
Submitted:
06 November 2023
Posted:
07 November 2023
You are already at the latest version
Abstract
An array of diverse processes and factors have been linked to the pathoetiology and pathophysiology of Parkinson’s disease (PD), including systemic CNS factors. This article reviews some of the many processes and factors linked to the emergence and progression of PD that culminates in the loss of substantia nigra pars compacts (SNpc) in PD patients. It is proposed that SNpc astrocytes may be a key hub, with numerous systemic and CNS factors acting to suppress the astrocyte tryptophan-melatonin pathway, leading to a loss of astrocyte melatonin and lactate for SNpc dopamine neurons. Consequently, dopamine neurons show an increase in α-synuclein, oxidative stress and suboptimal mitochondrial function, partly due to the loss of melatonin induced PTEN-associated kinase (PINK)1/parkin and mitophagy. This leads to an increase in the major histocompatibility (MHC)-1 and the chemoattraction of CD8+ T cells that destroy SNpc dopamine neurons in an ‘autoimmune’/’immune-mediated’ manner.
The upstream processes driving the end-point ‘chaos’ of SNpc dopamine neuron loss are proposed to be driven by the suppression of night-time pineal melatonin, gut microbiome derived butyrate and possibly bcl2-associated athanogene (BAG)-1, which all act to suppress the glucocorticoid receptor (GR) translocation to the nucleus in all systemic and CNS cells, thereby potentiating the effects of the rising levels of cortisol over the night and accelerated rise in the course of the cortisol awakening response (CAR). The potentiation of cortisol effects at the GR has consequences for the homeostatic regulation of the diverse array of systemic and CNS microenvironments, as well as a distinct regulation of different immune and glia cells. The morning CAR is classically proposed to ‘prepare the body for the coming day’. However, the differential regulation of the GR over the circadian rhythm at night would indicate that such preparation for the coming day may be powerfully determined by night-time factors and processes.
The article integrates this systemic, night-time pathoetiology with the ‘immune-mediated’ processes that ultimately drive SNpc dopamine neuron loss. This has a number of novel future research and treatment implications.
Keywords:
α-synuclein
; Parkinson’s disease
; mitochondria
; melatonin
; glucocorticoid receptor
; aryl hydrocarbon receptor
; TrkB
; gut microbiome
; circadian
; treatment
1. Introduction
The pathoetiology and pathophysiology of Parkinson’s disease (PD) is still the subject of intense investigation, with an array of diverse processes and factors, including systemic and CNS, proposed to play significant roles in driving the loss of dopamine neurons in the substantia nigra pars compacts (SNpc) in PD patients. Such pathophysiological processes include alterations in the gut microbiome, oxidative and nitrosative stress, the tryptophan-melatonin pathway, and the tryptophan-kynurenine pathway [1]. A suppressed capacity to produce melatonin in glia, neurons and gut cells has been proposed to underpin the CNS and systemic changes evident in PD pathophysiology [1], paralleling the systemic and CNS pathophysiology of Alzheimer’s disease [2,3]. As elevated α-synuclein level is a core aspect of PD pathophysiology, systemic and circadian processes may ultimately impact on α-synuclein regulation, with relevance to other synucleinopathies [4].
Heightened α-synuclein levels driven by systemic processes may ultimately drive neuronal loss via ‘immune-mediated’ processes, thereby overlapping PD pathophysiology with the processes classically attributed to ‘autoimmune’ disorders [5]. A growing number of diverse medical conditions are now proposed to be ‘autoimmune’/’immune-mediated’ disorders, including Alzheimer’s disease [3,6] and amyotrophic lateral sclerosis (ALS) [7], as well as PD [8]. Recent work indicates that alterations in mitochondrial interactions in a given microenvironment can leave one cell type susceptible to elimination by ‘immune-mediated’ processes, which is typically mediated by CD8+ T cells and to a lesser extent by natural killer (NK) cells [9]. Such cell elimination arises from a decrease in mitophagy in association with suppressed levels of PTEN-induced kinase (PINK)1 and parkin, leading to an increase in oxidative stress induction of major histocompatibility complex (MHC)-I, which triggers the chemoattraction of CD8+ T cells to eliminate SNpc dopamine neurons [9]. This is parsimonious with preclinical investigations showing intestinal Gram-negative bacteria, via toll-like receptor (TLR)4 activation, driving PD-like symptoms that can be successfully treated with L-DOPA in a PINK1 ko murine PD model [10].
The redefining of end-point cell loss as driven by immune-mediated processes opens avenues for more refined conceptualization of the data collected on PD pathoetiology and pathophysiology, including the processes underpinning the dysregulation of α-synuclein levels, fibrillization and effects. Recent work highlights the importance of a suppressed tryptophan-melatonin pathway in mediating the vulnerability of a particular cell type to elimination in a given microenvironment [11]., indicating the relevance of such microenvironment homeostatic regulation in the cells interacting with SNpc dopamine neurons Alterations in the levels of melatonin and its receptors have long been recognized in the SNpc of PD patients [12]. Given the growing appreciation of the importance of the melatonergic pathway in PD, the tryptophan-melatonin pathway will be briefly reviewed before detailing how this pathway may be an intricate aspects of PD pathophysiology, including in α-synuclein regulation and the initiation of ‘immune-mediated’ SNpc dopamine neuron cell destruction.
2. Tryptophan-melatonin pathway
Tryptophan is primarily derived from diet, although seems capable of being produced by the shikimate pathway of the mammalian gut microbiome. Tryptophan is taken up into cells by the large amino acids transporters (LAT), predominantly by LAT-1 present in astrocytic end-feet at the blood-brain barrier (BBB). Astrocyte tryptophan hydroxylase (TPH)2 converts tryptophan to 5-hydroxytryptophan (5-HTP), with TPH2 and body TPH1 needing to be stabilized by 14-3-3 isoforms, including 14-3-3ε, for 5-HTP to be produced. Aromatic-L-amino acid decarboxylase (AACD) then converts 5-HTP to serotonin (5-HT). The conversion of serotonin to N-acetylserotonin (NAS) is the first enzymatic stage of the melatonergic pathway, being mediated by aralkylamine N-acetyltransferase (AANAT), which also requires stabilization by 14-3-3 isoforms, including 14-3-3ζ, as well as the presence of acetyl-coenzyme A (acetyl-CoA). NAS is then converted by acetylserotonin methyltransferase (ASMT) to melatonin (reviewed in [7,9]). See Figure 1.
By requiring acetyl-CoA, the initial enzymatic reaction in the melatonergic pathway involving the AANAT conversion of serotonin to NAS may be intimately linked to mitochondrial function, given that the predominant source of acetyl-CoA arises from the conversion of pyruvate to acetyl-CoA by the pyruvate dehydrogenase complex (PDC). PDC activation increases ATP production by the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), whilst decreasing oxidant production by the electron transport complexes. As melatonin seems primarily produced in mitochondria, its production in cells seems intimately linked to the capacity of mitochondria to initiate the optimization of mitochondrial function by PDC. It is important to note that the mitochondrial melatonergic pathway is evident in all body cells so far investigated and is proposed to be present in all mitochondria-containing cells across the three kingdoms of multicellular life on planet Earth (animals, plants and fungi) [13].
3. Integrating the tryptophan-melatonin pathway in PD pathophysiology
The tryptophan-melatonin pathway is intimately linked to, and regulated by, factors and processes showing alterations in PD, including the gut microbiome. The gut microbiome derived short chain fatty acid, butyrate, is decreased in PD [14]. Butyrate induces sirtuin-3, as does pineal melatonin, thereby deacetylating and disinhibiting the PDC to increase the conversion of pyruvate to acetyl-CoA in the process of optimizing mitochondrial function [15]. Sirtuin-3 is dramatically reduced in the SNpc dopamine neurons of PD patients [16], with these authors also showing sirtuin-3 in preclinical PD models to negatively correlate with α-synuclein levels [16]. Sirtuin-3 is decreased over the course of aging and linked to many aging associated disorders, as well as human longevity [17], highlighting the importance of sirtuin-3 across different cell types in aging-associated medical conditions, such as PD. Another mitochondria and sirtuin-3 related change over the course of aging is the gradual 10-fold decrease in pineal melatonin production from adolescence to the ninth decade of life [18]. Melatonin significantly inhibits the induction and fibrillation of α-synuclein [19]. This would indicate that two of the major systemic inducers of sirtuin-3, namely butyrate and melatonin, are decreased in PD, with significant consequences for mitochondrial function and α-synuclein regulation. It is unknown whether the 10-fold decrease in pineal melatonin is also evident in other CNS and body cells, suggesting that local melatonin production [20], including in SNpc neurons and/or astrocytes, may also be significantly suppressed over the course of aging. If the attenuation of the tryptophan-melatonin pathway is evident across body and CNS cells, this would indicate not only the loss of the antioxidant, anti-inflammatory and mitochondria optimizing effects of melatonin, but also a suppressed capacity of local, cellular melatonin to induce sirtuin-3. Suppressed pineal and local melatonin production may therefore be significant aspects of wider PD pathophysiology arising from, and contributing to, suboptimal mitochondrial function.
Suboptimal mitochondrial function is widely recognized as a core aspect of PD pathophysiology. Most of the genes mutated in hereditary PD, including α-synuclein, PINK1, parkin, and DJ-1 impact on mitochondrial function, typically associated with mitochondrial oxidant upregulation [21], with raised α-synuclein levels and fibrilization upregulating Complex 1 oxidant production in SNpc neurons [22]. Suboptimal mitochondrial function in PD is therefore a crucial aspect of PD pathophysiology, with the suppression of pineal melatonin, local melatonin, sirtuin-3, and gut microbiome derived butyrate [16,23,24] contributing to suboptimal mitochondrial function in SNpc dopamine neurons in association with raised α-synuclein levels. Alterations in the gut microbiome and intestinal nervous system that increase gut permeability also allow for intestinal epithelial cell derived microRNAs (miRNAs) to enter the circulation, directly and/or within microvesicles/exosomes, and thereby impact on CNS mitochondrial function [25], including via the regulation of 14-3-3 and the mitochondrial melatonergic pathway, as in other medical conditions [26]. The changes in the SNpc mitochondrial function and the tryptophan-melatonin pathway are intimately linked to alterations in the gut microbiome given that butyrate not only increases sirtuin-3 but also increases the mitochondrial melatonergic pathway, as shown in intestinal epithelial cells [27]. This provides direct links of suppressed butyrate production in PD to suppressed levels of sirtuin-3 and mitochondrial melatonin that negatively regulate mitochondrial function, in association with raised α-synuclein levels in PD.
The tryptophan-melatonin pathway may therefore be intimately associated with core pathophysiological features of PD, including mitochondrial dysfunction, circadian dysregulation, α-synuclein upregulation, and gut dysregulation. Alterations in the tryptophan-melatonin pathway may also link wider bodies of data pertaining to PD pathophysiology.
4. Integrating wider Parkinson’s disease pathophysiology with the tryptophan-melatonin pathway
By conceptualizing the tryptophan-melatonin pathway as an integral aspect of core cellular function via its interactions with mitochondrial metabolism, and therefore in the regulation of oxidant/antioxidant ratio, ROS-driven microRNAs (miRNAs) and therefore patterned gene expression as well as ATP production, mitophagy regulation and α-synuclein levels/fibrillization, the tryptophan-melatonin pathway is intimately linked to wider aspects of PD pathophysiology.
4.1. Platelets and Erythrocytes
Importantly, wider systemic cells may be similarly dysregulated in PD, with relevance to α-synuclein production, including erythrocytes [28] and platelets [29]. Both erythrocytes and platelets are significant producers of α-synuclein, with erythrocyte α-synuclein proposed to seed the increase in gut α-synuclein in PD [28], thereby initiating alterations in the gut microbiome and gut permeability, whilst also providing α-synuclein to be retrogradely transported via the vagal nerve to the CNS [1]. There is a growing appreciation of the role of platelets in neurodegenerative disorders and other medical conditions, including cancer [30]. Importantly, like SNpc dopamine neurons, platelets are regulated by systemic processes, including circadian melatonin and gut microbiome-derived butyrate and lipopolysaccharide (LPS), with platelet-derived serotonin being an important serotonin source for the initiation of the melatonergic pathway across body and CNS microenvironments [30]. Platelet function may therefore be synchronized with wider changes in PD via regulation by systemic processes as well as genetic factors and epigenetic processes. Platelets are also a significant source of sphingosine-1-phosphate (S1P), which regulates enteric glial cell function and lymphocyte chemotaxis to the gut [30], as well as attenuating SNpc dopamine neuron loss [31]. The enteric nervous system [32], and especially enteric glial cells may be crucial regulators of the interface of the gut, immunity and vagal nerve in PD, as in other gut-linked medical conditions [9].
