
Submitted:
10 November 2023
Posted:
13 November 2023
You are already at the latest version
Abstract
Newcastle Disease (ND) remains a critical disease affecting poultry in sub-Saharan Africa. In some countries, repeated outbreaks have a major impact on local economies and food security. Recently, we developed an adenovirus-vectored vaccine encoding the Fusion protein from an Ethiopian isolate of Newcastle Disease Virus (NDV). The adenoviral vector was designed and a manufacturing process developed in the context of the Livestock Vaccine Innovation Fund initia-tive funded by the International Development Research Centre (IDRC) of Canada. The industrial-ly-relevant recombinant vaccine technology platform is being transferred to the National Veteri-nary Institute (Ethiopia) for veterinary applications. Here, we demonstrate that the instillation of the adenoviral vector through the nasal cavity can confer protection to chickens against a lethal challenge with NDV. A manufacturing process using HEK293 suspension cells cultured in stirred-tank bioreactor for the vaccine production is proposed. Taking into consideration supply chain limitations, options for serum-free media selection have been evaluated. A streamlined downstream process including a filtration, an ultrafiltration and a concentration step was de-veloped. With high volumetric yields (infectious titers up to 5 x 109 TCID50/mL) in the culture su-pernatant, the final formulations were prepared at 1010 TCID50/mL, either in liquid or lyophi-lized forms. The liquid formulation was suitable and safe for mucosal vaccination and was sta-ble for 1 week at 37˚C. Both liquid and lyophilized formulations were stable after 6 months of storage at 4˚C. Overall, a manufacturing process for adenovirus vectored vaccine was developed and protective doses were determined using a convenient route of delivery. Formulation and storage conditions were established, and quality control protocols were implemented.
Keywords:
Newcastle Disease virus
; adenovirus vaccine
; vaccine manufacturing
; intranasal vaccination
; mucosal protection
; HEK293 suspension cells
; bioreactor production
; downstream processing
; veterinary vaccine production platform
1. Introduction
Newcastle Disease (ND) is still considered one of the most prevailing infectious diseases with a major negative impact on poultry farming in Sub-Saharan Africa. It economically impacts producers at different scales and causes deterioration of food security in many of these regions [1,2,3,4]. In Sub-Saharan Africa, millions of chickens are farmed, with the majority being kept in villages and rural settings. In these settings, ND can have devastating effects at the level of small producers [5,6]. Immunization with inactivated or live-attenuated ND vaccines is the current prevention method; however, there are critical limitations with the supply chain of pathogen-free embryonated eggs and other disadvantages, such as possible virus shedding leading to disease in non-vaccinated animals [7]. The pathogen-free chicken embryonated eggs are imported from countries outside Africa, which significantly increases the cost of the manufacturing process [5,8,9,10]. Additionally, most available vaccines are not genotype-specific and are being prepared using Newcastle Disease virus (NDV) strains isolated decades ago with possible limited protection. Cell culture technologies are an alternative option for more effective vaccine production. Such technologies can also be adapted to rapid responses to other pathogens in case of newly emerging threats.
Considering the economic impact of NDV infections and the nature of the current production methods of NDV vaccines, we recently developed a streamlined and efficient technology platform that uses the human adenovirus serotype 5 and HEK293 cells for the production of a virus-vectored vaccine against NDV infection. The adenovirus encodes the Fusion antigen from a recent local isolate of NDV from Ethiopia [11]. It was produced in stirred tank bioreactors, purified by ultracentrifugation, and delivered through intramuscular injection for the immunization of chickens, which were fully protected against a lethal challenge with NDV. This viral vector was selected due to its extensive use in vaccination (including against COVID-19 for human populations), its “self-adjuvanting” properties and well-controlled and robust manufacturing process [12,13,14]. The system has a demonstrated safety; it supports high viral yields at relatively low production costs, and is of easy implementation for a veterinary product [15,16,17,18,19,20].
In this work, we present a further improved upstream process and develop a new downstream process for manufacturing the adenovirus-vectored vaccine. This resulted in a clarified product that was obtained mainly through filtration and ultrafiltration steps, being formulated in a liquid formulation that demonstrated remarkable stability. Stability assessments included thermally stressed storage conditions or long-term storage under standard conditions. Additionally, new formulations to sustain a lyophilization process were developed and further optimized. For the first time, mucosal immunization through the nasal cavity of chickens with the adenoviral vector encoding the F antigen from NDV was evaluated in one immunization and challenge experiment, in which different doses were assessed. Protection against a heterologous challenge was also evaluated.
2. Materials and Methods
2.1. Cells lines and culture media
HEK293A adherent cells were used for the Median Tissue Culture Infectious Dose (TCID50) assay. For this assay, HEK 293A cells were maintained in cell culture dishes in a humidified incubator at 5% CO2 and 370C in Dulbecco’s Modified Eagles Medium (DMEM) (Wisent, Canada), supplemented with 10% Fetal Bovine Serum (FBS) (Gibco, USA) without antibiotics. Cells were passaged twice a week and detached at confluence with Trypsin (Millipore Sigma, Canada). After centrifugation at 400g for 5 min, the cells were resuspended in fresh medium and seeded at a 1:10 dilution.
The HEK293SF cell line is derived from HEK293 cells, which were adapted for culture in suspension and serum-free medium. HEK293SF (clone 293SF-3F6) suspension cells were derived from the corresponding GMP master cell bank [21,22]. These cells were used for adenoviral vector production in bioreactors at a scale of 1 or 3L. HEK293SF cells were grown either in disposable polycarbonate vented cap shake flasks (TriForest Enterprises, Irvine, CA, USA) or bioreactors. For maintenance, cells were passaged twice per week by diluting to 2.5 x 105 viable cells per mL in fresh medium. They were grown either in HyClone HyCell TransFx-H medium (Cytiva, Logan, UT, USA) or Xell HEK-GM medium (Xell AG, USA). Both are chemically defined, animal-component-free and protein-free media, with no antibiotics added. HyCell TransFx-H was supplemented with 6 mM Glutamax (Gibco, USA) and 0.1% Kolliphor poloxamer 188 (Millipore Sigma, Canada). When grown in HEK-GM, it was supplemented with 6 mM GlutaMAX™ (Gibco, USA). Cell growth and viability were monitored by determining live cell density using 0.2% trypan blue exclusion dye (Thermo Fisher, Canada) in a Vi-CELL-XR Cell Viability Analyzer (Beckman Coulter, Canada).
2.2. Cell growth and virus production at small scale
Cell growth of HEK293SF cells was conducted in batch and fed-batch mode in HycellTransFx-H (Cytiva, Logan, UT, USA) and HEK-GM (Xell AG, Germany), initially using 250 mL shake flasks (TriForest Enterprises, Irvine, CA, USA) with 40 mL of initial working volume, maintained in an orbital shaker platform (Infor’s HT, Montréal, QC, Canada) at 135 revolutions per minute (rpm), in conditions of 75% humidity, 5% CO2 and 37°C. Cultures were seeded at a cell density of 0.25 x 106 cells/mL, and the cells were monitored daily. For fed-batch cultures, the addition of supplements was as follows: supplementing the HEK-GM basal media consisted of a bolus addition of HEK-FS (XELL AG, Germany) supplemented with 4 mM GlutaMAX™ (Gibco, Grand Island, NY, USA), at 3% (v/v), at 24h hours post-seeding and then every 48h until the harvest. Supplementing the HyCellTransFx medium was carried out with Cell Boost™ 5 (Cytiva, Logan, UT, USA), with the same schedule, adding a bolus at 5% (v/v) supplemented with 4 mM GlutaMAX™ (Gibco, Grand Island, NY, USA) until the end of the production phase. Two approaches were followed: i) supplementing until the time of infection and ii) supplementing during the complete process. Cell counts were performed daily. For infection and virus production characterization, HEK293SF cells were cultured until they reached densities of 2 x 106 cells/mL. Infections were performed at MOI equal to 0.35. Throughout the production, cells were monitored daily for cell density and viability. For every shake flask analyzed, 72 hpi, the harvest was performed, samples were collected, and the cells were lysed with three freeze/thaw cycles, alternating between incubations at -80°C and 30°C in a water bath. After cell lysis, DNase treatment with 5 U/mL of Benzonase (Millipore, Oakville, ON, Canada) was performed for 1 hour at 37°C. The lysate was centrifuged at 6000 × g for 15 min at 4°C to remove cell debris. Alternatively, at harvest, the cells were lysed with lysis buffer (10 mM HEPES, 0.5% w/v Tween-20, 2 mM MgCl2) for 1 hour under agitation, followed by the DNase treatment. The cell culture lysate containing the virus was quantified via TCID50 assay for infectious particles concentration. Measurements were conducted in duplicate.
2.3. Adenovirus production in bioreactors
Adenoviral production in 3L bioreactors (Applikon Biotechnologies, The Netherlands) was conducted in fed-batch mode as previously described for shake flasks), with the Ad-CMV-F adenoviral vector. The 3L bioreactor was equipped with a single marine impeller, a pH sensor, a temperature sensor, a dissolved oxygen (DO) concentration sensor, and a micro sparger with 100 um pore size. The 3L bioreactor (working volume 2.7L) was equipped with a double marine impeller and a capacitance probe (Aber Instruments Ltd., UK). The reactors were seeded at a viable cell density around 0.35 x 106 cells/mL in HyCellTransFx-H medium, and cell growth was allowed until the time of infection at approximately 2 x 106 cells/mL and MOI up to 3.5. The culture runs were supplemented with a bolus feed with 5% (v/v) Cell Boost 5 and 1 mM L-Glutamine during the production phase. The bioreactor control unit ensured controlled conditions with a DO concentration at 40% air-saturation by continuous surface aeration of 5 or 12.5 mL/min air (for the 1 or 3L units, respectively) and injection of pure oxygen when required. The pH was set to 7.15 and regulated by the injection of CO2 into the headspace or the addition of NaHCO3 (90 g/L) (Sigma, USA). Agitation was kept at 90 rpm and increased to 120 rpm in the last 24h of culture to avoid the formation of cell aggregates. Monitoring of the culture characteristics, such as cell growth and the infection process, was conducted through the analysis of capacitance values (uF/cm) in the 3L bioreactor using a capacitance probe (Aber Instruments Ltd., UK). The bioreactor control unit, equipped with a proportional-integral-derivative (PID) controller suite, ensured controlled conditions of process parameters throughout the run. Cells were harvested when cell viability reached around 66-78%, at about 44-94 hpi. The TCID50/mL was also determined.
2.4. Downstream processing
2.4.1. Cell harvest and lysis
To harvest the bioreactor production, the cells were either collected, centrifuged at 1000 × g for 15 min and lysed with three freeze/thaw cycles, alternating between incubations at -80°C and 30°C or were instead chemically lysed inside the bioreactor with 10 mM HEPES, 0.5% w/v Tween-20, 2 mM MgCl2 buffer. After cell lysis, in both cases, DNase treatment was performed with 5 to 10 U/mL of Benzonase (Millipore, Oakville, ON, Canada) for 1 hour at 37°C. Two approaches were followed for separation from the cell debris of lysed cells; the lysate was either centrifuged at 6000 × g for 15 min at 4°C to remove cell debris or subjected to a depth filtration step (no centrifugation) for clarification and separation from the cell debris generated. The cell culture lysate supernatant containing the virus was quantified via TCID50 assay for infectious particles concentration.
2.4.2. Purification by CsCl gradient ultracentrifugation or clarification through depth filtration and Tangential Flow Filtration steps
For purification by CsCl density gradient, cell culture lysate was loaded onto a step CsCl gradient and ultracentrifuged at 28,500 rpm (100,000 × g) at 4°C for 1 h and 30 min. The virus band was collected by side-puncturing the tube using an 18 gauge needle. Dialysis was immediately carried out using the Slide-A-Lyzer G2 Dialysis Cassettes (300 kDa cut-off) against the formulation buffer, as described by Farnos et al., [11].
