
Submitted:
08 November 2023
Posted:
08 November 2023
You are already at the latest version
Abstract
Dengue virus serotype 2 (DENV-2) is responsible for dengue epidemics on a global scale and is recognized for being associated with severe cases of the disease. This study conducted the phylogenetic investigation of DENV-2 in the Northern region of Brazil. DENV-2 isolates from 2017 to 2021, originating from Northern Brazilian states, were selected. Only one lineage (I) of the Asian-American genotype and one lineage (5) of the cosmopolitan genotype were observed. All sequences of the Asian-American genotype used in this study were segregated into a paraphyletic subgroup aligned with sequences from an American clade. These results indicate a probable geographic link starting from Puerto Rico. Additionally, sequences from Acre were identified and grouped into a monophyletic clade of the cosmopolitan genotype. These results indicate the cocirculation of two DENV-2 distinct lineages: Lineage 1, Asian-American genotype (probable origin from Puerto Rico); Lineage 5, cosmopolitan genotype (probable origin from Peru). Our results provide important data regarding the study of DENV genotypes and lineages distribution, as well as opening up possibilities for probable introduction and dissemination routes.
Keywords:
arboviruses
; dengue virus serotype 2
; cosmopolitan genotype
; asian-american genotype
1. Introduction
Dengue is an arboviral disease caused by dengue virus (DENV – Orthoflavivirus denguei) [1]. DENV belongs to the Flaviviridae family, Orthoflavivirus genus, in which member species have a single-stranded positive-sense RNA genome and a viral envelope. Virus transmission occurs primarily through Aedes mosquitoes bites [2]. DENV exhibits four antigenically distinct serotypes (DENV-1, DENV-2, DENV-3, and DENV-4), and each serotype is further divided into genotypes [3]. DENV infection can lead to a wide range of symptoms, from asymptomatic to severe. Infection caused by one serotype can induce specific long-lasting immunity to that particular serotype, providing short-lived non-protective cross-immunity (heterotypic) against the other serotypes [4].
DENV is spreading at an alarming rate worldwide, making it the fastest-expanding arbovirus. Over the past few decades, its incidence has increased in tropical and subtropical countries, turning it into an urgent global public health problem [5]. Besides being the most prevalent arbovirus, DENV is also the most widely disseminated and is endemic in practically all inhabited regions of the world, with an estimated 100 million symptomatic clinical cases of infection per year, 500,000 cases of severe dengue, and approximately 22,000 deaths. According to a World Health Organization (WHO) report [6], between Epidemiological Week (EW) 1 and EW 52 of 2022, a total of 2,809,818 dengue cases were reported, resulting in an accumulated incidence rate of 282.96 cases per 100,000 population. As of EW 10 of 2023, dengue remains the most predominant arbovirus, accounting for 75% of the cases (342,243) [6,7,8].
The cocirculation of multiple serotypes can increase the risk of severe infections. Serotypes DENV-1 and DENV-2 have been widely circulating in South America, and specifically DENV-2, has been contributing significantly to dengue-related mortality in countries like Brazil [7,8]. The genetic diversity, evolution, and transmission dynamics of the virus in regions conducive to its spread are widely discussed. In this context, the introduction of different serotypes and their ability to persist in the environment over time are factors that may be related to the unpredictability of clinical outcomes and viral dissemination capacity [9].
Among the four serotypes, DENV-2 is the most common cause of dengue epidemics on a global scale and, predominantly in American countries, is recognized for its association with cases of severe disease [10,11,12]. DENV-2 genotypes are constantly evolving due to their high mutation rate and rapid migration, which may lead to new lineages. The simultaneous circulation of different strains of DENV-2 in the country may be associated with a greater number of epidemic outbreaks and severe manifestations of the disease when compared to the other three dengue serotypes. The emergence and cocirculation of new DENV-2 lineages, replacing earlier ones, was previously reported [13,14,15,16,17,18].
There are five genotypes represented by strains reported in the Caribbean and South Pacific (genotype I or American); Taiwan, Philippines, New Guinea prototype virus, and the 1964 Thailand strain (genotype II or Cosmopolitan); strains from Vietnam, Jamaica, and Thailand (genotype III or Asian-American); Indonesia, Seychelles, Burkina Faso, and Sri Lanka (genotype IV or Asian I and II); and rural areas of Africa (genotype V or Sylvatic) [19,21].
Ideal conditions for vector proliferation characterize the Northern Region of Brazil. Additionally, due to its extensive coastal extension and bordering several countries such as Bolivia, Peru, Colombia, Venezuela, Guyana, Suriname, and French Guiana, it can significantly facilitate the introduction of diseases and the frequent occurrence of outbreaks through vector proliferation [20,21,22,23,24,25]. Monitoring the continuous spread of arboviruses worldwide, understanding the likely causes of reemergence in regions where the virus previously circulated, and conducting epidemiological surveillance in endemic regions are essential for prevention and control measures to reduce vector proliferation and minimize the risk of infection [21,26].
In this scenario, complete viral genome analysis allows the identification of DENV serotypes and genotypes, which is important for understanding the circulation of the virus, especially in endemic areas. This approach can help to determine the origin of isolated viruses, their introduction and dissemination routes within a region, as well as the identification of geographical provenance.
In this study, we conducted the phylogenetic investigation of DENV-2 in the Northern Region of Brazil, which contributes to the understanding of circulating genotypes and lineages, as well as elucidates the neighboring countries' influence on strain diversity and subsequent lineage replacement. The obtained information provides essential insights into DENV-2 genotypes and lineages distribution, while inferring contributions to probable introduction routes.
2. Materials and Methods
2.1. Study
This is an observational, descriptive, and retrospective study. The identification and processing of samples, laboratory techniques, and data registration and analysis procedures were conducted at the Department of Arbovirology and Hemorrhagic Fevers of the Evandro Chagas Institute. Experimental analysis procedures were carried out by norms and criteria required by the Internal Biosafety Committee. This study did not involve the use of animals, humans, or the environment as direct research subjects, thus excluding the need for submission to the research ethics committee. Only isolated samples of DENV, deposited and belonging to the institutional biorepository were used. Thus, DENV-2 samples, from Northern Brazilian States and collected between 2017 to 2021, were eligible.
2.2. Viral Sample
The used samples correspond to strains of DENV-2 isolated from previously collected biological samples. The samples are derived from the epidemiological surveillance of arboviruses at Evandro Chagas Institute. All biological samples were confirmed by molecular biology tests and subsequently isolated in cell cultures, stored in liquid nitrogen until use. Prior to analysis, the samples were registered and presented based on the following parameters: (I) Place of origin/Federal Unit (FU); Identification (ID); (II) Collection date (CD); (III) Serotype; (IV) Sample type (Table S1). Only DENV-2 samples from the Northern Region of Brazil (Acre, Tocantins, and Rondônia States).
2.3. RNA Extraction and Purification
For sample extraction, 140 µL of cell culture supernatant was used with the QIAamp® Viral RNA Mini Kit (Qiagen, Hilden, Germany), following the manufacturer's recommendations. Total RNA was quantified using the Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) with the Qubit® RNA HS Assay kit and the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) with the Agilent RNA 6000 Pico kit, following the respective manufacturer's protocols. Subsequently, the samples were stored in a freezer at -70 °C until further use.
