
Submitted:
10 November 2023
Posted:
14 November 2023
You are already at the latest version
Abstract
A picture is worth a thousand words. Many branches of Science have been historically benefited with plots and visual analyses (lately, image processing and deep learning) alongside with traditional number crunching. In Molecular Biophysics, one such problem is the structure validation problem in proteins which stands with a history of plot-tools being effectively serving the complex problem to its complete resolution. Spanning across six decades, validation of protein structures (from experimental to modeled) dates back to the legendary Ramachandran Plot (with its never ending growth and modern-day applications) to the relatively recent innovation of the Complementarity Plot (CP), establishing the dual nature of complementarity as the physical basis of both binding and folding of proteins. Lately, CP has been extended to serve as a trustworthy free energy predictor utilizing supervised learning in the form of a comprehensive web-server (EnCPdock: https://www.scinetmol.in/EnCPdock/) that can be directly used in the design of protein interfaces. The commentary recapitulate the history of structure validation with a special emphasis on plot tools, highlighting key features and important discoveries worth re-visiting.
Keywords:
Proteins
; Protein Science
; Structure validation
; Ramachandran Plot
; Complementarity Plot
There is a saying that “A picture is worth a thousand words” which has lately been made more realistic and (should we say) literal during setting of the de-facto standard for natural language processing tasks in image recognition and computer vision [1]. In that same spirit, Images or visual analyses have served overwhelmingly in different branches of science elegantly complementing all number crunching exercises. In molecular biophysics, the protein folding problem is one of the major unsolved (at least partially) problems branching out in a trifurcated way to (i) the thermodynamic problem, (ii) the kinetic problem and (iii) the structure prediction problem. Although the structure prediction problem (said to be the ‘holy grail’ of structural biology) have lately drawn much attention of the community through the success of deep learning and Alpha-Fold, the biophysical basis of protein folding still remain largely unexplained [2]. One of the related problems especially for protein crystallographers and other members of the experimental structural biology fraternity is the structure validation problem. India stands well in this field globally and historically, largely because of Prof. G.N. Ramachandran’s contribution to the field, for his extensive works on the collagen triple helix structures and for the legendary Ramachandran Plot [3], which yet after its 60 years of its initial proposition, remain invaluable and indispensable in protein conformational analyses [4,5]. While no full protein crystal structure was publicly available at the time of its proposition [3], the plot (or, in other words, the map) has swelled across its rigid boundaries (from the days of procheck [6] to Molprobilty [7], however retaining its original overall pattern) giving rise to a 4th layer (‘generously allowed regions’) over and above its three existing regions (‘allowed’, ‘partially allowed’, disallowed’), thereby accommodating more and more flexible conformations. Hence, new idiosyncratic members of the big protein family (such as the fold switch proteins [8] and intrinsically disordered proteins [9]), each and all, have found a place in the Ramchandaran map pointing out to its robustness and authenticity. The growing area of its innovative applications is still not at a halt, for example, exploring how the plot may be used to probe ‘disorder-to-order transitions’ of a protein region undergoing protease cleavage [10].
Motivated from the famous P-V diagram, Ramachandran developed his two dimensional Φ-Ψ plot to describe the stereo-chemistry of growing polypeptide chains, based on the principles of steric clashes. He took a poly-alanine model to determine the angular ranges of {Φ, Ψ} for which a di-peptide unit could give rise to steric clashes (and hence would be ‘disallowed’) [11]. The coordinates of relevant atoms upon each conformational twist were determined by the principles of third and forth atom fixations. The methyl group in alanine being a small and chemically inert side-chain simplified matters during the initial proposition of the plot, latter to be extended using the different specific side-chains attributed to the naturally occurring amino acids. A similar gradually step-up strategy can be found in the development of Conformation dependent library (CDL [12,13]) of ideal values (from the trivial uni-modal ideal values [14]) for main-chain bond angles and also in the side-chain prediction problem [15]. Even today, the most primary and fundamental parameter one has to validate against his/her experimentally solved protein structure is the Ramachandran Plot. However, today a vast array of parameters is available alongside the Ramachandran Plot complimenting it in protein structure validation. Indeed, the Ramachandran Plot is not all-inclusive, for example, a well packed folded globular protein with native side-chains (X-ray structure) and one with the same backbone with computationally randomized side-chain conformations (decoy) would map to an identical distribution in the Ramachandran Plot [16], thus emphasizing the demand of other exclusive and complementing measures. Deviations from ideality for bond lengths and angles [14], atomic short contacts (steric clash scores [7]), the distribution of the side-chain conformers (rotamers) [17] and hydrogen bonding parameters [18] are to name a few. The 1990’s decade was instrumental in putting forward a strong community of computational structural biologists dedicated to conformational analysis of proteins, given just marginally enough amount of protein crystal structures were available to carry forward meaningful statistics. Jannet Thornton was a pioneer figure in such analyses who laid the foundations of almost all structural biology problems (particularly in proteins) requiring and benefiting from the advent of modern computers and programming languages. Her works span across from solvent accessible surface area calculations [19] to structural validations in proteins (Procheck [6]), from probing the diversity of protein – ligand interactions in terms of reaction micro-environment [20] to the early work on salt-bridges [21] and disulphide bridges [22] within and between proteins. It was during this decade that the shape and electrostatic complementarities (Sc [23], EC [24]) were defined (Figure 1), demarcated and benchmarked at the protein – protein interface (by Peter Colman and co-workers). These calculations relied immensely on the advent of molecular surface (Connolly [25]) construction algorithms. The concept of complementarity with its dual nature and extensive sophistication were to be extended in the realm of protein structure validations [26] within a couple of forthcoming decades (2010’s).
