
Submitted:
10 November 2023
Posted:
13 November 2023
You are already at the latest version
Abstract
The surprisingly high catalytic activity of gold has been known
to the heterogeneous catalysis community since the mid-1980s.
Significant effort has been directed towards improving the reactivity of these
surfaces towards important industrial reactions. One such strategy is
the introduction of small amounts of other metals to create Au-based
surface alloys. In this work, we investigate the synergistic effect of
Pt doping of the Au(111) surface towards decreasing the activation
barrier of the methanol dehydrogenation elementary step, within
first-principles density functional theory. To this aim we construct
several models of \ce{Pt}-doped Au(111) surfaces, including a full
\ce{Pt} overlayer and monolayer. The effect of \ce{Pt} surface doping
is then investigated via the computation of adsorption energies of the
various chemical species involved in the catalytic step, and the
estimation of activation barriers of methanol dehydrogenation. Both
electronic and strain effects induced by \ce{Pt} surface doping concur
in substantially lowering the activation energy barrier of this
important reaction elementary step. Morever, in the presence of
preadsorbed atomic oxygen, \ce{Pt} surface doping can be used to
reduce the activation energy for methanol dehydrogenation to as low as
0.1 eV.
Keywords:
Density Functional Theory
; Methanol dehydrogenation reaction
; Nudged Elastic Band
; Activation Energy
; Reaction Energy
1. Introduction
Selective oxidation of alcohols is routinely used in the industrial synthesis of a large number of important chemicals, such as aldehydes, esthers and ketones [1], whose production can be selectively favored throughout the reaction steps leading to full oxidation [2]. In addition, oxidation of alcohols via electrochemical reactions is used in fuel cells [3]. A large number of metals including Pt [4], Pd [5], and Ag [6] and their alloys [7,8] have been used successfully as catalysts in the oxidation reactions. However, the most unexpected discovery in this field has been the catalytic properties of nanosized Au, whose reactivity in the form ultrafine particles was proven in 1987 by Haruta et al. [9]. Since then, the catalytic properties of Au both in bulk [3,7] and particle form [10,11] has been continuously investigated.
While making use of their low environmental cost [12], resistance to poisoning [13], and selectivity [14], the activity of Au catalysts can be improved by several methods including introduction of surface atomic oxygen [14] and tuning of the particle size [15,16]. Another method based on materials design is to utilize the ensemble effects that emerge through alloying the surface with other metals. There is evidence that both alloys [17] and mixtures [18] of Au with other metals outperform pure Au. Of particular interest is the work by Zhao et al. [18] who discovered, via measurements of conversion of 5-hydroxymethylfurfural that while AuPd alloys perform better than Au, a simple Au/Pd mixture gives an even better conversion rate than the alloy. In a detailed density functional theory (DFT) study by Li and Henkelman [19], the reactivity of a Au(111) surface decorated with Pd and Rh patterns of different shapes and sizes were investigated and a clear inverse relation between Pd or Rh surface concentration and ethanol dehydrogenation reaction barrier was discovered. Yi et al. synthesized NiAu nanoparticles of different size and structure, and observed a five-fold increase in benzylalcohol oxidation compared to supported Au particles [15].
Alloys of Au with many metals including Pd [7,18,20], Rh [19], Ag [21], Ni [15], Ru [22] and Ir [22] have been investigated experimentally and theoretically regarding their potential in the selective oxidation of alcohols. Conversely, although Pt is known to catalyze the C-H bond breaking, which tends to be a rate determining step for the partial oxidation of alcohols [23], Au-Pt alloys have received limited attention in this context. However, a noteworthy experimental study by Tenney et al. [24] revealed that the activity of bimetallic AuPt clusters (with Au content greater than 50%) display is similar to that of pure Pt. In another more recent experimental work, Stepanova et al. [25] observed that the introduction of Au into supported Pt clusters leads to an increase in the propane conversion without affecting the selectivity towards propylene. The atomistic origins of these interesting synergistic outcomes have yet to be uncovered.
In this paper, our objective is to explore, within planewave pseudopotential DFT framework and at the atomistic scale the catalytic activity of Pt atoms in the form of substitutional surface alloys with Au. Our model surface calculations aim to explore the methanol dehydrogenation reaction both in the presence and in the absence of surface atomic oxygen species. While atomic oxygen is known to drastically reduce the activation barrier for the dehydrogenation reactions, the pretreatment of the metal surface may prove difficult [26]. We perform this investigation by considering the activation energy for the reactions:
and
where the designation (ads) indicates that the species are all adsorbed on the surface.
Several different reaction pathways for methanol dehydrogenation on extended metallic surfaces and nanostructures are possible, depending on whether the initial bond activation involves the O–H bond (the alkoxy path) or the C–H bond (the hydroxyalkyl path). The question of whether H abstraction from OH- or CH3- groups represents the bottleneck at the beginning of the oxidation reactions must be addressed separately for each catalyst used [7,27]. In fact, depending on the catalyst, as demonstrated in the work by Wu and Wang [28], C–H bond breaking may have a larger activation barrier than that of O–H. In this work, we only focus on the O–H bond scission and compare different Pt-enhanced Au catalysts for this specific step of the reaction mechanism. Theoretical studies of O-assisted selective coupling of CH3OH on gold surfaces [29] and several DFT works on (100) and (310) Au surfaces [7,30] consider the alkoxy path; we concentrate on this elementary reaction step in this work, and use it as a probe reaction to understand the effect of Pt-surface doping on the Au (111) surface.
In addition to the bare Au(111) surface to serve as a benchmark, we consider surface alloys with Pt:Au ratios of 1:8 (1Pt/Au), 1:4 (2Pt/Au), 1:2 (3Pt/Au), and 1:1 (corresponding to a surface Pt monolayer), and a full subsurface Pt layer just below the topmost Au layer. These ratios refer not to the bulk but only to the nine surface atoms of our 3×3 surface slab. For each Pt concentration, we calculate the adsorption energies of CH3OH, CH3O, OH, and atomic O and H together with the energetics of CH3OH/H, CH3OH/O and CH3O/OH coadsorption. Understanding the adsorption and coadsorption energies are important since there may be synergistic stabilizing effects where the total adsorption energy may be greater than the sum of the individual energies [16]. Furthermore, we calculate the activation barrier of the reactions shown in Eqs. (1) and (2) on all model surfaces. The plan of the paper is the following: in Section 2 we give the computational details. Section 3 discusses the most important results of this study, while conclusions are reported in the final Section 4.