2.4. RT-qPCR and Viral Isolation
All samples were pre-tested for viral isolation confirmation using molecular biology techniques for DENV serotype 2. The assay was performed using the SuperScript III Platinum One-Step Quantitative RT-PCR System Rox Kit (Thermo Fisher Scientific), which included a set of oligonucleotide primers and dual-labeled fluorescent probes (Taqman) for in vitro qualitative detection of DENV-1-4. The 25 µL reaction consisted of 12.5 µL of a 2X SuperScript III Platinum RT-PCR master mix, 2.2 µL of nuclease-free water, 1.0 µL of forward and reverse primers (D1, F - 5', CAAAAGGAAGTCGYGCAATA, 3'; R - 5', CTGAGTGAATTCTCTCTGCTRAAC, 3', final concentration 1 µM), 0.5 µL (D2, F - 5', CAGGCTATGGCACYGTCACGAT, 3'; R - 5', CCATYTGCAGCARCACCATCTC, 3', final concentration 500 nM), 1.0 µL (D3, F - 5', GGACTRGACACACGCACCCA, 3'; R - 5', CATGTCTCTACCTTCTCGACTTGYCT, 3', final concentration 1 µM), 0.5 µL (D4, F - 5', TTGTCCTAATGATGCTRGTCG, 3'; R - 5', TCCACCYGAGACTCCTTCCA, 3', final concentration 500 nM), 0.45 µL of each Taqman probe (final concentration 180 nM), 0.25 µL of the D1-D4 probe, and 5 µL of extracted RNA.
In a 7500 Fast Real-Time PCR system (Thermo Fisher Scientific), the RT-qPCR assays were performed under the following cycling conditions: an initial RT step at 50 °C for 30 minutes, a denaturation step at 95 °C for 2 minutes, 45 cycles of 15 seconds at 95 °C, and a final extension step at 60 °C for 1 minute. Each sample was analyzed in duplicate and considered positive when the average Cycle Threshold (CT) value was below 37. DENV-2 positive samples with CT values below 20 were selected for viral isolation and genomic analysis of the supernatants. The assay was validated using positive controls (DENV-1-4) and negative controls (nuclease-free water) [27].
DENV-2 positive samples were isolated and stored until thawing at the Cell Culture Laboratory of the Department of Arbovirology and Hemorrhagic Fevers of the Evandro Chagas Institute. The processing and sample analysis were performed at the Molecular Biology Laboratory, Department of Arbovirology and Hemorrhagic Fevers, Evandro Chagas Institute. The Standard Operating Protocol for RT-qPCR from the same laboratory was followed [27]. DENV-2 positive samples were transferred for viral isolation and stored in liquid nitrogen until use. The results were evaluated, and the positive samples were further subjected to complementary DNA (cDNA) synthesis and genetic sequencing.
For the isolation, a cell line derived from Aedes albopictus, clone C6/36 (ATCC: CRL1 660), was used. The cells were seeded in 10 mL culture tubes containing 1.5 mL of modified Leibowitz medium with glutamine (L-15), supplemented with 2.95% triptose phosphate, non-essential amino acids, antibiotics (penicillin and streptomycin), and 2% fetal bovine serum. They were inoculated at a 1:10 ratio of serum to modified Leibowitz medium with glutamine (L-15) and observed daily for a period of 7 days or until cytopathic effect (CPE) was observed. Upon completion, the isolates were frozen until further processing for cDNA synthesis and genomic library preparation [27].
2.5. Synthesis of cDNA Double Strand and Genomic Library
The cDNA preparation from RNA for genomic sequencing started with the synthesis of the first and second cDNA strands using the SuperScriptTM VILOTM MasterMix kit (Thermo Fisher Scientific) and NEBNext® Second Strand Synthesis Module (New England BioLabs, Ipswich, MA, USA), respectively. The cDNA purification reaction was performed using the PureLink® PCR Purification Kit (Thermo Fisher Scientific). All steps followed the manufacturer's recommendations for the respective kits. The genomic library was prepared following the instructions of the Nextera XT DNA kit (Illumina, San Diego, CA, USA). Quantification and fragmentation level assessment were performed using the DNA HS Kit Assay on the Qubit 4.0 instrument and the Agilent RNA 6000 Pico kit in the Bioanalyzer 2100 instrument, respectively. Upon confirming that the library had the desired quantity and size of fragments, sequencing was carried out on the NextSeq 500 platform (Illumina) using the NextSeq 500/550 High Output Kit v2.5 (300 cycles) and paired-end methodology, as recommended by the manufacturer.
2.6. Bioinformatics and Phylogenetic Analysis
The data generated by NextSeq (Illumina) in .bcl format was converted to .fastq format using the bcl2fastq2 program [28], resulting in two files, R1 and R2. These files served as input for the initial data processing program. The generated data underwent an initial step of preprocessing the raw sequences, including adapter removal, filtering out low-quality reads (below Q20), and removing ambiguous bases using the Fastp v0.23.1 program [29]. The reads obtained from the previous step were assembled using the reference mapping methodology with the Bowtie2 v2.5.1 program [30]. These reads were mapped to the reference genome of DENV-2 (NC_001474) (available in the National Center for Biotechnology Information, NCBI, RefSeq database), using the Fastp v0.23.1 program to obtain consensus sequences. The Geneious v.9.1.8 software [31], the Samtools v1.16.1 [32], and Bamtools v2.5.2 [33] software, were used for analysis and extraction of relevant information through manipulation and analysis of SAM and BAM files, including extraction of read coverage information.
The obtained sequences, including complete and partial ones, were aligned with sequences from different genotypes of DENV-2 available in GenBank, using Mafft v.7 software [34]. Before constructing the phylogenetic tree, phylogenetic signal verification was performed to ensure the reliability of the analyzed data. TREE-PUZZLE v. 5.3 was used for this purpose, with the input file being the alignment of the sequence set in .phylip format (generated by Geneious v.9.1.8). The program employs the Maximum Likelihood methodology (ML) and produces a triangular diagram image for analysis [35].
After positive phylogenetic signal confirmation, the phylogenetic tree was built using the ML [36] phylogenetic inference with a fixed bootstrap value of 1000 replicates using IQ-TREE v.2 [37,38]. The best nucleotide substitution model was selected according to the ModelFinder incorporated into the IQ-TREE program v.2 [37,39]. The phylogenetic tree was rooted on midpoint [40]. Tree visualization and editing were performed using FigTree v.1.4.4 [41] and Inkscape v.1.1 [28] softwares.
3. Results
A total of 32 samples from DENV-2 isolates were analyzed, including 12 from Acre, 19 from Roraima, and one from Tocantins. The samples were tested, confirmed as DENV-2, and isolated in cell cultures. The obtained sequences were aligned through reference mapping with the DENV-2 reference genome (NC_001474). A set of 124 complete genome sequences available in GenBank, representing circulating genotypes worldwide, was aligned with the study sequences to perform the genotyping. All sequences included in the phylogenetic inference are listed in Supplementary Materials (Table S2). The highly resolved tree coordinates (consistent) concentrate at the tips of the triangle, equivalent to 97.4%; the partially resolved topology is represented in the side rectangles, accounting for 1.2%; and the unresolved tree topology is centered, amounting to 1.4% (Figure S1). Since the proportion of unresolved tree topology is below 30%, a positive phylogenetic signal is identified, indicating the high quality of the analyzed data.
Based on the obtained alignments, phylogenetic analysis was performed. The phylogenetic tree was constructed using maximum likelihood, with the GTR+F+I+G4 nucleotide substitution model identified as the best fit. For comparison purposes, the 124 sequences were included. The study sequences were grouped into a monophyletic clade belonging to the cosmopolitan genotype and a paraphyletic clade belonging to the Asian-American genotype (Figure 1). The names of the isolates used for comparison include the GenBank accession number, country of origin, and year of isolation.
