Submitted:
03 November 2023
Posted:
06 November 2023
You are already at the latest version
Abstract
Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis, is a rare form of necrotizing granulomatous inflammation that predominantly affects medium and small size vessels. Generally respiratory tracts and kidneys are the most affected organs. Neurological manifestations of GPA usually involve the peripheral nervous system causing mononeuritis multiplex or and cranial nerve palsies. Central nervous system (CNS) involvement is rare and characterized by cerebral vasculitis, meningeal inflammation, or mass lesions. The CNS involvement is often linked to Ear Nose Throat (ENT) system disease. In this report, we present two cases of patients diagnosed with GPA who exhibit CNS involvement as first symptom. In the first one with an ischemic stroke while in the second with an atypical headache. In the first patient, a positive medical history indicating involvement of the Ear, Nose, and Throat (ENT) facilitated a precise diagnosis through the prompt execution of autoimmune screening. This, in turn, enabled a timely commencement of treatment, resulting in neurological improvement. In the second case, the significance of ENT involvement was initially overlooked in the context of pathological definition, leading to a substantial delay in diagnosis and an ultimately grim prognosis for the patient. The ENT involvement could be an easy marker to suspect GPA in patient with atypical CNS presentation and could help to make a prompt diagnosis.
Keywords:
GPA
; Stroke
; headache
1. Introduction
Granulomatosis with polyangiitis (GPA), alternatively referred to as Wegener’s granulomatosis, is an uncommon condition characterized by necrotizing granulomatous inflammation that predominantly impacts medium and small-sized blood vessels. Its occurrence is estimated at around 1 in 25,000 people, with a higher occurrence among individuals of Caucasian ethnicity, and the typical age of onset ranges from 30 to 50 years. [1]. An environmental stimulus, typically an infection, would instigate an antibody-driven reaction in individuals with genetic predisposition. This response would result in the development of granulomas comprised of epithelioid cells and giant cells. [2]These granulomatous lesions impact both vascular and tissue structures. Virtually any organ or tissue may be implicated, although the kidneys, respiratory tract, and the ear-nose-throat (ENT) system are predominantly affected.[3]The condition is suspected when a significant elevation in c-ANCA (anti-neutrophil cytoplasmic antibodies) titers is observed in the serum. These antibodies specifically recognize proteinase 3 (PR3), an enzyme located within the primary granules of neutrophils. However, it is important to note that their presence is not considered obligatory for diagnostic purposes[4]. Additional diagnostic evaluations include assessments of renal function, CT scans of the chest and paranasal sinuses, and biopsies of clinically involved organs.
The therapeutic approach involves two distinct stages: the induction of remission and the maintenance. Corticosteroids (e.g., prednisone) serve as primary agents in the induction phase. They are usually administered and then gradually tapered over a period of 4 months. In the maintenance therapy the choice between Rituximab and Cyclosporine is determined by the clinical profile and the extent of disease activity [1]. Neurological involvement is documented in approximately 20-30% of GPA patients [5]. According to the Birmingham Vasculitis Activity Score for Wegener’s granulomatosis (BVAS-WG) [6], neurological manifestations are designated as major symptoms due to their potential life-threatening nature [7].GPA predominantly associates with peripheral nervous system (PNS) manifestations, including peripheral neuropathy, multiple mononeuritis, and cranial nerve palsy. In contrast, central nervous system (CNS) involvement is observed in only 10% of patients with neurological symptoms, presenting with manifestations such as headaches, sensorineural hearing loss, strokes, or meningitis [7].An increasingly supported hypothesis is that CNS involvement is a direct consequence of ENT involvement, with granulomas potentially spreading from these structures to adjacent cerebral areas [8]. We describe two cases of GPA in which CNS involvement presented as an initial clinical feature. In both instances, the patients initially exhibited primary ear-nose-throat (ENT) involvement, without displaying other characteristic symptoms, thus posing a significant diagnostic challenge. Patient A presented with an ischemic stroke, while Patient B presented with a headache accompanied by leptomeningitis and pachymeningitis. These two clinical presentations, despite being among the more common manifestations of GPA, are frequently misdiagnosed and not appropriately categorized as vasculitic symptoms. Nonetheless, the time gap between diagnosis and treatment initiation significantly impacts the prognosis of these patients.
The aim of our study is to emphasize that neurological manifestations of GPA should be thoroughly investigated, particularly in cases where patients have already displayed ENT involvement.
2. Clinical Presentation
1. Patient A
Patient A (Table 1) is a 67-year-old Caucasian male, with an history of smoking habit of approximately 40 pack-years. The patient suffered of high blood pressure, which had been managed with nebivolol 5mg/day. The patient engages in moderate physical activity and exhibits a Body Mass Index (BMI) of 25. He denies any personal history of type II Diabetes or dyslipidemia. Notably, there is a familiar predisposition for autoimmune thyroiditis and type II Diabetes. He contracted a SARS-CoV2 infection two months before admission and had two hospitalizations in the preceding month due to treatment-resistant left-sided otomastoiditis.
The patient presented to the general Emergency Room (E.R.) of our hospital, approximately at 3 p.m. Symptoms presented around 10 a.m. and were characterized by a leftward mouth deviation and difficulty in speaking. The vital signs upon admission are showed in Table 2. Upon general examination, the patient exhibited slight oedema in the soft tissues with no discernible swelling in the cheek or buccal mucosa. There was minor lymph node enlargement observed in the lateral cervical area. Cardiac examination revealed an arrhythmic pattern due to frequent extrasystoles. Pulmonary and abdominal physical examinations were within normal limits.
Neurological assessment revealed the patient to be conscious, temporo-spatially oriented, and cooperative (Glasgow Coma Scale 15[9]). Some walking difficulties were observed due to a strength deficiency in the right lower limb. The patient experienced slight difficulty in articulating speech and showed a leftward deviation of the mouth. There was mild atrophy in the lower limbs. Deep tendon reflexes were normal in the upper limbs but diminished in the lower limbs. Mild right hemiparesis was noted (score as 2-1-1 according to National Institute of Health Stroke Scale- NIHSS score[10] ), with dorsiflexion deficit in the right foot and weakness of the anterior tibial muscle (scored as 4 in Medical Resource Council -MRC[11]). Sensory deficit on the right side of the body, coupled with paresthesia in the distribution of the common peroneal nerve (CPN), was present. The Romberg test was negative. Cerebellar function tests were normal. No Babinski signs. The final NIHSS was 6.