2. Computational Details
The calculations were performed using the plane-wave pseudopotential density functional theory [31,32] method within the gradient-corrected approximation (GGA) as implemented in the open source Quantum Espresso [33] code suite. The Perdew-Wang (PW-91) exchange-correlation [34] functional was used to approximate electron-electron interactions. The open-source program XCrysDen [35] was used for visualization and to produce the figures. During BFGS (Broyden-Fletcher-Goldfarb-Shanno) geometry optimizations, a force threshold per atom of 0.001 eV/Å was used. The use of ultrasoft pseudopotentials [36] to model the interaction between atomic nuclei and electrons, allowed an affordable kinetic energy cutoff of 40 Ryd and a density cutoff of 400 Ryd. A Monkhorst-Pack k-point mesh of was used to compute the Brillouin zone integrals [37]. With these parameters, the fcc Au and Pt lattice constants were calculated to be 4.16 Å, and 3.99 Å in good agreement with the measured values of 4.08 Å [38] and 3.91 [38] respectively.
The Au(111) surface was represented with a slab model using a simulation cell of 3×3 unit cells consisting of four layers repeating in the ABCABC stacking. The two layers at the bottom were held fixed to mimic bulk behavior while the other two were allowed to move freely during geometry optimization. In order to reduce the interactions between successive slabs, the vacuum separation is set to be at least 14 Å in all calculations.
Adsorption energies of atomic or molecular species were calculated using
where is the total energy of the surface slab with the adsorbate, is the total energy of the surface slab and is the total energy of the isolated adsorbate, which was calculated in a large cubic simulation box at the Γ point. With this definition, negative adsorption energies correspond to stable adsorption configurations. For the coadsorbed species, the adsorption energy is calculated using
where the definition of , , and are the same as in Equation (3) while and are the energies of the two isolated adsorbates, calculated separately. All surfaces were geometry optimized prior to adsorption.
The transition states and the activation energies were calculated by using the climbing image nudged elastic band method (CI-NEB) [39]. The reaction path was divided into 7 images including the endpoints, however, in a few cases this number was increased to 9 to speedup convergence. Once the transition state was identified, no further optimizations were conducted. Finally, partial density of states (PDOSs) and Bader partial charge analyses as implemented by the Henkelman group [40] were used to elucidate our findings.
3. Results
The probe reaction chosen here for assessing the reactivity of Pt-doped Au surfaces is the dehydrogenation of CH3OH via O–H bond dissociation. Before focusing on this elementary reaction step, we first investigate the adsorption energies of all relevant chemical species on all Pt-free and Pt-enriched Au(111) surfaces, namely CH3OH, CH3O, OH, H and O as well as the coadsorption energies of CH3O/H, CH3O/OH and CH3OH/O pairs.
In the case of single atomic or molecular adsorption, we consider four possible high-symmetry adsorption sites as our starting points of the geometry optimization: face-centered cubic (fcc), hexagonal closed packed (hcp), bridge (b) and ontop (o) (see Figure 1). The fcc and hcp sites both refer to the centers of equilateral triangles on the Au(111) surface with the difference that the hcp site is located directly on top of a Au atom from the layer below while the fcc hollow site is aligned with another hollow site on the layer below. Bridge and ontop sites, on the other hand, refer to positions halfway between and directly on top of surface atoms, respectively. Often, an adsorbate placed on one of these adsorption sites will migrate to another site during geometry optimization. Figure 2 depicts the most stable geometries of all adsorbates on the surfaces investigated, while the corresponding adsorption energies are reported in Table 1. The labels on each figure refer to adsorption sites at the end of the structural relaxation. For completeness, adsorption energies for all starting sites of the adsorbates are reported in full in Tables S1-S5 of the accompanying supplementary material (SM).
From an analysis of Table 1 (and Tables S1-S5 of the SM) we can observe that, with the exception of CH3OH, which is rather weakly adsorbed on all surfaces, all species show strong affinity toward both the clean Au(111) and the various Pt-doped surfaces. For CH3OH only the ontop adsorption position is moderately stabilized by Pt-doping in the topmost layer (of about 0.3 eV), while on the clean Au(111) surface no adsorption site is strongly preferred over the other. For CH3O adsorption the two available high-symmetry sites (ontop an fcc) are equally stable on clean Au(111). As the Pt:Au ratio increases, an enhancement in stability is observed, with the Pt overlayer displaying a clear preference towards fcc and hcp sites over the ontop site. Conversely, the presence of a Pt sublayer has little effect on methoxy adsorption compared to the clean Au surface. The adsorption energies of OH and the atomic species are already quite large on Au(111). However, a clear trend of increasing stability with increasing surface Pt doping concentration in going from 1Pt/Au(111) to 3Pt/Au(111) can be easily pointed out, irrespective of the adsorption site. Pt doping in the form of a full Pt layer has the effect of either moderately decreasing the stability of the adsorbates (when in the form of an overlayer), or causes a noticeable decrease in the stability of the adsorbates (when in the form of a surface sublayer).
Figure 2.
From top to bottom panels, the most stable adsorption configurations of CH3OH, CH3O, OH, O, and H for all surfaces investigated in this work. In case of adsorption sites of similar stability (within the errors of the employed computational protocol), only one is arbitrarily reported.
Figure 2.
From top to bottom panels, the most stable adsorption configurations of CH3OH, CH3O, OH, O, and H for all surfaces investigated in this work. In case of adsorption sites of similar stability (within the errors of the employed computational protocol), only one is arbitrarily reported.

Figure 3.
