
Submitted:
11 November 2023
Posted:
14 November 2023
You are already at the latest version
Abstract
The present review explores the critical role of oxime and oxime ether moieties in enhancing the physicochemical and anticancer properties of structurally diverse molecular frameworks. Specific examples are carefully selected to illustrate the distinct contributions of these functional groups to general strategies for molecular design, modulation of biological activities, computational modeling, and structure-activity relationship studies. An extensive literature search was conducted across three databases, including Pub-Med, Google Scholar, and Scifinder, enabling us to create one of the most comprehensive overviews of how oximes and oxime ethers impact antitumor activities within a wide range of structural frameworks. This search focused on various combinations of keywords or their synonyms, related to the anticancer activity of oximes and oxime ethers, structure-activity relationships, mechanism of action, as well as molecular dynamics and docking studies. Each article was evaluated based on its scientific merit and the depth of the study, resulting in 268 cited references and more than 336 illustrative chemical structures carefully selected to support this analysis. As many previous reviews focus on one subclass of this extensive family of compounds, this report represents one of the rare and fully comprehensive assessments of the anti-cancer potential of this group of molecules across diverse molecular scaffolds.
Keywords:
Oxime and oxime ether moieties
; Anticancer activities
; Physicochemical properties
; SAR and docking studies
1. Introduction
Cancer is a generic term used to describe a broad spectrum of diseases characterized by the rapid formation of abnormal cells that grow uncontrollably, extending beyond their usual boundaries and invading adjacent parts of the body while destroying normal tissues [1,2,3]. This condition can affect any part of the body, with the 2020 estimates from the World Health Organization (WHO) indicating that the most common types in terms of new cases were breast cancer (2.26 million cases), lung cancer (2.21 million cases), colon and rectal cancer (1.93 million cases), prostate cancer (1.41 million cases), non-melanoma skin cancer (1.20 million cases), and stomach cancer (1.09 million cases), for an overall estimated total of 19.3 million new cancer cases [4,5,6,7]. In fact, as mortality rates for other leading causes of death such as stroke and coronary heart disease decline in many parts of the world, cancer is emerging as the deadliest disease worldwide, and according to WHO estimates, cancer ranks as the first or second leading cause of death before the age of 70 years in 112 out of 183 countries, and as the third or fourth leading cause in about 23 other countries [4,5,6,7,8,9]. In 2020, cancer was responsible for nearly one in six deaths, accounting for nearly 10 million deaths globally [4,5,6,7]. Compounding the issue, the global cancer burden is expected to reach 28.4 million cases by 2040, representing a 47% increase from 2020 [4,10,11]. One unique characteristic of cancer, unlike many other diseases, is its higher overall incidence in developed countries compared to transitioning or low-income countries, with rates ranging from 2 to 3 times higher [4,9,11,12]. This situation is further worsened by the prevailing trend of resistance to numerous available therapies, added to their pervasive side effects [13,14,15,16,17].
Fortunately, novel approaches to cancer treatment through a surgical targeting of tumors’ microenvironment, such as longitudinal single-cell profiling of chemotherapy [18,19] or targeted immunotherapies, including chimeric antigen receptor (CAR) cell therapy [20,21], immune checkpoint inhibitors [22,23,24], monoclonal antibodies [25,26,27], and immune system modulators [28,29,30], are demonstrating significant promise across various types of cancers. Nevertheless, small molecules have always been the bedrock of cancer therapy and are predicted to continue playing a critical role in the future. One such class of small molecules is oximes.
Oximes are nitrogen-containing molecules from the imine family, characterized by the general formula (RR’C=N–OH). They are available in the form of aldoximes or ketoximes, and are well-known since the 1960s to be widely distributed across various species in all realms of life [31,32], with the first synthetic member of the family (methylglyoxime, 1) reported in 1882 by Meyer and Janny [33]. In plants, they are viewed as critical metabolic bifurcation points between general and specialized pathways, with the majority of plant oximes originating from amino acids through processes catalyzed by the cytochrome P450 family of enzymes [31,32]. As a result, the structures of naturally occurring oximes are relatively simple, reflecting their parent amino acids. This can be observed with the first oximes from plant origin, namely isobutyraldoxime (2) [34] (derived from valine) and phenylacetaldoxime (3) [35] (derived from phenylalanine), which were identified in 1967 from Linum usitatissimum and Tropaeolum majus, respectively (Figure 1). Additionally, indole-3-acetaldehyde oxime (4) (derived from L-tryptophan) was isolated from Brassica oleracea just a year later [36]. In fact, plant metabolites such as auxin, cyanogenic glucosides, glucosinolates, and other bioactive volatile compounds were also shown to originate from oximes [32,36,37]. This family of compounds and their derivatives has also been suggested to play key roles in growth regulation, plant defense, pollinator attraction, and plant communication with the surrounding environment [38,39,40]. Furthermore, compounds derived from oximes can serve as quenchers for reactive oxygen species in plants or act as storage compounds for reduced nitrogen, which may be released on demand through the activation of endogenous turnover pathways [31,32,41].
As highly bioactive molecules, both naturally occurring and synthesized oximes have found versatile uses in many spheres of society, particularly in the agricultural and medical sectors. These hydroxy-imine derivatives are well-known for their antibacterial, antifungal, anti-inflammatory, antioxidant, and anticancer activities [42,43,44,45,46,47]. A recent review by Dhuguru et al. [48] provides extensive insights into the synthesis, mechanism of action, and pharmacokinetics of oxime-based FDA-approved drugs. The anticancer activity of this family of compounds is also well-documented, with several notable reviews published on the topic in recent years [45,49,50,51,52,53]. However, each of these reports has primarily focused on one subclass of this extensive family of compounds, thus failing to provide a clear view of the diversity of scaffolds encompassed within this group of molecules. The present review delves into the fundamental role of oxime and oxime ether moieties in enhancing the physicochemical and anticancer properties of a structurally diverse molecular frameworks. Specific selected examples are used to illustrate the distinct involvement of the oxime functional group in general strategies for molecular design, modulation of biological activities, computational modeling, and structure−activity relationships studies.