For a simplified clarification process, the cell culture lysate-containing virus was filtered using the Millistak+® Depth Filter in µPod® format (D0HC media series, 23 cm2 surface area) from Merck KGaA (Darmstadt, Germany) to remove the cell debris. Then, a tangential flow filtration step with hollow fibre cartridges MIDIKROS 20 cm 300kDa and 500KDa from Repligen (Rancho Dominguez, CA, USA) was performed to exchange the buffer and concentrate the product in the desired formulation buffer. For smaller volumes after TFF, an additional Amicon concentration step was used (Figure 1).
2.5. Assessment of product quality
2.5.1. Analytical assays for characterization of the recombinant adenovirus
2.5.1.1. Total particle quantification of the adenoviral vector
Digital Droplet Polymerase Chain Reaction (ddPCR): For quantification, viral DNA extraction was done for 20 µL of supernatant samples diluted with 180 µL PBS using the High Pure Viral Nucleic Acid kit (Roche, Basel, Switzerland). The DNA was diluted (between 1:10 and 1:10,000) to target the linear range of the ddPCR. Five µL of the template dilution were used with the QX200 ddPCR kit (Bio-Rad Laboratories, Hercules, CA, USA), using the EvaGreen master mix and primers to amplify the Fusion gene; Fw: TTAGCTGGTGGCAATATGGA and Rv: TCATGTCCTTGTAGTAGCTCTCATC. The manufacturer’s instructions were followed to prepare the reaction and generate droplets. As for the thermocycler program, after initial denaturation (5 min at 95°C), 34 cycles of the following steps were repeated: 30 s at 95°C, 1 min at 59°C, 30 s at 72°C. Then, the final elongation step happened for 5 min at 72°C.
Droplets are analyzed individually in the droplet reader, and the copies/µL of each sample is given. This output is corrected for the dilution and volumes used to determine the viral genomes/mL of the original sample with the following calculation:
Viral genomes/mL = I × J × K × (L/M) × (O/N)/P × Q × 1000
In which: I = Copies/µL (ddPCR output); J = volume of the ddPCR reaction; K = dilution of the cDNA template; L = volume of RT-PCR reaction; M = volume of the cDNA dilution added in the ddPCR reaction; N = volume of RNA added in the RT-PCR reaction; O = elution volume for RNA extraction; P = initial sample volume used for the RNA extraction; Q = dilution of the sample in RNA extraction.
2.5.1.2. Titration of infectious particles
Infectious titer (IVP/mL) was determined using HEK293A in 96-well plates. HEK 293A cells were seeded at 20,000 cells/well in DMEM supplemented with 2% FBS and allowed to grow for 24h. On the second day, serial dilutions of the virus samples were prepared in fresh growth medium that was added to the plate in duplicates, using two plates per sample to be analyzed (16 wells per sample). The starting dilutions were 10-3 or 10-4 depending on the expected titer. Subsequent 1:10 dilutions were performed horizontally in the plate, keeping one last column with no virus as a negative control. After 13-14 days, the cells were visualized for the development of a cytopathic effect in the inverted microscope. The TCID50/mL value was calculated according to the Reed and Muench method [23]. The average coefficient of variation (CV) was also calculated for the TCID50 assay, resulting in ± 27%. The average CV is the arithmetic mean of the coefficient of variation from the 7 independent runs, each with two or three replicates.
2.5.2. Host cell protein and total DNA determinations
Host cell protein content was determined in the final step of the clarification/purification process using the HEK 293 HCP ELISA Kit from Cygnus (Leland, NC, USA) to monitor the remaining host cell impurities in the products.
Total DNA quantifications were examined using the Quant–iT Picogreen dsDNA Determination Assay from Invitrogen (Eugene, OR, USA).
2.6. Liquid and solid formulations for Ad-CMV-F vaccine storage
2.6.1. Reagents and experimental design
Formulations for storage of Ad-CMV-F in liquid form and solid form were prepared and subjected to sterile filtration. After a series of preliminary assessments involving many different additives, the following excipients and reagents were included in the formulations for the stability assessments: Sucrose, magnesium chloride, kolliphor P188, and polysorbate-80 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium chloride and tris (hydroxymethy1)-aminomethane were purchased from BioShop (Burlington, ON, Canada). Trehalose and Sorbitol were purchased from Fisher Bioreagents (Fair Lawn, NJ, USA). Inulin was obtained from ThermoFisher (Belgium). Histidine was obtained from VWR (Mississauga, ON, Canada). Ethanol was obtained from Alcools de Commerce (Boucherville, QC, Canada). D-mannitol was obtained from BioBasic (Markham, ON, Canada).
For the liquid formulations, the concentrated Ad-CMV-F stock was thawed and mixed with excipient solutions, as shown in Table 1. Briefly, for the 5 liquid formulations, the adenoviral vector (at 5 × 109 TCID50/mL) was thawed, and a buffer exchange step was performed using the centrifugal filter unit Amicon 100 kDa (MilliporeSigma™, Darmstadt, Germany). The final virus concentration ranged from 2 x 109 to 5 x 109 TCID50/mL. Then, aliquots of 100 µL were transferred into 1.5 mL vials and stored at different temperatures. The five formulated products in liquid form were stored at room temperature (21.5°C), 37°C, 4°C, and -80°C for 1, 2, 4, 16, and 24 weeks for the stability study. A similar process was conducted for the sample preparation with the solid formulations (Table 2). Briefly, for the 3 solid formulations, the adenoviral vector (3 × 1010 TCID50/mL) was thawed and diluted with sterilized excipient solutions into the desired solid formulation. The final virus concentration ranged from 7 × 107 to 3 × 108 TCID50/mL. Then, aliquots of 1 mL were transferred into 2 mL glass vials for lyophilization and stored at different temperatures. The formulated products in solid form were stored at 21.5°C, 37°C, and 4°C for 1, 4, and 24 weeks for the stability assessment. After storage at the indicated temperature and the time mentioned, the Ad-CMV-F- vector was quantified vaccine in liquid and solid forms by TCID50 assays.
2.6.2. Lyophilization
Lyophilization was performed using an SP Virtis Advantage Pro Freeze Dryer (SP Scientific, USA). One milliliter of each formulated product containing the Ad-CMV-F was filled into glass vials and partially stoppered. Cycle parameters were extensively studied, optimized, and are tabulated in the Results section. These parameters were modified from those recently reported [24,25], with the addition of an annealing step with a temperature of -15°C for 2 hours during the freezing step, an extension of the primary drying phase to 35 hours at -50°C, and shortening of the tertiary drying phase to 15 hours at 20°C.
The lyophilized vaccine was reconstituted for infectivity titration with sterile ultrapure water and vortexed for 5 seconds. Residual moisture was calculated after the lyophilized samples were kept for 7 hours in the oven at 100°C for complete drying. Calculations were done as follows:
2.7. Immunization experiments and viral challenge
2.7.1. Ethics statement
The studies involving experimentation with animals were in accordance with guidelines and recommendations from the Guide for the Care and Use of Laboratory Animals and policies from the National Veterinary Institute, Ethiopia. When chickens were used for experimentation, they were housed in appropriate rooms with feeding, water supply and health monitoring. The experimental protocols were drafted by the authors and approved by the respective Institutional Committees of Ethics.
2.7.2. Vaccination and challenge with NDV strains
The Newcastle disease virus isolate NDV/Debre zeit/2018 [MN909678] was used for the administration of an intramuscular (homologous) challenge to chickens, consisting of a lethal dose of 0.5 × 106.5 ELD50.
Live thermostable vaccine based on the NDV I2 strain was prepared at the National Veterinary Institute (NVI, Debre zeit, Ethiopia) and used as positive control of vaccination in the immunization and challenge experiments.
A heterologous Newcastle disease virus isolate was used for the administration of an intramuscular heterologous challenge to chickens, at a lethal dose of 0.5 × 106.5 ELD50.
2.7.3. Immunization experiment in chickens
Different lots from two productions in 3L bioreactors were used for the immunization of chickens. CsCl purified lots and Ad-CMV-F purified by depth filtration and TFF steps were evaluated in immunizations by parenteral and mucosal routes. One to two-week old male and female chickens were randomly selected and segregated into 10 groups, each one composed of 10 chickens, which were reared in-house at NVI and maintained under standard conditions with ad libitum feed and water. Three groups of chickens were parenterally immunized. Other five groups of the animals were mucosally vaccinated by intranasal instillation of the liquid formulation in one nostril, using a pipette as the dispenser. Two of the three groups, parenterally vaccinated, received a dose of approximately 1 x 109 TCID50 (measured as 2.4 x 1011 Vg/mL) of the recombinant Ad-CMV-F purified by CsCl, formulated as previously described. These groups were immunized by i.m. and s.c. routes, with 100 µL total volume. A third group was intramuscularly vaccinated with a dose of CsCl purified adenovector at 1 x 108 TCID50 and challenged with a heterologous NDV strain. Five other groups were intranasally immunized by drop instillation into the nasal cavity with 100 or 200 µL of the Ad-CMV-F vaccine. Three of the i.n. immunized animals received the Ad-CMV-F vector in purified form and liquid formulation. The doses administered were 1 x 109, 1 x 108 and 0.5 x 107 TCID50. The two other groups received the Ad-CMV-F formulations obtained after clarification of the culture supernatant via depth filtration and TFF, as described 2.4.3, with doses of 1 x 109 and 5 x 108 TCID50. Finally, one positive control group was vaccinated by eye drops using the NDV live attenuated vaccine, following the manufacturer’s indications on dose and schedule, while a tenth group remained unvaccinated. The immunizations were administered 21 days apart, and the viral challenge was practiced on day 56 of the trial with the NDV isolate Debre zeit/2018 at a lethal dose of 0.5×106.5 ELD50, intranasally administered. All vaccination and challenge experiments were conducted at the National Veterinary Institute, Ethiopia.
3. Results
3.1. Upstream and downstream processing of the Ad-CMV-F adenovirus-based vaccine
3.1.1. Process media and additives for adenovirus production
The adenoviral vector carrying the F gene from a circulating NDV Ethiopian isolate (Ad-CMV-F) was produced in shake flasks and 3L bioreactors, following batch and fed-batch cultivation modes with two different media, as explained in Materials and Methods. After a series of preliminary experiments, it was shown that supplementing any of the two culture media assayed at the concentrations described provided advantages in terms of increased infectious titers measured in the culture supernatant after cell lysis. Addition of supplements to the culture during the cell growth and production phases resulted in the highest titers, in the range of 109 TCID50/mL (Table 3). Different fed-batch productions in shake flasks and 3L bioreactors, using HyCell TransFx-H medium and MOI up to 3.5, were run and are summarized in Table 4. These productions were achieved to support downstream processing studies, stability and formulation studies, and production of material for immunization trials in chicken. These runs yielded maximum titers of up to 5.10 × 109 TCID50/mL before any processing or concentration step.
3.1.2. Downstream processing
After harvest, chemical cell lysis, and DNAse treatment, two different methodologies were followed for the processing and separation of the adenoviral vector from the cell lysate supernatant. The ultracentrifugation in CsCl gradients was previously described [11]. It produces a highly concentrated and purified virus that demonstrated its protective capacity in chickens administered by a parenteral route. The second approach, as an alternative downstream method to ultracentrifugation, consisted of clarification of the cell lysate supernatant via depth filtration, followed by a tangential flow filtration step for a 10-20-fold concentration and buffer exchange. The depth filtration step with the DØCH membrane, which avoids the need for high-speed centrifugation for separation of cell debris, rendered recoveries of 100% of total and infectious particles after being repeatedly evaluated with different buffers to reach an efficient cell lysis. After this first clarification step and concentration with the tangential flow filtration (TFF) set-up, infectious viral particles with functional titers of up to 5.62 × 109 TCID50/mL were measured.
