolph, G. J. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol 2010, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Yona, S. & Gordon, S. From the Reticuloendothelial to Mononuclear Phagocyte System - The Unaccounted Years. Front Immunol 2015, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Donadon, M. et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y. & Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int Immunol 2016, 28, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. et al. Changes of T cells and cytokines TGF-β1 and IL-10 in mice during liver metastasis of colon carcinoma: implications for liver anti-tumor immunity. J Gastrointest Surg 2013, 17, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. et al. Increase in CD4FOXP3 regulatory T cell number and upregulation of the HGF/c-Met signaling pathway during the liver metastasis of colorectal cancer. Oncol Lett 2020, 20, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M. H. et al. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Neurath, M. F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev 2019, 45, 1–8. [Google Scholar] [CrossRef]
- Tian, Y. , Guo, X., Wu, T., Fei, K. & Wu, L. Identification of a novel cDC2-committed progenitor within mouse common dendritic cell progenitor population. Protein Cell 2022, 13, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y. S. et al. Human CD141 dendritic cells (cDC1) are impaired in patients with advanced melanoma but can be targeted to enhance anti-PD-1 in a humanized mouse model. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Cytlak, U. et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53. [Google Scholar] [CrossRef] [PubMed]
- Bourdely, P. et al. Transcriptional and Functional Analysis of CD1c Human Dendritic Cells Identifies a CD163 Subset Priming CD8CD103 T Cells. Immunity 2020, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. et al. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol 2021, 22, e358–e368. [Google Scholar] [CrossRef]
- Stine, Z. E. , Schug, Z. T., Salvino, J. M. & Dang, C. V. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Bansal, A. & Simon, M. C. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N. et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Niu, B. et al. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 2021, 277, 121110. [Google Scholar] [CrossRef]
- Li, X. et al. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef]
- Lee, J. C. et al. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol 2020, 5. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Different noncoding RNAs, transcription factors, and proteins affect EMT processes in colorectal cancer cells through different mechanisms.(↑:up-regulate; ↓:down-regulate; ←:promote; ⊢:inhibit.).
Figure 1.
Different noncoding RNAs, transcription factors, and proteins affect EMT processes in colorectal cancer cells through different mechanisms.(↑:up-regulate; ↓:down-regulate; ←:promote; ⊢:inhibit.).
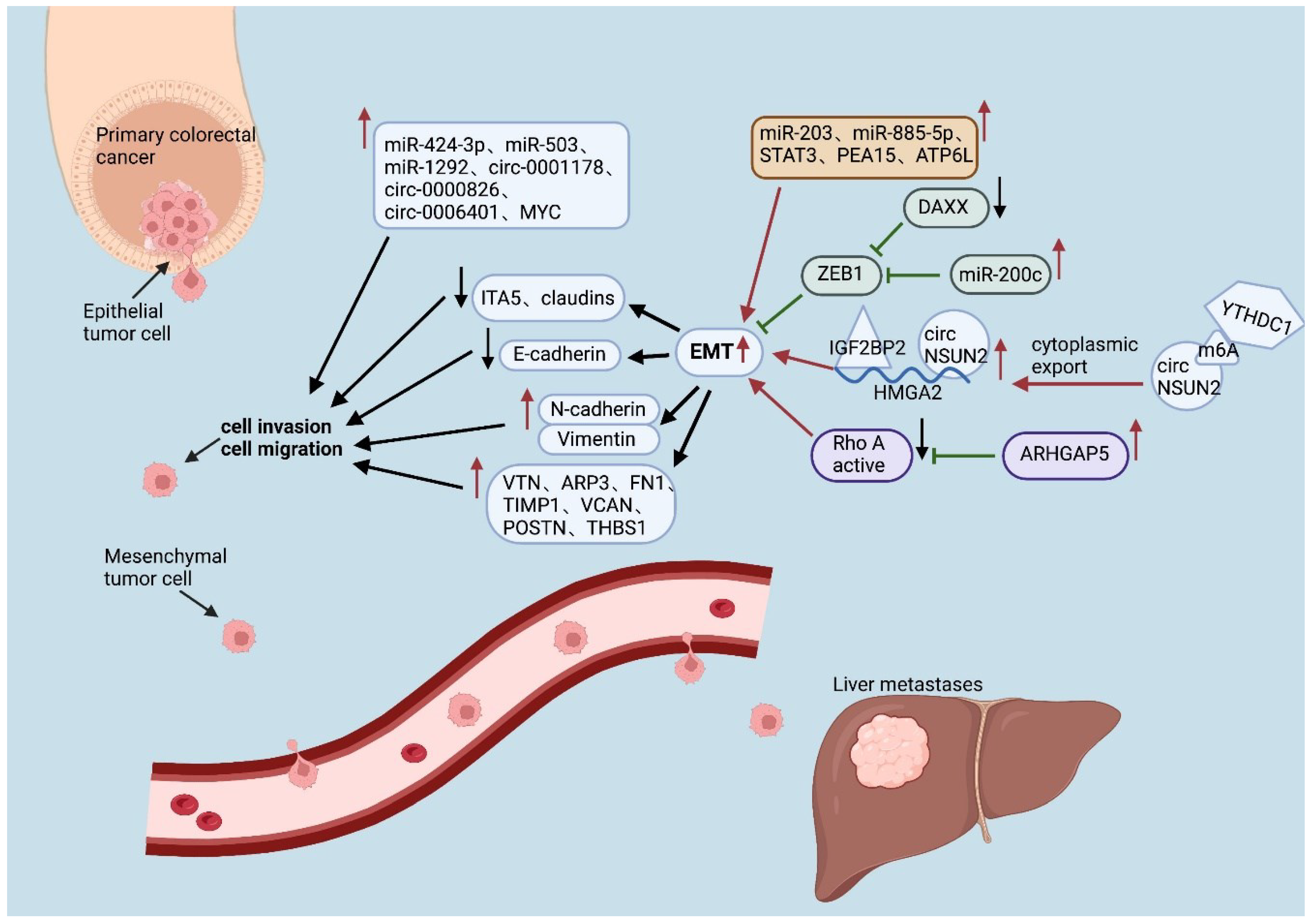
Figure 2.