4.2. Stress, HPA axis and morning cortisol awakening response
As with many other medical conditions, stress exacerbates PD pathophysiology, including via glucocorticoid receptor (GR) effects on motor and non-motor PD symptoms, as shown in preclinical PD models [33]. Stress induced hypothalamus-pituitary-adrenal (HPA) axis activation is classically associated with the emergence of psychiatric conditions, especially depression, in PD [34,35] and many other medical conditions [36,37]. However, there is a growing realization that such psychiatric presentations do not simply represent comorbidities. Rather these conditions emerge in association with pathophysiological changes relevant to classical PD pathophysiology, including from cortisol/GR effects [33]. Importantly, cortisol/GR effects do not arise solely from stress-linked HPA axis activation but also from the morning cortisol awakening response (CAR). The morning CAR surge starts just before awakening and lasts for 30 minutes. The morning CAR is an accentuation of the rise in plasma cortisol seen typically over sleep at night [38,39], see Figure 2. Although cortisol/GR activation regulates thousands of genes across all body cells, including immune cells, there is a surprising lack of investigation as to the role of CAR. Generally, CAR, along with the corelease of adrenal aldosterone, is proposed to increase respiration and blood pressure in ‘preparation for the coming day’ [40]. However, recent work indicates that CAR may be an important regulator of pathophysiology across diverse medical conditions, including neurodegenerative conditions and cancer, as well as aging [41,42].
The GR is typically held in a cytoplasmic complex with heat shock protein (hsp)90 and p23. Following GR activation by cortisol, the GR is translocated to the nucleus where it binds to the glucocorticoid response element (GRE) in the promotor of thousands of genes. The GR can also interact with other transcription factors in the nucleus to significantly regulate their transcription efficacy. Two of the factors shown to be decreased in PD, namely melatonin and butyrate, prevent the GR nuclear translocation, thereby suppressing the consequences of both stress and CAR driven GR activation [43,44]. Butyrate effects are mediated via its capacity of a HDACi, thereby acetylating the GR and/or hso90 to prevent GR nuclear translocation [45]. It has been proposed that many medical conditions may arise at night, involving aging-linked suppressed melatonin and therefore a decrease in the immune- and antioxidant-dampening effects of melatonin at night [46,47]. The suppression of night-time pineal and local melatonin as well as gut microbiome-derived butyrate will therefore have consequences for how night-time processes shape the nature of how morning CAR primes body and CNS cells/systems ‘for the coming day’. This may be of some importance given the dramatic impact of cortisol/GR on thousands of genes in all cells.
Another factor intimately associated with the regulation of the GR as well as providing protection in PD is bcl2-associated athanogene (BAG)-1. BAG-1 restores the function of the PARK7 gene protein, DJ-1, in an in vitro model of hereditary PD, whilst also attenuating α-synuclein toxicity in PD models [48]. Two common medications, lithium and sodium valproate, show some efficacy in PD clinical trials and models, with effects proposed to be mediated via increased antioxidants [49,50]. However, both lithium and valproate increase BAG-1 [51], suggesting a role of BAG-1 in their efficacy. Like melatonin and butyrate, BAG-1 also prevents GR nuclear translocation [51], with recent data indicating that BAG-1 may also translocate the GR to mitochondria, where it can cross into the mitochondrial matrix to bind with IL-6 and PDC and thereby impacting on mitochondrial ATP production [52]. As melatonin, via epigenetic processes and possibly directly, can increase BAG-1 [42], BAG-1 may be another factor to regulate stress-linked HPA axis activation and morning CAR, with relevance to PD pathoetiology and pathophysiology.
In the absence of an attenuation of GR nuclear translocation by melatonin, butyrate and BAG-1, no increase in cortisol production during stress or CAR would be necessary for an elevation in GR nuclear translocation. Such relative enhancement of HPA axis and CAR driven GR activation of thousands of genes with the GRE in their promotor will have significant wide-ranging consequences, including for the tryptophan-melatonin pathway. GR activation of the GRE increases tryptophan 2,3-dioxygenase (TDO), which is especially highly expressed in the liver and astrocytes, to drive the conversion of tryptophan to kynurenine and other kynurenine pathway products, thereby depleting the availability of tryptophan for the tryptophan-melatonin pathway. Circulating kynurenine is readily taken up over the blood-brain barrier into the CNS, like tryptophan, via LAT-1 (figure 1). 60% of CNS kynurenine is derived from the circulation, and therefore from cells out-with the CNS [53]. When taken up into the CNS, kynurenine can be converted to a number of powerfully neuroregulatory factors, including the excitatory picolinic acid and the excitotoxic quinolinic acid, as well as to kynurenic acid, which inhibits the N-methyl-d-aspartate (NMDA) receptor and activates the aryl hydrocarbon receptor (AhR). All of these factors are relevant regulators of PD pathophysiology [1]. Cortisol/GR/GRE effects therefore parallel the effects of the raised pro-inflammatory cytokines evident in PD, which increase indoleamine 2,3-dioxygenase (IDO), with similar consequence on the tryptophan/kynurenine ratio and kynurenine pathway products effects. There is considerable interest in the role of the kynurenine pathway in PD pathophysiology [54,55] and treatment [56]. Overall, the suppression of pineal melatonin, gut microbiome butyrate and BAG-1 in PD not only enhances stress-linked HPA activation of the GR, but also enhances the GR activation following the morning CAR, thereby differentially regulating the how the morning CAR primes body cells and systems (including immune and glial cells) ‘for the coming day’. Such differential priming includes increased kynurenine pathway products regulating patterned neuronal activity as well as kynurenine and kynurenic acid activation of the AhR, thereby regulating patterned immune responses [57].
4.3. Stress, α-synuclein, transcription factors and glutamatergic regulation
As well as suppressing pineal melatonin [58], stress, via CRH induction [59] and GR activation [60], coupled to raised pro-inflammatory cytokines [1], increases gut permeability/dysbiosis, thereby lowering butyrate levels and increasing circulating LPS, leading to enhanced activation of the transcription factors, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and yin yang 1 (YY1) [61]. Numerous studies link the TLR4/NF-kB pathway to PD pathophysiology [62], including via exogenous α-synuclein activation of astrocyte TLR4/NF-kB leading to released proinflammatory cytokines and oxidants [63]. TLR4 can also induce YY1, with astrocyte YY1 a significant driver of PD pathophysiology [64], partly mediated via the suppression of the excitatory amino acid transporter (EAAT)2, thereby increasing glutamate and excitatory neuronal damage [65]. Interestingly, erythrocyte extracellular vesicles containing α-synuclein can cross the blood-brain barrier, with α-synuclein accumulating in astrocyte end-feet, in association with suppressed EAAT2 function [66], being another route whereby systemic factors and processes modulate PD pathophysiology. Interestingly, both NF-kB and YY1 can increase the tryptophan-melatonin pathway and melatonin release thereby allowing melatonin to have autocrine and paracrine effects that dampen inflammation and oxidant induction, whilst optimizing mitochondrial function [67,68].
Astrocytes constitutively express and release melatonin, as first shown in 2007 [69], suggesting that the suppressed capacity of astrocytes to upregulate the tryptophan-melatonin pathway and melatonin efflux, may drive a maintained pro-inflammatory milieu, including from maintained YY1 suppression of EAAT2 [3]. As well as dysregulating glutamatergic activity in the SNpc, glutamatergic dysregulation will also significantly modulate cognition in PD via changes in other regions, including the hippocampal CA2 region [70]. Overall, the suppressed capacity of astrocytes to efflux melatonin will dramatically alter the nature of the interactions of SNpc dopamine neurons with their immediate microenvironment as well as in other CNS regions, coupled to the attenuation of melatonin’s suppression of α-synuclein, thereby heightening α-synuclein toxicity presynaptically and in mitochondria [71,72]. A number of studies suggest that α-synuclein spread in astrocytes and CNS cells may be dependent upon the heightened levels of hyperlipidemia, perhaps especially raised 27-hydroxycholesterol levels, and the 27-hydroxycholesterol modification of mitochondrial α-synuclein [72]. Notably, melatonin decreases intestinal lipid absorption, cholesterol synthesis and hyperlipidemia [73,74]. This may be of some importance to systemic alterations in PD, given that α-synuclein accumulates in all body organs in PD, including in the liver in association fatty liver disease [75]. Butyrate is also associated with the suppression of hyperlipidemia [76]. Overall, data indicate α-synuclein has effects in the liver, as well as in the gut, in the modulation of CNS α-synuclein spread, with the effects of melatonin and butyrate in other organs attenuating α-synuclein spread in the brain.
4.4. Astrocytes, α-synuclein, and neuronal mitochondrial metabolism
The suppression of astrocyte melatonin induction not only prolongs the pro-inflammatory milieu in the SNpc but also prevents melatonin from inducing lactate dehydrogenase, as shown in other cell types [77], thereby not only decreasing lactate as an energy substrate for SNpc dopamine neurons but also increasing the requirement for astrocytes to upregulate their antioxidant protection under challenge. The enhancement of astrocyte antioxidant protections seems to be mediated via cystine-glutamate antiporter (System Xc-) upregulation, whereby cystine is taken up in exchange for glutamate efflux in the course of glutathione (GSH) synthesis. System Xc- upregulation increases extracellular glutamate that contributes to excessive excitatory activity, especially when coupled to the α-synuclein/LPS/HMGB1-TLR4-YY1 suppression of EAAT2. The loss of astrocyte melatonin, coupled to suppressed EAAT2 levels, enhances extracellular glutamate and suppresses lactate for neuronal mitochondrial energy production making SNpc neurons vulnerable to a variety of challenge, especially the mitochondrial changes posed by various PD susceptibility genes. As the suppression of astrocyte melatonin prevents melatonin from suppressing α-synuclein levels and toxicity, SNpc neurons are significantly challenged by α-synuclein, irrespective of its source, as well as by the PD susceptibility genes that challenge mitochondrial function, including parkin, PINK1, and DJ-1, as well as α-synuclein [21,22]. Astrocytes, and the astrocyte tryptophan-melatonin pathway may therefore form an important hub for integrating CNS and systemic processes that ultimately drive PD pathophysiology, including from the melatonin regulation of astrocyte BMAL1 and BAG-3, which protects against both α-synuclein and hyperphosphorylated tau pathology [78].
The importance of the tryptophan-melatonin pathway, perhaps especially in astrocytes, in the pathophysiology of PD is given some support by wider data. A number of factors indicate suppressed melatonin production in the SNpc, including 1) decreased melatonin receptors in SNpc dopamine neurons, which may be indicative of suppressed melatonin, given that melatonin induces its own receptors [12]; 2) suppressed tryptophan levels and increased kynurenine pathway products in PD [1]; and 3) the efficacy of monoamine oxidase inhibitors on PD motor symptom treatment [79]. Importantly, decreased levels of different 14-3-3 isoforms significantly potentiate SNpc neuron loss with 14-3-3 isoforms attenuating α-synuclein aggregation and toxicity [80]. As noted, different 14-3-3 isoforms are important determinants of the tryptophan-melatonin pathway function (see Figure 1), with 14-3-3 isoforms proposed to modulate an array of processes linked to PD pathophysiology, including: 1) LRRK2 mutant effects [81]; 2) 14-3-3e stabilization decreasing α-synuclein [82]; 3) 14-3-3γ haploinsufficiency decreases SNpc dopamine production and induces motor deficits [83]; 4) 14-3-3ζ interacts with α-synuclein to maintain it in a monomeric form, thereby limiting α-synuclein toxicity [84]; 5) 14-3-3 interacts with, and regulates, parkin [85], thereby having relevance to the suppressed mitophagy, which is proposed to underpin the rise in MHC-1 and the chemoattraction of CD8+ T cells that drive SNpc dopamine neuron destruction, paralleling other ‘autoimmune’/’immune-mediated’ disorders [9].
The regulation of 14-3-3 isoforms may be intimately linked to the alterations in miRNAs evident in PD. A number of miRNAs, including miR-7, miR-375 and miR-451 can repress 14-3-3 isoforms, which is proposed to drive the suppression of the melatonergic pathway in intestinal epithelial cells, platelets and pinealocytes in autism [86]. As many miRNAs are induced by ROS, it will be interesting to determine whether the suboptimal mitochondrial function in PD drives ROS-regulated miRNAs that act to suppress 14-3-3 isoforms that are crucial in driving the enzymatic processes of the tryptophan-melatonin pathway (see Figure 1). The suppressed melatonergic pathway in astrocytes may, via decreased melatonin and lactate, raise mitochondrial ROS levels to drive miRNAs that act to suppress the melatonergic pathway in neurons. This is parsimonious with recent conceptualizations of cell elimination in a given microenvironment, where other cells act to suppress the melatonergic pathway in a cell to make it vulnerable to immune-mediated cell elimination [9,11].