When comparing the filtration performed with the 300 kDa and the 500 kDa cut-off columns, 100% recovery was achieved with the 300kDa column compared to 27.83% recovery with the 500kDa column (n=1). An excellent recovery capacity was seen for the TFF step, in which virus loss in the permeate fraction was quantified, and it was below 0.0053% in the case of the 300 kDa hollow fiber. The final product was always concentrated around 10-20 fold, subjected to sterile filtration, and quantified de novo. Table 5 shows different measurements at the depth filtration and TFFs steps, showing the step recovery levels in different fraction tests from the bioreactor runs. After the clarification and sterile filtration steps, maximum volumetric yields of up to 1012 IVP per liter of culture were measured. Table 6 summarizes the virus titers for the two productions processed which were further used for poultry vaccination.
In addition to measurements of infectious and total viral particles, the analysis on the content of contaminant DNA and host cell protein levels was performed as quality control on lots prepared by CsCl gradients or by depth filtration followed by tangential flow filtration. In the case of the clarified adenovirus, samples filtered by the 300 kDa or the 500 kDa hollow fiber cartridges were evaluated. The results showed lower amounts of DNA (in the range of 0.3 - 0.4 µg/mL) in the samples from clarified formulations (both 300 kDa and 500 kDa) compared to those subjected to ultracentrifugation (around 1.2-1.3 µg/mL), most likely due to variations in cell density at the time of harvest. The host cell protein levels showed a concentration of around 20 µg/mL in all the samples subjected to clarification while the Ad-CMV-F samples purified by CsCl exhibited a purity of around 0.2-0.3 µg per mL.
3.2. Stability of the Ad-CMV-F adenoviral vector
3.2.1. Stability study for storage in liquid formulations
The Ad-CMV-F adenoviral vector was subjected to different temperature stress treatments and storage conditions to evaluate the capacity of the final formulations under study to withstand potential adverse conditions during field applications. Adenoviral sample formulations were treated and quantified after storage at 37°C, 21.5°C, 4°C, and -80°C for 1, 4, and 24 weeks in five liquid formulations. The formulations assayed (see Materials and Methods) contained different key excipients with varied potential to provide long-term stability to the adenoviral vector. Notably, the titer was maintained after 1 week at 21.5°C across all the liquid formulations. However, after 1 week at 37°C, a significant functional titer loss was observed for formulations L1 and L3, resulting in a loss of infectivity of more than 54%. Formulation L5 had an average loss of 37.3 ± 21.1% after 1 week at 37°C (Figure 2A).
The formulation L2 maintained the virus activity after 1 week at 37°C with a minor 12.8 ± 33.8% loss of infectivity, which falls within the inherent assay variability of ± 27%, calculated as described in Materials and Methods. From a linear regression a half-life at 37°C was estimated to be 2 weeks. In line with this estimation, experimental results for L2 demonstrated an average loss of 66.9% ± 6.1% of infectivity after 14 days at 37°C (Figure 2B).
A prolonged storage time was also analyzed. A loss in titer of 50% is represented by the dashed line on the 0.301 log mark on the y-axis of Figure 2C. After 4 weeks at 37°C, a significant loss in titer, above 78.3%, was observed for all the formulations. After 4 weeks at 21.5°C, the functional titer decreased significantly in formulation L1, above 46.8% loss in titer. However, the infectious titer was maintained in the other 4 formulations at this temperature. Additionally, formulation L2 demonstrated, after 16 weeks at 21.5°C, its ability to preserve the virus titer (Figure 2B). Using an Arrhenius plot, it was estimated that the Ad-CMV-F stored for 4 weeks in formulation L2 at temperatures ≥ 23.6°C will have a functional titer loss above 0.14 log (represented by the dashed line on -1.96 ln mark on the y-axis of Figure 3), indicating a significant loss due to degradation rather than assay variability. Infectivity loss (k) was determined from Figure 2B, specifically at the 4-week storage time point at 4°C, 21.5°C, and 37°C.
At 4°C, formulations L2 to L5 maintained the titer after 4 and 24 weeks of storage. The formulation L1 lost 57.5% of its functional titer after 4 weeks (Figure 2C). A linear regression analyses of the data in Figure 3B, showed that the infectivity loss rate at 4°C for formulation L2 is 0.0031 log per month. Notably, the infectivity loss after 24 weeks of storage at 4°C remained ≤ 0.10 log. These data indicate that the half-life (t1/2) for Ad-CMV-F infectivity loss in L2 at 4°C is 94.7 months, i.e., 7.9 years. Furthermore, among all the formulations, only formulation L1 exhibited a significant loss in infectivity after storage at -80°C for 24 weeks, with a loss of 0.32 log (equivalent to a 52.6 ± 15.3% reduction in titer).
3.2.2. Stability study for adenovirus storage in solid formulations
3.2.2.1. Characterization and optimization of the lyophilization cycles
The formulations evaluated for the lyophilization process were described in Materials and Methods. Three solid formulations varying solely in their cryoprotectant component, sucrose (S1), trehalose (S2), and inulin (S3), were evaluated. The optimized cycles for the lyophilization of the Ad-CMV-F are detailed in Table 7. These cycles were reached after several series of tests for optimization, with assessments of the physical properties of the dried samples and their infectious titer after the process. With these cycles, cakes with satisfactory visual characteristics for the three proposed formulations were generated (Figure 4A). Residual moisture was quantified on samples from the three solid formulations in two separate experimental runs. In the case of the solid formulation S1, the residual moisture content post-freeze-drying was determined to be 4.74 ± 0.94%. For solid formulation S2, this value was measured at 5.66 ± 0.71%, while for solid formulation S3, it was 3.63 ± 1.64%; Notably, all these levels fell within the satisfactory range of 3 to 6% of residual moisture, indicative of a successful lyophilization process.
The reproducibility of the cycle was evaluated from three independent runs. For each experimental run, the Ad-CMV-F vector was quantified before and after the lyophilization process to measure any detrimental effect intrinsic to the process on the infectious titer. For the solid formulation S1, the average log loss was 0.11 log, representing around 23.4% loss in titer. This decrease falls within the expected assay variability. However, solid formulations S2 and S3 showed an average titer loss of 32.4% and 55.5%, respectively (Figure 4B).
The stability of the three solid formulations during prolonged storage was analyzed by quantifying the Ad-CMV-F vector after 1, 4, and 24 weeks of storage at different temperatures and comparing it to a control before lyophilization (i.e., considering in process and in storage losses) (Figure 5). After 1 week at 21.5°C, all formulations presented a significant functional titer loss, with storage in solid formulation S1 resulting in the lowest loss compared to the other formulations (around 43.6 ± 17.9%) (Figure 5A). Significant titer loss was observed for the 3 solid formulations after 1 and 4 weeks at 37°C. Nonetheless, after extended storage at 4°C, the three solid formulations were able to maintain the titer levels, demonstrating a significant capacity for preservation even over the course of 24 weeks.
Considering only the in storage loss, the three solid formulations maintained the functional titer after 1 week at 21.5°C (Figure 6). Only the solid formulation S2 presented a functional titer loss of 29.1 ± 42.4% (non-significant, p > 0.05).
3.3. Intranasal delivery of the Ad-CMV-F vaccine provides protection in poultry against a lethal challenge with NDV
The production lots were used for the mucosal immunization of chickens via drop instillation of 200 µL of the vaccine into the nasal cavity. The vaccines were prepared in liquid formulation L2 and delivered on days 0 and 21 of the trial (Figure 7A). The assay evaluated route (i.m. and s.c., doses from 2 x 109 to 0.5 x 107 TCID50), intranasal delivery, and the two purification methods. One heterologous strain of NDV was also used for the lethal challenge, followed by an observation period of two weeks.
Chickens immunized by intramuscular or subcutaneous injection were protected against NDV. The groups of animals mucosally immunized were all protected at different degrees in accordance with the dose administered. The groups vaccinated with the adenoviral vector highly purified by CsCl showed similar protection levels to those parenterally injected. One hundred percent of protection, without clinical disease, was observed at the highest dose evaluated, with 90% protection at lower doses. Remarkably, the group of animals vaccinated with the adenoviral vector subjected to the purification by filtration steps and concentration process, at the lower dose here assayed (5 x 108 TCID50 per animal), showed 100% protection (Figure 7B,C), with no animals showing clinical symptoms of the disease or evidence of typical tissue lesions after euthanasia and necropsy analyses. For clarity and simplicity, the vaccination groups and the results from the challenges are shown in two graphics grouped by the route of immunization employed. The survival curve also considers animals that did not die but showed clinical symptoms of the disease.
4. Discussion
The adenovirus Ad-CMV-F-vectored vaccine is a genotype-matched candidate against Newcastle Disease Virus, based on a non-replicative recombinant variant of the human type 5 adenovirus. It carries the transgene for the Fusion protein of the NDV Ethiopian isolate NDV/Debre zeit/2018 [MN909678], which was assigned to Genotype VI after phylogenetic analyses. The candidate vaccine was produced in stirred tank bioreactors using HEK293 suspension cells and serum-free media, purified in CsCl gradients, and demonstrated full protection against NDV in poultry immunized by intramuscular vaccination [11]. In this study, we further investigated cost-effective upstream and downstream process alternatives, resulting in the development of more streamlined protocols for production in bioreactors followed by a series of filtration, ultrafiltration and concentration steps. These procedures, with high recoveries, resulted in a highly immunogenic vaccine formulation inducing protective immunity when mucosally administered to poultry. Additionally, liquid and solid formulation of the adeno-vectored vaccine were developed showing remarkable stability of the vaccine when stored under standard conditions for prolonged time or under thermal stressing conditions.
The experiments conducted in shake flasks and the productions in bioreactors showed the suitability of the media evaluated and the fed-batch mode of production to effectively achieve volumetric yields of 1 to 5 x 1012 IVP per liter when MOI of 3 is used for cell culture infection at a cell density of around 2-2.5 x 106 cells/mL. Such yields are in the highest range of reports in the literature [26]. This process in bioreactors was previously studied, documented [11], and further optimized and validated by different productions of the Ad-CMV-F adenoviral vector product as described here. The methods described constitute an effective process for the rapid generation of the raw material to be processed and formulated for mucosal administration. After the adenovirus production, the clarification/purification method was established, using depth filtration and tangential flow filtration, as a scalable and suitable alternative for veterinary vaccines, in which the low cost of production plays a vital role for rapid implementation in the field and commercial success. Even though to our knowledge no specific limit of residual host cell DNA appears to be fixed or established to veterinary products, DNAse treatment was conducted to successfully reduce the DNA content and DNA length prior to the clarification steps. At the time of ultrafiltration, the hollow fiber columns evaluated, at 300 KDa and 500 KDa cut-off, resulted in similar levels of residual host cell protein and residual DNA left after filtration and buffer exchange. The 300 kDa cut-off column was far superior, with high recoveries and negligible virus loss in the permeate fraction.
