
Submitted:
07 November 2023
Posted:
08 November 2023
You are already at the latest version
Abstract
Analysis of telomere length is an important component of many studies aiming to characterize the role of telomere maintenance mechanisms in cellular lifespan, disease, or in general chromosome protection and DNA replication pathways. Several powerful methods to accurately measure telomere length from Southern blots have been developed, but their utility for large-scale genomic studies has not been previously evaluated. Here we performed comparative analysis of two recently developed programs, TeloTool and WALTER, for extracting mean telomere length values from Southern blots. Using both software packages, we measured telomere length in two extensive experimental datasets for the model plant Arabidopsis thaliana, consisting of 537 natural accessions and 65 T-DNA mutant lines in the Col-0 reference background. We report that TeloTool substantially overestimates telomere length in comparison to WALTER, especially for values over 4,500 bp. Importantly, TeloTool and WALTER-calculated telomere length values correlate the most in the 2,100-3,500 bp range, suggesting that telomeres in this size interval can be estimated by both programs equally well. We further show that genome-wide association studies using datasets from both telomere length analysis tools can detect the most significant SNP candidates equally well. However, GWAS analysis with the WALTER dataset consistently detects fewer significant SNPs than analysis with the TeloTool dataset, regardless of the GWAS method used. These results imply that telomere length data generated by WALTER may represent a more stringent approach to GWAS and SNP selection for downstream molecular screening of candidate genes. Overall, our work revealed the unanticipated impact of telomere length analysis method on the outcomes of large-scale genomic screens.
Keywords:
telomerase
; TeloTool
; WALTER
; telomere length
; SNP
; GWAS
1. Introduction
The ends of linear eukaryotic chromosomes are protected by telomeres, the evolutionarily conserved protein-DNA complexes, which are involved in genome maintenance and regulation of cellular lifespan. The critical functions of telomeres in chromosome protection were originally revealed by the classical Barbara McClintock’s studies in maize (McClintock, 1939, 1941). The mechanism of chromosome end deprotection was later predicted by Alexey Olovnikov to involve gradual attrition of telomere DNA over multiple cell divisions due to the intrinsic inability of conventional DNA polymerases to fully replicate linear chromosome ends (Olovnikov, 1973). Telomere shortening in human somatic cells was also shown to limit the number of cell divisions, contributing to the so-called Hayflick limit of cell proliferation (Hayflick, 1965; Hayflick and Moorhead, 1961). The discovery of the repeated nature of eukaryotic telomeres (Blackburn and Gall, 1978) opened the door for the analysis of telomere length, which in various organisms turned out to be incredibly varied, ranging from as little as 300 base pairs in yeast (Shampay et al., 1984) to as much as 160 kb in tobacco (Fajkus et al., 1995), while also varying up to 25-fold between different genotypes of the same species (Burr et al., 1992).
In recent years, many highly sensitive telomere length measuring assays have been developed, including Q-PCR, Q-FISH, STELA, TeSLA, among others (Lai et al., 2018). While most of these assays are very precise, many also have important drawbacks, including being very relative, expensive, or requiring sophisticated lab equipment. In the earlier years of telomere research, a simple method dubbed terminal restriction fragment analysis (TRF) emerged as one of the main and most accurate methods for telomere length analysis (Blackburn and Challoner, 1984; Ausubel et al., 1989; Lansdorp et al., 1996). The method is based on genomic DNA digestion using specialized restriction enzymes which cut frequently throughout the genome, but not inside telomeric sequences. Digested DNA is subsequently separated by molecular weight using agarose gel electrophoresis, transferred to a nylon membrane, and visualized using radioactive or fluorescently labeled probes (Fojtová et al., 2015; Mender and Shay, 2015). For many laboratories, telomere length measurement using the TRF protocol is still the gold standard method, and over the years various improvements have been made to enhance detection, applicability, and reproducibility, as well as to reduce associated costs (Jain et al., 2023; Nigmatullina et al., 2016).
The classical TRF method usually produces a smear representing the area of the Southern blot where the specific oligonucleotide probe hybridizes to telomeric DNA. This smear needs to be further quantified to obtain mean telomere length values for individual samples. Various software packages for estimating telomere length from Southern blots have been developed to assist in quantification of the telomere signals. One of the earlier specialized programs developed over 20 years ago to analyze TRF images was Telometric (Grant et al., 2001), which is still being extensively used in telomere research (Figure 1). However, Telometric calculates TRF values assuming a normally distributed intensity profile, often leading to substantially distorted median values. Furthermore, for very long telomeres over 12 kb, Telometric underestimates telomeres by up to 2 kb (Göhring et al., 2014), reducing its feasibility for analysis in long-telomere species. In 2014, a new software named TeloTool was developed, which fits the telomeric signal with a Gaussian function, making it particularly useful for samples with a unimodal distribution of the telomere-specific signal (Göhring et al., 2014). Finally, the most recently developed telomere length analysis tool, WALTER, is an online application that converts scanned TRF images into digital profiles consisting of telomere-specific signals and markers, allowing analysis of signals with a non-unimodal distribution of the telomere intensity profiles (Lyčka et al., 2021). The main features of TeloTool and WALTER telomere length analysis methods are summarized in Supplemental Table S1.
We have previously measured telomere length in 653 A. thaliana accessions and through a genome-wide association study (GWAS) discovered several significant SNPs, including one inside the TERT gene encoding the catalytic subunit of telomerase (Choi et al., 2021). The telomere length dataset for this study was generated using the TeloTool method (Göhring et al., 2014); however, recent comparison of telomere length analysis tools indicated that the WALTER program may provide a better way to analyze data for A. thaliana samples, as TeloTool tends to overestimate telomere length (Lyčka et al., 2021). Since our lab is currently engaged in several large-scale quantitative telomere length screens for which many samples were analyzed with TeloTool prior to publication of the WALTER method, it was necessary to evaluate the applicability of both methods to large-scale genomic screens. Specifically, we wanted to test if the two analysis tools produce similar results that can be used in combination with each other when 1) analyzing DNA samples from hundreds of Arabidopsis genotypes for GWAS screens, and 2) evaluating telomere length in individual T-DNA mutants of candidate genes as part of the follow-up molecular genetic tests.
Here we show that TeloTool substantially overestimates telomere length in comparison to WALTER, especially for values over 4,500 bp. However, both programs produce comparable results for telomeres in the 2,100-3,500 bp range, indicating that molecular analysis of most Arabidopsis T-DNA mutants of putative telomere biology genes can be performed equally well by both programs. Interestingly, the choice of telomere length analysis tools affects outcomes of GWAS screens: TeloTool dataset produces more significant SNPs than the WALTER dataset, though GWAS can identify the most significant hits using data from both datasets equally well. Collectively, these results indicate that data generated with different telomere length measurement tools can substantially influence downstream genomic and genetic screens.