2. Classes of anticancer oximes
2.1. Anticancer activity of indole based-oximes and oxime ethers
Among the indole-based oximes and oxime ethers, derivatives of indirubin (5) have been extensively investigated for their anticancer activity. This purplish-red bisindole alkaloid known as the key active ingredient in traditional Chinese medicine herbal formulas, was proven to be effective in treating human acute promyelocytic leukemia (APL) [54,55,56,57]. In fact, this Chinese concoction composed of Radix psudostellariae and Salvia miltiorrhiza, has demonstrated compelling synergistic effects in inducing differentiation of acute promyelocytic leukemia cells in vitro. Additionally, it has shown significant therapeutic activity in murine acute promyelocytic leukemia animal models [58,59] and thus has advanced into clinical trials for treating childhood acute promyeloid leukemia in China [55,57].
Numerous studies have also indicated that indirubin possesses a strong affinity for cyclin-dependent kinases (CDKs), which play an essential role in controlling cell cycle and proliferation [60,61]. This molecule was also shown to interact with glycogen synthase kinase-3β (GSK-3β) by binding to the ATP-binding site [62], with its anticancer activity arising through the induction of cell cycle arrest in the G1 or G2 phase of tumor progression, leading to apoptosis in various cancer cell types [60,63,64]. While CDKs are key enzymes governing cell cycle progression [65], GSK-3β has been reported to play important roles in transcription by regulating the activities of key factors, such as NFκB [ [66], transcriptional factor EB [67], and in cell signaling pathways, such as growth-factor signaling [68] and Wnt signaling [69].
However, indirubin's therapeutic effectiveness is hindered by its poor water solubility, inadequate pharmacokinetic properties, low absorption rate, and significant gastrointestinal toxicity [55,70]. These factors pose substantial obstacles to its clinical application. Consequently, multiple strategies have been employed to enhance the pharmacodynamic and pharmacokinetic properties of this family of compounds. One such strategy involves transforming the carbonyls from the indirubin's scaffold into oxime or oxime ether pharmacophores, as illustrated in Figure 2. This approach has led to the preparation of a wide variety of indirubin C-3’-oxime (6) derivatives, many of which exhibited superior properties compared to the naturally occurring parent alkaloid, including enhanced water solubility and better selectivity [55,61,70,71,72]. For example, while exploring the anticancer activity of a series of N1-alkylindirubin C-3’-oximes against MCF-7, LOVO, and LNCAP cells, Wang et al. [73] showed that many of these derivatives exhibited moderate cytotoxicity, with 7 being the most active across all these cell lines, with IC50 values ranging from 6.72 to 13.38 μM.
The effects of various substitution patterns on the activity of these compounds have also been extensively investigated. In fact, Lee et al. [74] demonstrated that indirubin-5-nitro-3’-oxime (8) exhibits superior antiproliferative activity against A549, HT-1080, and HL-60 cells compared to indirubin-3’-oxime, with IC50 values of 5.4, 5.9, and 9.2 µM, respectively. Further studies indicated that the presence of an electron-withdrawing group at the 5-position enhances the anticancer activity of these compounds. In fact, out of the 29 molecules synthesized and evaluated for their effects on MV4-11 cells (with FLT3/ITD mutation), compound 9 displayed the highest inhibition potency, with an impressive IC50 value of 0.072 μM [75]. Moreover, this molecule was found to selectively inhibit the activity of FLT3 kinase and induce cell cycle arrest at the G0/G1 phase in MV4-11 cells [75]. Additional structure-activity relationship studies revealed that a sulfonate group at the 5-position improved the inhibitory activity against FLT3 kinase. However, this substitution was found to be detrimental to antiproliferative activity due to the resulting molecule's low cell membrane permeability [75]. Nevertheless, 5-methoxyindirubin 3’-oxime (10) has been demonstrated to inhibit PDAC cell proliferation, with the administration of compound 10 leading to the inhibition of PDAC xenograft growth in BALB/c nu/nu mice, and further studies suggesting that this analog might exert its activity by inducing G2/M phase arrest in PDAC cells through the inhibition of CDK1/cyclin B1 levels, subsequently leading to apoptosis [76]. Furthermore, 5-diphenylacetamido-indirubin-3’-oxime (11), a mitochondria-targeting anti-leukemic agent, exhibited strong inhibitory activity against a panel of drug-sensitive and drug-resistant primary and malignant leukemia cell lines [77]. This compound rapidly collapses the mitochondrial membrane potential (MMP), followed by the release of cytochrome c into the cytosol, and a severe depletion of cellular ATP [77]. These findings indicate that this molecule induces leukemia cell death through a mechanism distinct from that of many other indirubin analogs [77].
As a member of the death-associated protein kinase (DAPK) family, DAP kinase-associated apoptosis-inducing kinase 2 (DRAK2) performs apoptosis-related functions [78]. Compelling evidence suggests that DRAK2 is involved in regulating the activation of T lymphocytes as well as pancreatic β-cell apoptosis in type I diabetes. In addition, DRAK2 has been shown to be involved in the development of tumor related diseases through a variety of mechanisms, including exacerbation of alcoholic fatty liver disease (NAFLD) through SRSF6-associated RNA selective splicing mechanism, regulation of chronic lymphocytic leukemia and acute myeloid leukemia, and progression of colorectal cancer [78]. A number of 5-amido-indirubin-3’-oxime derivatives have been shown to exhibit potent inhibitory activity against DRAK1 and DRAK2 [79]. Further structure activity relationship studies indicated that small aliphatic amide at the 5-position were better promotor of the inhibitory activity against DRAK2. Among them, 12 (IC50 3 nM and 51 nM), 13 (IC50 14nM and 24 nM), 14 (IC50 14 nM and 120 nM), and 15 (IC50 8 nM and 21 nM) displayed strong inhibitory activity against DRAK1 and DRAK2, respectively [79], with the molecular docking study revealing that compound 12 binds to the ATP-binding site in DRAK2 and forms three key H-bond interactions with Glu111, Ala113, and Glu117 [79].
Indirubin-3’-oxime analogs with carboxylic acid, ester, carbamide or sulfonamide at the 5-position have also been studied for their anticancer activity. In fact, a 5-methyl acetate analog of indirubin-3’-oxime (16) demonstrated potent inhibition of FLT3 kinase activity in vitro, with an IC50 of 7.89 nM [80]. In contrast, the corresponding free carboxylic acid (17) analog showed a relatively weak activity against FLT3 (IC50 of 3.19 μM) [80]. Interestingly, only the 5-methyl acetate analog exhibited a strong cytotoxic effect against MV4-11 cells, while the carboxylic acid analog was inactive. Further studies revealed that compound 16 inhibited the phosphorylation of downstream STAT5, promoted PARP cleavage in MV4-11 cells, and induced apoptosis and cell cycle arrest in the G1 phase [80]. On the other hand, Merz and coworkers designed and synthesized a series of indirubin-5-carboxamide oxime derivatives with an improved water solubility. These compounds were evaluated for their antiproliferative activity against LXFL529L cells, with 18 and 19 exhibiting noticeable inhibitory activity [81].
