Submitted:
10 November 2023
Posted:
10 November 2023
You are already at the latest version
Abstract
Soluble chaperones residing in the endoplasmic reticulum (ER) play vitally important roles in folding and quality control of newly synthesized proteins that transiently pass through the ER en route to their final destinations. These soluble residents of the ER are themselves endowed with an ER retrieval signal that enables the cell to bring the escaped residents back from the Golgi. Here, by using purified proteins, we showed that Nicotiana tabacum phytaspase, a plant aspartate-specific protease, introduces two breaks at the C-terminus of the N. tabacum ER resident calreticulin-3. These cleavages resulted in removal of either a dipeptide or a hexapeptide from the C-terminus of calreticulin-3 encompassing part or all of the ER retrieval signal. Consistently, expression of the calreticulin-3 derivative mimicking the phytaspase cleavage product in Nicotiana benthamiana cells demonstrated loss of the ER accumulation of the protein. Notably, upon its escape from the ER, calreticulin-3 was further processed by an unknown protease(s) to generate the free N-terminal (N) domain of calreticulin-3, which was ultimately secreted into the apoplast. Our study thus identified a specific proteolytic enzyme capable of precise detachment of the ER retrieval signal from a plant ER resident protein, with implications for the further fate of the escaped resident.
Keywords:
calreticulin-3
; endoplasmic reticulum
; phytaspase
; plant cell
; protein trafficking
; proteolytic processing
; retrieval signal
1. Introduction
Proteins are targeted to various specific locations within or outside the cell with the aid of localization signals – usually rather short amino acid sequences – within protein molecules. This notion holds true for soluble proteins which are ‘residents’ of the endoplasmic reticulum (ER). To fulfill their tasks related to protein folding and quality control within the ER, such proteins are usually equipped with two kinds of localization signals, each with a terminal location within the protein molecule. The first one is the N-terminal signal peptide (SP) that allows targeting of the newly synthesized protein to the ER and which is cleaved off in the process of protein translocation by signal peptidase [1,2]. The second one is the C-terminal ‘ER retrieval signal’ (usually KDEL in animals, HDEL in plants and yeast) [3,4,5] that is used to bring back these soluble chaperones when they move from the ER towards the Golgi apparatus, together with thousands of other proteins that are delivered through the ER to their final destinations. The ER retrieval signal within the escaped residents is recognized by the receptor ERD2 located at the cis-Golgi [6,7,8,9,10] to mediate retrograde transportation of these proteins to the ER.
In contrast to the cleavable SP, the ER retrieval signal is thought to be non-cleavable, or at least, no proteolytic enzyme, in any kingdom of life, is known that is able to specifically detach the ER retrieval signal from the protein. In this regard, it is surprising that for decades soluble chaperones of the ER have also been observed at other locations, both within and outside the cell (see, for example, [11,12]). Furthermore, experimental evidence suggested that these ‘escaped residents’ fulfill important tasks at these non-canonical locations [13,14,15,16]. An essential unresolved question was how these ER residents managed to escape despite the existence of an elaborated retrieval mechanism.
Here we present in vitro evidence that phytaspase (plant aspartate-specific protease), a member of the plant subtilisin-like protease (subtilase) family, is capable of precise detachment of the ER retrieval signal from N. tabacum calreticulin-3 (NtCRT3), an ER resident protein. Two features distinguish phytaspases from other members of the extensive family of plant subtilases. First, phytaspases possess rarely occurring aspartate cleavage specificity. Like animal apoptotic proteases, caspases [17], phytaspases hydrolyze the polypeptide chain after an Asp residue preceded by a characteristic, though degenerate, three amino acid-long motif [18,19,20]. This type of substrate recognition makes phytaspases highly selective enzymes that introduce one or two breaks in a limited number of protein substrates. Phytaspases were initially identified as plant proteases involved in the execution of programmed cell death induced by various biotic and abiotic insults [18,21,22,23], but were subsequently shown also to be instrumental in specific processing of precursors of the plant peptide hormones systemin and phytosulfokine to generate biologically active peptides [24,25].
Another peculiar feature of phytaspases is their dynamic localization. In healthy plant cells, the phytaspase precursor equipped with a SP is targeted to the ER, is constitutively and autocatalytically processed and thus activated on its way out of the cell, and the mature enzyme is then secreted into the apoplast [18]. In response to a number of biotic and abiotic stresses, phytaspase is internalized back into the cell by means of clathrin-mediated endocytosis to participate in execution of programmed cell death [18,26,27].
By using the BioID approach, the in vivo interaction of N. tabacum phytaspase (NtPhyt) with N. benthamiana CRT3 and several other ER resident chaperones was documented [28]. Notably, CRT3 from NtPhyt-overproducing plant cells displayed a slightly increased electrophoretic mobility (relatively to CRT3 from control cells), implying the existence of some kind of post-translational modification [28]. Here we demonstrate that NtCRT3 is not only the partner, but also the substrate of NtPhyt. We also show that the NtCRT3 derivative mimicking the phytaspase cleavage product escapes from the ER, is further processed to generate the N-domain of CRT3, the latter being secreted into the apoplast.
2. Results
2.1. Phytaspase-mediated Truncation of N. tabacum CRT3: Evidence with Purified Proteins
We assessed the hypothesis that CRT3 is a substrate of tobacco phytaspase using purified proteins. N. tabacum CRT3 bearing a His-tag instead of the native signal peptide was produced in E. coli cells and purified by means of Ni-NTA agarose affinity chromatography. N. tabacum phytaspase (NtPhyt) with a C-terminal His-tag was transiently overproduced in N. benthamiana leaves by agroinfiltration and isolated from the apoplast by affinity chromatography. Treatment of the recombinant CTR3 with NtPhyt resulted in a small, yet clearly detectable, shift in the electrophoretic mobility of CRT3 (Figure 1A), similar to that observed previously with extracts from NtPhyt-producing versus non-producing N. benthamiana leaves in our BioID experiments [28].
To verify that the truncation of CRT3 occurred due to phytaspase-specific proteolytic activity, purified recombinant NtPhyt was pre-treated with either Ac-VEID-CHO or Ac-DEVD-CHO peptide aldehyde prior to addition of the enzyme to CRT3. The former is the known inhibitor of NtPhyt [18], whereas the latter is a specific inhibitor of animal caspases-3 that is known not to interfere with the NtPhyt proteolytic activity [18,21]. As evidenced from Figure 1B, truncation of CRT3 was completely abolished by the NtPhyt-specific inhibitor, while pre-treatment with the control peptide aldehyde had no effect.
These results suggest that NtPhyt is capable of cleaving a short peptide from one of the CRT3 termini.
2.2. Identification of the NtPhyt Cleavage Site(s) in CRT3
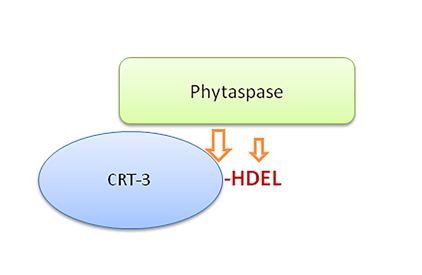
To identify the NtPhyt cleavage sites, CRT3 bands from phytaspase-treated and non-treated samples shown in Figure 1A were excised, treated with LysC protease, and the resultant peptides were characterized by MALDI TOF mass spectrometry analyses. While the majority of peptides, including the N-terminal, of both NtPhyt-treated and non-treated CRT3 displayed identical peaks in the mass spectra (Figure S1), the C-termini turned out to be different. In the untreated sample, the intact C-terminal semi-LysC peptide with the predicted m/z 2064.9 was readily identified, whereas in the NtPhyt-treated sample a shorter semi-LysC peptide with m/z 1292.6 was observed instead (Figure 1C; for the original MS data, see Figure S1). Identity of the characteristic peptides was confirmed by MS/MS fragmentation (Figure S2). These findings indicate that NtPhyt has cleaved off the C-terminal hexapeptide of CRT3, and that the hydrolysis occurred in accord with the NtPhyt specificity (that is, after an aspartate residue, D) in the DYMD420 motif.