The phylogenetic analysis of DENV-2 demonstrated the circulation of the Asian-American and cosmopolitan genotypes of the Northern Region (Acre, Rondônia, and Tocantins). We report an event of multiple lineages circulating in the Northern Region of Brazil. The choice of the comparison dataset, although restrictive in terms of global diversity, enabled a more in-depth and detailed investigation of the phylogenetic relationships within the local context, providing valuable insights into the evolution and spread of lineages in the region under study.
Although the dispersal route from the point of origin is unknown, it is possible to observe that the basal strain from Puerto Rico (EU687217) behaves as more evolutionarily ancient compared to the strains from Brazil (Acre, Roraima, and Tocantins). These sequences grouped into an American clade because it was the circulating genotype in Brazil until the year 2021 when the introduction of the cosmopolitan genotype (Acre) is believed to have occurred, possibly originating from strains from Madre de Dios (Peru). Previously, we hypothesized that the introduction of the cosmopolitan genotype (Acre) [55] occurred, possibly originating from strains from Madre de Dios, Peru (OM791800, OM791801).
Based on our analysis, the OR609399-OR609396 sequences, belonging to Lineage 1 of the Asian-American genotype, are in the same clade as the Puerto Rican sequence (EU687217). Although the dispersal route from the point of origin is unknown, it is evident that the basal Puerto Rican strain appears to be more evolutionarily ancient compared to the Brazilian strains (Acre, Roraima, and Tocantins). Figure 2 provides insights into a possible secondary center of dissemination of Lineage 1 of the Asian-American genotype of DENV-2, with the outbound route (from Puerto Rico) and the virus arrival through an unmapped route (Acre, Brazil / Roraima, Brazil / Tocantins, Brazil).
We can infer that there was a large-scale introduction of the cosmopolitan genotype of DENV-2 in South America. Our data allow us to hypothesize that our strains, as well as those from Peru (OM791800; OM791801), may have derived from those reported in Dhaka, Bangladesh. However, further studies are needed to determine where its introduction occurred and how long it has been circulating. Considering the sample dates, we can infer that this genotype has been circulating in Brazil for at least two years, highlighting the importance of retrospective studies to investigate the dispersal pattern of cosmopolitan genotype lineages of DENV-2.
The co-circulation of genotypes may have important implications for viral epidemic capacity. Considering the lack of retrospective studies evaluating the circulation period of different DENV-2 lineages, it is likely that multiple lineages are coexisting in the same region. To understand the dispersion route of both detected lineages from the point of origin, more information on sample collection and sequencing between Puerto Rico - Brazil for the Asian-American genotype (lineage 1) and Bangladesh - Brazil for the cosmopolitan genotype would be necessary. Consequently, a larger volume of data is needed to describe the evolutionary route.
4. Discussion
The emergence of arboviruses in previously unaffected areas poses significant challenges to public health [42]. Genomic surveillance plays a crucial role in monitoring the introduction of emerging infectious diseases and analyzing circulating viruses [6]. Therefore, efficient sequencing protocols and subsequent data sharing are essential, allowing sequence comparison. This approach enables the acquisition of information about viral introduction and dispersion routes, evolutionary implications, transmission pathways, and the design of effective epidemic containment measures. In this context, the advancement of sequencing technologies has had an unprecedented impact on virus studies. Research utilizing this approach contributes to the understanding of viral diversity, geographical distribution, and evolutionary inferences [43].
Dengue is endemic in all regions of Brazil, and frequent outbreaks represent a significant public health problem. Several dengue serotypes and genotype changes have been observed in recent decades [15,16,17,19,20]. More recently, the reemergence of serotype 3 has been reported in the Northern region of Brazil, associated with the emergence of a new lineage of the genotype III (Indian subcontinent) in the Americas [18]. Since dengue is also endemic in neighboring countries to Brazil, there is always a risk of virus importation, transmission of new serotypes, and an increased likelihood of genetic divergence. This highlights the need for early detection, surveillance, and continuous monitoring of DENV dissemination in Brazil and neighboring countries.
DENV-2 genotypes exhibit a wide genetic diversity, indicating continuous divergence and varied geographic distribution [44]. The Asian-American genotype was introduced to the Americas from the Asian continent in the early 1980s and spread widely throughout the continent [49]. From the 1990s onward, the virus was reintroduced to Brazil through strains originating from the Caribbean [49,50,51]. Reports describe the Caribbean as the main source of all DENV-2 lineages that were subsequently disseminated to continental regions, while Brazil, Venezuela, and Nicaragua were identified as major secondary centers of DENV-2 dissemination to other countries in the continent [49]. Therefore, the phylogeographic pattern of DENV-2 in the Americas can be attributed to a model of short-distance transmission [49]. In this model, the virus originated from a central point in the Caribbean and spread to nearby continental regions, which act as secondary centers of dissemination, transmitting the virus to neighboring countries in the continent [52].
Only one lineage of the Asian-American genotype was observed. All sequences of the Asian-American genotype used in this study segregate into a paraphyletic subgroup aligned with sequences from an American clade (Puerto Rico, Cuba, Nicaragua, Belize, Venezuela, and Brazil), well supported by high bootstrap values, with the Puerto Rico sequence supporting the diversity of DENV-2, Asian-American genotype. The phylogenetic inference also revealed that the American clade was positioned between the sequences of lineage 1 of the genotype, implying its likely origin from Puerto Rico. These results suggest the predominant circulation of lineage 1, as already documented in the recent history of DENV-2. We highlighted in our study the circulation of the Asian-American genotype in the states of Tocantins, Acre, and Rondônia from 07/2019 to 02/2020 (Figure 2) [53,54]. These cases may infer a geographical connection between the strain reported in Puerto Rico and parallel dispersion to the states of the Northern Region of Brazil, providing insights into the most likely dispersion route of this genotype in Brazil, originating from Puerto Rico and behaving as a secondary dissemination center.
The most significant finding of this study was the identification of sequences stemming from an outbreak that occurred in the state of Acre, Brazil, which phylogenetically clustered within the cosmopolitan genotype clade. This is noteworthy considering that previous studies have only documented the circulation of the Asian-American genotype in Brazil. The strain reported in Goiás represents the second official record of the circulation of this genotype in the Americas, following an outbreak that occurred in Madre de Dios, Peru, in 2019. Madre de Dios is an extensive territorial area that borders the Amazon Region [45,46,47]. Recent reports have revealed highlights about the widespread spread of the cosmopolitan genotype of DENV-2 in Brazilian territory. They indicate that, after several entries into the country, this genotype is already established in all geographic regions of Brazil [55,56].
The first report of a cosmopolitan strain of DENV-2 in the Americas occurred in the late 1990s in the Yucatán Peninsula, Mexico, although only one viral isolate was studied [45]. This genotype is in circulation in other regions of the world, replacing the genotypes that were predominant in several Asian regions [45]. Based on recent reports, it is possible to infer a probable large-scale introduction of the cosmopolitan genotype of DENV-2 from Asian regions to Madre de Dios, Peru, derived from strains reported in Dhaka, Bangladesh. The cosmopolitan genotype was recently identified in Madre de Dios and is globally circulating, showing wide dispersion of its lineages [45,46].