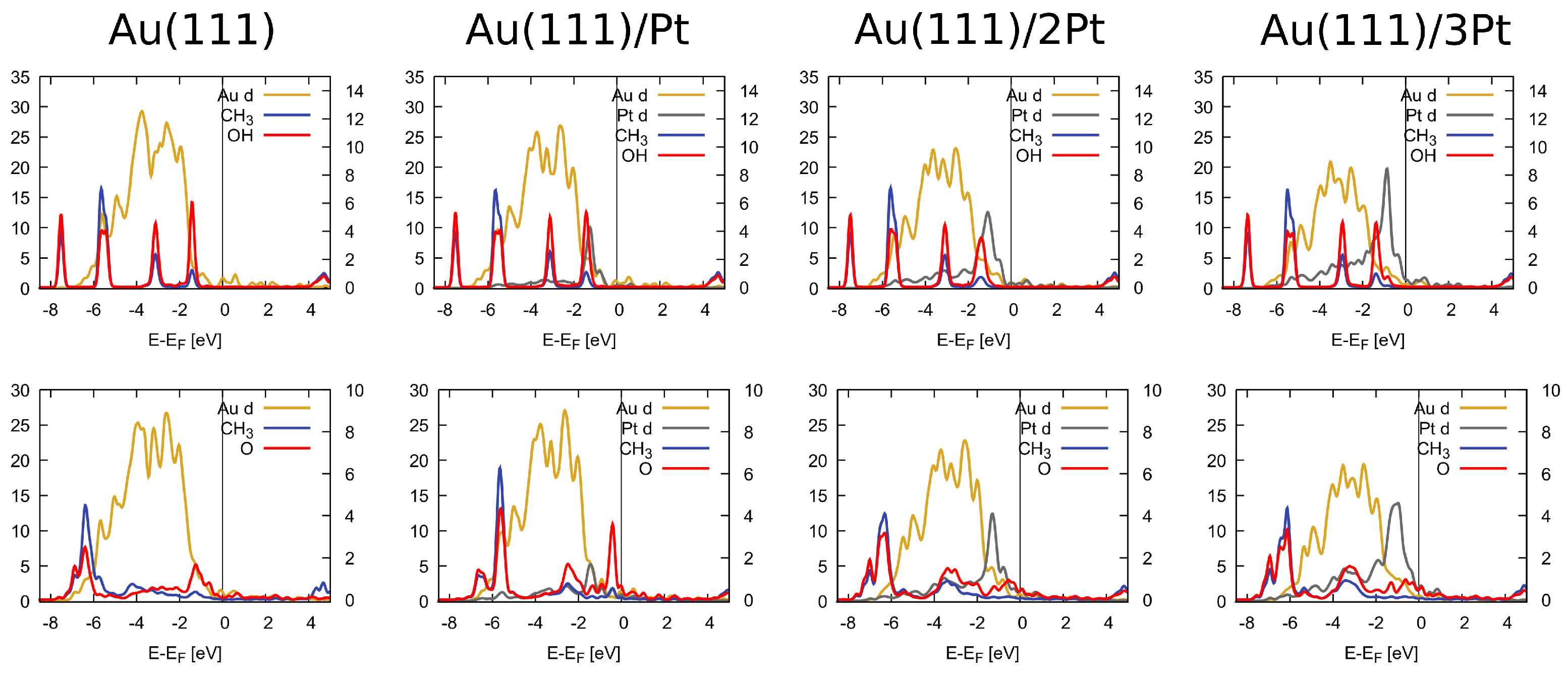
The partial density of states of CH3OH (top row), and CH3O (bottom row) adsorbed on Au(111), 1Pt/Au, 2Pt/Au, and 3Pt/Au. Only the d states of the metals are shown while all orbital contributions from the adsorbates are included. The vertical line indicates the position of the Fermi energy which is set to be at zero. The left vertical axis refers to the metallic (Au and Pt) DOS while the right vertical axis refers to the PDOS of the molecular fragments (CH3, OH, and O).
Figure 3.
The partial density of states of CH3OH (top row), and CH3O (bottom row) adsorbed on Au(111), 1Pt/Au, 2Pt/Au, and 3Pt/Au. Only the d states of the metals are shown while all orbital contributions from the adsorbates are included. The vertical line indicates the position of the Fermi energy which is set to be at zero. The left vertical axis refers to the metallic (Au and Pt) DOS while the right vertical axis refers to the PDOS of the molecular fragments (CH3, OH, and O).
We examine and rationalize the trends observed in the adsorption energies (and later on the activation barriers) based on both the characteristics of the two individual metals and the synergistic effects that emerge from their interaction as components of the surface alloy. For CH3OH adsorption on 1Pt/Au(111), 2Pt/Au(111), and 3Pt/Au(111) surfaces, the electronic effect due to alloying is particularly evident. Each substitutional Pt experiences a negative charge transfer of approximately -0.1 from the Au atoms, as can be seen from the total Bader charges on Pt listed in Table S1. In each instance where an adsorbate forms a bond directly with one or more Pt atoms, there is an electron transfer from the Pt atoms in direct contact with the adsorbate to the adsorbate itself (Tables S1-S5). The accumulated negative charge is therefore passed over to the adsorbate. This accumulation of negative charge suggests that the positively charged hydrogen atom of the OH functional group of methanol experiences a larger Coulomb attraction to the surface with increasing Pt surface concentration. The Coulomb interaction resulting from this arrangement along with the strong charge transfer can explain both the increased adsorption energies and destabilization of the O-H bond.
The electronic effect of Pt doping that enhances the surface affinity towards methanol and methoxy can be observed in the PDOS profiles of adsorbed methanol and methoxy, reported in Figure 3. In contrast to the disjoint appearance of the Au d and methanol states on the bare Au(111) surface, the Pt-doped surfaces exhibit some overlap between the Pt d and methanol states just below the Fermi level. In the case of methoxy, on the other hand, the overlapping states in the energy range 3-5 eV below the Fermi level are clearly visible.
A structural consequence of Pt doping is the strain effects on the Au lattice; this effect should be especially important in the case of the Pt overlayer. To isolate the effect of lattice strain from the others discussed above, we report in Table S6 of the SM the adsorption energies of methanol and methoxy on the Pt(111) surface constructed both at the Pt and Au lattice constants. While the adsorption energy of methanol remains largely unaffected (except for the ontop adsorption) by the added strain, the adsorption energy of methoxy is seen to increase by up to 0.5 eV for all adsorption sites, except for the ontop position, in the presence of lattice strain, which highlights the importance of strain in determining the stability of CH3O adsorption on the Pt overlayer. In the DFT study by Miao et al. [41], the reactivity of PdNi surface alloys towards ethanol dehydrogenation and oxidation was investigated in a similar manner to our work here. A Ni layer was placed both on top of the Pd(111) surface and as the second layer from the top. In that case, the sublayer of Ni was found to reduce the reactivity while the top layer was found to cause an increase. In this work as well, the changes in reactivity was rationalized by charge transfer and orbital overlap in the density of states.
Having developed an understanding for the alloying effects on the adsorption of single species, we next move on to investigate whether coadsorption of two species reveals any synergetic effects. The CH3OH/O, CH3O/OH and CH3O/H coadsorption energies were calculated assuming that CH3OH and CH3O adsorb at their most stable sites identified previously and placing O, OH and H adsorbates at the nearest four high symmetry locations (hcp, fcc, bridge, and ontop). The optimized geometries for the most stable coadsorption configurations considered are displayed in Figure S1 while the adsorption energies are listed in Table S7 of the SM. We also report the difference between the coadsorption energies calculated using Equation 4 and the sum of the individual most stable adsorption energies of the two species separately (each calculated using Equation 3).
For all coadsorption pairs considered, a clear trend of increasing coadsorption energy is evident as a function of increasing Pt number on the Au surface. For the CH3O/H pair, individual adsorption appears to be slightly more favored compared to coadsorption. For CH3O/OH, the presence of such an energy penalty depends on the surface. In contrast, for the CH3OH/O pair, there is a slight gain in energy upon coadsorption with the exception of the Pt overlayer.