With an initial clinical suspicion of ischemic stroke, the patient underwent an immediate cranial computer tomography (CT) scan. The scan showed evidence consistent with severe chronic microvascular leukoencephalopathy. To definitively exclude an acute ischemic event, the patient was subject to brain Magnetic Resonance Imaging (MRI). The diffusion- weighted imaging (DWI) at high b values (b=1000s/mm^2) indicated an area of restricted proton diffusivity within the left striatum, consistent with an acute ischemic stroke (Figure 1). Since there was no visible large vessel thrombotic occlusion, it was not possible to perform cerebral angiography with mechanical thrombectomy. Moreover, due to more than 4 and a half hours having elapsed since the onset of symptoms, the patient did not meet the eligibility criteria for intravenous thrombolysis [12]. In accordance with the American Heart Association/American Stroke Association guidelines for the Early Management of Patients With Acute Ischemic Stroke [13], the patient received a 40mg Omeprazole injection in 100 cc of saline solution during the acute phase, followed by a 300mg loading dose of Salicylic Acid. For secondary prevention, the patient’s treatment regimen was supplemented with Aspirin 100 mg and Atorvastatin 40 mg. The laboratory test values showed an increase in inflammation indices and renal function (Table 3).
During his hospital stay, the patient was subject to an exhaustive cardiac assessment to ascertain potential risk factors that could account for the acute ischemic event. To rule out a cardioembolic aetiology, electrocardiogram (EKG) and transthoracic echocardiography were conducted. The EKG revealed sinus bradycardia, with no significant arrythmias detected. The echocardiogram exhibited left atrial dilatation and grade I diastolic dysfunction (consistent with arterial hypertension), with an ejection fraction of 60%. Further, to dismiss an atherothrombotic origin of the stroke, a Doppler ultrasound of the supra-aortic vessels was performed. The investigation did not disclose any atheromatous plaques or any other significant alteration within the vessels. In the absence of substantial anomalies in the initial diagnostic investigations, the patient underwent screening for potential hypercoagulability determinant (Table 3). The screening revealed a high titer positivity for C-ANCA pr3 (1190 CU).
This discovery prompted consultation with Rheumatology department, who recommended a chest CT scan to rule out pulmonary involvement, and nephrological assessment to explore potential renal implications. The chest CT scans disclosed parenchymal consolidations of pseudo nodular morphology with finely irregular borders, findings consistent with initial lesion of GPA.A week post the admission, renal function was repeated with results indicating creatinine levels at 1.2 mg/dl, blood urea at 90 mg/dl, and a creatinine clearance of 26.5 ml/min. Further nephrological evaluation unveiled a 24-hour proteinuria of 640 mg/dl. Subsequent urinary sediment analysis exhibited the presence of red blood cells (228/µL) and numerous acanthocytes. To identify any additional renal abnormalities, an ultrasound of the urinary tract was performed. However, this only revealed the presence of renal calculi. The investigations conducted were consistent with GPA, satisfying three out of four diagnostic criteria of ACR/EULAR 2022 with a score exceeding 5 points.[14]Lastly, considering the potential PNS involvement in cases of GPA and the physical examination indicating the involvement of the CPN, the patient underwent a nerve conduction study and electromyography (ENG/EMG). Neurophysiological assessment revealed a multi-neuropathic pattern of peripheral nerve trunk impairment, with pronounced involvement of right CPN.
Considering the clinical-laboratory-instrumental picture, he underwent therapy with dexamethasone 12mg for day with some clinical benefit, such as improvement of paresthesia and right foot weakness. Considering the initial positive response to corticosteroid therapy, a decision was made to initiate with intravenous Rituximab. The regimen involved a dose of 375 mg/m^2 of body surface area administered once weekly for four weeks, in conjunction with oral corticosteroid 75 mg/die. Following the initial two weeks of treatment, an improvement in the ANCA values was observed.
A slight clinical improvement was noted, tough there was no significant enhancement in motor function. After first rituximab cycle, the daily dosage of cortisone was tapered from 75 mg to 57.5 mg. However, twenty days later, the patient experienced a recurrence of auditory disturbance and vertigo. Furthermore, hematological and biochemical investigations revealed a decline in renal function indices, prompting an intravenous administration of a steroid bolus. Meanwhile, the dosage of patient’s maintenance therapy was escalated to 67 mg/day. The patient also underwent a regiment of intravenous immunoglobulins (IVIg), resulting in a substantial reduction in c-ANCA levels (130 CU).
2. Patient B
Patient B is a 68-year-old Caucasian male with a history of smoking 40 pack-years. He was receiving treatment with oral hypoglycemic agents for Type II Diabetes Mellitus and Salicylic acid for chronic cerebral vasculopathy. He also used a hearing aid in his right ear. His family history included a positive association with intestinal neoplasia and hypertension. Six months before our observation, the patient had experienced a vertigo syndrome, which was diagnosed by an ENT specialist as equilibrium disturbance due to cupulolithiasis of the right semicircular canal. One month before admission, he suffered a sudden hearing loss in his left ear, with profound sensorineural hearing loss confirmed by tonal audiometry. In response, he received a ten-day treatment regimen of oral prednisone 40 mg and pentoxifylline 400 mg three times a day. Additionally, the patient reported frequent episodes of throbbing headaches, primarily localized in the temporal region, associated with dizziness. These headaches worsened in an upright position and were refractory to treatment with nonsteroidal anti-inflammatory drugs (NSAIDs). In suspicious of Giant Cell Arteritis (GCA), empirical therapy with methotrexate (MTX) 15 mg weekly and oral prednisone 40 mg daily for four weeks was initiated. The patient described experiencing some clinical improvement while on therapy. A brain MRI was also performed, revealing fluid material within the mastoid cells and the right tympanic cavity. He never went through confirmation biopsy because of on-going mastoiditis. About two weeks prior to our assessment, during a follow-up neurology appointment, the patient diagnosis was switched to tensive headache. Subsequently, the MTX and steroid regimen was ceased, and prophylactic treatment with Topiramate, Alprazolam, and Escitalopram was initiated.