The AhR and its activation by kynurenine is another factor that suppresses melatonin availability. The AhR induction of cytochrome P450 (CYP)1A2, CYP1B1 and CYP1A1 can suppress melatonin availability by two processes, namely: 1) the O-demethylation of melatonin to its immediate precursor NAS, which seems primarily mediated by CYP1A2 and CYP1B1; 2) the hydroxylation of melatonin to 6-hydroxymelatonin, which may be mediated predominantly by CYP1A1 and CYP1B1. The variable effects of these CYP1 isoforms arise from factors that bias the protein-protein interactions of melatonin with these CYP1 isoforms [87,88]. The plethora of endogenous and exogenous factors activating the AhR, such as air pollutants and kynurenine, may therefore contribute to the suppression of melatonin availability, The stress/HPA axis and morning CAR induction of TDO/kynurenine/AhR activation pathway allows heightened GR nuclear translocation and TDO induction to suppress melatonin availability by a number of means, including by decreasing the availability of tryptophan and by increasing melatonin hydroxylation and/or O-demethylation.
The role of brain-derived neurotrophic factor (BDNF) activation of the tyrosine receptor kinase (TrkB)-full length (FL) receptor is generally beneficial to neuronal survival and frequently recommended as a treatment target in neurodegenerative conditions, including PD [89]. However, TrkB also has a number of truncated forms, predominantly TrkB-T1, which generally has pro-apoptotic effects, and is linked to PD pathophysiology [90]. TrkB effects are further complicated by the expression of both TrkB-FL and TrkB-T1 on the plasma membrane and/or mitochondrial membrane [91]. This has consequences for AhR activation. As NAS is a BDNF mimic via TrkB activation [92], the AhR/CYP1A2/CYP1B1 O-demethylation of melatonin to NAS will activate TrkB, with differential consequences at the TrkB-FL vs TrkB-T1 and whether either form of TrkB is expressed on the mitochondrial and/or plasma membrane. NAS therefore has potentially negative consequences given the raised levels of the AhR evident in SNpc dopamine neurons and astrocytes as well as in the striatum, as shown in PD models [93], coupled to the raised levels of circulating and CNS kynurenine to activate the AhR [1]. As TrkB-T1 is induced under conditions of heightened ROS-driven miRNAs [94] the raised levels of AhR induced NAS may contribute to SNpc dopamine neuron loss in PD. Importantly, the heightened glutamatergic NMDAr activation evident in PD, including as induced YY1 suppression of astrocyte EAAT2 as well as by amyloid-β, upregulates TrkB-T1 levels [95].
The roles of TrkB and BDNF in neurodegenerative disorders may therefore also be intimately intertwined with the wider regulation of the melatonergic pathway, including by the AhR.
4.5. Melatonergic pathway regulation of amyloid-β and tau interactions with α-synuclein
There is a growing appreciation of the interactions of α-synuclein, with pathophysiological factors classically associated with Alzheimer’s disease, namely amyloid-β and hyperphosphorylated tau. Heightened amyloid-β levels that are evident in PD and Lewy Body diseases where they can increase α-synuclein aggregation [96,97], indicating a role for excessive amyloid-β and tau production in the pathophysiology of PD, especially when associated with dementia, and other tauopathies. Interestingly, melatonin affords powerful protection in dementia models where is it decreases the TRL4/NF-kB/YY1 induction of β-site amyloid precursor protein-cleaving enzyme (BACE)1 [98], whilst also preventing directly, and indirectly via decreased amyloid-β, tau hyperphosphorylation in neurons [99]. Melatonin also increases the non-amyloidogenic α-secretase activities of ‘A disintegrin and metalloproteinase domain-containing protein 10’ (ADAM10) and ADAM17, as well as suppressing the amyloidogenic β- and γ-secretases [100,101]. Such data would indicate that the attenuation of the melatonergic pathway in astrocytes and neurons may be an important commonality in the pathophysiological processes underpinning diverse neurodegenerative conditions [3]. See Figure 3. Notably, the suppression of melatonin and butyrate in PD allows plasma membrane GR activation to increase BACE1 and amyloid-β [102] whilst the GR also upregulates presenilin (PSEN)1 assembly on the endoplasmic reticulum (ER), which allows amyloid-β to accumulate on the ER mitochondrial associated membrane (MAM) [103]. It requires experimental investigation as to the nature of melatonin and butyrate’s modulation of such GR induced BACE1 and amyloid-β induction in the modulation of α-synuclein.
5. Integrating Parkinson’s disease pathoetiology and pathophysiology
Many PD prodromal symptoms, including REM sleep behavioral disorder and sleep/circadian dysregulation, are evident at night, indicative of alterations in night-time processes in PD pathoetiology [104,105]. As highlighted recently in other medical conditions, alterations in the night-time interactions of melatonin, butyrate and BAG-1 with GR activation in the course of CAR and stress-linked HPA axis activity may be important to the pathoetiology of diverse medical disorders, including cancer, Alzheimer’s disease and amyotrophic lateral sclerosis [30]. A growing body of data indicates the role of stress/trauma in the pathoetiology of REM sleep behavior disorder [106], with most REM sleep behavior disorder patients being diagnosed within 10 years with an α-synucleinopathy [107]. Notably, REM sleep behavior disorder is typically treated with melatonin [107], which, as highlighted throughout, significantly inhibits α-synuclein levels and toxicity. Cutting-edge research also indicates a significant role for decreased gut microbiome-derived butyrate in the pathoetiology of REM sleep behavior disorder [108], as well as in PD. Given the prodromal nature of REM sleep behavior disorder in PD susceptibility, such data strongly indicate a role for decreased melatonin and butyrate and increased stress in PD pathoetiology. The data reviewed above indicate that the pathoetiology of PD involves alterations in night-time processes, highlighting the importance of decreased melatonin and butyrate that increases GR nuclear translocation during the night and in the course of the morning CAR. The association of PD with aging would be parsimonious with this, given the 10-fold decrease in pineal melatonin over the course of aging (figure 2).
The data reviewed throughout indicates a progressive pathophysiological course that culminates with ‘immune-mediated’ SNpc dopamine neuron destruction in PD. The suppression of astrocyte (and SNpc neuronal) melatonin increases oxidative stress in SNpc dopamine neurons, which is exacerbated by an array of genetic and epigenetic susceptibility factors. The attenuation of astrocyte melatonin and lactate, coupled to increased glutamatergic excitatory activity suppresses SNpc dopamine neuron’s mitochondrial function, leading to heightened oxidative stress. Suboptimal mitochondrial function, enhanced excitatory activity and suppressed tryptophan-melatonin pathway induction in SNpc dopamine neurons contribute to the suppression of PINK1/parkin/mitophagy, thereby exacerbating mitochondria-derived oxidants and suboptimal mitochondrial metabolism. This provides a pathophysiological course whereby wider systemic processes (pineal melatonin, gut butyrate, cortisol/CAR GR) act on astrocytes to change their regulation of SNpc dopamine neurons. The failure of astrocytes to resolve the challenges posed by SNpc dopamine neuron dysfunction will lead to microglia chemoattraction and include microglia (expressing MHC-II [109]) as another important cells in the SNpc microenvironment. Although mitochondrial dysregulation driving MHC-1 upregulation in SNpc dopamine neurons may ultimately drive the chemoattraction of CD8+ T cells that destroy SNpc dopamine neurons, treatment of PD will require the monitoring and targeting of systemic processes and the modulation of the astrocyte tryptophan-melatonin pathway. This provides a conceptualization that may incorporate the roles of viral infections in PD susceptibility.
5.1. Integrating the role of herpes simplex virus-1 in Parkinson’s disease
Parsimonious with defining PD as an ‘immune-mediated’ disorder, there is a growing body of evidence implicating infections in PD pathophysiology, including cytomegalovirus (CMV), Epstein Barr virus (EBV), Borrelia burgdorferi (B. burgdorferi), Chlamydophila pneumoniae (C. pneumoniae) Helicobacter pylori (H. pylori) and especially herpes simplex virus type-1 (HSV-1) infection [110]. These authors also found infectious burden to be positively associated with heightened serum α-synuclein protein levels and the pro-inflammatory cytokines IL-1β and IL-6, indicating infection impacts on PD pathoetiology and pathophysiology [110]. HSV-1 is also significantly associated with Alzheimer’s disease risk and pathophysiology, including via increases in amyloid-β plaques and hyperphosphorylated (p)-tau tangles [111]. As indicated above, both amyloid-β and p-tau associate with PD and α-synuclein regulation as well as contributing to the cognitive loss in PD [112], including social cognition [70]. Notably, recent work indicates that the induction of both p-tau and amyloid-β is proposed to arise as an antimicrobial and anti-threat response, including to HSV-1 [11,113], indicating that, as in Alzheimer’s disease, amyloid-β and p-tau may be ‘too much of a good thing’, arising from the attenuated capacity of astrocytes to upregulate the tryptophan-melatonin pathway [2].
HSV-1 shows cross-reactivity with human α-synuclein peptides to drive an enhanced humoral response, indicating that HSV-1 stimulates immune cells against SNpc dopamine neurons expressing heightened levels of α-synuclein [114], including heightened CD8+ T cell and NK cell responses [115]. Such data has contributed to conceptualizations of PD as an ‘autoimmune’/’immune-mediated’ disorder, driven α-synuclein being presented by MHC-1 on SNpc dopamine (and perhaps by microglia MHC-I and –II [109]) leading to T cell activation and release of granzymes and proinflammatory cytokines that eventually result in neuron destruction [116]. This may be primed by a decrease in gut butyrate, leading to a decrease in the differentiation of T cells into regulatory T (Treg) cells, with consequent heightened enteric glial cell activation leading to the induction of α-synuclein (and prodromal PD gut symptomatology), eventually resulting in the retrograde transport of gut α-synuclein along the vagal nerve [117]. The administration of butyrate producing bacteria suppresses the impact of HSV-1 on gut-driven immune responses [118], suggesting a possible preventative role of an optimized gut microbiome in limiting α-synuclein production in the gut and its subsequent transport into the CNS. The importance of the gut microbiome is highlighted by data on the gut bacteria, Akkermansia muciniphila, which may be associated with the upregulation of the shikimate pathway in the gut microbiome [119] but is also capable of inducing mitochondrial dysfunction in enteroendocrine cells leading to α-synuclein upregulation [120]. As the A. muciniphila upregulation of the shikimate pathway would drive an increase in tryptophan, which is converted by tryptophan decarboxylase to tryptamine that activates the AhR to seal the gut [121], it would seem not unlikely that wider gut and gut microbiome dysregulation may prevent such protective A. muciniphila effects.
As melatonin can suppress HSV-1 symptoms [122], as well as increase gut microbiome derived butyrate in association with increased gut barrier integrity and decreased circulating LPS [123], the interactions of pineal and gut melatonin with butyrate producing bacteria may be relevant modulators of HSV-1 effects in PD, including HSV-1 effects in the gut and systemically, as well as in the CNS. The effects of butyrate and melatonin on HSV-1 may be important to place in the context of how these factors regulate the cortisol activation of the GR. Stress is a well-known mediator of HSV-1 reactivation [124], partly mediated by stress-linked GR activation [125]. GR induction of HSV-1 reactivation involves the promotor GRE, indicating that GR effects are mediated via GR nuclear translocation [126]. These authors show that the GR interacts with Slug, a stress-induced enhancer box (E-box) binding protein, in HSV-1 reactivation [126]. Such data strongly suggests that the inhibitory effects of melatonin and butyrate on HSV-1 reactivation and infection severity may be partly mediated via their suppression of GR nuclear translocation. Whether this is of particular relevance at night will be interesting to determine, with potential relevance to a wide array of other viral infections, especially given the growing appreciation of the role of viral infections in the pathoetiology of diverse medical conditions, including cancers and neurodegenerative disorders [127,128].
6. Future research directions
Is cellular melatonin decreased over the course of aging across body cells, in association with decreased pineal melatonin and sirtuin-3?
Does pineal and/or astrocyte melatonin induce lactate dehydrogenase in astrocytes, as shown in Sertoli cells [77], thereby allowing melatonin to regulate the availability of lactate as a necessary substrate for optimized neuronal mitochondrial metabolism?
Does suboptimal mitochondrial function in PD drive ROS-regulated miRNAs that inhibit 14-3-3 isoforms and therefore suppress the tryptophan-melatonin pathway in SNpc astrocytes and dopamine neurons?