Finally, we turn our attention to the effect of Pt doping on the activation barriers for the dehydrogenation reaction. Table 2 lists all the activation barriers and reaction energies calculated for the dissociation of the O-H bond in methanol, while Figure 4 illustrates the initial, transition, and final configurations of this elementary step on all Pt-doped surfaces investigated in this work. The PDOS profiles of the transition states are displayed in Figure S2 while the single imaginary frequency of the transition states are listed in Table S8 of the SM. The benchmark activation barrier of 1.76 eV calculated for the Au(111) surface is in agreement with previous work [42]. With the introduction of the single substitutional Pt atom on the gold surface, we immediately observe a drop in the activation energy of about 0.6 eV. This is also consistent with the observation of a Pt d peak appearing just below the Fermi level, overlapping with a molecular state in Figure S2. The activation barrier is not significantly affected in going from 1Pt/Au to 2Pt/Au (1.26 eV) and 3Pt/Au (1.28 eV), but a significant drop is seen for the full Pt overlayer (0.79 eV). This is mirrored in the similarity of the PDOS profiles of the 2Pt/Au and 3Pt/Au shown in Figure S2. An interesting observation is that this value of activation energy is lower than that calculated for the Pt(111) surface at the Au lattice constant (0.90 eV). This points towards a synergistic effect which can be seen in the PDOS of the transition state shown in Figure S3 of the SM where the PDOS profile displays clear Pt/Au hybrid states near the Fermi energy.
Our activation barriers compare favorably also with previously reported values in the literature. The calculated barrier for H elimination from the OH group of methanol on the Pd/Au(111) alloy surface was reported to be 1.41 eV by Zhang et al. [7]. Our activation barrier for the same two-Pt configuration is 0.13 eV lower. Calculated ethanol dehydrogenation barriers on Rh/Au(111) and Pd/Au(111) surfaces show a steady decrease of about 1 eV going from one to nine dopant atoms on the surface [19]. While more modest, our activation barriers also show a decrease with increasing dopant concentration. Finally, Zhong et al. [21] report a barrier for methanol dehydrogenation to methoxy of 0.20 eV on Ag doped Au.
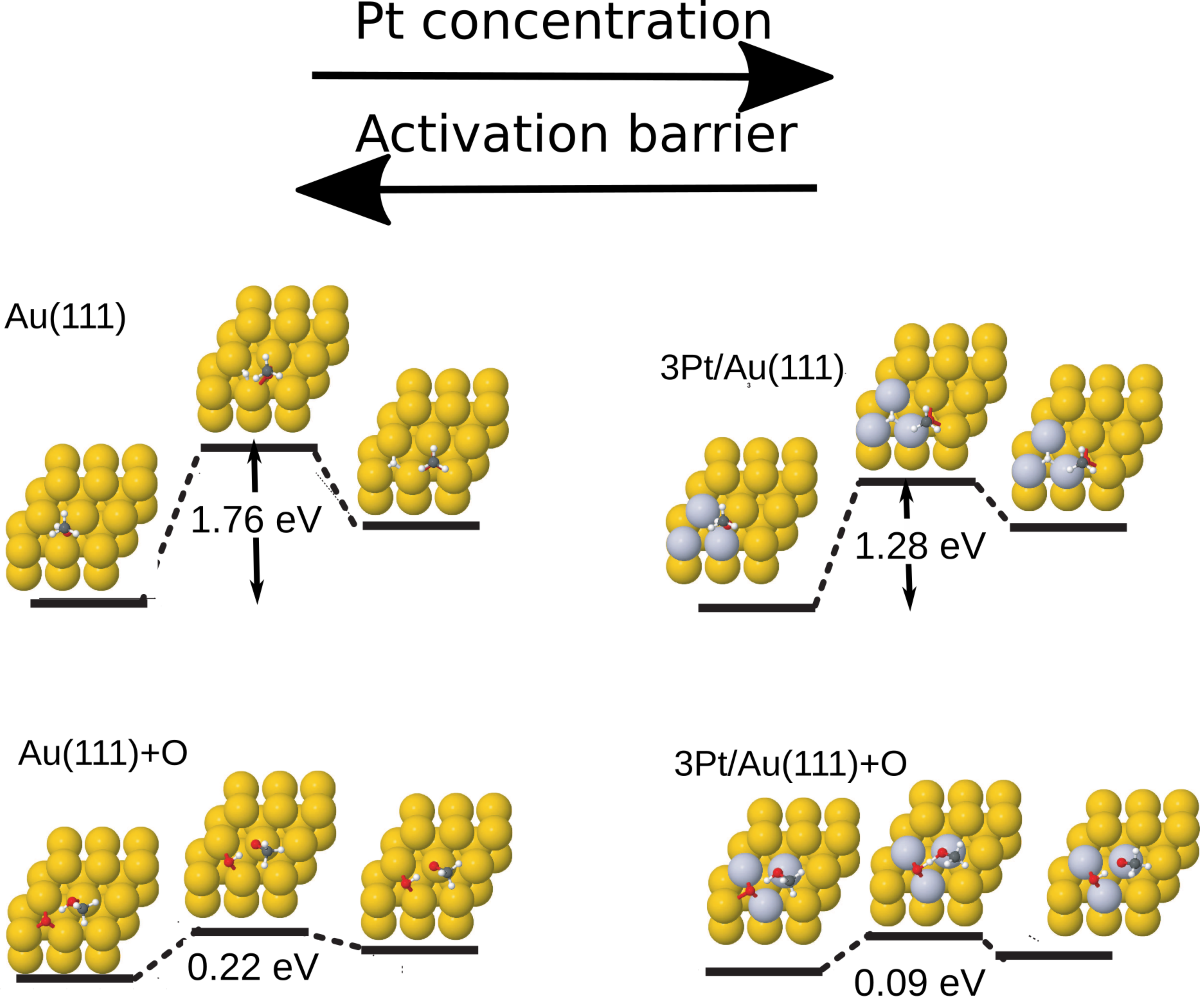
It is also important to note that Pt alloying can be used to further reduce the activation barrier of methanol dehydrogenation also on Au surfaces with pre-adsorbed atomic oxygen, as can be seen from Figure 5. In particular, the activation barrier is reduced to as low as 0.09 eV in the case of the 3Pt/Au surface with pre-adsorbed atomic oxygen.
Overall, our results are in agreement with the work with literature. For instance, Xu et al. [42] found that the adsorption energy of methanol on the pristine Au(111) surface was calculated to be 0.15 eV while in the presence of an oxygen, the adsorption energy is 0.44 eV. Furthermore, the O–H bond dissociation activation energies also agree closely. In Xu’s work, the activation barriers in the abscence and presence of pre-adsorbed O was calculated to be 1.58 eV and 0.41 eV, respectively. Finally, the thermodynamic reaction energy in the presence and absence of the surface O was calculated to be 1.33 eV and 0.27 eV, respectively, both cases being endothermic. Once again, these values are very close to our calculated thermodynamics energies of 1.34 eV and 0.16 eV.
