
Submitted:
06 November 2023
Posted:
06 November 2023
You are already at the latest version
Abstract
This work presents the synthesis and self-organization of the calamitic fluorinated mesogen, 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-4-iodobutoxy)ethanesulfonic acid, a potential model for perfluorosulfonic acid membranes (PFSA). The compound is derived in three steps from 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoethoxy)ethanesulfonyl fluoride, achieving an 78% overall yield. The resulting compound exhibits intricate thermal behavior. At 150 °C, a crystal-to-crystal transition is observed due to the partial disordering of calamitic molecules, which is followed by isotropization at 218 °C. Upon cooling, sample ordering occurs through the formation of large smectic liquid crystalline phase domains. This thermotropic state transforms into a layered crystal phase at lower temperatures, characterized by alternating hydrophilic and hydrophobic layers. Using X-ray diffraction, crystalline unit cell models at both room temperature and 170 °C were proposed. Computer simulations of the molecule across varying temperatures support the idea that thermal transitions correlate with a loss of molecular orientation. Importantly, the study underscores the pivotal role of precursor self-organization in aligning channels during membrane fabrication, ensuring controlled and oriented positioning.
Keywords:
DSC
; Fluorinated compounds
; liquid crystals
; melting point
; smectic
1. Introduction
In recent years, increasing concern has been raised about the growing environmental burden caused by the use of fossil fuels and the overall increase in energy consumption, especially in EU countries [1,2,3]. Searching for clean energy, particularly decarbonized sources, is relevant, and according to photovoltaics, PV [4,5], Lithium-ion batteries, LIB [6], wind mills [7], and fuel cells [8,9], these appear as innovating and useful means for overcoming such challenges. The fuel cell is a device that converts the electrochemical energy of a fuel’s oxidation-reduction into electricity, heat, and water. One of the most important components is the membrane, and among them, perfluorosulfonic acid ones (PFSA) as Nafion®, Flemion®, Aquivion®, and 3M ®membranes are commercially available from Chemours, Fumatech, Solvay, and 3M Innovative, respectively [10]. Many articles have been published on the synthesis, characterization, processing, and electrochemical application into portable, small, and bigger embedded devices (vehicles that are already produced from Toyota [11], Honda, or Hyundai). It has been found that proton transport properties of PFSA membranes strongly depend on their morphology [12]. The self-assembly of fluorinated side-groups determines the volume fraction, topology, and size of water channels. Despite numerous publications on the correlation between chemical structure, preparation conditions, and final supramolecular morphology of polymeric membranes, this question remains open due to polymer specificity, such as slow structure formation and polydispersity of model ionomers [13,14,15]. To better understand the self-assembly process in ion-transporting membranes, it would be of interest to propose models (i.e. small molecules) capable of self-assembling under conditions similar to those for fluorinated comb-like copolymers. Such semi-rigid amphiphilic compounds, known as calamitics, are known to form layered, cylindrical, or cubic superstructures as a function of molecular shape, temperature, humidity, etc. [16,17,18]. For example, Berrod et al. [19] suggested original perfluorosulfonic acid molecules that can reproduce phase-separated morphology of PFSA ionomer membranes used as electrolytes in fuel cells.
Investigation of thermotropic mesogens are also of great interest from the fundamental point of view as the peculiarities of structure and assembly mechanisms of liquid crystals is yet to be fully understood [20,21,22]. Specific details on molecular level arrangements, conditions of transitions between different phases and so on can be resolved through various simulations techniques [23,24], and comparison between experimental and theoretical data can be used for the refinements of force fields and models, leading to better understating of physical reasons of mentioned assemblies. Up today there are numerous modeling studies related to thermotropic mesogens [23,24,25,26,27] which show the high ability of modern methods to predict and reveal some features of the structuring of mesogens qualitatively as well as quantitatively.
2. Materials and Methods
Synthesis
1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoetoxy)ethanesulfonyl fluoride was supplied from Appollo while ethylene provided by Air Liquide, France; Perkadox 16S was purchased from Akzo Chemicals and tertbutanol and Li2CO3 from Sigma Aldrich.
Ethylenation of the iodofluorocompounds.
Ethylenation of 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoetoxy)ethanesulfonyl fluoride occurred in 50 mL Hastelloyl (HC276) autoclave equipped with inlet and outlet valves, a manometer, a rupture disk, a mechanical stirrer and a controller to check the speeding rate and the temperature. A solution composed of di(tert-butylcycloheylperoxy dicarbonate (Perkadox 16S) (0.247 g, 0.63 mmol), 28 mL of tert-butanol, and 16.82 g (39.4 mmol) of 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoetoxy)ethanesulfonyl fluoride was degassed by N2 bubbling for 30 min. During this time, the autoclave was put under vacuum (40 × 10−3 bar) for 30 min to remove the residual traces of oxygen. The above solution was transferred into the autoclave under vacuum via a funnel tightly connected to the introduction valve of the autoclave. Then, the vessel was cooled in a liquid nitrogen bath, and ethylene (2 g, 45.5 mmol) was introduced through the autoclave inlet valve while monitoring its amount by double weighing (i.e., the difference of masses before and after filling the autoclave with ethylene). The autoclave was then allowed to warm to ambient temperature and heated to the target reaction temperature (60 °C) under mechanical stirring and the evolutions of pressure and temperature were recorded. Suddenly, ca. after one hour-reaction at 60 °C, a fast high increase of pressure up to 70 bar occurred immediately followed by a drop of pressure as low as 5 bar, considering a complete reaction. The autoclave was then cooled in an ice bath for 30 minutes and no released of unreacted gas was noted, followed by the opening of the vessel. The total product mixture (yellow solid) was recovered from the autoclave and characterized by 19F NMR. The evidence by the absence of signal at -59 ppm assigned to CF2CF2I end-group indicated the quantitative conversion of 5-iodo-3-oxaperfluoropentane-1-sulfonyl fluoride. The crude product was then dissolved in acetone and precipitated from water and finally dried under vacuum (20 × 10−3 bar, 50 °C) for 8 h yielding 17.55 g of a yellowish powder (yield=94%).
Oxidation of the sulfonyl fluoride in I-CH2CH2CF2CF2OCF2CF2SO2F into SO3H was carried out in slightly basic conditions in presence of K2CO3 to lead to ICH2CH2CF2CF2OCF2CF2SO3Li (yield not determined). This latter was then acidified to chemically change the -SO3Li end-group into sulfonic acid, thus producing ICH2CH2CF2CF2OCF2CF2SO3H as a white powder. The overall yield from 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoetoxy)ethanesulfonyl fluoride was ca. 78%.