While evaluating the cytotoxicity of 32 5-sulfonamide indirubin oxime analogs with enhanced water solubility and improved physicochemical and pharmacological properties, Jautelat et al. [82] indicated that many of these analogs possess potent inhibitory activity toward CDK2. These molecules also exhibited significant antitumor activity against MCF-7 cells, with compounds 20 (IC50 0.04 and 0.4 μM) and 21 (IC50 0.04 and 0.1 μM) being the most active in both CDK2 inhibitory activity assay and tumor growth inhibition assay against MCF-7 cells [82].
The effects of other substitution patterns on the anticancer activity of this family of compounds have also been explored. 6-bromoindirubin-3′-oxime (22) was shown to inhibit the activity of glycogen synthase kinase 3β (GSK3β) [83]. This compound suppresses the proliferation, invasion, and migration of ovarian cancer cells (A2780 and OVCAR3), reduces lamellipodia formation, and induces G1 arrest of the cell cycle [83]. Exposure of these cells to compound 22 led to a significant downregulation of mRNA and protein expression of cyclin D1 and MMP9 compared with untreated control cells. Moreover, 6-bromoindirubin-3′-oxime strongly reduces the formation of lung metastasis in the well-established 4T1 mouse model of aggressive breast cancer [83]. Subtoxic concentrations of this analog affect several major hallmarks of the metastatic process, inhibiting adhesion, migration, and invasion of a variety of metastatic cell types in vitro [83]. Surprisingly, RNAi-mediated silencing of glycogen synthase kinase 3β and phosphoinositide-dependent protein kinase 1 (PDK1), both modulators of cellular metastasis targeted by 6-bromoindirubin-3′-oxime, did not affect invasive migration in this study [83]. Instead, the Jak/STAT3 signaling pathway appeared to play a major role by modulating downstream migration regulators such as C-terminal tensin-like protein and matrix metalloproteinase 2 [83]. However, PDK1 and GSK3β still contributed to the overall response to 6-bromoindirubin-3′-oxime, as silencing all three pathways resulted in almost a complete inhibition of migration, mimicking the response of compound 22 [83].
In contrast to its 5-bromo and 6-bromo isomers, 7-bromoindirubin-3′-oxime (23) exhibited only marginal inhibitory activity towards CDKs and GSK-3 [71,84,85]. However, this latter analog induced the appearance of large pyknotic nuclei, without exhibiting classical features of apoptosis such as chromatin condensation and nuclear fragmentation. The induced cell death was not accompanied by cytochrome c release or any measurable effector caspase activation [71,84]. This result indicates that, unlike other indirubin analogs, 7-bromoindirubin-3′-oxime triggers the activation of a non-apoptotic cell death pathway, possibly through necroptosis or autophagy. In this study however, the 7-fluoro derivative (24) appeared to have better inhibitory activity towards CDKs and GSK-3 than its 7-bromo counterpart [71,84]. Nevertheless, 7-bromoindirubin-3′-oxime significantly reduced cell viability in 14 thyroid carcinoma cell lines, with treated cells showing DNA fragmentation, cell cycle arrest, and lactate dehydrogenase release, but no LC3B cleavage [85]. In other words, this compound induced a non-classical kind of cell death that was caspase-independent and did not involve DNA fragmentation [85].
Several research groups have also investigated the anticancer activities of indirubin-3’-oxime bearing a diversity of substitution patterns on the bisindole scaffold. In a comparative analysis against indirubin-3’-oxime parent compound, Ichimaru et al. [86,87] prepared and evaluated a series of analogs with a variety of bromo- or dibromo-substituted patterns as illustrated by compounds 25 – 30 in Figure 2. This variation in substitution patterns did not appear to significantly affect the activity of the resulting compounds, with IC50 values ranging from 4.4 to 14 μM [86,87].
While exploring other substitution patterns, Kim and coworkers demonstrated the 5,5’-substituted derivatives of indirubin-3’-oxime are potent CDK inhibitors, as most of the prepared derivatives displayed potent inhibitory effects on CDK2 with over 90% inhibition potency at 1 μM concentration [88,89,90]. 5′-Hydroxy-5-nitro-indirubin oxime (31) and 5′-fluoro-5-nitro-indirubin oxime (32) not only displayed selective and significant inhibitory activities towards CDK1 and CDK2, but also exhibited good anticancer activity against several cancer cell lines [88,90]. Compound 31 was able to inhibit the phosphorylation of the retinoblastoma protein (Rb), which is a major substrate of CDK, as well as CDK2/cyclin E activity (IC50 = 2.16 nM) more efficiently when compared to its effect on the CDK1/cyclin B activity (IC50 = 13.8 nM) [75,89,90]. Docking studies indicated that compound 31 formed a new hydrogen between the 5’-hydroxy and Asp86 in the solvent-accessible region of CDK2, with the 3’-oxime moiety creating a hydrogen bond with Ile10 instead of Gln131 [75,88]. Further SAR studies revealed that small substituents at the 5’-position, such as -OH or -F, as well as electron withdrawing substituents at the 5-position, such as -NO2, significantly increased the inhibition potency against CDK2 [75,88,90].
Compounds 31 and 32 were able to induce apoptosis in Imatinib-resistant chronic myeloid leukemia cells, as well as efficiently decrease the viability of CML-derived drug-sensitive and Imatinib-resistant K562 cells, both in vitro and in vivo [90]. Park and co-workers further reported that compound 31 significantly decreased the viability of leukemia K562 cells with an IC50 value of 669 nM, whereas it inhibited imatinib-resistant K562R cells with a similar IC50 value of 783 nM [90]. A combination of 31 and Imatinib further increased the number of apoptotic K562R cells compared to that of either 31 or Imatinib, with the former (31, 15 mg/kg, but not Imatinib) notably inhibiting the K562R tumor growth in vivo [88,90,91].