To further substantiate this conclusion, we substituted D420 by E (glutamate) in CRT3 with the expectation of obtaining a phytaspase-resistant version of CRT3 (NtPhyt is a strictly D-specific protease). Upon isolation from bacterial overproducing strains, wild type and D420E mutant proteins were treated with NtPhyt and reaction products were compared by SDS gel-electrophoresis. Indeed, the D420E mutation abolished the characteristic mobility shift observed with the wild type CRT3 (Figure 2A). However, the CRT3 band in the D420E phytaspase-treated sample still appeared to migrate somewhat faster than that in the non-treated counterpart (Figure 2A, compare lanes 3 and 4). To clarify this issue, mass spectrometry analysis of LysC peptides generated from phytaspase-treated and untreated CRT3 D420E bands was performed. The intact C-terminal semi-LysC peptide of CRT3 D420E encompassing the D420E mutation (m/z 2078.9) was identified in the untreated sample (Figure 2B). However, in the NtPhyt-treated sample a shorter semi-LysC peptide (m/z 1836.8) was observed instead, corresponding to elimination of just two C-terminal amino acid residues of CRT3 D420E (Figure 2B; for the complete spectra of peptides from both samples, see Figures S1, S2). The cleavage therefore has occurred after the DYHD424 motif, and again with the characteristic NtPhyt specificity.
We therefore concluded that there are two rather similar NtPhyt cleavage sites (DYMD420 and DYHD424) positioned in very close proximity to the CRT3 C-terminus. NtPhyt-mediated cleavage appears to proceed more efficiently at D420 than at D424, as we failed to detect the elimination of only the two C-terminal residues unless the D420 was mutated. Also consistent with this interpretation, cleavage of the mutant protein at D424 was incomplete (Figure S1), in contrast to complete hydrolysis of the wild type protein at D420 (Figure 1A and Figure 2A) under similar conditions of hydrolysis.
2.3. Effect of NtCRT3 Truncation on the Protein Localization and Integrity
CRT3, like other soluble residents of the ER, contains an ‘ER retrieval signal’ represented by the HDEL sequence, at the very C-terminus of the molecule (Figure 3A). This signal allows the cell to return CRT3 that had escaped from the ER back from the Golgi, a feature actually making this soluble protein an ER resident. Phytaspase-mediated cleavage to detach of either a half or the entire ER retrieval signal would thus be predicted to alter at least the subcellular localization of CRT3. To test this assumption, we constructed two fluorescent NtCRT3 derivatives. The EGFP_CRT3_wt construct contained the full-length CRT3 protein with the EGFP tag inserted next to the N-terminal signal peptide in order not to interfere with the terminal localization signals in CRT3. The EGFP_CRT3_ΔC6 construct had a similarly positioned EGFP tag, yet it lacked 6 C-terminal amino acid residues of CRT3 encompassing the ER retrieval signal, thus mimicking the phytaspase cleavage product of CRT3 (see Figure 3A for a schematic representation). Both proteins were transiently produced in N. benthamiana leaves by agroinfiltration and their localization was compared using confocal fluorescence microscopy. The EGFP_CRT3_wt protein displayed characteristic ER localization, as expected (Figure 3B). Production of EGFP_CRT3_ΔC6 resulted in much lower fluorescence intensity (relatively to the wt construct) in the infiltrated leaves, with fluorescence shifting from the ER to the cell periphery (Figure 3C).
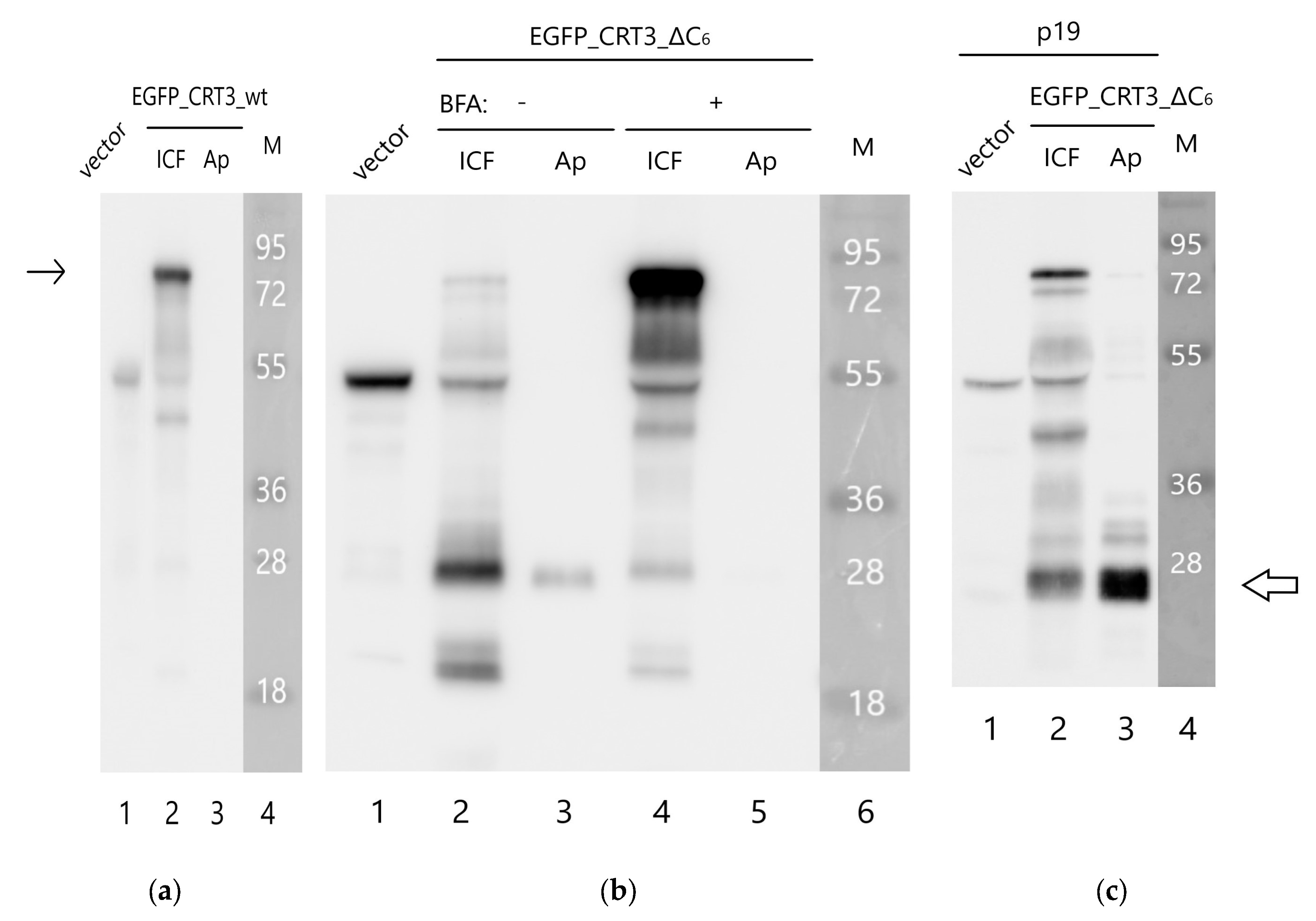
To characterize the behavior of these proteins in a greater detail, we separated proteins from the infiltrated leaves into intra- and extracellular (apoplastic) fractions and analyzed them by Western blotting using an anti-EGFP antibody. EGFP_CRT3_wt protein (calculated mass ~80 kDa) was largely intact and detected in the intracellular fraction (Figure 3D, left). Contrary to that, EGFP_CRT3_ΔC6 protein was degraded, forming a ~30 kDa product (Figure 3D, right), which is approximately the size of free EGFP. This product was observed in the extracellular fraction (Figure 3D).