Regarding the stability studies, five liquid formulations were evaluated under storage conditions at 21.5˚C (room temperature) and 37°C for 1 and 4 weeks. They were also assessed for stability at 4°C and -80°C for a prolonged storage time of 24 weeks. At 37°C, the L2 formulation maintained the virus activity for 1 week and presented a half-life (t1/2) of around 14 days, similar to a previous report [27] for an Ad5 vector formulated in A195 (containing sucrose as the cryoprotectant and polysorbate-80 as non-ionic surfactant). Even short-term stability at a high temperature, such as 37˚C, is considered important for manufacturing, handling, and distribution of vaccines in less developed areas, such as rural locations and home-based producers’ properties. These ones are the main source of poultry production in sub-Saharan countries [4,5,6]. Moreover, the functional or infectious titer was stable after storage for 16 weeks at 21.5°C and after 24 weeks at 4°C. Linear regression analyses of the formulation L2 data indicated that the t1/2 for Ad-CMV-F functional titer loss at 4°C is 94.7 months, i.e., 7.9 years, a slightly longer half-life than previous findings with the A195 formulation [27]. The overall data support the storage of the vaccine candidate in L2 formulation at 4°C, being also able to endure significant periods of time without refrigeration. The formulation described in the literature as A438, equivalent to the liquid formulation L3, was evaluated in our study, and provided poor stability at 37°C for 1 week (above 60% functional titer loss). Conversely, they found this formulation suitable for storage of Ad5 at 30°C for 1 month with a loss of 0.24 log [28]. Nevertheless, in the present study, the formulation L3 was stable at 21.5°C for 4 weeks, similar to what it was reported in 2021 for the Chimpanzee Adenovirus Vectored Vaccines ChAdOx1 and ChAdOx2 [24].
The only liquid formulation presenting functional titer loss after storage at -80°C for 24 weeks was L1, even though the formulation contained 2% of sucrose, a cryoprotectant agent. L2, also containing 2% sucrose, did not show infectious titer loss. The addition of a non-ionic surfactant in L2 might have helped the preservation of the virus. Formulations L4 and L5, containing other cryoprotectants, such as trehalose and sorbitol, were able to preserve the virus at -80°C for 24 weeks and after one freeze/thaw cycle. The addition of non-ionic surfactants, such as poloxamer 188 and polysorbate-80 in L2 and L4, had a clear positive impact in stability at 21.5°C after 4 weeks of storage, compared to formulation L1. Non-ionic surfactants are used in formulation to prevent adsorption and aggregation. The highest preservation in titer at 37°C after 4 weeks of storage was achieved with formulation L2, containing poloxamer 188 instead of polysorbate-80. In 2014, an investigation on surfactant’s mechanisms of action demonstrated that poloxamer 188 inhibits protein adsorption by forming protein-surfactant complexes of low adsorption affinity [29]. On the contrary, polysorbates 80 do not form protein–surfactant complexes; this non-ionic surfactant inhibits protein adsorption by their preferential location at an interface to which they show sufficient affinity. Also, at low concentrations, it has been reported that the stability of poloxamer 188 at 40°C was stable for up to 6 months in certain buffer conditions [30,31]. The different mechanisms of action of the two non-ionic surfactants evaluated and their stability at high temperatures might have influenced the preservation of the virus differently.
With regards to the lyophilized vaccine, it has been reported that the combination of mannitol and sucrose or inulin has a positive impact on the pH stabilization of Tris buffers during freezing [Chen 2012]. In the same line, trehalose is another cryoprotectant agent that can prevent pH shifts upon the freezing process of lyophilized viral vectors [32]. The parameters for the lyophilization process were optimized starting from those already reported [24,33]. No in process functional titer loss was identified for solid formulation S1, which is a noteworthy improvement compared to previous findings [33]. Nonetheless, we noticed a higher in process loss (above 55%) with formulation S3, which was still lower than the in process loss reported for ChAdOx1 and ChAdOx2 in 2021 [24] with the same solid formulation.
Notably, in storage functional titer loss after 1 week at 21.5°C in the 3 formulations was non-significant. The stability of the lyophilized formulations at 21.5°C may enable field distribution under such conditions. Also importantly, the infectious titer was maintained after 24 weeks at 4°C. Overall, the liquid formulations could maintain the infectious adenoviral titer for at least 4 weeks at 21.5°C, and for 24 weeks at 4°C. Specifically, we focused on the formulation L2, which maintains functional titers for at least 16 weeks at 21.5°C and has a half-life of 2 weeks at 37°C. We also described an optimized cycle for lyophilizing Ad-CMV-F without in process loss of functional titer and 3 solid formulations capable of maintaining Ad5 stability at 4°C for at least 24 weeks. The liquid formulation is seen as the most advantageous because it has satisfactory stability without the need for an additional lyophilization process. In addition, administration does not require reconstitution of the vaccine, and it can be distributed ready-to-use.
As per the immunogenicity, protective efficacy, and route of administration, we have demonstrated for the first time that a dose as low as 5 x 108 TCID50/mL administered by drop instillation into the animal’s nostril, with a second dose after 21 days, can provide complete protection against a lethal challenge of NDV. The protective capacity of intramuscular immunization had already been demonstrated. Now we corroborated that even doses as low as 5 x 107 can protect 70% of the animals, with the particularity that all the animals survived, although three of them presented clinical signs of the disease. The complete protection provided by immunization through the nasal cavity at a dose of 5 x 108 TCID50 support the general agreement in the vaccine community that the nasal route is the most straightforward and suitable mucosal delivery for vaccine administration as compared to oral, pulmonary, rectal or vaginal mucosal delivery routes [34]. The nasal-associated lymphoid tissue (NALT) is the primary inductive site for mucosal immunity in the nasopharyngeal tract [35]. In poultry, the NALT is located on the bottom of the nasal septum and both sides of the choanal cleft. Abundant lymphocytes appear to be distributed under the mucosal epithelium of the inferior nasal meatus. There are also diffuse lymphoid tissues under the epithelium of the concha nasalis media and the nasal cavity walls [36]. Recently, protection against genotype VII of the NDV was achieved by intranasal subunit vaccination based on bacterium-like particles bearing the F or HN antigen [37]. This mucosal subunit vaccine is based on a bacterium-like particles (BLPs) delivery platform derived from Lactococcus lactis. The NDV antigens F or HN were fused to protein anchor that was expressed by a recombinant baculovirus and loaded on the surface of BLPs. The vaccine provided as high as 90% protection rate against an intranasal NDV challenge.
For human pathogens, adenovirus vectored trivalent COVID-19 vaccines expressing the spike, nucleocapsid, and RdRp antigens used in single-dose intranasal immunization regimens have been recently evaluated in murine models [38]. In particular, the chimpanzee Ad-vectored COVID-19 vaccine via intranasal was superior to intramuscular immunization in the induction of the tripartite protective immunity, consisting of local and systemic antibody responses, mucosal tissue-resident memory T cells and mucosal trained innate immunity. Intranasal immunization provided protection against the ancestral SARS-CoV-2 strain and two more recent variants (VOC, B.1.1.7 and B.1.351), showing that mucosal vaccination with adenovirus-based vaccines is an effective vaccination strategy, with capacity to induce an extensive mucosal immunity. Other anti-COVID-19 vaccines based on human adenoviruses advanced to clinical trials and received approval for administration via intranasal in different countries [39]. They are expected to prevent milder cases of illness and block transmission to other people by prompting immune responses where SARS-CoV-2 first enters the body. Other vaccines based on adenoviral vectors from different species or human serotypes were developed against SARS-CoV-2 and were extensively used worldwide, with an exceptional contribution to saving millions of lives during the COVID-19 pandemic [40].
In summary, to prevent NDV infection in poultry and as an alternative to the current egg-based vaccines, we developed, produced and validated the Ad-CMV-F adenoviral-vectored vaccine. The vaccine is produced using HEK293 suspension cells cultured in serum-free media in stirred tanks bioreactors. At least two media commercially available can be used in the manufacturing process, yielding after clarification and sterile filtration volumetric yields over 1012 IVP per liter of culture. The clarification process is also streamlined and does not involve costly chromatography steps, rendering a purified material suitable for veterinary mucosal vaccination. Altogether, including the remarkable stability shown by one of the liquid formulations prepared, this vaccine product and the associated production process becomes a strong candidate for technology transfer and implementation on the manufacturing site of the National Veterinary Institute, Ethiopia, one of the partners in the project. The adenoviral production platform in stirred tank bioreactors remains an extremely valuable technology for rapid development and equitable delivery of vaccines against emerging or re-emerging pathogens affecting animal species for a One Health strategy.
Author Contributions
Conceptualization, O.F., B.C.M.F.P., E.G., A.A.K.; methodology, O.F., B.C.M.F.P., B.G., E.G., S.R., A.A.K.; validation, B.C.M.F.P., A.Ch.; formal analysis, O.F., B.C.M.F.P., E.G., A.A.K.; investigation, O.F., B.C.M.F.P., B.G., S.R., A.Ch., E.G., resources, E.G., A.A.K.; data curation, O.F., B.C.M.F.P.; writing—original draft preparation, O.F., B.C.M.F.P., A.A.K.; writing—review and editing, O.F., B.C.M.F.P., A.A.K.; supervision, A.A.K., funding acquisition, A.K.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the International Development Research Center (IDRC) Project Number 109618 – Phase II. A.A.K. is partially funded through Canada Research Chair- CRC-2021-00032.
Institutional Review Board Statement
The animal experiments approved by the Ethics Committee of the National Veterinary Institute, Ethiopia.