As butyrate production is increased by the gut microbiome during fasting [129], are there any alterations in butyrate levels in PD (and REM behavioral disorder) patients at night? Is late or mid-sleep feeding relevant to PD pathoetiology?
Given that melatonin increases gut butyrate, seals the gut barrier and suppresses circulating LPS [123], is the production of butyrate at night intimately linked to variations in pineal melatonin production and therefore to the dramatic decrease in pineal melatonin over aging [30]?
As both dopamine and melatonin at high doses can activate the AhR [130,131], how does this impact on the role of the AhR in PD pathophysiology?
Is there any overlooked role of the hypothalamus in PD? Recent data indicates that pineal melatonin is released via the pineal recess into the third ventricle, which is lined by tanycytes that interact with hypothalamic astrocytes to regulate hypothalamic functions and peptide releases. As the hypothalamus is a crucial site for the regulation of core processes, such as stress, metabolism, and reproduction, and subject to ‘local stress’ regulation, as proposed for polycystic ovary syndrome [132], alterations in the melatonin and butyrate regulation of the night-time cortisol/GR effects on hypothalamic function may be an interesting avenue of novel research on PD pathophysiology.
7. Treatment implications
Extracellular vesicles, regulated exosomal contents, cell-penetrating peptides and miRNAs will provide novel treatment approaches [133], especially when further investigation on more refined pathophysiological processes have been identified in PD. As the above would suggest astrocytes are a core hub in PD pathophysiology, more refined targeting of the astrocyte tryptophan-melatonin pathway, will be an important target for novel treatment approaches e.g., via the contents of astrocyte targeted exosomes.
The utilization of melatonin and butyrate, and factors acting to upregulate their production, may provide some prodromal utility for people with PD susceptibility genes.
8. Conclusions
The above integrates a wide array of diverse data on PD pathoetiology and pathophysiology, highlighting the importance of the systemic regulation of the tryptophan-melatonin pathway in astrocytes and SNpc dopamine neurons. It is proposed that an aging-linked decrease in pineal (and perhaps local) melatonin, coupled to suppressed gut microbiome derived butyrate and suppressed BAG-1, lead to a heightened nuclear translocation of the cortisol-activated GR at night that shapes how the microenvironments and systems across the body are primed for the ‘coming day’. This provides a conceptualization that integrates previously disparate data on PD pathoetiology, including the role of viral infections and prodromal night-time circadian and REM sleep behavioral disorder. The astrocyte tryptophan-melatonin pathway may be an important hub in coordinating the wide array of diverse data on PD pathophysiology, with the suppression of astrocyte melatonin and lactate leading SNpc dopamine neurons vulnerable to oxidant and metabolic challenges. The suppression of the tryptophan-melatonin pathway in SNpc neurons contributes not only to increased α-synuclein levels and toxicity but also to the suppression of PINK1/parkin/mitophagy, leading to MHC-1 upregulation that chemoattracts CD8+ T cells, which mediate SNpc dopamine neuron destruction in an ‘autoimmune’/’immune-mediated’ manner. This has a number of future research and treatment implications.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Not applicable
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| AANAT | aralkylamine N-acetyltransferase |
| AhR | aryl hydrocarbon receptor |
| ASMT | acetylserotonin methytransferase |
| BACE1 | beta-site amyloid precursor protein cleaving enzyme 1 |
| BDNF | rain-derived neurotrophic factor |
| Bmal1 | brain and muscle ARNT-Like 1 |
| CYP | cytochrome P450 |
| EAAT | excitatory amino acid transporter |
| HDAC | histone deacetylase |
| HMGB | high-mobility group box |
| hsp | heat shock protein |
| IDO | indoleamine 2,3-dioxygenase |
| LPS | lipopolysaccharide |
| MHC-1 | major histocompatibility complex, class 1 |
| NAS | N-acetylserotonin |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer cell |
| NMDA | N-methyl-d-aspartate |
| OXPHOS | oxidative phosphorylation |
| PD | Parkinson’s disease |
| PDC | pyruvate dehydrogenase complex |
| PINK1 | PTEN-induced kinase 1 |
| REM | rapid eye movement |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| TCA | tricarboxylic acid |
| TDO | tryptophan 2,3-dioxygenase |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TPH2 | tryptophan hydroxylase 2 |
| Treg | regulatory t cell |
| TrkB | tyrosine receptor kinase B |
References
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut Permeability and Microbiota in Parkinson's Disease: Role of Depression, Tryptophan Catabolites, Oxidative and Nitrosative Stress and Melatonergic Pathways. Curr Pharm Des. 2016, 22(40), 6142–6151. [Google Scholar] [CrossRef]
- Anderson, G. Why Do Anti-Amyloid Beta Antibodies not work? Time to reconceptualize Dementia Pathophysiology by incorporating astrocyte melatonergic pathway desynchronization from amyloid-beta production. Braz J Psychiatry. 2023, 45(2), 89–92. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. A more holistic perspective of Alzheimer’s disease: Roles of gut microbiome, adipocytes, HPA axis, melatonergic pathway and astrocyte mitochondria in the emergence of autoimmunity. Front Biosc (Landmark), In press. 19989.
- Battis, K.; Xiang, W.; Winkler, J. The Bidirectional Interplay of α-Synuclein with Lipids in the Central Nervous System and Its Implications for the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 13270. [Google Scholar] [CrossRef]
- de Fàbregues, O.; Sellés, M.; Ramos-Vicente, D.; Roch, G.; Vila, M.; Bové, J. Relevance of tissue-resident memory CD8 T cells in the onset of Parkinson's disease and examination of its possible etiologies: infectious or autoimmune? Neurobiol Dis. 2023, 187, 106308. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.F. Alzheimer's disease as an innate autoimmune disease (AD2 ): A new molecular paradigm. Alzheimers Dement. 2022, Sep 27. [CrossRef]
- Cui, C.; Longinetti, E.; Larsson, H.; Andersson, J.; Pawitan, Y.; Piehl, F.; Fang, F. Associations between autoimmune diseases and amyotrophic lateral sclerosis: a register-based study. Amyotroph Lateral Scler Frontotemporal Degener, 2021; 22, 3-4, 211–219. [Google Scholar] [CrossRef]
- Fan, H.H.; Cui, L.; Jiang, X.X.; Song, Y.D.; Liu, S.S.; Wu, K.Y.; Dong, H.J.; Mao, M.; Ovlyakulov, B.; Wu, H.M.; et al. Autoimmune Disease Associated CLEC16A Variants Convey Risk of Parkinson's Disease in Han Chinese. Front Genet. 2022, 13, 856493. [Google Scholar] [CrossRef]
- Anderson, G.; Almulla, A.F.; Reiter, R.J.; Maes, M. Redefining Autoimmune Disorders’ Pathoetiology: Implications for Mood and Psychotic Disorders’ Association with Neurodegenerative and Classical Autoimmune Disorders. Cells 2023, 12, 1237. [Google Scholar] [CrossRef]
- Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, Ducrot C, Laplante A, Bourque MJ, Zhu L, Cayrol R, Le Campion A, McBride HM, Gruenheid S, Trudeau LE, Desjardins M. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1-/- mice. Nature. 2019, 571(7766), 565-569. [CrossRef]
- Anderson, G. Type I Diabetes Pathoetiology and Pathophysiology: Roles of the Gut Microbiome, Pancreatic Cellular Interactions, and the 'Bystander' Activation of Memory CD8+ T Cells. Int J Mol Sci. 2023, 24(4), 3300. [Google Scholar] [CrossRef]
- Adi, N.; Mash, D.C.; Ali, Y.; Singer, C.; Shehadeh, L.; Papapetropoulos, S. Melatonin MT1 and MT2 receptor expression in Parkinson's disease. Med Sci Monit. 2010, 16(2), BR61–7. [Google Scholar] [PubMed]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin's primary function and evolution in eukaryotes. J Pineal Res. 2013, 54(2), 127–38. [Google Scholar] [CrossRef] [PubMed]
- Nuzum, N.D.; Szymlek-Gay, E.A. Loke, S.; Dawson, S.L.; Teo, W.P.; Hendy, A.M.; Loughman, A.; Macpherson, H. Differences in the gut microbiome across typical ageing and in Parkinson's disease. Neuropharmacology, 2023, 235, 109566. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr Top Med Chem. 2020, 20(7), 524–539. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.; Israwi, A.R.; Brar, H.; Villafuerte, J.E.A.; Laylo, R.; Patel, H.; Jafri, S.; Al Halabi, L.; Sinnathurai, S.; Reehal, K.; et al. Parkinson's disease pathology is directly correlated to SIRT3 in human subjects and animal models: Implications for AAV.SIRT3-myc as a disease-modifying therapy. Neurobiol Dis, 2023, 187, 106287. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Perico, L.; Macconi, D. Mitochondrial Dynamics Is Linked to Longevity and Protects from End-Organ Injury: The Emerging Role of Sirtuin 3. Antioxid Redox Signal. 2016, 25(4), 185–99. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M.; Reiter, R.J. Melatonin and aging. Neuro Endocrinol Lett. 2002, 23(Supp1), 14–6. [Google Scholar]
- Guo, Y.L.; Wei, X.J.; Zhang, T.; Sun, T. Molecular mechanisms of melatonin-induced alleviation of synaptic dysfunction and neuroinflammation in Parkinson's disease: a review. Eur Rev Med Pharmacol Sci. 2023, 27(11), 5070–5082. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. Local melatonin regulates inflammation resolution: a common factor in neurodegenerative, psychiatric and systemic inflammatory disorders. CNS Neurol Disord Drug Targets. 2014, 13(5), 817–27. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harbor perspectives in medicine, 2012, 2, (9), a009415. [Google Scholar] [CrossRef]
- Cookson, M.R. alpha-Synuclein and neuronal cell death. Mol Neurodegener, 2009, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.; Ozkan, A.; Aytac, G.; Agar, A.; Tanriover, G. Role of melatonin in TLR4-mediated inflammatory pathway in the MTPT-induced mouse model. Neurotoxicology. 2022, 88, 168–177. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, S.; Qian, Y.; Mo, C.; Ai, P.; Yang, X.; Xiao, Q. Sodium butyrate ameliorates gut dysfunction and motor deficits in a mouse model of Parkinson's disease by regulating gut microbiota. Front Aging Neurosci. 2023, 15, 1099018. [Google Scholar] [CrossRef]