After the Procheck [6] era, Molprobity [7] (along with Whatcheck [18]) had taken over the state-of-the-art in protein structure validations. In particular, the Protein Data Bank (PDB) [27] had high recommendations on these standard checks for any experimental structural biologists looking to submit his/her solved co-ordinates into the PDB. Molprobity has a highly sophisticated clash score involving hydrogen coordinates, geometrically fixed by REDUCE [28] while the hydrogen bond parameter in Whatcheck was apparently the only working electrostatic filter that may indirectly signal for unbalanced partial charges within the protein core. After the initial formulation of the complementarity measures [23,24] at the protein – protein interface, equivalent measures got formulated (as an extension) and characterized within the globular protein interior attributed to dedicated efforts in certain laboratories across the globe (e.g., Banerjee et. al.,) [26,29,30,31]. Folding was envisaged as self-docking of the embedded side-chains onto the polypeptide chain thereby conceptually bridging the gap between binding and folding in proteins based on complementarity [26]. The concept got well-supported by the results of other giant laboratories in the field working on the protein interior with similar (yet, alternative) approaches (e.g., anisotropic side-chain packing [32]). Overall, the combined effect of ‘packing and electrostatic harmony’ within the protein interior was realized to be an indispensable characteristic feature of native-like well-folded proteins and serves as a conjoint complex quality index rendering the authenticity of the (solved or built) atomic coordinates. Packages were also put forward in order to detect packing defects or holes [33] (often, short contacts too) within proteins.