4. Conclusions
In this work, by using first-principles density functional theory we have investigated the effect of Pt surface doping on the reactivity of the Au(111) surface towards methanol dehydrogenation The effect of the increase of Pt doping concentration of the surface on the activation barrier of this elementary step was systematically studied by substituting one, two and three surface Au atoms with Pt. In addition, Pt doping in the form of a full Pt overlayer and a full Pt sublayer were also investigated. The calculation of adsorption energies of CH3OH, CH3O, OH, O, and H on bare Au(111) and doped surfaces indicate that the affinity of the metallic surface towards these adsorbates increase with increasing Pt concentration on the surface. This effect of Pt doping is attributed, to a charge accumulation effect that steadily builds up on the surface Pt atoms. With increasing Pt concentration the d-center of the surface is seen to displace towards the Fermi level. The effect of induced strain due to the doping was also investigated and it was seen that lattice strain stabilizes the adsorbed CH3O species (by about 0.5 eV on a full Pt overlayer).
A key result of this work is the finding that the activation barrier for the dehydrogenation reaction is already reduced, by more than 30% compared to bare Au(111), for a small surface doping concentration of Pt. When the elementary step is studied on a full Pt overlayer, the activation barrier is further reduced to about 0.80 eV, which is lower than the activation barrier calculated on bare Pt(111). Therefore, even at the limit of full Pt coverage, the interaction between the Au and the Pt components act in concert to create a more reactive catalyst. This study also further points out the important role of preadsorbed atomic oxygen on further lowering the reaction barrier of the dehydrogenation step: when oxygen pre-treatment is coupled with Pt doping, the activation barrier of the elementary step reaches a value as low as 0.10 eV.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, H.U. and D.T.; methodology, M.D., H.U. and D.T.; software, M.D.; validation, M.D., H.U. and D.T.; formal analysis, M.D., H.U. and D.T.; investigation, M.D., H.U. and D.T.; resources, H.U. and D.T.; data curation, M.D., H.U. and D.T.; writing—original draft preparation, H.U. and D.T.; writing—review and editing, M.D., H.U. and D.T.; visualization, M.D. and H.U.; supervision, H.U. and D.T.; project administration, H.U.; funding acquisition, H.U. and D.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Scientific and Technological Research Council of Turkey (TÜBITAK), Grant No: 113F099. Computational resources were provided by the National Center of Academic Network and Information (TÜBITAK ULAKBIM), the National Center for High Performance Computing (UHEM) Grant No. 5003342014.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DFT | Density Functional Theory |
| GGA | Generalized Gradient Approximation |
| PW-91 | Perdew-Wang exchange Correlation potential |
| BFGS | Broyden-Fletcher-Goldfarb-Shanno |
| CI-NEB | Climbing Image Nudged Elastic Band method |
| PDOS | Partial Density of States |
| SM | Supplementary Material |
References
- Dijksman, A.; Marino-González, A.; Mairata i Payeras, A.; Arends, I.W.C.E.; Sheldon, R.A. Efficient and Selective Aerobic Oxidation of Alcohols into Aldehydes and Ketones Using Ruthenium/TEMPO as the Catalytic System. Journal of the American Chemical Society 2001, 123, 6826–6833. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhang, C.; Li, H.; Zhao, X.; Song, L.; Li, X. An Overview of Selective Oxidation of Alcohols: Catalysts, Oxidants and Reaction Mechanisms. Catalysis Surveys from Asia 2016, 20, 13–22. [Google Scholar] [CrossRef]
- Wang, B.; Tao, L.; Cheng, Y.; Yang, F.; Jin, Y.; Zhou, C.; Yu, H.; Yang, Y. Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts 2019, 9, 387. [Google Scholar] [CrossRef]
- Cheng, Z.; Fine, N.a.; Lo, C.S. Platinum Nanoclusters Exhibit Enhanced Catalytic Activity for Methane Dehydrogenation. Topics in Catalysis 2012, 55, 345–352. [Google Scholar] [CrossRef]
- Zhang, J.; She, Y. Mechanism of methanol decomposition on the Pd/WC(0001) surface unveiled by first-principles calculations. Frontiers of Chemical Science and Engineering 2020, pp. 1–13.
- Li, Z.; Xu, J.; Gu, X.; Wang, K.; Wang, W.; Zhang, X.; Zhang, Z.; Ding, Y. Selective Gas-Phase Oxidation of Alcohols over Nanoporous Silver. ChemCatChem 2013, 5, 1705–1708. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Yu, Y. The mechanism of methanol dehydrogenation on the PdAu(1 0 0) surface: A DFT study. Applied Surface Science 2020, 510, 145434. [Google Scholar] [CrossRef]
- Zhu, X.; Guo, Q.; Sun, Y.; Chen, S.; Wang, J.Q.; Wu, M.; Fu, W.; Tang, Y.; Duan, X.; Chen, D.; Wan, Y. Optimising surface d charge of AuPd nanoalloy catalysts for enhanced catalytic activity. Nature Communications 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0 °C. Chemistry Letters 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Schade, O.; Dolcet, P.; Nefedov, A.; Huang, X.; Saraçi, E.; Wöll, C.; Grunwaldt, J.D. The influence of the gold particle size on the catalytic oxidation of 5-(Hydroxymethyl)furfural. Catalysts 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Rucinska, E.; Pattisson, S.; Miedziak, P.J.; Brett, G.L.; Morgan, D.J.; Sankar, M.; Hutchings, G.J. Cinnamyl Alcohol Oxidation Using Supported Bimetallic Au–Pd Nanoparticles: An Optimization of Metal Ratio and Investigation of the Deactivation Mechanism Under Autoxidation Conditions. Topics in Catalysis 2020, 63, 99–112. [Google Scholar] [CrossRef]
- Shang, F.; Chu, Q.; Yang, H.; Yu, H.; Diao, T.; Wang, P.; Liu, H.; Wang, M. Double Catalyst-Catalyzed: An Environmentally Friendly Sustainable Process to Produce Methallyl Alcohol. Catalysis Letters 2020, 150, 2660–2673. [Google Scholar] [CrossRef]
