
Submitted:
01 November 2023
Posted:
07 November 2023
You are already at the latest version
Abstract
Constraint-based metabolic modeling approaches have enhanced our knowledge and understanding of the metabolism of prokaryotes and eukaryotes. This approach highly depends on the reconstruction process of genome-scale metabolic models (Mmodels). M-models can guide effective experimental design and yield new insights into the function and control of biological systems. Despite the recent advances in the automated generation of draft metabolic network reconstructions, the manual curation of these networks remains a labor-intensive and challenging task. Thus, these ten quick tips for the manual curation process are essential for optimizing high-quality metabolic model generation in less time. This collection of tips describes in great detail the resources and methods to ensure successful reconstruction. Furthermore, it increases the scope of other protocols of metabolic modeling by including resources to reconstruct eukaryotic organisms. Thus, all tips are applicable to a wide range of eukaryotic organisms. We believe this manuscript will interest a broad audience and researchers from different disciplines, spanning from microbiology and systems biology to biotechnology.
Keywords:
Genome-scale metabolic model reconstruction
; manual curation
; quick tips
; systems biology
Introduction
Systems biology tools integrate experimental and computational data to study the cellular and molecular biological interactions of organisms (1). The continuous development of sequencing methodologies and computational tools has improved the elucidation of interactions between different metabolic network components in complex biological systems (2–5). Constraint-based modeling involves formulating algorithmic protocols to create and simulate genome-scale metabolic models (M-models). M-models are comprehensive knowledge bases organized by gene-reaction, metabolite-reaction, and gene-protein-reaction (GPR) associations (6). These associations enable the in-silico simulation of growth phenotypes and metabolite production under a broad variety of conditions (7,8). Therefore, metabolic modeling aims to analyze physiological and big data (multi-omics information) to generate testable hypotheses (9). In addition, M-models are accompanied by the tools developed for metabolic engineering, which specialize in analyzing and modifying metabolic pathways to maximize the production of compounds of interest (10). Nowadays, evolution can be accelerated through the development of new metabolic engineering strategies aided by identifying metabolic targets using M-models (11).
In 2010, a 96-step detailed protocol for generating metabolic models was developed (6). It encompassed four stages: i) draft model generation, ii) model refinement/curation, iii) model conversion, and iv) model validation. The draft model can be generated automatically using one or more available pipelines (8,12–18), such as CarveMe, Model SEED, and Reconstruction, Analysis, and Visualization of Metabolic Networks Toolbox (RAVEN) (19–21). During model refinement, draft models are manually curated by verifying the metabolic pathways for the organism of interest (6). Manual curation allows the researcher flexibility in verifying the reactions, metabolites, and GPR associations. This step is critical to providing a high-quality model with specific metabolic details.
Despite advances in the automated generation of draft metabolic reconstructions, the manual curation of these networks remains a labor-intensive and challenging task. Hence, this paper will provide ten quick tips to guide and optimize the manual curation procedure for genome-scale metabolic modeling, ensuring the generation of high-quality M-models. Later, those models can be used to predict phenotypes accurately, contextualize big data, and be templates for expression and transcription (22,23), multi-strain, and community modeling (24,25).
Tip 1. Retrieve the genomic and proteomic information of the target organism.
The goal of creating an M-model is to define a metabolic network that connects each gene with its biochemical function. The process to obtain genomic and proteomic information depends on the accessibility of the data and the category of the organism (e.g., eukaryotic, prokaryotic, virus). If the genomic data is unavailable, it must be assembled using genome assembly tools (e.g., SPAdes (28), Velvet (29), Canu (30)). However, several public databases are available that store genome sequence information for various organisms (Table S1).
The PATRIC Database (31), now the Bacterial and Viral Bioinformatics Resource Center (BV-BCR), has been broadly used to retrieve comprehensive genomic, proteomic, and other omics information of a wide range of bacterial species for M-models reconstruction (16,32). Moreover, BV-BCR (35) also integrates data, tools, and infrastructure from the Influenza Research Database (IRD) and Virus Pathogen Resource (ViPR) databases containing an extensive amount of metadata of viruses.
The National Center for Biotechnology Information (NCBI) (36) is a prominent database that possesses a vast collection of biomedical and molecular biology data on prokaryotic and eukaryotic organisms. It hosts the Reference Sequence (RefSeq) (37) and GenBank (38) databases. The GenBank resource is fed by the public effort of independent laboratories that submit their novel or updated genome assemblies. RefSeq focused on curating the data in GenBank to provide well-annotated genomic sequences.
BioCyc (39) and The Kyoto Encyclopedia of Genes and Genomes (KEGG) (40) are bioinformatic repositories containing an extensive microbial genome collection. The data contained in BioCyc has been extensively curated from biological literature. KEGG analyzes the interaction of genes with their biological functions in a metabolic pathway within an organism. KEGG also provides genomic and proteomic information on prokaryotic and eukaryotic organisms.
Finally, single protein data can be retrieved instead of complete genome sequences. UniProt (41), BRENDA (42), and the Protein Data Bank (PDB) (43) provide information on amino acid sequences, three-dimensional structures, function, and enzymology of proteins.
Tip 2. Identify basic metabolic your microorganism of interest.
The genomic information of the target organism and a previously published model as a template is needed to start the reconstruction of an M-model. This first version of the metabolic network (draft model) must simulate as many metabolic capabilities of the target organism as possible. It is essential to select a template model that best matches the biological features of the target organism. Key characteristics such as phylogenetic relationship, protein homology, cell wall composition (gram-negative or gram-positive), growth mode (e.g., auto-, hetero-, mixotrophic, aerobic, anaerobic), and prokaryotic or eukaryotic features are critical when selecting the template organism (Figure 1).
