
Submitted:
08 November 2023
Posted:
09 November 2023
You are already at the latest version
Abstract
Cytochrome P450 oxidoreductase (POR) is an essential redox partner for steroid and drug-metabolizing cytochromes P450 located in the endoplasmic reticulum. Mutations in POR lead to metabolic disorders, including congenital adrenal hyperplasia, and affect the metabolism of steroids, drugs, and xenobiotics. In this study, we examined approximately 450 missense variants of the POR gene listed in the Genome Aggregation Database (gnomAD) using eleven different in silico prediction tools. We found that 64 novel variants were consistently predicted to be disease-causing by most tools. To validate our findings, we conducted population analysis and selected two variations in POR for further investigation. The human POR wild type, along with the R268W and L577P variants, were expressed in bacteria and subjected to enzyme kinetic assays using model substrate. We also examined the activities of several cytochrome P450 proteins in the presence of POR (WT or variants) by combining P450 and reductase proteins in liposomes. We observed a decrease in enzymatic activities (ranging from 35% to 85%) of key drug metabolizing enzymes, supported by POR variants R288W and L577P compared to WT-POR. These results validate our approach of curating a vast amount of data from genome projects and provide an updated and reliable reference for diagnosing POR deficiency.
Keywords:
Cytochrome P450 oxidoreductase
; POR
; gnomAD
; CAH
; drug metabolism
; steroid hormones
; SNV
; bioinformatics
1. Introduction
POR plays a fundamental role as the primary electron donor for all type II cytochrome P450 enzymes located in the endoplasmic reticulum [1,2,3,4] and is a central part of microsomal oxidase system [5,6]. These P450 enzymes include CP21A2, CYP17A1, and CYP19A1 responsible for steroid biosynthesis, as well as CYP51A1 and CYP26B1 contributing to bone formation [7,8,9]. Therefore, mutations in the human POR gene give rise to a diverse spectrum of clinical manifestations [7,10,11,12,13,14,15,16] [17,18]. The severest forms of POR deficiency (PORD) encompass congenital adrenal hyperplasia (CAH) [18,19,20,21], disruptions in sexual and pubertal development (DSD), and skeletal anomalies resembling Antley-Bixler syndrome (ABS) [22,23,24,25].
The pivotal role of POR in transferring electrons within the cell begins with the conversion of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to the cofactor flavin adenine dinucleotide (FAD) [2,26]. Subsequently, these electrons travel from FAD to another flavin cofactor, flavin mononucleotide (FMN). This relay of electrons between FAD and FMN is a key step in the electron transport process. These flavin cofactors, FAD and FMN, are vital components of the POR protein and serve as electron carriers [27]. The electron transfer from FMN further extends to its downstream redox partners, such as various cytochrome P450 enzymes [28,29]. POR is vital for many other metabolic processes beyond steroidogenesis and skeletal development. Among these functions are the metabolism of a wide range of substances, including drugs, xenobiotics, arachidonic acid, and eicosanoids, as well as involvement in the synthesis and degradation of vital compounds such as cholesterol, bile acids, and hemoglobin [1,2,12,14].
In the context of drug metabolism, both in vivo and especially in vitro studies consistently underscore the detrimental effects of various POR variants on drug metabolizing P450 enzymes [14,30,31]. Intriguingly, despite these findings, no clinically relevant adverse drug events have been directly associated with PORD patients to date [32,33]. Nevertheless, a detailed exploration of drug metabolism in a homozygous PORD patient and her heterozygous mother revealed reduced activities in several drug-metabolizing P450 enzymes [11,34,35,36]. Moreover, there is rather poor knowledge about how these variants could affect healthy or subclinical carriers. Thus, what we know about PORD could just be the surface of a complex phenomenon.
To move towards precise diagnosis and effective management of these complex metabolic disorders, we have designed a workflow. This approach rigorously filters and assesses the pathogenicity of missense variants within the POR gene. Furthermore, it advances our comprehension of POR-associated disorders and constitutes a valuable contribution to the field of precision medicine. In this regard, we examined a pool of 450 missense variants of the POR gene taken from the gnomAD database. Subsequently, we used multiple in silico prediction tools to identify potentially disease-causing variants from this pool. Population analysis and further investigation were conducted on selected variants to validate their pathogenicity. Finally, enzyme kinetics in a model substrate and activities of major drug metabolizing CYP450s (CYP3A4, CYP3A5, CYP2C9, and CYP2C19) were assessed on human POR wild type and selected variants.
2. Materials and Methods
Control Variants
The selection of control variants involved an exhaustive review of the existing literature to identify POR gene variants with well-documented experimental data [7,10,11,12,13,14,15,16,31]. These control variants were specifically chosen due to their established pathogenic or benign nature, supported by robust experimental evidence. By curating this set of control variants, we ensured a reliable benchmark for evaluating the performance of in silico prediction tools. The selected variants served as a reference group against which the predictions of our chosen in silico tools were compared. This meticulous approach not only bolstered the validity of our study but also enabled a rigorous assessment of the predictive accuracy of the computational tools in distinguishing between pathogenic and benign POR gene variants.
Database Collection
The POR gene analysis utilized gnomAD V2.1.1 (https://gnomad.broadinstitute.org), encompassing an extensive dataset of 125,748 exome sequences and 15,708 whole-genome sequences from diverse human populations [37]. This compilation includes individuals from European, Ashkenazi Jewish, East Asian, South Asian, African/African American, Latino/Ad-mixed American, and other ancestry groups. Genomic positions align with the GRCh37/hg19 assembly, while nucleotide positions adhere to the Homo sapiens POR mRNA sequence cataloged in the National Center for Biotechnology Information (NCBI) under the reference sequence NM_000941.3. Amino acid positions are denoted in concordance with NCBI's NP_000932.3 and UniProt's P16435 numbering systems. In this study, we focused on predicting the pathogenicity of missense variants.
Single Amino Acid Variant Prediction Tools
To assess the potential impact of amino acid substitutions within the POR gene, we employed multiple in silico prediction tools. These tools utilize diverse algorithms and databases to predict the functional consequences of genetic variants. Among them are single-predictors and meta-predictors, the last ones enhance accuracy by aggregating results from various sources, increasing the reliability of our predictions.
- 1.
- PANTHER
PANTHER (Protein Analysis Through Evolutionary Relationships) uses evolutionary conservation patterns to predict the functional impact of genetic variants. It's valuable for identifying variants that may disrupt protein structure and function based on evolutionary history [38].
- 2.
- PhD-SNP
PhD-SNP employs support vector machines and sequence-derived features to classify variants as disease-associated or benign. Known for its high accuracy, it's effective in discerning pathogenic mutations from benign ones [39].
- 3.
- SIFT
SIFT (Sorting Intolerant From Tolerant) uses sequence conservation to predict the impact of amino acid substitutions on protein function. It's widely used for identifying deleterious variants that may affect protein function [40].
- 4.
- SNAP
SNAP utilizes neural networks to classify variants based on their potential effect on protein function. It's valuable for distinguishing between tolerated and damaging variants [41].
- 5.
- MAPP
MAPP (Multivariate Analysis of Protein Polymorphism) combines sequence information, physicochemical properties, and evolutionary conservation to predict variant pathogenicity. It offers a multifaceted approach to predicting the functional consequences of genetic variants [42].
- 6.
- PolyPhen-1 and PolyPhen-2:
PolyPhen (Polymorphism Phenotyping) evaluates variants by considering sequence-based and structural features. These tools are known for their ability to predict the impact of variants on protein structure and function, with PolyPhen-2 being an improved version [43].
- 7.
- MetaSNP:
MetaSNP, a meta-predictor, combines the predictions from multiple existing tools, including PANTHER, PhD-SNP, SIFT, and SNAP, to generate a consensus prediction [44].
- 8.
- MutPred2:
MutPred2 utilizes a machine learning approach that considers sequence-based features, structure-based features, and functional annotations to evaluate the pathogenicity of variants. MutPred2 provides a holistic assessment, incorporating diverse data types to enhance prediction accuracy [45].
- 9.
- SNPs&Go:
SNPs&Go is a meta-predictor (PHANTHER, PhD-SNP) that integrates protein sequence information with Gene Ontology annotations to predict the functional impact of variants. This tool provides insights into how variants may disrupt protein function within the context of cellular processes [46].
- 10.
- PredictSNP2:
PredictSNP2 employs a machine learning-based approach, considering sequence-derived features, conservation scores, and structural information to make predictions. PredictSNP2's multi-modal approach (MAPP, PhD-SNP, SIFT, SNAP, Polyphen 1, Polyphen 2) enhances the robustness of predictions, particularly valuable for variants with complex effects [47].
Data Integration and Evaluation
The predictions generated by these diverse tools were systematically integrated and analyzed. First, to assess prediction accuracy, we utilized the control variants to compare these results with experimental data from the literature. Based on the performance, we selected tools that discriminated better between pathogenic and nonpathogenic variants to use in the analysis of the variants from the gnomAD database.
Variants consistently predicted as pathogenic for the selected tools were identified. Accordingly, we selected single nucleotide variants (SNVs) from the gnomAD dataset that met specific criteria: We chose variants that were consistently predicted as pathogenic by most of the integrated tools [37].