2. Results
TeloTool – WALTER Comparison: General Differences and Similarities in Analyzing a Large-Scale Dataset
We first assessed how much the mean telomere length (mean TRF) data calculated by the TeloTool and WALTER programs differ from each other. We utilized telomere length data obtained through TRF blots for 537 Arabidopsis accessions (584 individual TRF measurements) (Supplemental Data 1). This dataset is smaller than the one used in our earlier study (653 accessions, Choi et al., 2021), and only includes accessions for which TRF blots were performed in our lab. Distributions of mean TRF values for this 537-accession dataset as measured by TeloTool and WALTER were plotted as bar graphs (Figure 2). Mean TRF values as calculated by TeloTool ranged from 1,313 bp in Ak-1 accession to 12,546 bp in Mh-0 genotype, with a median length of 3,592 bp (Figure 2A). For the WALTER analysis, mean TRF values ranged from 1,451 bp in Hov1-10 to 9,359 bp in Wc-2 accession, with the median length of 3,305 bp (Figure 2B). The overall distribution of the mean TRF values in the WALTER dataset was narrower than in the TeloTool dataset (Figure 2). Overall, TeloTool calculations on average provide higher values (longer telomere length) than those calculated with WALTER.
To take a more detailed look at the TeloTool-WALTER comparison, we next calculated the overall correlations between the two datasets, which were relatively high: 0.9 (Spearman's rho) and 0.92 (Pearson's r) (Figure 3A). We then broke the data down to specific telomere length intervals, defined as short (≤ 2,100 bp), medium (2,101-3,500 bp range), long (3,501-4,500 bp) and very long (≥4,501 bp). The strongest correlations between TeloTool and WALTER values were observed in the medium telomere length interval 2,101-3,500 bp (Spearman's rho is 0.76 and Pearson's r is 0.74) and in the very long telomere interval ≥4,501 bp (Spearman's rho is 0.77, and Pearson's r is 0.84) (Supplemental Figure S1B,D). Correlation for the long telomere interval 3,501-4,500 bp was weaker (0.46 for Spearman and 0.41 for Pearson), while no statistically significant correlation between TeloTool and WALTER values was observed for the ≤ 2,100 bp interval (0.07 for Spearman and 0.08 for Pearson) (Supplemental Figure S1A,C).
We also compared absolute differences between TeloTool and WALTER values. In support of the notion that TeloTool tends to overestimate particularly long telomeres (in comparison to WALTER), for all analyzed telomeres above 4.5 kb (123 individual DNA samples for 111 accessions), TeloTool values were higher than WALTER values, with several TeloTool measurements being up to 91% higher than WALTER values (Figure 3B, Table 1). For the second group of DNA samples with telomere length in the long range (3,501-4,500 bp), the overestimation of telomere length by TeloTool is also observed, with the mean difference between values in this telomere length interval being at 12.01% (Table 1). For the telomere length data in the medium range (2,101-3,500 bp), we observed the most correlation between TeloTool and WALTER-calculated values, with the mean difference between the two datasets being only 7.40% (Figure 3B, Table 1). Finally, for the short (≤ 2,100 bp) telomere length range, WALTER values were on average slightly longer than the TeloTool values (mean difference is -10.94 %) (Table 1), though this effect is largely driven by 3 out of 16 DNA samples in this range (Figure 3B), and their removal from the analysis decreases the mean difference in this size range to -5.30%. Taken together, our data indicate that TeloTool- and WALTER-generated data overall correlate well, but display the most divergence in the shortest telomere length range ≤ 2,100 bp, while showing the most correlation in the 2,101-3,500 bp range.
Telomere Length Estimates by TeloTool or WALTER Methods Can Affect GWAS Outcomes
To evaluate how telomere length values extracted from TRF blots by different programs can influence results of large-scale genomic assays, we performed separate GWAS analyses using data generated by TeloTool and WALTER. GWAS was performed using the GWA-Portal (https://gwas.gmi.oeaw.ac.at), a user-friendly and interactive web application for running Arabidopsis GWAS studies (Seren, 2018). Data were analyzed through the standard pipeline using the following parameters: Imputed Fullsequence Dataset for Arabidopsis genotypes (TAIR 9) and LOG transformation for telomere length data.
To initiate our analysis, we first performed GWAS with TeloTool-generated data using the simple linear regression (LM) method, which revealed ten genomic regions with single-nucleotide polymorphisms (SNPs) significantly associated (after Bonferroni correction) with telomere length (Figure 4A). In contrast, analysis of the WALTER dataset using the same method revealed only 3 genomic regions significantly associated with telomere length (Figure 4B). Importantly, two of the significant SNPs with the highest p-values were detected in both analyses. One of these significant SNPs on chromosome 5 position 5538242 is located inside the TERT gene At5g16850 (Table 2) and represents the same SNP that was identified in our earlier study (Choi et al., 2021). The identification of TERT gene polymorphism in this and the previous studies, as well as when using both TeloTool and WALTER datasets, implies that both telomere length analysis tools are suitable for the identification of the most significant hits in large-scale genomic studies.
The second significant SNP detected with both TeloTool and WALTER datasets is located on chromosome 1 position 12379845 in intergenic region near promoter of gene At1g34042 (Table 2), which appears to be highly expressed in flowers, roots and seeds and encodes a small hypothetical protein. Other genes located in the vicinity of this SNP include Tryptophan Aminotransferase Related 3 (TAR3), Ribosomal protein S13, and UDP-RHA/UDP-GAL Transporter 6 (URGT6). The presence of the ribosomal protein S13 is particularly intriguing, as previous genome-wide assays implicated several ribosome biogenesis factors in telomere length control (Abdulkina et al., 2019).
Eight other significant SNPs identified in the TeloTool dataset have lower p-values and are distributed across all five Arabidopsis chromosomes, mostly in intergenic regions or inside protein-coding genes (Table 2). The three notable examples are SNPs located on: chromosome 4, position 2463357 (intron of At4g04870 gene, which encodes Cardiolipin synthase) and position 16935117 (missense nucleotide change aTg/aAg in At4g35733 gene, leading to M231K substitution in F-box SKIP23-like protein), and on chromosome 3, position 7407370 (missense nucleotide change Gga/Aga in At3g21120 gene, leading to G349R substitution in a little-characterized F-box protein).
One additional significant SNP identified using the WALTER dataset is located on chromosome 1 position 8743486 (intron of At1g24706 gene, which encodes THO2, a component of the putative Arabidopsis THO/TREX complex). Interestingly, with the exception of the polymorphism located inside the TERT gene, none of the other 10 significant SNPs from both GWAS analyses correspond to SNPs identified in our previous study using a larger dataset of 653 accessions (Choi et al., 2021). However, two significant SNPs (chromosome 2 positions 2422473 and 356311 from the TeloTool dataset) are located in a QTL interval on chromosome 2 that was previously identified in a telomere length mapping study using Pro-0/Col-0 recombinant inbred line population (Fulcher et al., 2015).
We next repeated our GWAS analyses with the accelerated linear mixed model (AMM), which is the only method available in the GWA-Portal that accounts for population structure and thus should work better for identifying loci of complex traits for species confounded by population structure, like Arabidopsis (Kang et al., 2008). With the AMM method, analysis of both TeloTool and WALTER datasets identified the same significant SNP on chromosome 5 position 5538242 that is located inside the TERT gene (Supplemental Figure S2). Additionally, GWAS analysis using the TeloTool dataset identified two more significant SNPs (Table 2). One is located very close to the telomere on chromosome 3 position 23295214 (promoter of At3g63030 gene, which encodes METHYL-CPG-BINDING DOMAIN 4 protein), and one is located in intergenic region on chromosome 1 position 21870431 between genes At1g59530 (encoding BASIC LEUCINE-ZIPPER 4 protein) and At1g59540 (encoding a kinesin-like protein). No other significant SNPs were discovered with the AMM method for either TeloTool or WALTER datasets.
