
Submitted:
10 November 2023
Posted:
13 November 2023
You are already at the latest version
Abstract
Myeloid neoplasms and acute leukemias include different entities that have been recently re-classify taking into account molecular and clinicopathological features. The myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) category comprises a heterogeneous group of hybrid neoplastic myeloid diseases characterized by the co-occurrence of clinical and pathologic features of both myelodysplastic and myeloproliferative neoplasms. The most frequent entity in this category is chronic myelomonocytic leukemia (CMML) which is, after acute myeloid leukemia (AML), the main myeloid disorder prone to develop cutaneous manifestations. Skin lesions associated with myelodysplastic and myeloproliferative neoplasms include a broad clinical, histopathological and molecular spectrum of lesions, poorly understood and without a clear-cut classification in the current medical literature. The aim of this review is to describe and classify the main clinical, histopathological and molecular patterns of cutaneous lesions in the setting of MDS/MPN in order to improve the diagnostic skills of dermatologists, hematologist and pathologist who deal with these patients.
Keywords:
myelodysplastic and myeloproliferative neoplasms
; CMML
; skin
1. Introduction
Myeloid neoplasms and acute leukemias include different entities that have been recently re-classify taking into account molecular and clinicopathological features. Two major articles have been published in 2022, the ICC and the WHO classifications1,2. Myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) category comprises a heterogeneous group of hybrid neoplastic myeloid diseases characterized by the co-occurrence of clinical and pathologic features of both myelodysplastic and myeloproliferative neoplasms. The most frequent entity in this category is chronic myelomonocytic leukemia (CMML) which is, after acute myeloid leukemia (AML), the main myeloid disorder prone to develop cutaneous manifestations3. Skin lesions associated with myelodysplastic and myeloproliferative neoplasms include a broad clinical and histopathological spectrum of lesions, poorly understood and without a clear-cut classification in the current medical literature.
The aim of this review is to describe and classify the main clinical and histopathological patterns of cutaneous lesions in the setting of MDS/MPN in order to improve the diagnostic skills of dermatologists, hematologist and pathologist who deal with these patients. Classically, these skin lesions have been divided into reactive or unspecific and neoplastic or specific ones. This traditional classification, was based on the histopathologic findings in cutaneous lesions4. However, within the past decade, the improvement in the molecular techniques to recognize gene mutations found in cutaneous lesions of these patients has changed the previous concept of the reactive nature of some of these dermatoses5. The term myelodysplasia cutis (MDS-cutis) was coined to name some of the previous considered reactive neutrophilic infiltrates clearly related with MDS, in which clonality of the myeloid infiltrate in the skin, common mutations in the cells of the cutaneous infiltrates and the hematological MDS/CMML clone found in blood and bone marrow (BM) and a close correlation between the course of the cutaneous lesions and the treatment and evolution of the disease, have been demonstrated. Some authors have adopted this term and also proposed the term CMML-cutis for the same scenario in CMML6. However, the literature is highly confuse with the terminology used to name the dermatoses related with MDS/MPN and the main goal of this review is clarify these terms, fundamentally differentiating between two main groups of cutaneous lesions in these patients: dermatosis of indolent clinical outcome (see Table 1).and cutaneous processes associated to MDS/MPN and aggressive biological behavior (see Table 2) On one hand, MDS/MPN dermatosis of indolent clinical outcome previously defined as “non-specific”, but in which it has been currently shown that they share specific mutations with myeloid neoplastic cells in the bone marrow (BM). The fact that some of these MDS/MPN dermatoses only respond to the specific treatment of the myeloid neoplasm has led us to change the previous “non-specific” term to this new one. On the other hand, we define cutaneous processes associated to MDS/MPN and aggressive biological behavior as the ones with a clearly neoplastic histopathologic appearance, formerly described as “specific” which include the ones with presence of blast leukemic cells in the cutaneous lesions. In addition, these cutaneous proliferations have been linked with a clearly worse prognosis. However, unlike the previous ones, these skin lesions have not such a poor prognosis and blast cells are usually not present in skin biopsies. This review structured along clinical, histopathological and molecular lines, has allow us to address conditions characterized by neutrophilic infiltrates, granulomatous infiltrates, mature plasmacytoid dendritic cell proliferations and others less frequently reported disorders in the group of indolent clinical outcome cutaneous infiltrates, and myeloid leukemia cutis or granulocytic sarcoma infiltrates, histiocytosis, histiocytic sarcoma and tumors of blastoid plasmacytoid dendritic cells in the group of processes associated to MDS/MPN and aggressive biological behavior. Finally, we include a small group of miscellaneous conditions less frequently reported in these patients, but with skin lesions which are worth-known, although they do not fulfill the criteria of the none of two previous major categories. All these cutaneous lesions demonstrate the plasticity of myeloid cells in the skin and BM and their recognition can help pathologists, hematologists and dermatologists to better diagnose and manage these patients.
2. Myelodysplastic/Myeloproliferative Related Dermatosis with Indolent Clinical Outcome
Different histopathologic patterns have been described in the classically considered reactive dermatoses in the setting of MDS/MPN. Since the description of shared molecular alterations, as well as a clinical course related with the hematological disease (persistence of the skin lesions and response to the treatment of the myeloid neoplasm) a new term, myelodysplasia cutis (MDS-cutis) has emerged7. Below we will detail the main clinical, histopathological and molecular characteristics of these dermatoses, including forms with rapid response to oral corticosteroids and more persistent forms with shared molecular alterations with BM that could be discussed in what is described as MDS-cutis. To date, the biological meaning of the different clinical and histopathological variants of these dermatoses is unknown.
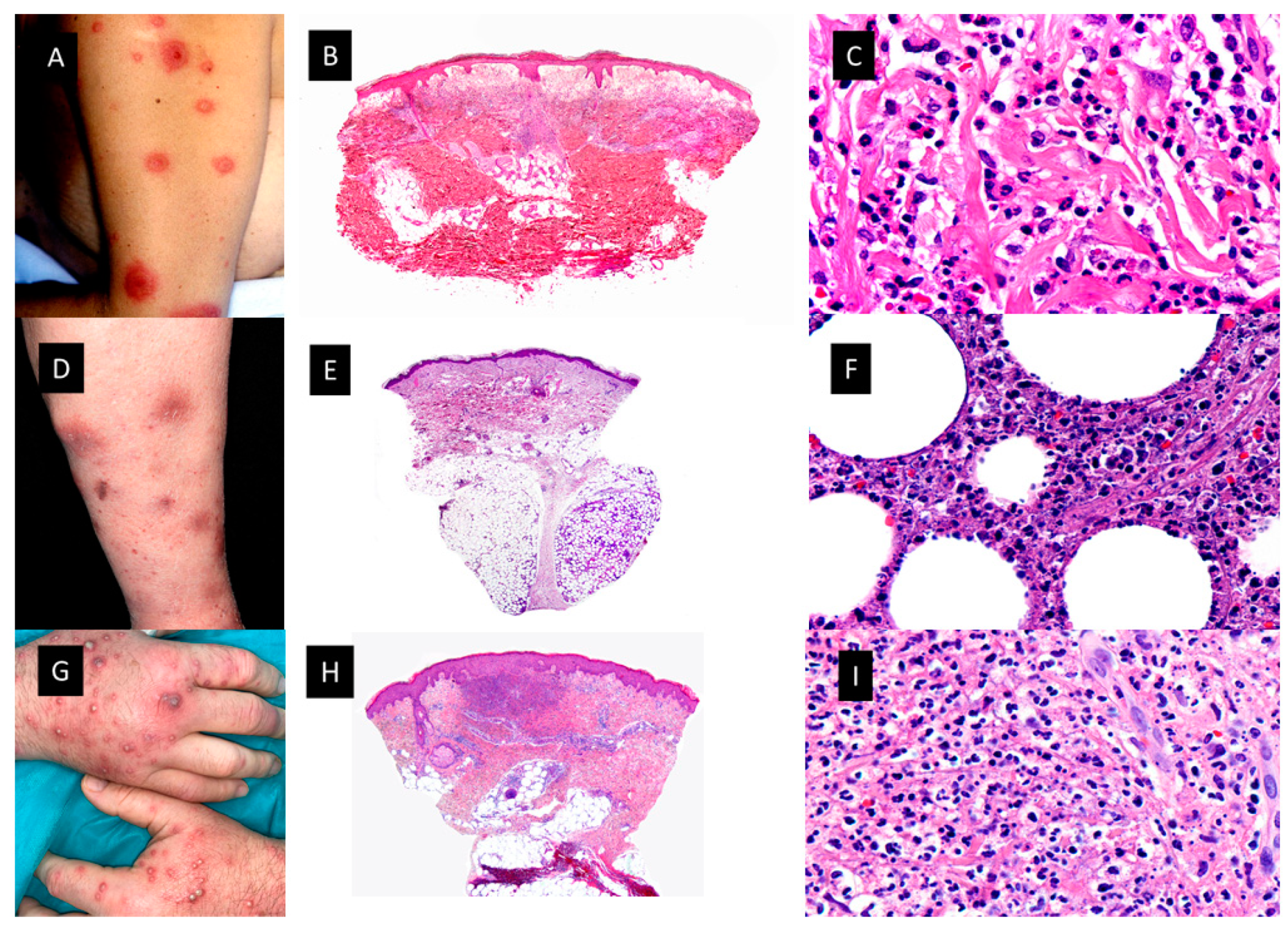
Figure 1.
Neutrophilic dermatoses A. Classic Sweet Syndrome (C-SS) presenting with edematous, erythematous and tender plaques located on the left arm. B. At low magnification intense edema of the papillary dermis and an underlying bandlike, dense, dermal, inflammatory infiltrate mostly composed of mature neutrophils, with leukocytoclasis (HE staining 4x) C. Detail of the neutrophils interstitially arranged in a linear way between collagen bundles (HE staining x40) D. Subcutaneous Sweet Syndrome (S-SS) with erythematous and violaceous skin nodules located on the leg E. Panoramic picture showing a mostly lobular subcutaneous infiltrate in the abscense of vascular changes (HE staining 4x). F. Higher magnification of the neutrophils located around fat lobules (HE staining 40x) G. Vexas Syndrome with violaceous edematous papules and pustules with erythematous halo located on the dorsum of both hands H. Dense dermal infiltrate located in superficial and middle dermis(HE staining 4x) I. On higher magnification dense infiltrate composed mainly by neutrophils (HE staining x40).
Figure 1.
Neutrophilic dermatoses A. Classic Sweet Syndrome (C-SS) presenting with edematous, erythematous and tender plaques located on the left arm. B. At low magnification intense edema of the papillary dermis and an underlying bandlike, dense, dermal, inflammatory infiltrate mostly composed of mature neutrophils, with leukocytoclasis (HE staining 4x) C. Detail of the neutrophils interstitially arranged in a linear way between collagen bundles (HE staining x40) D. Subcutaneous Sweet Syndrome (S-SS) with erythematous and violaceous skin nodules located on the leg E. Panoramic picture showing a mostly lobular subcutaneous infiltrate in the abscense of vascular changes (HE staining 4x). F. Higher magnification of the neutrophils located around fat lobules (HE staining 40x) G. Vexas Syndrome with violaceous edematous papules and pustules with erythematous halo located on the dorsum of both hands H. Dense dermal infiltrate located in superficial and middle dermis(HE staining 4x) I. On higher magnification dense infiltrate composed mainly by neutrophils (HE staining x40).
2.1. NEUTROPHILIC DERMATOSES
Neutrophilic dermatoses (ND) comprise a heterogeneous group of inflammatory skin disorders that share a similar histopathology, consisting of a sterile neutrophilic infiltrate. Clinical manifestations of the ND are diverse, even with variations in the same patient. Theoretically, the histopathologic location of the neutrophilic infiltrate (epidermis, dermis, subcutis), the disease course and the clinical appearance help to make the differential diagnosis among different ND, such as Sweet syndrome (SS), pyoderma gangrenosum (PG), Behçet syndrome (BS), erythema elevatum diutinum (EED) and neutrophilic eccrine hidradenitis (NEH)8.