In our selection, we focused on variants with low allele frequencies (less than 0.01), indicating their rarity within the population. This approach ensures that the chosen variants are not common background variations. Additionally, we prioritized variants that had not been previously described in the existing scientific literature.
Expression of POR in E. coli
As a proof of concept, we selected two variants (R268W and L577P) for further investigation to validate our methodology with functional assays in a recombinant system. The cDNAs of POR Wild type (WT), R268W, and L577P, each with 6xHis tag, were cloned into a pET15b vector and subsequently transformed into Escherichia coli BL21(DE3). Single colonies were selected by growth on LB-agar plates containing 100 μg/mL carbenicillin. Large-scale expression was facilitated through an auto-induction system, involving the growth of selected colonies in terrific broth with added glucose (0.05%), lactose (0.2%), succinate (20 mM), NaSO4 (5 mM), NH4Cl (50 mM), MgSO4 (2 mM), 0.05 mg/ml riboflavin, and 100 μg/ml carbenicillin. The initial growth occurred at 37°C until reaching an optical density (OD at 600nm) of 0.6. Subsequently, the temperature was lowered to 24°C, and the cultures were grown for an additional 16 hours with constant shaking. Bacterial cells were collected by centrifugation, washed with PBS, and suspended in a solution comprising 50 mM potassium phosphate (pH 7.6), 250 mM sucrose, 0.5 mM EDTA, 0.2 mg/ml lysozyme, 1 mM PMSF, and 20 U/ml Endonuclease. Spheroplasts were generated through one hour of slow stirring. After centrifugation at 5000×g for 20 minutes, the spheroplasts were suspended in a solution consisting of 50 mM potassium phosphate (pH 7.8), 6 mM MgOAc, 0.1 mM DTT, 20% (v/v) glycerol, and 1 mM PMSF. Disruption was achieved through sonication, yielding a clear lysate free of cellular debris. Membranes containing the over-expressed POR WT or variants were stored at -80°C [11,14,15].
Purification of Recombinant Human POR from Isolated E. coli Membranes
All steps were conducted at 4°C. The His-tagged proteins were solubilized at a concentration of 0.25 g of membrane per mL of 50 mM potassium phosphate (pH 7.4), 10% (v/v) glycerol, and 1% Triton X-100. The mixture was gently stirred for 16 hours and then centrifuged at 12,000 ×g for 15 minutes. The resulting supernatant was utilized for purification via ion-metal affinity chromatography (IMAC). The supernatant was diluted with buffer A to a final concentration of 50 mM potassium phosphate (pH 7.4), 30 mM imidazole, 0.1% Triton, 150 mM NaCl, and 10% glycerol. This mixture was loaded into 4 mL His60 Ni SuperflowTM Resin, and impurities were washed with buffer A, featuring an increasing concentration of imidazole up to 60 mM. The His-tagged proteins were subsequently eluted with the same buffer, incorporating increasing concentrations of imidazole up to 500 mM. The presence of POR was confirmed through Western blot analysis. Purified samples were concentrated, and the elution buffer was exchanged with 50 mM potassium phosphate (pH 7.4), 10% (v/v) glycerol, and 0.1% Triton X-100 using Amicon® Ultra Centrifugal Filters (20,000 MWCO). The protein concentration was measured using the Bradford assay [15].
Urea Denaturation Assay
POR Assays with Cytochrome c
The assays were conducted in triplicates in a 96-well format using a Spectramax M2e microplate reader (Molecular Devices, Sunnyvale, CA, USA). Each reaction well consisted of 50 nM POR in a solution containing 50 mM Tris-HCl (pH 7.8) and 150 mM NaCl. The concentration of cytochrome c was varied within the range of 2.5-60 μM. The reactions were initiated by adding 100 µM NADPH and monitored at 550 nm using the extinction coefficient (ε550= 21.1 mM-1 cm-1) for a duration of 10 minutes. The reaction rates were extrapolated from the linear range of the kinetic traces [13,48].
Reaction rates from all assays were determined by calculating the slope from the linear range of the kinetic traces. Vmax and Km values were obtained by fitting the data to the Michaelis-Menten equation and plotted using MATLAB (MathWorks, Natick, MA).
Assay of Cytochrome P450 (CYP) Activity in Reconstituted Liposomes
To assess the enzymatic activity of drug-metabolizing cytochrome P450s (CYP3A4, CYP3A5, CYP2C9, and CYP2C19) in the presence of wild-type (WT) or mutant POR, we employed specific fluorogenic substrates, BOMCC for CYP3A4, CYP3A5, and CYP2C9, and EOMCC for CYP2C19 (Invitrogen Corp, Carlsbad, CA, USA). Purified drug metabolizing cytochrome P450s (obtained from CYPEX, Dundee, Scotland, UK) were utilized to evaluate the activities of the POR variants, employing 20 μM BOMCC/EOMCC as the substrate.
For in vitro assays, we utilized a reconstituted liposome system consisting of pure WT/mutant POR, the respective CYP450s, and cytochrome b5 in a ratio of 5:1:1. The final assay mixture was composed of 20 µM DLPC (1,2-Dilauroyl-sn-glycero-3-phosphocholine)/DLPGV (1,2-Dilauroyl-sn-glycero-3-phosphoglycerol), proteins (100 nM POR, 20 nM CYP450s, 20 nM b5), 3 mM MgCl2, 20 μM BOMCC, in 50 mM Tris-HCl buffer with a pH of 7.4. The reaction volume was set at 100 µL.
To initiate the CYP3A5 reaction, NADPH was added to a final concentration of 1 mM. Subsequently, fluorescence measurements were performed using a Spectramax M2e plate reader (Molecular Devices, Sunnyvale, CA, USA), with excitation at a wavelength of 415 nm and emission at 460 nm for both BOMCC and EOMCC.
3. Results
3.1. Performance of the Prediction Tools
Our study involved a stringent selection process, focusing on 37 reference variants with well-documented experimental data. These data were primarily derived from functional assays measuring the impact of POR wild-type and its mutants on the activity of major steroid and drug-metabolizing CYP450s, as illustrated in Figure 1 [7,29,49,50].
Figure 1 demonstrates a correlation between the computational predictions and the empirical experimental findings. In most cases, variants deemed benign by most of the computational tools (e.g., P55L, R406H, G413S) exhibited over 50% of the redox partner activity. Conversely, variants classified as pathogenic (e.g., R457H, R550W, C569Y) displayed a significant reduction in activity across all redox partners. It is noteworthy that the pathogenic variants were discovered in individuals with severe clinical manifestations, underscoring the robust association between in silico predictions, experimental data, and the clinical phenotypes of carriers [51,52]. However, it is crucial to acknowledge that the impact of the same variant can fluctuate among different redox partners [53,54]. For instance, the case of D211N exhibited over 50% activity with most redox partners but only 6% activity with CYP2A1. A plausible explanation for this observation lies in the distinct interaction of POR with each partner, emphasizing the pivotal role of the specific amino acid alteration and its position within the protein [14,55,56].
As shown in Figure 2, to size the predictive performance of these tools, we conducted a Receiver Operating Characteristic (ROC) analysis with the reference group, which allowed us to quantify their ability to distinguish between pathogenic and benign variants. This analysis revealed the tools' proficiency in accurately classifying variants based on their pathogenic potential. The Area Under the Curve (AUC) is a metric used to assess the performance of classification models. Tools with AUC values ranging from 0.85 to 1 exhibited excellent predictive accuracy, suggesting their effectiveness in accurately classifying variants (Figure 2) [57,58]. On the other hand, SIFT, MAPP, and PolyPhen 1 and 2, with AUC values between 0.45 and 0.6, showed poorer performance and were consequently excluded from the analysis of the missense variants from gnomAD (Figure 2). These results underscore the importance of selecting and utilizing predictive tools with high AUC values for robust variant classification, particularly when evaluating the pathogenic potential of genetic variants in a clinical or research context.
3.2. Curating Variants from gnomAD
As it is shown in Figure 3, we conducted a comprehensive curation of variants in the POR gene retrieved from the gnomAD databases, which initially comprised a total of 1,419 variants. These variants encompassed a diverse range of mutation types, including intron variants, 3' prime variants, 5' variants, frameshift variants, in-frame deletions, splice site variants, start-lost variants, stop/gained mutations, synonymous variants, and the predominant category of interest, missense variants [37,59].
Our focus on missense variants was driven by their prevalence, mirroring the clinical scenario in patients with POR deficiency, where a significant portion of cases involves carriers with at least one missense variant [2,30,60]. This emphasis is due to the observation that missense variants often retain some residual enzymatic activity, which is critical for sustaining life [7,10]. In contrast, complete loss of POR enzyme, which is often the case with deletions or insertions, activity is incompatible with survival [61,62]. Hence, our decision to prioritize the analysis of missense variants underscores their clinical relevance and potential impact on patient outcomes. We further refined our analysis by applying a series of in silico tools that had previously shown robust performance in our control studies, namely PANTHER, PhD-SNP, SNAP, Meta-SNP, SNPs&GO, and MutPred2. Additionally, allele frequency played a role in our curation process.