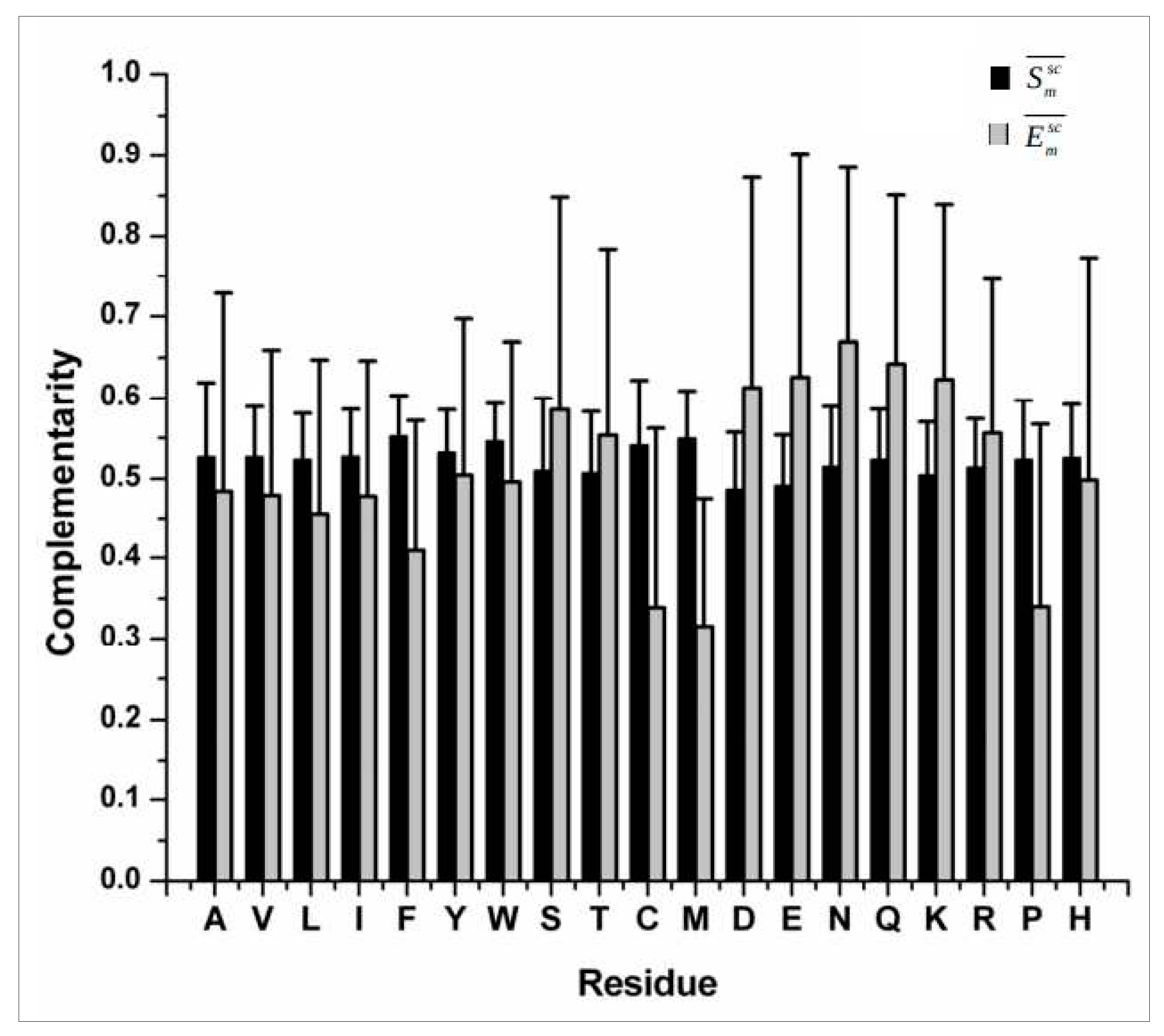
Several shape complementarity measures [30,34] (attributed to the three dimensional jig-saw puzzle model of protein packing) have been attempted to probe the geometric fit of buried and partially buried side-chains within natively folded globular protein interiors (e.g., Sm [29]), directly or indirectly adapting from the original Sc shape correlation statistic (proposed for protein – protein interfaces) [23] which is simple, effective and elegant. The complementarity of electrostatic potentials (Em) [24] on the side-chain van der Waals surface has also been surveyed across several publications which suggest that the surface potentials generated by the target and the neighboring set of atoms on a given surface trend to be anti-correlated. The long-range electric fields generated by polar main-chain atoms cast their shadow over the side-chain surface in a way that all residues, irrespective of their hydrophobicity and burial, attain a fairly uniform level of overall complementarity. Side-chain atoms of polar/charged residues additionally contribute to the elevated complementarity attained on their side-chain surfaces [26]. Overall, it was realized that the dual nature of complementarity with the elevated average thresholds in {Sm, Em} (Figure 2) could together serve as an additional non-redundant check-point in structural validations of proteins (alongside the Ramachandran Plot) based on interior packing and electrostatic harmony of side-chains within the native fold.
Figure 2.
Trends in shape and electrostatic complementarities (Figure reproduced from the Supporting Materials of [26]).