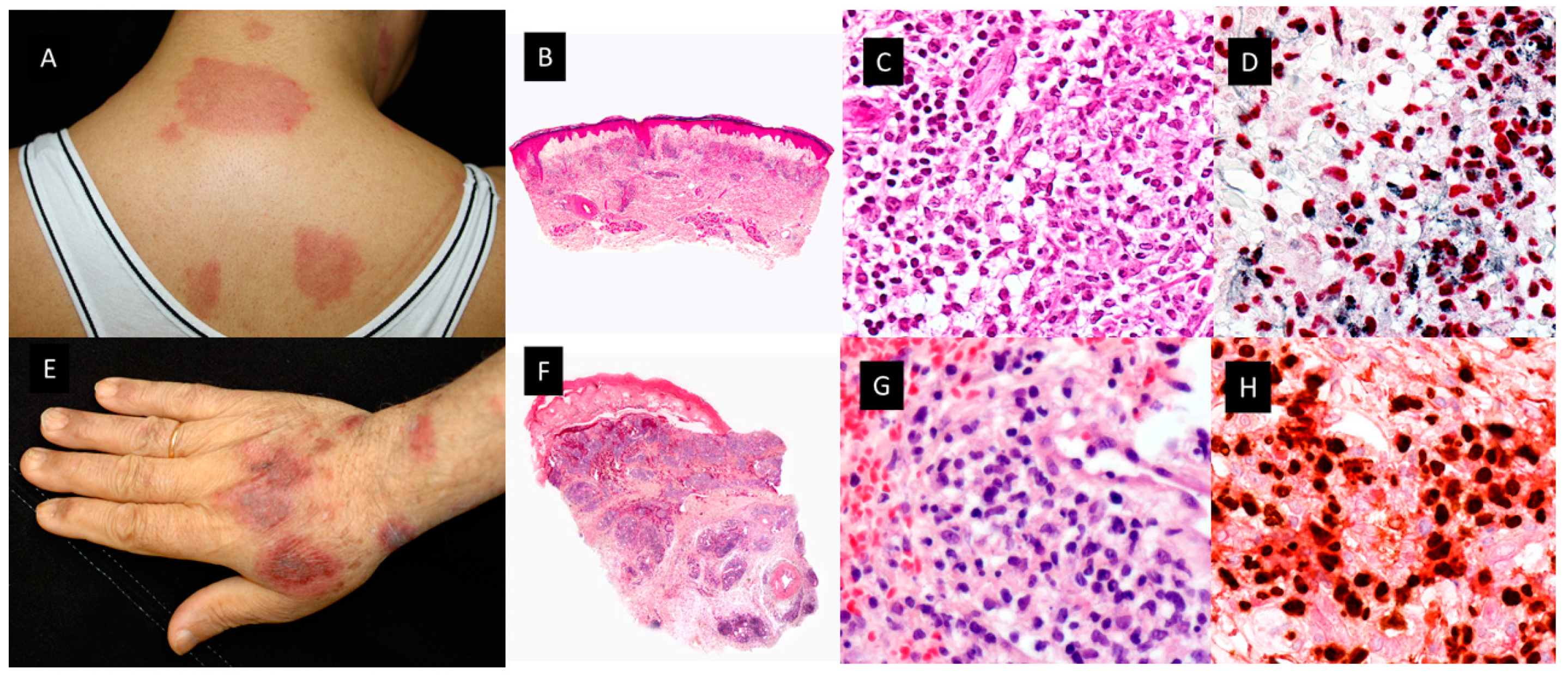
Figure 2.
Clinicopathological features of Histiocytoid Sweet Syndrome(H-SS) and Myelodysplasia cutis(MDS-cutis). A. H-SS Clinical picture showing erythematous and edematous plaques on the back and posterior region of the neck B. Scanning power shows edema and nodular infiltrates in the superficial dermis. (HE staining 4x) . C. The dermal nodules are mostly composed of mononuclear cells with twisted vesicular nuclei and scant eosinophilic cytoplasm. D. Double immunostained specimen with nuclear myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic myeloperoxidase [MPO, red]).(40x) E. MDS-cutis showing edematous plaques on the dorsum of the hand and arm, with raised erythematous border and a slightly depressed violaceous center. F. Histopathologic features consist of edema in the papillary dermis and multiple nodular infiltrates in the superficial and deep dermis; there are also perivascular infiltrates around deep dermal vascular plexus (HE 4x). G. On higher magnification, atypical, dermal hematolymphoid infiltrate and some red cells, the atypical cells demonstrate reniform nuclei and eosinophilic cytoplasm and include some pseudo–Pelger-Huet anomaly (HE 40x). H. Double immunostained specimen with myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic CD123 [CD123, red].
Figure 2.
Clinicopathological features of Histiocytoid Sweet Syndrome(H-SS) and Myelodysplasia cutis(MDS-cutis). A. H-SS Clinical picture showing erythematous and edematous plaques on the back and posterior region of the neck B. Scanning power shows edema and nodular infiltrates in the superficial dermis. (HE staining 4x) . C. The dermal nodules are mostly composed of mononuclear cells with twisted vesicular nuclei and scant eosinophilic cytoplasm. D. Double immunostained specimen with nuclear myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic myeloperoxidase [MPO, red]).(40x) E. MDS-cutis showing edematous plaques on the dorsum of the hand and arm, with raised erythematous border and a slightly depressed violaceous center. F. Histopathologic features consist of edema in the papillary dermis and multiple nodular infiltrates in the superficial and deep dermis; there are also perivascular infiltrates around deep dermal vascular plexus (HE 4x). G. On higher magnification, atypical, dermal hematolymphoid infiltrate and some red cells, the atypical cells demonstrate reniform nuclei and eosinophilic cytoplasm and include some pseudo–Pelger-Huet anomaly (HE 40x). H. Double immunostained specimen with myeloid nuclear differentiation antigen [MNDA, black] and cytoplasmic CD123 [CD123, red].
2.2. SWEET SYNDROME
In 1964, Robert Douglas Sweet reported 8 patients with an “acute febrile neutrophilic dermatosis” that afterwards was renamed SS9. Clinically, patients presented with fever and edematous, erythematous tender cutaneous plaques located on any area of the skin surface, but preferably involving the upper trunk. The usual histopathologic findings consisted of intense edema of the papillary dermis and an underlying bandlike, dense, dermal, inflammatory infiltrate mostly composed of mature neutrophils, with leukocytoclasis but without features of vasculitis. In rare instances, however, typical lesions of SS may show histopathologic features of leukocytoclastic vasculitis, which seems to be a secondary epiphenomenon. Approximately 21% of patients with SS have an associated malignancy10. Malignancy-associated SS is more commonly reported with hematologic malignancies and MDS/MPN compared to solid-organ malignancies. The most common associations are AML followed by MDS. SS may precede or follow a diagnosis of malignancy and it has even been described as a signal of cancer recurrence11. There are several clinical and histopathological variants of SS, including localized SS, histiocytoid SS (H-SS), subcutaneous SS, acute necrotizing SS, and xanthomatized neutrophilic dermatosis. One clinicopathological variant that deserves special attention in patients with myeloid neoplasms is H-SS.
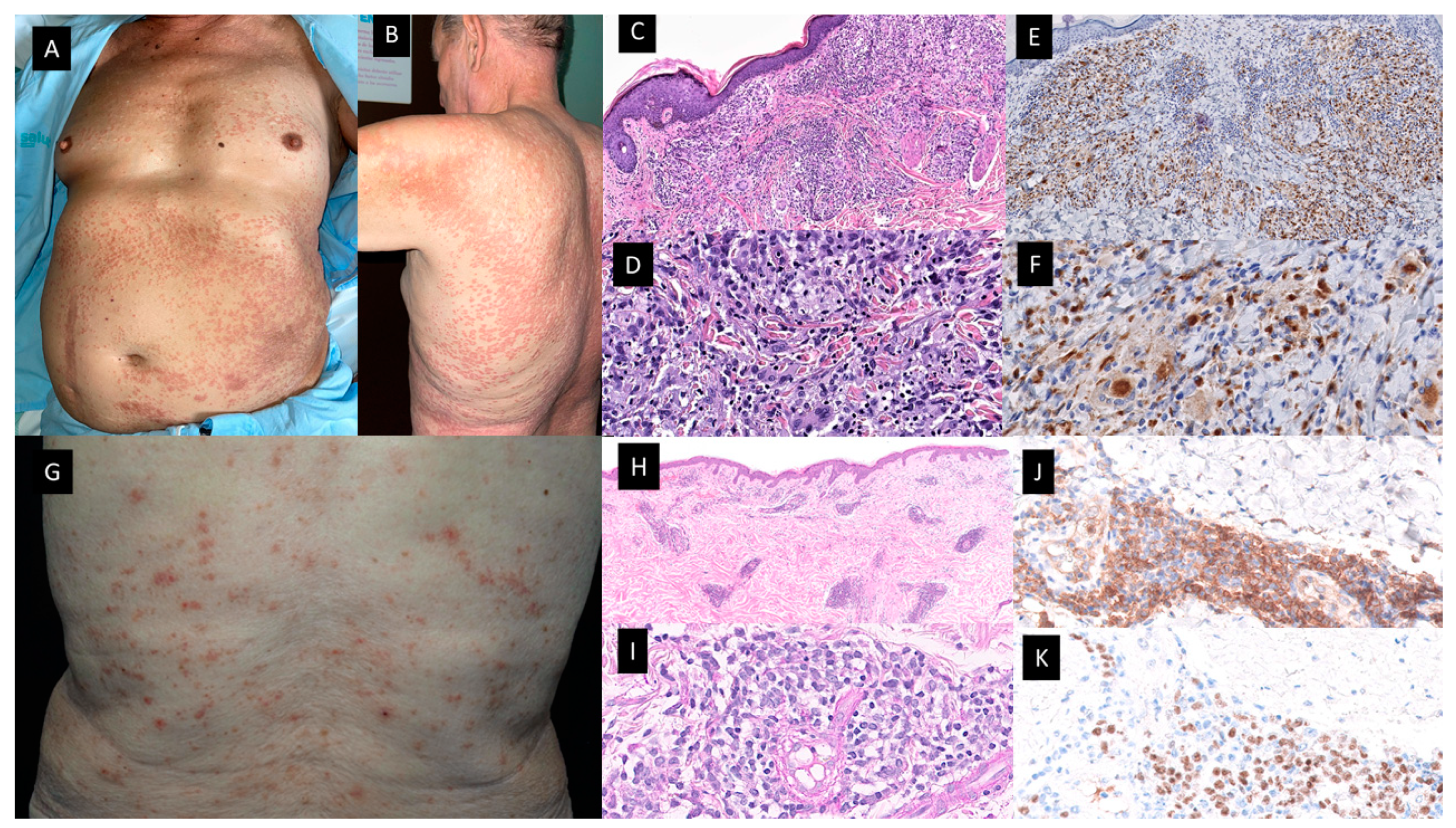
Figure 3.
Granulomatous dermatitis (A-F) and mature plasmacytoid dendritic cell dermatosis (G-K). Patient with a diagnosis of CMML presented with multiple 1-2 mm erythematous-brownish papules that coalesce on A. His anterior trunk and abdomen as well as on B. Back and upper arm and extremities (not-shown). C and D. Hematoxylin-eosin–stained specimen. C. Histopathologic features consist of a dermal inflammatory infiltrate composed of epithelioid histiocytes, giant cells, and lymphocytes, with rare neutrophils, configuring ill-defined granulomas (20x) D. On higher magnification detail of the granulomas with multinucleated giant cells (40x). E and F. On immunohistochemical study, CD68 stained the histiocytes [E (20x), F(40x)]. G. Multiple erythematous and purpuric papules on the back. H and I. Hematoxylin-eosin–stained specimen. H. Dermal nodular infiltrate with perivascular and periadnexal arrangement (20x) I. Detail of lymphocytic infiltration as well as interspersed larger, paler cells corresponding to mature PDC (40×). J. Cytoplasmic CD123 immunostaining highlithning clusters of plasmacytoid dendritic cells (PDC) (40x) K. Nuclear SPIB stain in PDC (40x).
Figure 3.
Granulomatous dermatitis (A-F) and mature plasmacytoid dendritic cell dermatosis (G-K). Patient with a diagnosis of CMML presented with multiple 1-2 mm erythematous-brownish papules that coalesce on A. His anterior trunk and abdomen as well as on B. Back and upper arm and extremities (not-shown). C and D. Hematoxylin-eosin–stained specimen. C. Histopathologic features consist of a dermal inflammatory infiltrate composed of epithelioid histiocytes, giant cells, and lymphocytes, with rare neutrophils, configuring ill-defined granulomas (20x) D. On higher magnification detail of the granulomas with multinucleated giant cells (40x). E and F. On immunohistochemical study, CD68 stained the histiocytes [E (20x), F(40x)]. G. Multiple erythematous and purpuric papules on the back. H and I. Hematoxylin-eosin–stained specimen. H. Dermal nodular infiltrate with perivascular and periadnexal arrangement (20x) I. Detail of lymphocytic infiltration as well as interspersed larger, paler cells corresponding to mature PDC (40×). J. Cytoplasmic CD123 immunostaining highlithning clusters of plasmacytoid dendritic cells (PDC) (40x) K. Nuclear SPIB stain in PDC (40x).