Overall, results of our GWAS analyses suggest that data obtained using the TeloTool or WALTER programs will provide partially overlapping but not identical results. Specifically, GWAS analyses with both datasets identified the same one (AMM) or two (LM) most significant SNPs with the highest p-values, but all additional SNPs with lower p-values differed between the two analyses. Furthermore, GWAS analysis performed with the TeloTool dataset revealed more significant SNPs than was the case for the WALTER dataset, regardless of the GWAS algorithm used. We conclude that both telomere length analysis tools can be used to identify the most significant hits, but less significant candidates will likely differ between the two datasets. We further suggest that the higher number of significant SNPs identified with the TeloTool dataset can likely be attributed to overestimation of telomere length by this program, in comparison to WALTER.
Comparison of TeloTool and WALTER Datasets Generated for the Arabidopsis T-DNA Mutant Screen
We next examined the utility of WALTER and TeloTool telomere length measurement methods for screening a large collection of A. thaliana T-DNA mutants generated in the reference Columbia genotype (Alonso et al., 2003). In total, TRF data for 205 individual DNA samples were analyzed, which included 22 replicates of the reference Col-0 accession, 51 individual T-DNA mutant lines with 2 or more biological replicates for each, and 14 T-DNA lines with only a single biological replicate (Supplemental Data 2). As was expected for T-DNA mutants generated in the same genetic background, the overall distribution of mean TRF values calculated using TeloTool was much narrower (Figure 5A) than that observed for natural Arabidopsis accessions and ranged from 2,017 to 3,574 bp, with the median length of 2,714 bp. This telomere length range effectively corresponds to the “medium” group of telomere lengths described for the GWAS samples. Analysis of the same TRF blots with WALTER produced a similar profile (Figure 5B), though the overall distribution of TRF values was shifted to the left in comparison to the TeloTool data and ranged from 1,776 to 3,315 bp, with the median length of 2,520 bp. The correlation coefficients between TeloTool and WALTER values for telomere length of T-DNA mutant lines were also relatively high, 0.73 for Spearman and 0.74 for Pearson (Figure 5C). Overall, similar to the situation with different Arabidopsis accessions, we observed a general trend for TeloTool to overestimate telomere length compared to WALTER. However, we infer from this analysis that when proper wild type controls are included in the study, either program can be used equally well to analyze telomere length phenotypes of mutants of Arabidopsis gene candidates discovered through GWAS or other large-scale genomic assays.
3. Discussion
Terminal restriction fragment analysis is a powerful and efficient way to measure telomere length in a number of species and populations. While different methods have been extensively used in the past to extract telomere length information from TRF gels, such as GelQuant software (Ungar et al., 2009) or Multi Gauge V3.0 package (Liu et al., 2010), the development of several specialized tools dedicated to telomere length quantification (Grant et al., 2001; Göhring et al., 2014; Lyčka et al., 2021) marked a major step forward towards normalizing and comparing results obtained from different gels and even from different research labs.
TeloTool and WALTER utilize contrasting approaches to calculating telomere length, and each method has its own advantages and drawbacks. Specifically, previous observations indicated that TeloTool can overestimate telomeres, while WALTER can potentially allow for a more subjective calculation (Lyčka et al., 2021). Using hundreds of individually analyzed DNA samples, we have compared the applicability of both methods to downstream large-scale genomic assays. Our findings using data for 537 natural Arabidopsis accessions confirmed previous observations (Lyčka et al., 2021) that TeloTool software can indeed overestimate telomere length, with the important additional clarification that this bias especially affects very long telomeres above 4,500 bp. For the shortest telomere range below 2,100 bp we also observed substantial variation between TeloTool- and WALTER-generated data, but it should be noted that due to the peculiarity of hybridization kinetics, short telomeres (2 kb or less) are generally difficult to measure with any quantification method, and especially with the TRF assays (Lai et al., 2018). Additionally, we noticed that samples in our analysis that show the greatest difference between TeloTool and WALTER values often come from TRF gels that are characterized by reduced quality of telomeric DNA signal (minor signs of degradation, weak signal, stains, bubbles). While some of these technical challenges are unavoidable when conducting large-scale screening of hundreds of natural accessions or T-DNA mutants, general improvements in the TRF technique will to some extent minimize differences in values calculated by the two programs.
Our findings also indicate that data generated with both TeloTool and WALTER can be used to detect the most significant SNPs in GWAS screens. The identification of the previously described (through a larger 653-accession study by Choi et al., 2021) significant SNP inside the TERT gene can be viewed as an internal positive control highlighting the general applicability of both methods to GWAS assays. However, the use of telomere length data generated by the TeloTool program resulted in detection of many more statistically significant SNPs than was obtained in the case of the WALTER dataset, regardless of the GWAS method used (AMM or LM). Only two overlapping significant SNP hits were identified using data from both TeloTool and WALTER, suggesting that WALTER provides a more conservative approach to GWAS mapping. We speculate that this substantial variability in GWAS results can be largely explained by differences in measuring the most extreme telomere length phenotypes (longer than 4.5 kb and below 2.1 kb).
For the T-DNA mutant screening experiment, our analysis indicated that Telo Tool and WALTER values in the size range of 2,100 – 3,500 bp are relatively similar to each other, suggesting that both programs can be used interchangeably for telomere length analysis, if all proper wild type controls are included. Since we also detected some degree of variation even between Col-0 wild type plants, we recommend that whenever possible TRF analysis of homozygous T-DNA mutant plants should be performed in comparison with their corresponding wild type siblings, and not with unrelated wild type plants. This is also important in the context of comparing data with studies from other labs, especially when telomere length calculation was carried out with a different program.
4. Materials and Methods
Plant Materials and Growth Conditions
For the GWAS experiment, seeds for the set of A. thaliana genotypes from the 1,001 Genome Project were purchased from the Arabidopsis Biological Resource Center (ABRC CS78942). A total of 584 DNA samples were run on TRF gels for telomere length analysis, representing 537 accessions from the CS78942 set, including 43 accessions with more than one biological replicate (individual plants). The 537-accession subset included previously published telomere length data for 424 A. thaliana accessions (Choi et al., 2021), as well as new data for 113 additional accessions included in this study (Supplemental Data 1).
For the T-DNA mutant screen, seeds for the wild type accession Col-0 (CS6673) and individual T-DNA mutants (Supplemental Data 2) were obtained from ABRC. A total of 205 DNA samples were run on TRF gels for telomere length analysis, representing one or more biological replicates of 65 individual T-DNA lines and the wild type control Col-0. Seeds were sown into a mixture of three parts Promix BX mycorrhizae soil, one part Profile Field and Fairway calcined clay, and one part Turface medium stabilizer, and plants were grown as described earlier (Choi et al., 2021). Plant tissue for TRF analysis was collected at the 5-week stage.
Telomere Length Measurement
Genomic DNA was extracted from individual whole plants and digested with the restriction enzyme Tru1I (Fermentas, Hanover, MD, USA) as previously described (Fitzgerald et al., 1999). [32P] 5’-end labeled or 5’-DIG-(T3AG3)4 oligonucleotides were used as probes (Nigmatullina et al., 2016; Abdulkina et al., 2019). Radioactive signals were scanned with a Pharos FX Plus Molecular Imager (Bio-Rad), and nonradioactive signals were scanned with a GBox-F3 Imager (Syngene). Images were visualized with Quantity One v.4.6.5 software (Bio-Rad), and mean telomere length values (mean TRF) were calculated using the TeloTool program (Göhring et al., 2014) or the WALTER program (Lyčka et al., 2021). TRF gels were run by different researchers, but all calculations with both TeloTool and WALTER were performed by the same person.