Methods of Characterization
The morphology and physical properties of the sample were studied through thermogravimetric analysis, differential scanning calorimetry, polarized optical microscopy, temperature-resolved small- (SAXS) and wide-angle X-ray scattering (WAXS), and computer simulation.
Thermogravimetric experiments were conducted using TGA Q500 from TA Instruments from 25 °C to 900 °C at a heating rate of 10 °C under a nitrogen flow 100 ml/min.
Differential scanning calorimetry (DSC) measurements were carried out using a Polyma 214, Netzsch. Samples were placed in pierced aluminum pans and subjected to two heating − cooling cycles from 20 to 260 °C at a rate of 10 K.min−1.
Microscopic images of thin films were captured using a Carl Zeiss AxioScope A1 POL optical microscope in polarized light with 100x objective. The films, prepared from the melt between two cover glasses, were positioned on a Linkam LTS heating stage. A small amount of the sample was heated to 260 °C, above melting temperature, and then crystallized at two different cooling rates: 1 and 10 K.min−1. Images were captured with a 5 MP CMOS camera.
Temperature-resolved SAXS and WAXS measurements were carried out at the ID02 beamline of the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). The measurements were performed in transmission geometry with X-ray photons at an energy of 11.9 keV. The calibration of the norm of the reciprocal vector s ( where θ is the Bragg angle and λ-wavelength) was done using several diffraction orders of silver behenate (for SAXS) and corundum (for WAXS).
All-atom molecular dynamics (AAMD) simulations of I(CH2)2(CF2)2O(CF2)2SO2F melt under different temperature conditions were carried out as follows. The initial unit cell configuration was established through Monte Carlo annealing among a set of most common symmetry groups, with geometry optimization using the COMPASS force field [28]. The elementary configuration with the lowest energy was then replicated to form a periodic box measuring 100 Å x 30 Å x 30 Å, containing 288 I(CH2)2(CF2)2O(CF2)2SO2F molecules. The COMPASS force field was utilized for the AAMD simulations, and electrostatic interactions were computed using the PPPM method [29].
The system was equilibrated in the isobaric-isothermal ensemble (NPT) for 10 ns with a Berendsen barostat [30] and a Nosé -Hoover thermostat [31], at a temperature of T = 25 °C and a pressure of P = 1 atm. Subsequently, the simulation box was heated sequentially by 5 °C every 10 ns up to 280 °C. Statistics were gathered over the last 1 ns of every heating step across three independent heating procedures.
Radial distribution function (RDF) and static structure factor S(k) were utilized for charactering the translational order of molecules within the box. Orientational ordering was monitored by calculating the first eigenvalue of nematic Q-tensor, [32], where γ is the angle between molecules' first principal axis and the director. The size of the molecules was estimated based on the calculation of the gyration radius Rg.
3. Results and discussion
1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-4-iodobutoxy)ethanesulfonic acid was synthesized in three steps from 1,1,2,2-tetrafluoro-2-(1,1,2,2-tetrafluoro-2-iodoetoxy)ethanesulfonyl fluoride (Scheme 1): i) by a quantitative ethylenation initiated by tert-butyl cyclohexyl peroxydicarbonate at 60 °C (that generates only the monoadduct as in previous works [33]. The vanishing of the signal at -59 ppm (highlighted by 19F NMR spectrum) and the presence of two complex multiplets at 2.3 and 3.0 ppm in the 1H NMR spectrum evidenced the formation of the ethylenated product (with the insertion of one ethylene unit only); ii) the second step enabled to modify the sulfonyl fluoride end group into SO3Li under mild basic conditions to avoid the dehydroiodination. iii) the final steps dealt with the acidification of that latter into sulfonic acid. These reactions were monitored by 19F, 1H and IR spectroscopies. The O-F frequencies (bending modes) centered at 1470 and 810 cm-1 (noted in the spectrum of the product bearing SO3Li end-group) were absent after the hydrolysis reaction while the -S(O)(O)OH stretching vibrations of the sulfonic acid groups at 3377 cm-1 and 1026 cm-1 were present, indicating the successful hydrolysis reaction. Additionally, the hydrolysis of the -SO2F groups was confirmed by the 19F NMR spectrum that does not display any signal at +45 ppm characteristic of the sulfonyl fluoride group [34]. The absence of this peak indicates that the hydrolysis-reaction yield was quasi quantitative.
The overall yield was 78 % from 5-iodo-3-oxaperfluoropentane-1-sulfonyl fluoride.