- Darby, M.T.; Sykes, E.C.H.; Michaelides, A.; Stamatakis, M. Carbon Monoxide Poisoning Resistance and Structural Stability of Single Atom Alloys. Topics in Catalysis 2018, 61, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Yang, X.F.; Long, B.; Li, J. A water-promoted mechanism of alcohol oxidation on a Au(111) surface: Understanding the catalytic behavior of bulk gold. ACS Catalysis 2013, 3, 1693–1699. [Google Scholar] [CrossRef]
- Yi, W.; Yuan, W.; Meng, Y.; Zou, S.; Zhou, Y.; Hong, W.; Che, J.; Hao, M.; Ye, B.; Xiao, L.; Wang, Y.; Kobayashi, H.; Fan, J. A Rational Solid-State Synthesis of Supported Au–Ni Bimetallic Nanoparticles with Enhanced Activity for Gas-Phase Selective Oxidation of Alcohols. ACS Applied Materials & Interfaces 2017, 9, 31853–31860. [Google Scholar] [CrossRef]
- Behravesh, E.; Melander, M.M.; Wärnå, J.; Salmi, T.; Honkala, K.; Murzin, D.Y. Oxidative dehydrogenation of ethanol on gold: Combination of kinetic experiments and computation approach to unravel the reaction mechanism. Journal of Catalysis 2021, 394, 193–205. [Google Scholar] [CrossRef]
- Itaciara, I.E.; de Sousa, S.A.; Laíse, L.N.; Oliveira, J.M.; Castro, K.P.; Costa, J.C.; de Moura, E.M.; de Moura, C.V.; Garcia, M.A. Au-Pd Selectivity-switchable Alcohol-oxidation Catalyst: Controlling the Duality of the Mechanism using a Multivariate Approach. ChemCatChem 2019, 11, 3022–3034. [Google Scholar] [CrossRef]
- Zhao, L.; Akdim, O.; Huang, X.; Wang, K.; Douthwaite, M.; Pattisson, S.; Lewis, R.J.; Lin, R.; Yao, B.; Morgan, D.J.; Shaw, G.; He, Q.; Bethell, D.; McIntosh, S.; Kiely, C.J.; Hutchings, G.J. Insights into the Effect of Metal Ratio on Cooperative Redox Enhancement Effects over Au- and Pd-Mediated Alcohol Oxidation. ACS Catalysis 2023, 13, 2892–2903. [Google Scholar] [CrossRef]
- Li, H.; Henkelman, G. Dehydrogenation Selectivity of Ethanol on Close-Packed Transition Metal Surfaces: A Computational Study of Monometallic, Pd/Au, and Rh/Au Catalysts. Journal of Physical Chemistry C 2017, 121, 27504–27510. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Veith, G.M.; Prati, L. Bismuth as a modifier of Au–Pd catalyst: Enhancing selectivity in alcohol oxidation by suppressing parallel reaction. Journal of Catalysis 2012, 292, 73–80. [Google Scholar] [CrossRef]
- Zhong, W.; Liang, J.; Hu, W.; Cao, X.; Jia, C.; Jiang, J. The O, OH and OOH-assisted selective coupling of methanol on Au-Ag(111). Physical Chemistry Chemical Physics 2016, 18, 9969–9978. [Google Scholar] [CrossRef]
- Nagy, G.; Gál, T.; Srankó, D.; Sáfrán, G.; Maróti, B.; Sajó, I.; Schmidt, F.P.; Beck, A. Selective aerobic oxidation of benzyl alcohol on alumina supported Au-Ru and Au-Ir catalysts. Molecular Catalysis 2020, 492, 110917. [Google Scholar] [CrossRef]
- Salciccioli, M.; Yu, W.; Barteau, M.A.; Chen, J.G.; Vlachos, D.G. Differentiation of O-H and C-H bond scission mechanisms of ethylene glycol on Pt and Ni/Pt using theory and isotopic labeling experiments. Journal of the American Chemical Society 2011, 133, 7996–8004. [Google Scholar] [CrossRef] [PubMed]
- a Tenney, S.; Shah, S.I.; Yan, H.; a Cagg, B.; Levine, M.S.; Rahman, T.S.; a Chen, D. Methanol Reaction on Pt-Au Clusters on TiO2(110): Methoxy- Induced Diffusion of Pt. The Journal of Physical Chemistry C 2013, 117, 26998–27006. [Google Scholar] [CrossRef]
- Stepanova, L.N.; Belskaya, O.B.; Trenikhin, M.V.; Leont’eva, N.N.; Gulyaeva, T.I.; Likholobov, V.A. Effect of Pt(Au)/MgAlOx catalysts composition on their properties in the propane dehydrogenation. Catalysis Today 2021, 378, 96–105. [Google Scholar] [CrossRef]
- Wang, J.; Voss, M.R.; Busse, H.; Koel, B.E. Chemisorbed Oxygen on Au(111) Produced by a Novel Route: Reaction in Condensed Films of NO2+H2O. The Journal of Physical Chemistry B 1998, 102, 4693–4696. [Google Scholar] [CrossRef]
- Wang, T.; Sha, J.; Sabbe, M.; Sautet, P.; Pera-Titus, M.; Michel, C. Identification of active catalysts for the acceptorless dehydrogenation of alcohols to carbonyls. Nature Communications 2021, 12, 5100. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, L. Vinyl alcohol formation via catalytic β-dehydrogenation of ethanol on Ir(100). Chemical Physics Impact 2021, 3, 100040. [Google Scholar] [CrossRef]
- Xu, B.; Haubrich, J.; Baker, T.A.; Kaxiras, E.; Friend, C.M. Theoretical Study of O-Assisted Selective Coupling of Methanol on Au(111). The Journal of Physical Chemistry C 2011, 115, 3703–3708. [Google Scholar] [CrossRef]
- Hussain, A.; Shah, S. MMENT>Computational study of complete methanol dehydrogenation on Au(100) and Au(310) surfaces: Dominant role of atomic oxygen. Surface Science 2014, 620, 30–37. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; others. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. Journal of physics: Condensed matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Physical review B 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Computational Materials Science 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Davey, W.P. Precision Measurements of the Lattice Constants of Twelve Common Metals. Phys. Rev. 1925, 25, 753–761. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. The Journal of chemical physics 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. Journal of Physics: Condensed Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Miao, B.; Wu, Z.P.; Zhang, M.; Chen, Y.; Wang, L. Role of Ni in Bimetallic PdNi Catalysts for Ethanol Oxidation Reaction. The Journal of Physical Chemistry C 2018, 122, 22448–22459. [Google Scholar] [CrossRef]
- Xu, B.; Haubrich, J.; Baker, T.A.; Kaxiras, E.; Friend, C.M. Theoretical study of O-assisted selective coupling of methanol on Au(111). Journal of Physical Chemistry C 2011, 115, 3703–3708. [Google Scholar] [CrossRef]
Figure 1.
High symmetry adsorption sites on Au(111) described in the text.