Acknowledgments
The authors would like to acknowledge the contribution from technical staff of the National Veterinary Institute, Ethiopia, involved in the animal experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grace, D.; Roesel, K.; Lore, T. Poverty and Gender Aspects of Food Safety and Informal Markets in Sub-Saharan Africa; ILRI (aka ILCA and ILRAD): Nairobi, Kenya, 2014. [Google Scholar]
- Dimitrov, K.M.; Afonso, C.L.; Yu, Q.; Miller, P.J. Newcastle disease vaccines—A solved problem or a continuous challenge? Vet. Microbiol. 2017, 206, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, C.; Adeyanju, A.T.; Owoade, A.A.; Couacy-Hymann, E.; Alkali, B.R.; Ottosson, U.; Muller, C.P. Genetic Diversity of Newcastle Disease Virus in Wild Birds and Pigeons in West Africa. Appl. Environ. Microbiol. 2013, 79, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Wodajo, W.; Mohammed, N.; Tora, E.; Seyoum, W. Sero-prevalence of Newcastle disease and associated risk factors in chickens at backyard chicken production Kindo Koisha, Wolaita zone, Southern Ethiopia. Front Vet Sci. 2023, 9, 1089931. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.T. Poultry production and performance in the Federal Democratic Republic of Ethiopia. World Poult. Sci. J. 2010, 66, 441–454. [Google Scholar]
- Tekelemariam, T.H.; Walkden-Brown, S.; Atire, F.A.; Tefera, D.A.; Alemayehu, D.H.; Gerber, P.F. Detection of Chicken Respiratory Pathogens in Live Markets of Addis Ababa, Ethiopia, and Epidemiological Implications. Vet Sci. 2022, 9, 503. [Google Scholar] [CrossRef]
- Miller, P.J.; Afonso, C.L.; El Attrache, J.; Dorsey, K.M.; Courtney, S.C.; Guo, Z.; Kapczynski, D.R. Effects of Newcastle Disease Virus Vaccine Antibodies on the Shedding and Transmission of Challenge Viruses. Dev. Comp. Immunol. 2013, 41, 505–513. [Google Scholar] [CrossRef]
- Wright, P.F. Vaccine preparedness—Are we ready for the next influenza pandemic? N. Engl. J. Med. 2008, 358, 2540–2543. [Google Scholar] [CrossRef] [PubMed]
- OIE. Newcastle disease. In OIE Manual for Diagnostic Tests and Vaccines for Terrestrial Animals, 5th ed.; OIE: Paris, France, 2004; Volume 1, pp. 270–283. [Google Scholar]
- Allan, W.H.; Lancaster, J.E.; Toth, B. Newcastle disease vaccines—Their production and use. FAO Anim. Prod. Ser. Rome 1978, 10, 1–163. [Google Scholar]
- Farnós, O.; Gelaye, E.; Trabelsi, K.; Bernier, A.; Subramani, K.; Kallel, H.; Yami, M.; Kamen, A.A. Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa. Vaccines 2020, 8, 338. [Google Scholar] [CrossRef]
- Joe, C.C.D.; Jiang, J.; Linke, T.; Li, Y.; Fedosyuk, S.; Gupta, G.; Berg, A.; Segireddy, R.R.; Mainwaring, D.; Joshi, A.; Cashen, P.; Rees, B.; Chopra, N.; Nestola, P.; Humphreys, J.; Davies, S.; Smith, N.; Bruce, S.; Verbart, D.; Bormans, D.; Knevelman, C.; Woodyer, M.; Davies, L.; Cooper, L.; Kapanidou, M.; Bleckwenn, N.; Pappas, D.; Lambe, T.; Smith, D.C.; Green, C.M.; Venkat, R.; Ritchie, A.J.; Gilbert, S.C.; Turner, R.; Douglas, A.D. Manufacturing a chimpanzee adenovirus-vectored SARS-CoV-2 vaccine to meet global needs. Biotechnol Bioeng. 2022, 119, 48–58. [Google Scholar] [CrossRef]
- Xiang, K.; Ying, G.; Yan, Z.; Shanshan, Y.; Lei, Z.; Hongjun, L.; Maosheng, S. Progress on adenovirus-vectored universal influenza vaccines. Hum. Vaccines Immunother. 2015, 11, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Kallel, H.; Kamen, A.A. Large-scale adenovirus and poxvirus-vectored vaccine manufacturing to enable clinical trials. Biotechnol. J. 2015, 10, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.F.; Lanthier, S.; Jacob, D.; Montes, J.; Beath, A.; Beresford, A.; Kamen, A. Process optimization and scale-up for production of rabies vaccine live adenovirus vector (AdRG1.3). Vaccine 2012, 30, 300–6. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.J.; Rosatte, R.C.; Fehlner-Gardiner, C.; Bachmann, P.; Ellison, J.A.; Jackson, F.R.; Taylor, J.S.; Davies, C.; Donovan, D. Oral vaccination and protection of red foxes (Vulpes vulpes) against rabies using ONRAB, an adenovirus-rabies recombinant vaccine. Vaccine 2014, 32, 984–9. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.T.; Johnson, S.R.; Nelson, K.M.; Chipman, R.B.; VerCauteren, K.C.; Algeo, T.P.; Rupprecht, C.E.; Slate, D. Field Trials of Ontario Rabies Vaccine Bait in the Northeastern USA, 2012-14. J. Wildl. Dis. 2018, 54, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Barrera, J.; Brake, D.A.; Kamicker, B.J.; Purcell, C.; Kaptur, R.Jr; Schieber, T.; Lechtenberg, K.; Miller, T.D.; Ettyreddy, D.; Brough, D.E.; Butman, B.T.; Colby, M.; Neilan, J.G. Safety profile of a replication-deficient human adenovirus-vectored foot-and-mouth disease virus serotype A24 subunit vaccine in cattle. Transbound Emerg Dis. 2018, 65, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Sitt, T.; Kenney, M.; Barrera, J.; Pandya, M.; Eckstrom, K.; Warner, M.; Pacheco, J.M.; LaRocco, M.; Palarea-Albaladejo, J.; Brake, D.; Rieder, E.; Arzt, J.; Barlow, J.W.; Golde, WT. Duration of protection and humoral immunity induced by an adenovirus-vectored subunit vaccine for foot-and-mouth disease (FMD) in Holstein steers. Vaccine 2019, 37, 6221–6231. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, Z.; Xiong, J.; Yang, K.; Chi, Y.; Tang, X.; Ma, L.; Zhang, R.; Deng, F.; Lan, K.; Zhou, D. A chimpanzee adenoviral vector-based rabies vaccine protects beagle dogs from lethal rabies virus challenge. Virology 2019, 536, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Garnier, A.; Massie, B.; Kamen, A. Serum-free production of recombinant proteins and adenoviral vectors by 293SF-3F6 cells. Biotechnol. Bioeng. 1998, 59, 567–575. [Google Scholar] [CrossRef]
- Stillman, B.W.; Gluzman, Y. Replication and supercoiling of simian virus 40 DNA in cell extracts from human cells. Mol. Cell. Biol. 1985, 5, 2051–2060. [Google Scholar]
- Reed, L.J.; Muench, H. A simple method of estimating fifty-percent endpoints. Am J Hyg. 1938, 27, 493–497. [Google Scholar]
- Berg, A.; Wright, D.; Dulal, P.; Stedman, A.; Fedosyuk, S.; Francis, M.J.; Charleston, B.; Warimwe, G.M.; Douglas, A.D. Stability of Chimpanzee Adenovirus Vectored Vaccines (ChAdOx1 and ChAdOx2) in Liquid and Lyophilised Formulations. Vaccines 2021, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guo, D.; Guo, B.; Liu, J.; Shen, Y.; Xu, X.; Huang, W.; Guo, S. Investigation on formulation and preparation of adenovirus encoding human endostatin lyophilized powders. Int. J. Pharm. 2012, 427, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Fedosyuk, S.; Merritt, T.; Peralta-Alvarez, M.P.; Morris, S.J.; Lam, A.; Laroudie, N.; Kangokar, A.; Wright, D.; Warimwe, G.M.; Angell-Manning, P.; Ritchie, A.J.; Gilbert, S.C.; Xenopoulos, A.; Boumlic, A.; Douglas, A.D. Simian adenovirus vector production for early-phase clinical trials: A simple method applicable to multiple serotypes and using entirely disposable product-contact components. Vaccine 2019, 37, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.K.; Nawrocki, D.K.; Isopi, L.A.; Williams, D.M.; Casimiro, D.R.; Chin, S.; Chen, M.; Zhu, D.M.; Shiver, J.W.; Volkin, D.B. Development of stable liquid formulations for adenovirus-based vaccines. J. Pharm. Sci. 2004, 93, 2458–2475. [Google Scholar] [CrossRef]
- Evans, R.; Volkin, D.; Isopi, L. Adenovirus Formulations. U.S. Patent 7456009B2, 25 November 2008. [Google Scholar]
- Kim, H.L.; McAuley, A.; Livesay, B.; Gray, W.D.; McGuire, J. Modulation of protein adsorption by poloxamer 188 in relation to polysorbates 80 and 20 at solid surfaces. J Pharm Sci. 2014, 103, 1043–9. [Google Scholar] [CrossRef]
- Wang, T.; Markham, A.; Thomas, S.J.; Wang, N.; Huang, L.; Clemens, M.; Rajagopalan, N. Solution stability of poloxamer 188 under stress conditions. J. Pharm. Sci. 2019, 108, 1264–1271. [Google Scholar] [CrossRef]
- Chen, W.; Stolz, S.; Wegbecher, V.; Parakkattel, D.; Haeuser, Ch.; Oltra, N.S.; Kishore, R.S.; Bond, S.; Bell, Ch.; Kopf, R. The degradation of poloxamer 188 in buffered formulation conditions. AAPS Open 2022, 8, 5. [Google Scholar] [CrossRef]
- Croyle, M.A.; Roessler, B.J.; Davidson, B.L.; Hilfinger, J.M.; Amidon, G.L. Factors that influence stability of recombinant adenoviral preparations for human gene therapy. Pharm. Dev. Technol. 1998, 3, 373–383. [Google Scholar] [CrossRef]
- Chen, S.; Guo, D.; Guo, B.; Liu, J.; Shen, Y.; Xu, X.; Huang, W.; Guo, S. Investigation on formulation and preparation of adenovirus encoding human endostatin lyophilized powders. Int. J. Pharm. 2012, 427, 145–152. [Google Scholar] [CrossRef]
- Zaman, M.; Chandrudu, S.; Toth, I. Strategies for intranasal delivery of vaccines. Drug Deliv Transl Res. 2013, 3, 100–9. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W.; Coffin, S.E.; Thurnheer, M.C.; Fundova, P.; Cebra, J.J. Nasal-associated lymphoid tissue is a mucosal inductive site for virus-specific humoral and cellular immune responses. J Immunol. 2002, 168, 1796–803. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Yan, M.; Yu, Q.; Yang, Q. Characteristics of nasal-associated lymphoid tissue (NALT) and nasal absorption capacity in chicken. PLoS One 2013, 8, e84097. [Google Scholar] [CrossRef]
- Wang, J.; Lan, Q.; Zong, X.; Zhu, G.; Yang, R.; Yang, G.; Jiang, Y.; Yang, W.; Huang, H.; Shi, C.; Zeng, Y.; Wang, N.; Cao, X.; Wang, C. Protection against genotype VII Newcastle disease virus by a mucosal subunit vaccination based on bacterium-like particles bearing the F or HN antigen. Int J Biol Macromol. 2023, 244, 125293. [Google Scholar] [CrossRef] [PubMed]
- Afkhami, S.; D'Agostino, M.R.; Zhang, A.; Stacey, H.D.; Marzok, A.; Kang, A.; Singh, R.; Bavananthasivam, J.; Ye, G.; Luo, X.; Wang, F.; Ang, J.C.; Zganiacz, A.; Sankar, U.; Kazhdan, N.; Koenig, J.F.E.; Phelps, A.; Gameiro, S.F.; Tang, S.; Jordana, M.; Wan, Y.; Mossman, K.L.; Jeyanathan, M.; Gillgrass, A.; Medina, M.F.C.; Smaill, F.; Lichty, B.D.; Miller, M.S.; Xing, Z. Respiratory mucosal delivery of next-generation COVID-19 vaccine provides robust protection against both ancestral and variant strains of SARS-CoV-2. Cell 2022, 185, 896–915. [Google Scholar] [CrossRef] [PubMed]
- Waltz, E. China and India approve nasal COVID vaccines - are they a game changer? Nature 2022, 609, 450. [Google Scholar] [CrossRef]
- Joe, C.C.D.; Jiang, J.; Linke, T.; Li, Y.; Fedosyuk, S.; Gupta, G.; Berg, A.; Segireddy, R.R.; Mainwaring, D.; Joshi, A.; Cashen, P.; Rees, B.; Chopra, N.; Nestola, P.; Humphreys, J.; Davies, S.; Smith, N.; Bruce, S.; Verbart, D.; Bormans, D.; Knevelman, C.; Woodyer, M.; Davies, L.; Cooper, L.; Kapanidou, M.; Bleckwenn, N.; Pappas, D.; Lambe, T.; Smith, D.C.; Green, C.M.; Venkat, R.; Ritchie, A.J.; Gilbert, S.C.; Turner, R.; Douglas, A.D. Manufacturing a chimpanzee adenovirus-vectored SARS-CoV-2 vaccine to meet global needs. Biotechnol Bioeng. 2022, 119, 48–58. [Google Scholar] [CrossRef]
Figure 1.
Downstream processes for the purification or clarification of the Ad-CMV-F adenoviral vector. Part of the productions were subjected to ultracentrifugation in CsCl gradients for purification of the virus while the other part was taken for a clarification streamlined process that involved a depth filtration step followed by a tangential flow filtration for ultrafiltration and 10-20x concentration purposes.
Figure 1.
Downstream processes for the purification or clarification of the Ad-CMV-F adenoviral vector. Part of the productions were subjected to ultracentrifugation in CsCl gradients for purification of the virus while the other part was taken for a clarification streamlined process that involved a depth filtration step followed by a tangential flow filtration for ultrafiltration and 10-20x concentration purposes.