2.2.1. HISTIOCYTOID SS (H-SS)
This histopathologic variant of SS, first described by Requena et al in 200512, is characterized by a dermal infiltrate of mononuclear cells of histiocytic appearance, but strongly positive for myelocytic and promyelocytic markers, as demonstrates the double immunostain for myeloperoxidase (MPO) and myeloid cell nuclear differentiation antigen (MNDA)13. As in classic SS, patients with H-SS present with tender erythematous plaques and nodules on the extremities and trunk accompanied by systemic symptoms such as fever and arthralgia12,13. Association with malignancy ranges from 30% to 61% and the most frequent associated neoplasia is MDS.
In the first 41-patient-series published by Requena et al, there were 7 patients with associated malignancies, including CMML (case 3) “lymphoma” (no more specific diagnosis could be obtained) (case 21), monoclonal gammopathy of undetermined significance (case 24), renal carcinoma (case 29), breast carcinoma (case 28), chronic B lymphocytic leukemia (case 38), and multiple myeloma (case 39)12. In a series of 9 patients, Vignon-Pennamen et al suggested for the first time that this histopathological variant could be more frequently associated with AML and MDS than classic SS14. Later, in 2015, Ghoufi et al published a larger series, which included 62 patients with a histopathologic diagnosis of SS, 22 cases with H-SS and 40 cases with the classical neutrophilic variant (N-SS)15. They found that MDS was diagnosed in 7 of 22 patients with H-SS and only in 1 of 40 patients with N-SS (p<0.001). In 3 H-SS patients with MDS, the cutaneous lesions preceded the hematological diagnosis by more than 6 months. Therefore, they proposed that a complete hematological assessment and close follow-up should be mandatory in patients with H-SS15. In contrast, Alegría et al found that H-SS was not more frequently related with hematologic malignancies than N-SS13. In their series of 33 patients with H-SS, 8 suffered from hematological malignancies, including 3 with MDS and 2 with CMML. In these patients CD163+/MPO+ population was absent in the infiltrate, whereas double immunostain with MNDA/MPO was intensely positive in most cells of the infiltrate, whereas a minor proportion of cells expressed the doble immunostain for MNDA/CD163. The skin lesions appeared 5 and 2 years before the diagnosis of the hematological condition13.
The initial presumption of the reactive nature of SS, including variants like H-SS, was supported by the acute onset of the skin lesions and the complete response to steroid therapy. However, the improvement in molecular techniques has demonstrated clonally related cells in both the skin and bone marrow of hematological patients with N-SS and H-SS5. In 2020, a molecular study including 10 patients with SS and AML, myelodysplastic syndrome, myeloproliferative neoplasm or MDS/MPN was published. 8 patients had N-SS and 2 H-SS. They performed Next Generation Sequencing (NGS) analysis of paired skin-lesion biopsies and detected 35 out of the 37 mutations found in hematopoietic samples, without additional mutations. Seven patients exhibited identical mutational profiles in both tissues. In all mutated cases (n 9), mutations of the major clone of the myeloid neoplasm were also found in the cells of cutaneous infiltrates. CD34 and myeloperoxidase immunohistochemical staining of skin biopsies revealed very few immature myeloid cells in only 2 out of 10 patients, ruling out leukemia cutis. Most mutations detected in skin-lesion samples were at relatively high allelic burden (median 20%, at least one mutation with variant allele frequencies 10% for each patient) indicating that they did not relate to rare infiltrating blast cells or blood contamination of skin biopsies. The authors assumed that the process of migration and infiltration of the skin in SS may be related to the tumoral phenotype of myeloid cells, rather than only triggered by cell extrinsic factors5.
These NGS studies in the skin of patients with MDS and ND, especially H-SS, also support the concept of MDS-cutis16.
Figure 4.
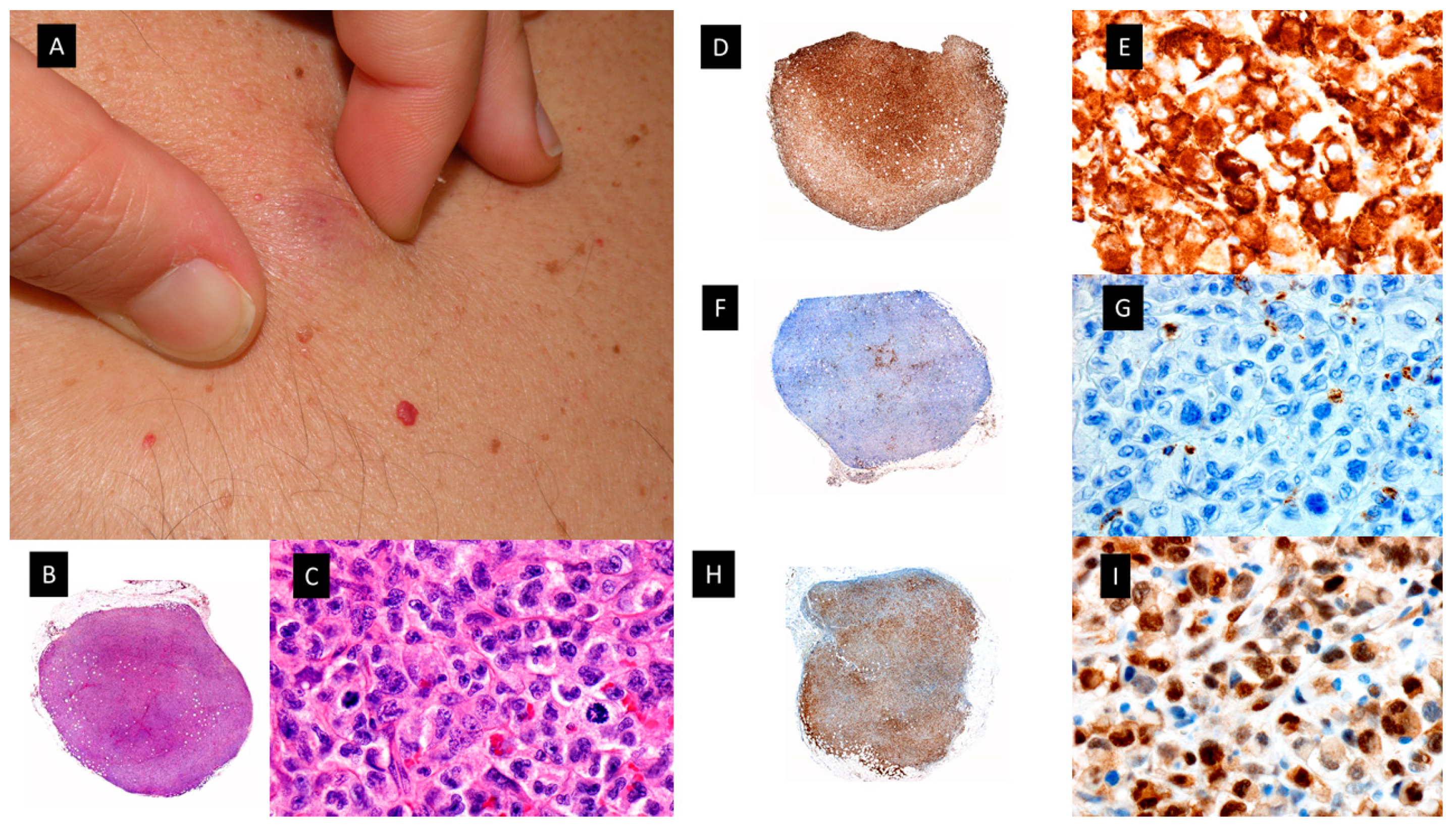
Plasmacytoid dendritic cell tumor. A. Multiple bruise-like plaques on the trunk and upper extremities. B. Scanning view showing diffuse tumoral infiltrate in dermis and subcutaneous tissue (HE staining 4x) C. CD4 positive diffuse staining in the tumoral infiltrate (x4). D. Higher magnification showing monomorphic proliferation of medium-sized blastoid cells and intratumoral hemorrhage (HE staining x40). E. Detail of the positivity of the majority of neoplastic cells for CD123 (40x) F. Tumoral cells showing also positivity for the cytoplasmic marker CD56 (40x).
Figure 4.
Plasmacytoid dendritic cell tumor. A. Multiple bruise-like plaques on the trunk and upper extremities. B. Scanning view showing diffuse tumoral infiltrate in dermis and subcutaneous tissue (HE staining 4x) C. CD4 positive diffuse staining in the tumoral infiltrate (x4). D. Higher magnification showing monomorphic proliferation of medium-sized blastoid cells and intratumoral hemorrhage (HE staining x40). E. Detail of the positivity of the majority of neoplastic cells for CD123 (40x) F. Tumoral cells showing also positivity for the cytoplasmic marker CD56 (40x).
2.2.2. OTHER MORPHOLOGICAL VARIANTS OF SWEET SYNDROME
There have been other clinicopathogical variants of reported in the literature in the setting of MDS/MPN mentioned above17. Among all these variants, it is worth highlighting the subcutaneous Sweet syndrome (S-SS) or neutrophilic lobular panniculitis18,19. Other names employed in the literature include “Sweet´s panniculitis”, “Sweet´s-like neutrophilic panniculitis” and “neutrophilic panniculitis associated with myeloid disorders”. Primary neutrophilic panniculitis, defined as neutrophilic infiltrates confined to the fat lobules of the subcutis as opposed to extension of a dermal neutrophilic infiltrate into the subcutis (secondary neutrophilic panniculitis) has been reported in the literature as a histologic pattern seen in a heterogeneous group of disorders including infectious panniculitis, “reactive panniculitis reaction”, which is associated with some bacterial antigens and usually has vascular changes, and leukemia cutis20. Some of these diseases may be readily diagnosed, but others are less distinct clinically and histopathologically. For example, it may be difficult to distinguish from S-SS and infectious panniculitis20.
S-SS in the setting of myeloid disorders have been described in scattered case reports and small series of cases as erythematous skin nodule(s) generally located in the extremities with the trunk and the face affected less frequently. Cutaneous lesions are usually accompanied by fever and they resolve within days or few weeks of oral steroid treatment like classic SS (C-SS). Histopathologically, these nodules are characterized by a dense neutrophilic infiltrate confined to the subcutis in the absence of vascular changes. Unlike C-SS, papillary dermal edema is minimal. Both septal and lobular panniculitic patterns have been reported, being the lobular one the commonest form. Interestingly, occasional large epithelioid cells consistent with reactive stromal fibroblasts and abnormal nuclear segmentation (hyposegmentation or hypersegmentation) of neutrophils has been described in these myeloid-related cases20. AML and MDS are the myeloid neoplasms more frequently related with S-SS in the reported cases20. A variant of subcutaneous H-SS has also been reported heralding transformation of MDS into AML21 in a relapsed myeloblastic leukemia22 and in MDS-refractory anemia23.