The growth mode of template organisms can affect the functionality of a newly reconstructed draft model. Some important growth modes of prokaryotic and eukaryotic organisms include aerobic, anaerobic, light-dependency, and nitrogen fixation conditions, among many others. Thus, the model template must be selected based on protein homology and metabolic capabilities. Figure 1 highlights common growth modes of microbes and suggests template models that have been extensively validated.
Tip 3. Semi-automatic reconstruction of a draft model
Semi-automatic reconstruction is an automated step that generates a draft model using a template model. Generating an initial good-quality draft model using automatic reconstruction methods and algorithms (19,20) reduces the time required during manual curation. For the semi-automatic reconstruction, the following inputs must be provided: i) the FASTA formatted proteome of the target organism, ii) the proteome and metabolic network of the template model, and ii) the minimal culture media. The algorithm performs bidirectional BLASTp to find homologous proteins between the target and template organisms. Subsequently, the reactions associated with the homologous proteins in the template model are added to the metabolic network generated for the target organism. The algorithm must ensure the connectivity and functionality of the model to perform growth rate simulations. Therefore, essential reactions are expected to be added to the network even if no homologous proteins are found. These reactions might be associated with no genes (orphan reactions) or genes belonging to the template organism (exogenous genes). Reactions associated with exogenous genes and orphan reactions are addressed through manual verification of GPR associations, as explained in Tip 4.
The algorithms that generate draft models can be designed by the researcher who aims to create a new M-model (13,14). Examples of those algorithms are currently available in The Constraint-Based Reconstruction and Analysis (COBRA) (33) and RAVEN (21) Toolboxes. Additionally, some automated reconstruction tools, such as CarveMe, PathwayTools, Agora, and ModelSEED, are available online (19,20,34,35).
Tip 4. Manual verification of GRP associations.
As mentioned in Tip 3, a draft model may contain issues related to exogenous genes and orphan reactions. These issues are addressed by ensuring reactions only correspond with genes from the target organism (verification of GPR associations).
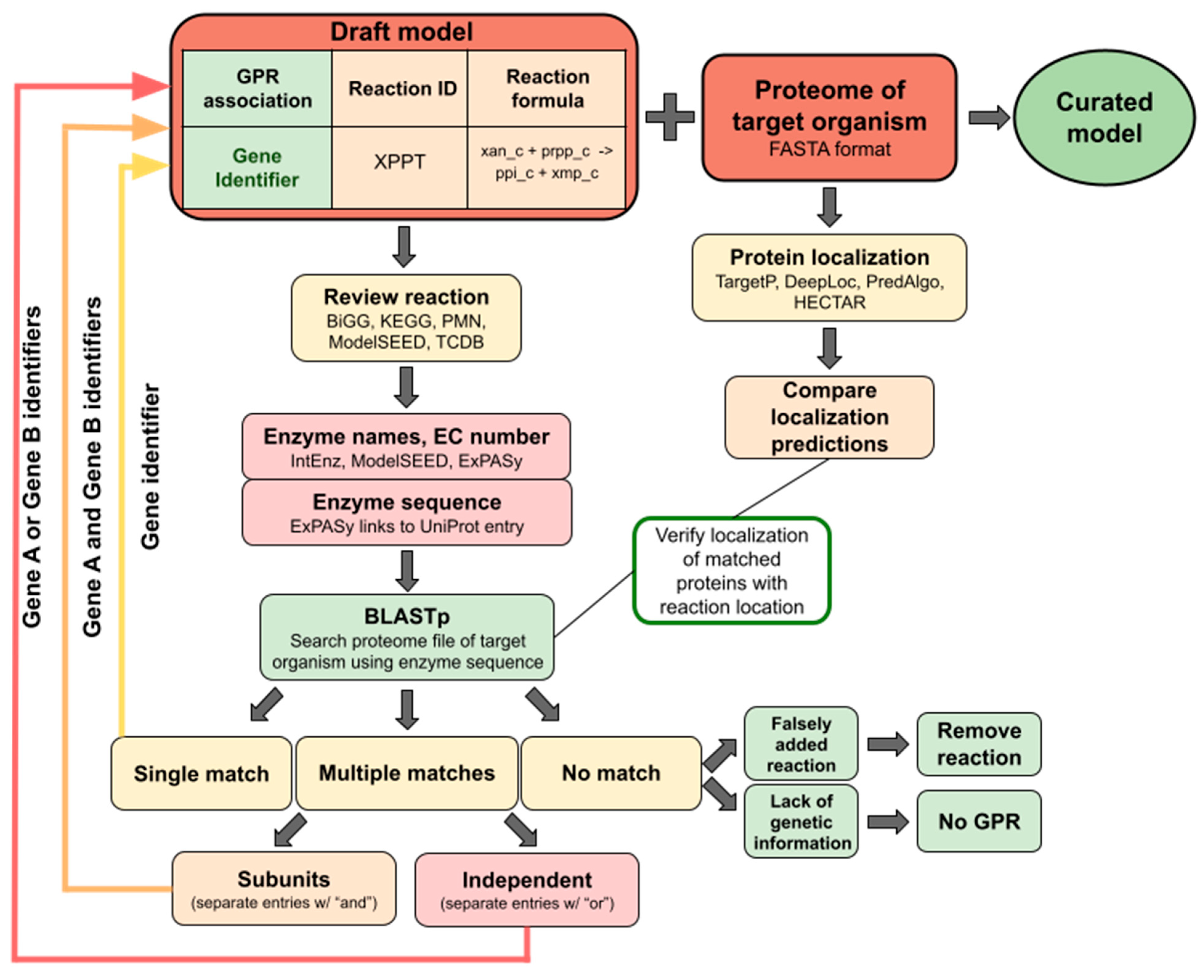
The quickest and most reliable way to verify a GPR is by searching for the assigned Enzyme Commission (EC) number or enzyme name of the reaction in the proteome FASTA file of the target organism. The genes found in the FASTA file are recorded to confirm that particular GPR is present. If multiple enzymes are found to catalyze the same reaction independently, then all gene identifiers are added to the GPR association using the operator "or" to separate entries. If multiple subunits for a particular enzyme are identified, then all gene identifiers are connected through the operator "and" (Figure 2).