To learn what happened to the CRT3 moiety of the fusion proteins, analogous Western blots were analyzed using anti-CRT3 antibodies. In accord with the previous analysis, the EGFP_CRT3_wt protein was found to be predominantly intact, although some degradation products were also observed, and it was localized intracellularly (Figure 4A). For the EGFP_CRT3_ΔC6 protein, although a minor amount of the intact (~80 kDa) protein was detected on the overexposed blot (Figure 4B, lane 2), the majority of this protein was degraded to produce a ~26 kDa protein fragment as the most intense band recognized by anti-CRT3 antibodies. Interestingly, this CRT3 fragment was found in the extracellular fraction as well (Figure 4B, lane 3). Also of note, endogenous CRT3 observed in the ‘vector only’ lane (Figure 4B, lane 1) was fairly intact.
The above results suggest that NtCRT3 lacking the ER retrieval signal is further processed within the plant cell, and a protein fragment is secreted into the apoplast. To obtain insights into where such an additional processing could occur, the effect of brefeldin A (BFA), a known inhibitor of protein transport from the ER to the Golgi complex [29], on EGFP_CRT3_ΔC6 behavior was studied. Treatment of the EGFP_CRT3_ΔC6-producing N. benthamiana leaves with BFA markedly impaired the formation of the 26 kDa CRT3 fragment and abolished its secretion, with the concomitant increase in the amount of the full-length protein (Figure 4B, lanes 4 and 5). These results are consistent with fragmentation of EGFP_CRT3_ΔC6 occurring within the plant cell upon protein release from the ER.
Finally, to characterize the secretion-competent 26 kDa CRT3 fragment, the EGFP_CRT3_ΔC6 construct was expressed in N. benthamiana leaves in the presence of p19 RNA silencing suppressor to maximize production. The rather abundant 26 kDa CRT3 fragment thus formed was isolated from the apoplastic fraction (Figure 4C) and, after trypsin or Glu-C digestion, identified by mass spectrometry analysis as the N-terminal domain of CRT3, in agreement with its recognition by anti-CRT3 antibodies (see Figure S3 for sequence coverage). Judging by the pattern of peptides identified, CRT3 cleavage both in the vicinity of the N-P domain border and at the N-terminus of the N-domain by an unknown protease(s) was required to generate this CRT3 fragment.
3. Discussion
In this study, we provide the in vitro evidence that NtPhyt efficiently and precisely removes the ER retrieval signal of NtCRT3 by introducing two breaks at the very C-terminus of the NtCRT3 polypeptide. We also demonstrate that the NtCRT3 derivative mimicking the phytaspase cleavage product loses the capacity for accumulation in the ER, is further processed by an unknown protease(s), and the stable N-domain of NtCRT3 thus formed is ultimately secreted into the apoplast. From the ‘functional’ point of view, the need for two similar and closely spaced phytaspase cleavage sites at the C-terminus of the NtCRT3 molecule is not straightforward. It would seem that one would be sufficient to destroy the ER retrieval signal completely. On the other hand, C-terminal decapeptides of, for example, Arabidopsis thaliana and N. tabacum CRT3 are identical in amino acid sequence, suggesting that such duplication is functionally important.
Another question is clearly of greater importance. Perhaps unsurprisingly, phytaspase-mediated trimming of CRT3 seems not to occur (or, at least, is inefficient) in N. benthamiana cells under the normal conditions. Otherwise the ER would lose its very important chaperone. It would be therefore essential to learn whether stress-inducing or some other conditions could trigger phytaspase-mediated cleavage of the ER retrieval signal from NtCRT3. An interaction of phytaspase with CRT3 was documented in vivo using the BioID approach [28]. In this regard, consideration of the complex trafficking pattern of phytaspase could suggest mechanisms for triggering the relocalization of NtCRT3.
In principle, there are two opportunities for phytaspase to encounter CRT3: either during export of phytaspase out of the cell (for the newly synthesized enzyme), or upon the stress-induced retrograde transportation (for the pre-existing phytaspase). For the first scenario, a caveat is that plant subtilases are believed to acquire activity not until they reach the Golgi apparatus [30,31,32,33], and proteolytically active phytaspase should therefore not be present in the ER. On the other hand, the ER retrieval signal endows soluble residents of the ER with the ability to shuttle between the ER and the Golgi [10]. It thus can be envisaged that proteolytic processing of CRT3 by phytaspase could occur in the Golgi, where phytaspase is already proteolytically competent, thus disabling CRT3 retrieval back to the ER. This regulatory option, if it turns out to be true, is achievable due to the difference (possibly, the advantage) of the retrieval mechanism for accumulation of the soluble residents in the ER over a simple retention mechanism.
The second scenario would predict that phytaspase, upon its stress-induced re-import, can process CRT3. The internalized phytaspase is known to retain its proteolytic activity [27], however the precise trafficking routes of the enzyme upon re-entering the cell are not known. Perhaps, some kind of a precedent could be provided by the plant protein toxin ricin and related toxins, which can reach the Golgi apparatus and the ER after being endocytosed [34]. Deciphering of the phytaspase trafficking pathways in stressed plant cells may thus contribute to understanding of the phytaspase-CRT3 interaction.
The formation and release into the extracellular space of the N-domain derived from CRT3_ΔC6 also deserves some comment. If phytaspase-mediated processing of CRT3 triggers formation of such a CRT3 fragment in vivo, a function of the secreted N-domain to serve as a signaling molecule can be envisioned. Notably, the N-domain of human calreticulin found in culture supernatants of human cells and named vasostatin was demonstrated to inhibit proliferation of endothelial cells and to suppress angiogenesis in vivo [35].
In conclusion, our study seems to provide an illustration related to the long considered problem: how can soluble residents of the ER equipped with the ER retrieval signal escape to new locations [14]? The presence of a highly specific dedicated protease (phytaspase) capable of precise detachment of the retrieval signal from the ER resident protein offers plants an intriguing opportunity.
4. Materials and Methods
4.1. Plant Growth Conditions
N. benthamiana plants were grown at 25o C in soil in a controlled environment under a 16/8 h day/night cycle. For transient protein production, Agrobacterium tumefaciens C58C1 cells transformed with the respective plasmid were infiltrated into leaves of six-week-old plants using a blunt syringe. Typically, leaves were harvested 2 days post-infiltration (dpi).
4.2. Plasmid Construction
Construction of the pSL1180_NtCRT3 and pET_His_LF CRT3 plasmids encoding full-length NtCRT3 or NtCRT3 with the SP substituted with the His tag, respectively, was described previously [28]. The D420E NtCRT3 mutant was created by site-directed mutagenesis via PCR on the pET_His_LF CRT3 plasmid using CRT3_Pst700_dir and CRT3_D420E_Sac_rev primers (Table S1), with the subsequent substitution of the original PstI-SacI DNA fragment of the pET_His_LF CRT3 plasmid with the mutant one.
For in planta production of EGFP_CRT3_wt, the SP-encoding DNA fragment (c. 110 bp) was amplified on the pSL1180_NtCRT3 using CRT_Kpn_Nco_dir and CRT_SP_Sal_rev primers, inserted into the SmaI site of the pUC19 vector, and further transferred, as the BamHI-SalI DNA fragment (c. 110 bp), into the pBluescript II vector to generate the pBS_SP plasmid. The EGFP gene (c. 750 bp) was amplified on the pEGFP-N1 vector (Clontech) using EGFP_Sal_dir and EGFP_Apa_rev primers and inserted downstream of and in frame with the SP-encoding sequence between the SalI and ApaI sites of the pBS_SP plasmid to generate the pBS_SP_EGFP plasmid. Finally, the SP-EGFP-encoding NcoI-ApaI DNA fragment (c. 850 bp) from the latter plasmid and the ApaI-SacI DNA fragment (c. 1200 bp) from the pET_His_LF CRT3 plasmid encoding CRT3 without the SP were ligated between the NcoI and SacI sites of the pCambia1300EX binary vector [27] to generate the pCambia_SP_EGFP_CRT3_wt plasmid. For production of EGFP_CRT3_ΔC6 protein, the mutated CRT3-encoding DNA fragment (c. 1200 bp) was obtained by PCR on the pET_His_LF CRT3 plasmid with CRT_LF_Apa_Nde_dir and CRT_1-420_Sac_rev primers. Substitution of the wild type CRT3-encoding ApaI-SacI DNA fragment within the pCambia_SP_EGFP_CRT3_wt plasmid for the mutant one generated the pCambia_SP_EGFP_CRT3_ΔC6 plasmid.