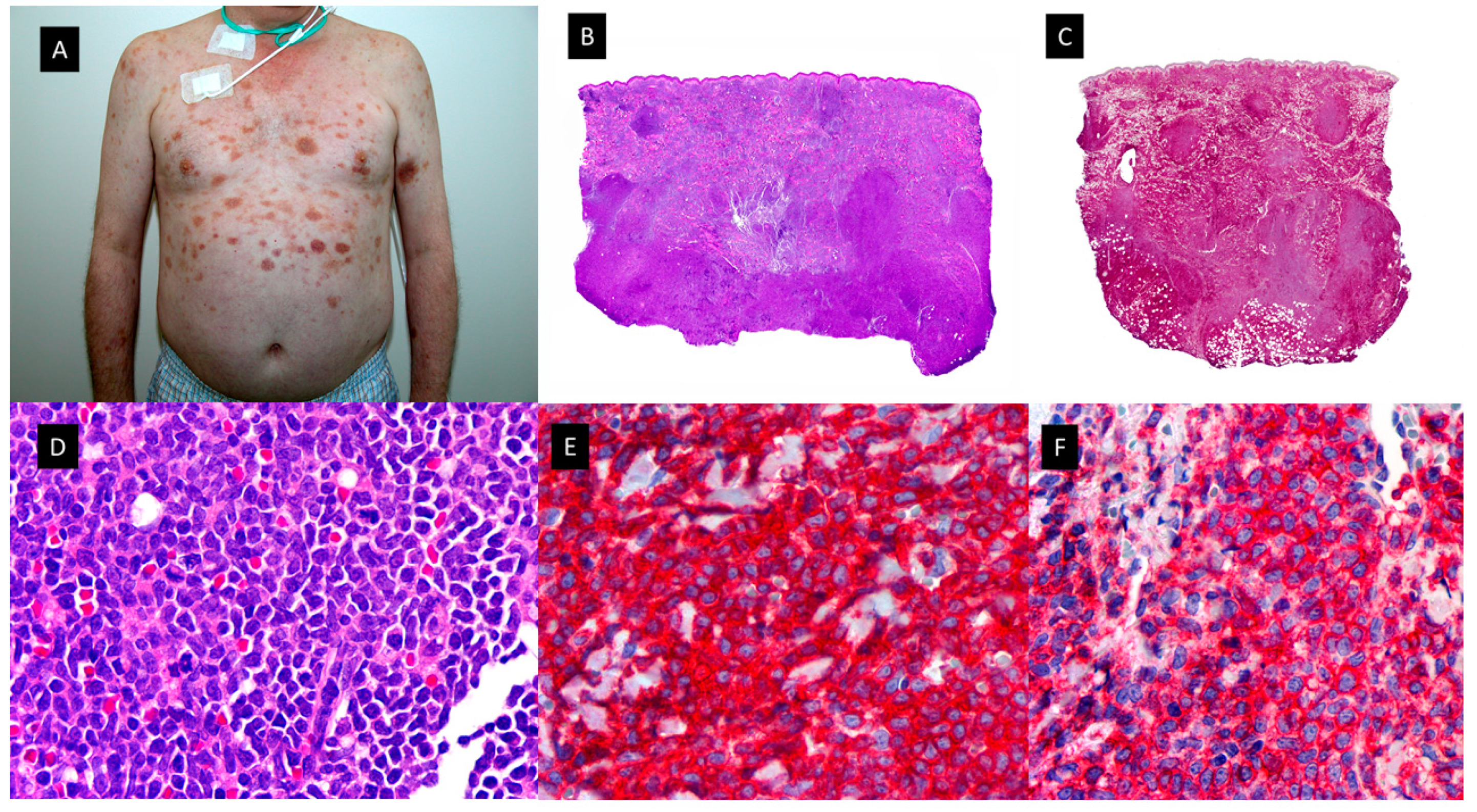
Figure 5.
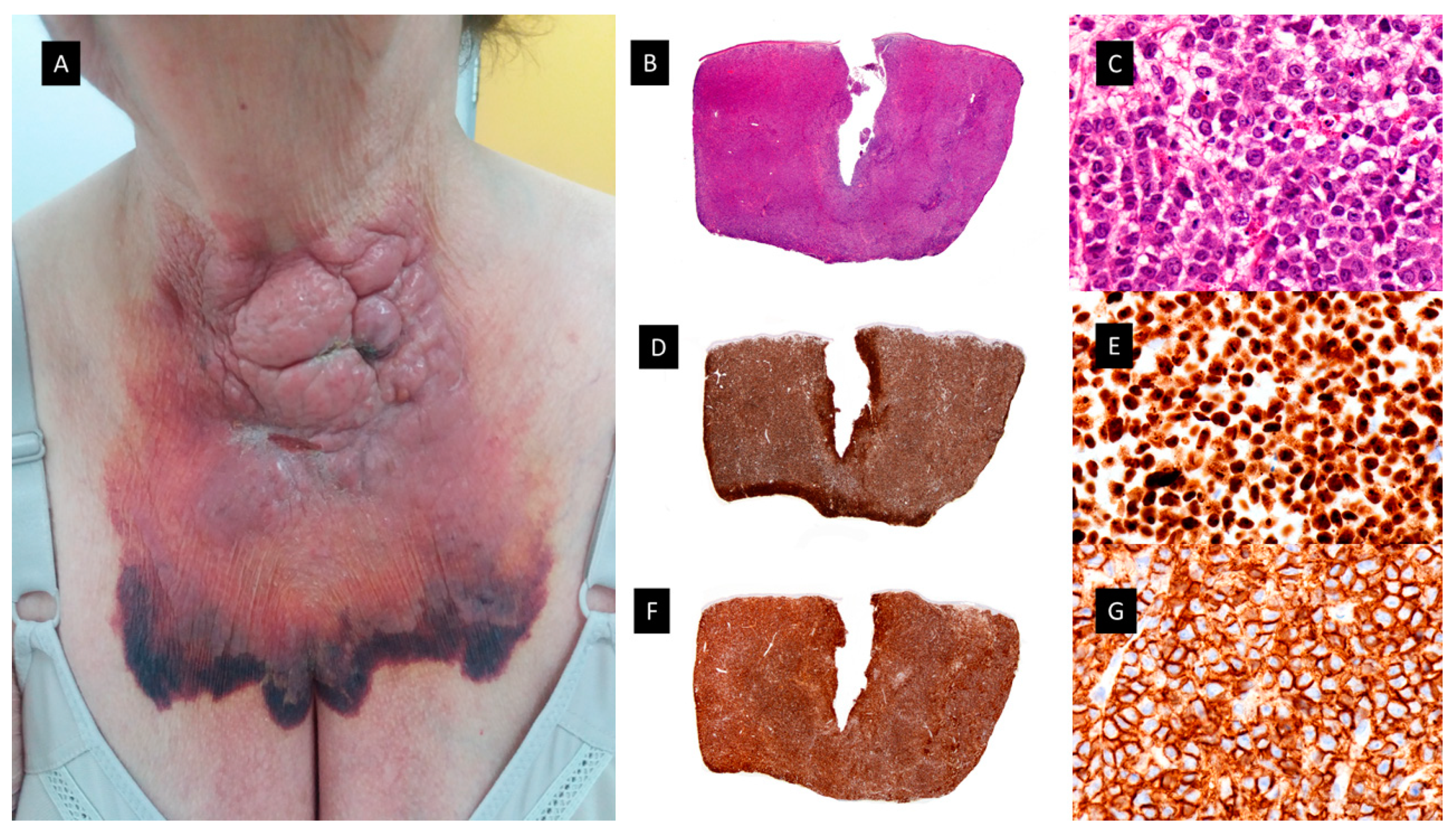
Granulocytic sarcoma. A. Clinical picture showing a large, hemorrhagic, violaceous polilobulated tumor on the anterior neck. B and C. Hematoxylin-eosin–stained specimen. B. Scanning view showing diffuse dermal infiltrate (4x). C. Higher magnification with medium-sized monomorphous atypical cells (40x). D and E. MNDA-stained specimen [D(4x) E (40x)]. F and G. CD56-stained specimen [F(4x), G(40x)].
Figure 5.
Granulocytic sarcoma. A. Clinical picture showing a large, hemorrhagic, violaceous polilobulated tumor on the anterior neck. B and C. Hematoxylin-eosin–stained specimen. B. Scanning view showing diffuse dermal infiltrate (4x). C. Higher magnification with medium-sized monomorphous atypical cells (40x). D and E. MNDA-stained specimen [D(4x) E (40x)]. F and G. CD56-stained specimen [F(4x), G(40x)].
2.3. VEXAS SYNDROME
In 2020, a new adult-onset autoinflammatory syndrome with overlapping features with MDS or multiple myeloma (MM) and inflammatory diseases such as relapsing polychondritis, SS, polyarteritis nodosa or giant-cell arteritis was described in 25 men with somatic mutations in p.Met41 of UBA1, the major E1 enzyme that initiates ubiquitylation24. This enzyme is encoded by the gene UBA1 located in the X chromosome.
The acronym VEXAS stands for vacuoles, E1 enzyme, X-linked, autoinflammatory and somatic syndrome. Somatic mutations, first acquired and then clonally selected, have been implicated as previously referred in neoplastic hematologic diseases25. With VEXAS syndrome the authors of the original description used a genotype-driven approach to identify a genetic cause of an autoinflammatory disease. All patients with UBA1 mosaic mutations had predominantly wild-type lymphocytes (T and B cells) and mutant myeloid cells (neutrophils and monocytes) in peripheral blood, although a patient with vacuolated lymphoid precursors has also been reported26. Hematopoietic stem cells and multipotent progenitors isolated from BM had abundant mutant cells. The patients began to develop the inflammatory syndromes in the fifth to seventh decade of life. Patients had progressive hematologic abnormalities, including macrocytic anemia, thrombocytopenia and myeloid dyspoiesis. Co-occurrence of VEXAS with hematologic neoplasms have been described with MDS and plasma cell dyscrasia and recently a male patient with chronic myeloid leukemia (CML) and VEXAS27 has also been reported. The dermatologic manifestations include neutrophilic dermatoses similar to SS with poor response to conventional therapies and frequent relapses, similar to the previously described MDS-cutis patients28.
Figure 6.
Histiocytic sarcoma. A Clinical picture showing a subcutaneous nodule of 4 cm on the chest. B and C, Hematoxylin-eosin–stained specimen. B. Scanning view of diffuse subcutaneous nodular infiltrate (4x) C. Higher magnification with atypical large cells with eosinophilic cytoplasm (40x). D and E. CD163-stained specimen showing diffuse positivity [D(4x) E (40x)]. F and G. MPO-stained specimen showing negative results [F(4x), G(40x)]. H and I. p-ERK-stained specimen showing diffuse positivity [H(4x) I (40x.
Figure 6.
Histiocytic sarcoma. A Clinical picture showing a subcutaneous nodule of 4 cm on the chest. B and C, Hematoxylin-eosin–stained specimen. B. Scanning view of diffuse subcutaneous nodular infiltrate (4x) C. Higher magnification with atypical large cells with eosinophilic cytoplasm (40x). D and E. CD163-stained specimen showing diffuse positivity [D(4x) E (40x)]. F and G. MPO-stained specimen showing negative results [F(4x), G(40x)]. H and I. p-ERK-stained specimen showing diffuse positivity [H(4x) I (40x.
2.4. MYELODYSPLASIA CUTIS
Osio et al identified in a series of 150 patients with MDS and cutaneous lesions 24 patients with non-blastic tumor cells in the dermal infiltrate, defined as medium-sized immature myeloid cells with abundant eosinophilic cytoplasm and twisted nuclei or pseudo-Pelger-Huet anomaly29. They found that these cells had a combined myeloid and monocytic immunophenotype, with expression of both MPO and CD163 or CD68 antigens and negativity for CD34, CD56 or CD117. The proliferative index was low in 56% of cases (<10% of positive Mib-1 cells) or intermediate in 44% of cases (10 to 66% positive Mib-1 cells). In addition, the cutaneous infiltrate was rich in mature neutrophils and normal CD3+ T-lymphocytes. The presence of edema in the superficial dermis was a frequent finding (67% of samples)29. Fluorescent in situ hybridization (FISH) analyses showed common cytogenetic abnormalities in the skin and the BM of 4/6 patients. Previously, only Sujobert et al had demonstrated a clonal relationship of this kind in neutrophilic dermatoses associated with AML30. These authors postulated that some cases previously reported as “H-SS” in the course of MDS, especially those with poor response to treatments such as hydroxychloroquine, dapsone, colchicine or thalidomide or patients with steroid dependence or in need of high dose oral prednisone could have MDS-cutis. Furthermore, they compared the survival rate and other clinicopathologic characteristics of patients with MDS-cutis and leukemia cutis and they found significant differences between the two groups29. Regarding overall survival (OS) after the skin diagnosis, it was significantly longer in MDS-cutis patients than in patients who suffered from leukemia cutis (62 vs. 5 months, (P<0.001)). In addition, there were other discriminant histopathologic features related with survival influence, such as positivity for CD34, CD56, or CD117 for shorter survival in patients with leukemia cutis (P<0.059), and presence of CD3+ T lymphocytes (P<0.001), edema (P<0.01), and a lower Mib-1 proliferative index (P<0.05) for longer survival in patients with MDS-cutis. Clinically, leukemia cutis patients usually have persistent skin nodules, whereas in MDS-cutis patients there were flares and relapses of cutaneous lesions. These lesions were nodules in leukemia cutis (P<0.01) and erythematous plaques (P<0.001), with a frequent annular pattern (P<0.05) accompanied by fever, or arthralgia (P<0.01) in MDS-cutis patients29.
Finally, there has been a recent report of 7 patients with MDS-cutis, one of them with UBA1 mutation (VEXAS syndrome), in which NGS analysis found 1 to 5 mutated genes both in the BM (with a median VAF of 30%) and in the cutaneous lesions of the same patients (with a median VAF of 11%)16. In addition, there were some mutations detected in one tissue but not in the other, supporting the possibility of clonal evolution in one of the tissues16. They suggested that MDS-cutis should be considered as differential diagnosis of leukemia cutis, classical SS, and MDS-associated vasculitis. MDS-cutis may potentially be underdiagnosed if a molecular investigation is not performed, especially in patients with H-SS. Unlike what happens in patients with N-SS, MDS-cutis patients seem to be more resistant to steroid therapy and they may respond to hypomethylating agents, like Azacytidine, although this sensitivity still must be confirmed in larger prospective series16.