GPRs that could not be located via EC number or enzyme name can be identified using BLASTp (36). First, the reaction ID must be located in the database used to create the draft model. Each database provides information about the target reaction and the protein that catalyzes it. For example, BiGG entries show the reaction formula, models containing the reaction, and external links to other databases with additional information (e.g., IntEnz, KEGG) (37). The goal is to retrieve a protein amino acid sequence from phylogenetically close organisms using the different enzyme names. TCDB (38) and ExPASy (39) are good resources for finding protein sequences. The retrieved amino acid sequence is compared against the proteome of the target organism using NCBI BLASTp. After obtaining the BLASTp results, gene identifiers are assigned to the GPR based on our discretion as researchers. A smaller E-value and higher query coverage and identity indicate a good match for the GPR (e.g., the E-value, identity, and query coverage cut-offs of Raven Toolbox are 1e-30, 40%, and 50%, respectively). The lack of a homologous might be due to missing genetic information (an empty GPR is added) or a falsely added reaction (the reaction is removed). Experimental or collected literature data is used to confirm the presence of the gene in the organism. Ultimately, the model will contribute to the update of the genome annotation. For example, the recent update of the B. subtilis model with up to 1,168 new genetic functions (40).
For eukaryotic cells, protein compartmentalization needs to be considered when assigning gene identifiers to GPR associations. It is recommended to complete the protein localization and comparison of the whole proteome before manually curating the draft model (Figure 2). Tools such as TargetP (41), HECTAR (42), DeepLoc (43) and PredAlgo (44) can determine signal peptides, chloroplast and mitochondria localization of the proteins. It is best to run multiple localization tools and compare outcomes. After a BLASTp search is run, the found gene identifiers can be compared to the predicted localization and added as the GPR association if the given reaction location matches. For example, this will prevent chloroplast-localized enzymes from being added to mitochondrion reactions, resulting in a more accurate model.
Tip 5. Addition of constraints to simulate basic metabolic capabilities, generating the QC/QA script
An M-model can estimate the growth rates of an organism for various environmental and genetic conditions using Flux Balance Analysis (FBA) (51). FBA calculates metabolic fluxes while constrained for an objective function and substrate uptake rates (51). These constraints are defined as mathematical equations or inequalities that limit the range of possible solutions for the simulated metabolic fluxes and can be identified through experimental data (6,51). For example, the constraints associated with nutrient uptake or enzyme activities (e.g., gene expression) limit biomass formation during computational simulations (52).
Changes in the architecture of the model while following Tip 4, can result in changes in stoichiometric constraints and affect the functionality of the model (11). A Quality Control and Quality Assurance (QC/QA) script is generated to assess the energetic feasibility and the mass and charge balance of the model. The energetic feasibility test verifies that the metabolic fluxes adhere to the principles of thermodynamics, ensuring that no matter or energy is generated without mass input (53,54). The mass balance test verifies the total consumption of each metabolite produced within the metabolic network (6). Finally, the charge balance test evaluates that the sum of the reagent and product charges of each biochemical equation equals zero (6).
QC/QA scripts help identify and correct errors in the metabolic model to ensure the reconstruction of a high-quality M-model. Open-source software, such as MEMOTE (55), offers a QC/QA script that automatically evaluates the quality of M-models. However, organism-specific growth simulations are out of the scope of M-models. Hence, it is recommended to build your own QC/QA script. There are example protocols available for organisms like E. coli (51) and Chlamydomonas reinhardtii (56), and other photosynthetic organisms (57) that use The COBRA Toolbox.
Tip 6. Determination of the biomass objective function.
An M-model is a network of interconnected biochemical reactions that can predict growth rates through the sum of individual fluxes of biomass metabolites. The biomass components (i.e., carbohydrates, lipids, proteins, nucleotide triphosphates, and RNA) are integrated into the metabolic network through an artificial modeling reaction defined as the Biomass Objective Function (BOF) (58). The stoichiometric coefficients of each metabolite in the BOF reaction represent the molar composition of the structural components of the cell in units of mmol per gram of cell dry weight. Therefore, the stoichiometric coefficient values can be experimentally calculated as previously described by Lanchance et al., 2019 (59). For the model functionality, at least one BOF is needed. Nevertheless, several BOFs can be generated for unconventional organisms that dramatically change their biomass composition depending on environmental conditions (e.g., phototrophs, yeast) (14,17) or the BOF can be split for easier model manipulation (60).
Available computational tools, such as BOFdat (59), use experimental measurements of structural macromolecule compositions to generate BOFs automatically. However, when the experimental determination of the proportional contribution of biomass components is not feasible, a BOF from a previously reconstructed M-model can be imported (13,19).
Tip 7. Addition of new metabolites and pathways based on untargeted metabolomics data
Untargeted metabolomics is an analytical approach to determine as many metabolites as possible in the biomass of the target organism (61). In addition to biomass composition compounds, organism-specific metabolites are usually identified through untargeted metabolomics data, depending on the growth conditions (61–63). Therefore, the template model might not contain the biosynthesis reactions of the whole metabolome of the target organism. In those cases, the metabolic pathways are manually added to the draft model to allow simulation of the production of those molecules (see Tip 8). This process is widespread during the reconstruction of lipid-producing organism M-models. Since the lipid profile varies among organisms, researchers manually add new pathways for lipid production to their M-models (14).