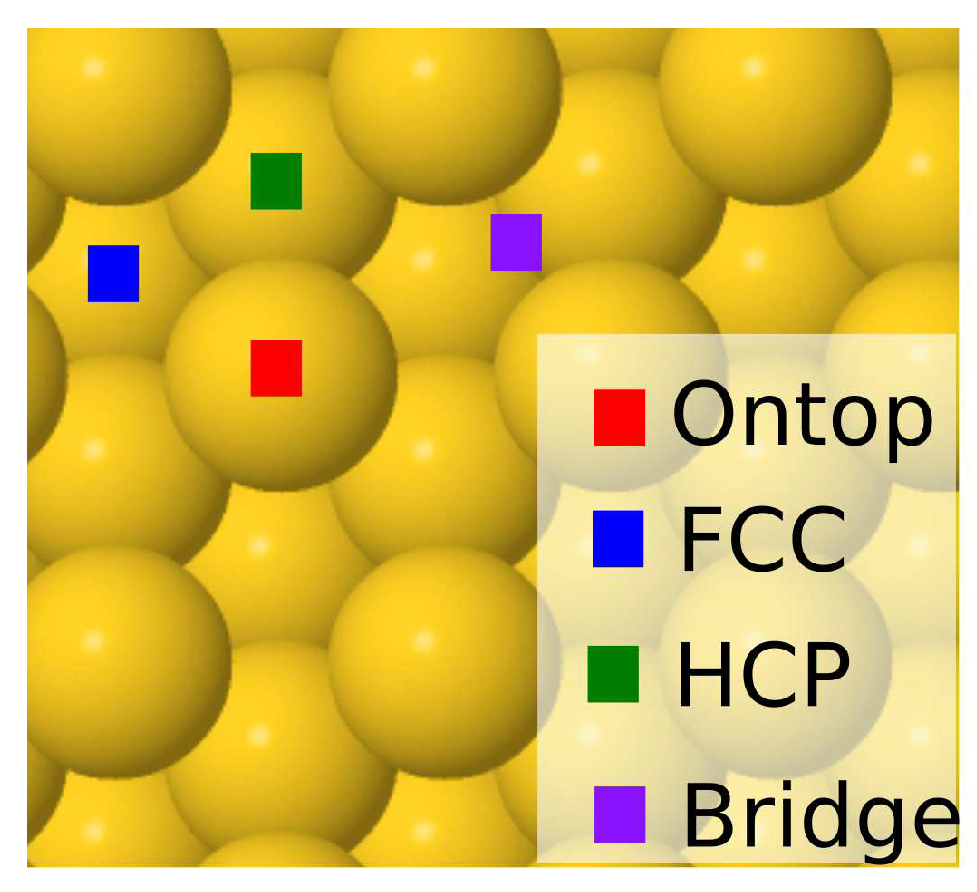
Figure 4.
Initial, transition and final states of the methanol dehydrogenation reaction on Au(111), Pt/Au(111), 2Pt/Au(111), 3Pt/Au(111), 9Pt/Au(111) (overlayer), subsurface Pt layer and Pt(111).
Figure 4.
Initial, transition and final states of the methanol dehydrogenation reaction on Au(111), Pt/Au(111), 2Pt/Au(111), 3Pt/Au(111), 9Pt/Au(111) (overlayer), subsurface Pt layer and Pt(111).
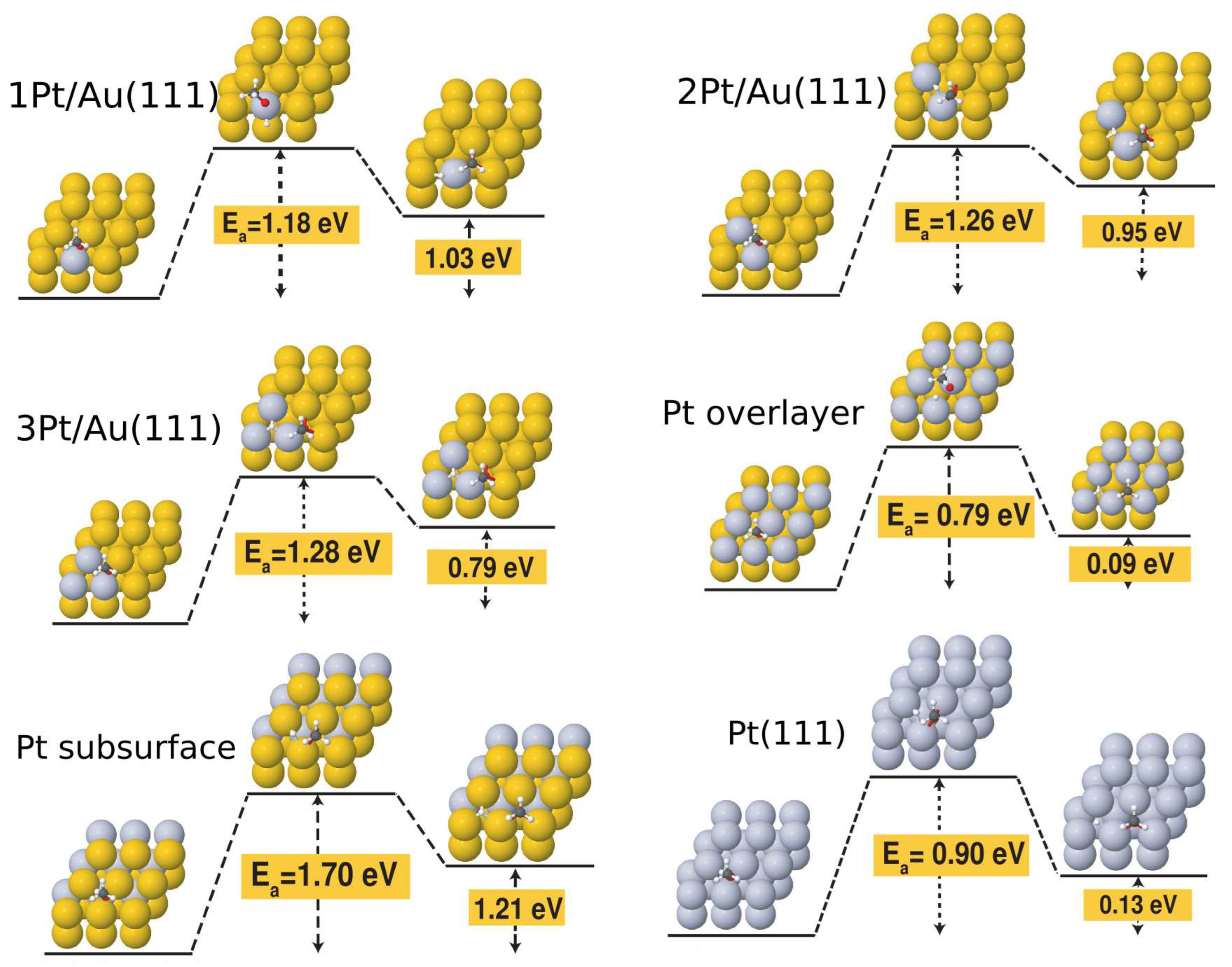
Figure 5.