The enhanced metabolic and immunosuppressive microenvironment in the liver provides better survival conditions for CRLM. (↑:up-regulate; ↓:down-regulate; ←:promote; ⊢:inhibit.).
Figure 2.
The enhanced metabolic and immunosuppressive microenvironment in the liver provides better survival conditions for CRLM. (↑:up-regulate; ↓:down-regulate; ←:promote; ⊢:inhibit.).
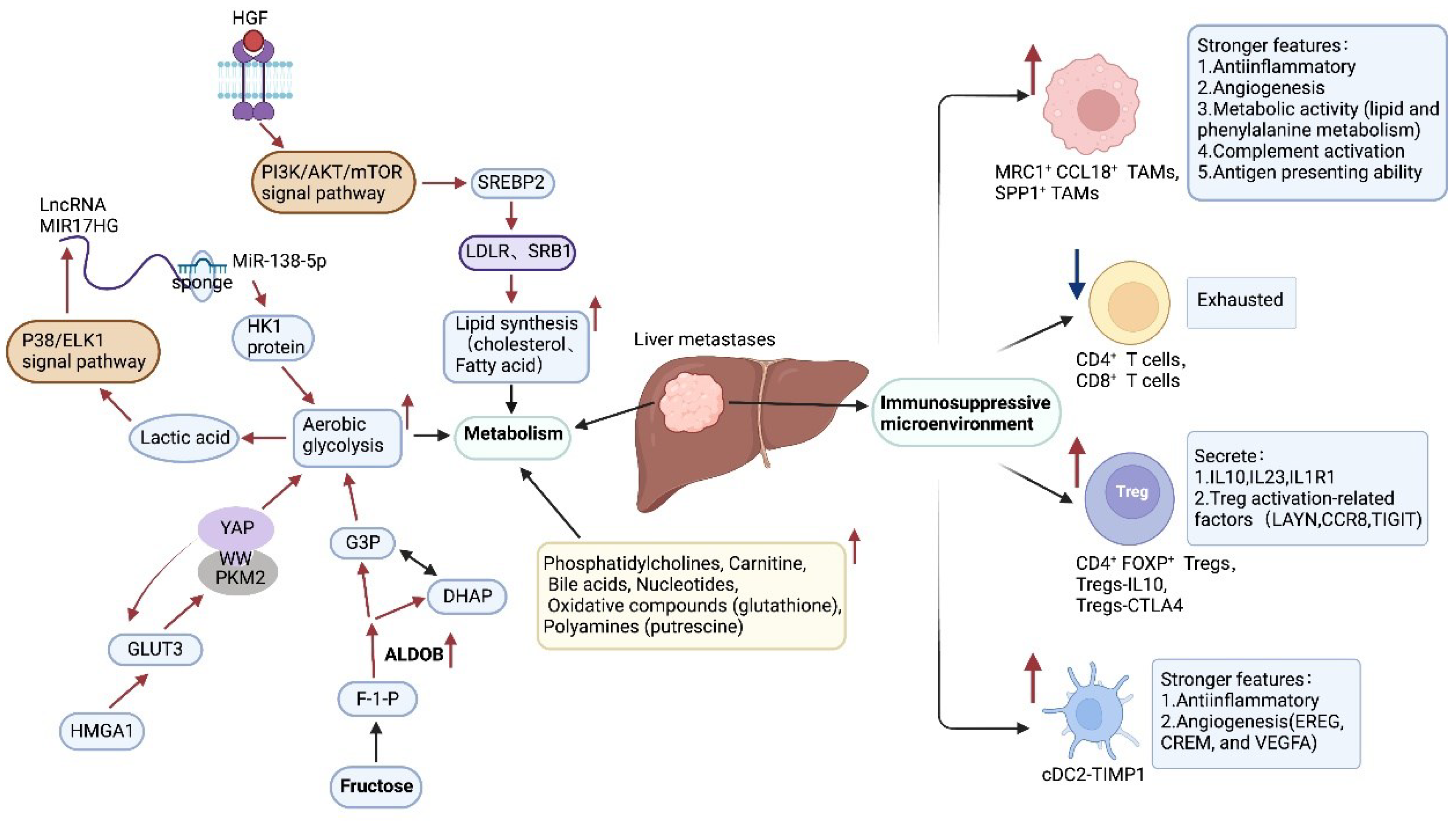

Table 1.