Genome-Wide Association Study
The GWA-Portal (https://gwas.gmi.oeaw.ac.at) web application was utilized for running Arabidopsis GWAS studies (Seren, 2018). Analysis was conducted with the “Imputed Fullsequence” genotype dataset (2,029 genotypes with ~ 10 million SNPs), which represents a combined dataset of the 250K SNP dataset and the 1001 genomes dataset using imputation. Upon performing LOG transformation of telomere length for both TeloTool and WALTER datasets, GWAS was conducted with the linear regression (LM) method and with the accelerated linear mixed model (AMM).
Statistical Analysis
Statistical analysis was carried out with GraphPad Prism 8 software (San Diego, USA). Trend line analysis was performed using linear regression parameters. Number of DNA samples, minimum and maximum differences, median and mean values of differences were calculated using the descriptive statistics parameters. Correlation coefficients for Pearson and Spearman methods along with their statistical significance were calculated using R software (cor.test() function).
5. Conclusions
In conclusion, our study provides several important recommendations for evaluating large-scale telomere length datasets for genomic studies and for the analysis of individual mutants with deregulated telomere length homeostasis. First, when analyzing DNA samples from hundreds of Arabidopsis accessions, TeloTool and WALTER should not be used in combination with each other for telomere length calculations, as each program calculates telomere length differently, especially in the very long range of over 4,500 bp. If a more conservative approach to GWAS is desired, one should choose WALTER, but if a more extended candidate list is expected, TeloTool may be the more appropriate program to choose. However, when evaluating telomere length in individual T-DNA mutants of candidate genes following the extensive genomic screens, either program will be efficient in analyzing telomere length. Although both TeloTool and WALTER programs were developed by research groups primarily working with the model plant Arabidopsis thaliana (Göhring et al., 2014; Lyčka et al., 2021), these programs quickly gained popularity for telomere length analysis in many other classical and emerging plant models (Gutiérrez et al., 2023), as well as for the analysis of non-plant telomeres, including those in human cells, cancer cell lines, green algae and Trypanosomes (Jain et al., 2023; Schreglmann et al., 2023; Kato et al., 2021; Fajkus et al., 2021; Poláková et al., 2021; Mannherz and Agarwal, 2023). Thus, our results may be relevant for telomere biology studies and functional analysis of candidate genes in other systems, including large-scale genomic screens in other models and in humans.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
All authors contributed significantly to this work. L.R.A., I.A.A., and E.V.S. designed the research. L.R.A, I.A.A., L.R.V. and E.V.S. performed the research. L.R.A, I.A.A., O.S.K., M.R.S., and E.V.S. analyzed the results. L.R.A. measured telomere length with both TeloTool and WALTER, and I.A.A. created the figures. L.R.A, I.A.A., and E.V.S. wrote the article with contributions from all other authors.
Data Availability
The data that support the findings of this study are available in the supplementary material of this article. Correspondence and requests for materials should be addressed to E.V.S.
Acknowledgements
We thank Dr. Jae Choi (University of Kansas) for insightful discussions. This work was supported in part by the National Institutes of Health (R01 GM127402 to E.V.S.) and the Kazan Federal University Strategic Academic Leadership Program. Russian Science Foundation project 21-14-00147 to L.R.A. supported genetic analysis of Arabidopsis T-DNA mutant lines. The authors declare no competing financial interests.
References
- Abdulkina, L.R.; Kobayashi, C.; Lovell, J.T.; Chastukhina, I.B.; Aklilu, B.; Agabekyan, I.A.; Valeeva, L.R.; Nyamsuren, C.; Aglyamova, G.V.; Sharipova, M.R.; Shippen, D.E.; Juenger, T.J.; Shakirov, E.V. Components of the ribosome biogenesis pathway underlie establishment of telomere length set point in Arabidopsis. Nature Communications. 2019, 10, 5479. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.M.; Stepanova, A.N.; Leisse, T.J.; Kim, C.J.; Chen, H.; Shinn, P.; Stevenson, D.K.; Zimmerman, J.; Barajas, P.; Cheuk, R.; Gadrinab, C.; Heller, C.; Jeske, A.; Koesema, E.; Meyers, C.C.; Parker, H.; Prednis, L.; Ansari, Y.; Choy, N.; Deen, H.; Geralt, M.; Hazari, N.; Hom, E.; Karnes, M.; Mulholland, C.; Ndubaku, R.; Schmidt, I.; Guzman, P.; Aguilar-Henonin, L.; Schmid, M.S.; Weigel, D.; Carter, D.E.; Marchand, T.; Risseeuw, E.; Brogden, D.; Zeko, A.; Crosby, W.L.; Berry, C.C.; Ecker, J.R. Genome-Wide Insertional Mutagenesis of Arabidopsis thaliana. Science. 2003, 301, 653. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current protocols in molecular biology; John Wiley & Sons, Inc.: Media, PA, USA, 1989; pp. 1–146. [Google Scholar]
- Blackburn, E.H.; Challoner, P.B. Identification of a telomeric DNA sequence in Trypanosoma brucei. Cell. 1984, 36, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Burr, B.; Burr, F.A.; Matz, E.C.; Romero-Severson, J. Pinning down loose ends: Mapping telomeres and factors affecting their length. Plant Cell. 1992, 4, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Abdulkina, L.R.; Yin, J.; Chastukhina, I.B.; Lovell, J.T.; Agabekian, I.A.; Young, P.G.; Razzaque, S.; Shippen, D.E.; Juenger, Th.E.; Shakirov, E.V.; Purugganan, M.D. Natural variation in plant telomere length is associated with flowering time. The Plant Cell. 2021, 33, 1118–1134. [Google Scholar] [CrossRef] [PubMed]