When adding a new pathway not in the database used to create your model, new reaction and metabolite identifiers must be created. Additionally, compartmentalization, GPR association, reversibility, directionality, and the mass and charge balance of each reaction must be defined (6). Furthermore, it is essential to verify the stoichiometric coefficients and the charged formulas of the metabolites in the growth condition in which the model is being reconstructed.
Tip 8. Gap-filling using high-throughput experimental data.
During an M-model reconstruction, high-throughput data is added (e.g., omics, phenotyping) to increase the feasible simulations of growth phenotypes under known physiological states. To achieve this goal, the concept of gap-filling was introduced (64). Gap-filling utilizes manual methods and algorithms to detect missing reactions of a specific pathway likely to be present in the metabolism of the target organism (64). These gaps exist in metabolic networks due to incomplete organism knowledge and the lack of genomic and functional annotations. Therefore, the gap-filling process will cover missing reactions, unknown pathways, unannotated genes, and promiscuous enzymes in the M-model (65). Gap-filling can be performed manually (guided by literature and bioinformatic databases) or automatically with the help of computer algorithms (65,66) such as Fastgapfill and Globalfit (67,68).
The prediction capabilities of an M-model can be determined from the Matthews Correlation Coefficient (MCC). This is a common metric used to evaluate the accuracy of M-models. MCC calculation can be performed for gene essentiality and growth phenotypes by comparing in-vitro and in-silico analysis (69). The MCC is computed from a confusion matrix of true positive (TP, positive growth in-vitro and in-silico), true negative (TN, negative growth in-vitro and in-silico), false positive (FP, negative growth in vitro and positive growth in-silico), and false negative (FN, positive growth in-vitro and negative growth in-silico) simulations (59). With this approach, Equation 1 can be used to estimate the MMC.
Tip 9. Addition of metadata to metabolites and reactions is critical to ensure compatibility.
While reconstructing an M-model, different databases and tools are used to find detailed information about reactions, metabolites, genes, etc (Table S1). In order to facilitate the exchange of information between M-models reconstructed based on different databases, an additional mapping of elements must be carried out. Standardization tools are also available to facilitate the mapping process (e.g., MetaboAnnotator) (70–73). This process consists of connecting the specific identifiers from one model to another as described in the following steps: a) Determine if the reaction/enzyme has an associated Enzyme Commission (EC) number. EC numbers are usually common "threads" between all databases. b) If no EC number exists or is outdated, search for the reaction/enzyme name in the Integrated Relational Enzyme database (IntEnz) (37). A reaction could have more than one name. c) Identify the different reaction IDs in the databases of interest. It is recommended to consider information from Rhea (74), BiGG (45), KEGG (46), MetaNetX (75), BioCyc (76), ModelSEED (20) and Reactome (77). d) Confirm the reaction is the same by verifying the stoichiometric coefficients and metabolites involved. e) Add the identifiers and links to the model. f) If a reaction is not found in a database, it can be skipped.
Tip 10. Sharable format JSON, MAT, SBML, XML, and visualization
M-models must be ready to simulate, user-friendly, shareable, open-access, and compatible with different programming languages. Remarkable progress has been made in this front of constraint-based modeling (72). Table S2 shows the most common formats in which M-models are publicly available.
The Systems Biology Markup Language (SBML) format is a widely adopted standardized format that facilitates the sharing of models (78). It is highly encouraged to follow the SBML XML Schema format, such as XML format to ensure that SBML Models adhere to their specified structures and data types (79). XML Schema format allows for compatibility and consistency in SBML models across various software applications.
M-models can also be stored in JSON (JavaScript Object Notation) format (80). This format includes the necessary components of an M-model, such as reactions, proteins, metabolites, genes, compartments, and their respective properties (45). Moreover, The JSON format is compatible with Constraint-Based Reconstruction and Analysis for Python (COBRApy) (81) and the M-models visualization software Escher (82).
Another essential format is the MATLAB binary file format "mat”. The "mat" format is compatible with the COBRA Toolbox (33) which has the same applications as COBRApy but runs in the MATLAB environment.
Finally, the YAML format (YAML Ain’t Markup Language) (83) is a human-readable data-serialization format designed to provide simple readability that promotes sharing and collaboration. Researchers can edit the format without reliance on specialized tools or software, facilitating the communication and exchange of biological models.