3.3. Analysis of the Missense Variants
From the pool of 450 missense variants within the POR gene, the majority exhibited a minor allele frequency (MAF) of less than 0.01, classifying them as rare variants. Notably, two variants, A503V (MAF 0.3), and T201M (MAF 0.01), stood out as polymorphisms frequently observed in large genome databases. The remaining variants with higher allele frequencies include P228L (MAF 0.003), P181L (MAF 0.002), V472M (MAF 0.002), and V631I (MAF 0.001) [15,37,63]. This distribution highlights the predominantly rare nature of the missense variants within the POR gene, with only a select few displaying higher frequencies, thus underscoring their significance in the context of genetic diversity and disease association.
Our stringent filtering process led to the identification of 64 novel missense variants (Figure 4). These variants demonstrated consistent classification as pathogenic by our selected in silico tools. As mentioned, they exhibited low allele frequencies, indicating their rarity in the population, which is often associated with more severe clinical phenotypes. Crucially, our literature review confirmed that these 64 variants had not been previously described in patients, further emphasizing their novelty [7,64,65].
These 64 variants are primarily distributed across the co-factor binding domains of the POR protein. Notably, a significant cluster of these variants is situated within the FAD and NADPH binding domain, which has been classified as highly pathogenic (Figure 4). This observation is in concordance with findings in the existing literature, as depicted in Figure 1, where variants within these specific regions consistently exhibit a severe reduction in their activity with redox partners [12,31]. Various factors contribute to the pathogenicity of an amino acid change. Among these factors, the position of the amino acid within the protein is pivotal, with buried amino acids being more likely to induce disease than those located on the protein's surface. Additionally, the chemical nature of the changes, such as the transition from hydrophobic to hydrophilic amino acids or vice versa, significantly influences their impact on protein function [66].
In our extensive dataset, we identified a significant portion of disease-causing mutations primarily involving Arginine (R) and Tyrosine (Y) amino acids, contributing 27% and 11%, respectively [66,67]. In the literature, Arginine substitutions, including changes to Cysteine, Proline, and Tryptophan, formed a major subset of prevalent disease-causing mutations. We observed variants like R202C, R268W, R293W, R427P, and others (Figure 4). Regarding Arginine, its significance lies in its positive charge and participation in important functional and structural roles within proteins [68,69]. Substituting Arginine, particularly with amino acids that have distinct chemical characteristics, can disrupt these roles and result in protein malfunction or misfolding, thus contributing to disease. Glycine substitutions to Arginine, Aspartic Acid, Glutamic Acid, and Valine were also common in our dataset, with variants like G39D, G535D, and G37R. As we can see in our dataset with variants G39D, G535D, G37R and others [70]. Glycine, being the smallest among the 20 amino acids, is especially sensitive to substitutions with other residues, potentially causing profound alterations to protein structure [66].
Changes in Tyrosine residues, as seen in variants like Y43D, Y87C, and Y181N, are harmful due to their involvement in maintaining protein stability, structure, and function. Tyrosine's chemical properties, including hydrogen bonding capacity and roles in active sites or binding pockets, make it essential. Substituting Tyrosine with amino acids of varying properties can disturb the protein's structure and interactions, potentially leading to functional loss and disease [71,72].
Mutations that change Leucine to Proline (L454P and L577P, Figure 4) can be detrimental due to the significant differences in their chemical properties. Leucine is a hydrophobic amino acid, while Proline has a unique cyclic structure that disrupts protein secondary structures. Substituting Leucine with Proline can lead to structural distortions, potentially impacting protein function and contributing to disease [66,71].
Of note, our analysis revealed that all the 64 pathogenic missense variants were present in a heterozygous state, suggesting that they occur in conjunction with a wild-type copy of the gene or in combined heterozygosity with another variant. This observation raises intriguing possibilities regarding the potential clinical impact of these variants. We postulate that individuals who carry two copies of pathogenic variants, either in homozygous or combined heterozygous form, may manifest a severe phenotype akin to congenital adrenal hyperplasia (CAH). In less severe cases, these variants could still exert a substantial influence on drug metabolism, potentially resulting in altered drug responses. These findings underscore the importance of further investigations to elucidate the precise clinical ramifications of these newly identified POR variants [34,73,74].
3.4. Functional Assay of Selected Variants
In our study, we focused on two specific variants, R268W (MAF 0.0002) and L577P (MAF 0.0006), as they were the most frequent among the 64 variants. To assess their impact on POR protein stability, we expressed the wild-type (WT) and variant forms in a bacterial system and performed urea denaturation assays. The fluorescence, which corresponds to flavin co-factor release, served as a proxy for protein structural stability [11,15]. Our results revealed that WT POR steadily released flavins as urea concentration increased, with a peak at 4 M urea (Figure 5). In contrast, R268W and L577P variants exhibited a rapid increase in fluorescence at a very low urea concentration (0.25 M), which was sustained at higher urea levels. This suggests that these variants are less stable than WT. Additionally, we observed reduced flavin content in the variants compared to WT, implying a significant reduction in function (Figure 5).
Furthermore, kinetic assays using Cytochrome c as a substrate indicated that R268W retained 113% of the WT activity, while L577P exhibited a more substantial reduction with only 51% activity. These results were corroborated by different Km values, where L577P showed a higher Km of 6.4 µM, and R268W displayed a slightly lower Km of 3.9 µM compared to the WT's Km of 4.6 µM (Figure 6, Table 1). The kinetic parameter of POR WT is similar to what was described previously in the literature a KM around 5 μM [13,14,15].
We also conducted functional assays with major drug metabolizing CYP450s (Figure 7). In CYP3A4 assays, POR variants L577P and R268W exhibited 51% and 62% of WT activity, respectively. In CYP3A5 assays, these variants displayed 63% and 33% of WT activity, and in CYP2C9 assays, the variants demonstrated 52% and 10% of WT activity for L577P and R268W, respectively. For CYP2C19, L577P and R268W showed 30% and 15% of WT activity, highlighting their varying effects on drug metabolism enzymes [14,15,75,76].
4. Discussion
Our study delves into the intricate landscape of POR (P450 oxidoreductase) gene variants, shedding light on the clinical implications and predictive potential of these genetic alterations. The comprehensive evaluation of POR gene variants within the gnomAD database underscores the remarkable rarity of missense variants within this gene [77]. We emphasize that this rarity is a pivotal observation, given the essential role of POR in the function of cytochrome P450 enzymes, which are central players in steroid biosynthesis, drug metabolism and various physiological processes.
The lack of known polymorphisms in the POR gene is a critical finding. In the broader context of pharmacology and drug metabolism, POR is a backbone in the catalytic cycle of numerous cytochrome P450 enzymes, including CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5, which are vital for the metabolism of a multitude of drugs. In particular, CYP3A4, one of the most abundant CYP450 enzymes in the human liver, is involved in the metabolism of approximately 50% of all marketed drugs [75,78,79]. Consequently, the scarcity of polymorphisms in the POR gene emphasizes its stability and evolutionary conservation. This knowledge can be harnessed to improve the predictability and safety of drug responses by considering POR genetic variability in drug development and administration [77].
Our focused analysis of 64 novel pathogenic missense variants within the POR gene further highlights the clinical relevance of our findings. These variants, primarily clustered within the FAD and NADPH binding domains of the POR protein, have a substantial impact on POR's ability to transfer electrons to cytochrome P450 enzymes, ultimately affecting drug metabolism [12,80]. We observed that these variants often result in significantly reduced protein stability and function, leading to altered drug responses. Such observations offer insights into the potential clinical consequences for individuals carrying these variants, particularly in relation to drug dosing and efficacy [72]. Notably, our findings highlight the predominance of arginine and tyrosine substitutions among the identified disease-causing mutations, underscoring the clinical significance of these amino acid changes [14,72]. Moreover, our analysis underscores the substantial impact of glycine alterations, emphasizing the importance of specific amino acid characteristics in protein structure and function [81].
Furthermore, our study goes beyond mere variant curation. We link computational predictions with experimental data, corroborating the pathogenicity of these variants [59]. The strong association we observed between in silico predictions, empirical experimental findings, and clinical phenotypes in patients further validates the effectiveness of our methods. This multifaceted approach can serve as a paradigm for future research in the pharmacogenomics field, where understanding genetic variations and their functional impact on drug-metabolizing enzymes is of paramount importance.
The clinical importance of our findings cannot be overstated [13,82]. As we identify these novel pathogenic variants, it raises the intriguing possibility that individuals carrying two copies of such variants, whether in a homozygous or combined heterozygous form, may manifest severe clinical phenotypes, akin to congenital adrenal hyperplasia (CAH). Even in less severe cases, the presence of such variants can significantly influence drug metabolism, potentially resulting in adverse drug reactions or altered therapeutic outcomes [13,75,78]. Our study underscores the necessity for further investigations into the clinical consequences of these newly identified POR variants, paving the way for precision medicine and personalized drug therapies.