2.1. First Admission
The patient presented to our E.R. with severe throbbing headache, dizziness and diplopia developed three days before. The headache was unresponsive to treatment with Indomethacin 50 mg and Tramadol 100 mg. Family members reported an episode of transient disorientation lasting approximately 3 hours.
Vital signs upon admission are reported in Table 4. On neurological examination there were spontaneous nystagmus with slight rotary oscillations in all gaze positions, particularly to the left; brisk deep tendon reflex in all four limbs; left-sided hemiparesis; bilateral symmetric hypoesthesia with a stocking-glove distribution and Babinski sign on the left. Cerebellar tests were within normal limits. No other pyramidal signs were present.
A brain CT scan revealed bilateral hypodense lesion foci with flattening of adjacent cerebral and cerebellar convolutions. Subsequently, a brain MRI revealed hyperintense lesion foci in FLAIR imaging in the right occipital lobe, temporal lobe and left cerebellar hemisphere. After gadolinium administration an intense diffuse enhancement of the pachymeninges and leptomeninges was observed (Figure 2a).
Laboratory tests at admission showed an ongoing inflammatory process [Table 3]. A lumbar puncture was performed. The cerebrospinal fluid chemistry revealed a protein level of 122 mg/dl, chloride level of 108 mEq, glucose level of 49 mg/dl, and a cell count of 19 mm2. Initial microbiological examinations yielded negative results [Table 3].The Department of Infectious Diseases initiated empiric antibiotic and antiviral therapy for acute meningitis as per the IDSA guidelines [15]: Ceftriaxone 2g IV every 12 hours for 5 days, Vancomycin 20 mg/kg IV every 12 hours, Ampicillin 2g IV every 4 hours for 12 days, Acyclovir 10 mg/kg IV every 8 hours for 5 days, and Dexamethasone 0.15 mg/kg IV every 6 hours. Bronchoalveolar lavage (BAL) was performed to search for Galactomannan, revealing a bronchitis pattern. In suspicion of Aspergillosis, a chest CT scan was conducted, highlighting pseudo-nodular consolidations, some with excavated components within (Figure 2b). Galactomannan was positive only in BAL but not in serum, thus, deferring antifungal treatment.
Five days into the therapy, there was a resolution of inflammatory markers. A follow-up brain MRI showed a reduction in the size of all lesion foci; however, contrast enhancement persisted (Figure 2c). The patient was discharged with a diagnosis of "Meningoencephalitis with probable pulmonary Aspergillosis. Suspected Horton's Arteritis" with instructions for a gradual taper of corticosteroids.
2.1. Second Admission
Three weeks after discharge, the patient underwent a follow-up neurological examination. The exam revealed a deterioration in neurological status: marked drowsiness, incongruent speech, and gait difficulties.
The patient was readmitted to neurology department. A new chest CT scan showed an increase in the size of cavitating lung lesions (Figure 2d). Despite the negative serum Galactomannan result, it was decided to initiate therapy with Voriconazole at a dosage of mg/kg every 12 hours. Given the multisystem involvement, a comprehensive panel for autoimmunity and onconeural markers [Table 5] was requested, which revealed positivity for c-ANCA (192.6 CU).
In suspicion of GPA, the Rheumatology Department was consulted. The patient had sensorineural hearing loss, c-ANCA positivity, lung nodules, and cavitations on chest CT, with a cumulative score of 9 in the EULAR/ACR classification criteria, allowing for a diagnosis of Granulomatosis with Polyangiitis[14].Nephrological evaluation was then performed, with a 24-hour proteinuria of 552 mg/24 hours and a creatinine clearance of 76.8 ml/min. Urine sediment showed severe haematuria (>100 red blood cells per high-power field at 40x magnification. To determine the disease activity and prognosis, the BVAS [6] was applied: Persistent Score of 7 points and New/Worse Score of 23 points. N light of the disease activity, it was decided to initiate treatment with methylprednisolone 1 mg in 100 ml of saline solution twice a day. Immunosuppressive therapy or monoclonal antibodies were not pursued due to a strong suspicion of Aspergillosis. In two weeks, chest CT showed a reduction in the size of all lung lesions and inflammatory markers become negative.
Neurological examination was normal despite the brain MRI remained unchanged compared to the previous one (Figure 2e). The patient was discharge with 25 mg of oral Prednisone twice a day and 240 mg of IV Voriconazole every 12 hours in addition to his baseline therapy.
2.3. Third Admission
Three months after the discharge, the patient presented to our E.R. with a sudden loss of strength and hypoesthesia in the lower limbs. Neurologically, the patient exhibited paraplegia and hypoesthesia from the level of the umbilicus downward.
In suspicion of a spinal cord injury, a spinal CT scan was performed, followed by lumbosacral MRI. Findings were consistent with multiple fractures at the levels of D7, D11-L1, and L2, associated with adjacent edematous changes in the spinal cord parenchyma (Figure 2f). Given the complete spinal cord injury and the patient's overall condition, a conservative approach was chosen.
The patient was readmitted to neurology department. On the first day of hospitalization, he presented an episode of severe hypotension. A consultation with the intensive care unit was conducted, which recommended dopamine therapy at 5 mcg/kg/min to achieve hemodynamic stabilization of the patient. After consultation with Rheumatologist Department, the patient started rescue therapy with Rituximab 1g, due to the worsening disease activity [BVAS v3: Persistent score 15, New/Worse score 36]. To prevent hypogammaglobulinemia and further infectious complications, intravenous immunoglobulins (IV Ig) were administered at 5 g per vial, one vial per day, before starting monoclonal antibody therapy. In addition, Clodronate 200 mg and Prednisone 50 mg were prescribed.
Two days after the initiation of IV Ig infusion, due to the persistent deterioration of the patient's clinical conditions and the worsening of kidney and hepatic function, a decision was made to transfer the patient to the intensive care unit, where the patient unfortunately passed away approximately 7 days later.