Figure 2.
Trends in shape and electrostatic complementarities (Figure reproduced from the Supporting Materials of [26]).
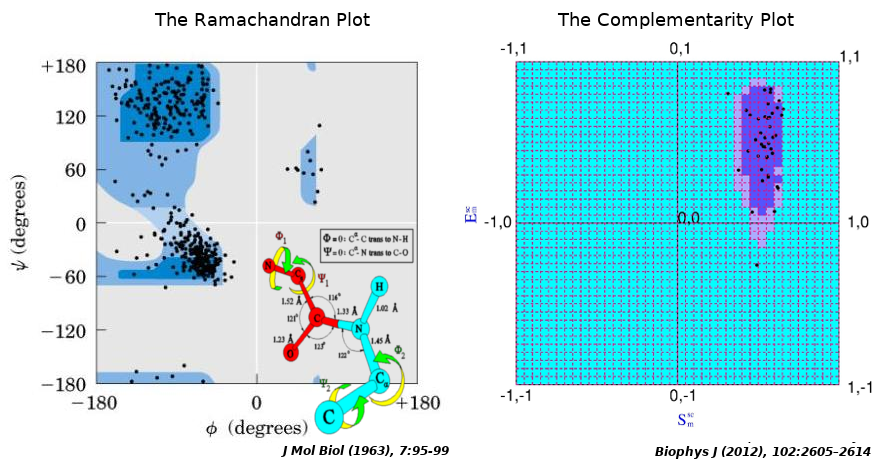
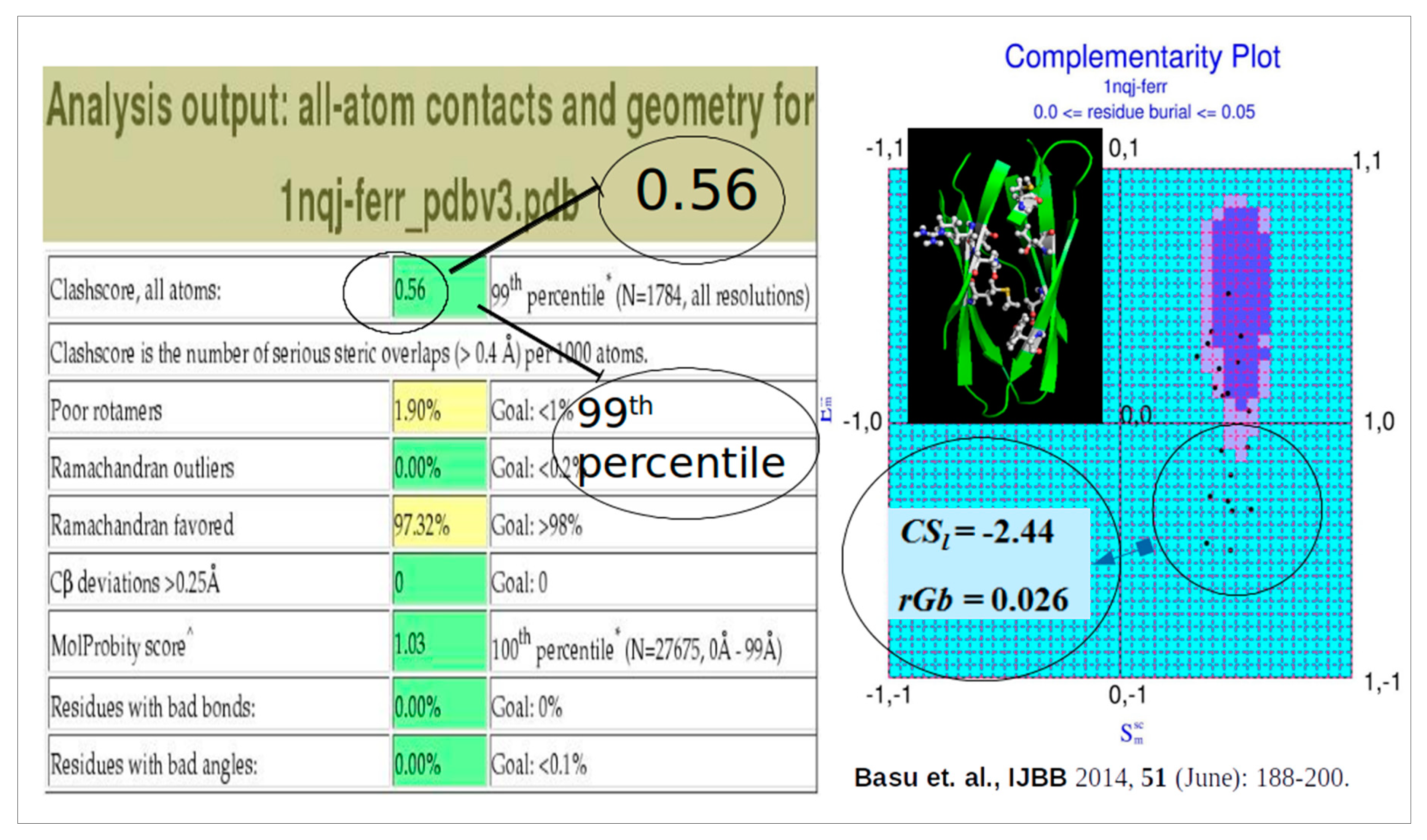
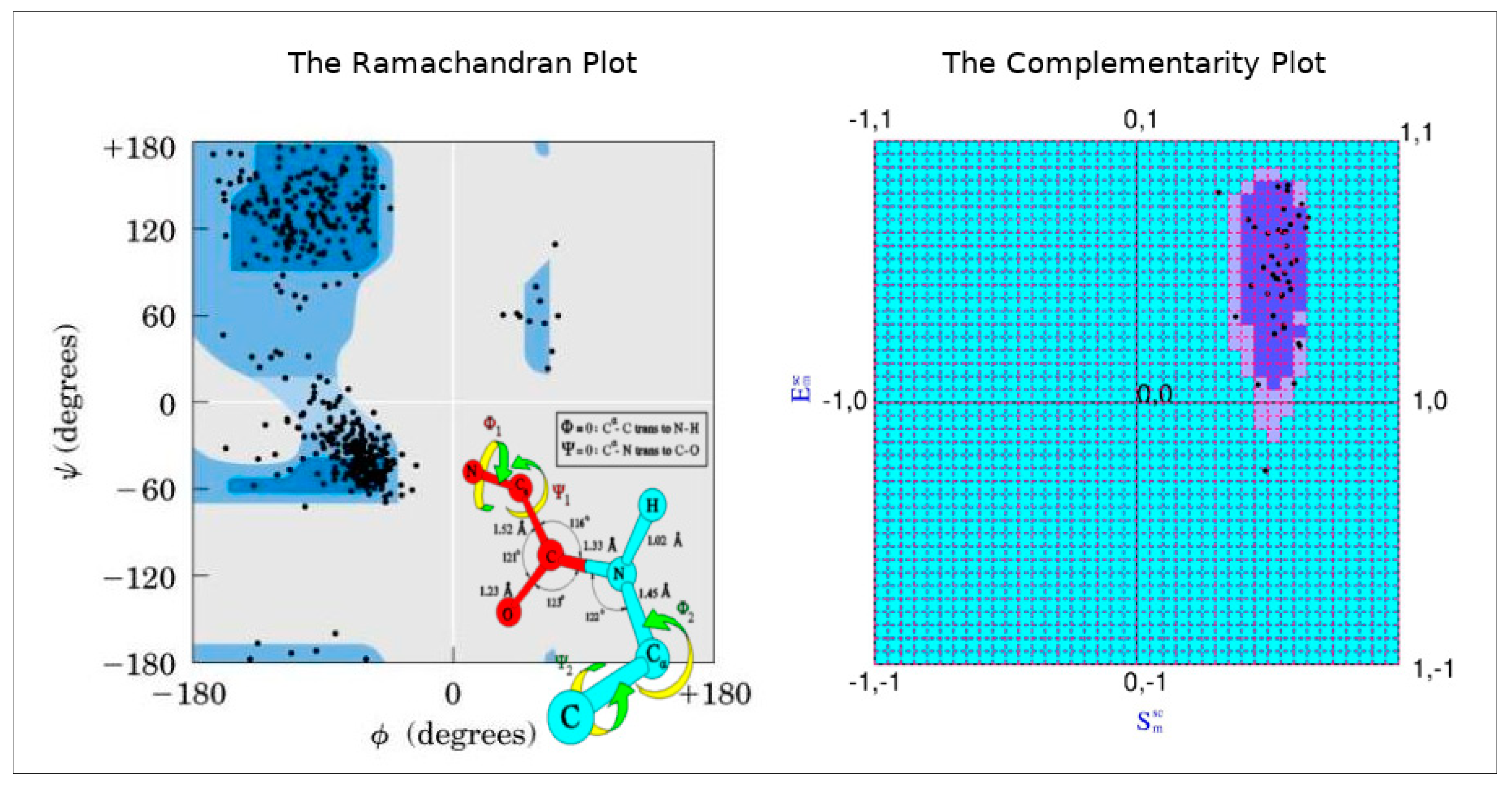
To that end, the Complementarity Plot (CP) was proposed, largely being inspired by the Ramachandran Plot in its design (but, of course, not in its physicochemical attributes). The Complementarity Plot has shape and electrostatic complementarity along its X- and Y-axes analogous to the two main-chain torsion angles Φ and Ψ in the Ramachandran Plot. Both Sm and Em are correlation functions with theoretical ranges of -1 to +1 (likewise, {Φ, Ψ} → [–180, 180]) (Figure 3). These measures (Sm, Em) are computed over completely / partially buried residue (van der Waal’s) surfaces with respect to their environment constituted by rest of the polypeptide chain, and, together as an ordered pair, is a sensitive indicator of the harmony or disharmony of interior residues with regard to the short and long range forces sustaining the native fold. The term 'Complementarity Plot' (CP) is perhaps a misnomer as practically there are three plots: CP1, CP2, CP3 serving for plotted residues with different degree of solvent exposure. To that end metrices have been designed to numerically report the CP-results based on a log odd score (Complementarity Score: CSl) and an accessibility score (rGb) [31] as a negative-check to confirm that the score is authentic (i.e., not being attained by a few residues in a partially unfolded structure by chance). Analogous to the Ramachandran Plot, CPs are demarcated by two contours thereby giving rise to three disjoint regions: 'probable', 'less probable' and 'improbable' (analogous to the ‘allowed’, ‘partially allowed’ and 'disallowed' regions of the Ramachandran Plot).