Based on the understanding of the evolutionary dynamics and global spread of DENV-2 in the Americas, we can infer that the cosmopolitan genotype shows more significant heterogeneity compared to other DENV-2 genotypes. Our findings reinforce what we proposed in our partial report regarding the introduction of a new genotype, raise awareness about its dissemination, and provide insights into the co-circulation of multiple lineages in the Americas [56]. Although a phylogenetic relationship was observed between the only case of the cosmopolitan genotype in the Midwest region of Brazil and the cases described in Madre de Dios, Peru [45,46] no geographical linkage has been established between these regions that do not share borders [46,47,48].
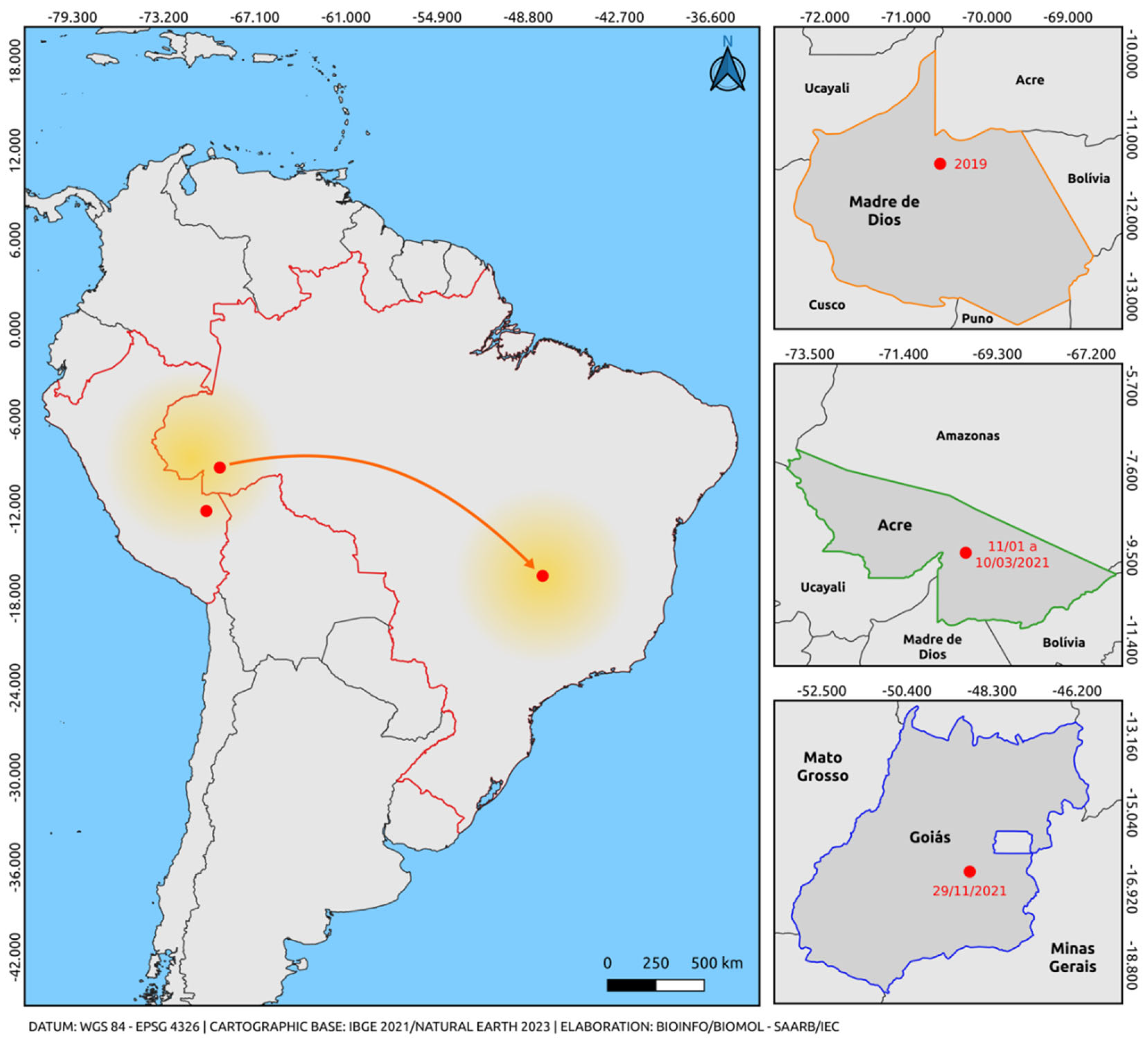
Ten sequences obtained in this study clustered within a well-supported South American clade, supported by high bootstrap values, including two Peruvian sequences and the only sequence previously described in Brazil. Our previous report revealed the first evidence of the cosmopolitan genotype in the Northern region of Brazil, during a DENV-2 outbreak in the state of Acre, Northern Region, in early 2021, months before the single detection case in Goiás [46,56]. Samples in this study were collected between January and March 2021, showing the circulation of this genotype prior to the reported case in Goiás. Phylogenetic inference also revealed that the South American clade was positioned among sequences belonging to lineage 5 of the genotype. These cases establish a closer geographic connection between findings in Peru and the Central-West region of Brazil, providing insights into the most likely route of introduction of this genotype into Brazil from the border with Peru, in the Madre de Dios Region (Figure 3).
Although the persistence and local evolution of some strains are possible, the crucial element for the dynamics of DENV-2 epidemics in the state seems to have been the introduction of Peruvian strains (OM791800; OM791801). The cosmopolitan genotype of DENV-2 is currently in circulation throughout Brazil, expanding upon the findings of previous research [46,55,56]. A recent study identified multiple introductions of this genotype in Brazil, originating from Peru, resulting in several clusters that have spread throughout the country with different levels of dissemination. These results emphasize the need to monitor the introduction of new genotypes/lineages in a specific region. Rapid changes in these genotypes or lineages can occur and are attributed to fitness advantages, resulting in a greater ability to reach higher levels of viremia in humans. This leads to an increased rate of transmission between humans and mosquitoes, which is also associated with the circulation of different genotypes in the same region.
The population sampled in our dataset represents a small fraction of DENV-2 cases since the first report of the cosmopolitan genotype in Brazil. This makes it impossible to define the introduction route and the time of virus circulation. To investigate the time of virus circulation in the region, retrospective studies with samples from other states are needed. However, our approach has proven robust for studying the spread of DENV-2 at a regional level, as long as the results are interpreted with caution within a solid biological framework. This study emphasizes the relevance of molecular research in the context of the origin and evolution of the dengue virus. The results point to the promising use of these studies as a tool capable of predicting epidemics in the context of genomic surveillance.
5. Conclusions
The DENV-2 serotype isolated from the period of 2017 to 2021 in the Northern region of Brazil, specifically the states of Acre, Roraima, and Tocantins, belong to the Asian-American and Cosmopolitan genotypes. During this period, two distinct lineages of DENV-2 circulated: lineage 1 from the Asian-American genotype (with a probable origin from Puerto Rico) and lineage 5 from the cosmopolitan genotype (with a probable origin from Peru).
However, starting in 2021, the cosmopolitan genotype began to circulate in the state of Acre, with a reported strain in the state of Goiás in the same year. The discovery of the circulation of a new genotype in Brazilian territory strengthens previous findings in South America regarding the circulation of the cosmopolitan genotype in the Americas. The occurrence of two distinct genotypes in the same region and in bordering areas suggests the co-circulation of genotypic lineages, primarily due to the detection period analyzed.