Structural Characteristics of I(CH2)2(CF2)2O(CF2)2SO2OH
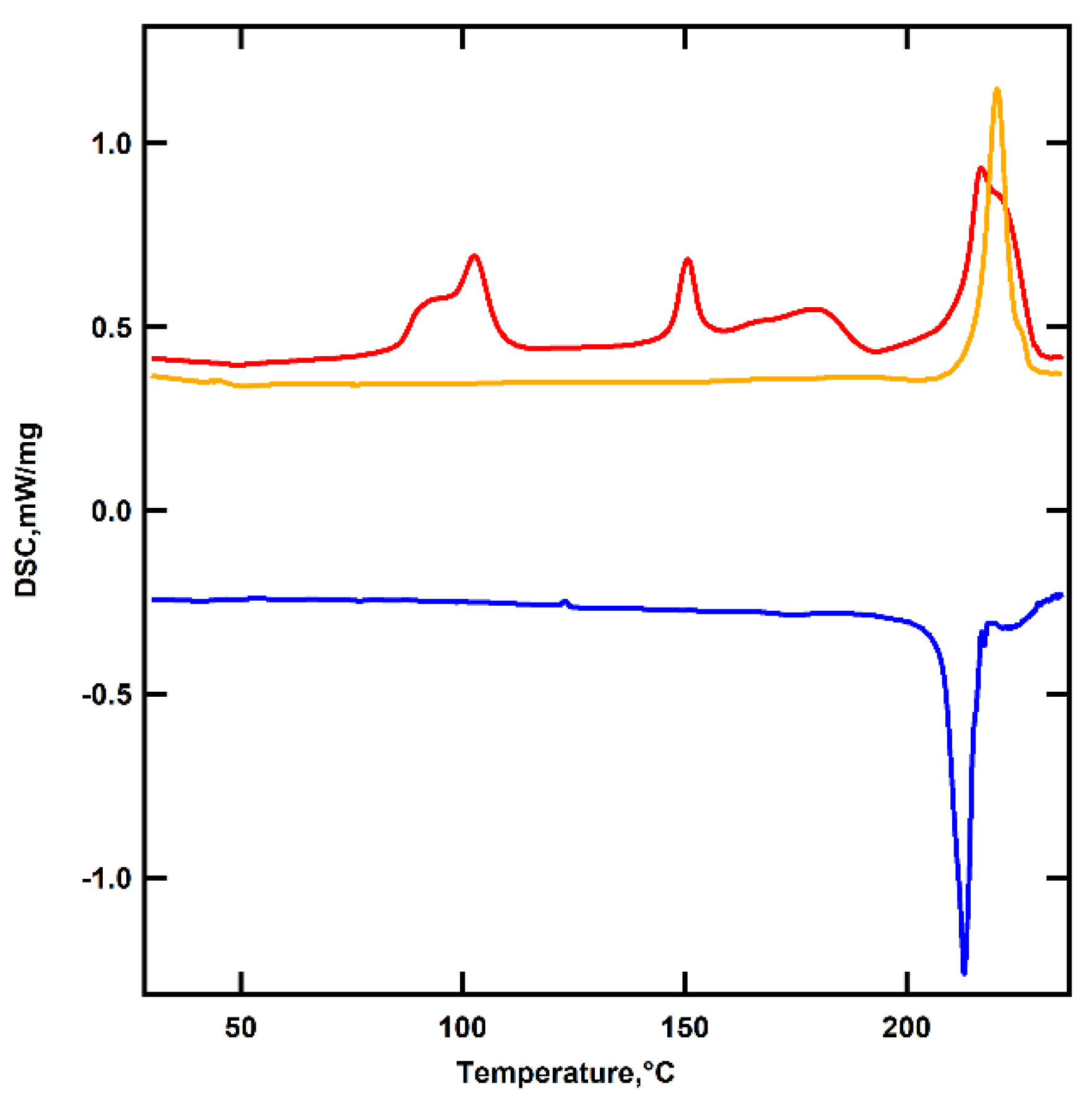
The first DSC heating thermogram shows a double endothermic peak within the temperature range of 80 to 110 °C, which is attributed to the sample’s dehydration or dehydroiodination. The peak at 150 °C is likely due to a crystal-to-crystal transition, typical for amphiphilic calamitic molecules [35]. An intense peak at 218 °C corresponds to the sample’s isotropization [36]. The second heating curve shows only isotropization, indicating irreversibility of the solid-to-solid transition.
Figure 1.
DSC thermograms of I(CH2)2(CF2)2O(CF2)2SO2OH: 1st heating (yellow), cooling (blue) and 2nd heating (red). Exo processes are down.
Figure 1.
DSC thermograms of I(CH2)2(CF2)2O(CF2)2SO2OH: 1st heating (yellow), cooling (blue) and 2nd heating (red). Exo processes are down.
Polarized optical microscopy of thin films of I(CH2)2(CF2)2O(CF2)2SO2OH at 216 °C reveals crystalline spherulites and rod-like domains of the smectic liquid crystalline (LC) phase (Figure 2a). It is plausible that the LC phase is thermotropic, which signifies that it is in equilibrium in a certain temperature range. During cooling, the nucleation rate increases, leading to the emergence of a grainy polycrystalline texture at room temperature (Figure 2b).
Structural analysis was performed using temperature-resolved small- (SAXS) and wide-angle (WAXS) X-ray scattering. At room temperature, powder diffraction indicates a highly ordered crystalline structure with numerous peaks in the WAXS region (Figure 3). The SAXS region shows peaks typical for a layered structure. The complete table of the peaks is presented in Table S1. According to SAXS, the longest inter-layer distance is 2.72 nm. The high-intensity peaks at s=4.3 and 4.9 nm-1 are associated with aluminum foil used as a sample container.
The crystalline structure observed can be assigned to a monoclinic unit cell with lattice parameters: a = 27.5 Å, b = 5.2 Å, c = 5.0 Å, and angles α = 81°, β = 90°, and γ = 90°. Temperature-dependent SAXS and WAXS measurements are presented in Figure 4(a) and Figure 4(b), respectively. During heating, distinct changes in both peak positions and intensities are noticeable in the SAXS and WAXS regions. Specifically, shifts observed at 90 °C and 150 °C are attributed to dehydration and a crystal-to-crystal transition, respectively. The isotropization process at 220 °C becomes evident with the fading of sharp crystalline peaks.
Indexation of the high-temperature phase indicates the formation of a triclinic unit cell with the following parameters: a=25.6 Å, b=5.12 Å, c=4.96 Å, α=95°, β=97°, γ=88°.
To gain insights into the arrangement of I(CH2)2(CF2)2O(CF2)2SO2OH at different temperatures, all-atom molecular dynamics simulations of a box sized 100 Å x 30 Å x 30 Å, filled with 288 molecules, were conducted at room temperature, followed by a heating procedure with a temperature increase up to 280 °C. A detailed description of the modeling methodology is provided in the Materials and Methods section.
Initially, after system equilibration at T=25 °C, the molecules are elongated and retain crystal ordering. Approximation of the lattice parameters, obtained through identification of the minimal symmetric parallelepiped [37] and averaged over the system, yielded the following values: a = 27.2 Å, b = 5.0 Å, c = 5.0 Å, 𝛼 =81.1°, 𝛽 = 89.6°, 𝛾 = 90.4°. These parameters correspond to a cell with monoclinic symmetry and are in good agreement with experimental data (Figure S1).