- Guedes, B.F.S; Cardoso, S.M.; Esteves, A.R. The Impact of microRNAs on Mitochondrial Function and Immunity: Relevance to Parkinson's Disease. Biomedicines. 2023, 11(5), 1349. [Google Scholar] [CrossRef]
- Seo, M.; Anderson, G. Gut-Amygdala Interactions in Autism Spectrum Disorders: Developmental Roles via regulating Mitochondria, Exosomes, Immunity and microRNAs. Curr Pharm Des. 2019, 25(41), 4344–4356. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: role of melatonin and lipid peroxidation. Br J Nutr. 2016, 116(10), 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Stewart, T.; Zhang, C.; Wang, P.; Xu, Z.; Jin, J.; Huang, Y.; Liu, Z.; Lan, G.; Liang, X.; Sheng, L.; Shi, M.; Cai, Z.; Zhang, J. Erythrocytic α-Synuclein and the Gut Microbiome: Kindling of the Gut-Brain Axis in Parkinson's Disease. Mov Disord. 5. [CrossRef]
- Beura, S.K.; Panigrahi, A.R.; Yadav, P.; Singh, S.K. Role of platelet in Parkinson's disease: Insights into pathophysiology & theranostic solutions. Ageing Res Rev. 2022, 80, 101681. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Gut Microbiome and Circadian Interactions with Platelets Across Human Diseases, including Alzheimer's Disease, Amyotrophic Lateral Sclerosis, and Cancer. Curr Top Med Chem. 2023, Oct 2. [CrossRef]
- Pépin, É.; Jalinier, T.; Lemieux, G.L.; Massicotte, G.; Cyr, M. Sphingosine-1-Phosphate Receptors Modulators Decrease Signs of Neuroinflammation and Prevent Parkinson's Disease Symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Front Pharmacol. 2020, 11, 77. [Google Scholar] [CrossRef]
- Montanari, M.; Imbriani, P.; Bonsi, P.; Martella, G.; Peppe, A. Beyond the Microbiota: Understanding the Role of the Enteric Nervous System in Parkinson's Disease from Mice to Human. Biomedicines. 2023, 11(6), 1560. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hao, Y.; Shabanpoor, A.; Metz, G. A. Stress and corticosterone alter synaptic plasticity in a rat model of Parkinson's disease. Neurosc Lett. 2017, 651, 79–87 doiorg/101016/jneulet201704063. [Google Scholar] [CrossRef] [PubMed]
- Soares, N. M., Pereira, G. M., Altmann, V., de Almeida, R. M. M., & Rieder, C. R. M. Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson's disease: a systematic review. J Neural Transm (Vienna). 2019, 126(3), 219-232. [CrossRef]
- Anderson, G.; Maes, M. TRYCAT pathways link peripheral inflammation, nicotine, somatization and depression in the etiology and course of Parkinson's disease. CNS Neurol Disord Drug Targets. 2014, 13(1), 137–49. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Anderson, G.; Maes, M. Hypothalamic-Pituitary-Adrenal Hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a Consequence of Activated Immune-Inflammatory and Oxidative and Nitrosative Pathways. Mol Neurobiol. 2017, 54(9), 6806–6819. [Google Scholar] [CrossRef]
- Muntsant, A.; Giménez-Llort, L. Crosstalk of Alzheimer's disease-phenotype, HPA axis, splenic oxidative stress and frailty in late-stages of dementia, with special concerns on the effects of social isolation: A translational neuroscience approach. Front Aging Neurosci. 2022, 14, 969381. [Google Scholar] [CrossRef]
- Hickie, I.B.; Naismith, S.L.; Robillard, R.; Scott, E.M.; Hermens, D.F. Manipulating the sleep-wake cycle and circadian rhythms to improve clinical management of major depression. BMC Med. 2013, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Robertson-Dixon, I.; Murphy, M.J.; Crewther, S.G.; Riddell, N. The Influence of Light Wavelength on Human HPA Axis Rhythms: A Systematic Review. Life (Basel). 2023, 13(10), 1968. [Google Scholar] [CrossRef] [PubMed]
- Law, R.; Clow, A. Stress, the cortisol awakening response and cognitive function. Int Rev Neurobiol. 2020, 150, 187–217. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Melatonin, BAG-1 and cortisol circadian interactions in tumor pathogenesis and patterned immune responses. Explor Target Antitumor Ther. 2023, 4, 962–93. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Why are aging and stress associated with dementia, cancer, and other diverse medical conditions? Role of pineal melatonin interactions with the HPA axis in mitochondrial regulation via BAG-1. Melatonin Res. 2023, 6(3), 345–371. [Google Scholar] [CrossRef]
- Quiros, I.; Mayo, J.C.; Garcia-Suarez, O.; Hevia, D.; Martin, V.; Rodríguez, C.; Sainz, R.M. Melatonin prevents glucocorticoid inhibition of cell proliferation and toxicity in hippocampal cells by reducing glucocorticoid receptor nuclear translocation. J Steroid Biochem Mol Biol. 2008; 110, 1-2, 116–24. [Google Scholar] [CrossRef]
- Kim, M.; Lee, H.A.; Cho, H.M.; Kang, S.H.; Lee, E.; Kim, I.K. Histone deacetylase inhibition attenuates hepatic steatosis in rats with experimental Cushing's syndrome. Korean J Physiol Pharmacol. 2018, 22(1), 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Zhang, Y.; Zhang, J.; Zhou, F.; Zhang, L.; Wang, S.; Zhu, Q.; Liu, Q.; Wang, X.; Zhou, L. Acetylation of Hsp90 reverses dexamethasone-mediated inhibition of insulin secretion. Toxicol Lett. 2020, 320, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Daytime orexin and night-time melatonin regulation of mitochondria melatonin:roles in circadian oscillations systemically and centrally in breast cancer symptomatology. Melatonin Res. 2019, 2(4) 1-8. [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: a proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–19. [Google Scholar] [CrossRef]
- Kermer, P.; Köhn, A.; Schnieder, M.; Lingor, P.; Bähr, M.; Liman, J.; Dohm, C.P. BAG1 is neuroprotective in in vivo and in vitro models of Parkinson's disease. J Mol Neurosci. 2015, 55(3), 587–95. [Google Scholar] [CrossRef]
- Meka, S.T.; Bojja, S.L.; Kumar, G.; Birangal, S.R.; Rao, C. M. Novel HDAC inhibitors provide neuroprotection in MPTP-induced Parkinson's disease model of rats. Eur J Pharmacol. 2023, 959, 176067. [Google Scholar] [CrossRef]
- Guttuso, T.; Jr, Shepherd, R. ; Frick, L.; Feltri, M.L.; Frerichs, V.; Ramanathan, M.; Zivadinov, R.; Bergsland, N. Lithium's effects on therapeutic targets and MRI biomarkers in Parkinson's disease: A pilot clinical trial. IBRO Neurosci Rep. 2023, 14, 429–434. [Google Scholar] [CrossRef]
- Zhou, R.; Gray, N.A.; Yuan, P.; Li, X.; Chen, J.; Chen, G.; Damschroder-Williams, P.; Du, J.; Zhang, L.; Manji, H.K. The anti-apoptotic, glucocorticoid receptor cochaperone protein BAG-1 is a long-term target for the actions of mood stabilizers. J Neurosci. 2005, 25(18), 4493–502. [Google Scholar] [CrossRef]
- Luo, S.; Hou, Y.; Zhang, Y.; Feng, L.; Hunter, R.G.; Yuan, P.; Jia, Y.; Li, H.; Wang, G.; K Manji, H.; S McEwen, B.; Xiao, C.; Bao, H.; Du, J. Bag-1 mediates glucocorticoid receptor trafficking to mitochondria after corticosterone stimulation: Potential role in regulating affective resilience. J Neurochem. 2021, 158(2), 358–372. [Google Scholar] [CrossRef]
- Németh, H.; Toldi, J.; Vécsei, L. Role of kynurenines in the central and peripheral nervous systems. Curr Neurovasc Res. 2005, 2(3), 249–60. [Google Scholar] [CrossRef]
- Qin, W.; Shi, Y.; Chen, W.; Jia, X.; Asakawa, T. Can kynurenine pathway be considered as a next-generation therapeutic target for Parkinson's disease? An update information. Biosci Trends. 2022, 16(4), 249–256. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Neurodegeneration in Parkinson's disease: interactions of oxidative stress, tryptophan catabolites and depression with mitochondria and sirtuins. Mol Neurobiol. 2014, 49(2), 771–83. [Google Scholar] [CrossRef]
- Chen, P.; Geng, X. Research progress on the kynurenine pathway in the prevention and treatment of Parkinson's disease. J Enzyme Inhib Med Chem. 2023, 38(1), 2225800. [Google Scholar] [CrossRef]
- Anderson, G.; Carbone, A.; Mazzoccoli, G. Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress. Biology (Basel). 2020, 9(9), 249. [Google Scholar] [CrossRef]
- Fernandes, P.A.; Tamura, E.K.; D'Argenio-Garcia, L.; Muxel, S.M.; da Silveira Cruz-Machado, S.; Marçola, M.; Carvalho-Sousa, C.E.; Cecon, E.; Ferreira, Z.S.; Markus, R.P. Dual Effect of Catecholamines and Corticosterone Crosstalk on Pineal Gland Melatonin Synthesis. Neuroendocrinology. 2017, 104(2), 126–134. [Google Scholar] [CrossRef]
- Vanuytsel, T.; van Wanrooy, S.; Vanheel, H.; Vanormelingen, C.; Verschueren, S.; Houben, E.; Salim Rasoel, S.; Tόth, J.; Holvoet, L.; Farré, R.; Van Oudenhove, L.; Boeckxstaens, G.; Verbeke, K.; Tack, J. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut, 63(8), 2014, 1293–1299. [CrossRef]
- Shukla, P.K.; Meena, A.S.; Pierre, J.F.; Rao, R. Central role of intestinal epithelial glucocorticoid receptor in alcohol- and corticosterone-induced gut permeability and systemic response. FASEB J. 2022, 36(1), e22061. [Google Scholar] [CrossRef]
- Khan, H.N.; Perlee, D.; Schoenmaker, L.; van der Meer, A.J.; Franitza, M.; Toliat, M.R.; Nürnberg, P.; Zwinderman, A.H.; van der Poll, T.; Scicluna, B. P. Leukocyte transcriptional signatures dependent on LPS dosage in human endotoxemia. J Leukoc Biol. 2019, 106(5), 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Fan, L.; Yue, B.H.; Lou, Z. Saikosaponin A mitigates the progression of Parkinson's disease via attenuating microglial neuroinflammation through TLR4/MyD88/NF-κB pathway. Eur Rev Med Pharmacol Sci. 2023, 27(15), 6956–6971. [Google Scholar] [CrossRef]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.; Kodi, T.; Satarker, S.; Gurram, P.C.; Fayaz, S.M.; Nampoothiri, M. Astrocytic transcription factors REST, YY1, and putative microRNAs in Parkinson's disease and advanced therapeutic strategies. Gene. 2023, Oct 11, 892:147898. [CrossRef]
- Karki, P.; Webb, A.; Smith, K.; Johnson, J.; Jr, Lee, K.; Son, D S.; Aschner, M.; Lee, E.Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol Cell Biol. 2014, 34(7), 1280-9. [CrossRef]
- Sheng, L.; Stewart, T.; Yang, D.; Thorland, E.; Soltys, D.; Aro, P.; Khrisat, T.; Xie, Z.; Li, N.; Liu, Z.; Tian, C.; Bercow, M.; Matsumoto, J.; Zabetian, C.P.; Peskind, E.; Quinn, J.F.; Shi, M.; Zhang, J. Erythrocytic α-synuclein contained in microvesicles regulates astrocytic glutamate homeostasis: a new perspective on Parkinson's disease pathogenesis. Acta Neuropathol Commun. 2020, 8(1), 102. [Google Scholar] [CrossRef] [PubMed]