3. Discussion
The text continues here.
GPA is a systemic vasculitis associated with autoimmune reaction against small and medium sized blood vessels. [1] The granulomatous and necrotizing lesions affects mainly the ENT system, the airways and the kidney (Figure 3).The mortality rate linked to this condition is 80% [16], attributed to delays in diagnosis and the challenges of promptly initiating the appropriate treatment.
Throughout the progression of the disease, around 33% of patients exhibit CNS involvement or the peripheral nervous system (PNS) in the form of multiple mononeuritis or sensorimotor neuropathy. CNS involvement, particularly at the disease's onset, is notably less common, occurring in approximately 9% of cases (Figure 3) [17]. The most common manifestations encompass pachymeningitis, strokes, seizures, and headaches. [1]
CNS involvement in GPA manifests itself through three distinct histological patterns: isolated granulomatous lesions within the brain or meninges, vasculitis affecting the small vessels in the brain or spinal cord, and the invasion of the CNS originating from extracranial regions [17,18]. Among these mechanisms, the latter is typically the most frequently observed. This process initiates with an initial engagement of the paranasal sinuses or regions within the ENT system, leading to the formation of granulomatous lesions. These lesions subsequently extend into neighboring regions, resulting in damage to blood vessels or meninges. The proportion of individuals presenting with ENT system involvement followed by CNS impairment varies, ranging from approximately 10% to 45% [19].
Despite the heightened disease activity, CNS involvement serves as a favorable prognostic indicator. These patients exhibit a more favorable response to treatment, even though approximately 50% may develop neurological sequelae. In light of the potential for effective intervention, it becomes imperative to swiftly detect cases of GPA featuring CNS engagement. This entails recognizing early diagnostic cues or suspicious factors.
In the case of Patient, A, GPA screening was conducted as a second-level screening due to the negative results of first-level investigations. This allowed us to promptly identify the condition and initiate early treatment, ensuring a better prognosis for the patient. In the case of Patient B, who presented with only headaches, autoimmune testing was not performed despite a prior history of ENT symptoms. This led to a significant diagnostic delay, postponing the initiation of immunosuppressive therapy and resulting in an unfavorable prognosis for the patient.
The cases of the two patients we have presented spurred us to reflect on the connection between ENT and CNS involvement and how this interplay might aid in disease identification. In response, we conducted a concise review of the existing literature. Our primary objectives were to ascertain the frequency of GPA patients presenting with an initial CNS manifestation, to determine how many of them had prior ENT system involvement, and to explore potential solutions to mitigate the diagnostic delays frequently encountered in cases with unconventional presentations.
Concerning the first aspect, we unearthed multiple studies highlighting the notable prevalence of both manifestations. In the study conducted by Huang et al. [7], an impressive 80% of the patients with initial CNS involvement also exhibited concurrent ENT system involvement. Interestingly, two of these individuals presented an ENT symptom as their sole additional symptom at the outset (Patients 1 and 9), while two others subsequently developed ENT system involvement (Patients 2 and 8).In the research conducted by Nishino et al. [20], a striking 98% of the 39 patients they examined with CNS involvement displayed concurrent ENT manifestations.
Similarly, Fragoulis et al. [21] identified 9 patients with CNS manifestations in their patient cohort, with a substantial 77.8% of them presenting concurrent ENT system involvement.
Furthermore, Gul et al. [22] conducted a study involving 12 patients with pituitary gland involvement, and intriguingly, every single one of them exhibited simultaneous ENT system involvement.
Although the association is evidently established, the primary challenge revolves around patients who initially present with exclusive CNS involvement but have a documented history of only ENT involvement. Regrettably, we have limited case studies that shed light on this particular scenario.
In the investigation conducted by Fragoulis et al. [20], Patient 1 presented frequent otitis concurrent with CNS symptoms, devoid of other characteristic manifestations. In the study by Parisot et al. [24], Patients 2, 3, and 4 exhibited exclusive ENT and CNS involvement at the time of diagnosis. Similarly, Yaijina et al. [25] documented sinusitis followed by subarachnoid haemorrhage [23].
Exploring the genuine prevalence of patients exclusively grappling with both CNS and ENT involvement through a dedicated study is an intriguing proposition, considering the paucity of contemporary data. However, our clinical insights, reinforced by corroborating cases found in the medical literature, emphasize the importance of conducting GPA screening in individuals exhibiting suspected neurological symptoms and a positive ENT history. This screening approach remains minimally invasive, economically advantageous, and straightforward to implement, particularly given the potential life-threatening consequences or substantial disability associated with GPA in cases where timely diagnosis is lacking.
Author Contributions
M.D.L. writing—original draft preparation, review and editing; M.R. writ-ing—original draft preparation, review and editing; O.P. data curation; M.S. review; S.M. supervi- sion.