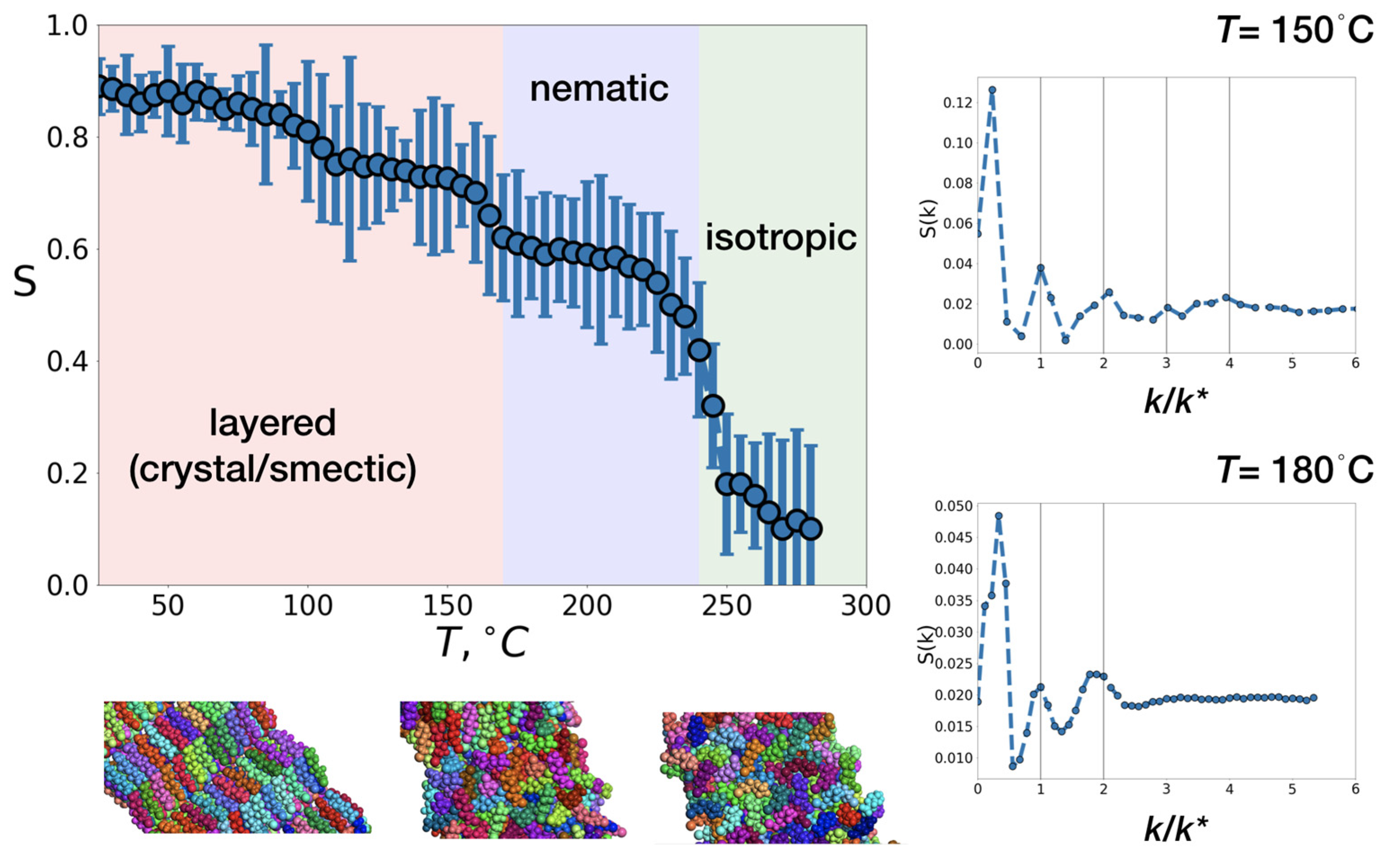
After equilibration at room temperature, the box of I(CH2)2(CF2)2O(CF2)2SO2OH was slowly heated in the NPT (isobaric-isothermal ensemble) ensemble from T=25 °C to T = 280 °C. Within this temperature range, the system undergoes a series of transitions, analyzed using nematic order parameter S [32], which indicates whether the molecules’ main axis is oriented in one direction (S=1) or their orientations are distributed isotropically (S=0). The presence of a layered structure was monitored through the calculation of the static structure factor S(k/k*).
Figure 5 shows the dependency of the nematic order parameter S on temperature, the insets provide typical snapshots of the system under different states and the normalized static structure factor for the chosen T. The latter was plotted against k/k*, where k* is the location of the first maximum. For the layered structure, positions of consequent peaks k(1):k(2):k(3): k(4) should satisfy the relations 1:2:3:4 [38], indicating the periodicity of layers. This relation breaks when layered ordering is disrupted. Positions of these peaks are marked with vertical lines.
Overall, the dependency of the nematic order parameter on temperature is qualitatively similar to what is expected for ellipsoidal mesogens [39,40], with S close to 1 at low temperatures (crystal) and decreasing monotonically as T increases. Sharp changes in the order parameter S indicate transitions between different states (crystal, smectic, nematic, isotropic), with the most pronounced drop at the nematic-isotropic transition.
In the interval between 25 °C and ~ 170 °C, the molecules maintain a layered arrangement (peaks at k/k* = 1, 2, 3, 4 at T = 150 °C, Figure 5 upper right, Figure S2(left)) and retain a high orientational ordering parameter, which decreases from S = 0.9 to S = 0.64 upon heating. At T ~ 170 °C, layers begin to overlap, leading to the destruction of the smectic phase - at T = 180 °C, only the first two peaks of S(k/k*) remain (Figure 5 bottom right, Figure S2(middle)). However, the system maintains high orientational order for I(CH2)2(CF2)2O(CF2)2SO2OH (S ~ 0.64). Thus, at T ~ 170 °C the smectic to nematic transition can be observed. The model temperature for this transition to the nematic phase was found to lie within the interval between 170 and 245 °C, with a monotonic decrease of S upon heating from 0.64 to 0.42. A subsequent increase in T leads to a sharp drop in the nematic order parameter to 0.19 at T = 250 °C, with the nematic-to-isotropic transition temperature occurring approximately at T ~ 245 °C, Figure S2 (right).
Therefore, within the all-atom molecular dynamics framework, two pronounced transition temperatures were identified: smectic-to-nematic at T ~170 °C and nematic-to-isotropic at T ~ 245 °C, both of which are in good agreement with experimental data (Figure 3). The fact that the values derived from simulations are slightly higher than those observed experimentally (i.e., 150 and 230 °C, respectively) can be attributed to finite-size effect or to inaccuracies within the chosen force field.