The dataset used in this study emphasizes the need for retrospective studies investigating the origin and introduction route of DENV-2, as well as possible nucleotide and amino acid alterations in the genome of isolates associated with mutation or particularities in the clinical manifestation of the disease or type of infection.
The detection of the Asian-American and cosmopolitan genotypes alerts to the emergence of DENV-2 in neighboring regions with intense population exchange, where the inability to adhere to public policies for arbovirus control hinders the establishment of truly effective genomic surveillance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.T.A. and A.C.R.C.; methodology, L.H.A.H. and S.P.d.S.; validation, B.T.D.N. and S.P.d.S.; investigation, M.T.A, C.C.S.O and L.H.A.H.; resources, A.C.M.S.; data curation, F.S.d.S., T.Y.B.P and E.V.P.d.S.; writing—original draft preparation, M.T.A, L.H.A.H., A.L.N.Q.; writing—review and editing, A.C.R.C. and F.G.N.; visualization, A.L.M.W. and B.T.D.N.; supervision, A.C.R.C.; project administration, A.C.R.C.; funding acquisition, A.C.R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Coordination for the Improvement of Higher Education Personnel (CAPES) and the Evandro Chagas Institute, Health and Environment Surveillance Secre- tariat, Ministry of Health, Brazil. Grant number “88887.612806/2021-00”. The manuscript payment was funded by PROPESP/UFPA(PAPQ).
Institutional Review Board Statement
Ethical review and approval were waived for this study due to the use of extracted RNAs.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequences generated in this study were deposited in GenBank under the following accession numbers: OR609396 - OQ511274.
Acknowledgments
We thank the State Health Departments and Central Laboratories in the states of Acre, Tocantins and Rondônia,
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization (WHO). Fact sheet, 2013; N. 117.
- Gubler, D.J.; Kuno, G.; Markoff, L. Flaviviruses. In: Knipe, D.M.; Howley, P.M.; Griffin, D.E.; Lamb, R.A, eds. Fields Virology. 5th ed. Philadelphia, PA, USA: Lippincott-Raven Publishers, 2007; pp. 1497-1526.
- Sabin, A. Research on Dengue during World War II. The American Journal of Tropical Medicine and Hygiene, 1952; Volume 1, Number 1, pp. 30-50.
- Guzmán, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martínez, E.; Nathan, M.B.; Pelegrino, J.L.; Simmons, C.; Yoksan, S.; Peeling, R.W. Dengue: a continuing global threat. Nature Reviews Microbiology, 2010; Volume 8, Supplementer 2, S7-S16. [CrossRef]
- World Health Organization- WHO. Dengue and Dengue haemorhagic fever. WHO Report on Global Surveillance of Epidemic-prone Infectious Disease. Geneva, 2000.
- World Health Organization- WHO. Global genomic surveillance strategy for pathogens with pandemic and epidemic potential, 2022-2032. Geneva, 2022.
- Pan-American Health Organization - PAHO. Strategy for the prevention and control of arboviroses: Final Report, 58 Board of Directors, 72nd Session of the WHO Regional Committee for the Americas. 2020.
- Pan-American Health Organization - PAHO. Dengue Epidemiological Update in the Region of the Americas. World Health Organization-2022. 2023.
- Yenamandra, S.P.; Koo, C.; Chiang, S.; Lim, H.S.J.; Yeo, Z.Y.; Ng, L.C.; Hapuarachchi, H.C. Evolution, heterogeneity and global spread of the cosmopolitan genotype of Dengue virus type 2. Sci. Rep. 2021, Volume 11, Number 1, pp 13496.
- Cologna, R.; Armstrong, P.M.; Rico-Hesse, R. Selection for virulent dengue viruses occurs in humans and mosquitoes. Journal of Virology. 2005, Volume 79, Number 2, pp 853-859. [CrossRef]
- Rico-Hesse R. Dengue Virus Evolution and Virulence Models. Clin. Infect. Dis. 2007, Volume 44, Number 8, pp 1462-1466. [CrossRef]
- Waman, V.P.; Kasibhatla, S.M.; Kale, M.; Kulkarni-Kale, U. Population genomics of dengue virus serotype 4: insights into genetic structure and evolution. Archives of Virology. 2016, Volume 161, Number 4, pp 1091-1104. [CrossRef]
- Holmes, E.C.; Twiddy, S.S. A origem, emergência e genética evolutiva do vírus da dengue. Infect. Genet. Evol. 2003, Volume 3, Number 1, pp 19-28.
- Foster, J.E.; Bennet, S.N.; Carrington, C.V.; Vaughan, H.; McMillan, W.O. Phylogeography and molecular evolution of dengue 2 in the Caribbean basin, 1981-2000. Virology. 2004, Volume 324, Number 1, pp 48-59. [CrossRef]
- Hang, V.T.; Holmes, E.C.; Veasna, D.; Quy, N.T.; Hien, T.T.; Quail, M.; Churcher, C.; Parkhill, J.; Cardosa, J.F.; Wills, B. Emergence of the Asian 1 genotype of dengue virus serotype 2 in Vietnam: in vivo fitness advantage and lineage replacement in South-East Asia. PLoS Neglected Tropical Diseases. 2010, Volume 4, Number 7, e2326.
- Carneiro, A.R.; Cruz, A.C.R.; Vallinoto, M.; Melo, D.V.; Ramos, R.T.J.; Medeiros, D.B.A.; da Silva, E.V.P.; Vasconcelos, P.F.C. Caracterização molecular do vírus da Dengue 1 revela substituição de linhagem durante a circulação em território brasileiro. Memórias do Instituto Oswaldo Cruz. 2012, Volume 107, Number 6, pp 805-812.
- Drumond, B.P.; Mondini, A.; Schmidt, D.J.; De Morais-Bronzoni, R.V.; Bosch, I.; Nogueira, M.L. Circulation of Different Lineages of Dengue Virus 2, Genotype American/Asian in Brazil: Dynamics and Molecular and Phylogenetic Characterization. PLoS ONE. 2013, Volume 8, Number 3, e58501. [CrossRef]
- Naveca, F.G.; Santiago, G.A.; Maito, R.M.; Meneses, C.A.R.; Do Nascimento, V.A.; Silva, D.; Mejía, M.; Gonçalves, L.; De Figueiredo, R.M.P.; Cruz, A.C.R.; Nunes, B.T.D.; Oresibella, M.M.; Marques, N.F.Q.; Riediger, I.N.; De Mendonça, M.C.L.; Bruycker-Nogueira, F.; Sequeira, P.C.; De Filippis, A.M.B.; Resende, P.; Campos, T.; Wallau, G.; Graf, T.; Delatorre, E.; Kopp, E.; Morrison, A.; Munoz-Jordán, J.L.; Bello, G. Reemergence of Dengue Virus Serotype 3, Brazil, 2023. Emerg. Infect. Dis. 2023, Volume 9, Number 7, pp 1482-1484, 2023. [CrossRef]
- Rico-Hesse, R. Molecular Evolution and Distribution of Dengue Viruses Type 1 and 2 in Nature. Virology. 1990, Volume 174, Number 2, pp 479-493. [CrossRef]
- Rico-Hesse, R.; Harrison, L.M.; Salas, R.A.; Tovar, D.; Nisalak, A.; Ramos, C.C.; Boshell, J.; de Mesa, M.T.; Nogueira, R.M.; Rosa, A.T. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology. 1997, Volume 230, pp 244-251.
- Rico-Hesse, R. Microevolution and Virulence of Dengue Viruses. Advances in Virus Research. 2003, Volume 59, pp 315-341.