Based on the X-ray structure of 5-bromoindirubin and CDK2, Yan et al. [92], prepared various 5’,6’-difluoro-indirubin derivatives, with compounds 33 – 35 showing potent inhibitory effects on both CDK2/cyclin E1 and CDK9/cyclin T1 at nanomolar or low micromolar levels. Docking results indicated that compound 35 was inserted into the ATP pocket and exhibited similar bindings with hinge region residues in CDK9 and CDK2, with this 5-cholro analog unexpectedly forming two additional halogen bonds with Lys48 and Asp167 residues in CDK9, whereas, such halogen bond was absent in CDK2, indicating that the selectivity exhibited by 35 towards CDK9 might be related to the formation of these halogen bonds. Yet, while compounds 33 and 34 showed a significant cytotoxic activity against five human cancer cell lines with IC50 values at low micromolar levels, 35 displayed only a marginal activity, a situation that might be attributable to its poor solubility [92].
Dual-specificity tyrosine phosphorylation-regulated kinases (DYRKs) are well known to be involved in multiple cellular functions, including intracellular signaling, mRNA splicing, chromatin transcription, DNA damage repair, cell survival, cell cycle control, differentiation, endocytosis, and synaptic plasticity [93], to name just a few. Myrianthopoulos et al. reported the synthesis of indirubin-3’-oxime-based selective DYRK inhibitors [94]. They revealed that a carboxylate substitution at the 5’-position in combination with a bromine at the 7-position significantly promoted selective DYRK inhibition, as illustrated with compounds 36 (IC50 0.31 and 0.35 μM) and 37 (IC50 0.21 and 0.13 μM) [94].
To rationalize the observed affinity and selectivity profiles, docking studies of compound 37 to a DYRK1a-derived homology model of DYRK2 were conducted, revealing two distinct binding modes characterized by a 180° flip of the indirubin core in relation to either its primary or secondary axis [94]. In both cases, the typical hydrogen bond triplet between the indirubin pharmacophore and the kinase hinge was either disrupted (mode I) or inverted (mode II), with the 5′-carboxylate forming a salt bridge either with Lys165 (mode I) or with the catalytic Lys178 (mode II) [94]. This binding mode appears notably different from the indirubin-kinase binding mode experimentally established in previous studies [95].
The crystal structure of 37 bound to DYRK2 (Figure 3) at a resolution of 2.28 Å confirmed that the binding mode was indeed inverted compared to the geometric arrangement of all previously reported indirubin-kinase complexes [95], consistent with the predicted binding mode II [94]. Notably, the crystal structure also revealed not only the predicted salt bridge anchoring the anionic 5′-carboxylate to the side chain of Lys178, but also an additional hydrogen bond between the 5′-carboxylate and the NH backbone of Asp295, with three water molecules participating in the hydrogen bond network by bridging the inhibitor carboxylate and Lys178 ammonium groups to the side chains of Asp295 and Glu193 [94].
Despite the numerous substitutions performed on the core bisindole skeleton, indirubin C-3’-oxime derivatives still exhibited solubility issues, particularly in vivo within live tissue. These poor solubility and inadequate pharmacokinetic properties are primarily attributed to the high polarity and hydrophilic nature of the oxime functional group. In order to modulate the hydrophilic/hydrophobic balance of this family of compounds, various substituents, including glycosides, polyhydric alcohols, epoxides, amide chains or their corresponding salts, were added to the 3’-oxime functional group, effectively transforming them into an oxime ether.
As such, Nam et al. [96] synthesized several indirubin-3’-oxime ethers by introducing diverse polyether or polyhydric alcohol chains to the hydroxyl group of the 3’-oxime. They demonstrated that compounds 38 and 39 strongly inhibited Stat3 signaling in four human prostate and breast cancer cell lines [96]. Additionally, 39 was found to directly inhibit c-Src kinase activity in vitro (IC50 = 0.43 μM) [96]. Treatment with the same compound appeared to suppress the tyrosyl phosphorylation of c-Src kinase’s downstream target protein Stat3, while concurrently downregulating the Stat3-associated proteins Mcl-1 and Survivin, triggering MDA-MB-468 breast cancer cell apoptosis [96].
Owing to the fact that many epoxides can be further metabolized by epoxide hydrolase in the cellular metabolic environment to generate the corresponding polyhydric alcohols, Miyairi and co-workers prepared a series of indirubin-3’-(O-oxiran-2-ylmethyl)-oxime derivatives designed to enhance cell permeability, and thus serving as prodrugs [86,87]. The anticancer activity of each molecule was evaluated on HepG2 cells through a comparative analysis against their respective indirubin-3’-oxime parent compounds with appropriate substituents [76,86,87]. This study indicated that all indirubin 3’-(O-oxiran-2-yl)oxime derivatives consistently outperformed their corresponding indirubin-3’-oxime intermediates [86,87].
A docking study with 40 at the ATP-binding site of GSK-3β designed to evaluate the positioning of this molecule within the site, including the interactions between the oxirane and amino acid residues, suggested that the oxirane group of 40 is positioned closely enough to Cys-199 to form a covalent linkage between them [86,87]. It should be mentioned that Cys-199 is a critical amino acid for GSK-3β kinase activity [62]. Given that halogen atoms in organic compounds have exhibited strong affinity for the sulfur atoms in cysteine and methionine residues in a number of proteins [97], the participation of Cys-199 in enhancing binding affinity through sulfur-halogen interaction was anticipated. The authors concluded that the sulfur-halogen interaction forces the oxirane moiety close to Cys-199, thereby accelerating the covalent bond formation between the oxirane and the thiol [76,86,87]. In addition, Kurita et al. [98] demonstrated that these indirubin-3’-(O-oxiran-2-ylmethyl)-oxime derivatives possess potent cytotoxicity against human neuroblastoma IMR-32 and SK-N-SH cell lines, with IC50 values of 0.16 and 0.07 µM, respectively. These molecules induce caspase-independent apoptosis by inhibiting DNA repair and causing DNA fragmentation in IMR-32 cells [76,98].
While exploring the effect of different substitution patterns on the behavior of these analogs, Ichimaru et al. [86,87] found that many indirubin-3’-oxime derivatives were potential anticancer agents, with compounds 41 – 44 exhibiting significant inhibitory activity against HepG2 cells, with IC50 values of 0.62, 1.7, 1.3, and 1.6 μM, respectively [86]. Notably, compound 41 (with a 6-bromo substituent) displayed the most promising anticancer profile, whereas the activity of compounds 42 and 44 was not superior to that of their corresponding unsubstituted analog 40 [86,87,98].