5. Conclusions
The rarity of missense variants in the POR gene, the clinical significance of pathogenic variants, and the profound impact on drug metabolism underscore the importance of our study. These findings not only enhance our understanding of POR gene variations but also offer a valuable framework for future pharmacogenomic research, with the potential to revolutionize drug development, prescription, and dosing strategies, ultimately improving patient outcomes and drug safety.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.N.R.V and A.V.P.; POR assays, M.N.R.V.; molecular modeling and bioinformatics analysis M.N.R.V. and S.T.; writing—original draft preparation, M.N.R.V.; writing—review and editing, M.N.R.V, S.T., and A.V.P.; project administration A.V.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by SWISS NATIONAL SCIENCE FOUNDATION, grant number 310030M_204518 and The APC was funded by SWISS NATIONAL SCIENCE FOUNDATION. M.N.R.V was funded by the SWISS GOVERNMENT EXCELLENCE SCHOLARSHIP (ESKAS) grant number 2020.0557.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data are available in manuscript text or in supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Pandey, A.V. and C.E. Flück, NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Ther, 2013. 138(2): p. 229-54. [CrossRef]
- Xia, C. , et al., Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc Natl Acad Sci U S A, 2011. 108(33): p. 13486-91. [CrossRef]
- Pandey, A.V. and P. Sproll, Pharmacogenomics of human P450 oxidoreductase. Front Pharmacol, 2014. 5: p. 103. [CrossRef]
- Riddick, D.S. , et al., NADPH-cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metab Dispos, 2013. 41(1): p. 12-23. [CrossRef]
- Lu, A.Y.H. and M.J. Coon, Role of Hemoprotein P-450 in Fatty Acid Omega-Hydroxylation in a Soluble Enzyme System from Liver Microsomes. Journal of Biological Chemistry, 1968. 243(6): p. 1331-+. [CrossRef]
- Lu, A.Y.H., K. W. Junk, and M.J. Coon, Resolution of Cytochrome P-450-Containing Omega-Hydroxylation System of Liver Microsomes into 3 Components. Journal of Biological Chemistry, 1969. 244(13): p. 3714-+. [CrossRef]
- Burkhard, F.Z. , et al., P450 Oxidoreductase deficiency: Analysis of mutations and polymorphisms. J Steroid Biochem Mol Biol, 2017. 165(Pt A): p. 38-50. [CrossRef]
- Flück, C.E., M. N. Rojas Velazquez, and A.V. Pandey, Chapter 12 - P450 oxidoreductase deficiency, in Genetic Steroid Disorders (Second Edition), M.I. New, Editor. 2023, Academic Press: San Diego. p. 239-264. [CrossRef]
- Flück, C.E. and A.V. Pandey, Human P450 Oxidoreductase Deficiency, in Encyclopedia of Endocrine Diseases (Second Edition), I. Huhtaniemi and L. Martini, Editors. 2019, Academic Press: Oxford. p. 431-443. [CrossRef]
- Parween, S. , et al., Molecular basis of CYP19A1 deficiency in a 46, XX patient with R550W mutation in POR: Expanding the PORD phenotype. J Clin Endocrinol Metab, 2020. [CrossRef]
- Parween, S. , et al., P450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation. J Clin Endocrinol Metab, 2016. 101(12): p. 4789-4798. [CrossRef]
- Parween, S. , et al., Differential effects of variations in human P450 oxidoreductase on the aromatase activity of CYP19A1 polymorphisms R264C and R264H. J Steroid Biochem Mol Biol, 2020. 196: p. 105507. [CrossRef]
- Udhane, S.S. , et al., Altered CYP19A1 and CYP3A4 Activities Due to Mutations A115V, T142A, Q153R and P284L in the Human P450 Oxidoreductase. Front Pharmacol, 2017. 8: p. 580. [CrossRef]
- Velazquez, M.N.R. , et al., Variability in human drug metabolizing cytochrome P450 CYP2C9, CYP2C19 and CYP3A5 activities caused by genetic variations in cytochrome P450 oxidoreductase. Biochem Biophys Res Commun, 2019. 515(1): p. 133-138. [CrossRef]
- Rojas Velazquez, M.N., M. Noebauer, and A.V. Pandey, Loss of Protein Stability and Function Caused by P228L Variation in NADPH-Cytochrome P450 Reductase Linked to Lower Testosterone Levels. Int J Mol Sci, 2022. 23(17). [CrossRef]
- Flück, C.E. , et al., Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet, 2004. 36(3): p. 228-30. [CrossRef]
- Williamson, L. , et al., Linking Antley-Bixler syndrome and congenital adrenal hyperplasia: a novel case of P450 oxidoreductase deficiency. Am J Med Genet A, 2006. 140A(17): p. 1797-803. [CrossRef]
- Krone, N. , et al., Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf), 2007. 66(2): p. 162-72. [CrossRef]
- Shackleton, C. , et al., Prenatal diagnosis of p450 oxidoreductase deficiency (ORD): A disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome. American Journal of Medical Genetics Part A, 2004. 129A(2): p. 105-112. [CrossRef]
- Miller, W.L. , Disorders of androgen synthesis--from cholesterol to dehydroepiandrosterone. Med Princ Pract, 2005. 14 Suppl 1: p. 58-68. [CrossRef]
- Baronio, F. , et al., 46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features. Int J Mol Sci, 2019. 20(18). [CrossRef]
- Yauy, K. , et al., B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genetics in Medicine, 2018. 20(2): p. 269-274. [CrossRef]
- Xie, M. , et al., [Advance in clinical research on Antley-Bixler syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2018. 35(2): p. 280-283.
- Tomkova, M. , et al., [Antley-Bixler syndrome or POR deficiency?]. Cas Lek Cesk, 2008. 147(5): p. 261-5.
- Shackleton, C. , et al., Biochemical diagnosis of Antley-Bixler syndrome by steroid analysis. Am J Med Genet A, 2004. 128A(3): p. 223-31. [CrossRef]
- Xia, C. , et al., Structural and Kinetic Studies of Asp632 Mutants and Fully Reduced NADPH- Cytochrome P450 Oxidoreductase Define the Role of Asp632 Loop Dynamics in the Control of NADPH Binding and Hydride Transfer. Biochemistry, 2018. 57(6): p. 945-962. [CrossRef]
- Rwere, F. , et al., Mutants of Cytochrome P450 Reductase Lacking Either Gly-141 or Gly-143 Destabilize Its FMN Semiquinone. J Biol Chem, 2016. 291(28): p. 14639-61. [CrossRef]
- Pandey, A.V. , et al., Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol Endocrinol, 2007. 21(10): p. 2579-95. [CrossRef]
- Nicolo, C. , et al., Restoration of mutant cytochrome P450 reductase activity by external flavin. Mol Cell Endocrinol, 2010. 321(2): p. 245-52. [CrossRef]
- Pandey, A.V. , Biochemical analysis of mutations in P450 oxidoreductase. Biochem Soc Trans, 2006. 34(Pt 6): p. 1186-91. [CrossRef]
- Parween, S. , et al., Variability in Loss of Multiple Enzyme Activities Due to the Human Genetic Variation P284T Located in the Flexible Hinge Region of NADPH Cytochrome P450 Oxidoreductase. Front Pharmacol, 2019. 10: p. 1187. [CrossRef]
- Soneda, S. , et al., Proximal promoter of the cytochrome P450 oxidoreductase gene: identification of microdeletions involving the untranslated exon 1 and critical function of the SP1 binding sites. J Clin Endocrinol Metab, 2011. 96(11): p. E1881-7. [CrossRef]
- Fukami, M. , et al., Cytochrome P450 oxidoreductase deficiency: identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients. J Clin Endocrinol Metab, 2009. 94(5): p. 1723-31. [CrossRef]
- Idkowiak, J. , et al., Concomitant Mutations in the P450 Oxidoreductase and Androgen Receptor Genes Presenting with 46, XY Disordered Sex Development and Androgenization at Adrenarche. Journal of Clinical Endocrinology & Metabolism, 2010. 95(7): p. 3418-3427. [CrossRef]
- Huang, N. , et al., Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet, 2005. 76(5): p. 729-49. [CrossRef]
- Dean, B. , et al., P450 Oxidoreductase Deficiency: A Systematic Review and Meta-analysis of Genotypes, Phenotypes, and Their Relationships. J Clin Endocrinol Metab, 2020. 105(3). [CrossRef]
- Gudmundsson, S. , et al., Variant interpretation using population databases: Lessons from gnomAD. Hum Mutat, 2022. 43(8): p. 1012-1030. [CrossRef]
- Thomas, P.D. , et al., PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci, 2022. 31(1): p. 8-22. [CrossRef]
- Capriotti, E. and P. Fariselli, PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res, 2017. 45(W1): p. W247-W252. [CrossRef]
- Ng, P.C. and S. Henikoff, SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res, 2003. 31(13): p. 3812-4. [CrossRef]
- Hecht, M., Y. Bromberg, and B. Rost, Better prediction of functional effects for sequence variants. BMC Genomics, 2015. 16 Suppl 8(Suppl 8): p. S1. [CrossRef]