To create the NtPhyt-His fusion, the NtPhyt cDNA from the previously described NtPhyt-GST construct [18] within the pLEX7000 backbone [24] was PCR amplified using pLH_seq_dir and NtPhyt-His_rev primers and inserted between the XhoI and SacI sites of the pLEX7000 expression vector downstream of the dual CaMV 35S promoter to generate the pLEX_NtPhyt_His plasmid.
The identity of all constructs was confirmed by sequence analysis.
4.3. Production, Purification, and Treatment of the His-tagged NtCRT3 with NtPhyt
Production of the His-tagged NtCRT3 in E. coli BL21(DE3) cells and purification of the recombinant protein using metal chelate affinity chromatography on Ni-NTA agarose (Qiagen) was performed as described previously [28]. The protein was eluted from the beads with 100 mM EDTA in B1 buffer (50 mM MES, pH 5.7, 50 mM NaCl, 2mM DTT, 0.1% Tween 20, 5% glycerol) and stored at -28℃. The purification procedure was also performed with the lysate of E. coli BL21 (DE3) cells transformed with the empty pET28a(+) vector (Novagen), to use the obtained eluate as a control.
Purified His-CRT3 (wt or D420E), in 0.5-1 μg aliquots, was mixed with 15 ng of NtPhyt-His (see section 4.5 below for NtPhyt-His isolation) in the total volume of 40 μL of B1 buffer containing 500 mM NaCl, chymostatin (6 μg/mL), AEBSF (25 μg/mL), and E-64 (2 μg/mL) proteinase inhibitors. The reaction mixture was incubated at 28 oC for 1h. To terminate the reaction, 10 μL of 5xSample buffer (250 mM Tris-HCl, pH 6.8, 5 mM EDTA, 10% SDS, 30% glycerol, 10 mM DTT) was added to the reaction tube, and the sample was immediately heated for 5 min at 98℃. As a control, an equal aliquot of His-CRT3 without the addition of NtPhyt was incubated identically. The protein samples were analyzed by 10% SDS-PAGE. The gel was run at low voltage (150V) until the 35 kDa protein marker reached the edge of the gel, and stained with Coomassi Blue R-250.
To test the effect of peptide aldehyde inhibitors, NtPhyt-His (in approx. 6 ng aliquots) was pre-incubated with 200 μM of Ac-VEID-CHO, Ac-DEVD-CHO (from stock solutions in DMSO), or with the equivalent amount of DMSO (mock control) in the total volume of 20 μL of B1 buffer containing 500 mM NaCl at 28℃ for 30 min. Then 0.5-1 μg of His-CRT3 in 20 μL of the same buffer was added to each pre-incubated NtPhyt aliquot, and the samples were processed as described above.
4.4. MS Analyses
The pieces of about 2 mm3 of the Coomassie stained protein containing gel were destained twice with 20 mM NH4HCO3, pH 7.5, 40% aqueous acetonitrile solution; dehydrated with 200 μL of 100% acetonitrile and rehydrated with 5 μL of the digestion solution containing 15 µg/mL sequencing grade Lys-C or Glu-C protease or trypsin (Promega) in 20 mM NH4HCO3, pH 7.5 aqueous solution. Digestion was carried out at 37 °C for 3 h. The resulting peptides were extracted with 7 μL of 0.5% TFA, 50% acetonitrile solution. A 1-μL aliquot of in-gel digest extract was mixed with 0.5 μL of 2,5-dihydroxybenzoic acid solution (40 mg/mL in 30% acetonitrile, 0.5% TFA) and left to dry on the stainless steel target plate. MALDI-TOF MS analysis was performed on an UltrafleXеtreme MALDI-TOF-TOF mass spectrometer (Bruker Daltonik, Germany). The MH+ molecular ions were measured in a reflector mode; the accuracy of monoisotopic mass peak measurement was within 100 ppm. Spectra of fragmentation were obtained in a LIFT mode, the accuracy of daughter ions measurement was within 1 Da range. Mass-spectra were processed with the FlexAnalysis 3.2 software (Bruker Daltonik, Germany). Protein identification was carried out by MS+MS/MS ion search with the use of Mascot software version 2.3.02 (Matrix Science) through the Home protein database. One missed cleavage, Met oxidation and Cys-propionamide were permitted. Mass spectra of fragmentation of C-terminal peptides were labeled manually.
4.5. Transient Expression in N. benthamiana and Protein Fractionation
The obtained binary plasmids encoding EGFP-NtCRT3 derivatives were introduced into A. tumefaciens C58C1 cells, and transformed agrobacteria were infiltrated into N. benthamiana leaves. Agrobacteria carrying the empty vector were used as a control. Where indicated, leaves were infiltrated with BFA (15 μg/mL) 1 day after agroinfiltration of the EGFP_CRT3 construct and harvested 24 h later. At 2 dpi, leaves were examined by confocal fluorescence microscopy. In parallel, leaves were subject to protein fractionation, essentially as described previously [27], with some modifications. Briefly, the leaf discs were weighed and vacuum infiltrated with water. Apoplastic washes were obtained by low-speed (2 000 x g) centrifugation at 4oC for 10 min, the liquid being collected directly into 5xSample Buffer placed at the bottom of a centrifuge tube. After the separation, the residual leaf material was frozen in liquid nitrogen and disrupted in Minilys homogenizer (Bertin Instruments) using 1.6 mm ceramic beads by three 10 s bursts. After the addition of 5xSample Buffer, homogenates were boiled for 5 min, debris was eliminated by 10 min centrifugation at 10 000 x g, and the protein samples were fractionated by SDS-polyacrylamide gel electrophoresis. Separated proteins were electrophoretically transferred onto polyvinylidene fluoride (PVDF) membrane. Western blot analyses were performed using mouse monoclonal anti-EGFP 3A9 antibody [36] and rabbit polyclonal anti-NtCRT3 antibodies. Rabbit anti-NtCRT3 antibodies were raised against the synthetic NtCRT3 peptide GSMYTDNDILPPRKIK by GenScript USA. Chemiluminescence detection was performed with ECL Western Lightning Plus reagent (PerkinElmer) using the ChemiDoc Imaging System (Bio-Rad Laboratories).
To enhance protein yield when desirable (e.g., for production of NtPhyt-His or for the experiment shown in Figure 4C), transformed agrobacteria were mixed with an equal amount of Agrobacterium cells bearing the p19 suppressor of silencing prior to infiltration. Leaves were harvested at 3 dpi.
To isolate proteolytically active NtPhyt-His, apoplastic washes were obtained from approx. 3 g of leaves by vacuum infiltration with MES100 buffer (20 mM MES, pH 5.7, 100 mM NaCl), as described [37]. Apoplastic liquid was then 10 x fold diluted with 50 mM Na-phosphate buffer, pH 8.0, supplemented with aprotinin (2 μg/mL), leupeptin (6 μg/mL), AEBSF (25 μg/mL), and chymostatin (6 μg/mL) proteinase inhibitors, and applied onto Ni-NTA agarose (~ 200 μL suspension) pre-washed with the same buffer. After incubation with rotation at 4 oC for 1.5 h, the beads were separated by centrifugation at 2000 x g for 10 min, washed with Na-phosphate buffer, pH 8.0, containing 1 M NaCl and 10% glycerol (10 x 1 mL), and finally with 1 mL of Na-phosphate buffer. NtPhyt-His was then eluted from the beads with 20 mM MES, pH 6.5, 100 mM EDTA (3 x 100 μL). The eluate was transferred into B1 buffer containing 50 mM NaCl or into 0.1 M HEPES, pH 8.0 buffer using a centrifugal concentrator with 50,000 molecular weight cut-off (Amicon Ultra-4) and stored at -28 oC. Phytaspase proteolytic activity was evaluated using fluorogenic peptide substrate Ac-VEID-AFC, as described [38].