- Fajkus, J.; Kovarik, A.; Kralovics, R.; Bezdek, M. Organization of telomeric and subtelomeric chromatin in the higher plant Nicotiana tabacum. Mol. Gen. Genet. 1995, 247, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Fajkus, P.; Kilar, A.; Nelson, A.D.L.; Holá, M.; Peška, V.; Goffová, I.; Fojtová, M.; Zachová, D.; Fulnečková, J.; Fajkus, J. Evolution of plant telomerase RNAs: farther to the past, deeper to the roots. Nucleic Acids Res. 2021, 49, 7680–7694. [Google Scholar] [CrossRef]
- Fitzgerald, M.S.; Riha, K.; Gao, F.; Ren, S.; McKnight, T.D.; Shippen, D.E. Disruption of the telomerase catalytic subunit gene from Arabidopsis inactivates telomerase and leads to a slow loss of telomeric DNA. Proc Natl Acad Sci U S A. 1999, 96, 14813–14818. [Google Scholar] [CrossRef]
- Fojtová, M.; Fajkus, P.; Polanská, P.; Fajkus, J. Terminal Restriction Fragments (TRF) Method to Analyze Telomere Lengths. Bio-protocol. 2015, 5, e1671. [Google Scholar] [CrossRef]
- Fulcher, N.; Teubenbacher, A.; Kerdaffrec, E.; Farlow, A.; Nordborg, M.; Riha, K. Genetic architecture of natural variation of telomere length in Arabidopsis thaliana. Genetics. 2015, 199, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Göhring, J.; Fulcher, N.; Jacak, J.; Riha, K. TeloTool: a new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction. Nucleic Acids Research. 2014, 42, e21. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.D.; Broccoli, D.; Muquit, M.; Manion, F.J.; Tisdall, J.; Ochs, M.F. Telometric: a tool providing simplified, reproducible measurements of telomeric DNA from constant field agarose gels. Biotechniques. 2001, 31, 1314–1316. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, M.L.; Rodríguez-González, R.; Pascual-Díaz, J.P.; Fuentes, I.; Garcia, S. Online Resources Useful for Plant Cytogenetics and Cytogenomics Research. In Plant Cytogenetics and Cytogenomics. Methods in Molecular Biology; Heitkam, T., Garcia, S., Eds.; Humana: New York, NY, USA, 2023; Volume 2672, pp. 549–560. [Google Scholar]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The Limited in vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Madeka, S.; Khattar, E. Optimization of Performance Parameters of the TAGGG Telomere Length Assay. J. Vis. Exp. 2023, 194, e65288. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics. 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Kawaguchi, A.; Nagata, K. Template activating factor-I epigenetically regulates the TERT transcription in human cancer cells. Sci Rep. 2021, 11, 17726. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos Trans R Soc Lond B Biol Sci. 2018, 373, 20160451. [Google Scholar] [CrossRef]
- Lansdorp, P.M.; Verwoerd, N.P.; van de Rijke, F.M.; Dragowska, V.; Little, M.T.; Dirks, R.W.; Raap, A.K.; Tanke, H.J. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 1996, 5, 685–691. [Google Scholar] [CrossRef]
- Liu, N.N.; Han, T.X.; Du, L.L.; Zhou, J.Q. A genome-wide screen for Schizosaccharomyces pombe deletion mutants that affect telomere length. Cell Res. 2010, 20, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Lyčka, M.; Peska, V.; Demko, M.; Spyroglou, I.; Kilar, A.; Fajkus, J.; Fojtová, M. WALTER: an easy way to online evaluate telomere lengths from terminal restriction fragment analysis. BMC Bioinformatics. 2021, 22, 145. [Google Scholar] [CrossRef] [PubMed]
- Mannherz, W.; Agarwal, S. Thymidine nucleotide metabolism controls human telomere length. Nat Genet. 2023, 55, 568–580. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc. Natl. Acad. Sci. 1939, 25, 405–416. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941, 26, 234–282. [Google Scholar] [CrossRef] [PubMed]
- Mender, I.; Shay, J.W. Telomere Restriction Fragment (TRF) Analysis. Bio Protoc. 2015, 5, e1658. [Google Scholar] [CrossRef] [PubMed]
- Nigmatullina, L.R.; Sharipova, M.R.; Shakirov, E.V. Non-radioactive TRF assay modifications to improve telomeric DNA detection efficiency in plants. BioNanoScience. 2016, 6, 325–328. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Poláková, E.; Albanaz, A.T.S.; Zakharova, A.; Novozhilova, T.S.; Gerasimov, E.S.; Yurchenko, V. Ku80 is involved in telomere maintenance but dispensable for genomic stability in Leishmania mexicana. PLoS Negl Trop Dis. 2021, 15, e0010041. [Google Scholar] [CrossRef]
- Schreglmann, S.R.; Goncalves, T.; Grant-Peters, M.; Kia, D.A.; Soreq, L.; Ryten, M.; Wood, N.W.; Bhatia, K.P.; Tomita, K. Age-related telomere attrition in the human putamen. Aging Cell. 2023, 22, e13861. [Google Scholar] [CrossRef]
- Seren, Ü. GWA-Portal: genome-wide association studies made easy. Methods Mol Biol. 2018, 1761, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature. 1984, 310, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Ungar, L.; Yosef, N.; Sela, Y.; Sharan, R.; Ruppin, E.; Kupiec, M. A genome-wide screen for essential yeast genes that affect telomere length maintenance. Nucleic Acids Res. 2009, 37, 3840–3849. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Citations per year for the three telomere length measurement tools: Telometric, TeloTool and WALTER. The number of citations for the three original articles describing each method (Grant et al., 2001; Gohring et al., 2014; Lyčka et al., 2021) was obtained from the Google Scholar database (accessed on August 29, 2023), and each of the papers was manually verified for using the corresponding method in calculating telomere length from TRF blots.
Figure 1.
Citations per year for the three telomere length measurement tools: Telometric, TeloTool and WALTER. The number of citations for the three original articles describing each method (Grant et al., 2001; Gohring et al., 2014; Lyčka et al., 2021) was obtained from the Google Scholar database (accessed on August 29, 2023), and each of the papers was manually verified for using the corresponding method in calculating telomere length from TRF blots.
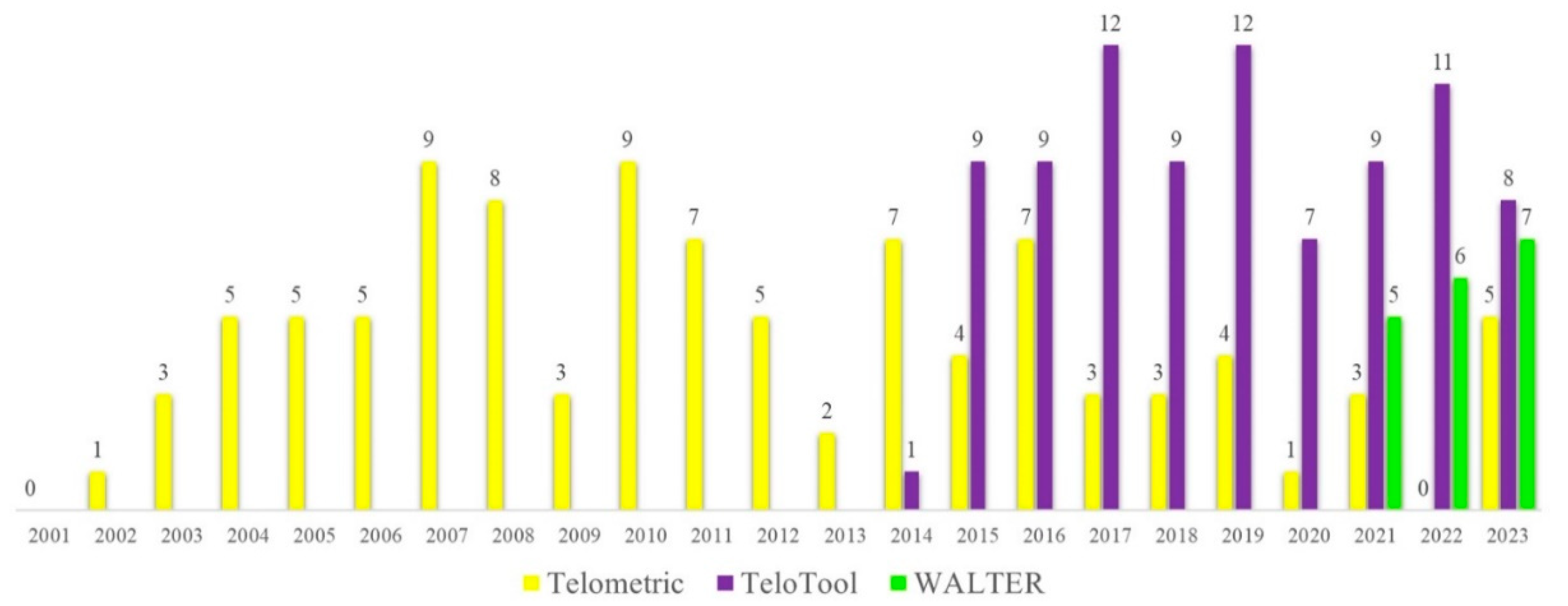
Figure 2.