Conclusion
The semi-automatic reconstruction of an M-model involves generating a draft model using automatic tools followed by applying manual curation to improve the model prediction accuracy. Despite several recent advances in the automated generation of draft metabolic reconstructions, the manual curation of these networks remains a labor-intensive and challenging task. Rigorous manual curation of genome-scale metabolic models is a high-work-high-reward process. An M-model with high accuracy will enable building on top of it as a template for future reconstructions or advanced modeling approaches such as multi-strain modeling (84), metabolism and gene expression models (ME-models) (22,85), community models (CM-models) (24,25,86,87), and multi-scale models (7).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgements
This material is based upon work supported by the National Science Foundation, Directorate for Biological Sciences (Grant No.DBI-2313313), and the start-up funds of Cristal Zuniga provided by the College of Sciences of San Diego State University. Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
References
- Tavassoly I, Goldfarb J, Iyengar R. Systems biology primer: The basic methods and approaches. Vol. 62, Essays in Biochemistry. Portland Press Ltd; 2018. p. 487–500. [CrossRef]
- Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, Mi H. PANTHER: Making genome-scale phylogenetics accessible to all. Vol. 31, Protein Science. John Wiley and Sons Inc; 2022. p. 8–22. [CrossRef]
- Montagud A, Ponce-de-Leon M, Valencia A. Systems biology at the giga-scale: Large multiscale models of complex, heterogeneous multicellular systems. Vol. 28, Current Opinion in Systems Biology. Elsevier Ltd; 2021. [CrossRef]
- Ngo RJK, Yeoh JW, Fan GHW, Loh WKS, Poh CL. BMSS2: A Unified Database-Driven Modeling Tool for Systematic Biomodel Selection. ACS Synth Biol. 2022 Aug 19;11(8):2901–6. [CrossRef]
- Erdem C, Birtwistle MR. MEMMAL: A tool for expanding large-scale mechanistic models with machine learned associations and big datasets. Frontiers in Systems Biology. 2023 Mar 9;3. [CrossRef]
- Thiele I, Palsson B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010 Jan;5(1):93–121. [CrossRef]
- Li CT, Eng R, Zuniga C, Huang KW, Chen Y, Zengler K, et al. Optimization of nutrient utilization efficiency and productivity for algal cultures under light and dark cycles using genome-scale model process control. NPJ Syst Biol Appl. 2023 Dec 1;9(1). [CrossRef]
- Tec-Campos D, Zuñiga C, Passi A, Del Toro J, Tibocha-Bonilla JD, Zepeda A, et al. Modeling of nitrogen fixation and polymer production in the heterotrophic diazotroph Azotobacter vinelandii. Metab Eng Commun. 2020 Dec 1;11. [CrossRef]
- Passi A, Tibocha-Bonilla JD, Kumar M, Tec-Campos D, Zengler K, Zuniga C. Genome-scale metabolic modeling enables in-depth understanding of big data. Vol. 12, Metabolites. MDPI; 2022. [CrossRef]
- Gudmundsson S, Nogales J. Recent advances in model-assisted metabolic engineering. Vol. 28, Current Opinion in Systems Biology. Elsevier Ltd; 2021.
- Garcia-Albornoz MA, Nielsen J. Application of Genome-Scale Metabolic Models in Metabolic Engineering. Industrial Biotechnology [Internet]. 2013 Aug 1;9(4):203–14. [CrossRef]
- Norena-Caro DA, Zuniga C, Pete AJ, Saemundsson SA, Donaldson MR, Adams AJ, et al. Analysis of the cyanobacterial amino acid metabolism with a precise genome-scale metabolic reconstruction of Anabaena sp. UTEX 2576. Biochem Eng J. 2021 Jul 1;171. [CrossRef]
- Canto-Encalada G, Tec-Campos D, Tibocha-Bonilla JD, Zengler K, Zepeda A, Zuñiga C. Flux balance analysis of the ammonia-oxidizing bacterium Nitrosomonas europaea ATCC19718 unravels specific metabolic activities while degrading toxic compounds. PLoS Comput Biol. 2022 Feb 1;18(2). [CrossRef]
- Zuñiga C, Li CT, Huelsman T, Levering J, Zielinski DC, McConnell BO, et al. Genome-scale metabolic model for the green alga Chlorella vulgaris UTEX 395 accurately predicts phenotypes under autotrophic, heterotrophic, and mixotrophic growth conditions. Plant Physiol. 2016 Sep 1;172(1):589–602.
- Zuñiga C, Peacock B, Liang B, McCollum G, Irigoyen SC, Tec-Campos D, et al. Linking metabolic phenotypes to pathogenic traits among “Candidatus Liberibacter asiaticus” and its hosts. NPJ Syst Biol Appl. 2020 Dec 1;6(1). [CrossRef]
- Seif Y, Monk JM, Mih N, Tsunemoto H, Poudel S, Zuniga C, et al. A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types. PLoS Comput Biol. 2019;15(1). [CrossRef]
- Tibocha-Bonilla JD, Kumar M, Richelle A, Godoy-Silva RD, Zengler K, Zuñiga C. Dynamic resource allocation drives growth under nitrogen starvation in eukaryotes. NPJ Syst Biol Appl. 2020 Dec 1;6(1). [CrossRef]
- Tec-Campos D, Posadas C, Tibocha-Bonilla JD, Thiruppathy D, Glonek N, Zuñiga C, et al. The genome-scale metabolic model for the purple non-sulfur bacterium Rhodopseudomonas palustris Bis A53 accurately predicts phenotypes under chemoheterotrophic, chemoautotrophic, photoheterotrophic, and photoautotrophic growth conditions. PLoS Comput Biol [Internet]. 2023 Aug 9;19(8):e1011371-. [CrossRef]
- Machado D, Andrejev S, Tramontano M, Patil KR. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018 Sep 6;46(15):7542–53. [CrossRef]
- Henry CS, Dejongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010 Sep;28(9):977–82. [CrossRef]
- Wang H, Marcišauskas S, Sánchez BJ, Domenzain I, Hermansson D, Agren R, et al. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput Biol. 2018 Oct 1;14(10). [CrossRef]
- Tibocha-Bonilla JD, Zuñiga C, Lekbua A, Lloyd C, Rychel K, Short K, et al. Predicting stress response and improved protein overproduction in Bacillus subtilis. NPJ Syst Biol Appl. 2022 Dec 1;8(1). [CrossRef]
- O’Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson B. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Syst Biol. 2013;9. [CrossRef]
- Zuñiga C, Li T, Guarnieri MT, Jenkins JP, Li CT, Bingol K, et al. Synthetic microbial communities of heterotrophs and phototrophs facilitate sustainable growth. Nat Commun. 2020 Dec 1;11(1). [CrossRef]
- Zuñiga C, Li CT, Yu G, Al-Bassam MM, Li T, Jiang L, et al. Environmental stimuli drive a transition from cooperation to competition in synthetic phototrophic communities. Nat Microbiol. 2019;4(12):2184–91.
- Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, et al. iML1515, a knowledgebase that computes Escherichia coli traits. Vol. 35, Nature Biotechnology. Nature Publishing Group; 2017. p. 904–8. [CrossRef]
- Oh YK, Palsson BO, Park SM, Schilling CH, Mahadevan R. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. Journal of Biological Chemistry. 2007 Sep 28;282(39):28791–9. [CrossRef]
- Lu H, Li F, Sánchez BJ, Zhu Z, Li G, Domenzain I, et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nat Commun. 2019 Dec 1;10(1).
- Brunk E, Sahoo S, Zielinski DC, Altunkaya A, Dräger A, Mih N, et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat Biotechnol. 2018 Mar 1;36(3):272–81. [CrossRef]
- Hefzi H, Ang KS, Hanscho M, Bordbar A, Ruckerbauer D, Lakshmanan M, et al. A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism. Cell Syst. 2016 Nov 23;3(5):434-443.e8. [CrossRef]
- Nagarajan H, Sahin M, Nogales J, Latif H, Lovley DR, Ebrahim A, et al. Characterizing acetogenic metabolism using a genome-scale metabolic reconstruction of Clostridium ljungdahlii. Microb Cell Fact [Internet]. 2013 Nov 25;12(118). Available online: http://www.microbialcellfactories.
- Broddrick JT, Rubin BE, Welkie DG, Du N, Mih N, Diamond S, et al. Unique attributes of cyanobacterial metabolism revealed by improved genome-scale metabolic modeling and essential gene analysis. Proc Natl Acad Sci U S A. 2016 Dec 20;113(51):E8344–53. [CrossRef]
- Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nature Protocols 2019 14:3 [Internet]. 2019 Feb 20 [cited 2023 Jun 4];14(3):639–702. Available online: https://www.nature.com/articles/s41596-018-0098-2.
- Karp PD, Midford PE, Billington R, Kothari A, Krummenacker M, Latendresse M, et al. Pathway Tools version 23.0 update: Software for pathway/genome informatics and systems biology. Vol. 22, Briefings in Bioinformatics. Oxford University Press; 2021. p. 109–26. [CrossRef]
- Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017 Jan 1;35(1):81–9. [CrossRef]
- Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41(Web Server issue). [CrossRef]
- Fleischmann A, Darsow M, Degtyarenko K, Fleischmann W, Boyce S, Axelsen KB, et al. IntEnz, the integrated relational enzyme database. Nucleic Acids Res. 2004 Jan 1;32(DATABASE ISS.). [CrossRef]
- Saier MH, Tran C V., Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34(Database issue).
- Duvaud S, Gabella C, Lisacek F, Stockinger H, Ioannidis V, Durinx C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021 Jul 2;49(W1):W216–27. [CrossRef]
- Bremer E, Calteau A, Danchin A, Harwood C, Helmann JD, Médigue C, et al. A model industrial workhorse: Bacillus subtilis strain 168 and its genome after a quarter of a century. Vol. 16, Microbial Biotechnology. John Wiley and Sons Ltd; 2023. p. 1203–31. [CrossRef]
- Emanuelsson O, Nielsen H, Brunak S, Von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000 Jul 21;300(4):1005–16. [CrossRef]
- Gschloessl B, Guermeur Y, Cock JM. HECTAR: A method to predict subcellular targeting in heterokonts. BMC Bioinformatics. 2008 Sep 23;9. [CrossRef]
- Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017 Nov 1;33(21):3387–95.
- Tardif M, Atteia A, Specht M, Cogne G, Rolland N, Brugière S, et al. Predalgo: A new subcellular localization prediction tool dedicated to green algae. In: Molecular Biology and Evolution. 2012. p. 3625–39. [CrossRef]
- King ZA, Lu J, Dräger A, Miller P, Federowicz S, Lerman JA, et al. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44(D1):D515–22. [CrossRef]
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000 Jan 1;28(1):27–30.
- Hawkins C, Ginzburg D, Zhao K, Dwyer W, Xue B, Xu A, et al. Plant Metabolic Network 15: A resource of genome-wide metabolism databases for 126 plants and algae. Vol. 63, Journal of Integrative Plant Biology. John Wiley and Sons Inc; 2021. p. 1888–905. [CrossRef]
- Seaver SMD, Liu F, Zhang Q, Jeffryes J, Faria JP, Edirisinghe JN, et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021 Jan 8;49(D1):D575–88.
- Bairoch, A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28(1):304–5. [CrossRef]
- Bateman A, Martin MJ, Orchard S, Magrane M, Ahmad S, Alpi E, et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023 Jan 6;51(D1):D523–31.
- Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Vol. 28, Nature Biotechnology. 2010. p. 245–8.
- Gustafsson J, Anton M, Roshanzamir F, Jörnsten R, Kerkhoven EJ, Robinson JL, et al. Generation and analysis of context-specific genome-scale metabolic models derived from single-cell RNA-Seq data. Proc Natl Acad Sci U S A. 2023 Feb 7;120(6).
- Hamilton JJ, Dwivedi V, Reed JL. Quantitative assessment of thermodynamic constraints on the solution space of genome-scale metabolic models. Biophys J. 2013 Jul 16;105(2):512–22. [CrossRef]
- Fritzemeier CJ, Hartleb D, Szappanos B, Papp B, Lercher MJ. Erroneous energy-generating cycles in published genome scale metabolic networks: Identification and removal. PLoS Comput Biol. 2017 Apr 1;13(4). [CrossRef]
- Lieven C, Beber ME, Olivier BG, Bergmann FT, Ataman M, Babaei P, et al. MEMOTE for standardized genome-scale metabolic model testing. Nat Biotechnol [Internet]. 2020;38(3):272–6. [CrossRef]
- Chang RL, Ghamsari L, Manichaikul A, Hom EFY, Balaji S, Fu W, et al. Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol Syst Biol. 2011;7. [CrossRef]
- Tibocha-Bonilla JD, Kumar M, Zengler K, Zuniga C. Integrating Metabolic Modeling and High-Throughput Data to Characterize Diatoms Metabolism. In: The Mathematical Biology of Diatoms. Wiley; 2023. p. 165–91.