2.5. GRANULOMATOUS DERMATOSES
Interstitial granulomatous dermatitis (IGD) was first described by Gottlieb et al in 1995 as a granulomatous dermatitis with interstitially disposed dermal infiltrates composed mainly of histiocytes in a palisaded arrangement around foci of degenerated collagen bundles, with scattered neutrophilis and eosinophilis31. Shortly after, Chu et al proposed the term palisaded neutrophilic and granulomatous dermatitis (PNGD) for the papular eruption on the extensor surface of extremities described in the context of collagen vascular diseases32. Both terms, IGD and PNGD as well as the overlapping between IGD and granuloma annulare, are now included by some authors in the spectrum of the so-called reactive granulomatous dermatitis33.
These cutaneous lesions have been mainly related to rheumatologic diseases and drug reactions. However, the association of PNGD or IGD with hematological malignancies, including CMML and MDS has been reported firstly as a reactive dermatitis in the setting of the hematological condition and lately as a possible skin manifestation of the disease34,35.
Federmann et al published in 2017 three patients with widespread papular eruptions resistant to conventional skin-directed therapies, which histopathologically showed interstitial granulomas composed of epithelioid histiocytes in the reticular dermis arranged around foci of degenerated collagen and variable neutrophilic inflammation consistent with PNGD36. The granulomas showed a similar immunohistochemical profile, with positivity for CD14, CD68 (PG-M1), and CD123, whereas only very few cells were positive for TCL1 and CD303. CD56, S100 protein and CD1a were negative. The most interesting contribution of this study was the finding of SRSF2 P95 hotspot mutations, found in 40%-50% of patients suffering from CMML, which were retrospectively detected in both skin and BM biopsies of the three patients. In addition, in one of them these mutations were already found 5 years before the diagnosis of the hematologic neoplasia36. Since then, only few more cases have been reported35,37,38. All but one were patients with CMML, in all cases where molecular techniques were performed, SRSF2 mutations were found in the BM and in the cutaneous infiltrates of the patients. For that reason, Enescu et al proposed that this kind of dermatitis is specific for CMML patients with this molecular alteration38. The molecular technique used for the study of mutations in BM and skin of these patients was variable, including NGS, pyrosequencing or RFLP analysis by BsaJI digestion and/or sequencing of SRSF2 polymerase chain reaction products containing the hotspot region in codon 95 (2 p.Pro95His;1 p.Pro95Leu)35,36,38.
Before the description of the Federmann cases in 2017, this type of granulomatous reaction had been published in association with MDS and the authors argued that it could be a manifestation of the disease, because the cutaneous lesions did not respond to conventional treatments for reactive dermatoses, but they improved with the specific chemotherapy treatment for the myeloid neoplasia34.
2.6. MATURE PLASMACYTOID DENDRITIC CELL DERMATOSES
Plasmacytoid dendritic cells (PDC) represent a subset of cells within the immune system with unique morphology, immunophenotype and specific functions39. Mature PDC are a small normal component of the BM, representing less than 1% of all nucleated cells. An increase in their number has been described in inflammatory conditions such as tuberculosis, sarcoidosis, hyaline-vascular subtype of Castleman disease, lupus erythematosus and Kikuchi-Fujimoto lymphadenopathy40. In addition, it is known that the number of PDC increases in both CMML and MDS BM biopsies39,41,42. Some authors found CD123-positive monocyte nodules only in CMML, whereas they were not present in BM samples from CML and atypical CML. Besides, their amount has been related to prognosis in previous publications43. Two extremely rare clinicopathological forms of PDC neoplasm in hematological patients have been reported so far: mature PDC proliferations and blastic plasmacytoid dendritic cells neoplasms (BPDCN). Myeloid neoplasms that have been related to both mature PDC proliferations and BPDCN in previous literature are again MDS, CMML and acute leukemia with monocytic differentiation39,41-43.
In 2012, Vitte et al published an article describing 42 patients with CMML and skin infiltrates44. They divided their patients in 4 different groups, (1) tumors of myelomonocytic cells; (2) tumors of mature PDC; (3) blastic PDC tumors (BPDCT); and (4) tumors of blastic indeterminate dendritic cells. They included 16 patients with tumors of mature PDC. This group was composed by 15 males and 1 female, whose median age at presentation was 76,35 years. Clinically, they presented with erythematous and itching maculo-papules. Generally, cutaneous lesions took place at the same time or after the diagnosis of CMML diagnosis
In 2022, Machan et al published 6 patients with pruritic papular eruptions histopathologically characterized by dermal infiltrates of mature T-lymphocytes with large clusters of CD123+ and SPIB+ cells and with a lack of myeloid cell nuclear differentiation antigen (MNDA). Four patients have been diagnosed of CMML and 2 had MDS (AREB-I and MDS with 5q deletion). The skin lesions developed in all cases coincidentally with either progression or full-establishment of their hematological disease45. Clinically, the skin lesions were more heterogeneous than in the case of the granulomatous dermatoses previous described and unlike the previous ones, most cutaneous lesions disappeared spontaneously or after oral or topical corticosteroid treatment. There was one patient with disseminated erythematous macules and papules in the same stage of development with occasional paler halos in which a clinical diagnosis of drug eruption versus vasculitis was established. Another patient presented with isolated erythematous nodules located on both left flank and breast, which were clinically interpreted as suggestive of primary cutaneous lymphoma. Finally, small papules on extremities mimicking insect bites were also described. Biopsy samples in these patients showed focal epidermotropism in 3 patients, with vacuolar degeneration of dermo-epidermal interface and folliculotropism of the infiltrate, but without mucin deposition. This histopathology with features mimicking lupus erythematosus was previously reported in a patient with CMML type 2 diagnosed simultaneously with the appearance of the cutaneous lesions46. In this study, PDC aggregates were positive for CD4, CD68, granzyme B, CD123, BDCA-2/CD303, TCL1 and the IFN-α inducible protein myxovirus A (MxA). In addition, PDC were aberrantly positive for CD5 and CD7, two antigens usually not found on normal PDC. CD56 stain was negative. They did not find abnormal number of Langerhans and dermal dendritic cells expressing CD1a, S100 protein or langerin/CD207. Besides, the patient shared the same chromosomal abnormality (chromosome 13 trisomy) detected by Fluorescence in situ hybridization (FISH) analysis in his cutaneous lesions and BM. The cutaneous lesions resolved with chemotherapy for the hematological neoplasm and the patient died one year later after progression to AML without recurrence of the cutaneous rash45.
The phenotype of PDC found by Machan et al showed that CD123+ cells were negative for CD4, myeloperoxidase, MNDA, CD56, TDT, BCL2, and cytotoxic markers and positive for SPIB. Neither CD34 nor CD117 positive cells were found. Interestingly, they found an increased number of S-100 protein and CD1a positive cells, mainly localized in the epidermis and superficial dermis. Only scattered double positive cells for S-100 protein and CD1a were identified45. From a molecular point of view, patient number 2 was diagnosed with CMML, and mutations in TET2, SRSF2, and ASXL1 genes were found in both BM and skin specimens by NGS techniques. The allele frequency of TET2 gene mutations were 90.3% and 15.4% in BM and skin, respectively; 45.8% and 7.3% for SRSF2 gene in BM and skin, respectively; and 39.9% and 5.9% for the ASXL1 gene in either BM and skin, respectively. The combination of TET2, SRSF2, and ASXL1 mutation is highly specific (98%) for the diagnosis of CMML and has prognostic implications47. Furthermore, their patient number 3 was diagnosed as AREB-I and mutations of genes DNMT3A and IDH1 with an allele frequency of 38.5% and 32.9%, respectively, were found in BM by NGS. The same IDH1 gene mutation using pyrosequencing was found on paraffin-embedded tissue from cutaneous lesions of this patient.45 Both TET2 and DNMT3A gene mutations have been related to clonal hematopoiesis of indeterminate potential (CHIP), in myeloid neoplasms48 and IDH1 mutations have been reported to be related with a poor outcome in MDS49.
Finally, if we compare histopathologically the Vitte et al. reported cases with the cases of Machan et al, clusters of mature PDC, lymphocytes, plasma cells, and indeterminate dendritic cells were present in the dermis in a similar proportion in both series44,45. However, cutaneous lesions of Vitte et al series either did not respond or showed only partial response in 12.5% and 37.5% of the patients, respectively, whereas in Machan et al series, all cutaneous lesions disappeared spontaneously or after topical or oral corticosteroid treatment. In addition, in all cases published by Machan et al, the skin lesions developed coincidentally with either progression of full-establishment of their hematological condition. Despite that, in the study published by Vitte et al the group of patients with mature PDC proliferations seemed to have better prognosis compared to the other ones, which were more consistent with leukemia cutis (see below)44.
2.7. OTHERS MYELODYSPLASTIC/MYELOPROLIFERATIVE DERMATOSIS
To conclude with the discussion about MDS/MPN related dermatoses with indolent clinical course, it is necessary to mention a rare clinical form of MLC that can be seen in CMML patients. It has been described as chilblain-like eruptions that can be a diagnostic clue to the blast crisis or the relapse of AL50 of chronic myelocytic leukemia51. Clinically, it presented as bluish erythematous macules with mild itching on the toes of both feet. Immunohistochemistry of the cutaneous lesions showed that infiltrating cells were positive for CD68 and partially positive for CD14. These cells were negative for CD13, CD33, CD34, and CD5651. Similar waxing and waning chilblain-like lesions of 8 months of duration were described in a 6-year-old girl diagnosed from juvenile myelomonocytic leukemia. Her skin lesions were histopathologically characterized by dense superficial perivascular dermal infiltrate, which was CD3-, CD43+ and lysozyme +, suggesting a monocytic origin52.
3. CUTANEOUS PROCESSES RELATED TO EITHER MDS/MNP AND AGGRESSIVE CLINICAL BEHAVIOR
Disease progression is a major source of mortality in myeloid neoplasms, including MDS/MPN. Apart for transformation to AML and overgrowth of pre-existing hematopoiesis by the myeloid neoplasm without an additional transforming event, there are other recurrent less well-known scenarios that exhibit a propensity for extramedullary sites like the skin. These scenarios include (1) acquisition of MDS features in MPN (2) acquisition of MPN features in MDS (3) progressive myelofibrosis (MF), (4) acquisition of CMML-like features, (5) development of granulocytic sarcoma/leukemia cutis (6) lymphoblastic transformation, and (7) histiocytic and dendritic cell overgrowth53. Here, we are going to describe the cutaneous lesions more frequently found in MDS/MPN neoplasms with disease progression and how the gain of distinct mutations/mutational patterns seem to be responsible or at least concomitant with these clinical scenarios.
3.1. LEUKEMIA CUTIS /MYELOID LEUKEMIA CUTIS
Leukemia-specific skin infiltrations are often named leukemia cutis (LC) in the literature, which includes both lymphoid and myeloid leukemias. In the case of myeloid disorders (including AMML, CMML, myelodysplasias, myeloproliferative syndromes such as polycythemia vera, essential thrombocythemia, and idiopathic myelofibrosis), myeloid leukemia cutis (MLC) seems to be a more appropriate term. Other names used to designate MLC are chloroma, myeloid sarcoma, and granulocytic sarcoma, which all mainly signify extramedullary localization of a myelomonocytic malignancy54. Most of these myeloid disorders represent AMLs, particularly those of monocytic differentiation. However, other myeloid disorders, less frequently represented as CMML, refractory anemia (RA), and MPS, may also involve the skin at the time of their blastic transformation. The frequency of LC is reported at 2.1–30 % depending on the underlying form of leukemia4 and it can reach 50% in some series of AML of monocytic subtypes55,56. Regardless of the type of myelogenous leukemia, the onset of specific skin manifestations clearly correlates with an aggressive course and short survival56.
3.2. GRANULOCYTIC SARCOMA
In 2011 Mathew et al performed a retrospective analysis exploring the role of LC in disease progression of CMML57. Of 108 patients in their series with CMML, 11 patients (10.2%) had LC including one patient with multicentric reticulohistiocytosis (MCRH) and one patient with BPDCN, which they define as its equivalent (2 patients). Four of these patients developed AML within 0–4 months. The remaining 7 patients demonstrated increased monocytes (<20% blasts), with 3 of them developing extramedullary involvement. They also showed that the overall survival (OS) from MLC to disease progression was 7.8 months. In addition, the OS from diagnosis to the last follow-up in patients with MLC was 28.2 months, shorter than patients without MLC (44 months). They postulated that MLC and its equivalent could predict disease progression to AML57.