Distribution of mean TRF values in Arabidopsis accessions used for GWAS analysis. Telomere length values as calculated by TeloTool (A) and WALTER (B) programs are grouped into 50 bp intervals, and the number of individual DNA samples falling into each interval are plotted against telomere length, sorted from shortest to longest. Red lines indicate median telomere length for each dataset. Dotted lines indicate limits of telomere length intervals selected for further analysis.
Figure 2.
Distribution of mean TRF values in Arabidopsis accessions used for GWAS analysis. Telomere length values as calculated by TeloTool (A) and WALTER (B) programs are grouped into 50 bp intervals, and the number of individual DNA samples falling into each interval are plotted against telomere length, sorted from shortest to longest. Red lines indicate median telomere length for each dataset. Dotted lines indicate limits of telomere length intervals selected for further analysis.
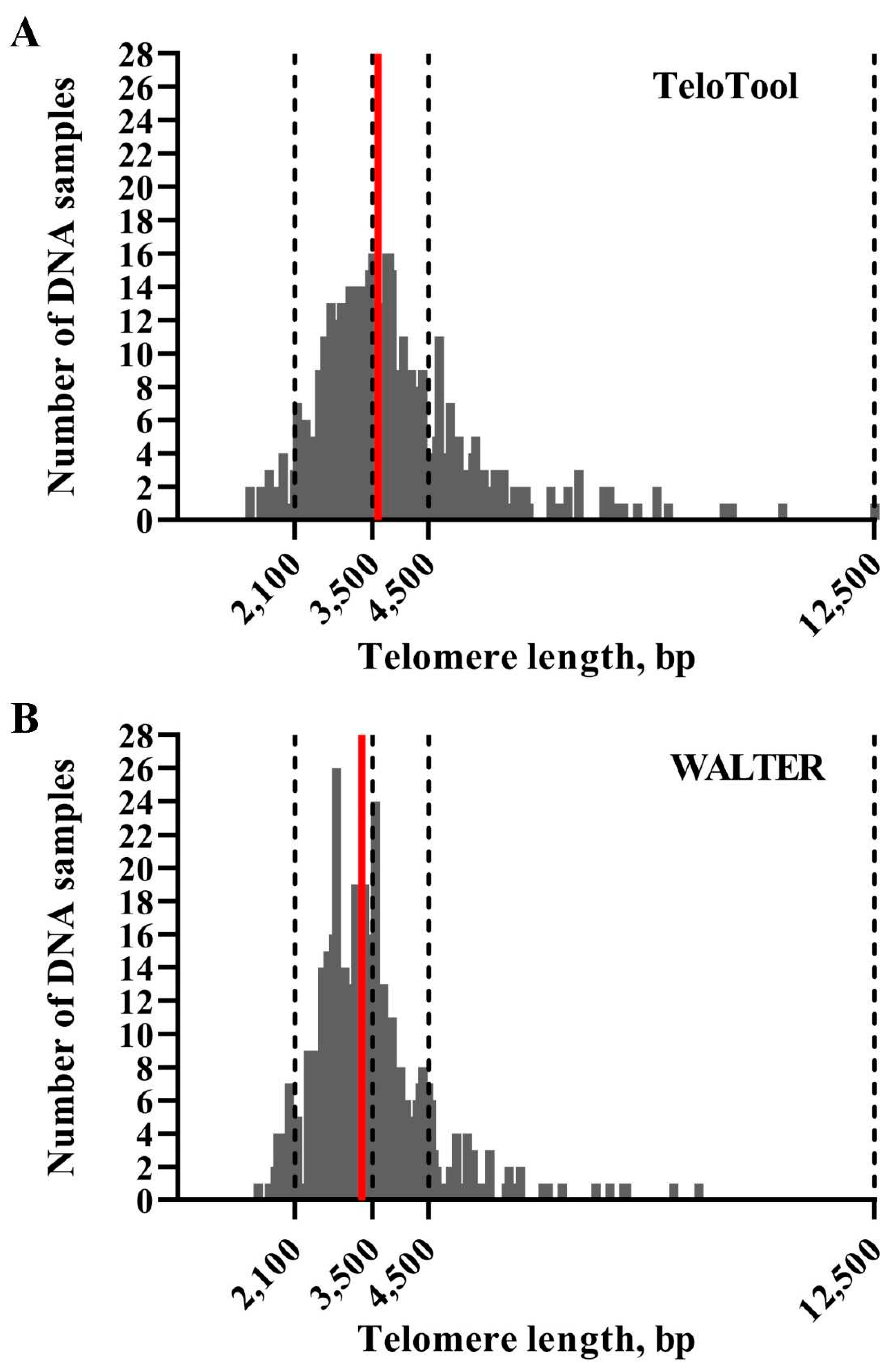
Figure 3.
Analysis of differences in telomere length measurements between TeloTool and WALTER programs. (A) Length values for TeloTool and WALTER measurements are plotted for each DNA sample. Dotted lines indicate limits of selected telomere length intervals. The trend line is shown in red. (B) Box plots of the percent differences between TeloTool data and WALTER data for each telomere length range, determined as (TeloTool-WALTER)/WALTER *100%. Whiskers indicate min to max range; points indicate the percent difference for each individual sample.
Figure 3.
Analysis of differences in telomere length measurements between TeloTool and WALTER programs. (A) Length values for TeloTool and WALTER measurements are plotted for each DNA sample. Dotted lines indicate limits of selected telomere length intervals. The trend line is shown in red. (B) Box plots of the percent differences between TeloTool data and WALTER data for each telomere length range, determined as (TeloTool-WALTER)/WALTER *100%. Whiskers indicate min to max range; points indicate the percent difference for each individual sample.