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.; Cecon, E.; Tamura, E K.; Floeter-Winter, L.M.; Markus, R.P. NF-κB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS One. 2012, 7(12), e52010. [CrossRef]
- Bernard, M.; Voisin, P. Photoreceptor-specific expression, light-dependent localization, and transcriptional targets of the zinc-finger protein Yin Yang 1 in the chicken retina. J Neurochem. 2008, 105(3), 595–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Zhuang, J.; Zhu, H.Y.; Shen, Y.X.; Tan, Z.L.; Zhou, J.N. Cultured rat cortical astrocytes synthesize melatonin: absence of a diurnal rhythm. J Pineal Res. 2007, 43(3), 232–8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Behnisch, T. The Enigmatic CA2: Exploring the Understudied Region of the Hippocampus and Its Involvement in Parkinson's Disease. Biomedicines. 2023, 11(7), 1996. [Google Scholar] [CrossRef] [PubMed]
- Melachroinou, K.; Divolis, G.; Tsafaras, G.; Karampetsou, M.; Fortis, S.; Stratoulias, Y.; Papadopoulou, G.; Kriebardis, A.G.; Samiotaki, M.; Vekrellis, K. Endogenous Alpha-Synuclein is Essential for the Transfer of Pathology by Exosome-Enriched Extracellular Vesicles, Following Inoculation with Preformed Fibrils in vivo. Aging Dis. 2023, Aug 4. [CrossRef]
- Dai, L.; Wang, J.; Zhang, X.; Yan, M.; Zhou, L.; Zhang, G.; Meng, L.; Chen, L.; Cao, X.; Zhang, Z.; Wang, G.; Zhang, Z. 27-Hydroxycholesterol Drives the Spread of α-Synuclein Pathology in Parkinson's Disease. Mov Disord. 2023, Aug 18. [CrossRef]
- Ku, H.; Kim, Y.; Kim, A.L.; Lee, G.; Choi, Y.; Kim, B. Protective Effects of Melatonin in High-Fat Diet-Induced Hepatic Steatosis via Decreased Intestinal Lipid Absorption and Hepatic Cholesterol Synthesis. Endocrinol Metab (Seoul). 2023, 38(5), 557–567. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.H.; Tung, Y.T.; Yang, T.H.; Chien, Y.W. Melatonin Improves Fatty Liver Syndrome by Inhibiting the Lipogenesis Pathway in Hamsters with High-Fat Diet-Induced Hyperlipidemia. Nutrients. 2019, 11(4), 748. [Google Scholar] [CrossRef]
- Kakimoto, T.; Hosokawa, M.; Ichimura-Shimizu, M.; Ogawa, H.; Miyakami, Y.; Sumida, S.; Tsuneyama, K. Accumulation of α-synuclein in hepatocytes in nonalcoholic steatohepatitis and its usefulness in pathological diagnosis. Pathol Res Pract. 2023, 247, 154525. [Google Scholar] [CrossRef]
- Xiao, Y.; Guo, Z.; Li, Z.; Ling, H.; Song, C. Role and mechanism of action of butyrate in atherosclerotic diseases: a review. J Appl Microbiol. 2021, 131(2), 543–552. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.P.; Matzkin, M.E.; Riviere, E.; Martinez, G.; Ponzio, R.; Levalle, O.; Terradas, C.; Calandra, R.S.; Frungieri, M.B. Melatonin improves oxidative state and lactate metabolism in rodent Sertoli cells. Mol Cell Endocrinol. 2023, 576, 112034. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, P.W.; Nadarajah, C.J.; Kanan, M.F.; Patterson, J.N.; Novotny, B.; Lawrence, J.H.; King, M.W.; Brase, L.; Inman, C.E.; Yuede, C.M.; Lee, J.; Patel, T.K.; Harari, O.; Benitez, B.A.; Davis, A A.; Musiek, E.S. An astrocyte BMAL1-BAG3 axis protects against alpha-synuclein and tau pathology. Neuron. 2023, 111(15), 2383-2398.e7. [CrossRef]
- Espinoza-Vinces, C.; Villino-Rodríguez, R.; Atorrasagasti-Villar, A.; Martí-Andrés, G.; Luquin, M.R. Impact of Safinamide on Patient-Reported Outcomes in Parkinson's Disease. Patient Relat Outcome Meas. 2023, 14, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Underwood, R.; Gannon, M.; Pathak, A.; Kapa, N.; Chandra, S.; Klop, A.; Yacoubian, T.A. 14-3-3 mitigates alpha-synuclein aggregation and toxicity in the in vivo preformed fibril model. Acta Neuropathol Commun. 2021, 9(1), 13. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, R; Petit, C.M.; Yacoubian, T.A. 14-3-3 phosphorylation inhibits 14-3-3θ's ability to regulate LRRK2 kinase activity. bioRxiv [Preprint]. 2023, 2023.05.27.542591. [CrossRef]
- Vinueza-Gavilanes, R.; Bravo-González, J.J.; Basurco, L.; Boncristiani, C.; Fernández-Irigoyen, J.; Santamaría, E.; Marcilla, I.; Pérez-Mediavilla, A.; Luquin, M.R.; Vales, A.; González-Aseguinolaza, G.; Aymerich, M.S.; Aragón, T.; Arrasate, M. Stabilization of 14-3-3 protein-protein interactions with Fusicoccin-A decreases alpha-synuclein dependent cell-autonomous death in neuronal and mouse models. Neurobiol Dis. 2023, 183, 106166. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Park, J.; Hwang, E.M.; Kim, H.W.; Park, J.Y. 14-3-3γ haploinsufficiency leads to altered dopamine pathway and Parkinson's disease-like motor incoordination in mice. Mol Brain. 2023, 16(1), 2. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Thorn, D.C.; Dobson, C.M.; Meehan, S.; Jackson, S.E.; Woodcock, J.M.; Carver, J.A. The Amyloid Fibril-Forming β-Sheet Regions of Amyloid β and α-Synuclein Preferentially Interact with the Molecular Chaperone 14-3-3ζ. Molecules. 2021, 26(20), 6120. [Google Scholar] [CrossRef] [PubMed]
- Giusto, E.; Yacoubian, T.A.; Greggio, E.; Civiero, L. Pathways to Parkinson's disease: a spotlight on 14-3-3 proteins. NPJ Parkinsons Dis. 2021, 7(1), 85. [Google Scholar] [CrossRef] [PubMed]
- Pagan, C.; Goubran-Botros, H.; Delorme, R.; Benabou, M.; Lemière, N.; Murray, K.; Amsellem, F.; Callebert, J.; Chaste, P.; Jamain, S.; Fauchereau, F.; Huguet, G.; Maronde, E.; Leboyer, M.; Launay, J.M.; Bourgeron, T. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci Rep. 2017, 7(1), 2096. [Google Scholar] [CrossRef]
- Mokkawes, T.; De Visser, T.; Cao, Y.; De Visser, S. P. Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes. Molecules. 2023, 28(19), 6961. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab Dispos. 2005, 33(4), 489–94. [Google Scholar] [CrossRef] [PubMed]
- Fenner, M.E.; Achim, C.L.; Fenner, B.M. Expression of full-length and truncated trkB in human striatum and substantia nigra neurons: implications for Parkinson's disease. J Mol Histol. 2014, 45(3), 349–61. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson's Disease. J Clin Med. 2020, 9(1), 257. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Siemen, D.; Mawrin, C.; Horn, T.F.; Dietzmann, K. The neurotrophin receptor TrkB is colocalized to mitochondrial membranes. Int J Biochem Cell Biol. 2006, 38(4), 610–20. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W. , Liu, X., Pradoldej, S., Tosini, G., Chang, Q., Iuvone, P.M., Ye, K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc Natl Acad Sci U S A. 2010, 107(8), 3876–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, W.J.; Quan, W.; Qiao, C.M.; Cui, C.; Hong, H.; Shi, Y.; Niu, G.Y.; Zhao, L.P.; Shen, Y.Q. Dynamic changes of activated AHR in microglia and astrocytes in the substantia nigra-striatum system in an MPTP-induced Parkinson's disease mouse model. Brain Res Bull. 2021, 176, 174–183. [Google Scholar] [CrossRef]
- Maussion, G.; Yang, J.; Yerko, V.; Barker, P.; Mechawar, N.; Ernst, C.; Turecki, G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by Hsa-miR-185* in frontal cortex of suicide completers. PLoS One. 2012, 7(6), e39301. [Google Scholar] [CrossRef] [PubMed]
- Tanqueiro, S.R.; Ramalho, R.M.; Rodrigues, T.M.; Lopes, L.V.; Sebastião, A.M.; Diógenes, M.J. Inhibition of NMDA Receptors Prevents the Loss of BDNF Function Induced by Amyloid β. Front Pharmacol. 2018, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Vadukul, D.M.; Papp, M.; Thrush, R.J.; Wang, J.; Jin, Y.; Arosio, P.; Aprile, F.A. α-Synuclein Aggregation Is Triggered by Oligomeric Amyloid-β 42 via Heterogeneous Primary Nucleation. J Am Chem Soc. 2023, 145(33), 18276–18285. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, K.; Fernando, S.; Fisher, P.R.; Annesley, S.J. Interactions and Cytotoxicity of Human Neurodegeneration- Associated Proteins Tau and α-Synuclein in the Simple Model Dictyostelium discoideum. Front Cell Dev Biol. 2021, 9, 741662. [Google Scholar] [CrossRef]
- Sun, C. , Qiu, X., Wang, Y., Liu, J., Li, Q., Jiang, H., Li, S., & Song, C. (2020). Long-term oral melatonin alleviates memory deficits, reduces amyloid-β deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice. Neurosci Lett. 2020, 735, 135192. [Google Scholar] [CrossRef]
- Das, R. , Balmik, A. A., & Chinnathambi, S. Melatonin Reduces GSK3β-Mediated Tau Phosphorylation, Enhances Nrf2 Nuclear Translocation and Anti-Inflammation. ASN Neuro. 2020, 12, 1759091420981204. [Google Scholar] [CrossRef]
- Shukla, M.; Htoo, H.H.; Wintachai, P.; Hernandez, J.F.; Dubois, C.; Postina, R.; Xu, H.; Checler, F.; Smith, D.R.; Govitrapong, P; Vincent, B. Melatonin stimulates the nonamyloidogenic processing of βAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J Pineal Res. 2015, 58(2), 151-65. [CrossRef]
- Panmanee, J.; Nopparat, C.; Chavanich, N.; Shukla, M.; Mukda, S.; Song, W.; Vincent, B.; Govitrapong, P. Melatonin regulates the transcription of βAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J Pineal Res. 2015 Oct;59(3):308-20. [CrossRef]
- Choi, G.E.; Lee, S.J.; Lee, H.J.; Ko, S.H.; Chae, C.W.; Han, H.J. Membrane-Associated Effects of Glucocorticoid on BACE1 Upregulation and Aβ Generation: Involvement of Lipid Raft-Mediated CREB Activation. J Neurosci. 2017, 37(35), 8459–8476. [Google Scholar] [CrossRef]
- Choi, G.E.; Park, J.Y.; Park, M.R.; Yoon, J.H.; Han, H.J. Glucocorticoid enhances presenilin1-dependent Aβ production at ER's mitochondrial-associated membrane by downregulating Rer1 in neuronal cells. Redox Biol. 2023, 65, 102821. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; Titze-de-Almeida, S.S.; Ferreira, G.G.; Brito Silva, A.P.; de Paula Brandão, P.R.; Oertel, W.H.; Schenck, C.H.; Delgado Rodrigues, R.N. microRNA signatures in prodromal REM sleep behavior disorder and early Parkinson's disease as noninvasive biomarkers. Sleep Med. 2021, 78, 160–168. [Google Scholar] [CrossRef]