- Feist AM, Palsson BO. The biomass objective function. Vol. 13, Current Opinion in Microbiology. 2010. p. 344–9.
- Lachance JC, Lloyd CJ, Monk JM, Yang L, Sastry A V., Seif Y, et al. BOFDAT: Generating biomass objective functions for genome-scale metabolic models from experimental data. PLoS Comput Biol. 2019;15(4). [CrossRef]
- Broddrick JT, Welkie DG, Jallet D, Golden SS, Peers G, Palsson BO. Predicting the metabolic capabilities of Synechococcus elongatus PCC 7942 adapted to different light regimes. Metab Eng [Internet]. 2019;52:42–56. Available online: https://www.sciencedirect.com/science/article/pii/S1096717618303288.
- Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J Am Soc Mass Spectrom. 2016 Dec 1;27(12):1897–905. [CrossRef]
- Zhou F, Zuo J, Gao L, Sui Y, Wang Q, Jiang A, et al. An untargeted metabolomic approach reveals significant postharvest alterations in vitamin metabolism in response to LED irradiation in pak-choi (Brassica campestris L. ssp. chinensis (L.) Makino var. communis Tsen et Lee). Metabolomics. 2019 Dec 1;15(12). [CrossRef]
- Lommen A, van der Weg G, van Engelen MC, Bor G, Hoogenboom LAP, Nielen MWF. An untargeted metabolomics approach to contaminant analysis: Pinpointing potential unknown compounds. Anal Chim Acta. 2007 Feb 12;584(1):43–9. [CrossRef]
- Bernstein DB, Sulheim S, Almaas E, Segrè D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol [Internet]. 2021;22(1):64. [CrossRef]
- Pan S, Reed JL. Advances in gap-filling genome-scale metabolic models and model-driven experiments lead to novel metabolic discoveries. Vol. 51, Current Opinion in Biotechnology. Elsevier Ltd; 2018. p. 103–8. [CrossRef]
- Karp PD, Weaver D, Latendresse M. How accurate is automated gap filling of metabolic models? BMC Syst Biol. 2018 Jun 19;12(1).
- Thiele I, Vlassis N, Fleming RMT. FASTGAPFILL: Efficient gap filling in metabolic networks. Bioinformatics. 2014 Sep 1;30(17):2529–31. [CrossRef]
- Hartleb D, Jarre F, Lercher MJ. Improved Metabolic Models for E. coli and Mycoplasma genitalium from GlobalFit, an Algorithm That Simultaneously Matches Growth and Non-Growth Data Sets. PLoS Comput Biol. 2016 Aug 1;12(8). [CrossRef]
- Boughorbel S, Jarray F, El-Anbari M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS One. 2017 Jun 1;12(6). [CrossRef]
- Leonidou N, Fritze E, Renz A, Dr¨ager AD. SBOannotator: a Python Tool for the Automated Assignment of Systems Biology Ontology Terms. 2023. Available online: https://uni-tuebingen.de/en/216529.
- Anton M, Almaas E, Benfeitas R, Benito-Vaquerizo S, Blank LM, Dräger A, et al. standard-GEM: standardization of open-source genome-scale metabolic models. bioRxiv [Internet]. 2023. [CrossRef]
- Carey MA, Dräger A, Beber ME, Papin JA, Yurkovich JT. Community standards to facilitate development and address challenges in metabolic modeling. Mol Syst Biol. 2020 Aug;16(8). [CrossRef]
- Thiele I, Preciat G, Fleming RMT. MetaboAnnotator: an efficient toolbox to annotate metabolites in genome-scale metabolic reconstructions. Bioinformatics. 2022 Oct 14;38(20):4831–2. [CrossRef]
- Bansal P, Morgat A, Axelsen KB, Muthukrishnan V, Coudert E, Aimo L, et al. Rhea, the reaction knowledgebase in 2022. Nucleic Acids Res. 2022 Jan 7;50(D1):D693–700. [CrossRef]
- Moretti S, Tran VDT, Mehl F, Ibberson M, Pagni M. MetaNetX/MNXref: Unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 2021 Jan 8;49(D1):D570–4. [CrossRef]
- Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2018 Mar 27;20(4):1085–93. [CrossRef]
- Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022 Jan 7;50(D1):D687–92.
- Hucka M, Bergmann FT, Hoops S, Keating SM, Sahle S, Schaff JC, et al. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 1 Core. J Integr Bioinform. 2015;12(2):266.
- Hucka M, Bergmann FT, Dräger A, Hoops S, Keating SM, Le Novère N, et al. Systems Biology Markup Language (SBML) Level 2 Version 5: Structures and Facilities for Model Definitions. J Integr Bioinform. 2015;12(2):271.
- JSON [Internet]. [cited 2023 Jul 18]. Available online: https://www.json.org/json-en.html.
- Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst Biol. 2013 Aug 8;7. [CrossRef]
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: The United States department of energy systems biology knowledgebase. Vol. 36, Nature Biotechnology. Nature Publishing Group; 2018. p. 566–9. [CrossRef]
- Ben-Kiki O, Evans C, Ingerson B, Oren Ben-Kiki by. YAML Ain’t Markup Language (YAMLTM) Version 1.1 Working Draft 2005-01-18-CVS XSL • FO RenderX YAML Ain’t Markup Language (YAMLTM) Version 1.1 Working Draft 2005-01-18-CVS [Internet]. 2001. Available online: http://www.unicode.org/.