Funding
This research received no external funding.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Comarmond, C.; Cacoub, P. Granulomatosis with Polyangiitis (Wegener): Clinical Aspects and Treatment. Autoimmunity Reviews 2014, 13, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Lutalo, P.M.K.; D’Cruz, D.P. Diagnosis and Classification of Granulomatosis with Polyangiitis (Aka Wegener’s Granulomatosis). Journal of Autoimmunity 2014, 48–49, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Hagen, E.C.; Daha, M.R.; Hermans, J.; Andrassy, K.; Csernok, E.; Gaskin, G.; Lesavre, P.; Lüdemann, J.; Rasmussen, N.; Sinico, R.A.; et al. Diagnostic Value of Standardized Assays for Anti-Neutrophil Cytoplasmic Antibodies in Idiopathic Systemic Vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int 1998, 53, 743–753. [Google Scholar] [CrossRef]
- VENNING, M.C.; QUINN, A.; BROOMHEAD, V.; BIRD, A.G. Antibodies Directed Against Neutrophils (C-ANCA and P-ANCA) Are of Distinct Diagnostic Value in Systemic Vasculitis. QJM: An International Journal of Medicine 1990, 77, 1287–1296. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, G.; Shi, Q.; Zhang, X.; Zeng, X.-F.; Zhang, F.-C. Clinical Analysis of Nervous System Involvement in ANCA-Associated Systemic Vasculitides. Clin Exp Rheumatol 2009, 27, S65–69. [Google Scholar]
- Mukhtyar, C.; Lee, R.; Brown, D.; Carruthers, D.; Dasgupta, B.; Dubey, S.; Flossmann, O.; Hall, C.; Hollywood, J.; Jayne, D.; et al. Modification and Validation of the Birmingham Vasculitis Activity Score (Version 3). Annals of the Rheumatic Diseases 2009, 68, 1827–1832. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Ro, L.-S.; Lyu, R.-K.; Chang, H.-S.; Wu, Y.-R.; Chang, K.-H.; Kuo, H.-C. Wegener’s Granulomatosis with Nervous System Involvement: A Hospital-Based Study. European Neurology 2015, 73, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, S.B.; Rahimian, A.; Ahadi, M.S.; Tavakolpour, S.; Alesaeidi, S. Neurologic Involvement in Granulomatosis with Polyangiitis: A Comparative Study. Biology and Life Sciences Forum 2023, 19, 19. [Google Scholar] [CrossRef]
- Sternbach, G.L. The Glasgow Coma Scale11Medical Classics Is Coordinated by George Sternbach, MD, of Stanford University Medical Center, Stanford, California. The Journal of Emergency Medicine 2000, 19, 67–71. [Google Scholar] [CrossRef]
- Ortiz, G.A.; Sacco, R.L. National Institutes of Health Stroke Scale (NIHSS). In Wiley Encyclopedia of Clinical Trials; John Wiley & Sons, Ltd, 2008; pp. 1–9. ISBN 978-0-471-46242-2. [Google Scholar]
- Dyck, P.J.; Boes, C.J.; Mulder, D.; Millikan, C.; Windebank, A.J.; Dyck, P.J.B.; Espinosa, R. History of Standard Scoring, Notation, and Summation of Neuromuscular Signs. A Current Survey and Recommendation. Journal of the Peripheral Nervous System 2005, 10, 158–173. [Google Scholar] [CrossRef]
- Berge, E.; Whiteley, W.; Audebert, H.; De Marchis, G.; Fonseca, A.C.; Padiglioni, C.; Pérez de la Ossa, N.; Strbian, D.; Tsivgoulis, G.; Turc, G. European Stroke Organisation (ESO) Guidelines on Intravenous Thrombolysis for Acute Ischaemic Stroke. European Stroke Journal 2021, 6, I–LXII. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef] [PubMed]
- Robson, J.C.; Grayson, P.C.; Ponte, C.; Suppiah, R.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Watts, R.A.; Merkel, P.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Granulomatosis with Polyangiitis. Ann Rheum Dis 2022, 81, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Tunkel, A.R.; Hasbun, R.; Bhimraj, A.; Byers, K.; Kaplan, S.L.; Scheld, W.M.; van de Beek, D.; Bleck, T.P.; Garton, H.J.L.; Zunt, J.R. 2017 Infectious Diseases Society of America’s Clinical Practice Guidelines for Healthcare-Associated Ventriculitis and Meningitis. Clin Infect Dis 2017, 64, e34–e65. [Google Scholar] [CrossRef] [PubMed]
- Panupattanapong, S.; Stwalley, D.L.; White, A.J.; Olsen, M.A.; French, A.R.; Hartman, M.E. Epidemiology and Outcomes of Granulomatosis with Polyangiitis (GPA) in Pediatric and Working-Age Adults Populations in the United States: Analysis of a Large National Claims Database. Arthritis Rheumatol 2018, 70, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Mahr, A.; Ramanoelina, J.; Pagnoux, C.; Cohen, P.; Guillevin, L. Central Nervous System Involvement in Wegener Granulomatosis. Medicine 2006, 85, 53. [Google Scholar] [CrossRef]
- De Luna, G.; Terrier, B.; Kaminsky, P.; Le Quellec, A.; Maurier, F.; Solans, R.; Godmer, P.; Costedoat-Chalumeau, N.; Seror, R.; Charles, P.; et al. Central Nervous System Involvement of Granulomatosis with Polyangiitis: Clinical–Radiological Presentation Distinguishes Different Outcomes. Rheumatology 2015, 54, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Holle, J.U.; Gross, W.L. Neurological Involvement in Wegener’s Granulomatosis. Curr Opin Rheumatol 2011, 23, 7–11. [Google Scholar] [CrossRef]
- Nishino, H.; Rubino, F.A.; DeRemee, R.A.; Swanson, J.W.; Parisi, J.E. Neurological Involvement in Wegener’s Granulomatosis: An Analysis of 324 Consecutive Patients at the Mayo Clinic. Ann Neurol 1993, 33, 4–9. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Lionaki, S.; Venetsanopoulou, A.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M.; Tzioufas, A.G. Central Nervous System Involvement in Patients with Granulomatosis with Polyangiitis: A Single-Center Retrospective Study. Clin Rheumatol 2018, 37, 737–747. [Google Scholar] [CrossRef]
- Gu, Y.; Sun, X.; Peng, M.; Zhang, T.; Shi, J.; Mao, J. Pituitary Involvement in Patients with Granulomatosis with Polyangiitis: Case Series and Literature Review. Rheumatol Int 2019, 39, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Garefis, K.; Marini, K.; Skliris, J.P.; Tarazis, K.; Nikolaidis, V.; Poutoglidis, A.; Tsetsos, N.; Tsikopoulos, A.; Markou, K. Granulomatosis with Polyangiitis: Otorhinolaryngological Manifestations and Meningeal Involvement. Ear Nose Throat J 2022, 01455613221078180. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A) The diffusion- weighted imaging (DWI) at high b values (b=1000s/mm^2) indicated an area of restricted proton diffusivity within the left striatum, consistent with an acute ischemic stroke B) Parenchymal consolidations of pseudo nodular morphology with finely irregular borders.
Figure 1.
A) The diffusion- weighted imaging (DWI) at high b values (b=1000s/mm^2) indicated an area of restricted proton diffusivity within the left striatum, consistent with an acute ischemic stroke B) Parenchymal consolidations of pseudo nodular morphology with finely irregular borders.