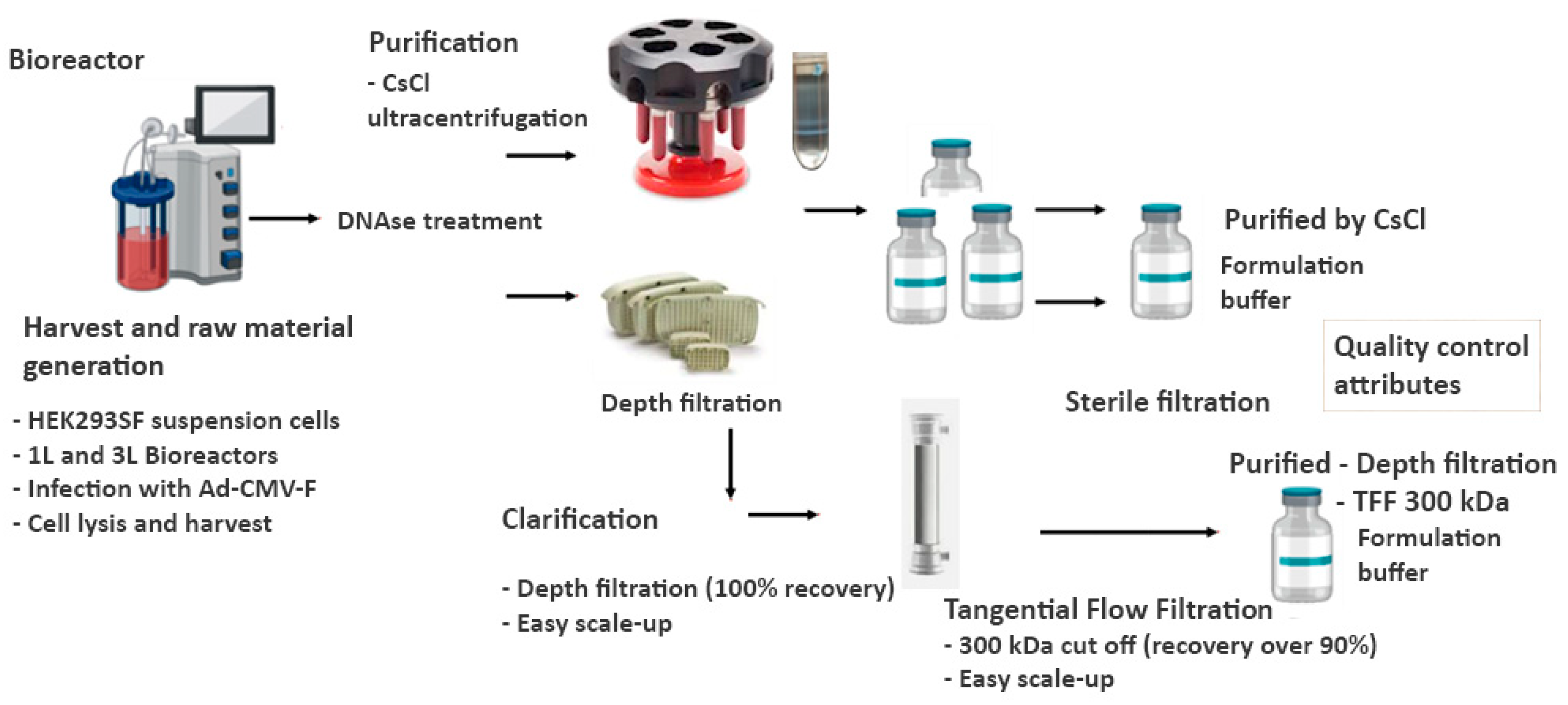
Figure 2.
Functional titer loss after storage at 37°C, 21.5°C, and 4°C in five different liquid formulations. A) Virus stored for 1 week at 21.5°C and 37°C. B) Virus stored in formulation 2 for 2 weeks at 37°C and 21.5°C and 16 weeks at 21.5°C. C) Storage up to 24 weeks. Values represent mean ± standard deviation (n = 5), ***p < 0.001, **p < 0.01, *p < 0.05, by analysis of variance (ANOVA) followed by a Dunnett’s post test. A Student’s t-test was also conducted, depending on the number of groups under analysis. The dashed line represents the threshold for a 50% loss in titer, equivalent to 0.301 log.
Figure 2.
Functional titer loss after storage at 37°C, 21.5°C, and 4°C in five different liquid formulations. A) Virus stored for 1 week at 21.5°C and 37°C. B) Virus stored in formulation 2 for 2 weeks at 37°C and 21.5°C and 16 weeks at 21.5°C. C) Storage up to 24 weeks. Values represent mean ± standard deviation (n = 5), ***p < 0.001, **p < 0.01, *p < 0.05, by analysis of variance (ANOVA) followed by a Dunnett’s post test. A Student’s t-test was also conducted, depending on the number of groups under analysis. The dashed line represents the threshold for a 50% loss in titer, equivalent to 0.301 log.
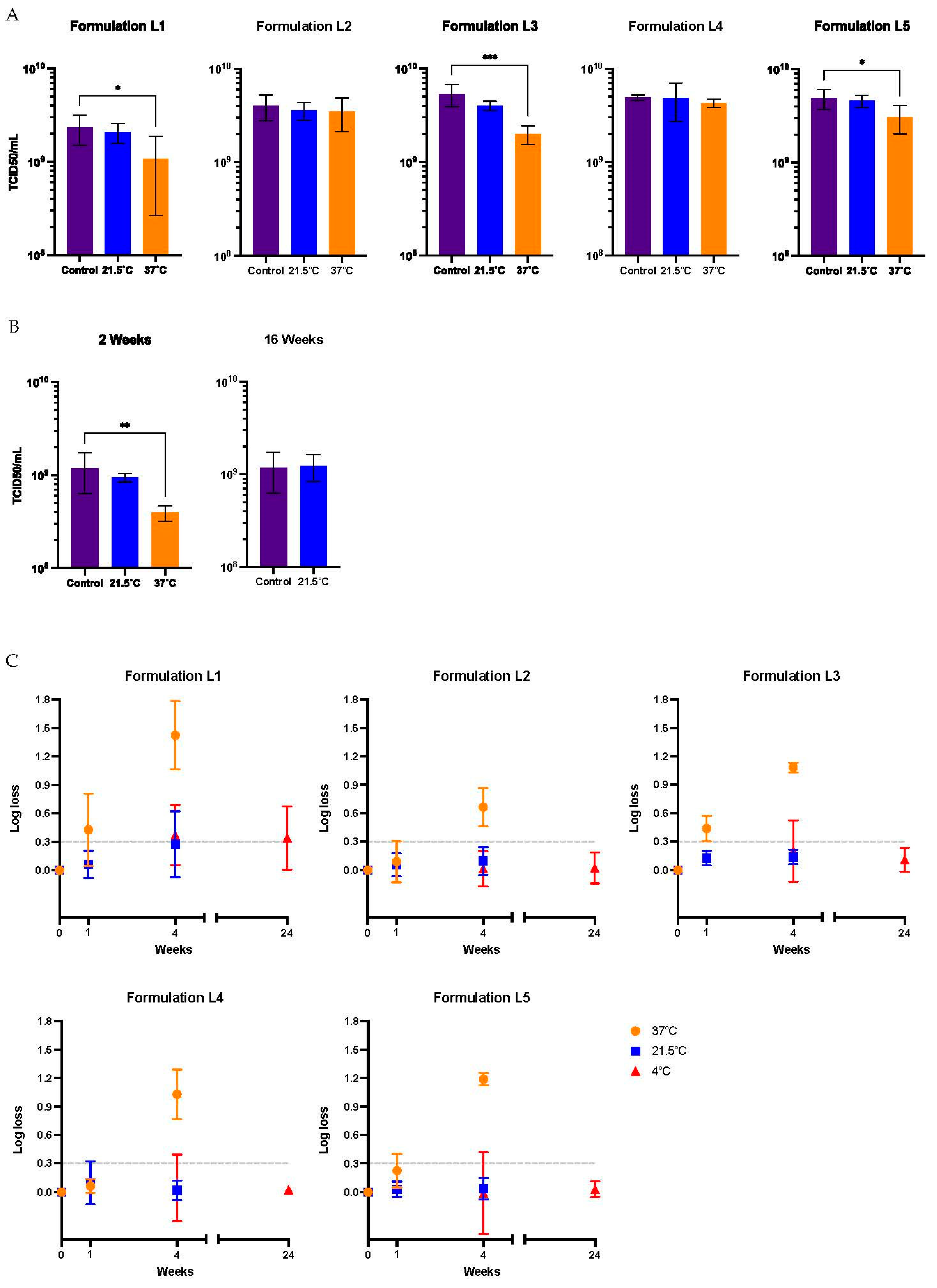
Figure 3.
Arrhenius plot of Ad-CMV-F stability data in formulation L2. The y-axis shows the natural log (LN) of the infectivity loss (k), where k is expressed as log loss (base 10). The data are shown as the mean. T = absolute temperature in Kelvins. The dashed line represents a titer loss of 0.14 log (i.e., 27%), equivalent to the average variability of the TCID50 assay.
Figure 3.
Arrhenius plot of Ad-CMV-F stability data in formulation L2. The y-axis shows the natural log (LN) of the infectivity loss (k), where k is expressed as log loss (base 10). The data are shown as the mean. T = absolute temperature in Kelvins. The dashed line represents a titer loss of 0.14 log (i.e., 27%), equivalent to the average variability of the TCID50 assay.
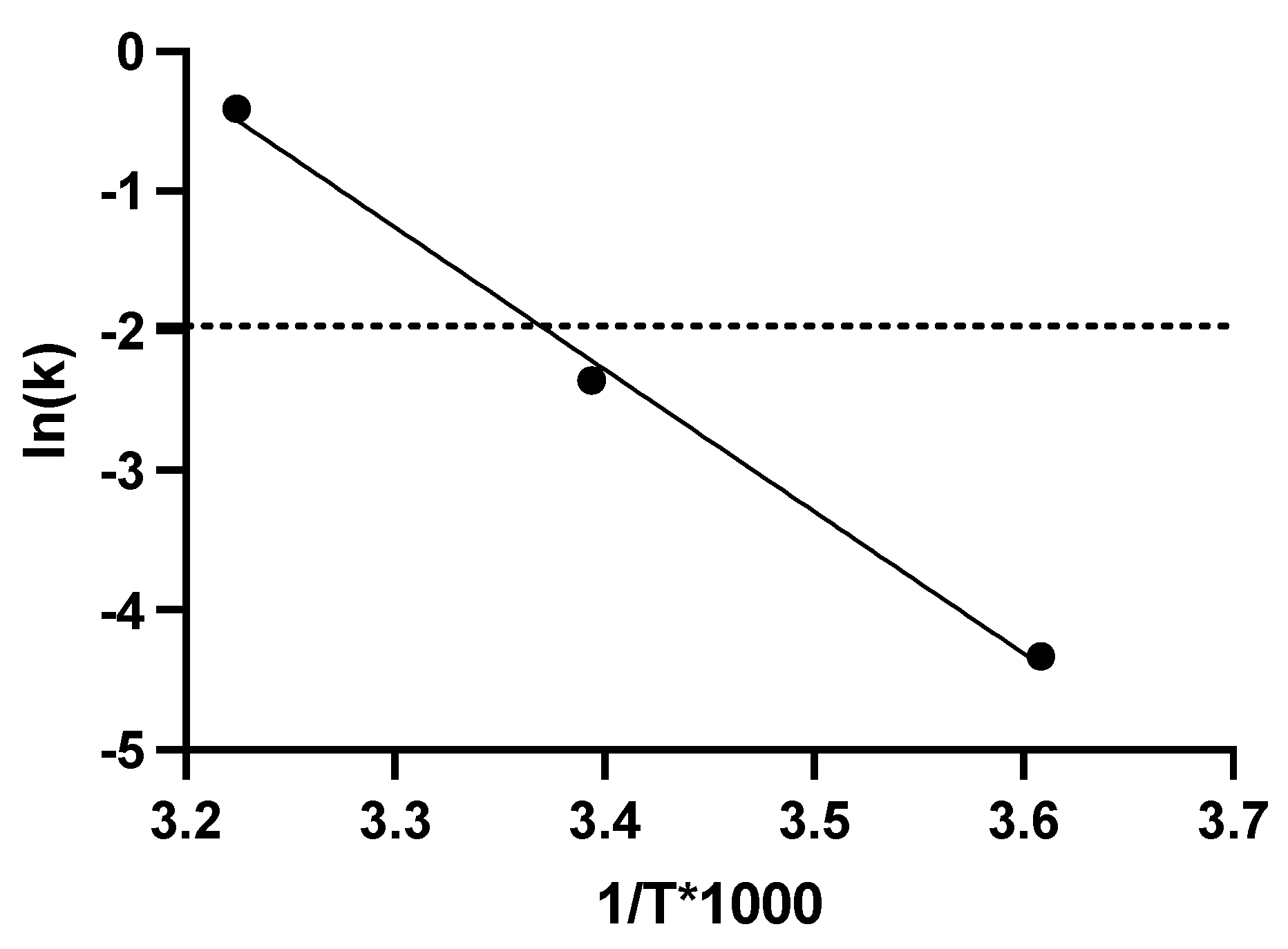
Figure 4.