Lately, in the study previously mentioned from Vitte et al the authors recognized 4 main subtypes of cutaneous involvement by CMML, one of them, as previously discussed, was the mature PDC tumors. The most frequent group was myelomonocytic cell tumors (MMCT) (n=18), exhibiting a proliferation of granulocytic or monocytic blast cells, which were CD68 and/or MPO positive, but negative for dendritic cell markers, similar to what it is known classically as granulocytic sarcoma44. According to the criteria of Vitte et al, 9 cases from Mathew’s et al series were diagnosed as MMCT. The median age in this group was 71,5 years with male to female ratio of 5:1. The lesions were most often papulonodular and multiple. The skin tumors consisted of a proliferation of medium-sized and/or large sized cells, mainly or entirely blast cells. A reactive infiltrate was present in 70% of the cases, predominantly lymphocytes. Tumor cells displayed a myelomonocytic phenotype, expressing CD68 (17/18), MPO (13/18), CD33 (13/18) and CD13 (4/18). PDC markers were negative in tumor cells. In addition, all cases were negative for CD1a and Langerin and all but one case were also negative for S100 protein. Ten cases were CD4 positive and 5 were CD56 positive. Four cases (22%) were both CD4 and CD56 positive. The proliferation index with Mib-1 antibody was highly variable and generally low. The OS was worse than in the group of mature PDC tumors, but better than the other two groups44. Taking all these findings together and including the series of Mathew et al and Vitte et al, these MMCT could be considered as the most frequent form of MLC in CMML patients.
ALEUKEMIC LC
Another interesting concept worth discussing when talking about LC is aleukemic LC, which has also been described in MDS/MPN58-60. It is characterized by the infiltration of the skin by leukemic cells before their appearance in peripheral blood or BM. The prompt recognition with a high suspect index is of crucial relevance, in order to make a correct diagnosis and distinguish it from other entities such as cutaneous large cell lymphoma. The prognosis is also poor, as in common MLC58-60. Cutaneous lesions initially suggestive of ND may actually represent LC, but it must not be confused with myelodysplasia cutis, previously discussed within neutrophilic dermatoses, because the evolution, clinical and histopathological characteristics are different, having MLC a much worse prognosis29. Some authors have coined the term aleukemic cutaneous myeloid sarcoma or aleukemic granulocytic sarcoma when there is an isolated atypical cutaneous infiltrate, reserving the term aleukemic LC for multiple skin lesions61, but there are authors which use both terms interchangeably62.
3.3. BLASTIC PLASMACYTOID DENDRITIC CELL TUMOR
This category of cutaneous disorder related to MDS/MPN and aggressive clinical behavior includes the third group mentioned by Vitte et al in their article as blastic PDC tumors (BPDCT). They included in this group 9.5% of the patients of their study (n=4). The median age of the patients was 71.5 years, with a male to female ratio of 3:1. Skin lesions were multiple and disseminated in 3 cases and solitary in 1 case. They comprised infiltrated plaques or violaceous nodules. Histopathologically, these lesions showed dermal infiltrates of medium-sized blast cells with slightly irregular nuclei and small nucleoli. Immunohistochemical studies revealed that CD68 was positive in 3 cases, whereas MPO, CD13, and CD33 were all negative. The 4 cases were CD4 positive, but only 3 cases showed positivity for CD56. All cases were CD1a negative, but 1 was S100 protein positive. The 4 cases were positive for the PDC markers CD123 and TCL1. Two cases were also positive for the PDC marker CD303. All 4 cases were negative for granzyme B44. In 2006, after the recognition of BPDCN as a separate entity, Dijkman et al carried out an integrated genomic analysis using expression profiling and array-based comparative genomic hybridization (CGH) on lesional skin biopsy samples of patients with CD4+CD56+ BPDCN and cutaneous myelomonocytic leukemia63. Their results demonstrated that CD4+CD56+ BPDCN and cutaneous myelomonocytic leukemia show distinct gene-expression profiles and distinct patterns of chromosomal aberrations. CD4+CD56+ BPDCN and cutaneous myelomonocytic leukemia split into 2 distinct groups consisting of a group of 5 CD4+CD56+ BPDCN and a group of 3 cutaneous myelomonocytic leukemia. CD4+CD56+BPDCN is characterized by recurrent deletion of regions on chromosome 4 (4q34), chromosome 9 (9p13-p11 and 9q12-q34), and chromosome 13 (13q12-q31) that contain several tumor suppressor genes with diminished expression (Rb1, LATS2). Elevated expression of the oncogenes HES6, RUNX2, and FLT3 was found, but it was not associated with genomic amplification. They found high expression of various plasmacytoid dendritic-cell (pDC)-related genes in BPDCN, pointing to the cell of origin of this malignancy63.
Recently, Khanlari et al have published a study showing that bone marrow clonal hematopoiesis (BMCH) is highly prevalent in BPDCN and frequently sharing a clonal origin in elderly patients. They assessed a potential clonal relationship between BPDCN and BMHC by comparing mutation profiles. A clonal association of BPDCN with BMCH was confirmed in 13 of their patients (54%) by demonstrating shared common mutations, being TET2, ASXL1 and ZRSR2 the most common ones. Although about 30-40% carry a diagnosis of MDS, CMML or MPN, many patients had no clinical manifestations of a myeloid neoplasm. In around 50-60% of cases, BPDCN share a clonal origin with these pre-leukemic clones but with additional molecular cytogenetic abnormalities64.
Case reports on the common clonal origins of BPDCN and MDS are rare. Recently, Yamada et al have reported a patient diagnosed with BPDCN limited to the skin and MDS with increased blasts 1 (MDS-IB1) in the BM65. They performed targeted NGS and copy number analyses (CNA) that revealed that both originated from the same clonal origin which subsequently evolved into BPDCN by acquiring multiple CNVs. The higher VAFs of the common mutations compared to the other mutations suggest that the genetic events occurred in the early phase and could have originated from CHIP65.
3.4. HISTIOCYTOID DISORDERS/HISTIOCYTOSIS
The histiocytosis are uncommon disorders of children and adults characterized by the neoplastic proliferation of dendritic cells, macrophages, or monocyte-derived cells in various tissues and organs, including the skin. There have been more than 100 different subtypes described in the literature. with a revised classification based on histopathology, immunophenotype, molecular alterations, and clinical and imaging characteristics published in 201666.
The last group included by Vitte et al in their study, which was the less frequent and the one with a worse prognosis, included histiocytic proliferations. They mentioned it as a putatively novel category of tumor that they named blastic indeterminate dendritic cell tumors (BIDCT) (n= 4), distinguished by a proliferation of large blast cells that not only exhibited monocytic markers but also the dendritic markers CD1a and S100 protein. The median age of the patients in this group was 65.5 years with a male to female ratio of 1:1. Tumors comprised a dermal infiltration of large blast cells with round, oval, or irregular nuclei and several nucleoli. Immunohistochemically, the 4 cases were positive for CD68, CD13, CD33, and CD1a.Three cases were positive for S100 protein, and none of the cases stained for langerin. Two cases were CD4 positive, and 3 cases were also CD56 positive. Interestingly, 1 case (case 42) also expressed the PDC marker CD303, whereas all other PDC markers were negative in the 4 cases. Mib-1 was available for 3 cases. In 2 of them (67%) Mib-1 index was high, being over 66%. The clinical characteristics of skin lesions consisted of a single nodule in 2 cases, multiple nodules in 1 case, and a large and ulcerated tumor of the scalp in 1 case44.
3.4.1. HISTIOCYTIC SARCOMA
Histiocytic sarcoma (HS) is an extremely rare histiocytosis with an aggressive course and limited treatment options. Clinically, the disease usually presents with single or multiple extranodal tumors, frequently located in the skin, intestines or soft tissues. The histopathology shows large atypical cells with eosinophilic cytoplasm and immunopositivity to CD163, CD68 and lysozyme. The overlap with some forms of non-Hodgkin´s lymphomas (NHL) have resulted in controversy about this diagnosis, and in this regard a number of earlier cases describing HS are most likely forms of high grade NHL67. In the literature there have been published cases of HS as secondary event following the diagnoses of other malignancies including acute monocyte leukemia68 and CMML69. In addition, Pérez-Sáenz et al published a patient who initially had CMML and HS on the skin (two nodules on the chest), who subsequently developed a therapy-related AML with monoblastic differentiation. They reported mutations on KRAS, TET2 and SRSF2 shared by the three neoplasms, suggesting a possible common clonal origin. The HS and AML showed additional molecular features70.
3.4.2. OTHER HISTIOCYTOSIS
A recently published multi-institutional cohort of 189 patients with Erdheim-Chester disease (ECD) and ECD overlapping with Langerhans cell histiocytosis [so-called mixed histiocytosis, (MH)] showed that 10,1% of patients (19/189) with ECD have an overlapping myeloid neoplasm, most commonly occurring as MPN, MDS, or mixed MDS/MPN overlap syndrome (including CMML)71. Molecular analysis frequently detected hallmark driver mutations typical of myeloid neoplasms (such as JAK2V617F and CALR mutations) coexisting with those characteristic of histiocytosis (such as BRAFV600E and MAP2K1 mutations). In addition, histiocytosis patients diagnosed with a concomitant myeloid malignancy were significantly older at diagnosis and more commonly presented with MH than those without a myeloid malignancy. The authors postulated that the presence of distinct kinase mutations in the histiocytosis and myeloid neoplasm resulted in discordant and adverse responses to kinase-directed targeted therapies71.
Later, a Dutch retrospective population-based study identified three patients with different histiocytic neoplasms and additional hematological malignancies bearing identical oncogenic mutations72. One patient had concomitant KRAS p.A59E mutated histiocytic sarcoma and CMML, one patient with synchronous NRAS p.G12V mutated indeterminate cell histiocytosis and CMML and finally, one patient with subsequent NRAS p.Q61R mutated ECD and AML72.
Furthermore, other case reports and series have published the association of cutaneous xanthomatous tumors73 and xanthelasma-like lesions fulfilling clinical, histopathological and molecular criteria for ECD associated with CMML, with a proven clonal relationship between the skin lesions and CMML cells based on molecular analyses using NGS techniques74, testicular Rosai-Dorfman disease associated with CMML sharing a KRAS variant c 0.35 G>A / p.G12D in both75 and also Langerhans cell histiocytosis (LCH) associated with MDS and other hematological neoplasms76,77. There have been some publications which pointed out that the cell of origin of one third of patients with systemic histiocytosis resides in a CD34+ hematopoietic progenitor cell prior to committed monocytes/macrophages78,79. Taking this into account, two main biologic mechanisms could explain the link between CMML and ECD. On one hand, the first one would be a differentiation from a clonal CMML cell into foamy macrophages in the skin through an additional genetic hit or a transforming event. On the other hand, the second would be a mutant common progenitor cell giving rise to the two distinct neoplasms via divergent differentiation.
In conclusion, these above mentioned studies, highlight the clinical importance of evaluating adults with histiocytosis for a concomitant myeloid neoplasm.
4. OTHER CUTANEOUS DISORDERS IN MDS/MPN PATIENTS
4.1. CUTANEOUS MANIFESTATIONS SECUNDARY TO MICROCIRCULATION ABNORMALITIES
Pernio and chilblain lupus in the setting of myeloid neoplasms, specially CMML are well-known cutaneous disorders described for the first time in four elderly patients with a hematologic disorder whose clinical features were identifiable as CMML80. The occurrence of florid pernio in an elderly man with CMML is unusual. In the reported cases, pernio usually preceded the diagnosis of the hematologic disorder by a period between 6 months and 3 years81. The histopathologic features of these lesions consisted of a superficial and deep lymphohistiocytic dermal infiltrate associated with lymphocytic vasculitis. Some authors postulated three possible mechanisms for these lesions: (1) Presence of malignant monocytic cells, because of their rigidity, large diameter and difficulty in passing through the vascular lumen may cause modifications in vascular flow in the microcirculation; (2) Polyclonal activation of B lymphocytes with polyclonal hypergammaglobulinemia with autoantibodies secretion; and finally, (3) The role of cold, which has an influence in blood viscosity82. The immunohistochemical demonstration of the lymphoid nature of the infiltrate and ruling out specific leukemic infiltrates allows the histopathologic differential diagnosis with the lesions of chilblain-like leukemia cutis described before83.