While Ramachandran Plot is deterministic in nature, CP is probabilistic. While, the Ramachandran plot deals with main-chain torsion angles with locally restricted errors, CP deals with geometric, electrostatic fit of the interior side-chains with their local, non-local neighborhood. Disharmony (misfit) in these conjugated parameters ({Sm, Em}) may arise due to a vast range of errors coming from bond angles and/or dihedrals from effectively the whole folded protein chain. Alongside detecting local errors in atomic coordinates, CP also correctly matches an amino acid sequence to its native three dimensional fold situated amid decoys. Astonishingly somewhat, among its different applications, CP could vitally signal for unbalanced partial charges embedded within protein cores that have been computationally altered with miss-identified buried side-chains [31] which could pass all standard checks even in Molprobity [7]. In this computational exercise, the hydrophobic characters were altered while the shape and size of the side-chains were retained as much as possible (Ala → Ser, Ser ↔ Cys, Thr ↔ Val, Phe ↔ Tyr, Leu → Asn, Leu → Asp, Ile → Met etc.), the side-chains were fit by SCWRL4.0 [15] while the decoy structures were energy minimized by CHARMM [35] to remove all steric clashes and hence Molprobity could not find anything wrong. This clearly emphasized the importance of adding an electrostatic filter in the recommended validation filters in the PDB [27] but to the best of our knowledge, this has not yet been addressed till date.