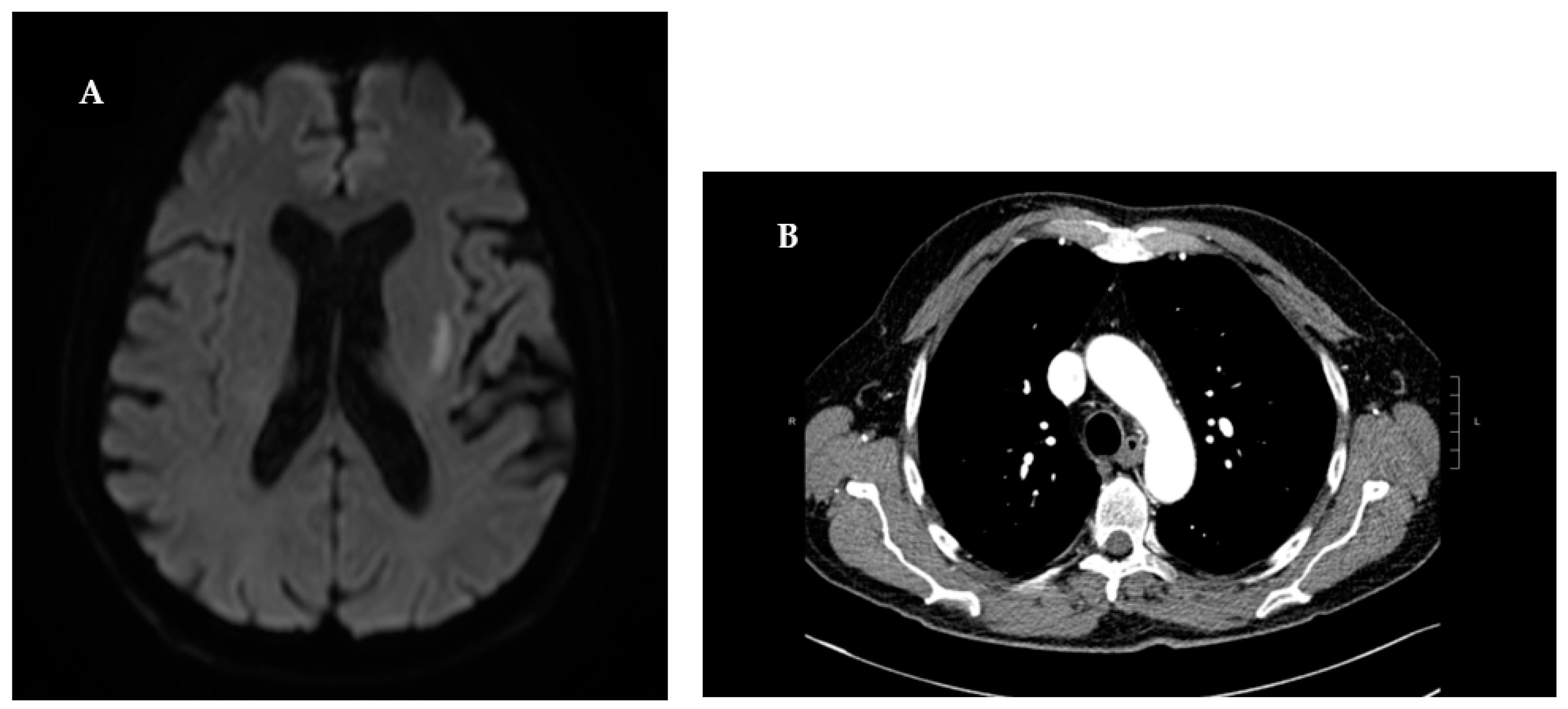
Figure 2.
A) Hyperintense lesion foci in FLAIR imaging in the right occipital lobe, temporal lobe and left cerebellar hemisphere. Diffuse enhancement of the pachymeninges and leptomeninges. B) Pseudo-nodular consolidations, some with excavated components within. C) A follow-up brain MRI showed a reduction in the size of all lesion foci; however, contrast enhancement persisted. D) Increase in the size of cavitating lung lesions. E) MRI remained unchanged compared to the previous one. F) Multiple fractures at the levels of D7, D11-L1, and L2, associated with adjacent oedematous changes in the spinal cord parenchyma.
Figure 2.
A) Hyperintense lesion foci in FLAIR imaging in the right occipital lobe, temporal lobe and left cerebellar hemisphere. Diffuse enhancement of the pachymeninges and leptomeninges. B) Pseudo-nodular consolidations, some with excavated components within. C) A follow-up brain MRI showed a reduction in the size of all lesion foci; however, contrast enhancement persisted. D) Increase in the size of cavitating lung lesions. E) MRI remained unchanged compared to the previous one. F) Multiple fractures at the levels of D7, D11-L1, and L2, associated with adjacent oedematous changes in the spinal cord parenchyma.