Other chronic myeloproliferative disorder associated with livedoid and purpuric cutaneous lesions due to microcirculation abnormalities is essential thrombocythemia84,85. Due to the elevated platelet count, the main histopathologic finding in these lesions consists of luminal obliteration by homogeneous eosinophilic material of medium sized blood vessels. Immunohistochemistry demonstrates positivity for the platelet marker CD61 in the eosinophilic material, without features vasculitis, characteristic of occlusive vasculopathy84,85.
4.2. OTHER INFLAMMATORY/ AUTOIMMUNE DISEASES
There are still some others cutaneous manifestations more rarely reported, which cannot be included into one of the previously described categories. The most important are the ones related to autoimmunity. Autoimmune paraneoplastic syndromes are commonly encountered in patients with MDS and CMML. A review of case reports and small series suggest as many as 10% of MDS patients might experience several autoimmune syndromes. Clinical manifestations of these syndromes include an acute systemic vasculitic syndrome, cutaneous vasculitis, fever, arthritis, pulmonary infiltrates, peripheral polyneuropathy, inflammatory bowel disease, glomerulonephritis, and even classical connective tissue disorders, such as relapsing polychondritis, posting sometimes a diagnostic challenge with VEXAS syndrome86.
Other very rare reported cutaneous lesions related with these myeloid neoplasms are the ones derived from extramedullary hematopoiesis87.
5. CONCLUSION
Interestingly, in recent literature there are increasing numbers of papers based on FISH or next-generation sequencing studies suggesting that the skin infiltrate of dermatoses that arise in the context of myeloid malignancies carry the same molecular alterations than the malignant clone. This has been particularly demonstrated in H-SS, MDS-cutis, PNGD, plasmacytoid dendritic cell dermatosis and in rare instances for erythema nodosum and pyoderma gangrenosum. Moreover, this relationship has also been established for histiocytosis.
In conclusion, the reason for MDS/MPN presenting with this broad spectrum of clinical and histopathologic cutaneous manifestations and its biological meaning is not known. In some cases, cutaneous manifestations, like granulomatous dermatoses, could represent an additional criterion for early diagnosis of CMML/MDS. These studies suggest a high degree of plasticity between myeloid, monocytic and PDC cells that originates from common precursors and this review article summarizes the cutaneous manifestations of these rare myeloid neoplasms, highlighting the importance they have in the diagnosis and management of these challenging patients.
Acknowledgments
LP: LR and SMR performed the research and wrote the manuscript, all contributed to the design and implementation of the research, to the analysis of the results and to the writing of the manuscript. All authors discussed the results and contributed to the final manuscript.
References
- Khoury, J.D.; Solary, E.; Abla, O.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Orazi, A.; Germing, U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008, 22, 1308–19. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Fenchel, K.; Back, W.; Schulz, A.; Sachse, M.M. Leukemia cutis - epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012, 10, 27–36. [Google Scholar] [CrossRef]
- Passet, M.; Lepelletier, C.; Vignon-Pennamen, M.D.; et al. Next-Generation Sequencing in Myeloid Neoplasm-Associated Sweet's Syndrome Demonstrates Clonal Relation between Malignant Cells and Skin-Infiltrating Neutrophils. J Invest Dermatol. 2020, 140, 1873–1876. [Google Scholar] [CrossRef]
- Martin de Frémont, G.; Hirsch, P.; Gimenez de Mestral, S.; et al. Myeloid Clonal Infiltrate Identified With Next-Generation Sequencing in Skin Lesions Associated With Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: A Case Series. Front Immunol. 2021, 12, 715053. [Google Scholar] [CrossRef] [PubMed]
- Whittington, C.P.; Ross, C.W.; Ramirez, J.A.; Lowe, L.; Brown, N.; Hristov, A.C. Myelodysplasia Cutis. Arch Pathol Lab Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.H.; Ko, C.J.; Leung, T.H.; et al. Neutrophilic Dermatoses: a Clinical Update. Curr Dermatol Rep. 2022, 11, 89–102. [Google Scholar] [CrossRef]
- Sweet, RD. AN ACUTE FEBRILE NEUTROPHILIC DERMATOSIS. Br J Dermatol. 1964, 76, 349–56. [Google Scholar] [CrossRef]
- Ferea, C.R.; Mihai, S.N.; Balan, G.; Badescu, M.C.; Tutunaru, D.; Tatu, A.L. Sweet Syndrome Associated with Myelodysplastic Syndrome-A Review of a Multidisciplinary Approach. Life (Basel) 2023, 13. [Google Scholar] [CrossRef]
- Cohen, P.R. Sweet's syndrome--a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Requena, L.; Kutzner, H.; Palmedo, G.; et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005, 141, 834–42. [Google Scholar] [CrossRef]
- Alegría-Landa, V.; Rodríguez-Pinilla, S.M.; Santos-Briz, A.; et al. Clinicopathologic, Immunohistochemical, and Molecular Features of Histiocytoid Sweet Syndrome. JAMA Dermatol. 2017, 153, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Vignon-Pennamen, M.D.; Juillard, C.; Rybojad, M.; et al. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia: a report of 9 cases. Arch Dermatol. 2006, 142, 1170–6. [Google Scholar] [CrossRef] [PubMed]
- Ghoufi, L.; Ortonne, N.; Ingen-Housz-Oro, S.; et al. Histiocytoid Sweet Syndrome Is More Frequently Associated With Myelodysplastic Syndromes Than the Classical Neutrophilic Variant: A Comparative Series of 62 Patients. Medicine (Baltimore). 2016, 95, e3033. [Google Scholar] [CrossRef] [PubMed]
- Delaleu, J.; Kim, R.; Zhao, L.P.; et al. Clinical, pathological, and molecular features of myelodysplasia cutis. Blood. 2022, 139, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, A.; Yanagisawa, H.; Tsunemi, Y.; et al. Normolipemic xanthomatized Sweet's syndrome: A variant of Sweet's syndrome with myelodysplastic syndrome. J Dermatol. 2021, 48, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Sutra-Loubet, C.; Carlotti, A.; Guillemette, J.; Wallach, D. Neutrophilic panniculitis. J Am Acad Dermatol. 2004, 50, 280–5. [Google Scholar] [CrossRef]
- Jagdeo, J.; Campbell, R.; Long, T.; Muglia, J.; Telang, G.; Robinson-Bostom, L. Sweet's syndrome--like neutrophilic lobular panniculitis associated with all-trans-retinoic acid chemotherapy in a patient with acute promyelocytic leukemia. J Am Acad Dermatol. 2007, 56, 690–3. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.P.; Duncan, L.M.; Nazarian, R.M. Subcutaneous Sweet syndrome in the setting of myeloid disorders: a case series and review of the literature. J Am Acad Dermatol. 2013, 68, 1006–15. [Google Scholar] [CrossRef] [PubMed]
- Srisuttiyakorn, C.; Reeve, J.; Reddy, S.; Imaeda, S.; Lazova, R. Subcutaneous histiocytoid Sweet's syndrome in a patient with myelodysplastic syndrome and acute myeloblastic leukemia. J Cutan Pathol. 2014, 41, 475–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cornejo, K.M.; Rork, J.; Rothman, K.; Deng, A. Subcutaneous Histiocytoid Sweet Syndrome in a Patient With Relapsed Acute Myeloblastic Leukemia. Am J Dermatopathol. 2018, 40, 459–462. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, Q.; Chen, M. Subcutaneous histiocytoid Sweet's syndrome in a patient associated with myelodysplastic syndrome-refractory anemia. J Dermatol. 2012, 39, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef]
- Cooper, J.N.; Young, N.S. Clonality in context: hematopoietic clones in their marrow environment. Blood. 2017, 130, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- 26. Haines P, Pullarkat S, Said J. VEXAS syndrome: Vacuoles in myeloid, erythroid, and lymphoid lineages. Int J Lab Hematol. [CrossRef]
- Djerbi, N.; Zimmermann, K.; Balabanov, S.; Manz, M.G.; Becker, M.O.; Roncador, M. Intra-patient competition of VEXAS syndrome and CML clones. Blood Adv. 2023. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Routledge, D.; van Der Weyden, C.; et al. VEXAS syndrome: A dermatological perspective. Australas J Dermatol. 2022, 63, 488–492. [Google Scholar] [CrossRef]
- Osio, A.; Battistella, M.; Feugeas, J.P.; et al. Myelodysplasia Cutis Versus Leukemia Cutis. J Invest Dermatol. 2015, 135, 2321–2324. [Google Scholar] [CrossRef]
- Sujobert, P.; Cuccuini, W.; Vignon-Pennamen, D.; et al. Evidence of differentiation in myeloid malignancies associated neutrophilic dermatosis: a fluorescent in situ hybridization study of 14 patients. J Invest Dermatol. 2013, 4, 1111–4. [Google Scholar] [CrossRef]
- Gottlieb GJ DR, Ackerman AB. 1:3-6. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: linear subcutaneous bands in rheumatoid arthritis revisited. Dermatopathol Pract Concept. 1995, (1):3-6.
- Chu, P.; Connolly, M.K.; LeBoit, P.E. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994, 130, 1278–83. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach M, English JC, 3rd. Reactive Granulomatous Dermatitis: A Review of Palisaded Neutrophilic and Granulomatous Dermatitis, Interstitial Granulomatous Dermatitis, Interstitial Granulomatous Drug Reaction, and a Proposed Reclassification. Dermatol Clin. 2015, 33, 373–87. [CrossRef]
- Yoneta, K.; Fujimoto, N.; Teramura, K.; Takayama, S.; Tanaka, T. Disseminated granulomatous skin lesions associated with myelodysplastic syndrome treated successfully with tranilast: a case report and review of the literature. Eur J Dermatol. 2016, 26, 398–400. [Google Scholar] [CrossRef]
- Prieto-Torres, L.; Santonja, C.; Chamizo, C.; et al. Granulomatous Dermatitis Heralding Myelodisplastic/Myeloproliferative Neoplasms. Neoplastic or Reactive Cells? A Study of 2 Cases. Am J Dermatopathol. 2022, 44, 456–460. [Google Scholar] [CrossRef]
- Federmann, B.; Bonzheim, I.; Yazdi, A.S.; Schmidt, J.; Fend, F.; Metzler, G. Generalized palisaded neutrophilic and granulomatous dermatitis-a cutaneous manifestation of chronic myelomonocytic leukemia? A clinical, histopathological, and molecular study of 3 cases. Hum Pathol. 2017, 64, 198–206. [Google Scholar] [CrossRef]
- Kyriakou, A.; Patsatsi, A.; Papadopoulos, V.; Kioumi, A.; Efstratiou, I.; Lazaridou, E. A case of palisaded neutrophilic granulomatous dermatitis with subsequent development of chronic myelomonocytic leukemia. Clin Case Rep. 2019, 4, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Enescu, C.D.; Patel, A.; Friedman, B.J. Unique Recognizable Histopathologic Variant of Palisaded Neutrophilic and Granulomatous Dermatitis that Is Associated With SRSF2-Mutated Chronic Myelomonocytic Leukemia: Case Report and Review of the Literature. Am J Dermatopathol. 2022, 44, e33–e36. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Hebeda, K.; Kremer, M.; et al. Plasmacytoid dendritic cell proliferations and neoplasms involving the bone marrow : Summary of the workshop cases submitted to the 18th Meeting of the European Association for Haematopathology (EAHP) organized by the European Bone Marrow Working Group, Basel 2016. Ann Hematol. 2017, 5, 765–777. [Google Scholar]
- Wang, P.; Feng, Y.; Deng, X.; et al. Tumor-forming plasmacytoid dendritic cells in acute myelocytic leukemia: a report of three cases and literature review. Int J Clin Exp Pathol. 2017, 10, 7285–7291. [Google Scholar] [PubMed]