Characterization of the three solid formulations after lyophilization cycle optimization A) Representative images depicting cake formation in the three solid formulations. B) Functional titer loss after lyophilization in three different runs. Values represent mean ± standard deviation (n = 2 for runs #1 and #3, n = 4 for run #2).
Figure 4.
Characterization of the three solid formulations after lyophilization cycle optimization A) Representative images depicting cake formation in the three solid formulations. B) Functional titer loss after lyophilization in three different runs. Values represent mean ± standard deviation (n = 2 for runs #1 and #3, n = 4 for run #2).
Figure 5.
Functional titer after storage at 37°C, 21.5°C, and 4°C for 1, 4, and 24 weeks in different solid formulations. A) Solid formulation S1. B) Solid formulation S2. C) Solid formulation S3. Values represent mean ± standard deviation (n=5), ****p < 0.0001, ***p < 0.001, **p < 0.01, by analysis of variance (ANOVA) followed by Dunnett’s test.
Figure 5.
Functional titer after storage at 37°C, 21.5°C, and 4°C for 1, 4, and 24 weeks in different solid formulations. A) Solid formulation S1. B) Solid formulation S2. C) Solid formulation S3. Values represent mean ± standard deviation (n=5), ****p < 0.0001, ***p < 0.001, **p < 0.01, by analysis of variance (ANOVA) followed by Dunnett’s test.
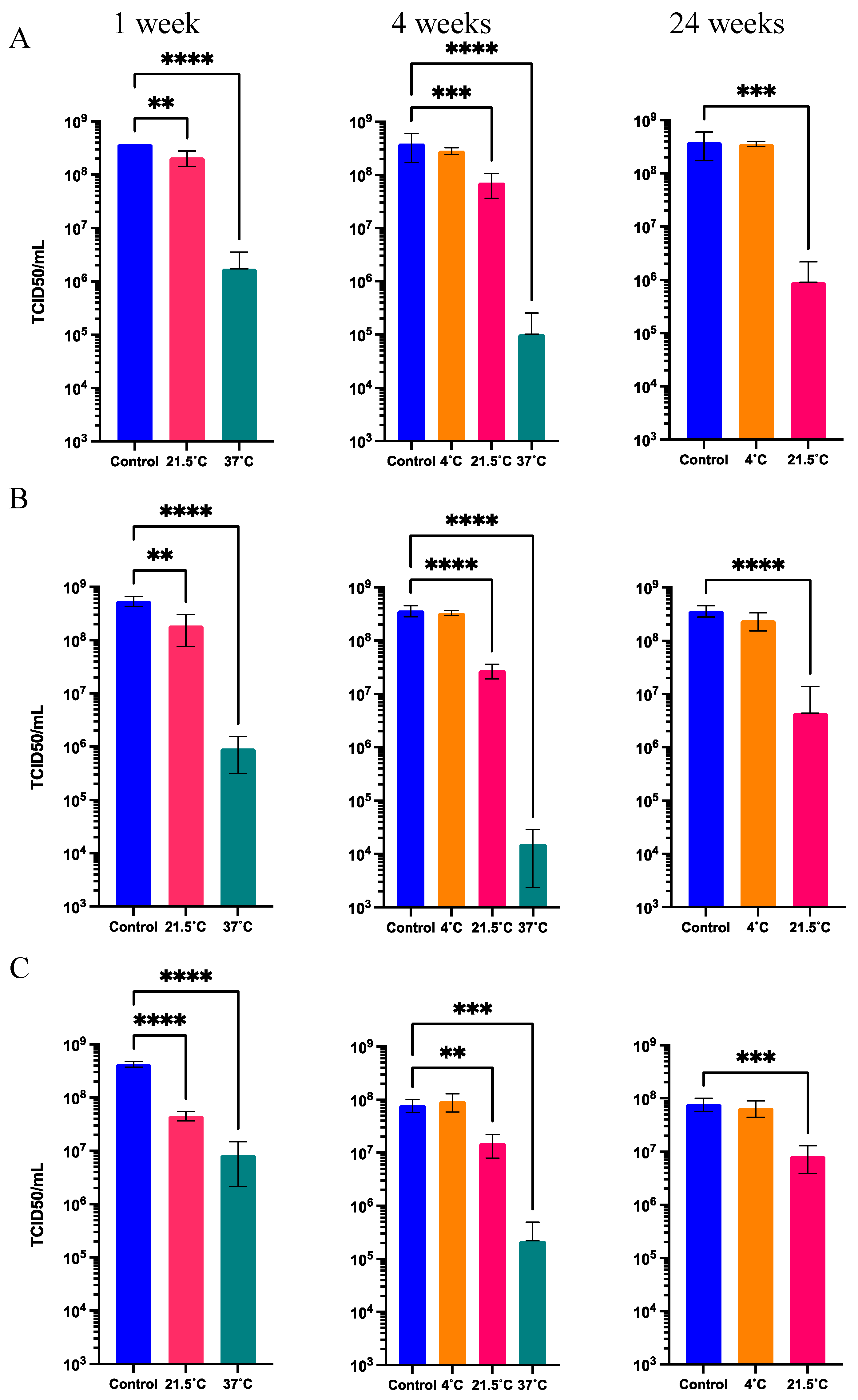
Figure 6.
Functional titer after storage at 21.5°C for 1 week in the three solid formulations. Values represent mean ± standard deviation (n=5).
Figure 6.
Functional titer after storage at 21.5°C for 1 week in the three solid formulations. Values represent mean ± standard deviation (n=5).
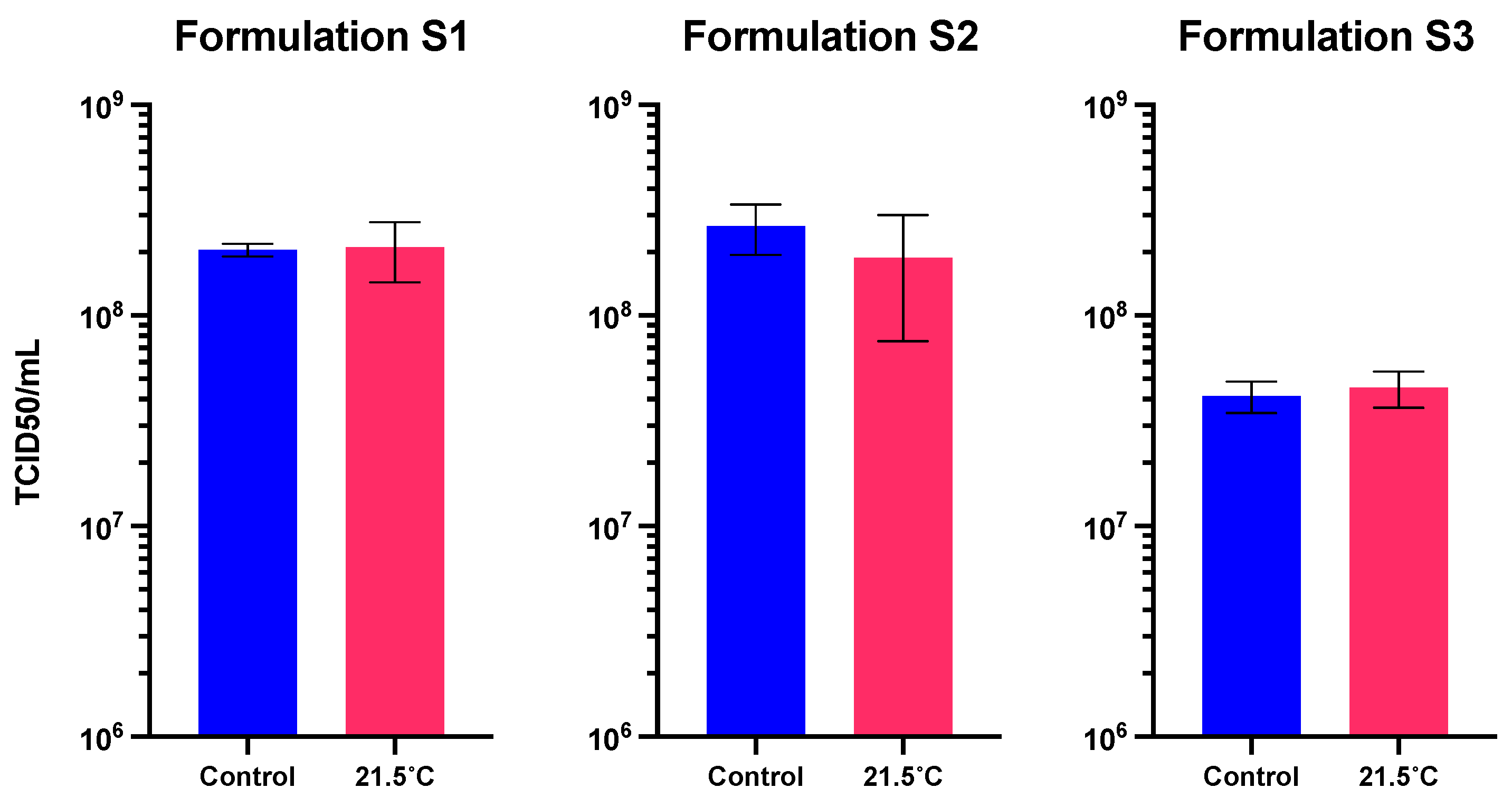
Figure 7.
Schematic representation (A) and results of the immunization experiment, in which chickens were vaccinated by parenteral (B) injection of the CsCl purified Ad-CMV-F adenovirus or were mucosally immunized (C) by intranasal delivery of the virus clarified by filtration steps. All animals were challenged 56 days after the primary immunization with the homologous strain. One of the groups vaccinated by intramuscular injection with purified adenovirus was challenged with a heterologous NDV strain. The doses evaluated ranged from 1 x 109 to 5 x 107 TCID50 viral particles per animal, and a high degree of protection was seen in all groups. Remarkably, the group i.n immunized with 5 x 108 TCID50 infectious particles per animal reached 100% protection to the NDV challenge. For each animal group, the dose is shown between parentheses and the percent of protection is shown.
Figure 7.