Further investigations by other research groups have indicated that compound 39 possesses a broad range of in vitro and in vivo activities against numerous cancer cell lines. For instance, 39 was found to significantly inhibit the growth of CT-26 allografts in syngeneic BALB/c mice without affecting body weight [99,100]. Immunohistochemistry revealed that 39 could induce apoptosis and hinder tumor angiogenesis, leading to substantial impediment of cell proliferation, migration, and tube formation in vascular endothelial growth factor (VEGF)-treated HUVECs [99,100]. In vivo studies showed that this analog has the potential to inhibit the formation of subintestinal vessels in a zebrafish model in a dose-dependent manner [100]. In a different study, Nam et al. [101] further indicated that 39 significantly blocked tyrosyl phosphorylation of Stat5, while inhibiting Stat5 DNA-binding activity in human K562, KCL-22M and primary CML cell lines, with treatments with 39 strongly decreasing the autophosphorylation of Src and SFKs in K562 and KCL-22M cells at 5 μM, and in primary CML cells. This compound also induced apoptosis in chronic myelogenous leukemia cells by downregulating the expression levels of Stat5 target proteins Mcl-1 and Bcl-xL, highlighting the potential of 39 to inhibit SFK/Stat5 signaling in leukemia cells [101]. During an investigation into the effects of different substitution patterns on the anticancer activity of compound 39, Cheng et al. [102] noted that compound 45, a 5-methoxy derivative of 39, could significantly block TGFb/BMP signaling by inducing ubiquitin-proteasome-mediated depletion of total R-Smads. This molecule also exhibited a strong inhibition of cell viability across several pancreatic cancer cell lines (Panc-1, MIA-PaCa2, BxPC3, and AsPC1), with IC50 values ranging from 0.76 to 2.2 μM [103].
Nevertheless, while further investigating the physicochemical and cell permeability properties of these molecules, Heshmati et al. [104] demonstrated that water solubility remained a significant hurdle for drug permeation, although a Calcein-AM uptake assay also indicated that 39 was not a good substrate for P-glycoprotein, suggesting that this compound could be a promising candidate for oral delivery due to its high membrane permeability [104]. It is worth mentioning that Jakobs et al. [64,105], while investigating some glycoside derivatives of indirubin C-3’-oxime, showed that 39, 46 and 47 exhibited potent cytotoxicity effects against LXFL529L, MCF-7, and HCT-116 cell lines at low micromolar concentrations [105], with compound 46 proven to block pRb phosphorylation in LXFL-529L cells [64].
Another strategy used to improve the physicochemical properties of this family of compounds is to attach a quaternary ammonium group to the oxime moiety. In line with this approach, Ginzinger et al. [106] prepared a series of carnitine oxime ether and choline oxime ether prodrugs. They not only investigated the activities of these compounds against three human cancer cell lines (A549, CH1, and SW480), but also assessed their suitability for clinical administration [106]. Compounds 48 (IC50 7.6, 1.8, and 5.6 μM), 49 (IC50 13, 1.3, and 2.5 μM), and 50 (IC50 1.0, 0.28, and 0.52 μM) exhibited low micromolar activity against A549, CH1, and SW480, respectively, with 50 being the most potent [106]. Furthermore, a 5-nitro derivative (51) of these analogs exhibited a significant anticancer activity against HCT-116 and MDA-MB-231 cell lines, with IC50 values of 0.302 and 0.738 μM, respectively [107]. This compound also significantly inhibited cell invasion and induced apoptosis in MDA-MB-231 cells, while blocking cell metastasis in a zebrafish human tumor xenograft model without any apparent toxicity [107]. Further assays indicated that compound 51 acts as a dual inhibitor of GSK-3β (IC50 5.46 nM) and Aurora A (IC50 0.22 μM) [107].
2-(N-methylpiperazin-1-yl)ethyl and 2-(N-methylpiperazinium)ethyl hydrochloride derivatives of these analogs, also exhibited some anticancer activity against LXFL and MCF7 cells in vitro, with compounds 52 (IC50 of 0.86 and 0.80 μM) and 53 (IC50 of 1.1 and 0.85 μM) displaying the highest potency [72]. Notably, compound 53 exhibited greater water solubility than its counterpart (52), and also exhibited selective inhibitory activity on IGF-1R, with an IC50 value of 169 nM [72]. This compound also induced apoptosis and caused cell cycle arrest, primarily at the G2/M phase in MCF-7 cells [72].
Jeong et al. [108,109,110] further demonstrated that these piperazinium hydrochloride derivatives of indirubin-3’-oxime were potent inhibitors of FLT3 kinase and MV4-11 cells, with compounds 53 and 54 exhibiting the highest potency. Docking studies suggested that compound 53 bound tightly to the ATP binding pocket in FLT3, surrounded by residues Leu616, Leu818, Cys828, and Tyr693, and forming approximately five hydrogen bonds with these residues [108,109]. While the indirubin core formed two significant hydrogen bonds with the backbone Cys694 residue, the tertiary amine of the piperazine moiety formed a hydrogen bond with Asn816, and the terminal amino group established two hydrogen bonds with both Asn816 and Asp829 [108,109]. Moreover, compound 53 effectively inhibited cell growth in vivo in an MV4-11 xenograft model following a daily oral administration of 20 mg/kg, without inducing significant hERG-related cardiotoxicity [108,109,110].
Conversely, compound 52 inhibited the growth of TT thyroid carcinoma cells by suppressing cell proliferation and inducing apoptosis [110]. This compound was also found to suppress RET signaling pathway via a downregulation of the phosphorylation of RET kinase as well as the downstream Shc and ERK1/2 [108,109].
Dicationic piperazinium dihydrochloride derivatives of indirubin-3’-oxime have also been extensively studied for their anticancer activities. In fact, compound 55 demonstrated activity against Abl1 and T315I mutant Abl1 leukemia cells, with IC50 values of 0.87 and 9.4 μM, respectively [111]. This compound also exhibited inhibitory effects on both c-Src and Abl kinases, with SAR studies indicating that the presence of a bromine atom at the 6-position and a flexible alkylamino chain on the 3'-oxime functional group enhances cytotoxicity [111]. Conversely, the trifluoromethyl or bromine group at the 7-position in compound 56 seems to be detrimental to its cytotoxic activity [111]. However, this latter compound showed activity against SH-SY5Y, while displaying only marginal inhibitory activity towards CDKs and GSK-3α/β kinases [112]. Furthermore, compound 56 has been shown to significantly suppress the viability of four human melanoma cell lines (A2058, A375, G361, and MeWo) in vitro in a dose-dependent manner [113]. However, the 5-methyl acetate derivative (57) exhibited the highest potency among these analogs, displaying an excellent in vitro FLT3 inhibition, with an IC50 value of 3 nM, while blocking the phosphorylation of downstream STAT5 [110]. This same compound exhibited highly selective anticancer activity against leukemia MV-4-11 cells, with an IC50 value of 1.2 nM, achieved through G2/M phase arrest and apoptosis induction. Additionally, it inhibited c-Met kinase and blocked c-Met phosphorylation, along with downstream signaling pathways (Erk1/2, STAT3, STAT5, and Akt) in a dose-dependent manner [114]. In an in vivo study, this compound demonstrated a significant inhibitory activity in MV4-11 tumor xenograft model, without causing alterations in the mice's body weight [110]. Further studies by Vougogiannopoulou et al. [115] indicated that compound 58, the 6-bromo derivative, exerted strong activity against neuroblastoma SH-SY5Y cells, while also significantly inhibiting GSK-3 activity, with an IC50 value of 1.3 nM.