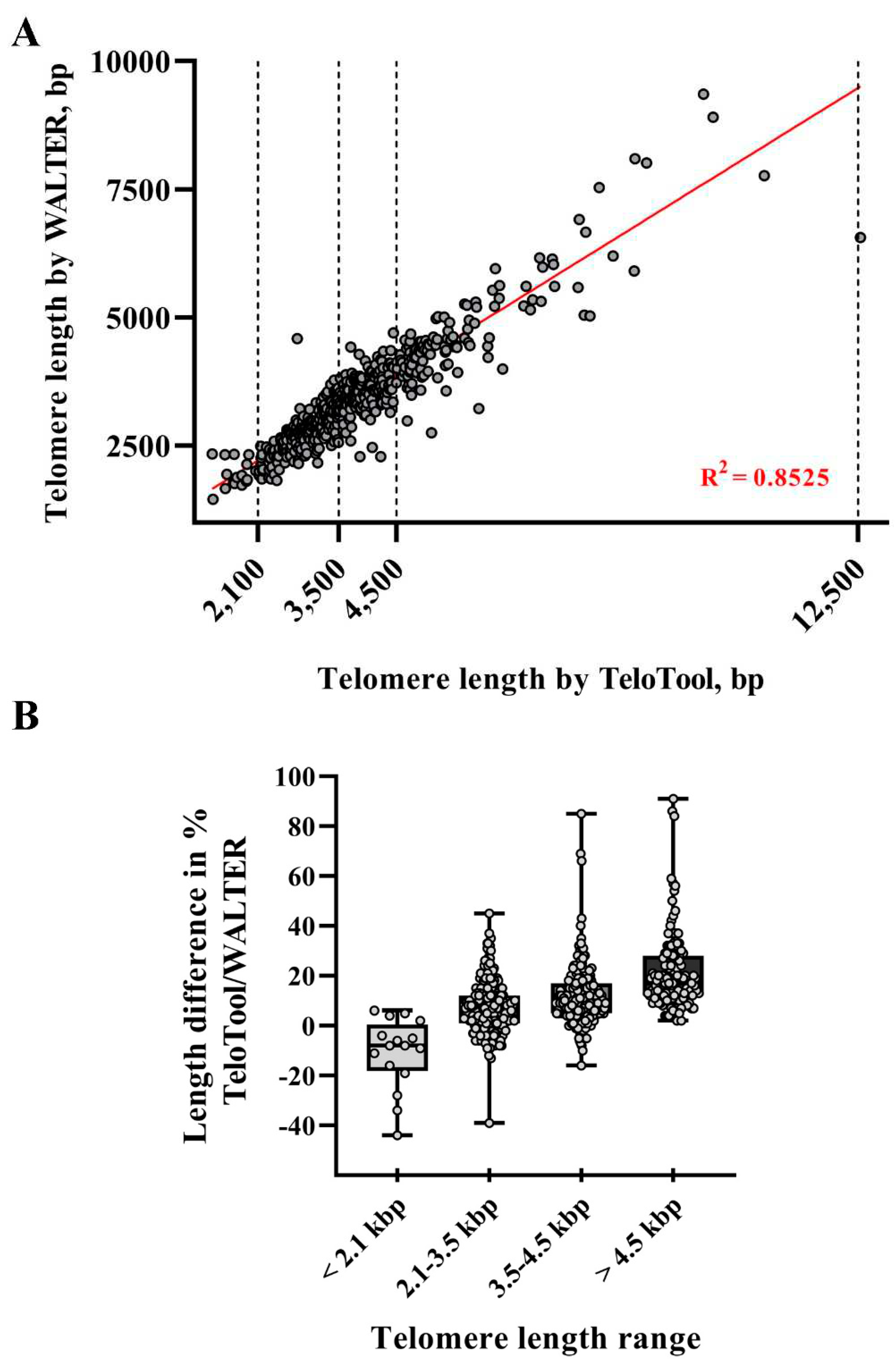
Figure 4.
GWAS of A. thaliana telomere length variation using the linear regression (LM) method. Manhattan plot of the genome-wide P-values indicating the strongest associations between the five Arabidopsis chromosomes and telomere length data obtained from TeloTool (A) and WALTER (B). Red dotted lines indicate the Bonferroni-corrected significance threshold (α = 0.05). The GWAS-significant regions discovered with both the TeloTool and WALTER datasets are indicated by red arrows.
Figure 4.
GWAS of A. thaliana telomere length variation using the linear regression (LM) method. Manhattan plot of the genome-wide P-values indicating the strongest associations between the five Arabidopsis chromosomes and telomere length data obtained from TeloTool (A) and WALTER (B). Red dotted lines indicate the Bonferroni-corrected significance threshold (α = 0.05). The GWAS-significant regions discovered with both the TeloTool and WALTER datasets are indicated by red arrows.
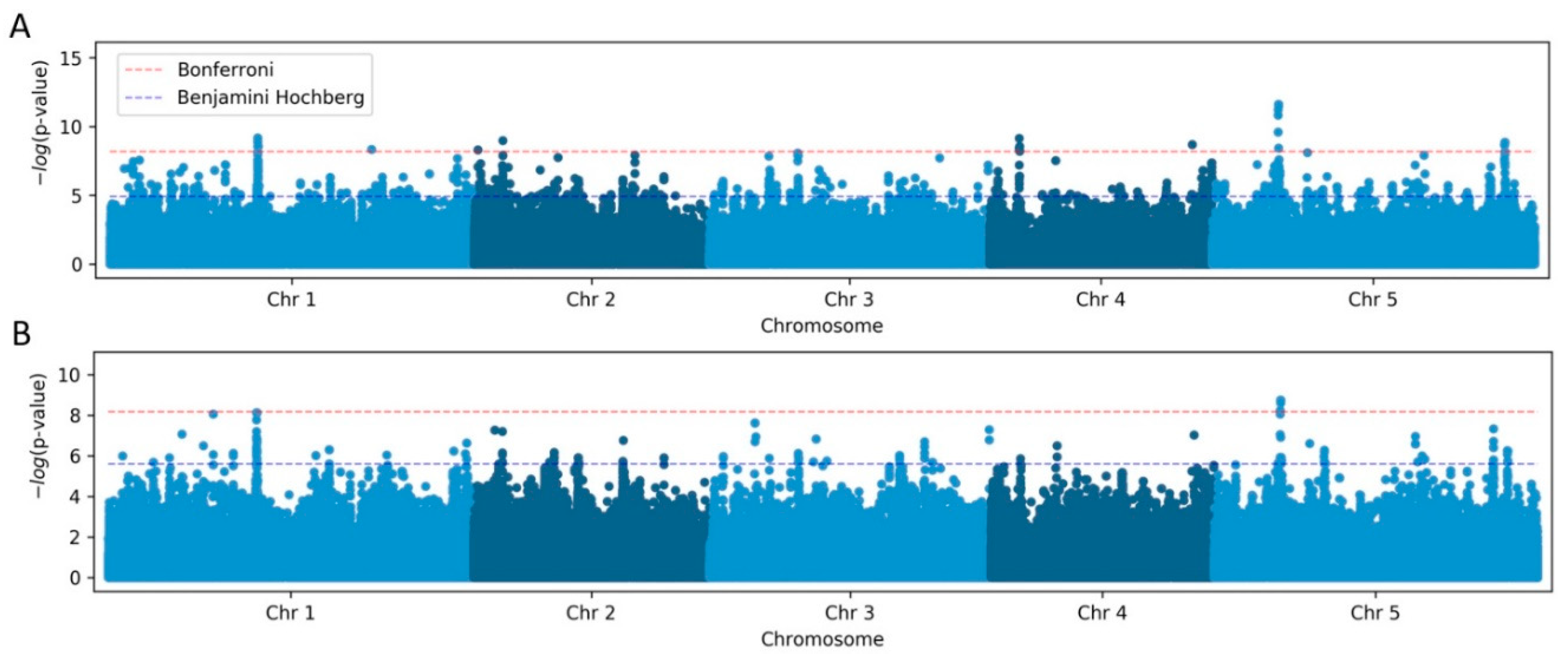
Figure 5.
Distribution of mean TRF values in Arabidopsis T-DNA mutant plants. Telomere length values as calculated by TeloTool (A) and WALTER (B) programs are grouped into 50 bp intervals, and the number of individual DNA samples falling into each interval are plotted against telomere length, sorted from shortest to longest. Red lines indicate median telomere length for each dataset. Dotted lines indicate the medium telomere length range (2,100-3,500 bp) characteristic of most Arabidopsis T-DNA mutants. (C) Length values for TeloTool and WALTER measurements are plotted for each DNA sample. The trend line is shown in red.
Figure 5.