- Pinheiro, F.P. Dengue in the Americas, 1980-1987. Epidemiol. Bull. 1989, Volume 10, Number 1, pp 1-8.
- Travassos-da-Rosa, A.P.A.; Travassos-da-Rosa, J.F.S.; Vasconcelos, P.F.C.; Pinheiro, F.P. Arboviroses. In: Leão, R.N.Q, editor. Doenças Infecciosas e Parasitárias. Belém: Cejup: UEPA: Instituto Evandro Chagas. 1997. pp 207-226.
- Câmara, T.N.L. Arboviroses emergentes e novos desafios para a saúde pública no Brasil. Revista de Saúde Pública. 2016, Volume 50, pp 36-46.
- Lopes, N.; Nozawa, C.; Linhares, R.E.C. Características gerais e epidemiologia dos arbovírus emergentes no Brasil. Rev. Pan-Amaz. Saúde. 2015, Volume 5, Number 3, pp 55-64.
- Anez, G.; Chancey, A.; Rios, M. Dengue vírus and other arboviruses: a global view of risks. ISBT: Science Series. 2012, Volume 7, pp 274-282. [CrossRef]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and clinical performance of the CDC real-time RT-PCR assay for detection and typing of dengue virus. PLoS Negl Trop Dis. 2013, Volume 11, Number 7, e2311.
- ILLUMINA. bcl2fastq2 conversion software v2.20. Available at: https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html. 2022, Accessed: August 12, 2022.
- Chen, S.; Zhou, Y.; Chen, Y.; GU, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, Volume 34, Number 17, pp 884-890. [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012, Volume 9, Number 4, pp 357-359, 2012. [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Havas-Stones, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; Thierer, T.; Ashton, B.; Meintjes, P.; Drummond, A. Geneious Basic: an integrated and extensible software platform for organizing and analyzing sequence data. Bioinformatics. 2012, Volume 28, Number 12, pp 1647-1649.
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009, Volume 15, Number 16, pp 2078-2079. [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Strömberg, M.P.; Marth, G.T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011, Volume 15, Number 12, pp 1691-1692. [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, Volume 30, Number 4, pp 772-780. [CrossRef]
- Strimmer, K.; von Haeseler, A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA. 1997, Volume 94, Number 13, pp 6815-6819.
- Myung J. Tutorial on maximum likelihood estimation. J. Math. Psychol. 2003, Volume 47, pp 90-100.
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Haeseler, A.V.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, Volume 37, Number 5, pp 1530-1534.
- Felsenstein, J. Phylogenies and the Comparative Method. Am. Nat. 1985, Volume 125, Number 1. [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, Volume 14, pp 587-589. [CrossRef]
- Hess, P.N.; Russo, C.A.M. An empirical test of the midpoint rooting method. Biol. J. Linn. Soc. 2007, Volume 92, Number 4, pp 669-674. [CrossRef]
- Rambaut, A. FigTree. Disponível em: http://tree.bio.ed.ac.uk/software/figtree/. Accessed: June. 2023.
- Câmara, T.N.L. Arboviroses emergentes e novos desafios para a saúde pública no Brasil. Ver. Saude Publica. 2016, Volume 50, pp 36-46.
- Ardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, Volume 19, Number 1, pp 9-20.
- Weaver, S.C.; Vasilakis, N. Molecular evolution of dengue viruses: Contributions of phylogenentics to understand the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 2009, Volume 9, pp 523-540.
- García, M.P.; Padilla, C.; Figueroa, D.; Manrique, C.; Cabezas, C. Emergence of the Cosmopolitan genotype of dengue virus serotype 2 (DENV2) in Madre de Dios, Peru, 2019. Rev. Peru. Med. Exp. Salud Publica. 2022, Volume 39, pp 126-128.
- Giovanetti, M.; Pereira, L.A.; Santiago, G.A.; Fonseca, V.; Mendoza, M.P.G.; de Oliveira, C.; de Moraes, L.; Xavier, J.; Tosta, S.; Fristch, H.; de Castro Barbosa, E.; Rodrigues, E.S.; Figueroa-Romero, D.; Padilla-Rojas, C.; Cáceres-Rey, O.; Mendonça, A.F.; de Bruycker Nogueira, F.; Venancio da Cunha, R.; de Filippis, A.M.B.; Freitas, C.; Peterka, C.R.L.; de Albuquerque, C.F.C, Franco, L.; Méndez Rico, J.A.; Muñoz-Jordán, J.L.; Lemes da Silva, V.; Alcantara, L.C.J. Emergence of Dengue Virus Serotype 2 Cosmopolitan Genotype, Brazil. Emerg. Infect. Dis. 2022, Volume 28, Number 8, pp 1725-1727. [CrossRef]
- Williams, M.; Mayer, S.V.; Johnson, W.L.; Chen, R.; Volkova, E.; Vilcarromero, S.; Widen, S.G.; Wood, T.G.; Suarez-Ognio, L.; Long, K.C.; Hanley, K.A. Lineage II of Southeast Asian/American DENV-2 is associated with a severe dengue outbreak in the Peruvian Amazon. Am. J. Trop. Med. Hyg. 2014, Volume 91, Number 3, pp 611-620. [CrossRef]
- Falconi-Agapito, F.; Selhorst, P.; Merino, X.; Torres, F.; Micheils, J.; Fernandez, C.; Talledo, M.; Ariën, K.K. A new genetic variant of dengue serotype 2 virus circulating in the Peruvian Amazon. Int. J. Infect. Dis. 2020, Volume 96, pp 136-138. [CrossRef]
- Mir, D.; Romero, H.; de Carvalho, L.M.F.; Bello, G. Spatiotemporal dynamics of DENV-2 Asian-American genotype lineages in the Americas. PLoS One. 2014, Volume 9, e98519. [CrossRef]
- Figueiredo, L.B.; Sakamoto, T.; Coelho, L.F.L.; Rocha, E.S.O.; Cota, M.M.G.; Ferreira, G.P.; Oliveira Rocha, E.S.; Gomes Cota, M.M.; Ferreira, G.P.; de Oliveira, J.G.; Kroon, E.G. Dengue virus 2 American-Asian genotype identified during the 2006/2007 outbreak in Piauí, Brazil reveals a Caribbean route of introduction and dissemination of dengue virus in Brazil. PLoS One. 2014, Volume 9, e104516. [CrossRef]
- Faria, N.R.D.C.; Nogueira, R.M.R.; de Filippis, A.M.B.; Simões, J.B.S.; Nogueira, F.D.B.; Lima M.R.Q.; Dos Santos, F.B. Twenty Years of DENV-2 Activity in Brazil: Molecular Characterization and Phylogeny of Strains Isolated from 1990 to 2010. PLoS Negl. Trop. Dis. 2013, Volume 7, e2095. [CrossRef]
- Allicock, O.M.; Lemey, P.; Tatem, A.J.; Pybus, O.G.; Bennett, S.N.; Carrington, C.V.F.; Soldan, V.P.; Oliveira, L.F.; Watts, D.M.; Sequeda, M.M.; Guzman, M.G.; Austin, C.C.; Rupprecht, C.E.; Sarkar, S.; Harrison, L.J. Phylogeography and Population Dynamics of Dengue Viruses in the Americas. Mol. Biol. Evol. 2012, Volume 29, Number 6, pp 1533-1543.