- Stone, E.A. and A. Sidow, Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res, 2005. 15(7): p. 978-86. [CrossRef]
- Adzhubei, I., D. M. Jordan, and S.R. Sunyaev, Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, 2013. Chapter 7: p. Unit7 20. [CrossRef]
- Capriotti, E., R. B. Altman, and Y. Bromberg, Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics, 2013. 14 Suppl 3(Suppl 3): p. S2. [CrossRef]
- Pejaver, V. , et al., Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun, 2020. 11(1): p. 5918. [CrossRef]
- Capriotti, E. , et al., WS-SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics, 2013. 14 Suppl 3(Suppl 3): p. S6. [CrossRef]
- Bendl, J. , et al., PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol, 2014. 10(1): p. e1003440. [CrossRef]
- Fluck, C.E. and A.V. Pandey, Impact on CYP19A1 activity by mutations in NADPH cytochrome P450 oxidoreductase. Journal of Steroid Biochemistry and Molecular Biology, 2017. 165: p. 64-70. [CrossRef]
- Fan, L. , et al., Novel phenotypes and genotypes in Antley-Bixler syndrome caused by cytochrome P450 oxidoreductase deficiency: based on the first cohort of Chinese children. Orphanet J Rare Dis, 2019. 14(1): p. 299. [CrossRef]
- Wudy, S.A. , et al., A male twin infant with skull deformity and elevated neonatal 17-hydroxyprogesterone: a prismatic case of P450 oxidoreductase deficiency. Endocr Res, 2004. 30(4): p. 957-64. [CrossRef]
- Tzetis, M. , et al., Compound heterozygosity of a paternal submicroscopic deletion and a maternal missense mutation in POR gene: Antley-bixler syndrome phenotype in three sibling fetuses. Birth Defects Res A Clin Mol Teratol, 2016. 106(7): p. 536-41. [CrossRef]
- Sandee, D. , et al., Effects of genetic variants of human P450 oxidoreductase on catalysis by CYP2D6 in vitro. Pharmacogenet Genomics, 2010. 20(11): p. 677-86. [CrossRef]
- Miller, W.L. , P450 oxidoreductase deficiency: a disorder of steroidogenesis with multiple clinical manifestations. Sci Signal, 2012. 5(247): p. pt11. [CrossRef]
- Sahakitrungruang, T. , et al., Clinical, Genetic, and Enzymatic Characterization of P450 Oxidoreductase Deficiency in Four Patients. Journal of Clinical Endocrinology & Metabolism, 2009. 94(12): p. 4992-5000. [CrossRef]
- Miller, W.L. , Minireview: regulation of steroidogenesis by electron transfer. Endocrinology, 2005. 146(6): p. 2544-50. [CrossRef]
- Guaragna-Filho, G. , et al., 46,XX DSD and Antley-Bixler syndrome due to novel mutations in the cytochrome P450 oxidoreductase gene. Arq Bras Endocrinol Metabol, 2012. 56(8): p. 578-85. [CrossRef]
- Zou, K.H., A. J. O'Malley, and L. Mauri, Receiver-operating characteristic analysis for evaluating diagnostic tests and predictive models. Circulation, 2007. 115(5): p. 654-7. [CrossRef]
- Prado, M.J. , et al., Variant predictions in congenital adrenal hyperplasia caused by mutations in CYP21A2. Front Pharmacol, 2022. 13: p. 931089. [CrossRef]
- Pio, M.G. , et al., Curating the gnomAD database: Report of novel variants in the thyrogobulin gene using in silico bioinformatics algorithms. Mol Cell Endocrinol, 2021. 534: p. 111359. [CrossRef]
- Turesky, R.J. , et al., Effect of Cytochrome P450 Reductase Deficiency on 2-Amino-9H-pyrido[2,3-b]indole Metabolism and DNA Adduct Formation in Liver and Extrahepatic Tissues of Mice. Chem Res Toxicol, 2015. 28(12): p. 2400-10. [CrossRef]
- Flück, C.E. , et al., Deletion of P399_E401 in NADPH cytochrome P450 oxidoreductase results in partial mixed oxidase deficiency. Biochem Biophys Res Commun, 2011. 412(4): p. 572-7. [CrossRef]
- Fluck, C.E. , Congenital Adrenal Hyperplasia Owing to 17 alpha-Hydroxylase/17,20 Lyase and P450 Oxidoreductase Deficiencies, in Hormonal and Genetic Basis of Sexual Differentiation Disorders and Hot Topics in Endocrinology, M.I. New and J.L. Simpson, Editors. 2011, Springer-Verlag Berlin: Berlin. p. 3-5. [CrossRef]
- Xiao, X. , et al., Functional POR A503V is associated with the risk of bladder cancer in a Chinese population. Sci Rep, 2015. 5: p. 11751. [CrossRef]
- Camats, N., C. E. Fluck, and L. Audi, Oligogenic Origin of Differences of Sex Development in Humans. Int J Mol Sci, 2020. 21(5). [CrossRef]
- Fluck, C.E., C. Nicolo, and A.V. Pandey, Clinical, structural and functional implications of mutations and polymorphisms in human NADPH P450 oxidoreductase. Fundam Clin Pharmacol, 2007. 21(4): p. 399-410. [CrossRef]
- David, A. and M.J. Sternberg, The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J Mol Biol, 2015. 427(17): p. 2886-98. [CrossRef]
- Petukh, M., T. G. Kucukkal, and E. Alexov, On human disease-causing amino acid variants: statistical study of sequence and structural patterns. Hum Mutat, 2015. 36(5): p. 524-534. [CrossRef]
- Iijima, S., A. Ohishi, and T. Ohzeki, Cytochrome P450 oxidoreductase deficiency with Antley-Bixler syndrome: steroidogenic capacities. J Pediatr Endocrinol Metab, 2009. 22(5): p. 469-75. [CrossRef]
- Hosoe, J. , et al., Structural Basis and Genotype-Phenotype Correlations of INSR Mutations Causing Severe Insulin Resistance. Diabetes, 2017. 66(10): p. 2713-2723. [CrossRef]
- Lim, W.A., D. C. Farruggio, and R.T. Sauer, Structural and energetic consequences of disruptive mutations in a protein core. Biochemistry, 1992. 31(17): p. 4324-33. [CrossRef]
- de Beer, T.A. , et al., Amino acid changes in disease-associated variants differ radically from variants observed in the 1000 genomes project dataset. PLoS Comput Biol, 2013. 9(12): p. e1003382. [CrossRef]
- Vacic, V. , et al., Disease-associated mutations disrupt functionally important regions of intrinsic protein disorder. PLoS Comput Biol, 2012. 8(10): p. e1002709. [CrossRef]
- Oh, J. , et al., A Case of Antley-Bixler Syndrome With a Novel Likely Pathogenic Variant (c.529G>C) in the POR Gene. Ann Lab Med, 2017. 37(6): p. 559-562. [CrossRef]
- Scott, R.R. , et al., Apparent manifesting heterozygosity in P450 oxidoreductase deficiency and its effect on coexisting 21-hydroxylase deficiency. J Clin Endocrinol Metab, 2007. 92(6): p. 2318-22. [CrossRef]
- Yamaguchi, Y. , et al., Topological changes in the CYP3A4 active site probed with phenyldiazene: effect of interaction with NADPH-cytochrome P450 reductase and cytochrome b5 and of site-directed mutagenesis. Drug Metab Dispos, 2004. 32(1): p. 155-61. [CrossRef]
- Si, S. , et al., Impact of single nucleotide polymorphisms on P450 oxidoreductase and peroxisome proliferator-activated receptor alpha on tacrolimus pharmacokinetics in renal transplant recipients. Pharmacogenomics J, 2019. 19(1): p. 42-52. [CrossRef]
- Goswami, C., A. Chattopadhyay, and E.Y. Chuang, Rare variants: data types and analysis strategies. Ann Transl Med, 2021. 9(12): p. 961. [CrossRef]
- Zhang, H. , et al., Polymorphic variants of cytochrome P450 2B6 (CYP2B6.4-CYP2B6.9) exhibit altered rates of metabolism for bupropion and efavirenz: a charge- reversal mutation in the K139E variant (CYP2B6.8) impairs formation of a functional cytochrome p450-reductase complex. J Pharmacol Exp Ther, 2011. 338(3): p. 803-9. [CrossRef]
- Tomkova, M. , et al., Identification of six novel P450 oxidoreductase missense variants in Ashkenazi and Moroccan Jewish populations. Pharmacogenomics, 2012. 13(5): p. 543-54. [CrossRef]
- Miller, W.L. , et al., P450 oxidoreductase deficiency. Lancet, 2004. 364(9446): p. 1663. [CrossRef]
- Hershkovitz, E. et al., Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J Clin Endocrinol Metab, 2008. 93(9): p. 3584-8. [CrossRef]
- Flück, C.E. and A.V. Pandey, Clinical and biochemical consequences of p450 oxidoreductase deficiency. Endocr Dev, 2011. 20: p. 63-79. [CrossRef]
Figure 1.
Comparison between in silico tools and experimental data from the literature. To assess the accuracy of the tools, we used known disease-causing and benign variants of human POR as control samples. Our analysis involved comparing the predictions of the tools with the experimental data. In the figure, the prediction of the tools is to be interpreted as red squares for disease-causing variants, while benign variants are denoted by green squares. The variants in red were found in patients with POR deficiency. Created with BioRender.com.