Figure 4.
CP signals for deliberately incorporated unbalanced partial charges in protein cores which even Molprobity can’t detect.
Figure 4.
CP signals for deliberately incorporated unbalanced partial charges in protein cores which even Molprobity can’t detect.
Over the years (since, 2012 [26]) CP (and its variants: CPint [36], CPdock [16], EnCPdock [37]) has found its way as an established structural validation tool for proteins and protein complexes [16] and has its demonstrated applications across several areas of protein science spanning from homology modeling [38], docking scoring and optimization [10,39] to protein, epitope and interface design [40,41,42]. During these applications, it was realized that complementarity in terms of shape and electrostatics (Sc, EC) together forms the physical basis of protein-protein interaction. Also, these are indirect probabilistic estimates of affinity and stability of the interactions, mapped to their locations in the complementarity plot (CPdock [16]) amid different regions (‘probable’, ‘less-probable’, ‘improbable’).
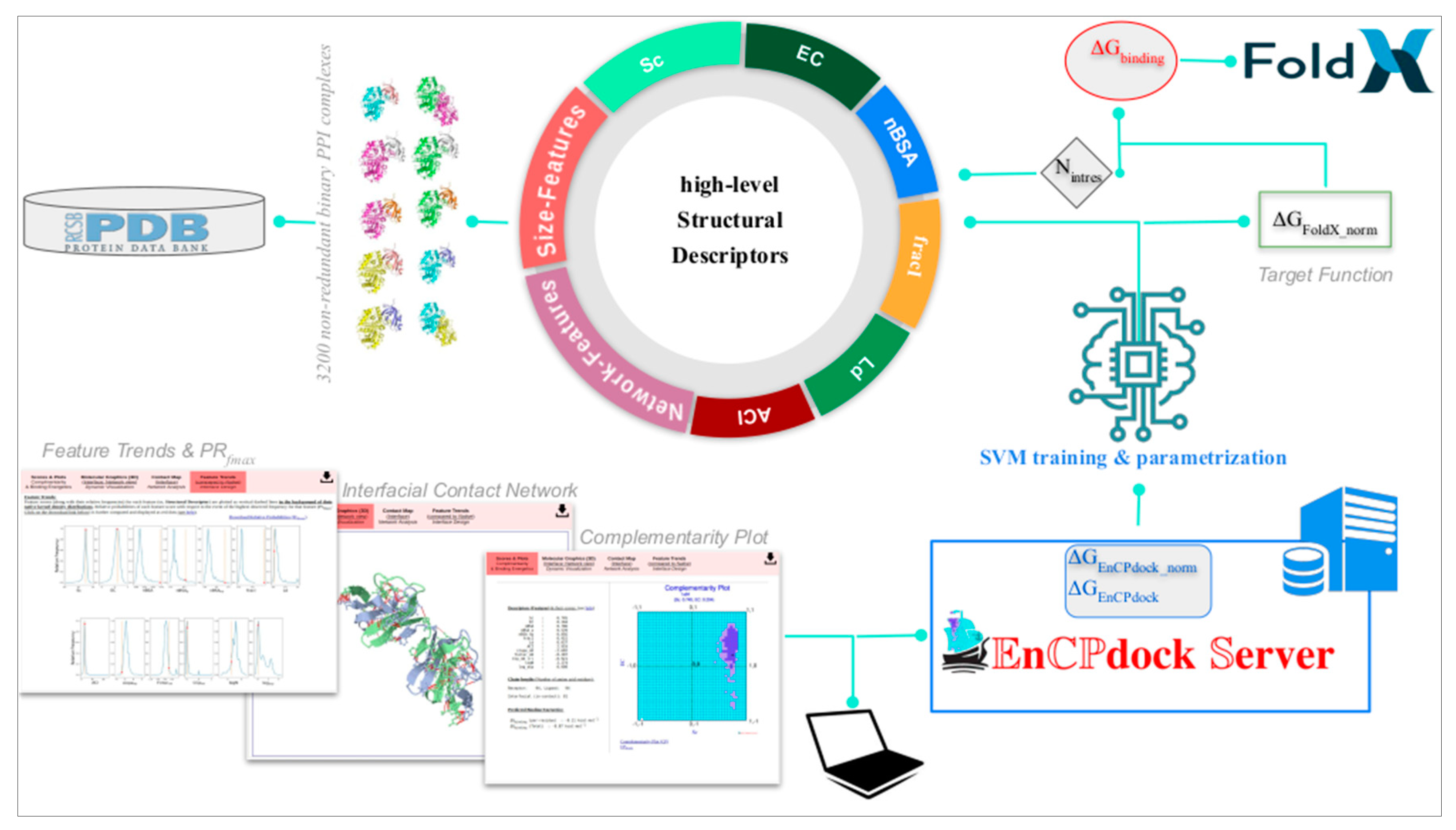
Lately, a more direct approach has been adapted to convert these indirect probabilistic estimates of affinity and stability to actual free energies of binding (∆Gbinding) by a structure based thermodynamics approach implementing Artificial Intelligence (AI). This has been presented as a unique comprehensive user-friendly web-interface (or, web-server), namely, EnCPdock (https://www.scinetmol.in/EnCPdock/) to be used for the direct conjoint comparative analyses of complementarity and binding energetics in proteins. EnCPdock returns an AI-predicted ∆Gbinding computed by combining complementarity (Sc, EC) and other high-level structural descriptors (input feature vectors) [37], and, renders a prediction accuracy comparable to the state-of-the-art. EnCPdock further locates a PPI complex in terms of its {Sc, EC} values (taken as an ordered pair) in CPdock (the docking – version of the two-dimensional Complementarity Plot). In addition, it also generates mobile molecular graphics of the interfacial atomic contact network for further analyses. Perhaps, most importantly, EnCPdock also furnishes individual feature trends along with the relative probability estimates (Prfmax) of the obtained feature-scores with respect to the events of their highest observed frequencies. Together, these functionalities are of real practical use for structural tinkering and intervention as might be relevant in the design of targeted protein-interfaces. Thus, as it has been for the Ramachandran Plot, the Complementarity Plot also extends across different distant yet related applications in protein science, being originated primarily as a structure validation tool.