4. Conclusions
Initially, a model of the membrane was proposed based on ethylenation and chemical modification. The formation of the structure of ionomeric comb-like polymer precursors was carried out, and the dependencies of their self-organization on thermal history were established through both experimental and theoretical considerations. It was discovered that at room temperature, the compound exhibits a highly ordered crystal structure with a monoclinic lattice formed by almost extended molecules. Heating to 150 °C results in partial disordering of fluorinated tails and a transition to a triclinic unit cell. During cooling from the isotropic state, the molecules form large domains of the smectic LC structure. This smectic phase acts as a pre-ordered state for crystallization into a layer-like monoclinic phase at room temperature. Experimental findings on the formation of the crystalline phase and crystal-to-crystal phase transitions were validated by computer modeling using molecular mechanics approaches. The emergence of a smectic phase for relatively short linear fluorinated molecules is significant for the synthesis of self-organizing comb-like ionomers with controlled morphology of ion-conductive channels. The formation of the smectic phase was confirmed through all-atom molecular dynamic simulations, and the temperatures of smectic-nematic and nematic-isotropic transitions upon heating were determined. It was demonstrated that theoretical results are in good agreement with experimental findings. The insights presented in this work can be utilized for the development of new types of fuel cells with enhanced control over their morphology and improved performance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Possible symmetries of unit cell of I(CH2)2(CF2)2O(CF2)2SO2OH used in simulation; Figure S2: Model of P1 unit cell of I(CH2)2(CF2)2O(CF2)2SO2OH at different temperatures; Table S1: Experimental and theoretical peak positions for I(CH2)2(CF2)2O(CF2)2SO2OH at room temperature; Figure S3: 19F-NMR spectrum of ICF2CF2OCF2CF2SO2F recorded in d6-acetone; Figure S4: 1H-NMR spectrum of ICH2CH2CF2CF2OCF2CF2SO3H recorded in d6-acetone. Figure S5: 19F-NMR spectrum of ICH2CH2CF2CF2OCF2CF2SO3H recorded in d6-acetone. Figure S6: FT-IR spectra of ICH2CH2CF2CF2OCF2CF2SO3H.
Author Contributions
Conceptualization, D.V.A. and B.M.A.; methodology, D.V.A.; formal analysis, A.V.M. and A.I.B.; investigation, D.V.A.; data curation, A.V.M.; writing—original draft preparation, D.V.A.; writing—review and editing, B.A.M., D.A.I and UUO.; visualization, A.I.B.; supervision, D.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation within State Contract 075-15-2022-1117 from June 30, 2022.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors acknowledge perfect technical support from the personnel of the ID02 beamline of the ESRF in Grenoble (France).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Sample Availability
Samples of the compounds are available from the authors.
References
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner, R. Gorini, The Role of Renewable Energy in the Global Energy Transformation, Energy Strategy Reviews, 2019, 24, 38-50. [CrossRef]
- M. Z. Jacobson, The Health and Climate Impacts of Carbon Capture and Direct Air Capture, Energy & Environmental Science, 2019, 12, 3567-3574. [CrossRef]
- D. Lew, N. Miller, K. Clark, G. Jordan, Z. Gao, Impact of High Solar Penetration in the Western Interconnection, National Renewable Energy Agency, 2010, 1-9.
- Inganäs O. Organic Photovoltaics over Three Decades. Adv Mater. 2018,30(35), 1800388. [CrossRef]
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Jen, H. L. Yip, Renewed Prospects for Organic Photovoltaics, Chem Rev., 2022, 122(18), 14180-14274. [CrossRef]
- M. Armand, J.-M. Tarascon, Building better batteries, Nature, 2008, 451, 652–657. [CrossRef]
- P. K. Sharma, D. A. Kumar, P. William, D. Obulesu, P. M. Pandian, T. K. H. Khan, G. Manikandan, Energy Storage System Based on Hybrid Wind and Photovoltaic Technologies, Measurement: Sensors, 2023, 30, 100915, ISSN 2665-9174. [CrossRef]
- Okonkwo, Paul C.; Belgacem, I.B.; Emori, W.; Uzoma, P.C., Nafion degradation mechanisms in proton exchange membrane fuel cell (PEMFC) system: A review; Intern. J. Hydrogen Energy, 2021, 46, 27956-27973. [CrossRef]
- A. Kusoglu, A. Z. Weber, New Insights into Perfluorinated Sulfonic-Acid Ionomers, Chemical Reviews, 2017, 117, 987-1104. [CrossRef]
- L. Fan, Z. Tu, S. H. Chan, Recent development of hydrogen and fuel cell technologies: A review, Energy Reports, 2021, 7, 8421-8446. [CrossRef]
- Y. Tanaka, Development of the MIRAI Fuel Cell vehicle, In Hydrogen Energy Engineering, A Japanese Perspective; K. Sasaki, H. W. Li, A. Hayashi, J. Yamabe, T. Ogura, S. M. Lyth, Eds; Springer: Japan, Tokyo, 2016, 34, 461-476. [CrossRef]
- Jinnouchi, R.; Kudo, K.; Kodama, K.; Kitano, N.; Suzuki, T.; Minami, S.; Shinozaki, K.; Hasegawa, N., Shinohara, A., The role of oxygen-permeable ionomer for polymer electrolyte fuel cells, Nature Comm.; 2021, 12, 4956. [CrossRef]
- T. Suzuki, S. Tsushima, S. Hirai, Effects of Nafion® Ionomer and Carbon Particles on Structure Formation in a Proton-Exchange Membrane Fuel Cell Catalyst Layer Fabricated by the Decal-Transfer Method, International Journal of Hydrogen Energy, 2011, 36, 19, 12361–12369. [CrossRef]
- S. Liu, R. Lin, J. Lu, Y. Wang, X. Cai, The Impact of Different Side Chain Ionomer on Membrane Electrode Assembly Performance and Durability, Chemical Engineering Journal, 2023, 472, 145050. [CrossRef]