- Norsigian CJ, Fang X, Seif Y, Monk JM, Palsson BO. A workflow for generating multi-strain genome-scale metabolic models of prokaryotes. Nat Protoc. 2020 Jan 1;15(1):1–14. [CrossRef]
- Domenzain I, Sánchez B, Anton M, Kerkhoven EJ, Millán-Oropeza A, Henry C, et al. Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0. Nat Commun. 2022 Dec 1;13(1). [CrossRef]
- Heinken A, Thiele I. Microbiome Modelling Toolbox 2.0: efficient, tractable modelling of microbiome communities. Bioinformatics. 2022 Apr 15;38(8):2367–8. [CrossRef]
- Heinken A, Basile A, Thiele I. Advances in constraint-based modelling of microbial communities. Curr Opin Syst Biol [Internet]. 2021;27:100346. Available online: https://www.sciencedirect.com/science/article/pii/S2452310021000317.
Figure 1.
Template organisms with their model IDs used for M-models reconstruction. Organisms are organized depending on the carbon source they consume (organic carbon, CO2, CO+H2, CO+H2+CO2, and organic carbon+CO2), their metabolisms (A, aerobic; An, anaerobic, NF, nitrogen-fixing; AO, ammonia-oxidizing; LU, light uptake) and their category (gram-positive rod, gram-negative rod, mammal cell, yeast, green microalga, cyanobacterium). Organisms highlighted in blue and green mean prokaryote or eukaryote, respectively. References in parentheses. (8,13,14,26–32.).
Figure 1.
Template organisms with their model IDs used for M-models reconstruction. Organisms are organized depending on the carbon source they consume (organic carbon, CO2, CO+H2, CO+H2+CO2, and organic carbon+CO2), their metabolisms (A, aerobic; An, anaerobic, NF, nitrogen-fixing; AO, ammonia-oxidizing; LU, light uptake) and their category (gram-positive rod, gram-negative rod, mammal cell, yeast, green microalga, cyanobacterium). Organisms highlighted in blue and green mean prokaryote or eukaryote, respectively. References in parentheses. (8,13,14,26–32.).
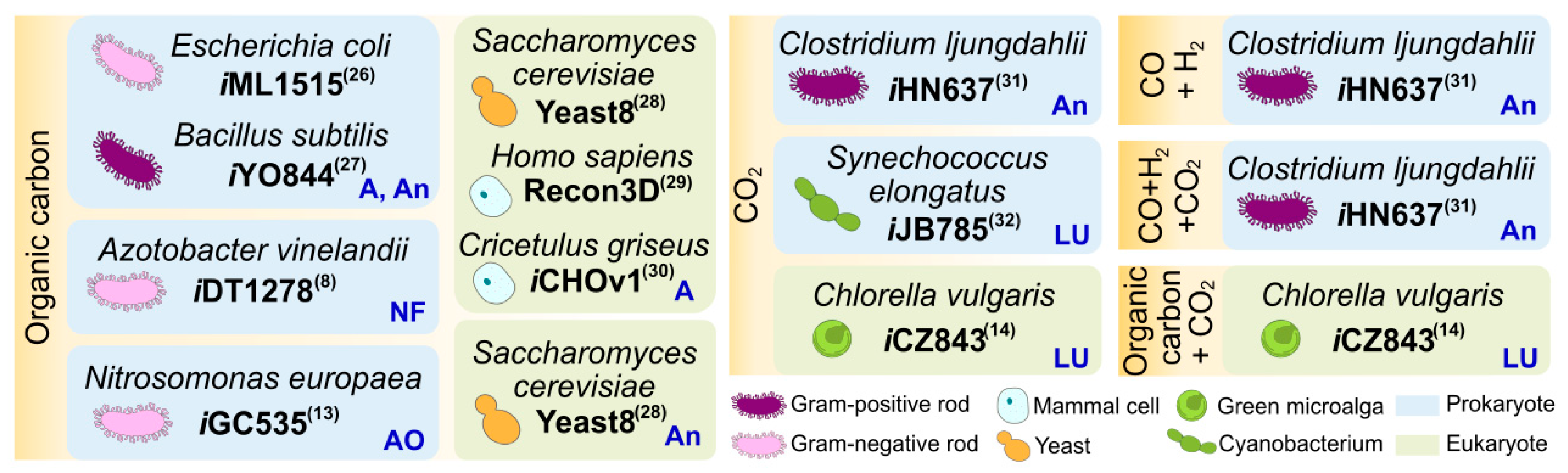
Figure 2.
Collecting information for manual curation. Workflow of GPR associations for a target organism. Several resources are used during the manual curation phase, such as primary literature and the databases BiGG (45), KEGG (46), IntEnz (37), PMN (47), ModelSEED (48), ENZYME@ExPASy (49), and UniProt (50). Information regarding transport proteins are obtained from TCDB (38). Subcellular protein localizations are predicted using TargetP (41), DeepLoc (43), HECTAR (42), and PredAlgo (44).
Figure 2.
Collecting information for manual curation. Workflow of GPR associations for a target organism. Several resources are used during the manual curation phase, such as primary literature and the databases BiGG (45), KEGG (46), IntEnz (37), PMN (47), ModelSEED (48), ENZYME@ExPASy (49), and UniProt (50). Information regarding transport proteins are obtained from TCDB (38). Subcellular protein localizations are predicted using TargetP (41), DeepLoc (43), HECTAR (42), and PredAlgo (44).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