Several indirubin C-3’-oxime ethers bearing a 2-(pyrrolidin-1-yl)ethyl substituent at the oxime functional group have also been shown to strongly inhibit the growth of a series of leukemia cells, while simultaneously blocking the activity of both c-Src and Abl kinases [111]. Among these compounds, 59 and 60 exhibited potent cytotoxicity against wild-type KCL-22 cells and T315I mutant KCL-22 cells in the sub-micromolar range [111]. Additionally, compounds 61 and 62 exhibited significant growth inhibition in SH-SY5Y, HCT116, F1, and Huh7 cell lines [112]. These compounds only displayed marginal inhibitory activity towards CDKs and GSK-3α/β kinases, suggesting that, unlike many indirubin derivatives, their anticancer activities might not be primarily mediated through a direct effect on CDK1, CDK5, and GSK-3α/β kinases [112].
Ultimately, Van et al. synthesized a number indirubin-3’-oxime ethers by attaching polyhydric alcohol chains with bulky 1,3,4-thiadiazole substituents to the hydroxyl of the oxime functional group [116]. Among these compounds, 63 and 64 exhibited only a moderate activity, albeit comparable to that of the parent indirubin-3’-oxime [116]. Further SAR studies indicated that the introduction of bulky substituents to the 3'-oxime group mostly resulted in a decrease in the anticancer activity of the resulting molecule [116]. The structure of all these indirubin-3’-oxime ether derivatives are summarized in Figure 4.
While investigating the effect of different substituents at N1 of the bisindole core structure on the anticancer activity of the resulting indirubin-3’-oxime derivatives, Anh et al. [117] prepared a series of indirubin-based conjugates by attaching a range of hydroxyamides at that position, as illustrated in Figure 5. The inhibitory potential of the resulting molecules on the proliferation of cancer cell lines was investigated, with many of these conjugates exhibiting cytotoxic activity several magnitudes higher than that of the parent indirubin-3’-oximes [117]. Furthermore, compounds 65 – 67 significantly inhibited the proliferation of SW620, PC3, and NCI-H23 cells, with IC50 values in the sub-micromolar range. These compounds also displayed strong histone deacetylases (HDACs) inhibitory activity, with IC50 values as low as 22 nM. The inhibitory activity of 65 towards HDAC6 (IC50 0.007 μM) was 29-fold higher compared to its effect towards the HDAC2 isoform (IC50 0.205 μM) [117]. Docking studies revealed that 65 positioned into the HDAC6 pocket with the lowest energy and formed three hydrogen bonds with key residues, namely Tyr782, His610, and Asp649. However, it did not display any such hydrogen bond-type interaction within the HDAC2 pocket [117].
The activity of a series of indirubin-3’-oxime conjugates, prepared by attaching different chalcone units at the N1-position of the core indirubin skeleton via 1,3,4-thiadiazole click chemistry, was also evaluated against four human cancer cell lines (HepG2, LU-1, SW480, and HL-60), with the human normal kidney cell line (HEK-293) used as a control [118]. Among them, compounds 68 – 70 displayed significant growth inhibitory properties on all cell lines, with the most potent compound 68 exhibiting IC50 values of 2.01, 1.30, 2.54, and 0.98 μM against HepG2, LU-1, SW480, and HL-60 cell lines, respectively [118]. Docking studies indicated that compound 68 forms three hydrogen bonds towards GSK-3β via two residues, namely Val135 and Thr138 [118]. Further SAR studies indicated that bulky and rigid substituents are not suitable for the 5′-position, as they led to reduced cytotoxicity and poor water solubility. In the NCI60 cell line panel, these compounds generally inhibited the growth of malignant tumor cells at low micromolar concentrations [118].
Other types of bisindole derivatives bearing an oxime moiety, not directly related to indirubin, have also undergone investigation for their anticancer activities. In fact, the anti-proliferative activities of a series of 1'H,3H-[2,3'-biindol]-3-ones, designed and synthesized by Qu et al. [119], were explored for their inhibitory effects on seven human cancer cell lines, using HUVEC normal cells as a control. Initial structure-activity relationship (SAR) studies indicated that the presence of an oxime group at position 2 was crucial for the cytotoxic effects. Furthermore, the introduction of a polar substituent at C-5 and C-5' significantly enhanced the anti-cancer activities of the resulting compounds [119]. Among these compounds, 71 exhibited the highest potency, with IC50 values of 6.09, 7.66, 9.92, and 4.31 μM against A529, MGC-803, HepG2, and T24 cell lines, respectively [119].
Subsequent investigations revealed that this compound induced apoptosis in T24 cells by elevating intracellular reactive oxygen species (ROS) levels and altering the balance of anti- and pro-apoptotic proteins, leading to mitochondrial dysfunction and activation of caspase-9 and caspase-3 [119]. Furthermore, both cell cycle analysis and Western blotting indicated that compound 71 effectively arrest the growth of T24 cells in the G1 stage, possibly influencing cell cycle regulatory proteins, particularly cyclin D1, while also causing a significant increase in the activity of p53, p21, and p16 [119].