- X. Yan, Z. Xu, S. Yuan, A. Han, Y. Shen, X. Cheng, Y. Liang, S. Shen, J. Zhang, Structural and Transport Properties of Ultrathin Perfluorosulfonic Acid Ionomer Film in Proton Exchange Membrane Fuel Cell Catalyst Layer: A Review, Journal of Power Sources, 2022, 536, 231523. [CrossRef]
- I. Antipin, M. Alémov, V. Arslanov, V. Burilov, S. Vatsadze, Y. Voloshin, K. Volcho, V. Gorbatchuk, Y. Gorbunova, S. Gromov, et al., Functional supramolecular systems: Design and applications, Russ. Chem. Rev. 2021, 90, 895–1107. [CrossRef]
- A. Alfutimie, R. Curtis, G. J. T. Tiddy, Lyotropic Surfactant Liquid Crystals: Micellar Systems, In Handbook of Liquid Crystals, 2nd ed.; J. Goodby, P. Collings, T. Kato, C. Tschierske, H. Gleeson, P. Raynes, V. Vill, Eds.; Wiley-VCH: Weinheim, Germany, 2014; Volume 6. [CrossRef]
- N. Illy, D. Urayeneza, A.V. Maryasevskaya, L. Michely, S. Boileau, B. Brissault, E.A. Bersenev, D.V. Anokhin, D.A. Ivanov, J. Penelle. Synthesis and Solid-State Properties of PolyC3 (Co)polymers Containing (CH2-CH2-C(COOR)2) Repeat Units with Densely Packed Fluorocarbon Lateral Chains, Macromolecules, 2019, 52(23), 9199-9207. [CrossRef]
- Q. Berrod, S. Lyonnard, A. Guillermo, J. Ollivier, B. Frick, A. Manseri, B. Ameduri, G. Gebel, Nanostructure and transport properties of proton conducting self-assembled perfluorinated surfactants: a bottom-up approach towards PFSA fuel cell membranes, Macromolecules, 2015, 48, 6166–6176. [CrossRef]
- Y. Wang, J. Liu, S. Yang, Multi-functional liquid crystal elastomer composites, Applied Physics Reviews, 2022, 9(1). [CrossRef]
- A. Doostmohammadi, B. Ladoux, Physics of liquid crystals in cell biology, Trends in cell biology, 2022, 32(2), 140-150. [CrossRef]
- N. Kapernaum, A. Lange, M. Ebert, M. A. Grunwald, C. Haege, S. Marino, A. Zens, A. Taubert, F. Giesselmann, S. Laschat, Current topics in ionic liquid crystals, ChemPlusChem, 2022, 87(1), e202100397. [CrossRef]
- A. V. Komolkin, A. Laaksonen, A. Maliniak, Molecular dynamics simulation of a nematic liquid crystal, The Journal of chemical physics, 1994, 101(5), 4103-4116. [CrossRef]
- F. Affouard, M. Kröger, S. Hess, Molecular dynamics of model liquid crystals composed of semiflexible molecules, Physical Review E, 1996, 54(5), 5178. [CrossRef]
- M. R. Wilson, Molecular simulation of liquid crystals: progress towards a better understanding of bulk structure and the prediction of material properties, Chem. Soc. Rev., 2007,36, 1881-1888. [CrossRef]
- X. Wei, J. B. Hooper, D. Bedrov, Influence of electrostatic interactions on the properties of cyanobiphenyl liquid crystals predicted from atomistic molecular dynamics simulations, Liquid Crystals, 2017, 44(2), 332-347. [CrossRef]
- P. Ahmadpour-Samani, P. Zahedi, An investigation on nematic-isotropic phase transition, viscosity, and diffusion coefficient of liquid crystalline elastomers at different temperatures using molecular dynamics simulation, Journal of Molecular Liquids, 2022, 367, 120403. [CrossRef]
- Sun, H., Ren, P., & Fried, J. R. The COMPASS force field: parameterization and validation for phosphazenes. Computational and Theoretical Polymer Science, 1998, 8(1-2), 229-246. [CrossRef]
- Beckers, J. V. L., Lowe, C. P., & De Leeuw, S. W. An iterative PPPM method for simulating Coulombic systems on distributed memory parallel computers. Molecular simulation, 1998, 20(6), 369-383. [CrossRef]
- Berendsen, H. J. C., Postma,J. P. M., van Gunsteren, W. F., DiNola, A., Haak, J. R. Molecular dynamics with coupling to an external bath. Journal of Chemical Physics , 1984, 81, 3684-3690. [CrossRef]
- Shuichi N. Constant temperature molecular dynamics methods, Progress of Theoretical Physics Supplement, 1991, 103, 1-46. [CrossRef]
- Ball, J. M., Majumdar, A. Nematic liquid crystals: from Maier-Saupe to a continuum theory. Molecular crystals and liquid crystals, 2010, 525, 1-11. [CrossRef]
- A. Manseri, B. Ameduri, B. Boutevin, G. Caporiccio, M. Kotora, M. Hajek, Synthesis of Telechelic Dienes from Fluorinated α,ω-Diiodoperfluoroalcanes. Part I. Divinyl and Diallyl Derivatives from Model I(C2F4)nI Compounds, J. Fluorine Chem., 1995, 73, 151-158. [CrossRef]
- L. Sauguet, B. Ameduri, B. Boutevin, Fluorinated Copolymers and Terpolymers Based on Vinylidene Fluoride and Bearing Sulfonic Acid Side-Group, J. Polym. Sci. Part A: Polym. Chem., 2007, 45, 1814-1834. [CrossRef]
- J. W. Goodby. Materials and Phase Structures of Calamitic and Discotic Liquid Crystals. Handbook of Visual Display Technology, Springer, Berlin, Heidelberg, 2012, 1243–1287. [CrossRef]
- J.-M. Corpart, S. Girault, D. Juhué, Structure and Surface Properties of Liquid Crystalline Fluoroalkyl Polyacrylates: Role of the Spacer, Langmuir, 2001, 17, 7237–7244. [CrossRef]
- F. Vivien, N. Wicker, Minimal enclosing parallelepiped in 3D, Comput. Geom, 2004, 29, 177–190. [CrossRef]
- A.A. Glagoleva, V.V. Vasilevskaya, A.R. Khokhlov, Microphase separation in the melts of diblock copolymers composed of linear and amphiphilic blocks, Polymer Science Series A, 2010, 52, 182-190. [CrossRef]
- I. Dierking, S. Al-Zangana, Lyotropic liquid crystal phases from anisotropic nanomaterials, Nanomaterials, 2017, 7(10), 305. [CrossRef]
- M.R. Wilson, G. Yu, T.D. Potter, M. Walker, S. J. Gray, J. Li, N. J. Boyd, Molecular simulation approaches to the study of thermotropic and lyotropic liquid crystals. Crystals, 2022, 12(5), 685. [CrossRef]
Scheme 1.
General reaction mechanism.