4.6. Confocal Fluorescence Microscopy
Confocal microscopy was performed using a Nikon C2plus confocal microscope on a Nikon Eclipse Ti inverted stand (Nikon, Japan) equipped with VC PlanApo 60x NA1.2 water immersion lens. A 486 nm laser was used for excitation of EGFP. Stacks of optical sections of the apical half of the epidermal cells were acquired at 0.8 μm z-step. The obtained data are presented as maximum intensity projection images.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Mass spectra of Lys-C peptides originated from CRT3; Figure S2: MS/MS fragmentation spectra of the characteristic C-terminal semi-LysC peptides of CRT3; Figure S3: Sequence coverage of the apoplastic 26 kDa CRT3 fragment with mass spectrometry-identified peptides. Table S1: List of the primers used in this study.
Author Contributions
Conceptualization, A.B.V. and N.V.C.; Supervision, A.B.V. and N.V.C.; Investigation, A.D.T., A.A.P., M.V.S., S.A.G., R.A.G. and N.V.C.; Formal Analysis, A.D.T., A.A.P., M.V.S., S.A.G., R.A.G., N.V.C. and A.B.V.; Writing—Original Draft Preparation, A.B.V.; Writing—Review and Editing, A.D.T., A.A.P., M.V.S. and N.V.C.; funding acquisition, A.B.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Russian Science Foundation, grant numbers 19-14-00010 and 22-14-00071.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Acknowledgments
We thank C.U.T. Hellen for critically reading the manuscript. MALDI MS facility and CLSM became available to us in the framework of the Moscow State University Development Programs PNG 5.13 and PNR 5.13.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Paetzel, M.; Karla, A.; Strynadka, N.C.; Dalbey, R.E. Signal peptidases. Chem Rev. 2002, 102, 4549–4580. [Google Scholar] [CrossRef] [PubMed]
- Liaci, A.M.; Steigenberger, B.; Telles de Souza, P.C.; Tamara, S.; Gröllers-Mulderij, M.; Ogrissek, P.; Marrink, S.J.; Scheltema, R.A.; Förster, F. Structure of the human signal peptidase complex reveals the determinants for signal peptide cleavage. Mol. Cell 2021, 81, 3934–3948. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Pelham, H.R. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem. Sci. 1990, 15, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Denecke, J.; De Rycke, R.; Botterman, J. Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 1992, 11, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Pelham, H.R. A human homologue of the yeast HDEL receptor. Nature 1990, 348, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M. .J.; Pelham, H.R. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell 1992, 68, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.I.; Gal, S.; Newman, T.C.; Raikhel, NV. The Arabidopsis endoplasmic reticulum retention receptor functions in yeast. Proc. Natl. Acad. Sci. U S A 1993, 90, 11433–11437. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao-Hui, C.; Batoux, M.; Nekrasov, V.; Roux, M.; Chinchilla, D.; Zipfel, C.; Jones, J.D. Specific ER quality control components required for biogenesis of the plant innate immune receptor EFR. Proc. Natl. Acad. Sci. U S A 2009, 106, 15973–15978. [Google Scholar] [CrossRef]
- Alvim, J.C.; Bolt, R.M.; An, J.; Kamisugi, Y.; Cuming, A.; Silva-Alvim, F.A.L.; Concha, J.O.; daSilva, L.L.P.; Hu, M.; Hirsz, D.; Denecke, J. The K/HDEL receptor does not recycle but instead acts as a Golgi-gatekeeper. Nat. Commun. 2023, 14, 1612. [Google Scholar] [CrossRef]
- Arosa, F.A.; de Jesus, O.; Porto, G.; Carmo, A.M.; de Sousa, M. Calreticulin is expressed on the cell surface of activated human peripheral blood T lymphocytes in association with major histocompatibility complex class I molecules. J. Biol. Chem. 1999, 274, 16917–16922. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Holaska, J.M.; Black, B.E.; Love, D.C.; Hanover, J.A.; Leszyk, J.; Paschal, B.M. Calreticulin is a receptor for nuclear export. J. Cell Biol. 2001, 152, 127–140. [Google Scholar] [CrossRef]
- Johnson, S.; Michalak, M.; Opas, M.; Eggleton, P. The ins and outs of calreticulin: from the ER lumen to the extracellular space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell, 2005, 123, 321-334. [CrossRef]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J., 2010, 24, 665–683. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; Chapman, K.T.; Nicholson, D.W. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [PubMed]
- Chichkova, N.V.; Shaw, J.; Galiullina, R.A.; Drury, G.E.; Tuzhikov, A.I.; Kim, S.H.; Kalkum, M.; Hong, T.B.; Gorshkova, E.N.; Torrance, L.; Vartapetian, A.B.; Taliansky, M. Phytaspase, a relocalisable cell death promoting plant protease with caspase specificity. EMBO J. 2010, 29, 1149–1161. [Google Scholar] [CrossRef]
- Galiullina, R.A.; Kasperkiewicz, P.; Chichkova, N.V.; Szalek, A.; Serebryakova, M.V.; Poreba, M.; Drag, M.; Vartapetian, A.B. Substrate specificity and possible heterologous targets of phytaspase, a plant cell death protease. J. Biol. Chem. 2015, 290, 24806–24815. [Google Scholar] [CrossRef]
- Reichardt, S.; Repper, D.; Tuzhikov, A.I.; Galiullina, R.A.; Planas-Marquès, M.; Chichkova, N.V.; Vartapetian, A.B.; Stintzi, A.; Schaller, A. The tomato subtilase family includes several cell death-related proteinases with caspase specificity. Sci. Rep. 2018, 8(1):10531. [CrossRef]
- Chichkova, N.V.; Kim, S.H.; Titova, E.S.; Kalkum, M.; Morozov, V.S.; Rubtsov, Y.P.; Kalinina, N.O.; Taliansky, M.E.; Vartapetian, A.B. A plant caspase-like protease activated during the hypersensitive response. Plant Cell 2004, 16, 157–171. [Google Scholar] [CrossRef]
- Vartapetian, A.B.; Tuzhikov, A.I.; Chichkova, N.V.; Taliansky, M.; Wolpert, T.J. A plant alternative to animal caspases: subtilisin-like proteases. Cell Death Differ. 2011, 18, 1289–1297. [Google Scholar] [CrossRef]