Summary of genetic heterogeneity in CRLM (↑: up-regulated, ↓: down-regulated, -: no change).
Table 1.
Summary of genetic heterogeneity in CRLM (↑: up-regulated, ↓: down-regulated, -: no change).
| levels | items | factors | change | references |
|---|---|---|---|---|
| Genetic level | APC, RAS, BRAF, PIK3CA, TP53, MSI status | - | [18,19,20,21,22,38,39,40,41] | |
| DNA copy numbers | -/↑ | [28,29,30,31,34,35] | ||
Table 2.
Summary of transcriptomic heterogeneity in CRLM (↑: up-regulated, ↓: down-regulated, -: no change).
Table 2.
Summary of transcriptomic heterogeneity in CRLM (↑: up-regulated, ↓: down-regulated, -: no change).
| levels | items | factors | change | references |
|---|---|---|---|---|
| Transcriptomic level | MiRNAs | MiR-122, MiR-122*, MiR-885-5p ,MiR-203, MiR-200c, MiR-424-3p, MiR-503, MiR-1292 | ↑ | [46,47,48,55,56,59] |
| MiR-143, MiR-10b, MiR-28-5p | ↓ | [46,47,48] | ||
| CircRNAs | Circ0001178, Circ0000826, CircNSUN2, Circ0006401 | ↑ | [67,68,69] | |
| LncRNAs | LncRNA GAL, LncRNA MIR17HG | ↑ | [71,72] | |
| Transcription factors | STAT1, STAT3, MYC, HIF1α | ↑ | [78,82] | |
| DAXX, STAT4, STAT5 | ↓ | [77,78] |
Table 3.
Summary of protein heterogeneity in CRLM (↑: up-regulated,↓: down-regulated,-: no change).
| levels | items | factors | change | references |
|---|---|---|---|---|
| Protein level | EMT-related proteins | Migration-associated proteins: VTN, ARP3, FN1, TIMP1, VCAN, POSTN, THBS1, IGFBP7 | ↑ | [85,86,92] |
| Adhesion protein: claudins, ITA5 | ↓ | [85,95,96] | ||
| ARHGAP5, PEA15, ATP6L, FILIP1L | ↑ | [97,100,103] | ||
| Other proteins | PLG, Serpin A1, APOA1A, CA1, SDHA | ↑ | [103,104] | |
| the mitochondrial matrix, the mitochondrial intermembrane space, the proteasome complex, and the actin cytoskeleton | ↓ | [104] |
Table 4.
Summary of metabolic heterogeneity in CRLM (↑: up-regulated,↓: down-regulated,-: no change).
Table 4.
Summary of metabolic heterogeneity in CRLM (↑: up-regulated,↓: down-regulated,-: no change).
| levels | items | factors | change | references |
|---|---|---|---|---|
| Metabolic level | Aerobic glycolysis | GLUT3, HMGA1, PKM2, DKK2, Pyruvate carboxylase, Fructose-bisphosphate aldolase B, Fructose-1,6-bisphosphatase 1 | ↑ | [111,115,116,117,118] |
| Fructose metabolism | ALDOB | ↑ | [119] | |
| Cholesterol metabolism | SREBP2, LDLR, SRB1 | ↑ | [120] | |
| Fatty acids, acylcarnitines, oxidative compounds, polyamines | GSH, putrescine | ↑ | [121] |
Table 5.
Summary of immune heterogeneity in CRLM (↑: up-regulated,↓: down-regulated,-: no change).
| levels | items | factors | change | references |
|---|---|---|---|---|
| Immune cells level | TAMs | MRC1+ CCL18+ TAMs, SPP1+ TAMs | ↑ | [3,128,130] |
| T cells | Treg-IL10, Treg-CTLA4, CD4+FOXP3+ Tregs | ↑ | [137,138] | |
| CD4+ T cells, CD8+ T cells | ↓ | [137,138] | ||
| DCs | cDC2-TIMP1 | ↑ | [128] | |
| cDC2-C1QC(DC3s) | ↓ | [128] | ||
| Neutrophils | ↑ | [3] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