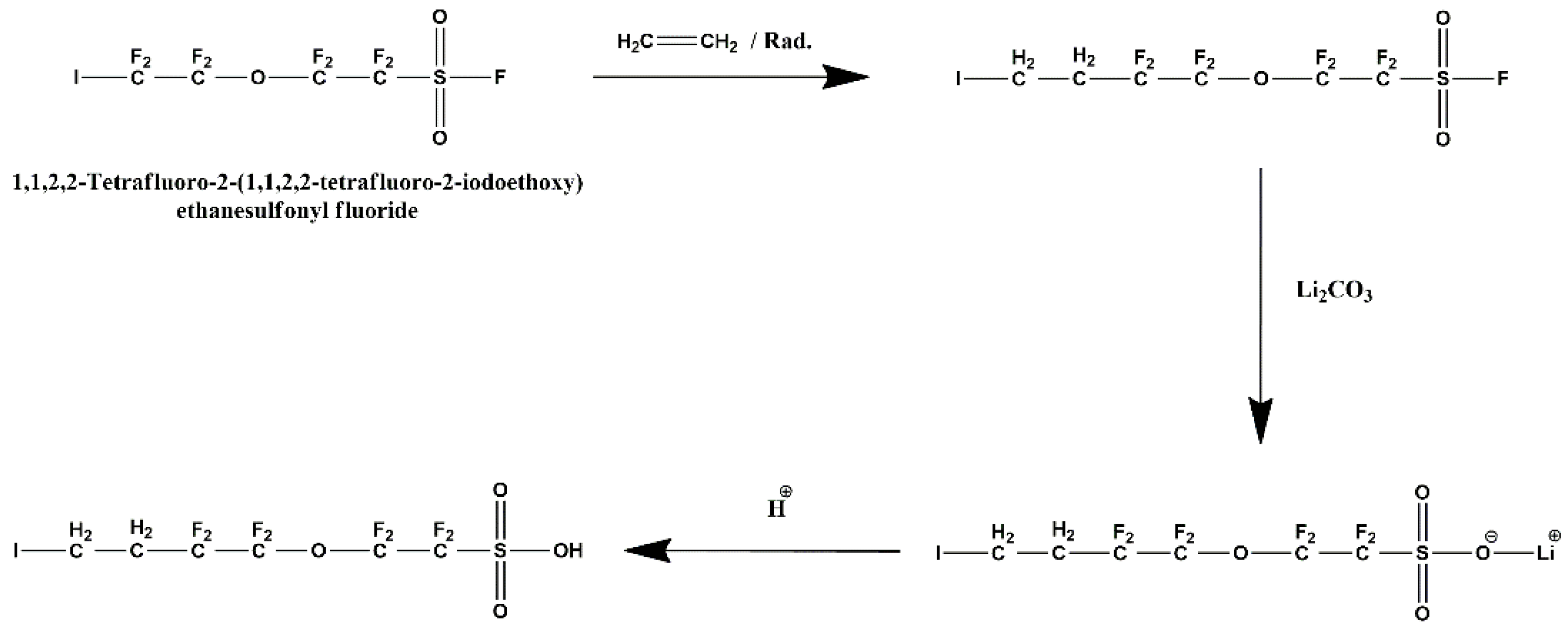
Figure 2.
POM images of the studied sample during isothermal crystallization 216 °C (a) and after cooling to room temperature (b) Scale bar is 50 µm.
Figure 2.
POM images of the studied sample during isothermal crystallization 216 °C (a) and after cooling to room temperature (b) Scale bar is 50 µm.

Figure 3.
SAXS (red) and WAXS (blue) 1D diffractograms measured at room temperature. Several orders of 100 peak are denoted on the SAXS curve (* peaks at 4.3 and 4.9 nm-1 are assigned to Al foils).
Figure 3.
SAXS (red) and WAXS (blue) 1D diffractograms measured at room temperature. Several orders of 100 peak are denoted on the SAXS curve (* peaks at 4.3 and 4.9 nm-1 are assigned to Al foils).
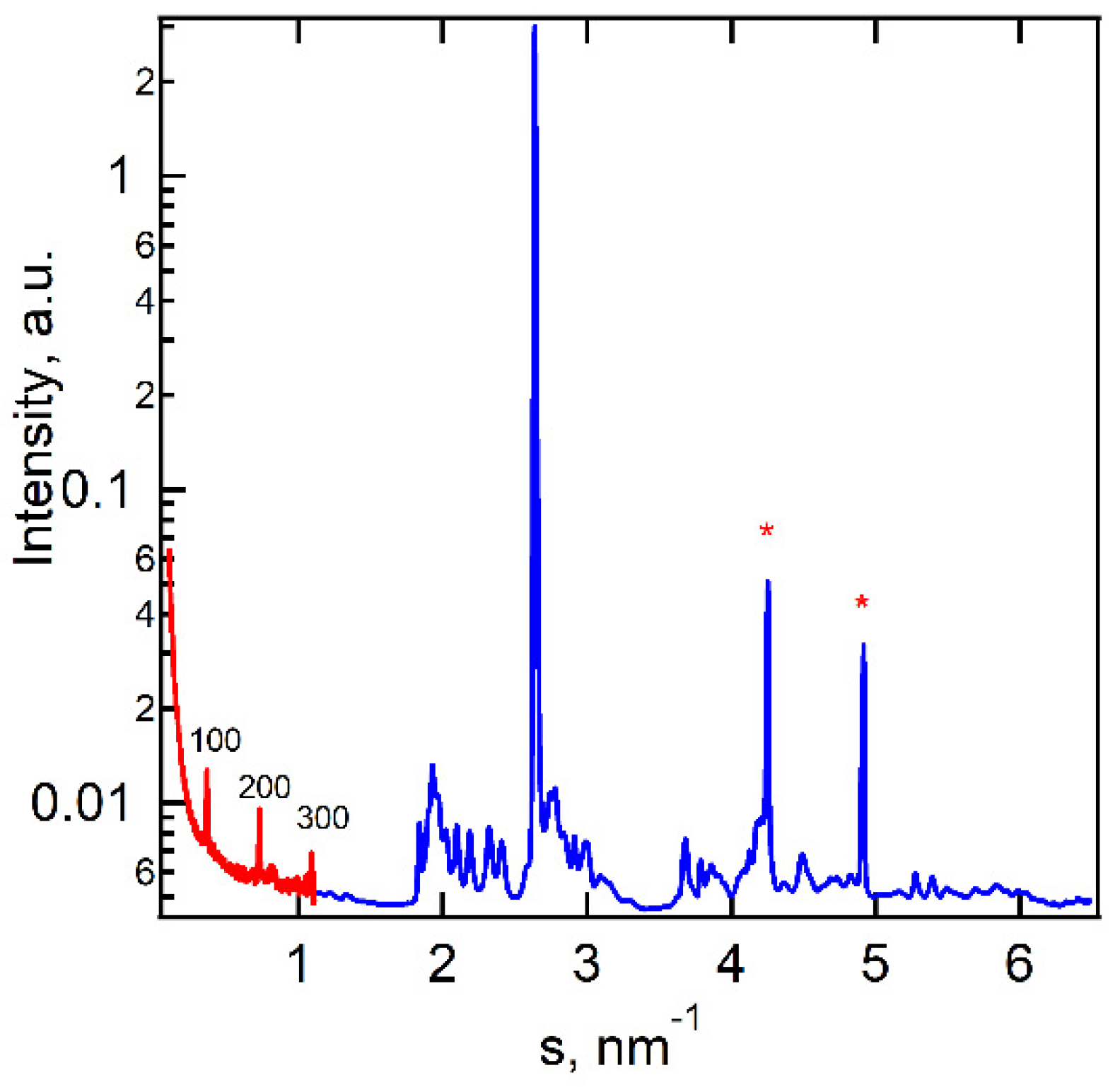
Figure 4.