- Chichkova, N.V.; Tuzhikov, A.I.; Taliansky, M.; Vartapetian, A.B. Plant phytaspases and animal caspases: structurally unrelated death proteases with a common role and specificity. Physiol Plant. 2012, 145, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Beloshistov, R.E.; Dreizler, K.; Galiullina, R.A.; Tuzhikov, A.I.; Serebryakova, M.V.; Reichardt, S.; Shaw, J.; Taliansky, M.E.; Pfannstiel, J.; Chichkova, N.V.; Stintzi, A.; Schaller, A.; Vartapetian, A.B. Phytaspase-mediated precursor processing and maturation of the wound hormone systemin. New Phytol. 2018, 218, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.; Piepho, H.P.; Stintzi, A.; Schaller, A. Peptide signaling for drought-induced tomato flower drop. Science 2020, 367, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Trusova, S.V.; Golyshev, S.A.; Chichkova, N.V.; Vartapetian, A.B. Sometimes they come back: endocytosis provides localization dynamics of a subtilase in cells committed to cell death. J. Exp. Bot. 2019, 70, 2003–2007. [Google Scholar] [CrossRef]
- Trusova, S.V.; Teplova, A.D.; Golyshev, S.A.; Galiullina, R.A.; Morozova, E.A.; Chichkova, N.V.; Vartapetian, A.B. Clathrin-mediated endocytosis delivers proteolytically active phytaspases into plant cells. Front. Plant Sci. 2019, 10:873. [CrossRef]
- Teplova, A.D.; Serebryakova, M.V.; Galiullina, R.A.; Chichkova, N.V.; Vartapetian, A.B. Identification of phytaspase interactors via the proximity-dependent biotin-based identification approach. Int. J. Mol. Sci. 2021, 22(23):13123. [CrossRef]
- Nebenführ, A.; Ritzenthaler, C.; Robinson, D.G. Brefeldin A: deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002, 130, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Cedzich, A.; Huttenlocher, F.; Kuhn, B.M.; Pfannstiel, J.; Gabler, L.; Stintzi, A.; Schaller, A. The protease-associated domain and C-terminal extension are required for zymogen processing, sorting within the secretory pathway, and activity of tomato subtilase 3 (SlSBT3). J. Biol. Chem. 2009, 284, 14068–14078. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Leptihn, S.; Welz, M.; Schaller, A. Functional characterization of propeptides in plant subtilases as intramolecular chaperones and inhibitors of the mature protease. J. Biol. Chem. 2016, 291, 19449–19461. [Google Scholar] [CrossRef] [PubMed]
- Schaller, A.; Stintzi, A.; Rivas, S.; Serrano, I.; Chichkova, N.V.; Vartapetian, A.B.; Martínez, D.; Guiamét, J.J.; Sueldo, D.J.; van der Hoorn, R.A.L.; Ramírez, V.; Vera, P. From structure to function - a family portrait of plant subtilases. New Phytol. 2018, 218, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Stührwohldt, N.; Scholl, S.; Lang, L.; Katzenberger, J.; Schumacher, K.; Schaller, A. The biogenesis of CLEL peptides involves several processing events in consecutive compartments of the secretory pathway. Elife 2020, 9:e55580. [Google Scholar] [CrossRef]
- Lord, J.M.; Spooner, R.A. Ricin trafficking in plant and mammalian cells. Toxins 2011, 3, 787–801. [Google Scholar] [CrossRef]
- Pike, S.E.; Yao, L.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Teruya-Feldstein, J.; Wirth, P.; Gupta, G.; Tosato, G. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J. Exp. Med. 1998, 188, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Sukhacheva, E.A.; Evstafieva, A.G.; Fateeva, T.V.; Shakulov, V.R.; Efimova, N.A.; Karapetian, R.N.; Rubtsov, Y.P.; Vartapetian, A.B. Sensing prothymosin alpha origin, mutations and conformation with monoclonal antibodies. J. Immunol. Methods 2002, 266, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Galiullina, R.A.; Dyugay, I.A.; Vartapetian, A.B.; Chichkova, N.V. Purification of phytaspases using the biotinylated peptide inhibitor. Methods Mol. Biol. 2023, in press. [Google Scholar]
- Galiullina, R.A.; Chichkova, N.V.; Safronov, G.G.; Vartapetian, A.B. Characterization of phytaspase proteolytic activity using fluorogenic peptide substrates Methods Mol. Biol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
NtCRT3 is the substrate of NtPhyt. (a) Treatment of isolated recombinant His-calreticulin-3 (CRT) with phytaspase (Phyt) caused a small increase in the electrophoretic mobility of CRT3 (arrows). Vector, protein fraction obtained upon Ni-NTA agarose affinity chromatography from E. coli cells transformed with empty vector. M, MW protein markers. (b) Hydrolysis was suppressed by Ac-VEID-CHO but not by Ac-DEVD-CHO, both inhibitors were used at a concentration of 200 μM. In (a) and (b), reaction products were fractionated by 10% SDS-gel electrophoresis and stained with Coomassie Blue. (c) Schematic representation of the phytaspase cleavage product of CRT3. Mass spectrometry (MS) analysis revealed characteristic semi-LysC peptides with m/z 2064.9 and 1292.6 derived from untreated and Phyt-treated CRT3 bands, respectively (arrows in (a)).
Figure 1.
NtCRT3 is the substrate of NtPhyt. (a) Treatment of isolated recombinant His-calreticulin-3 (CRT) with phytaspase (Phyt) caused a small increase in the electrophoretic mobility of CRT3 (arrows). Vector, protein fraction obtained upon Ni-NTA agarose affinity chromatography from E. coli cells transformed with empty vector. M, MW protein markers. (b) Hydrolysis was suppressed by Ac-VEID-CHO but not by Ac-DEVD-CHO, both inhibitors were used at a concentration of 200 μM. In (a) and (b), reaction products were fractionated by 10% SDS-gel electrophoresis and stained with Coomassie Blue. (c) Schematic representation of the phytaspase cleavage product of CRT3. Mass spectrometry (MS) analysis revealed characteristic semi-LysC peptides with m/z 2064.9 and 1292.6 derived from untreated and Phyt-treated CRT3 bands, respectively (arrows in (a)).
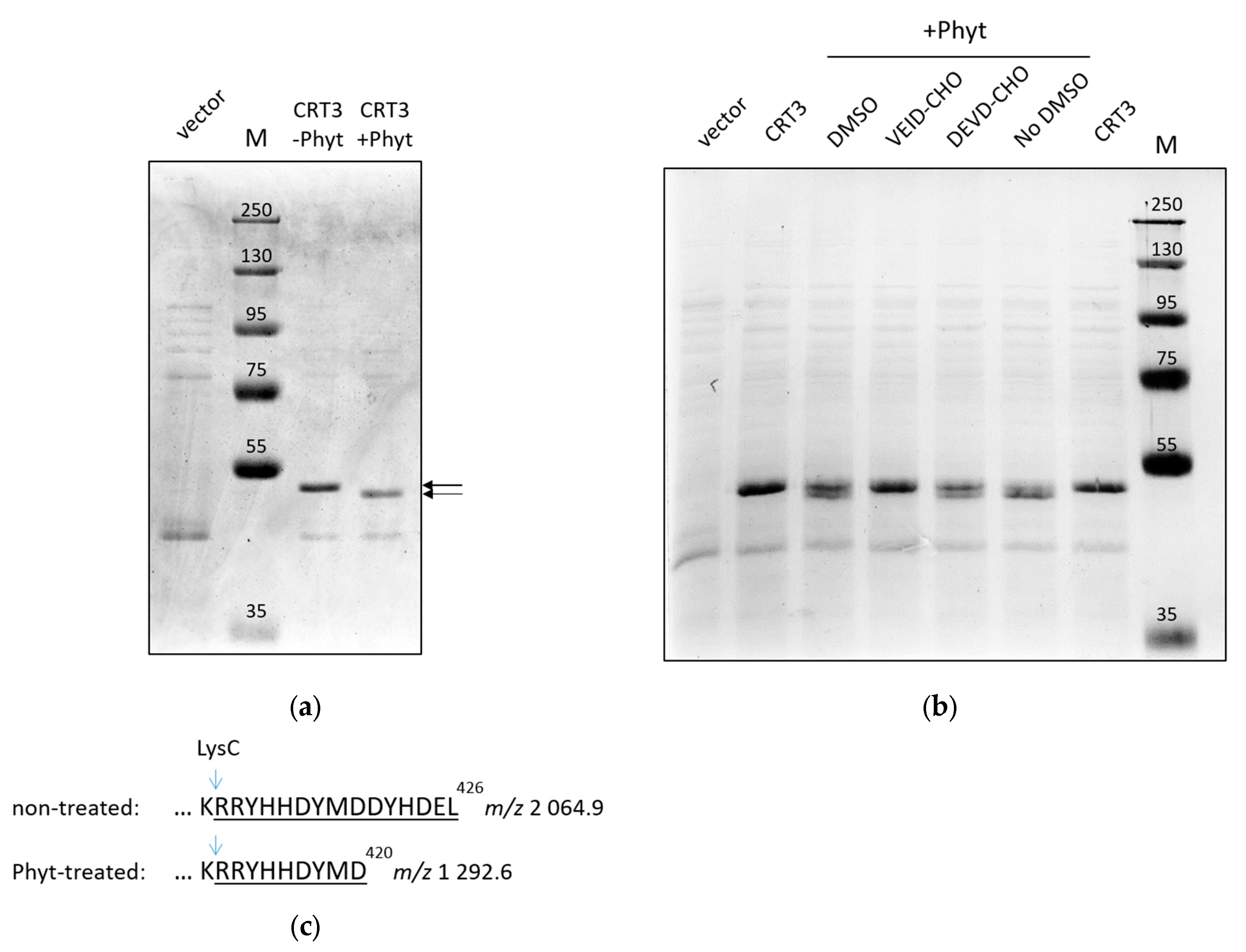
Figure 2.