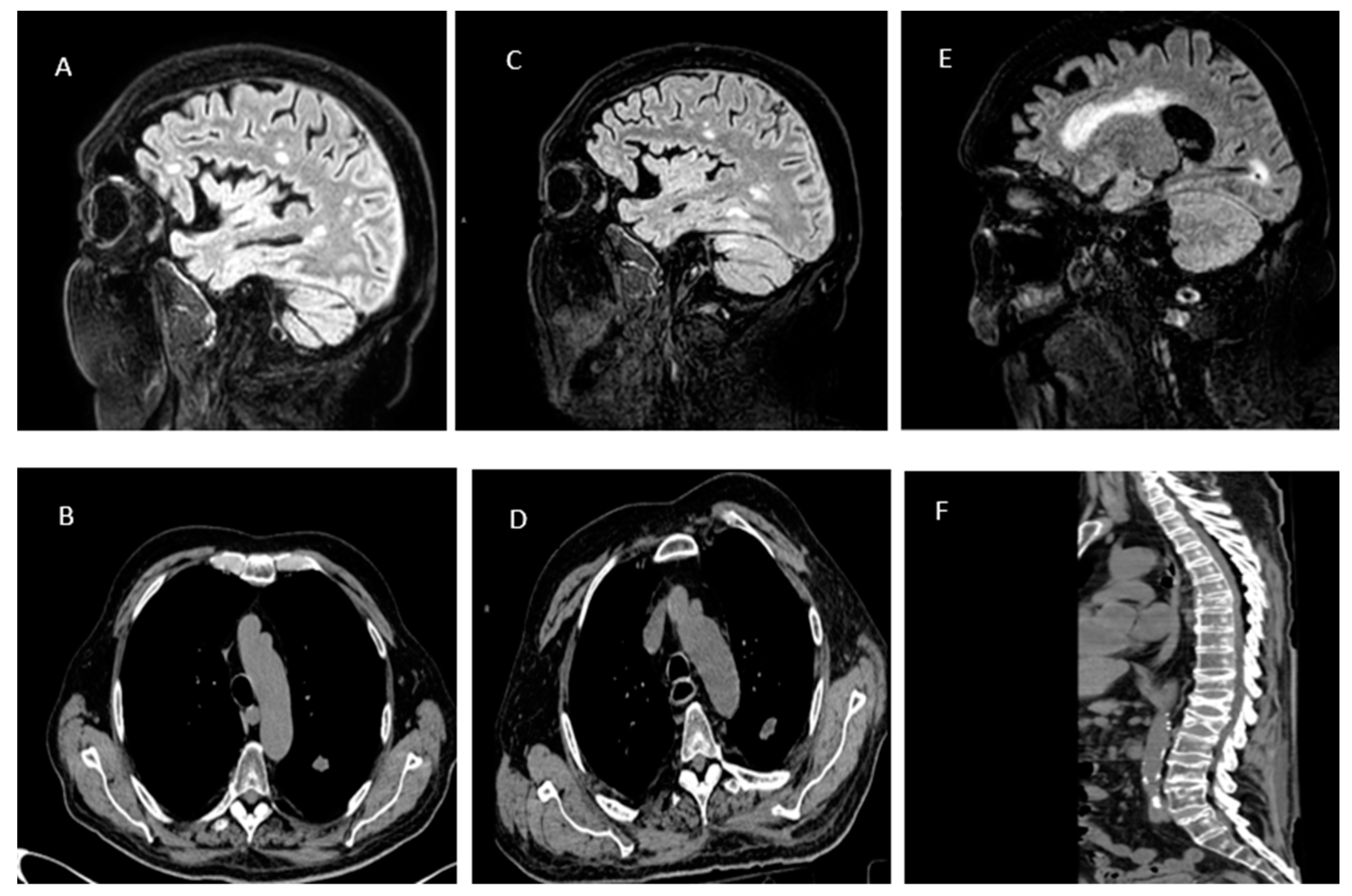
Figure 3.
Distribution of symptoms among GPA patients. In figure are shown the incidence of respiratory (blue), kidney (orange), ENT (grey), PNS (yellow) e CNS (red) involvement.
Figure 3.
Distribution of symptoms among GPA patients. In figure are shown the incidence of respiratory (blue), kidney (orange), ENT (grey), PNS (yellow) e CNS (red) involvement.
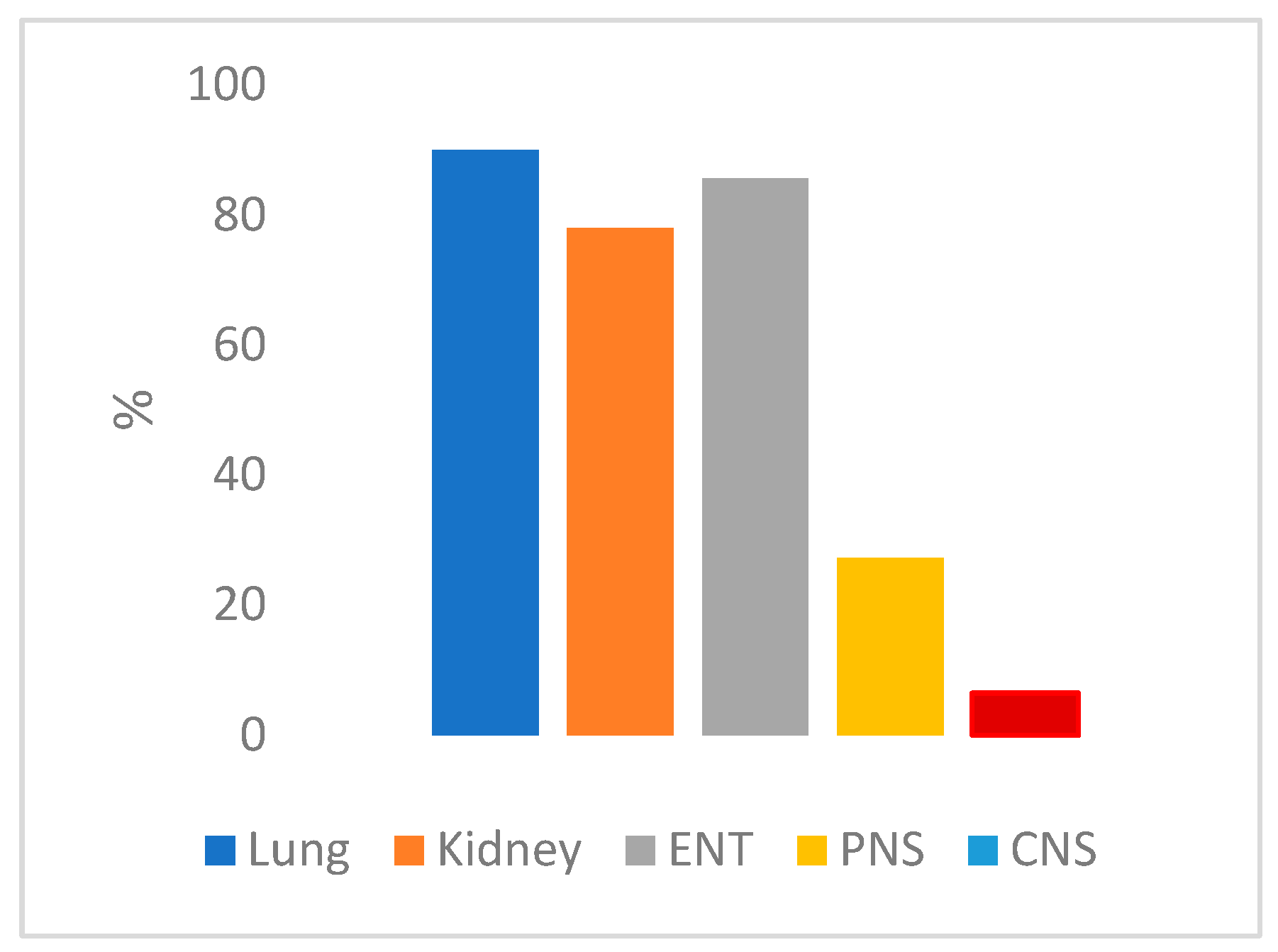

Table 2.