Figure 1.
Comparison between in silico tools and experimental data from the literature. To assess the accuracy of the tools, we used known disease-causing and benign variants of human POR as control samples. Our analysis involved comparing the predictions of the tools with the experimental data. In the figure, the prediction of the tools is to be interpreted as red squares for disease-causing variants, while benign variants are denoted by green squares. The variants in red were found in patients with POR deficiency. Created with BioRender.com.
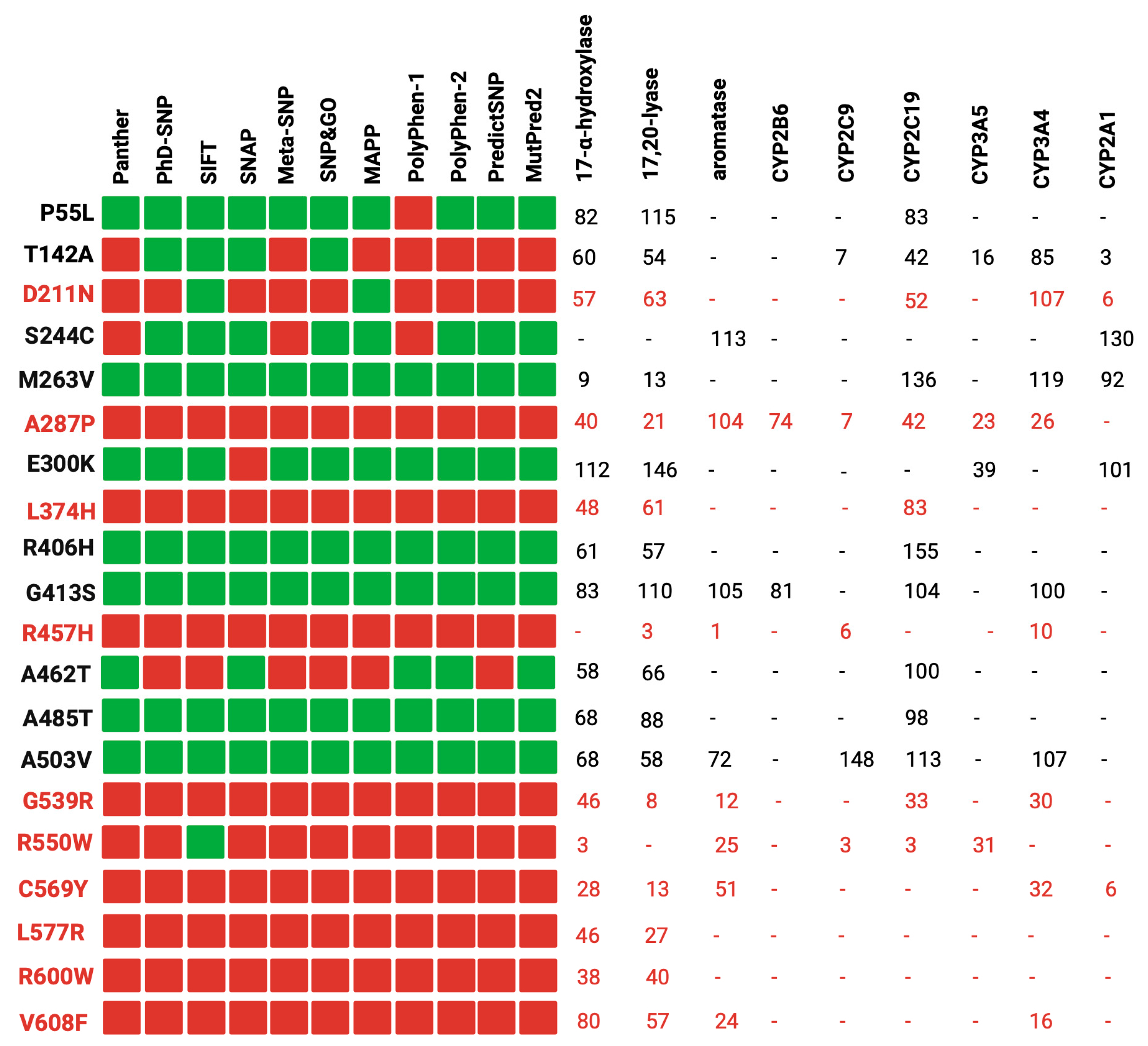
Figure 2.
Receiver Operating Characteristic (ROC) curve illustrating the performance of prediction tools in distinguishing between pathogenic and benign variants. The Area Under the Curve (AUC) values reflect the tools' accuracy in classifying variants, aiding in the assessment of their pathogenic potential. Data was plotted using MATLAB (MathWorks, Natick, MA).
Figure 2.
Receiver Operating Characteristic (ROC) curve illustrating the performance of prediction tools in distinguishing between pathogenic and benign variants. The Area Under the Curve (AUC) values reflect the tools' accuracy in classifying variants, aiding in the assessment of their pathogenic potential. Data was plotted using MATLAB (MathWorks, Natick, MA).
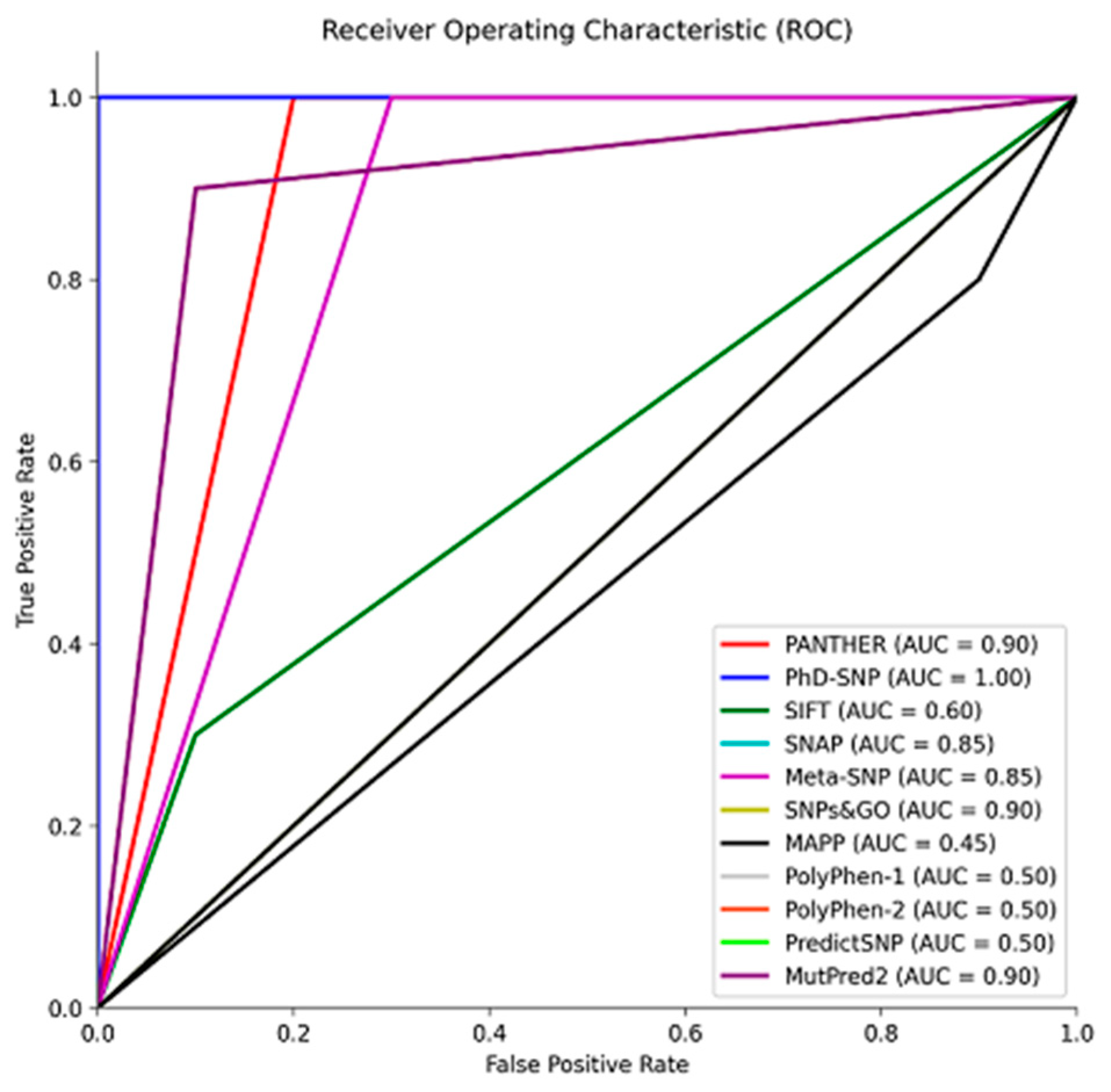
Figure 3.
Workflow outlining the curation process of 1,419 variants initially extracted from the gnomAD databases, focusing on the subsequent analysis of 450 missense variants. Notably, in silico tools highlighted in red were excluded from the study, as they showed suboptimal performance in the analysis. Created with BioRender.com.
Figure 3.