While assessing a series of bis(indolyl)methane oximes against several tumoral cell lines, namely HepG2 (hepatocellular carcinoma), MDA-MB-468 (human breast carcinoma), RAW 264.7 (murine leukaemic monocyte macrophages), THP1 (human acute monocytic leukaemia), U937 (human leukaemic monocytic lymphoma), and EL4 cells (murine T-lymphoma), Grosso et al. [120] demonstrated that compounds 72 and 73 exhibited potent cytotoxic effects across all tested cells. The IC50 values varied between 1.62 (THP1) and 23.9 μM (RAW) for 72, and between 10.7 (MDA) and 34.1 μM (U937) for 73. Notably, compound 72 displayed pronounced activity against non-adherent cell lines, with IC50 values ranging from 1.62 in THP1 to 1.65 μM in EL4 [120]. In contrast, 74 exhibited a considerably lower cytotoxicity in tumoral cell lines compared to the other two compounds, with IC50 values ranging from 35.7 (HepG2) to 124 μM (THP1), without displaying any selectivity. The difference in activity between the N-unsubstituted 72 and the N-methyl substituted derivative 74 was quite significant, underscoring the potential importance of a free amine interaction within the binding pocket through hydrogen bonds [120]. Conversely, the notably lower IC50 values observed for 72 in non-adherent cell lines in comparison to those obtained for 73 demonstrated that the presence of a bromine substituent contributes to higher cytotoxic activity. Unfortunately, these compounds exhibited only a marginal selectivity, with only about 1 to 2-fold difference compared to their activity on non-tumoral cells [120].
Dandu et al. [121] prepared and screened a series of dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole oximes against recombinant human VEGF-R2 and TIE-2 receptor tyrosine kinases. Compounds 75, 76, and 77 exhibited activity against both TIE-2 (with IC50 values of 30, 25, and 26 nM, respectively) and VEGF-R2 (with IC50 values of 7, 4, and 4 nM, respectively), while also demonstrating favorable pharmacokinetic properties in rats [121].
Homology modeling and docking experiments revealed that 75 binds effectively within the ATP pocket of the TIE-2 model, with the lactam NH/CO moiety forming a bidentate donor/acceptor interaction with Glu903/Ala905 at the hinge region, with the anti-oxime orientation serving as an acceptor for the Asp982 backbone amide [121]. The hydrophobic cavity that accommodates the O-alkyl oxime is defined by Leu888, Leu976, Phe983, Gly984, Leu985, and Ile902. Indeed, a single crystal structure solved for 75, confirmed both the trans-oxime orientation and the regio-chemistry of the lactam carbonyl, as well as the orientation of the indazole N2 -methyl group [121].
A number of indeno[1,2-b]quinoxaline derivatives were also synthesized and evaluated for their antiproliferative effects. Among them, compounds 78 (IC50 0.87, 5.78, 0.82, and 0.64 μM), 79 (IC50 0.67, 6.88, 0.89, and 0.83 μM), 80 (IC50 0.45, 5.69, 0.68, and 0.82 μM), 81 (IC50 0.90, 0.81, 0.78, and 0.75 μM), 82 (IC50 0.68, 0.92, 0.78, and 0.32 μM), and 83 (IC50 0.66, 0.83, 0.91, and 0.63 μM) demonstrated the ability to inhibit the growth of MDA-MB231, H1299, PC-3, and Huh-7 cancer cell lines, respectively [122]. Compound 78 was found to be inactive against the growth of the normal human fetal lung fibroblast cell line (MRC-5), with an IC50 value of 31.51 μM. This study also demonstrated that compounds 78 – 80 (Figure 6) exhibited comparable inhibitory activities against topoisomerase I and topoisomerase II [122]. Mechanistic studies indicated that compound 78 induced cell cycle arrest at the S phase through the activation of caspase-3, -7, an increase in the protein expression of Bad and Bax, and a decrease in the expression of Bcl-2 and PARP, ultimately resulting in cell death [122]. Furthermore, compound 78 attenuated the levels of phosphorylated Src, Akt-1, and Akt-2 proteins, without affecting the total protein expression of Akt. Additional studies involving human hepatocellular carcinoma cells in a zebrafish xenograft assay confirmed the anti-tumor effect of 78 in vivo [122].
A series of diarylmethyloxime derivatives containing 5-indolyl moieties were also found to be potent inhibitors of tubulin polymerization [123]. Their inhibitory activity was assessed against several cancer cell lines, including HeLa human cervix epithelioid carcinoma, A-549 human lung carcinoma, HL-60 human leukemic, and HT-29 human colon adenocarcinoma. Among these compounds, 84 exhibited the highest activity against HeLa, HL-60, and HT-29 cell lines, with IC50 values of 0.36, 0.33, and 0.11 μM, respectively. However, this compound showed no activity against the A-549 cell line [123].
A number of indoles psammaplin A derivatives were synthesized by replacing the o-bromophenol unit with an indole ring. Biochemical and cellular characterizations conducted on U937 and MCF-7 cells confirmed that many of these analogs exhibited more potent activities compared to the original natural product [124]. Furthermore, in addition to the well-documented dual HDAC and DNMT epigenetic inhibitory profile of the parent compound, some analogs, notably compound 85, also demonstrated inhibition of NAD+-dependent SIRT deacetylase enzymes. The structure-activity relationship (SAR) study provided insights into the mechanism of action underlying these multiple epigenetic ligands, and sets the stage for further structural exploration aimed at optimizing their pharmacological profiles [124]. In fact, enzymatic inhibition studies of HDAC1 and HDAC4 indicated that the C5-Br moiety could be substituted with other halogens (F, Cl, I) or oxygen-containing substituents (OMe, OBn), as these analogs displayed approximately equal potency. However, the absence of a substituent was found to be detrimental to the inhibitory activity [124]. Pharmacokinetic analyses revealed that compound 85 functions as a prodrug that rapidly transforms into a glutathione conjugate. The anticancer effects mediated by 85 seemed to involve the activation of distinct apoptosis pathways in cancer cells due to synergism between its inhibitory activities [124]. Notably, compound 85 demonstrated good tolerability in experimental mouse models, with a maximal tolerable dose higher than that of well-known HDAC inhibitors [124].
2.2. Anticancer activity of small nitrogen-containing hetorocyclic based oximes
Small heterocyclic-based oximes bearing a triazole, pyrazole, imidazole, pyrimidine, xanthine, or quinazoline core structure have also been widely investigated for their anticancer properties, with some illustrative examples provided in Figure 7 and Figure 8.