Mutating D420 in CRT3 revealed the second Phyt cleavage site located within the ER retrieval signal (HDEL426). (a) Recombinant N-terminally His-tagged wild type (wt) and mutant (D420E) CRT3 were isolated from E. coli cells, treated with NtPhyt (+Phyt) or left untreated (-Phyt), and reaction products were fractionated by 10% SDS-gel electrophoresis and stained with Coomassie Blue. Note a slight mobility shift of the D420E mutant protein caused by NtPhyt treatment. Vector, an analogous protein fraction from E. coli cells transformed with empty vector. Positions of MW protein markers are indicated on the left. (b) Schematic representation of the phytaspase cleavage product of CRT3 D420E mutant. The mutation is shown in red. Characteristic semi-LysC peptides with m/z 2078.9 (untreated sample) and 1836.8 (Phyt-treated) were identified by MS analyses.
Figure 2.
Mutating D420 in CRT3 revealed the second Phyt cleavage site located within the ER retrieval signal (HDEL426). (a) Recombinant N-terminally His-tagged wild type (wt) and mutant (D420E) CRT3 were isolated from E. coli cells, treated with NtPhyt (+Phyt) or left untreated (-Phyt), and reaction products were fractionated by 10% SDS-gel electrophoresis and stained with Coomassie Blue. Note a slight mobility shift of the D420E mutant protein caused by NtPhyt treatment. Vector, an analogous protein fraction from E. coli cells transformed with empty vector. Positions of MW protein markers are indicated on the left. (b) Schematic representation of the phytaspase cleavage product of CRT3 D420E mutant. The mutation is shown in red. Characteristic semi-LysC peptides with m/z 2078.9 (untreated sample) and 1836.8 (Phyt-treated) were identified by MS analyses.
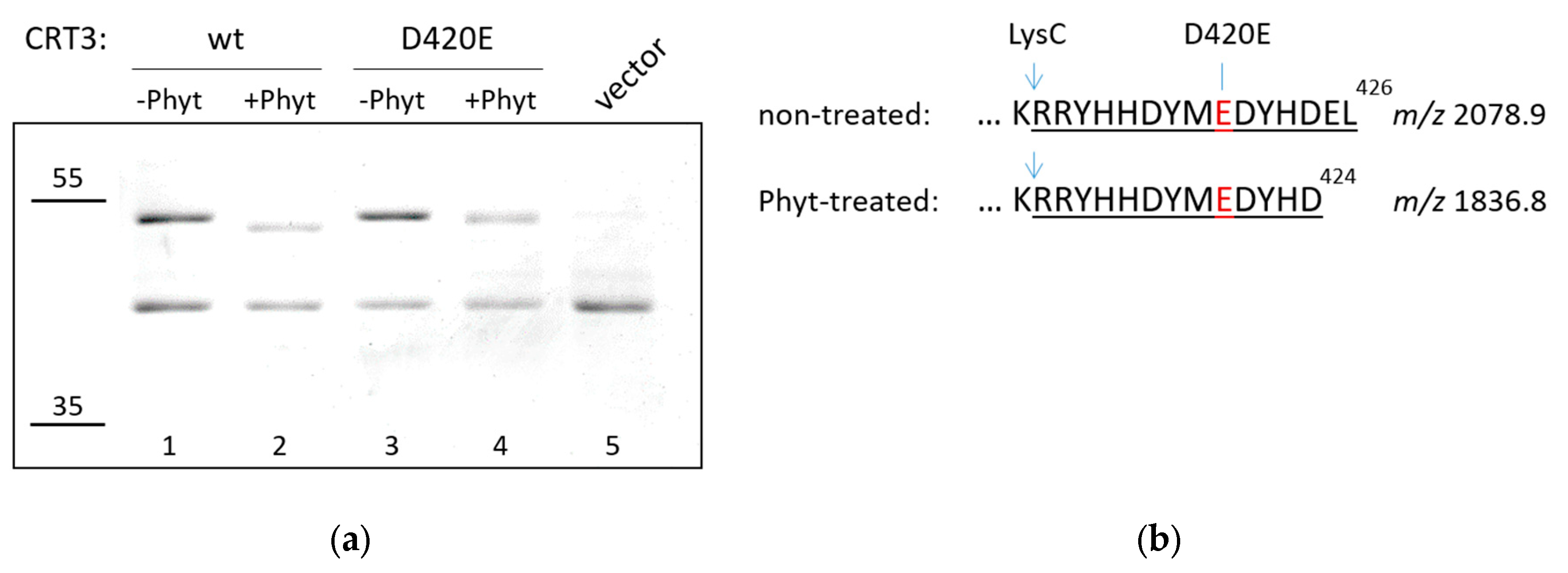
Figure 3.
EGFP-CRT3 derivative mimicking the major phytaspase cleavage product loses the ER accumulation and is further processed. (a) Schematic representation of NtCRT3 structure and features addressed in the current study. SP, signal peptide. Position of the insertion of the EGFP moiety next to the SP is indicated (for details, see Materials and Methods section). Phyt cleavage sites, as determined in Figure 1 and Figure 2, are shown with red arrows. The ER retrieval signal is marked with the red bottom bracket. Blue bottom bracket, position of the epitope at the boundary of N-domain (hatched) and P-domain recognized by the generated anti-CRT3 antibodies (see below). (b) and (c), confocal fluorescence microscopy images of N. benthamiana leaf epidermal cells transiently producing EGFP_CRT3_wt (b) or EGFP_CRT3_ΔC6 (c) proteins. A characteristic reticular localization of the wild type protein has shifted to the more peripheral localization for the truncated one. Images were obtained 2 days post infiltration (dpi) with agrobacteria carrying the respective plasmid. Equal exposure conditions were used in (b) and (c). (d) Western blot analysis with anti-EGFP antibody of the intracellular (ICF) and apoplastic (Ap) protein fractions obtained from equal weight amounts of leaves transiently producing EGFP_CRT3_wt or EGFP_CRT3_ΔC6, as indicated. Protein fractionation was performed at 2 dpi. Proteins were separated by 12% SDS-gel electrophoresis. M, MW protein markers.
Figure 3.
EGFP-CRT3 derivative mimicking the major phytaspase cleavage product loses the ER accumulation and is further processed. (a) Schematic representation of NtCRT3 structure and features addressed in the current study. SP, signal peptide. Position of the insertion of the EGFP moiety next to the SP is indicated (for details, see Materials and Methods section). Phyt cleavage sites, as determined in Figure 1 and Figure 2, are shown with red arrows. The ER retrieval signal is marked with the red bottom bracket. Blue bottom bracket, position of the epitope at the boundary of N-domain (hatched) and P-domain recognized by the generated anti-CRT3 antibodies (see below). (b) and (c), confocal fluorescence microscopy images of N. benthamiana leaf epidermal cells transiently producing EGFP_CRT3_wt (b) or EGFP_CRT3_ΔC6 (c) proteins. A characteristic reticular localization of the wild type protein has shifted to the more peripheral localization for the truncated one. Images were obtained 2 days post infiltration (dpi) with agrobacteria carrying the respective plasmid. Equal exposure conditions were used in (b) and (c). (d) Western blot analysis with anti-EGFP antibody of the intracellular (ICF) and apoplastic (Ap) protein fractions obtained from equal weight amounts of leaves transiently producing EGFP_CRT3_wt or EGFP_CRT3_ΔC6, as indicated. Protein fractionation was performed at 2 dpi. Proteins were separated by 12% SDS-gel electrophoresis. M, MW protein markers.