Vital signs and laboratory tests. HR: heart rate; RR respiratory rate; T: body temperature; BP: blood pressure; OS: oxygen saturation; BS: blood sugar.CR: creatinine levels; N: blood nitrogen. R.C.P.: reactive C protein; E.S.R: erythrocyte sedimentation rate.
Table 2.
Vital signs and laboratory tests. HR: heart rate; RR respiratory rate; T: body temperature; BP: blood pressure; OS: oxygen saturation; BS: blood sugar.CR: creatinine levels; N: blood nitrogen. R.C.P.: reactive C protein; E.S.R: erythrocyte sedimentation rate.
| Vital signs upon admission: | BP: 165/75 mmHg. HR 70 bpm RR 18. T 36.4°C. OS: 97%. BS: 925 mg/dl |
| Renal function | Cr: 1.4 N: 88 |
| Inflammatory indices | R.C.P. 11 mg/dl, E.S.R.93 1^ora, White blood count 15,000/mm2 |
Table 3.
Hypercoagulability screening. ANA: antinuclear antibody; ASMA: anti-smooth muscle; ANCA: anti-cytoplasm; ENA: anti-nuclear antigen.
Table 3.
Hypercoagulability screening. ANA: antinuclear antibody; ASMA: anti-smooth muscle; ANCA: anti-cytoplasm; ENA: anti-nuclear antigen.
| C Protein | Negative |
| S Protein | Negative |
| AT III | Negative |
| Factor II | Negative |
| Factor V | Negative |
| Factor XII | Negative |
| Anti-cardiolipin | Negative |
| ANA | Negative |
| A.S.M.A. | Negative |
| c-ANCA | 1190.7 CU |
| p- ANCA | Negative |
| ENA | Negative |
Table 4.
Vital signs and laboratory tests during I, II and III hospitalizations. HR: heart rate; RR respiratory rate; T: body temperature; BP: blood pressure; OS: oxygen saturation; BS: blood sugar. N: nitrogen; Cr: creatinine; Na: sodium; K: potassium; Mg: magnesium; Ca: calcium. WBC: white blood cells; R.C.P.: Reactive C Protein; E.S.R.: erythrocyte sedimentation rate.
Table 4.
Vital signs and laboratory tests during I, II and III hospitalizations. HR: heart rate; RR respiratory rate; T: body temperature; BP: blood pressure; OS: oxygen saturation; BS: blood sugar. N: nitrogen; Cr: creatinine; Na: sodium; K: potassium; Mg: magnesium; Ca: calcium. WBC: white blood cells; R.C.P.: Reactive C Protein; E.S.R.: erythrocyte sedimentation rate.
| I | II | III | |
|---|---|---|---|
| Vital signs upon admission: | HR 70 bpm. RR 18. T 37.4°C. OS: 98%. BS: 95 mg/dl |
HR 89bpm. RR 18. T 36.4°C. OS: 98%. |
|
| Renal function | N: 16 mg/dl, Cr: 0.7 mg/dl, | N:54 mg/dl Cr: 0.8mg/dl | N:73 mg/dl Cr: 2.5 mg/dl |
| Liver function | |||
| Inflammatory indices | WBC: 15.800; R.C.P.: 20 | WBC: 21.800 | WBC: 20.360 |
| E.S.R: 53 | R.C.P.: 2.95 E.S. R: 56 | R.C.P.: 7.25 E.S. R: 75 | |
| Blood cultures | Negative | K. Pneumoniae | Negative |
| Quantiferon test | Negative | Negative | Negative |
| Beta-glucan | Negative | Negative | Negative |
| Serum galactomannan | Negative | Negative | Negative |
Table 5.
A) Antibodies. ANA: ; ENA: anti-nuclear antigen; ANCA: anti-cytoplasm; ASMA: anti-smooth muscle; nDNA: anti-nuclear DNA; LAC: lupus like anticoagulant; C3: complement fraction C3; C4: complement fraction C4; R.F.: Rheumatoid factor. B) Oncomarkers. NSE: neuronal specific enolase; CEA: carcinoembryonic antigen; AFP: alpha fetoprotein.
Table 5.
A) Antibodies. ANA: ; ENA: anti-nuclear antigen; ANCA: anti-cytoplasm; ASMA: anti-smooth muscle; nDNA: anti-nuclear DNA; LAC: lupus like anticoagulant; C3: complement fraction C3; C4: complement fraction C4; R.F.: Rheumatoid factor. B) Oncomarkers. NSE: neuronal specific enolase; CEA: carcinoembryonic antigen; AFP: alpha fetoprotein.
| (A) | ||
| Antibodies | Result | |
| ANA | Negative | |
| ENA screening | Negative | |
| Anti-LMK | Negative | |
| p-ANCA | Negative | |
| c-ANCA | Positive | |
| nDNA | Negative | |
| ASMA | Negative | |
| L.A.C. | Negative | |
| C3 | Negative | |
| C4 | Negative | |
| R.F. | Negative | |
| Anti-cardiolipin | Negative | |
| Anti-Jo1 | Negative | |
| Anti-phospholipid | Negative | |
| (B) | ||
| Oncomarkers | Results | |
| NSE | 14.3 | |
| CEA | 0.74 | |
| Ca 19.9 | 10.5 | |
| Ca 15.3 | 22.10 | |
| AFP | 2 | |
| Calcitonin | 0.67 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