Distribution of mean TRF values in Arabidopsis T-DNA mutant plants. Telomere length values as calculated by TeloTool (A) and WALTER (B) programs are grouped into 50 bp intervals, and the number of individual DNA samples falling into each interval are plotted against telomere length, sorted from shortest to longest. Red lines indicate median telomere length for each dataset. Dotted lines indicate the medium telomere length range (2,100-3,500 bp) characteristic of most Arabidopsis T-DNA mutants. (C) Length values for TeloTool and WALTER measurements are plotted for each DNA sample. The trend line is shown in red.
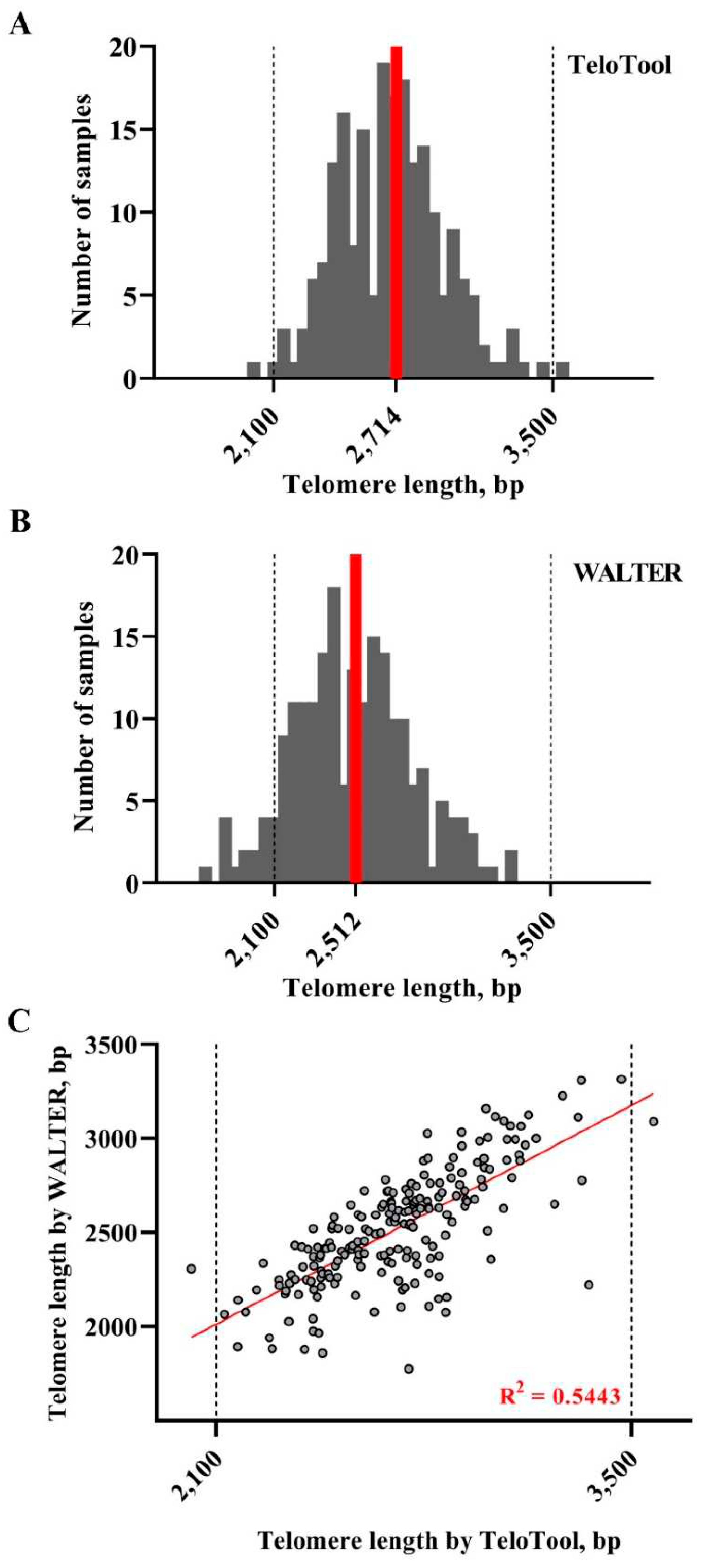

Table 1.
Statistical differences between TeloTool and WALTER datasets for Arabidopsis accessions.
| Telomere length range | ≤2,100 bp | 2,101–3,500 bp | 3,501–4,500 bp | ≥ 4,501 bp |
|---|---|---|---|---|
| Number of DNA samples | 16 | 250 | 195 | 123 |
| Number of accessions | 15 | 232 | 179 | 111 |
| Minimum difference, % | -44.00 | -39.00 | -16.00 | 2.00 |
| 25% Percentile | -18.25 | 1.000 | 5.000 | 12.00 |
| Median difference, % | -8.00 | 7.000 | 10.00 | 17.00 |
| 75% Percentile | 0.50 | 12.00 | 17.00 | 28.00 |
| Maximum difference, % | 6.00 | 45.00 | 85.00 | 91.00 |
| Mean difference, % | -10.94 | 7.40 | 12.01 | 21.68 |
*Telomere length ranges are defined based on the TeloTool dataset. Percent differences between TeloTool and WALTER values calculated as (TeloTool-WALTER)/WALTER *100%.
Table 2.
GWAS-significant SNPs for telomere length in the TeloTool and Walter datasets.
| SNP | Chromosome | Position | P-value | Major Allele | Minor Allele | maf | Effect |
|---|---|---|---|---|---|---|---|
| TeloTool (LM) | |||||||
| 5:5538242* | 5 | 5538242 | 11.61 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 1:12379845* | 1 | 12379845 | 10.46 | G | A | 0.028 | Intergenic |
| 4:2463357 | 4 | 2463357 | 9.13 | C | T | 0.046 | INTRON At4g04870 |
| 2:2422473 | 2 | 2422473 | 8.98 | G | A | 0.059 | Intergenic |
| 5:24491588 | 5 | 24491588 | 8.86 | G | A | 0.055 | Intergenic |
| 4:18303636 | 4 | 18303636 | 8.78 | C | T | 0.028 | Intergenic |
| 4:16935117 | 4 | 16935117 | 8.7 | T | A | 0.04 | MISSENSE aTg/aAg M231K At4g35733 |
| 1:21870431 | 1 | 21870431 | 8.32 | T | G | 0.038 | Intergenic |
| 2:356311 | 2 | 356311 | 8.30 | T | A | 0.036 | Intergenic |
| 3:7407370 | 3 | 7407370 | 8.06 | G | A | 0.104 | MISSENSE Gga/Aga, G349R At3g21120 |
| WALTER (LM) | |||||||
| 5:5538242* | 5 | 5538242 | 8.73 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 1:12379845* | 1 | 12379845 | 10.01 | G | A | 0.028 | Intergenic |
| 1:8743486 | 1 | 8743486 | 8.06 | A | G | 0.04 | INTRON At1g24706 |
| TeloTool (AMM) | |||||||
| 5:5538242* | 5 | 5538242 | 11.61 | T | C | 0.19 | 3’UTR_3 At5g16850 |
| 3:23295214 | 3 | 23295214 | 8.66 | T | G | 0.03 | Promoter of At3g63030 |
| 1:21870431 | 1 | 21870431 | 8.08 | T | G | 0.038 | Intergenic |
| WALTER (AMM) | |||||||
| 5:5538242* | 5 | 5538242 | 9.0 | T | C | 0.19 | 3’UTR_3 At5g16850 |
*Asterisks indicate significant SNPs identified with both datasets.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