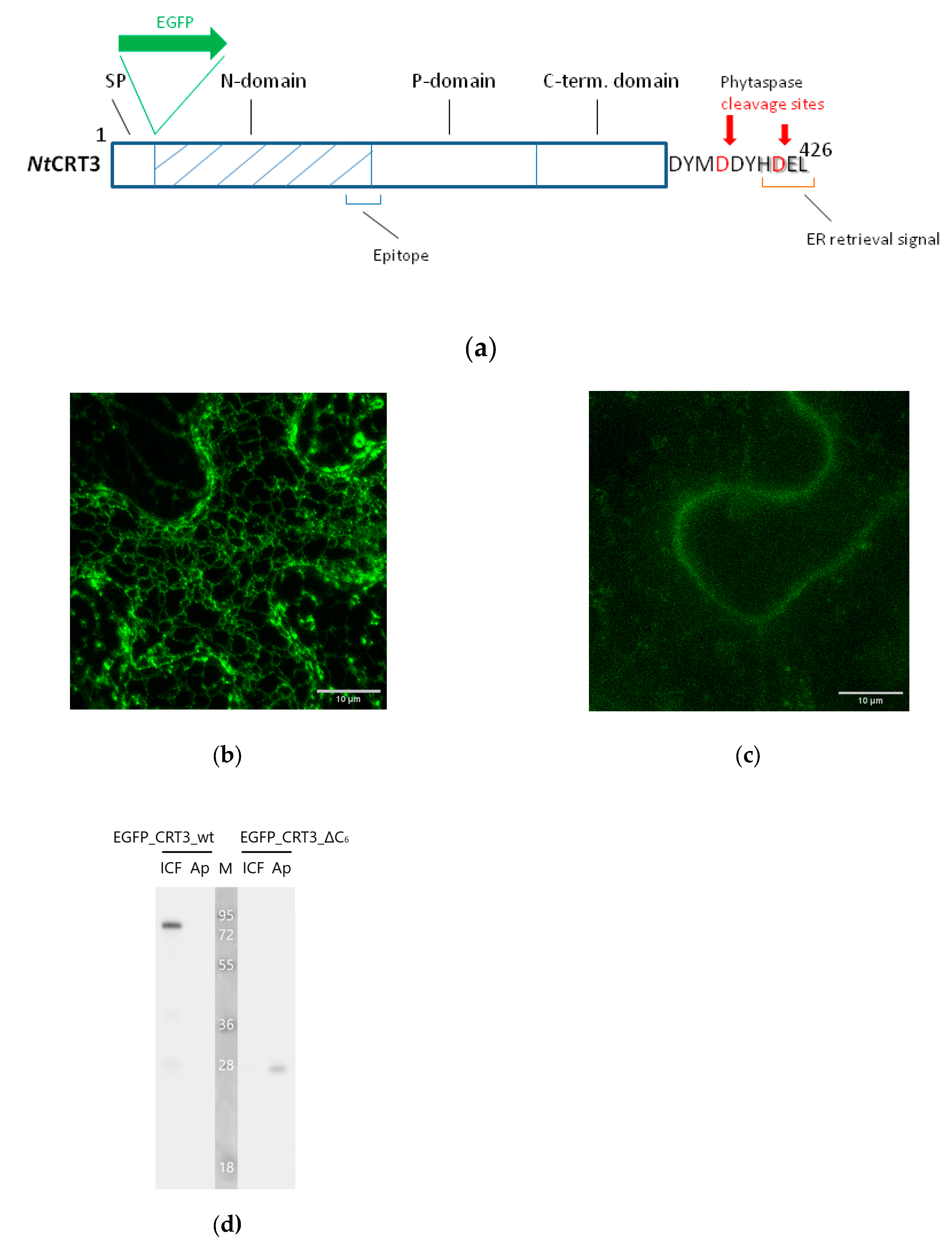
Figure 4.
Upon release from the ER, the EGFP_CRT3_ΔC6 protein is processed to generate a 26 kDa fragment capable of secretion into the apoplast. Western blot analyses with anti-CRT3 antibodies of the intracellular (ICF) and apoplastic (Ap) protein fractions from N. benthamiana leaves producing EGFP_CRT3_wt (a) or EGFP_CRT3_ΔC6 (b) and (c). Proteins were separated by 12% SDS-gel electrophoresis. Whereas the EGFP_CRT3_wt protein was largely intact and intracellular (arrow in (a)), the full-length EGFP_CRT3_ΔC6 protein was barely detectable even on overexposed blots due to its extensive degradation (b, lane 2). The major ~26 kDa CRT3 fragment thus formed was capable of being exported from the cell (lane 3 in (b) and (c)). Treatment of EGFP_CRT3_ΔC6-producing leaves with BFA (15 μg/mL) applied 1 dpi for 24 h markedly suppressed fragmentation of EGFP_CRT3_ΔC6 and secretion of the 26 kDa protein fragment (compare lanes 2 and 3 with lanes 4 and 5 in (b)). (c) To obtain sufficient material for identification of the 26 kDa CRT3 fragment, EGFP_CRT3_ΔC6 protein was produced in N. benthamiana leaves in the presence of the p19 suppressor of silencing for 3 dpi. From the apoplastic fraction, a gel band with electrophoretic mobility of the 26 kDa CRT3 fragment was excised (arrow in (c)), and its protein content was characterized by MS analysis (see Figure S3). In (a-c), vector, total protein from leaves infiltrated with agrobacteria carrying the empty vector. Note that intensities of the endogenous CRT3 bands (~50 kDa) in these lanes allow evaluation of relative exposure of each blot. M, MW protein markers. Data were reproducible over three independent experiments.
Figure 4.
Upon release from the ER, the EGFP_CRT3_ΔC6 protein is processed to generate a 26 kDa fragment capable of secretion into the apoplast. Western blot analyses with anti-CRT3 antibodies of the intracellular (ICF) and apoplastic (Ap) protein fractions from N. benthamiana leaves producing EGFP_CRT3_wt (a) or EGFP_CRT3_ΔC6 (b) and (c). Proteins were separated by 12% SDS-gel electrophoresis. Whereas the EGFP_CRT3_wt protein was largely intact and intracellular (arrow in (a)), the full-length EGFP_CRT3_ΔC6 protein was barely detectable even on overexposed blots due to its extensive degradation (b, lane 2). The major ~26 kDa CRT3 fragment thus formed was capable of being exported from the cell (lane 3 in (b) and (c)). Treatment of EGFP_CRT3_ΔC6-producing leaves with BFA (15 μg/mL) applied 1 dpi for 24 h markedly suppressed fragmentation of EGFP_CRT3_ΔC6 and secretion of the 26 kDa protein fragment (compare lanes 2 and 3 with lanes 4 and 5 in (b)). (c) To obtain sufficient material for identification of the 26 kDa CRT3 fragment, EGFP_CRT3_ΔC6 protein was produced in N. benthamiana leaves in the presence of the p19 suppressor of silencing for 3 dpi. From the apoplastic fraction, a gel band with electrophoretic mobility of the 26 kDa CRT3 fragment was excised (arrow in (c)), and its protein content was characterized by MS analysis (see Figure S3). In (a-c), vector, total protein from leaves infiltrated with agrobacteria carrying the empty vector. Note that intensities of the endogenous CRT3 bands (~50 kDa) in these lanes allow evaluation of relative exposure of each blot. M, MW protein markers. Data were reproducible over three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