Figure 5.
Schematic diagram of EnCPdock web server workflow. Figure reproduced from [37].
Figure 5.
Schematic diagram of EnCPdock web server workflow. Figure reproduced from [37].
Acknowledgment
The author acknowledges the research committee of Asutosh college for the invitation to write this book chapter (commentary).
References
- Dosovitskiy A, Beyer L, Kolesnikov A, et al (2021) An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale.
- Basu S, Chakravarty D, Hou Q, Uversky V (2023) Editorial: From the Hydrophobic Core to the Globular-Disorder Interface: New Challenges and Insights into Protein Design. Frontiers in Molecular Biosciences 10. [CrossRef]
- Ramachandran GN, Ramakrishnan C, Sasisekharan V (1963) Stereochemistry of polypeptide chain configurations. J Mol Biol 7:95–99. 10.1016/s0022-2836(63)80023-6. [CrossRef]
- Kleywegt GJ, Jones TA (1996) Phi/Psi-chology: Ramachandran revisited. Structure 4:1395–1400. [CrossRef]
- Ramachandran Plot - an overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ramachandran-plotAccessed 5 Nov 2021.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst, J Appl Crystallogr 26:283–291. [CrossRef]
- Chen VB, Arendall WB, Headd JJ, et al (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [CrossRef]
- Bryan PN, Orban J (2010) Proteins that switch folds. Curr Opin Struct Biol 20:482–488. [CrossRef]
- Basu S, Bahadur RP (2021) Conservation and coevolution determine evolvability of different classes of disordered residues in human intrinsically disordered proteins. Proteins. [CrossRef]
- Roy S, Ghosh P, Bandyopadhyay A, Basu S (2022) Capturing a Crucial ‘Disorder-to-Order Transition’ at the Heart of the Coronavirus Molecular Pathology—Triggered by Highly Persistent, Interchangeable Salt-Bridges. Vaccines 10:301. [CrossRef]
- Ramachandran GN, Sasisekharan V (1968) Conformation of Polypeptides and Proteins**The literature survey for this review was completed in September 1967, with the journals which were then available in Madras and the preprinta which the authors had received.††By the authors’ request, the publishers have left certain matters of usage and spelling in the form in which they wrote them. In: Anfinsen CB, Anson ML, Edsall JT, Richards FM (eds) Advances in Protein Chemistry. Academic Press, pp 283–437.