Schematic representation (A) and results of the immunization experiment, in which chickens were vaccinated by parenteral (B) injection of the CsCl purified Ad-CMV-F adenovirus or were mucosally immunized (C) by intranasal delivery of the virus clarified by filtration steps. All animals were challenged 56 days after the primary immunization with the homologous strain. One of the groups vaccinated by intramuscular injection with purified adenovirus was challenged with a heterologous NDV strain. The doses evaluated ranged from 1 x 109 to 5 x 107 TCID50 viral particles per animal, and a high degree of protection was seen in all groups. Remarkably, the group i.n immunized with 5 x 108 TCID50 infectious particles per animal reached 100% protection to the NDV challenge. For each animal group, the dose is shown between parentheses and the percent of protection is shown.
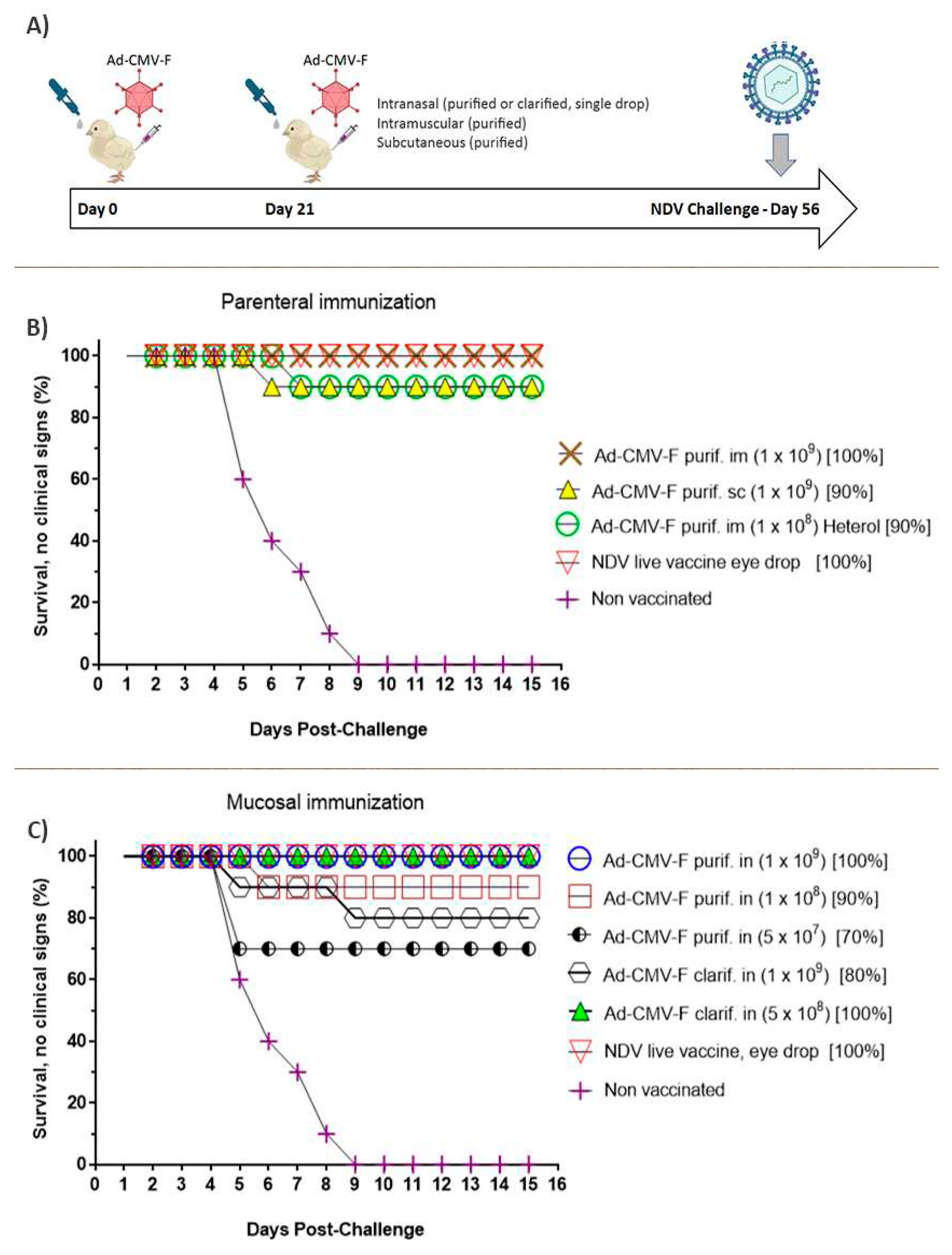

Table 1.
Composition of the five liquid formulations assessed for stability of the Ad-CMV-F adenoviral vector.
Table 1.
Composition of the five liquid formulations assessed for stability of the Ad-CMV-F adenoviral vector.
| Liquid formulations | ||||
|---|---|---|---|---|
| Formulation L1 | Formulation L2 | Formulation L3 | Formulation L4 | Formulation L5 |
| 100mM NaCl | 25mM NaCl | 35mM NaCl | 100mM NaCl | 25mM NaCl |
| 10mM Tris | 20mM Tris | 10mM Histidine | 10mM Tris | 10mM Tris |
| 2mM MgCl2 | 2mM MgCl2 | 1mM MgCl2 | 2mM MgCl2 | 2mM MgCl2 |
| 2% Sucrose | 2% Sucrose | 7.5% Sucrose | 5% Trehalose | 5% Trehalose |
| 0.005% w/v Poloxamer 188 | 0.1% w/v Polysorbate-80 | 0.5% v/v Polysorbate-80 | 0.5% v/v Polysorbate-80 | |
| 0.1mM EDTA | 3% Sorbitol | |||
| 0.5% Ethanol | ||||
| pH 7.5 | pH 7.5 | pH 6.6 | pH 7.5 | pH 7.5 |
Table 2.
Composition of the three solid formulations assessed for the stability of the Ad-CMV-F adenoviral vector.
Table 2.
Composition of the three solid formulations assessed for the stability of the Ad-CMV-F adenoviral vector.
| Solid formulations | |||
|---|---|---|---|
| Formulation S1 | Formulation S2 | Formulation S3 | |
| 100mM NaCl | 100mM NaCl | 100mM NaCl | |
| 10mM Tris | 10mM Tris | 10mM Tris | |
| 1mM MgCl2 | 1mM MgCl2 | 1mM MgCl2 | |
| 5% Mannitol | 5% Mannitol | 5% Mannitol | |
| 5% Sucrose | 5% Trehalose | 5% Inulin | |
| pH 8.2 | pH 8.2 | pH 8.2 | |
Table 3.
Evaluation in shake flask experiments of two different culture media and a streamlined fed-batch cultivation mode for the production of the Ad-CMV-F adenoviral vector. Infectious titers of ~109 TCID50/mL were measured in culture supernatant of lysed cells. The mean titers of duplicate measurements plus the standard deviations are shown.
Table 3.
Evaluation in shake flask experiments of two different culture media and a streamlined fed-batch cultivation mode for the production of the Ad-CMV-F adenoviral vector. Infectious titers of ~109 TCID50/mL were measured in culture supernatant of lysed cells. The mean titers of duplicate measurements plus the standard deviations are shown.
|
Medium |
Approach |
Supplement | TCID50/mL |
|---|---|---|---|
| Harvest (mean ± SD) | |||
| HyCell Trans-Fx-H #1 | No additives | 2.78 x 108 ± 2.90 x 107 | |
| HyCell Trans-Fx-H #2 | Supplementing during the cell growth phase only | Cell Boost™ 5 bolus at 5% (v/v) |
9.77 x 108 ± 8.53 x 107 |
| HyCell Trans-Fx-H #3 | Supplementing during the complete process | Cell Boost™ 5 bolus at 5% (v/v) |
2.11 x 109 ± 1.49 x 108 |
| HEK-GM #4 | No additives | 3.82 x 108 ± 1.20 x 107 | |
| HEK-GM #5 | Supplementing during the cell growth phase only | HEK-FS bolus at 3% (v/v) | 7.22 x 108 ± 5.80 x 107 |
| HEK-GM #6 | Supplementing during the complete process | HEK-FS bolus at 3% (v/v) | 1.83 x 109 ± 1.77 x 108 |
Table 4.
Different bioreactor production runs were conducted, and the infectious titers shown were calculated in Tissue Culture Infectious Dose 50 assays, before any concentration or purification steps.
Table 4.
Different bioreactor production runs were conducted, and the infectious titers shown were calculated in Tissue Culture Infectious Dose 50 assays, before any concentration or purification steps.
| Production | MOI | Titer before purification steps (TCID50/mL) |
|---|---|---|
| Ad-CMV-F Production 1 | 0.05 - 1 | 5.62 x 108 |
| Ad-CMV-F Production 2 | 1 | 3.89 x 108 |
| Ad-CMV-F Production 3 | 3.5 | 2.01 x 109 |
| Ad-CMV-F Production 4 | 3.5 | 5.10 x 109 |
Table 5.
Adenoviral titers and step recovery values from fractions before and after the depth filtration and Tangential Flow Filtration steps. The samples from before and after these steps were quantified by TCID50 and ddPCR assays. Different bioreactor runs and faction volumes were used in the experiment.
Table 5.
Adenoviral titers and step recovery values from fractions before and after the depth filtration and Tangential Flow Filtration steps. The samples from before and after these steps were quantified by TCID50 and ddPCR assays. Different bioreactor runs and faction volumes were used in the experiment.
| Test 1 | Fraction Volume (mL) | Titer (IVP/mL) | IVP total | Step Recovery (%) |
|---|---|---|---|---|
| Harvest | 100 | 5.60E+09 | 5.60E+11 | |
| After Depth Filter + 0.22µm | 200 | 3.10E+09 | 6.20E+11 | 100 |
| Test 2 | ||||
| Harvest | 875 | 3.70E+08 | 3.24E+11 | |
| After Depth Filter + 0.22µm | 875 | 5.60E+08 | 4.90E+11 | 100 |
| Test 3 | ||||
| Before TFF | 480 | 4.64E+08 | 2.22E+11 | |
| After TFF (300 kDa) | 70 | 4.64E+09 | 3.24E+11 | 100 |
| Test 4 | ||||
| Before TFF | 875 | 5.60E+08 | 4.90E+11 | |
| After TFF (300 kDa) | 190 | 3.53E+09 | 6.71E+11 | 100 |
Table 6.
Adenoviral titers at different time points of the process are shown for the two production lots used for animal experiments. Analytical quantifications by TCID50 and ddPCR assays were run for the samples under study.
Table 6.
Adenoviral titers at different time points of the process are shown for the two production lots used for animal experiments. Analytical quantifications by TCID50 and ddPCR assays were run for the samples under study.
|
Production |
Infectious Titer (TCID50/mL) in supernatant | Downstream processing | Starting Volume (mL) | Final Volume after DSP (mL) | Infectious Titer (TCID50/mL) | Total particles (VG/mL) |
|---|---|---|---|---|---|---|
| Ad-CMV-F Production 1 |
5.62 x 108 | CsCl purified, sterile filtration | 1050 | 11.5 | 1.58 x1010 | 7.60 x1010 |
| Ad-CMV-F Production 4 |
3.06 x 109 | Depth filtration, TFF, final Amicon concentration, sterile filtration | 540 | 16.5 | 3.10 x1010 | 2.71 x1010 |
Table 7.
Optimized lyophilization cycle.
| Step | Shelf temp (°C) | Pressure (mTorr) | Time (min) |
|---|---|---|---|
| Loading | 5 | atm | 1 |
| Freezing | -50 | atm | 55 |
| Freezing | -50 | atm | 120 |
| Annealing | -15 | atm | 120 |
| Freezing | -50 | atm | 120 |
| Vacuum | -50 | 22 | 30 |
| 1st drying | -50 | 22 | 2100 |
| 2nd drying ramp | 0 | 22 | 50 |
| 2nd drying | 0 | 22 | 240 |
| 3rd drying ramp | 20 | 22 | 20 |
| 3rd drying | 20 | 22 | 900 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