Temperature-resolved SAXS (a) and WAXS (b) curves corresponding to a heating ramp. Color table indicates the peak intensity of 1D diffractograms as a function of the norm of the scattering vector s. Two intense reflections marked by arrows are attributed to aluminum foil.
Figure 4.
Temperature-resolved SAXS (a) and WAXS (b) curves corresponding to a heating ramp. Color table indicates the peak intensity of 1D diffractograms as a function of the norm of the scattering vector s. Two intense reflections marked by arrows are attributed to aluminum foil.
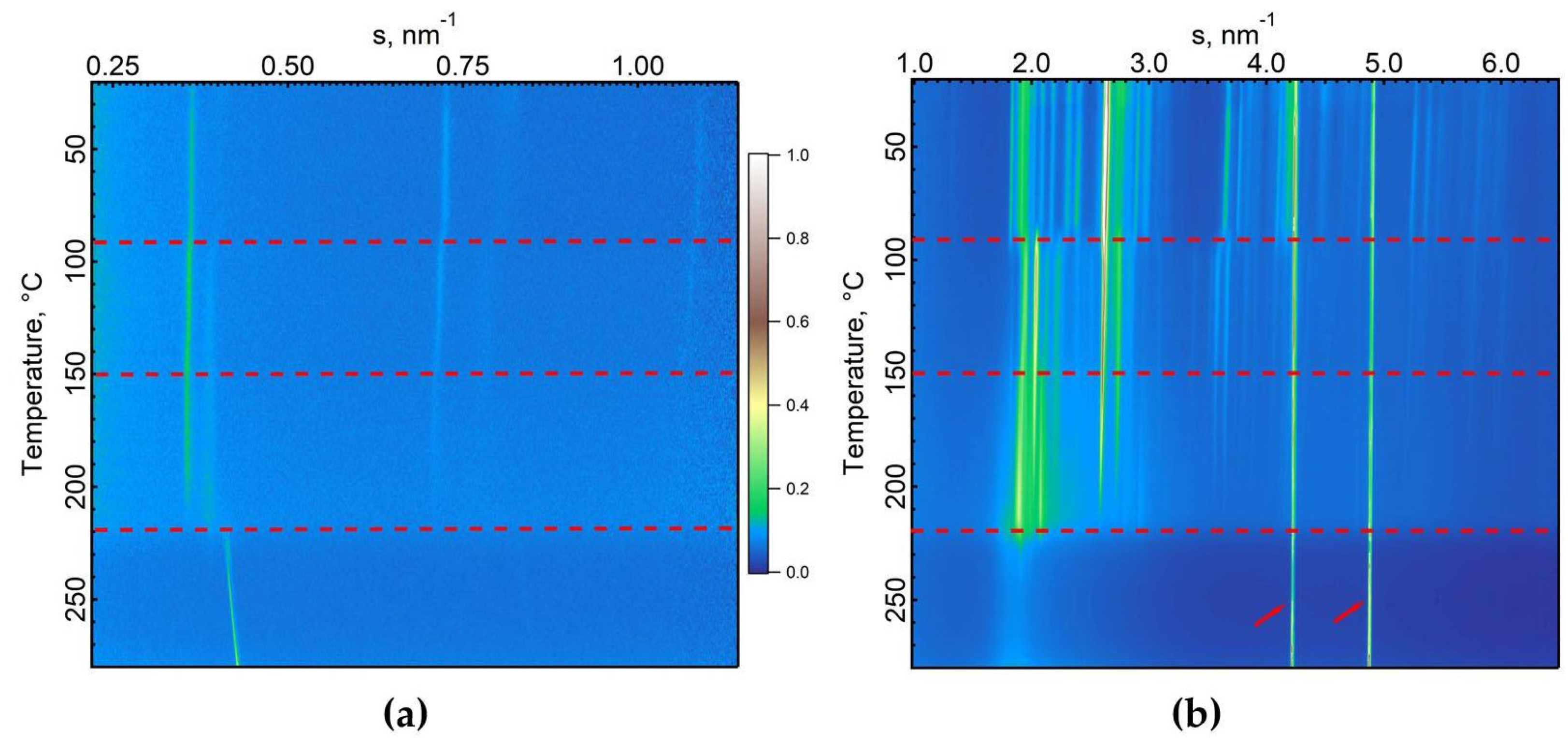
Figure 5.
Orientational order parameter S as a function of temperature T, obtained from simulations; snapshots of the system, corresponding to layered, nematic and isotropic state; and normalized structure factor, plotted against k/k*, where k* is the location of the first meaningful peak at the vicinity of smectic - nematic transition. Within every molecule atoms are colored in the same way, neighboring molecules are colored differently.
Figure 5.
Orientational order parameter S as a function of temperature T, obtained from simulations; snapshots of the system, corresponding to layered, nematic and isotropic state; and normalized structure factor, plotted against k/k*, where k* is the location of the first meaningful peak at the vicinity of smectic - nematic transition. Within every molecule atoms are colored in the same way, neighboring molecules are colored differently.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