- Facchetti, F.; Cigognetti, M.; Fisogni, S.; Rossi, G.; Lonardi, S.; Vermi, W. Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol. 2016, 29, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Orazi, A.; Chiu, R.; O'Malley, D.P.; et al. Chronic myelomonocytic leukemia: The role of bone marrow biopsy immunohistology. Mod Pathol. 2006, 19, 1536–45. [Google Scholar] [CrossRef]
- Lucas, N.; Duchmann, M.; Rameau, P.; et al. Biology and prognostic impact of clonal plasmacytoid dendritic cells in chronic myelomonocytic leukemia. Leukemia. 2019, 33, 2466–2480. [Google Scholar] [CrossRef] [PubMed]
- Vitte, F.; Fabiani, B.; Bénet, C.; et al. Specific skin lesions in chronic myelomonocytic leukemia: a spectrum of myelomonocytic and dendritic cell proliferations: a study of 42 cases. Am J Surg Pathol. 2012, 36, 1302–16. [Google Scholar] [CrossRef] [PubMed]
- Machan, S.; Alonso-Dominguez, J.M.; Sánchez García, F.J.; et al. Plasmacytoid Dendritic Cell Dermatosis Associated to Myeloproliferative/Myelodysplastic Neoplasms. Am J Surg Pathol. 2022, 46, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Dargent, J.L.; Henne, S.; Pranger, D.; et al. Tumor-forming plasmacytoid dendritic cells associated with myeloid neoplasms. Report of a peculiar case with histopathologic features masquerading as lupus erythematosus. J Cutan Pathol. 2016, 43, 280–6. [Google Scholar] [CrossRef]
- Jian, J.; Qiao, Y.; Li, Y.; Guo, Y.; Ma, H.; Liu, B. Mutations in chronic myelomonocytic leukemia and their prognostic relevance. Clin Transl Oncol. 2021, 23, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.M.; Nazha, A.; Kneen, K.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia. 2018, 32, 1751–1761. [Google Scholar] [CrossRef]
- Wang N, Wang F, Shan N, Sui X, Xu H. IDH1 Mutation Is an Independent Inferior Prognostic Indicator for Patients with Myelodysplastic Syndromes. Acta Haematol. 2017, 138, 143–151. [CrossRef] [PubMed]
- Brazão, C.; Mancha, D.; Antunes-Duarte, S.; Kempf, W.; Soares-de-Almeida, L. Chilblain-Like Eruption Unveiling Cutaneous Aleukemic Relapse of Acute Myeloid Leukemia. Am J Dermatopathol. Sep 13 2023. [CrossRef]
- Yazawa, H.; Saga, K.; Omori, F.; Jimbow, K.; Sasagawa, Y. The chilblain-like eruption as a diagnostic clue to the blast crisis of chronic myelocytic leukemia. J Am Acad Dermatol. 2004, 50 (2 Suppl), S42–4. [Google Scholar] [CrossRef]
- Affleck, A.G.; Ravenscroft, J.C.; Leach, I.H. Chilblain-like leukemia cutis. Pediatr Dermatol. 2007, 24, 38–41. [Google Scholar] [CrossRef]
- Faria, C.; Tzankov, A. Progression in Myeloid Neoplasms: Beyond the Myeloblast. Pathobiology. 2023, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bénet, C.; Gomez, A.; Aguilar, C.; et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: a study of 173 cases. Am J Clin Pathol. 2011, 135, 278–90. [Google Scholar] [CrossRef] [PubMed]
- Cho-Vega, J.H.; Medeiros, L.J.; Prieto, V.G.; Vega, F. Leukemia cutis. Am J Clin Pathol. Jan 2008, 129, 130–42. [Google Scholar] [CrossRef] [PubMed]
- Kaddu, S.; Zenahlik, P.; Beham-Schmid, C.; Kerl, H.; Cerroni, L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol. 1999, 40 Pt 1, 966–78. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.A.; Bennett, J.M.; Liu, J.J.; et al. Cutaneous manifestations in CMML: Indication of disease acceleration or transformation to AML and review of the literature. Leuk Res. 2012, 36, 72–80. [Google Scholar] [CrossRef]
- Ohno, S.; Yokoo, T.; Ohta, M. Aleukemic leukemia cutis. J Am Acad Dermatol. Feb 1990;22(2 Pt 2):374-7. [CrossRef]
- Heskel, N.S.; White, C.R.; Fryberger, S.; Neerhout, R.C.; Spraker, M.; Hanifin, J.M. Aleukemic leukemia cutis: juvenile chronic granulocytic leukemia presenting with figurate cutaneous lesions. J Am Acad Dermatol. 1983, 9, 423–7. [Google Scholar] [CrossRef]
- Gil-Mateo, M.P.; Miquel, F.J.; Piris, M.A.; Sánchez, M.; Martin-Aragonés, G. Aleukemic "leukemia cutis" of monocytic lineage. J Am Acad Dermatol. 1997, 36 (5 Pt 2), 837-40. [CrossRef]
- Aboutalebi, A.; Korman, J.B.; Sohani, A.R.; et al. Aleukemic cutaneous myeloid sarcoma. J Cutan Pathol. 2013, 40, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Iliadis, A.; Koletsa, T.; Georgiou, E.; Patsatsi, A.; Sotiriadis, D.; Kostopoulos, I. Bilateral Aleukemic Myeloid Sarcoma of the Eyelids With Indolent Course. Am J Dermatopathol. Apr 2016, 38, 312–4. [Google Scholar] [CrossRef]
- Dijkman, R.; van Doorn, R.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Gene-expression profiling and array-based CGH classify CD4+CD56+ hematodermic neoplasm and cutaneous myelomonocytic leukemia as distinct disease entities. Blood. 2007, 109, 1720–7. [Google Scholar] [CrossRef]
- Khanlari, M.; Yin, C.C.; Takahashi, K.; et al. Bone marrow clonal hematopoiesis is highly prevalent in blastic plasmacytoid dendritic cell neoplasm and frequently sharing a clonal origin in elderly patients. Leukemia. 2022, 36, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Hiramoto, N.; Mori, T.; et al. Coincidence of cutaneous blastic plasmacytoid dendritic cell neoplasm and myelodysplastic syndrome derived from clonal hematopoiesis. Blood Cancer J. 2023, 1, 119. [Google Scholar] [CrossRef] [PubMed]
- Emile, J.F.; Abla, O.; Fraitag, S.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016, 127, 2672–81. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Naqash, A.R.; Munker, R.; et al. Histiocytic sarcoma as a secondary malignancy: pathobiology, diagnosis, and treatment. Eur J Haematol. 2016, 97, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Niu, X.; Wang, Z.; Lu, H.; Lin, X.; Lu, Q. Histiocytic sarcoma combined with acute monocytic leukemia: a case report. Diagn Pathol. 2015, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Matsushita, A.; Takiuchi, Y.; et al. Histiocytic sarcoma and underlying chronic myelomonocytic leukemia: a proposal for the developmental classification of histiocytic sarcoma. Int J Hematol. 2010, 92, 168–73. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sáenz, M.A.; Rodriguez-Pinilla, S.M.; Salgado, R.N.; et al. Three monocytic neoplasms in a single patient. Leuk Lymphoma. 2020, 61, 2523–2526. [Google Scholar] [CrossRef] [PubMed]
- Papo, M.; Diamond, E.L.; Cohen-Aubart, F.; et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017, 130, 1007–1013. [Google Scholar] [CrossRef]
- Kemps, P.G.; Hebeda, K.M.; Pals, S.T.; et al. Spectrum of histiocytic neoplasms associated with diverse haematological malignancies bearing the same oncogenic mutation. J Pathol Clin Res. 2021, 7, 10–26. [Google Scholar] [CrossRef]
- Miralles, E.S.; Escribano, L.; Bellas, C.; Núñez, M.; Ledo, A. Cutaneous xanthomatous tumours as an expression of chronic myelomonocytic leukaemia? Clin Exp Dermatol. 1996, 21, 145–7. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, P.; Chasset, F.; Moguelet, P.; et al. Erdheim-Chester disease associated with chronic myelomonocytic leukemia harboring the same clonal mutation. Haematologica. 2019, 104, e530–e533. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, A.; Dirnhofer, S.; Juskevicius, D.; et al. Testicular Rosai-Dorfman disease clonally related to CMML - Case report and literature review. Pathol Res Pract. Jul 2023, 247, 154548. [Google Scholar] [CrossRef] [PubMed]
- Kiavash, K.; Malone, J.C. Langerhans Cell Histiocytosis Associated With Underlying Hematolymphoid Disorders in Adults: Report of 2 Cases and Review of the Literature. Am J Dermatopathol. 2018, 40, 588–593. [Google Scholar] [CrossRef]
- Billings, S.D.; Hans, C.P.; Schapiro, B.L.; et al. Langerhans cell histiocytosis associated with myelodysplastic syndrome in adults. J Cutan Pathol. 2006, 33, 171–4. [Google Scholar] [CrossRef]
- Durham, B.H.; Roos-Weil, D.; Baillou, C.; et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017, 130, 176–180. [Google Scholar] [CrossRef]
- Milne, P.; Bigley, V.; Bacon, C.M.; et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017, 130, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Marks, R.; Lim, C.C.; Borrie, P.F. A perniotic syndrome with monocytosis and neutropenia--a possible association with a preleukaemic state. Br J Dermatol. 1969, 81, 327–32. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W.; Dowling, J.P. Pernio. A possible association with chronic myelomonocytic leukemia. Arch Dermatol. 1985, 121, 1048–52. [Google Scholar] [CrossRef]
- Dreno, B.; Gandon, P.; Bureau, B.; Milpied, N.; Barrière, H. Skin lesions from hypersensitivity to cold during chronic myelomonocytic leukaemia. Br J Dermatol. 1986, 115, 607–9. [Google Scholar] [CrossRef]
- Nazzaro, G.; Genovese, G.; Marzano, A.V. Idiopathic chilblains in myelomonocytic leukemia: not a simple association. Int J Dermatol. 2018, 57, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Pielasinski, U.; Haro, R.; Santonja, C.; Kutzner, H.; Requena, L. Essential thrombocythemia presenting as localized livedo reticularis. Am J Dermatopathol. 2013, 35, e22–5. [Google Scholar] [CrossRef] [PubMed]
- Lapeña Casado, A.; Santonja, C.; García-García, M.; et al. Essential thrombocythemia manifesting as livedoid and purpuric skin lesions: Report of two cases and literature review. J Cutan Pathol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Hopkins, J.L.; Gore, S.D. Autoimmune phenomena in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Lymphoma. 2002, 43, 2083–92. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Horiuchi, M.; Ueda, H.; et al. [Cutaneous extramedullary hematopoiesis associated with myelodysplastic/myeloproliferative neoplasm, unclassifiable]. Rinsho Ketsueki. 2015, 56, 911–4. [Google Scholar] [CrossRef] [PubMed]

Table 1.
MDS/MPN related dermatoses with indolent clinical course with main immunohistochemical characteristics of myeloid cells.
Table 1.
MDS/MPN related dermatoses with indolent clinical course with main immunohistochemical characteristics of myeloid cells.
| 1. NEUTROPHILIC DERMATOSES 1.1 SWEET SYNDROME (SS) 1.1.1 HISTIOCYTOID SWEET SYNDROME (H-SS) CD15+, CD43+, CD45+, Lysozyme+, MPO+ CD68/KP1+>CD68/PGM1+, MNDA+ (Double IS MNDA/MPO+) 1.1.2 OTHER MORPHOLOGICAL VARIANTS OF SS 1.2 VEXAS SYNDROME MPO+, MNDA+ |
| 1. MYELODYSPLASIA CUTIS CD33+, CD163+, CD14+, CD68/PGM1+, MPO+(Double IS CD123+/MNDA+) CD34- CD117-, CD56-, low ki67 |
| 3. GRANULOMATOUS DERMATOSES CD14+, CD68/PGM1+, CD123+ CD56-, S100-, CD1a- |
| 4. MATURE PLASMACYTOID DENDRITIC CELL DERMATOSES Double IS CD123+/SPIB + CD56-, CD34-, CD117-, MPO-, TDT-, Bcl2-, MNDA- |
| 5. OTHERS |
MDS/MPN: Myelodysplastic/Myeloproliferative, MPO: Myeloperoxidase, IS: Immunostain.
Table 2.
Cutaneous processes related to either MDS/MPN and aggressive clinical behavior with main immunohistochemical characteristics of tumor cells.
Table 2.
Cutaneous processes related to either MDS/MPN and aggressive clinical behavior with main immunohistochemical characteristics of tumor cells.
| 1. LEUKEMIA CUTIS/MYELOID LEUKEMIA CUTIS 1.1 GRANULOCYTIC SARCOMA CD68+, MPO+, CD33+, CD13+, high ki67 More likely to express CD34, CD117 1.2. ALEUKEMIC LEUKEMIA CUTIS No systemic involvement at diagnosis |
| 2. BLASTIC PLASMACYTOID DENDRITIC CELL TUMOR CD4+, CD56+ high Ki67, SPIB+, CD123+ MPO-, CD13-, CD33-, MNDA- |
| 3. HISTIOCYTOID DISORDERS/HISTIOCYTOSES 3.1 HISTIOCYTIC SARCOMA CD163+, CD68+, Lysozyme+, pERK+, MPO- 3.2 OTHERS ECD:CD68+, CD163+, CD1a- LCH: S100+, CD1a+, Langerin/CD203+ |
MDS/MPN: Myelodysplastic/Myeloproliferative, MPO: Myeloperoxidase, ECD Erdheim-Chester disease, LCH: Langerhans cell histiocytoses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