Initial, transition-state and final configurations for the methanol dehydrogenation step on the following surfaces with preadsorbed atomic O: (a) bare Au(111) surface; (b) 1Pt/Au surface; (c) 2Pt/Au surface; (d) 3Pt/Au surface
Figure 5.
Initial, transition-state and final configurations for the methanol dehydrogenation step on the following surfaces with preadsorbed atomic O: (a) bare Au(111) surface; (b) 1Pt/Au surface; (c) 2Pt/Au surface; (d) 3Pt/Au surface
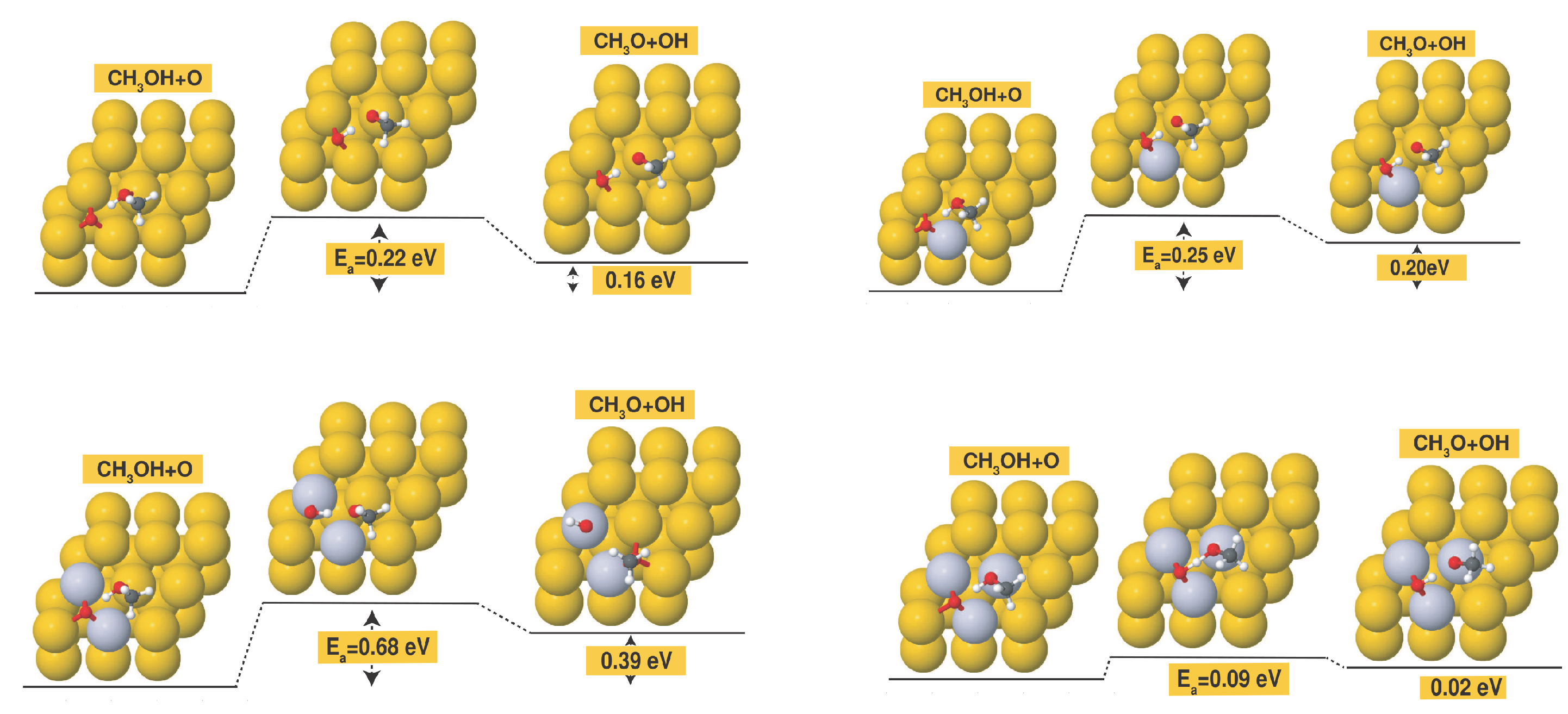

Table 1.
Adsorption energies (in eV) corresponding to the most stable adsorption site (reported in parenthesis) for CH3OH, CH3O, OH, O, and H adsorbates on bare Au(111) surface and on all Pt-doped surfaces investigated in this work.
Table 1.
Adsorption energies (in eV) corresponding to the most stable adsorption site (reported in parenthesis) for CH3OH, CH3O, OH, O, and H adsorbates on bare Au(111) surface and on all Pt-doped surfaces investigated in this work.
| Au(111) | 1Pt/Au | 2Pt/Au | 3Pt/Au | Pt overlayer | Pt sublayer | |
| CH3OH | -0.11 (o) | -0.12 (fcc) | -0.13 (o) | -0.27 (o) | -0.38 (o) | -0.12 (o) |
| CH3O | -1.75 (fcc) | -1.88 (o) | -1.87 (o) | -1.92 (o) | -2.24 (fcc) | -1.77(fcc) |
| OH | -2.66 (fcc) | -2.73 (o) | -2.80 (b) | -2.82 (b) | -2.65 (fcc) | -2.19(fcc) |
| O | -5.07 (fcc) | -5.24 (fcc) | -5.43 (fcc) | -5.40 (fcc) | -4.67 (fcc) | -3.46(fcc) |
| H | -3.18 (fcc) | -3.46 (b) | -3.74 (b) | -3.84 (hcp) | -3.04 (fcc) | -2.26(fcc) |
Table 2.
Activation barriers (, in eV) and reaction energies (, in eV) for the methanol dehydrogenation step on the surfaces investigated in this work.
Table 2.
Activation barriers (, in eV) and reaction energies (, in eV) for the methanol dehydrogenation step on the surfaces investigated in this work.
| surface | ||
| Au(111) | 1.76 | 1.34 |
| 1Pt/Au | 1.18 | 1.03 |
| 2Pt/Au | 1.26 | 0.95 |
| 3Pt/Au | 1.28 | 0.79 |
| Pt overlayer | 0.79 | 0.08 |
| Pt sublayer | 1.70 | 1.21 |
| Pt(111)a | 0.90 | 0.13 |
| aat the Au lattice constant. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