- Kunz, D.; Oster, H.; Rawashdeh, O.; Neumann, W.J.; Münte, T.; Berg, D. Sleep and circadian rhythms in α-synucleinopathies-Perspectives for disease modification. Acta Physiol (Oxf). 2023, 238(1), e13966. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, K.; So, H.S.; Choi, J.H.; Yoon, I.Y.; Choi, H. REM Sleep Behavior Disorder among Veterans with and without Post-Traumatic Stress Disorder. Psychiatry Investig. 2020, 17(10), 987–995. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Garay, A. Melatonin as a Chronobiotic/Cytoprotective Agent in REM Sleep Behavior Disorder. Brain Sci. 2023, 13(5), 797. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Chau, S.W.H.; Liu, Y.; Chan, J.W.Y.; Wang, J.; Ma, S.L.; Zhang, J.; Chan, P.K.S.; Yeoh, Y.K.; Chen, Z.; Zhou, L.; Wong, S.H.; Mok, V.C.T.; To, K.F.; Lai, H.M.; Ng, S.; Trenkwalder, C.; Chan, F.K.L.; Wing, Y.K. Gut microbiome dysbiosis across early Parkinson's disease, REM sleep behavior disorder and their first-degree relatives. Nat Commun. 2023, 14(1), 2501. [Google Scholar] [CrossRef]
- Liang, S.Q.; Li, P.H.; Hu, Y.Y.; Zhao, J L. ; Shao, F.Z.; Kuang, F.; Ren, K.X.; Wei, T.X.; Fan, F.; Feng, L.; Han, H.; Qin, H.Y. Myeloid-specific blockade of notch signaling alleviates dopaminergic neurodegeneration in Parkinson's disease by dominantly regulating resident microglia activation through NF-κB signaling. Front Immunol. 2023, 14, 1193081. [Google Scholar] [CrossRef]
- Bu, X.L.; Wang, X.; Xiang, Y.; Shen, L.L.; Wang, Q.H.; Liu, Y.H.; Jiao, S.S.; Wang, Y.R.; Cao, H.Y.; Yi, X.; Liu, C.H.; Deng, B.; Yao, X.Q.; Xu, Z.Q.; Zhou, H.D.; Wang, Y.J. The association between infectious burden and Parkinson's disease: A case-control study. Parkinsonism Relat Disord. 2015, 21(8), 877–81. [Google Scholar] [CrossRef] [PubMed]
- Powell-Doherty, R.D.; Abbott, A.R.N.; Nelson, L.A.; Bertke, A.S. Amyloid-β and p-Tau Anti-Threat Response to Herpes Simplex Virus 1 Infection in Primary Adult Murine Hippocampal Neurons. J Virol. 2020, 94(9), e01874–19. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; He, Z. Concomitant protein pathogenesis in Parkinson's disease and perspective mechanisms. Front Aging Neurosci. 2023, 15, 1189809. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Depression Pathophysiology: Astrocyte Mitochondrial Melatonergic Pathway as Crucial Hub. Int J Mol Sci. 2022, 24(1), 350. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Arru, G.; Piredda, R.; Sechi, G.P.; Sechi, L.A. Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson's disease, a triggering role in the disease? J Neuroimmunol. 2016, 291, 110–4. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Galleri, G.; Arru, G.; Manetti, R.; Sechi, G.P.; Sechi, L.A. Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson's disease. J Neuroimmunol. 2017, 310, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in Parkinson's Disease: The Role of α-Synuclein-Specific T Cells. Front Immunol. 2019, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Campos-Acuña, J.; Elgueta, D.; Pacheco, R. T-Cell-Driven Inflammation as a Mediator of the Gut-Brain Axis Involved in Parkinson's Disease. Front Immunol. 2019, 10, 239. [Google Scholar] [CrossRef]
- Joubert, M.; André, M.; Barnich, N.; Billard, E. Microbiome and Behçet's disease: a systematic review. Clin Exp Rheumatol. 2023, Jun 7. [CrossRef]
- Mesnage, R.; Antoniou, M.N. Computational modelling provides insight into the effects of glyphosate on the shikimate pathway in the human gut microbiome. Curr. Res. Toxicol. 2020, 1, 25–33. [Google Scholar] [CrossRef]
- Amorim Neto, D.P.; Bosque, B.P.; Pereira de Godoy, J.V.; Rodrigues, P.V.; Meneses, D.D.; Tostes, K.; Costa Tonoli, C.C.; Faustino de Carvalho, H.; González-Billault, C.; de Castro Fonseca, M. Akkermansia muciniphila induces mitochondrial calcium overload and α -synuclein aggregation in an enteroendocrine cell line. iScience. 2022, 25(3), 103908. [Google Scholar] [CrossRef]
- Dopkins, N.; Becker, W.; Miranda, K.; Walla, M.; Nagarkatti, P.; Nagarkatti, M. Tryptamine Attenuates Experimental Multiple Sclerosis Through Activation of Aryl Hydrocarbon Receptor. Front Pharmacol. 2021, 11, 619265. [Google Scholar] [CrossRef]
- Nunes Oda, S.; Pereira Rde, S. Regression of herpes viral infection symptoms using melatonin and SB-73: comparison with Acyclovir. J Pineal Res. 2008, 44(4), 373–8. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.l Cao, J.l Dong, Y.; Chen, Y. Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome. 2023, 11(1), 17. [CrossRef]
- Goswami, P.; Ives, A.M.; Abbott, A.R.N.; Bertke, A.S. Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons. Viruses. 2022, 14(5), 1115. [Google Scholar] [CrossRef]
- Harrison, K.S.; Wijesekera, N.; Robinson, A.G.J.; Santos, V.C.; Oakley, R.H.; Cidlowski, J.A.; Jones, C. Impaired glucocorticoid receptor function attenuates herpes simplex virus 1 production during explant-induced reactivation from latency in female mice. J Virol. 2023, Oct 12, e0130523. [Google Scholar] [CrossRef]
- Santos, V.C.; Ostler, J.B.; Harrison, K.S.; Jones, C. Slug, a Stress-Induced Transcription Factor, Stimulates Herpes Simplex Virus 1 Replication and Transactivates a cis-Regulatory Module within the VP16 Promoter. J Virol. 2023, 97(4), e0007323. [Google Scholar] [CrossRef]
- Ogarek, N.; Oboza, P.; Olszanecka-Glinianowicz, M.; Kocelak, P. SARS-CoV-2 infection as a potential risk factor for the development of cancer. Front Mol Biosci. 2023, 10, 1260776. [Google Scholar] [CrossRef]
- Cohen, M.; Austin, E.; Bradu, S.; Jagdeo, J. The Association Between Herpes Simplex Virus and Alzheimer's Disease: A Systematic Review. J Drugs Dermatol. 2023, 22(10), 1046–1052. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; Zale, E.; Wang, Y.; Huang, Y.; Theriault, B.; Dinner, A.R.; Musch, M.W.; Kudsk, K.A.; Prendergast, B.J.; Gilbert, J.A.; Chang, E.B. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015, 17(5), 681–9. [Google Scholar] [CrossRef]
- Park, H.; Jin, U.H.; Karki, K.; Jayaraman, A.; Allred, C.; Michelhaugh, S.K.; Mittal, S.; Chapkin, R.S.; Safe, S. Dopamine is an aryl hydrocarbon receptor agonist. Biochem J. 2020, 477(19), 3899–3910. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Slominski, R.M.; Song, Y.; Qayyum, S.; Placha, W.; Janjetovic, Z.; Kleszczyński, K.; Atigadda, V.; Song, Y.; Raman, C.; Elferink, C.J.; Hobrath, J.V.; Jetten, A.M.; Reiter, R.J. Melatonin and Its Metabolites Can Serve as Agonists on the Aryl Hydrocarbon Receptor and Peroxisome Proliferator-Activated Receptor Gamma. Int J Mol Sci. 2023, 24(20), 15496. [Google Scholar] [CrossRef]
- Milutinović, D. V., Macut, D., Božić, I., Nestorov, J., Damjanović, S., & Matić, G. (2011). Hypothalamic-pituitary-adrenocortical axis hypersensitivity and glucocorticoid receptor expression and function in women with polycystic ovary syndrome. Exp Clin Endocrinol Diabetes. 2011, 119(10), 636-43. [CrossRef]
- Keighron, C.N.; Avazzadeh, S.; Goljanek-Whysall, K.; McDonagh, B.; Howard, L.; Ritter, T.; Quinlan, L.R. Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson's and Alzheimer's Disease. Biomedicines. 2023, 11(3), 728. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Shows the tryptophan-melatonin pathway. Gut and shikimate derived tryptophan is taken up from the circulation by the astrocyte large amino acids transporter (LAT)-1. Astrocyte tryptophan hydroxylase (TPH)2, stabilized by 14-3-3, converts tryptophan to 5-hydroxytryptophan (5-HTP), with 5-HTP converted by Aromatic-L-amino acid decarboxylase (AACD) to serotonin (5-HT). Serotonin is converted to N-acetylserotonin (NAS) by 14-3-3 stabilized aralkylamine N-acetyltransferase (AANAT) in the presence of acetyl-coenzyme A (acetyl-CoA). NAS is then converted by acetylserotonin methyltransferase (ASMT). Abbreviations: 5-HT: 5-hydroxytryptamine; 5-HTTP: 5-hydroxytryptophan; AACD: aromatic-L-amino acid decarboxylase; AANAT: aralkylamine N-acetyltransferase; acetyl-CoA: acetyl-coenzyme A; ASMT: acetylserotonin methyltransferase; LAT: large amino acids transporter; NAS: N-acetylserotonin.
Figure 1.
Shows the tryptophan-melatonin pathway. Gut and shikimate derived tryptophan is taken up from the circulation by the astrocyte large amino acids transporter (LAT)-1. Astrocyte tryptophan hydroxylase (TPH)2, stabilized by 14-3-3, converts tryptophan to 5-hydroxytryptophan (5-HTP), with 5-HTP converted by Aromatic-L-amino acid decarboxylase (AACD) to serotonin (5-HT). Serotonin is converted to N-acetylserotonin (NAS) by 14-3-3 stabilized aralkylamine N-acetyltransferase (AANAT) in the presence of acetyl-coenzyme A (acetyl-CoA). NAS is then converted by acetylserotonin methyltransferase (ASMT). Abbreviations: 5-HT: 5-hydroxytryptamine; 5-HTTP: 5-hydroxytryptophan; AACD: aromatic-L-amino acid decarboxylase; AANAT: aralkylamine N-acetyltransferase; acetyl-CoA: acetyl-coenzyme A; ASMT: acetylserotonin methyltransferase; LAT: large amino acids transporter; NAS: N-acetylserotonin.
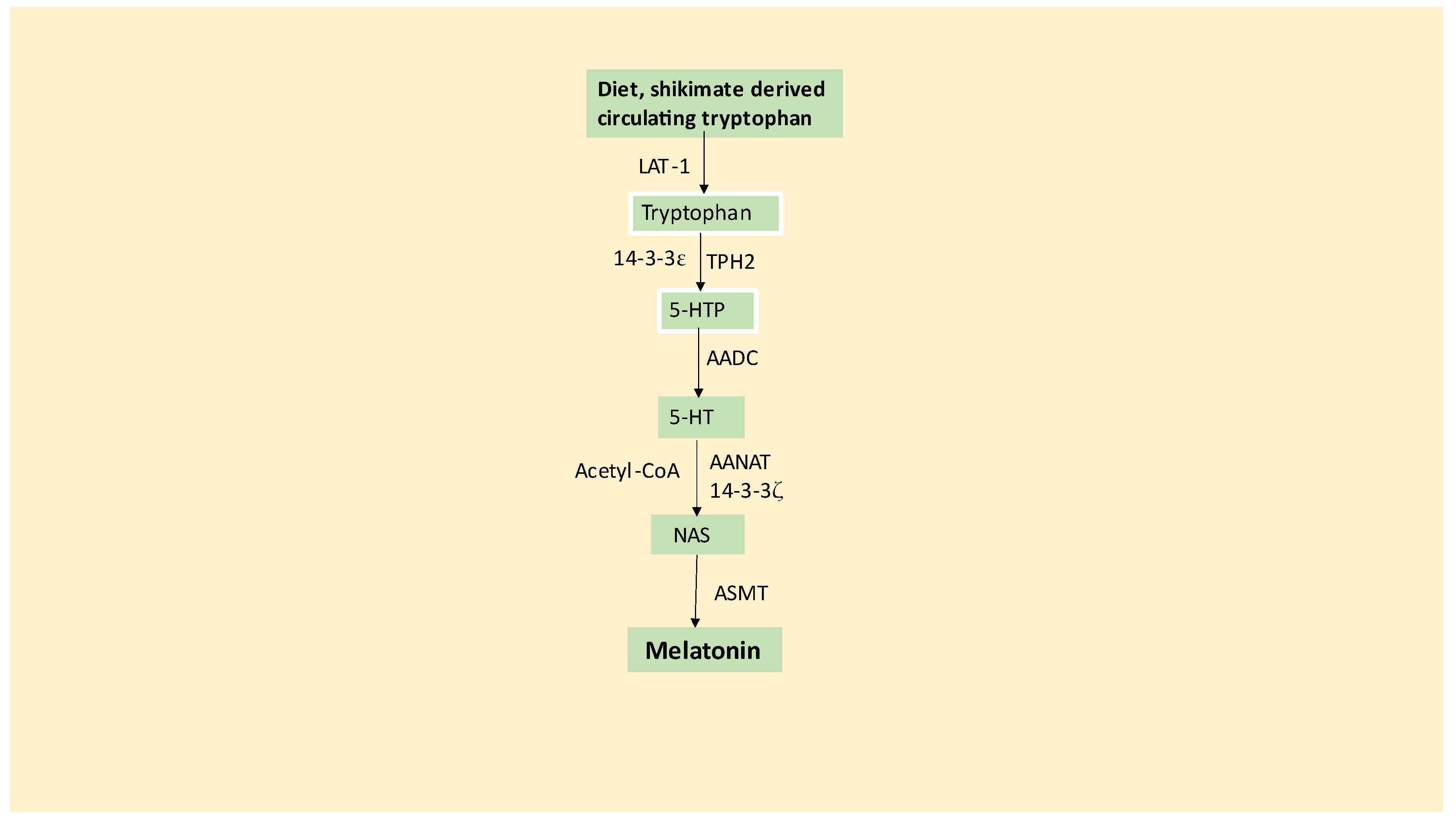
Figure 2.
Shows typical variations in cortisol and melatonin over the evening, night (sleep) and morning periods. Plasma cortisol gradually rises over the sleep period, with an accelerated rise around wakening, known as the cortisol awakening response (CAR). Plasma melatonin rapidly rises at night, slowly decreasing over the night and morning periods. The dramatic decrease in pineal melatonin in the elderly is not matched by significant changes in the cortisol rhythm and CAR. The suppression of melatonin in the elderly attenuates melatonin’s suppression of the glucocorticoid receptor nuclear translocation, thereby altering the consequences of cortisol effects at night and during CAR.
Figure 2.
Shows typical variations in cortisol and melatonin over the evening, night (sleep) and morning periods. Plasma cortisol gradually rises over the sleep period, with an accelerated rise around wakening, known as the cortisol awakening response (CAR). Plasma melatonin rapidly rises at night, slowly decreasing over the night and morning periods. The dramatic decrease in pineal melatonin in the elderly is not matched by significant changes in the cortisol rhythm and CAR. The suppression of melatonin in the elderly attenuates melatonin’s suppression of the glucocorticoid receptor nuclear translocation, thereby altering the consequences of cortisol effects at night and during CAR.