- Cunha, M.P.; Duarte-Neto, A.N.; Pour, S.Z.; Hajjar, L.A.; Frassetto, F.P.; Dolhnikoff, M.; Saldiva, P.H.N.; Zanotto, P.M.A. Systemic dengue infection associated with a new dengue virus type 2 introduction in Brazil – a case report. BMC Infect. Dis. 2021, Volume 21, pp 311. [CrossRef]
- De Jesus, J.G.; Dutra, K.R.; Sales, F.C.S.; Claro, I.M.; Terzian, A.C.; Candido, D.S.; Hill, S.C.; Thézé, J.; Torres, C.; D'Agostini, T.L.; Felix, A.C.; Reis, A.F.N.; Alcantara, L.C.J.; de Abreu, A.L.; Croda, J.H.R.; Oliveira, W.K.D.; de Filippis, A.M.B.; Camis, M.D.; C.R.S.; Romano, C.M.; Loman, N.J.; Pybus, O.G.; Sabino, E.C.; Nogueira, M.L.; Faria, N.R. Genomic detection of a virus lineage replacement event of dengue virus serotype 2 in Brazil, 2019. Mem. Inst. Oswaldo Cruz. 2020, Volume 115, e190423.
- Gräf, T.; Ferreira, C. D. N.; Lima, G. B.; Lima, R. E.; Machado, L. C.; Campos, T. L.; ... Naveca, F. Multiple introductions and country-wide spread of DENV-2 genotype II (Cosmopolitan) in Brazil. Virus Evolution, 2023, Volume 9, e. 2, vead059. [CrossRef]
- Amorim, M.T.; Hernández, L.H.A.; Naveca, F.G.; Essashika Prazeres, I.T.; Wanzeller, A.L.M.; Silva, E.V.P.D.; Casseb, L.M.N.; Silva, F.S.D.; da Silva, S.P.; Nunes, B.T.D.; Cruz, A.C.R. Emergence of a New Strain of DENV-2 in South America: Introduction of the Cosmopolitan Genotype through the Brazilian-Peruvian Border. Trop. Med. Infect.Dis.2023, Volume 8, pp 325. [CrossRef]
Figure 1.
Maximum likelihood phylogenetic tree based on the alignment of the dataset comprising 156 DENV-2 genome sequences. The scale bar indicates the evolutionary distance in the number of nucleotide substitutions per site, and the bootstrap values are indicated on the main branches, with a focus on the cosmopolitan genotype (represented in green) and the Asian-American genotype (blue). Lineage 5 - cosmopolitan genotype - is represented in red, and lineage 1 - Asian-American genotype - is represented in cyan. The name and information of the isolates characterized in this study are highlighted, including the sample identification number, country, state of origin, and collection date.
Figure 1.
Maximum likelihood phylogenetic tree based on the alignment of the dataset comprising 156 DENV-2 genome sequences. The scale bar indicates the evolutionary distance in the number of nucleotide substitutions per site, and the bootstrap values are indicated on the main branches, with a focus on the cosmopolitan genotype (represented in green) and the Asian-American genotype (blue). Lineage 5 - cosmopolitan genotype - is represented in red, and lineage 1 - Asian-American genotype - is represented in cyan. The name and information of the isolates characterized in this study are highlighted, including the sample identification number, country, state of origin, and collection date.
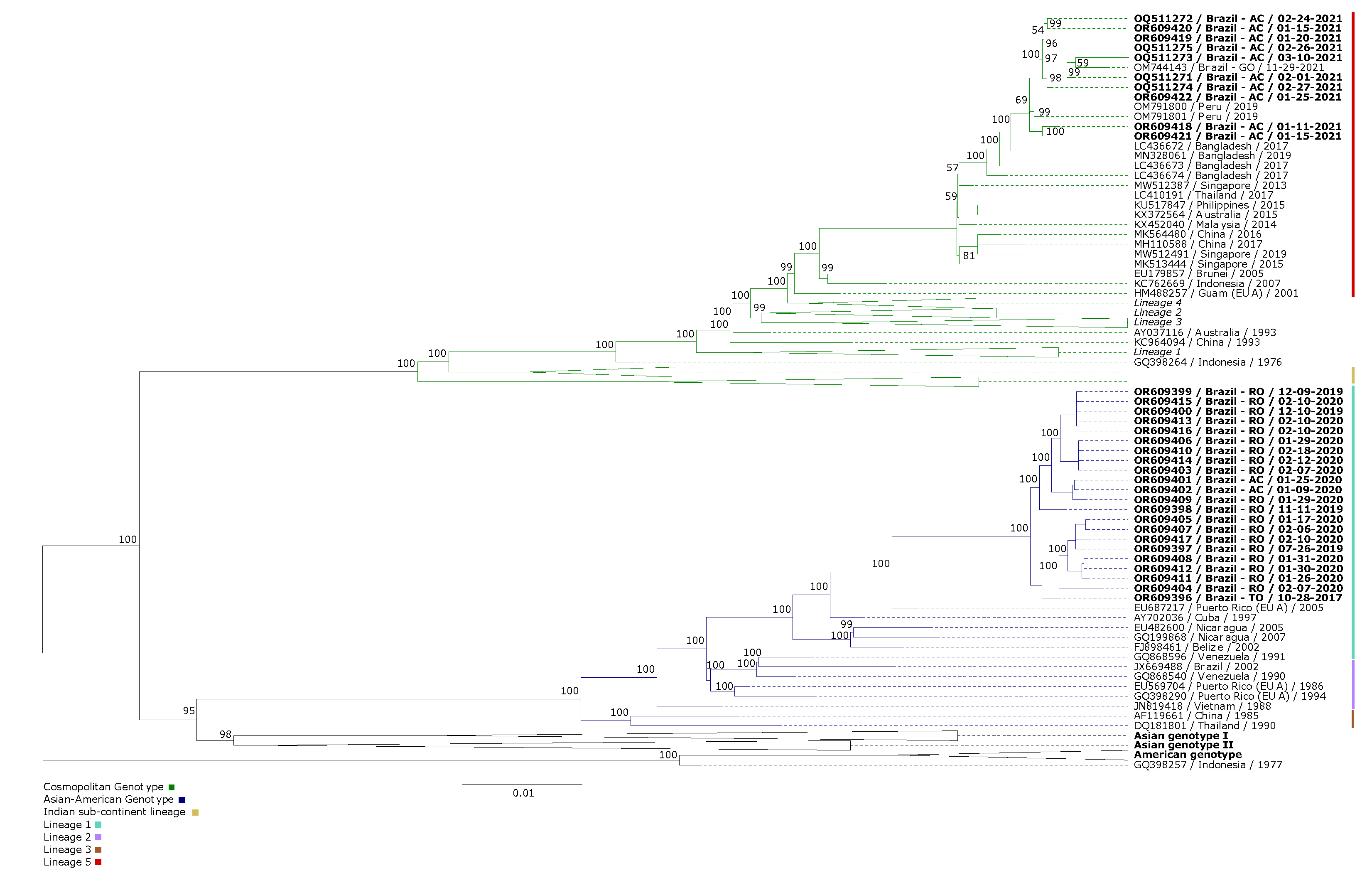
Figure 2.