Workflow outlining the curation process of 1,419 variants initially extracted from the gnomAD databases, focusing on the subsequent analysis of 450 missense variants. Notably, in silico tools highlighted in red were excluded from the study, as they showed suboptimal performance in the analysis. Created with BioRender.com.

Figure 4.
Novel diseases causing variants of human POR. A comprehensive analysis was conducted on approximately 450 missense variants obtained from the Genome Aggregation Database (gnomAD). Utilizing distinct in silico prediction tools, we assessed the potential pathogenicity of these variants. A filtering process was applied to focus exclusively on variants classified as disease causing. Intriguingly, this rigorous selection yielded 64 variants characterized by remarkably low allele frequencies, with no prior description within the scientific literature. Created with BioRender.com.
Figure 4.
Novel diseases causing variants of human POR. A comprehensive analysis was conducted on approximately 450 missense variants obtained from the Genome Aggregation Database (gnomAD). Utilizing distinct in silico prediction tools, we assessed the potential pathogenicity of these variants. A filtering process was applied to focus exclusively on variants classified as disease causing. Intriguingly, this rigorous selection yielded 64 variants characterized by remarkably low allele frequencies, with no prior description within the scientific literature. Created with BioRender.com.
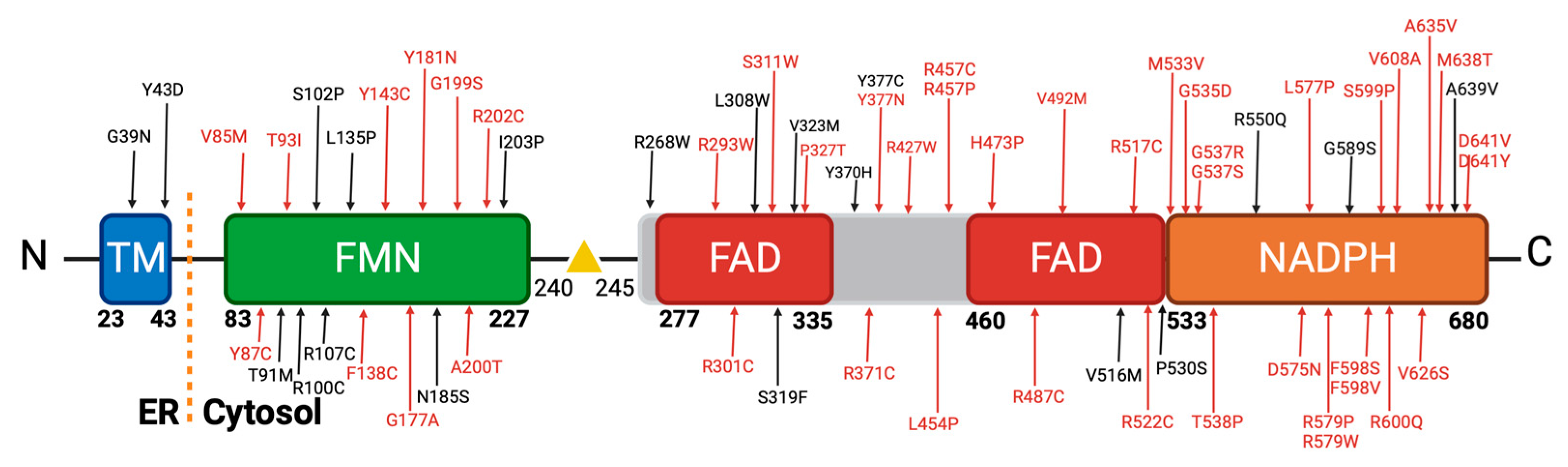
Figure 5.
Urea denaturation assay to assess protein stability. Flavin co-factor release was measured through fluorescence, with POR WT showing a gradual release with increasing urea concentration. In contrast, variants R268W and L577P exhibited rapid increases in fluorescence at low urea concentrations, indicating reduced stability. The fluorescence of released FMN and FAD was measured excitation at 450 nm, and emission at 535 nm to determine the Flavin content. Data was plotted using MATLAB (MathWorks, Natick, MA).
Figure 5.
Urea denaturation assay to assess protein stability. Flavin co-factor release was measured through fluorescence, with POR WT showing a gradual release with increasing urea concentration. In contrast, variants R268W and L577P exhibited rapid increases in fluorescence at low urea concentrations, indicating reduced stability. The fluorescence of released FMN and FAD was measured excitation at 450 nm, and emission at 535 nm to determine the Flavin content. Data was plotted using MATLAB (MathWorks, Natick, MA).
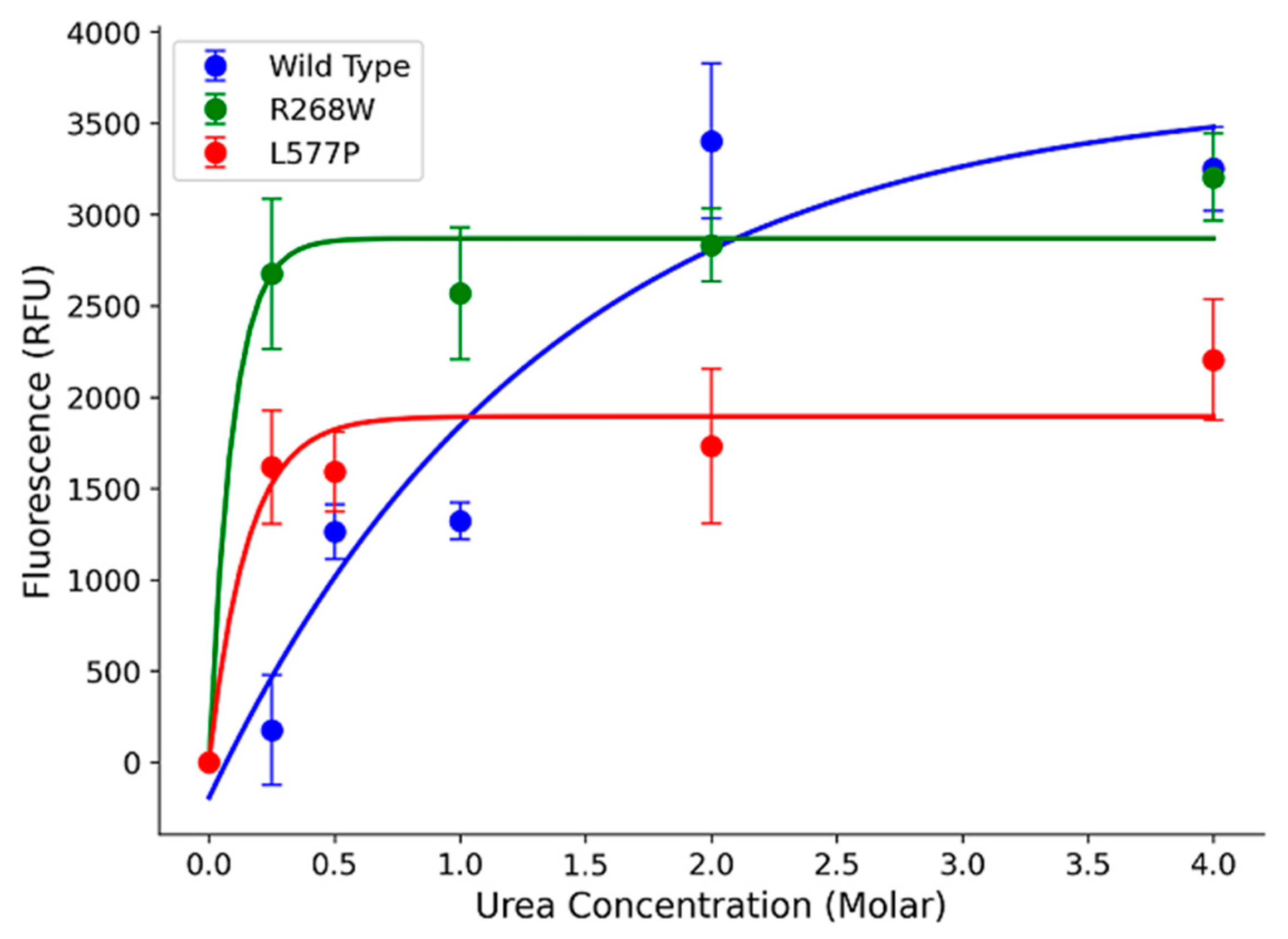
Figure 6.
Kinetics assays using Cytochrome c as a substrate. POR variants R268W and L577P exhibited varying effects on enzymatic activity compared to WT POR. R268W retained 113% of WT activity, while L577P displayed a more substantial reduction with only 51% activity. Data was plotted using MATLAB (MathWorks, Natick, MA).
Figure 6.
Kinetics assays using Cytochrome c as a substrate. POR variants R268W and L577P exhibited varying effects on enzymatic activity compared to WT POR. R268W retained 113% of WT activity, while L577P displayed a more substantial reduction with only 51% activity. Data was plotted using MATLAB (MathWorks, Natick, MA).