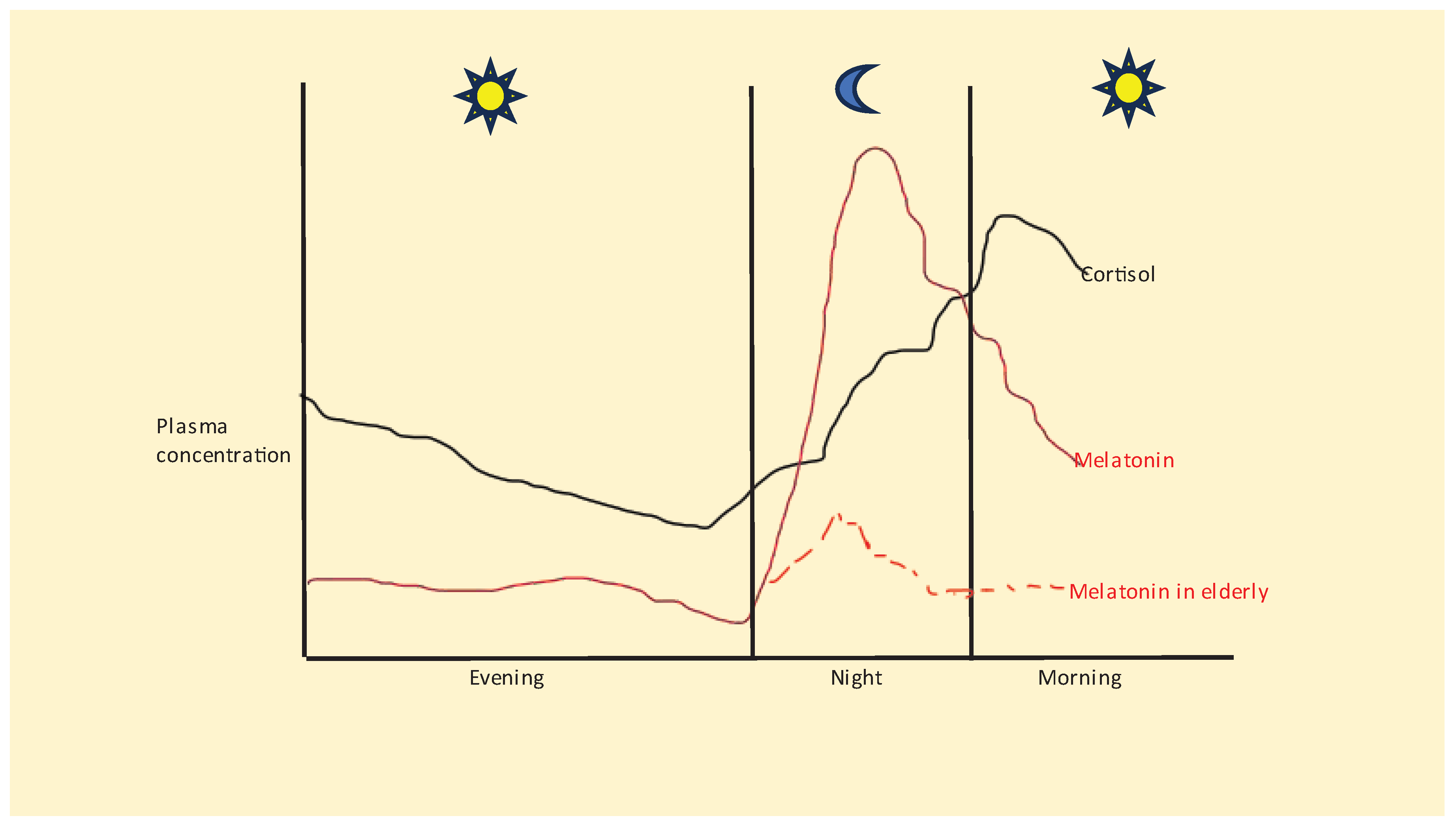
Figure 3.
Shows how many of the factors associated with PD pathophysiology may be intimately linked to the tryptophan-melatonin pathway (green shade), with relevance for PD pathophysiology in SNpc dopamine neurons but especially in SNpc astrocytes. The tryptophan-melatonin pathway is regulated by a wide array of systemic and CNS processes, including: 1) Tryptophan availability from diet and gut microbiome shikimate pathway. Cortisol-induced TDO and pro-inflammatory cytokine-induced IDO suppress tryptophan availability by converting tryptophan to kynurenine, which has a number of consequences including activating the AhR as directly and via other kynurenine pathway products regulating the neuronal activity. The AhR (blue shade) O-demethylates melatonin to NAS via CYP1A2 and CYP1B1, with NAS, like BDNF, activating TrkB receptors, with differential effects at TrkB-FL, versus TrkB-T1, as well as when TrkB is on the mitochondrial, versus plasma, membrane. Kynurenine activation of the AhR can therefore deplete cellular melatonin, whilst driving diverse effects of NAS. 2) GR activation also suppresses pineal melatonin and increases gut permeability, thereby increasing circulating LPS, whilst also inducing gut dysbiosis-linked decreases in butyrate. The suppression of pineal melatonin and gut butyrate attenuates sirtuin-3 induction and therefore inhibits mitochondrial ATP and acetyl-CoA production by the PDC. As acetyl-CoA is a necessary cosubstrate for the initiation of the mitochondrial melatonergic pathway, this is one route whereby heightened cortisol can suppress the melatonergic pathway, whilst also potentiating GR nuclear translocation during morning CAR and stress-linked HPA axis activation. 3) Given the decrease in a number of 14-3-3 isoforms in PD, it is notable that 14-3-3 isoforms are crucial to the regulation of the tryptophan-melatonin pathway, via 14-3-3 stabilizing astrocyte TPH2 and AANAT. By suppressing melatonin, such factors increase α-synuclein and contribute to wider PD pathophysiology, such as decreased mitophagy. The suppressed levels of melatonin will also modulate the consequences of GR and LPS as well as the association of increased NF-kB and YY1 induced BACE1 and amyloid-β as well as hyperphosphorylated tau in PD pathophysiology, including dementia. Abbreviations: 5-HT: serotonin; 5-HTP: 5-hydroxytryptophan; AADC: aromatic-L-amino acid decarboxylase; AANAT: Aralkylamine N-acetyltransferase; AhR: aryl hydrocarbon receptor; ASMT: N-acetylserotonin O-methyltransferase; BACE1: β-site amyloid precursor protein-cleaving enzyme 1; BDNF: brain-derived neurotrophic factor; CRH: corticotrophin releasing hormone; CYP: cytochrome P450: GR: glucocorticoid receptor; HPA: hypothalamus-pituitary-adrenal; LAT-1: large amino acid transporter 1; LPS: lipopolysaccharide; NAS: N-acetylserotonin; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; TLR: toll-like receptor; TrkB-FL: tyrosine receptor kinase B-full length; TrkB-T1: tyrosine receptor kinase B-truncated.
Figure 3.
Shows how many of the factors associated with PD pathophysiology may be intimately linked to the tryptophan-melatonin pathway (green shade), with relevance for PD pathophysiology in SNpc dopamine neurons but especially in SNpc astrocytes. The tryptophan-melatonin pathway is regulated by a wide array of systemic and CNS processes, including: 1) Tryptophan availability from diet and gut microbiome shikimate pathway. Cortisol-induced TDO and pro-inflammatory cytokine-induced IDO suppress tryptophan availability by converting tryptophan to kynurenine, which has a number of consequences including activating the AhR as directly and via other kynurenine pathway products regulating the neuronal activity. The AhR (blue shade) O-demethylates melatonin to NAS via CYP1A2 and CYP1B1, with NAS, like BDNF, activating TrkB receptors, with differential effects at TrkB-FL, versus TrkB-T1, as well as when TrkB is on the mitochondrial, versus plasma, membrane. Kynurenine activation of the AhR can therefore deplete cellular melatonin, whilst driving diverse effects of NAS. 2) GR activation also suppresses pineal melatonin and increases gut permeability, thereby increasing circulating LPS, whilst also inducing gut dysbiosis-linked decreases in butyrate. The suppression of pineal melatonin and gut butyrate attenuates sirtuin-3 induction and therefore inhibits mitochondrial ATP and acetyl-CoA production by the PDC. As acetyl-CoA is a necessary cosubstrate for the initiation of the mitochondrial melatonergic pathway, this is one route whereby heightened cortisol can suppress the melatonergic pathway, whilst also potentiating GR nuclear translocation during morning CAR and stress-linked HPA axis activation. 3) Given the decrease in a number of 14-3-3 isoforms in PD, it is notable that 14-3-3 isoforms are crucial to the regulation of the tryptophan-melatonin pathway, via 14-3-3 stabilizing astrocyte TPH2 and AANAT. By suppressing melatonin, such factors increase α-synuclein and contribute to wider PD pathophysiology, such as decreased mitophagy. The suppressed levels of melatonin will also modulate the consequences of GR and LPS as well as the association of increased NF-kB and YY1 induced BACE1 and amyloid-β as well as hyperphosphorylated tau in PD pathophysiology, including dementia. Abbreviations: 5-HT: serotonin; 5-HTP: 5-hydroxytryptophan; AADC: aromatic-L-amino acid decarboxylase; AANAT: Aralkylamine N-acetyltransferase; AhR: aryl hydrocarbon receptor; ASMT: N-acetylserotonin O-methyltransferase; BACE1: β-site amyloid precursor protein-cleaving enzyme 1; BDNF: brain-derived neurotrophic factor; CRH: corticotrophin releasing hormone; CYP: cytochrome P450: GR: glucocorticoid receptor; HPA: hypothalamus-pituitary-adrenal; LAT-1: large amino acid transporter 1; LPS: lipopolysaccharide; NAS: N-acetylserotonin; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; TLR: toll-like receptor; TrkB-FL: tyrosine receptor kinase B-full length; TrkB-T1: tyrosine receptor kinase B-truncated.
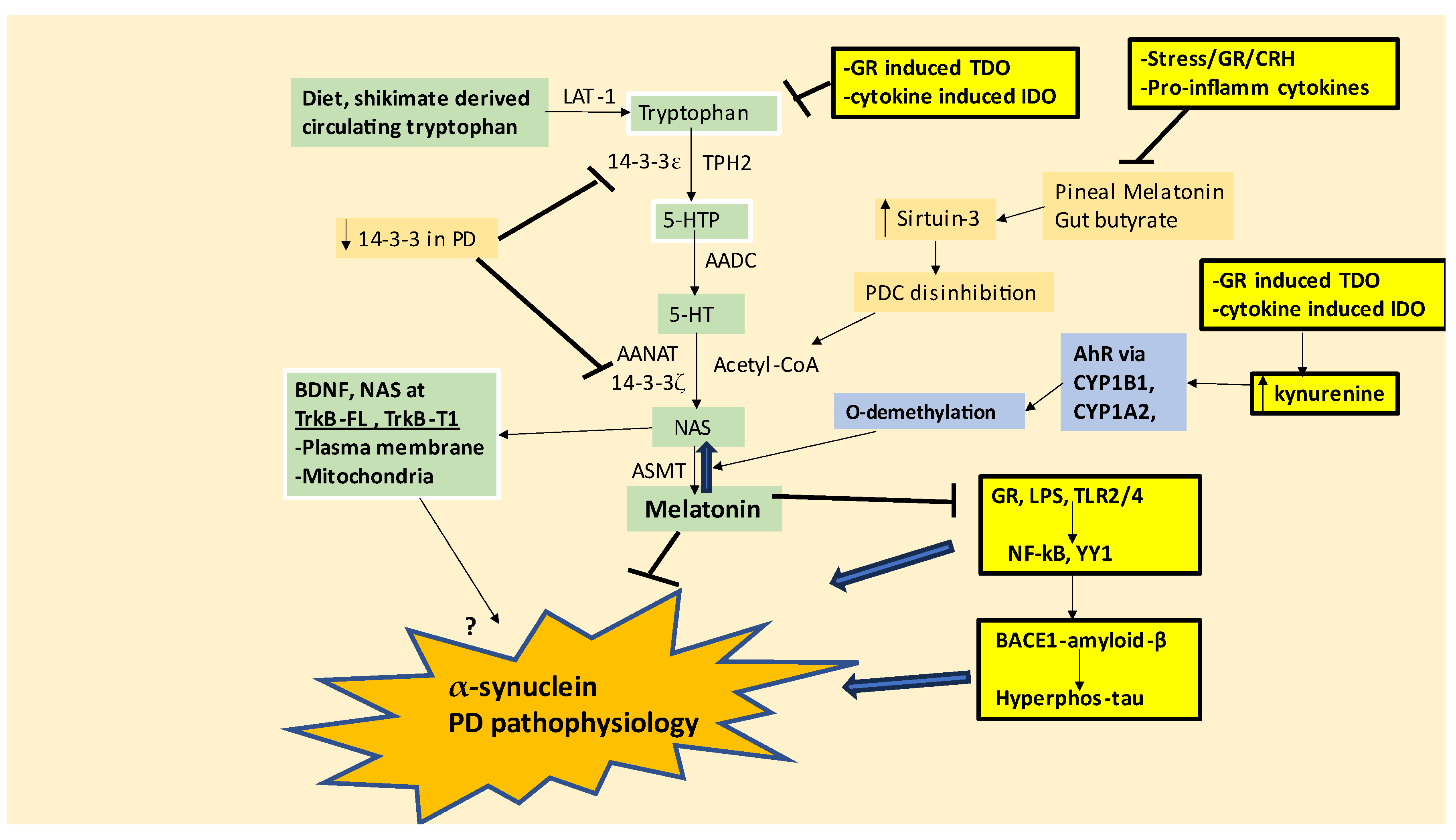
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