- Map illustrating the possible route of introduction and dispersal of the Asian-American genotype of DENV-2 inferred in this study. The orange arrow indicates parallel dispersion from Puerto Rico through an unmapped route (Puerto Rico-Brazil/Tocantins, Acre, and Rondônia). The bifurcation region indicates only the intersection corresponding to parallel dispersion for the states of Acre, Tocantins, and Rondônia, and not that the dispersion route necessarily includes the state in which the bifurcation region is located. In other words, it suggests that the strains recorded in Brazilian states until 2020 likely originated from Puerto Rico. The colors delineate the states and the intersections between the regions of the countries (Blue: Tocantins, Brazil; Green: Acre, Brazil; Orange: Rondônia, Brazil; Red: border region with Brazilian states). The yellow gradient circle indicates the occurrence of potential strain dispersion routes to regions in the Americas and neighboring countries, originating from Puerto Rico as a secondary dissemination center. The map was created using QGIS software v.3.28, available at https://qgis.org/pt_BR/site/ (accessed on 09/06/2023).
Figure 2.
- Map illustrating the possible route of introduction and dispersal of the Asian-American genotype of DENV-2 inferred in this study. The orange arrow indicates parallel dispersion from Puerto Rico through an unmapped route (Puerto Rico-Brazil/Tocantins, Acre, and Rondônia). The bifurcation region indicates only the intersection corresponding to parallel dispersion for the states of Acre, Tocantins, and Rondônia, and not that the dispersion route necessarily includes the state in which the bifurcation region is located. In other words, it suggests that the strains recorded in Brazilian states until 2020 likely originated from Puerto Rico. The colors delineate the states and the intersections between the regions of the countries (Blue: Tocantins, Brazil; Green: Acre, Brazil; Orange: Rondônia, Brazil; Red: border region with Brazilian states). The yellow gradient circle indicates the occurrence of potential strain dispersion routes to regions in the Americas and neighboring countries, originating from Puerto Rico as a secondary dissemination center. The map was created using QGIS software v.3.28, available at https://qgis.org/pt_BR/site/ (accessed on 09/06/2023).
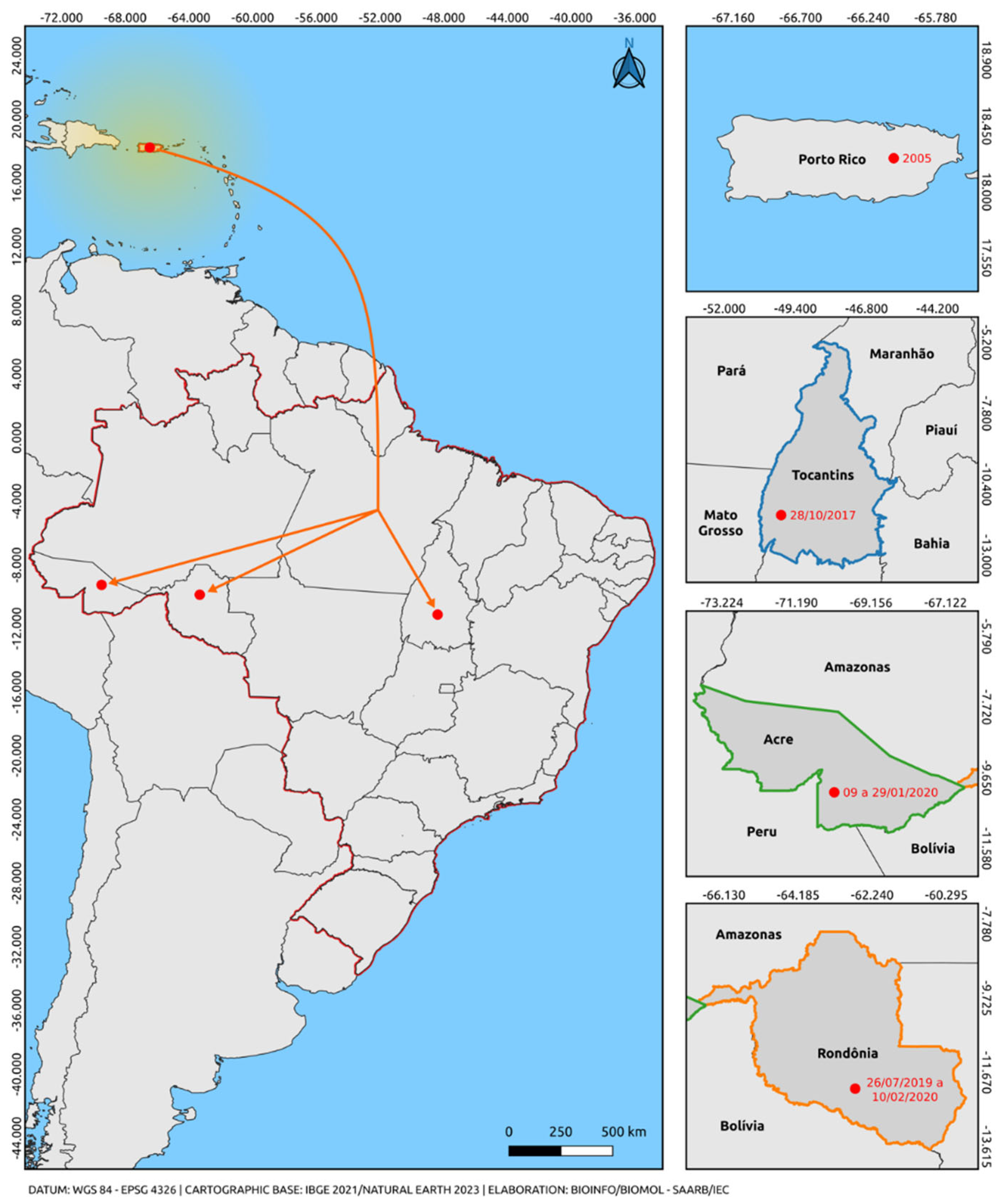
Figure 3.
Map illustrating the possible route of introduction and dispersal of the DENV-2 cosmopolitan genotype. The orange arrow indicates only the departure from the point of origin and arrival through an unmapped route (Madre de Dios, Peru), (Acre/Brazil-Goiás/Brazil), based on the analysis of sequences obtained from reports in the last five years and the sequences from this study. The highlighted arrow is not related to the virus dispersion route; it only indicates the departure from the point of origin (Madre de Dios, Peru and Acre, Brazil) and the destination (via an unmapped route) to Goiás, Brazil. The colors represent the states and intersections between regions of the countries (Orange: Madre de Dios, Peru; Green: Acre, Brazil; Blue: Goiás, Brazil; Red: border region between Brazil and Peru). The yellow gradient circle indicates the potential spread of the strain to other regions of Brazil and neighboring countries. The map was created using QGIS software v.3.28, available at https://qgis.org/pt_BR/site/ (accessed on 09/06/2023).
Figure 3.
Map illustrating the possible route of introduction and dispersal of the DENV-2 cosmopolitan genotype. The orange arrow indicates only the departure from the point of origin and arrival through an unmapped route (Madre de Dios, Peru), (Acre/Brazil-Goiás/Brazil), based on the analysis of sequences obtained from reports in the last five years and the sequences from this study. The highlighted arrow is not related to the virus dispersion route; it only indicates the departure from the point of origin (Madre de Dios, Peru and Acre, Brazil) and the destination (via an unmapped route) to Goiás, Brazil. The colors represent the states and intersections between regions of the countries (Orange: Madre de Dios, Peru; Green: Acre, Brazil; Blue: Goiás, Brazil; Red: border region between Brazil and Peru). The yellow gradient circle indicates the potential spread of the strain to other regions of Brazil and neighboring countries. The map was created using QGIS software v.3.28, available at https://qgis.org/pt_BR/site/ (accessed on 09/06/2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