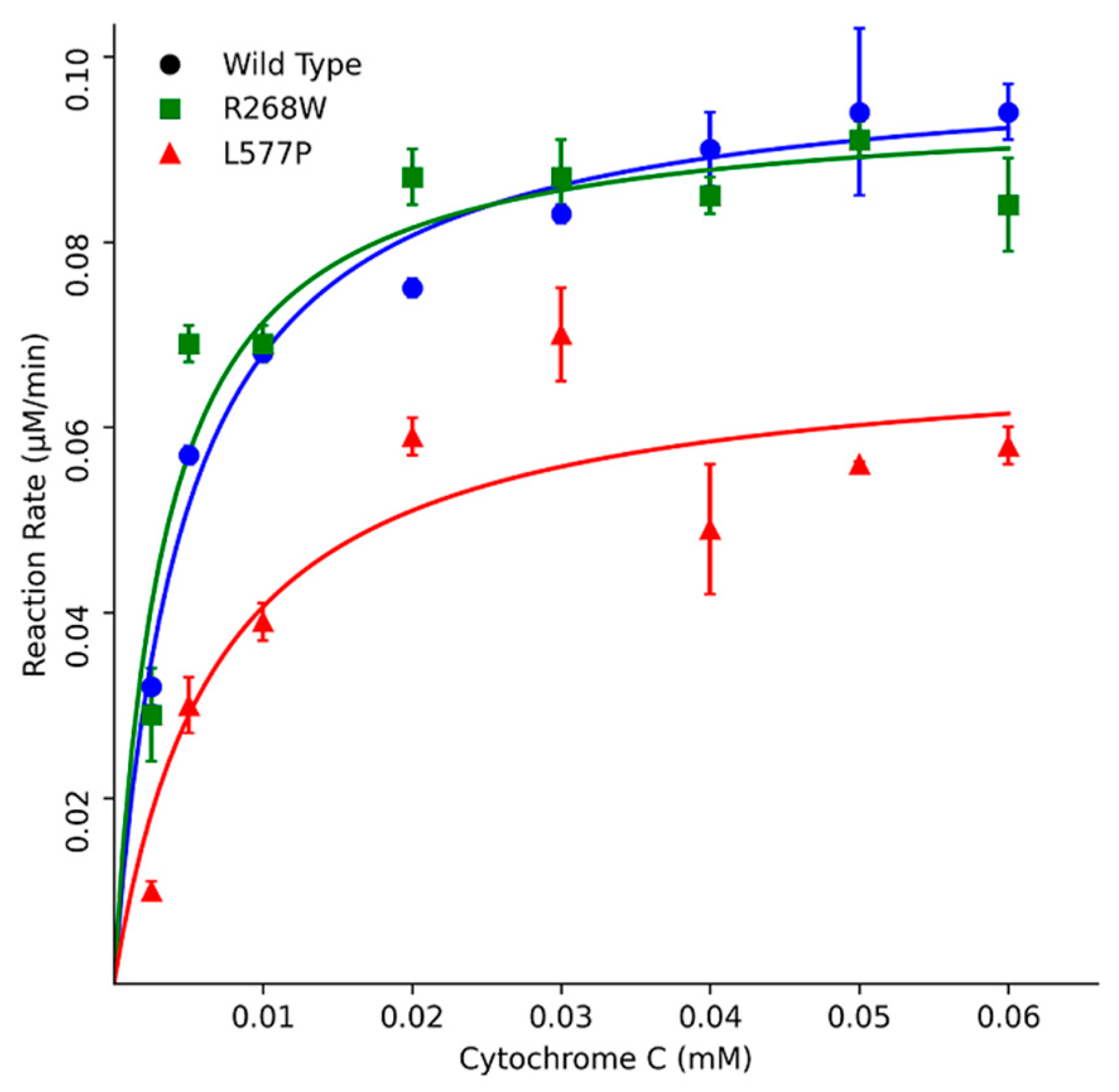
Figure 7.
CYP3A4, CYP3A5, CYP2C9, and CYP2C19 activities promoted by WT, L577P R268W POR. The activities promoted by the WT POR were set as a hundred percent, and results are shown as a percentage of WT activity. (a) In CYP3A4 assays POR variants L577P and R268W showed 51% and 62% of WT activity while (b) In CYP3A5 assays POR variants L577P and R268W showed 63% and 33% of WT activity and (c) In CYP2C9 assays POR variants L577P and R268W showed 52% and 10% of WT activity (d) In CYP2C9 assays POR variants L577P and R268W showed 30% and 15 % of WT.
Figure 7.
CYP3A4, CYP3A5, CYP2C9, and CYP2C19 activities promoted by WT, L577P R268W POR. The activities promoted by the WT POR were set as a hundred percent, and results are shown as a percentage of WT activity. (a) In CYP3A4 assays POR variants L577P and R268W showed 51% and 62% of WT activity while (b) In CYP3A5 assays POR variants L577P and R268W showed 63% and 33% of WT activity and (c) In CYP2C9 assays POR variants L577P and R268W showed 52% and 10% of WT activity (d) In CYP2C9 assays POR variants L577P and R268W showed 30% and 15 % of WT.
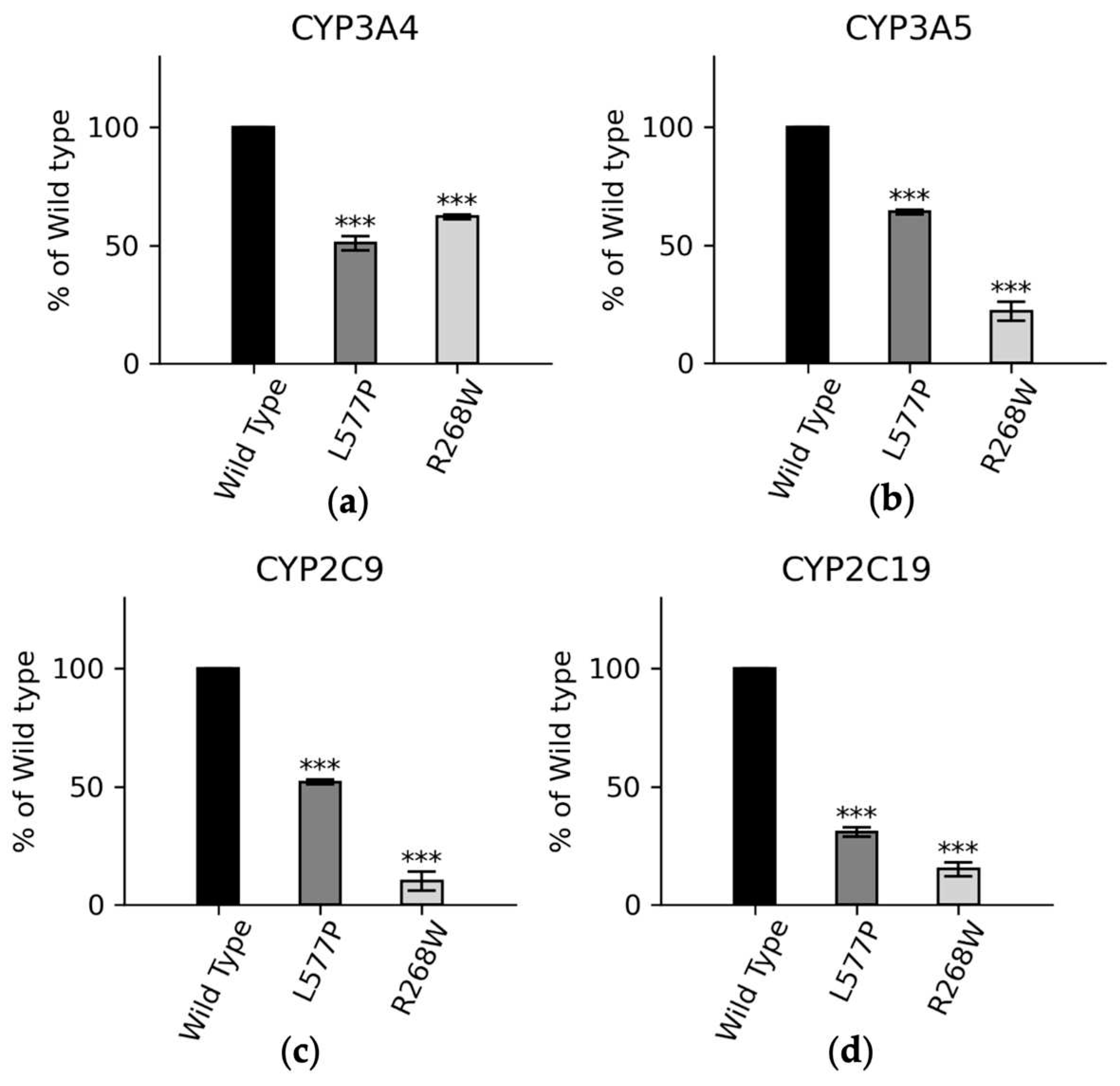

Table 1.
Kinetics parameters using the substrate model Cytochrome c.
| Wild Type | R268W | L5770 | |
|---|---|---|---|
| Vmax (μM/min) | 78 | 75 | 55 |
| KM ((μM) | 4.6 | 3.9 | 6.4 |
| Vmax/KM | 17 | 19 | 8.6 |
| % | 100 | 113 | 51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