- Tronrud DE, Berkholz DS, Karplus PA (2010) Using a conformation-dependent stereochemical library improves crystallographic refinement of proteins. Acta Crystallogr D Biol Crystallogr 66:834–842. [CrossRef]
- Berkholz DS, Shapovalov MV, Dunbrack RL, Karplus PA (2009) Conformation Dependence of Backbone Geometry in Proteins. Structure 17:1316–1325. [CrossRef]
- Engh RA, Huber R (2006) Structure quality and target parameters. In: Rossmann MG, Arnold E (eds) International Tables for Crystallography Volume F: Crystallography of biological macromolecules. Springer Netherlands, pp 382–392. [CrossRef]
- Krivov GG, Shapovalov MV, Dunbrack RL (2009) Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77:778–795. [CrossRef]
- Basu S (2017) CPdock: the complementarity plot for docking of proteins: implementing multi-dielectric continuum electrostatics. J Mol Model 24:8. [CrossRef]
- Dunbrack RL, Cohen FE (1997) Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci 6:1661–1681. [CrossRef]
- Hooft RWW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272–272. [CrossRef]
- Hubbard S, Thornton J (1993) NACCESS. Computer Program, Department of Biochemistry and Molecular Biology, University College London. - Open Access Library. Available online: http://www.oalib.com/references/5299711Accessed 1 Mar 2017.
- Gr S, Jm T (2005) Conformational diversity of ligands bound to proteins. J Mol Biol 356:928–944. [CrossRef]
- Barlow DJ, Thornton JM (1986) The distribution of charged groups in proteins. Biopolymers 25:1717–1733. [CrossRef]
- Thornton JM (1981) Disulphide bridges in globular proteins. Journal of Molecular Biology 151:261–287. [CrossRef]
- Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234:946–950. [CrossRef]
- McCoy AJ, Chandana Epa V, Colman PM (1997) Electrostatic complementarity at protein/protein interfaces. J Mol Biol 268:570–584. [CrossRef]
- Connolly ML (1983) Analytical molecular surface calculation. Journal of Applied Crystallography 16:548–558. [CrossRef]
- Basu S, Bhattacharyya D, Banerjee R (2012) Self-Complementarity within Proteins: Bridging the Gap between Binding and Folding. Biophys J 102:2605–2614. [CrossRef]
- Berman HM, Westbrook J, Feng Z, et al (2000) The Protein Data Bank. Nucl Acids Res 28:235–242. [CrossRef]
- Word JM, Lovell SC, Richardson JS, Richardson DC (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285:1735–1747. [CrossRef]
- Banerjee R, Sen M, Bhattacharya D, Saha P (2003) The jigsaw puzzle model: search for conformational specificity in protein interiors. J Mol Biol 333:211–226. [CrossRef]
- Basu S, Bhattacharyya D, Banerjee R (2011) Mapping the distribution of packing topologies within protein interiors shows predominant preference for specific packing motifs. BMC Bioinformatics 12:195. [CrossRef]
- Basu S, Bhattacharyya D, Banerjee R (2014) Applications of complementarity plot in error detection and structure validation of proteins. Indian J Biochem Biophys 51:188–200.
- Misura KMS, Morozov AV, Baker D (2004) Analysis of anisotropic side-chain packing in proteins and application to high-resolution structure prediction. J Mol Biol 342:651–664. [CrossRef]
- Sheffler W, Baker D (2009) RosettaHoles: Rapid assessment of protein core packing for structure prediction, refinement, design, and validation. Protein Sci 18:229–239. [CrossRef]
- Word JM, Lovell SC, LaBean TH, et al (1999) Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol 285:1711–1733. [CrossRef]
- Liu H, Song D, Zhang Y, et al (2019) Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins. Phys Chem Chem Phys 21:21918–21931. [CrossRef]
- Basu S, Bhattacharyya D, Wallner B (2014) SARAMAint: The Complementarity Plot for Protein–Protein Interface. Journal of Bioinformatics and Intelligent Control 3:309–314. [CrossRef]
- Biswas G, Mukherjee D, Dutta N, et al (2023) EnCPdock: a web-interface for direct conjoint comparative analyses of complementarity and binding energetics in inter-protein associations. J Mol Model 29:239. [CrossRef]
- Basu S, Bhattacharyya D, Banerjee R (2014) Applications of complementarity plot in error detection and structure validation of proteins. Indian J Biochem Biophys 51:188–200.
- Williams G (2018) Shape complementarity at protein interfaces via global docking optimisation. Journal of Molecular Graphics and Modelling 84:69–73. [CrossRef]
- Basu S, Chakravarty D, Bhattacharyya D, et al (2021) Plausible blockers of Spike RBD in SARS-CoV2—molecular design and underlying interaction dynamics from high-level structural descriptors. J Mol Model 27:191. [CrossRef]
- Biswas G, Ghosh S, Basu S, et al (2022) Can the jigsaw puzzle model of protein folding re-assemble a hydrophobic core? Proteins. [CrossRef]
- Michel-Todó L, Reche PA, Bigey P, et al (2019) In silico Design of an Epitope-Based Vaccine Ensemble for Chagas Disease. Frontiers in Immunology 10:. [CrossRef]

Figure 3.
The Ramachandran Plot and the peptide unit inspires the design on the Complementarity Plot.
Figure 3.
The Ramachandran Plot and the peptide unit inspires the design on the Complementarity Plot.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