A series of tubulin polymerization inhibitor hybrid molecules, prepared by replacing the 'ethylene-bridge' of Combretastatin A-4 with isooxazoline or triazole heterocyclic pharmacophores, was evaluated for their cytotoxic properties [125]. Many of these compounds were found to possess potent antiproliferative activity comparable to that of Colchicine against breast cancer cell line (MDA-MB-231) and lung cancer cell line (A-549). Among them, compounds 86 (IC50 0.50 and 0.42 mM), 87 (IC50 0.70 and 0.60 mM), 88 (IC50 0.80 and 0.567 mM), and 89 (IC50 0.80 and 0.76 mM) displayed the best potency against MDA-MB-231 and A-549 cancer cell lines, respectively [125]. These compounds also exhibited significant tubulin depolymerization effects, as indicated by western blot analysis and confocal staining assay, with compound 89 also strongly inhibiting tubulin polymerization in vitro [125]. Docking and molecular dynamics studies confirmed the binding interaction patterns of these molecules at the Colchicine binding site of tubulin and convincingly correlated the observed experimental results with the different structural variations. The docking scores and molecular dynamic simulations also correlated with the cytotoxic activity displayed against A-549 and MDA-MB-231 cancer cell lines [125].
A series of 5-(4-pyridyl)-1,2,4-triazole hybrids bearing acetophenone-oxime moieties rationally designed and synthesized were evaluated for their properties as epidermal growth factor receptor (EGFR) kinase inhibitor [126]. Some of these compounds moderately inhibited the growth of the NCI-60 human cancer cell lines when screened in vitro. The most potent oxime analog (90) was further evaluated for its EGFR enzyme inhibition activity. This compound exhibited an IC50 value of 0.18 µM, in comparison with Gefitinib (IC50 0.06 µM) used as a reference. Docking and molecular dynamic simulations indicated that this compound tightly binds to the EGFR tyrosine kinase binding site [126].
Another series of 1,2,4-triazole/pyrazole hybrids linked to oxime moieties demonstrated potent cytotoxic activities against A-549, MCF-7, HCT-116, and PC-3 cancer cell lines [127]. Among them, the sulphamoyl derivatives with internal oxime, 91 and 92, were the most potent derivatives against all tested cell lines, especially PC-3 (IC50 = 1.48 and 0.33 µM, respectively), showing roughly 12- and 39-fold selectivity towards PC-3 over F180 fibroblasts, respectively [127]. Mechanistic investigations of 91 and 92 revealed that both compounds arrested the cell cycle at the G2/M phase, up-regulated Bax expression, and down-regulated the expression of the Bcl-2 gene. These two compounds also proved to be good inhibitors of p38MAPK (IC50 0.65 µM for 91 and 0.58 µM for 92) and VEGFR-2 (IC50 0.39 µM for 91 and 0.54 µM for 92), in comparison with PC-3 control cells [127].
Abdel-Aziz et al. [128,129] also revealed that a group of nitric oxide-donating 1,2,4-triazole-oxime hybrids were able to moderately inhibit the growth of several cancer cell lines. Compound 93 achieved cell growth inhibition against both leukemia HL-60(TB) and renal cancer UO-31, while 94 exhibited moderate cell growth inhibition activity against non-small cell lung cancer HOP-92 and renal cancer A498 cell lines, while also exhibiting weak cell growth inhibition activity against both leukemia K-562 and renal cancer UO-31 cell lines. Additionally, 95 was active against leukemia MOLT-4, RPMI-8226, SR, and renal cancer A498 cell lines, respectively [128,129,130]. As for compound 96, it displayed moderate cell growth inhibition activity against leukemia HL-60(TB) and RPMI-8226 cell lines, while 97 showed moderate potency against leukemia RPMI-8226, SR, renal cancer CAKI-1, and UO-31 cell lines [128,129,130]. This study demonstrated that analogs with N-allyl or N-ethyl-substituted triazoles exhibited weak antiproliferative activity, while those with N-phenyl-substituted triazoles revealed moderate to weak activity against most of the tested cell lines. Notably, the unsubstituted 1,2-diphenyl triazole (98) exhibited the most significant activity among the tested compounds against renal cancer A498 cell lines[128,129,130].
A number of pyrazole oxime ether derivatives have also been studied for their growth-suppressing properties against human solid tumor cell lines. In fact, compounds 99 (IC50 0.12, 0.21, 13.21, 0.04, and 0.02 µg/mL), 100 (IC50 0.10, 0.28, 18.78, 0.02, and 0.01 µg/mL), and 101 (IC50 0.13, 0.92, 4.91, 0.28, and 0.26 µg/mL), all bearing a phenyl residue at position 3 of the core pyrazole, show relatively potent activity against A549, SKOV-3, SKMEL-2, XF 498, and HCT15 cell lines, respectively [131]
Xiong et al. [132] also prepared a series of bis-pyrazole oximes by conjugating thiazolyl-substituted pyrazoles with pyrazoxime, and screened them for their antiproliferative activity against four cancer cell lines in an MTT assay. Compound 102 was the most active across all four cancer cell lines in vitro, with potencies superior to that of 5-fluorouracil (5-FU). Furthermore, this compound selectively promoted intracellular ROS accumulation in HCT116, believed to be a key component of its mechanism of action [132]. This compound also dose-dependently induced cancer cell apoptosis through the regulation of apoptotic protein expression and DNA damage [132].
To further the SAR study for this family of compounds, the same research group designed and synthesized other pyrazole derivatives, and investigated their antitumor effects both in vitro and in vivo [47,133]. Several of these compounds displayed good antiproliferative activity, with IC50 values in the low-micromolar range against three human cancer cell lines in vitro. The most potent compound (103) selectively inhibited the proliferation of SMMC-7721 (IC50 0.76 μM), SGC7901 (IC50 2.01 μM), and HCT116 (IC50 1.26 μM), respectively, without affecting the proliferation of non-tumor LO2 cells [133]. Compound 104 was a close second and even more active against HCT116 (IC50 1.18 μM) than 103. Additionally, 103 exhibited significant induction of cell apoptosis through marked cleavage of both PARP and caspase-3, with its effects on cell growth inhibition and apoptosis believed to be related to DNA damage and activation of the p53 signaling pathway [133]. This compound also displayed low acute cytotoxicity and significant growth inhibition of cancer cells in vivo [133].
A computer-aided design led to the synthesis of a series of nitroimidazole-oxime derivatives as potential PLK1-PBD inhibitors. Among them, 105 (IC50 0.73, 0.76, and 0.76 µg/mL), 106 (IC50 0.82, 0.71, and 0.79 µg/mL), 107 (IC50 0.03, 0.92, and 0.89 µg/mL), 108 (IC50 0.01, 0.73, and 0.78 µg/mL), 109 (IC50 0.02, 0.81, and 0.81 µg/mL), and 110 (IC50 0.05, 0.75, and 0.72 µg/mL) exhibited potent activity against MGC-803, HepG-2, and MCF-7 tumor cell lines, respectively [134]. Compound 108 exhibited a better biological profile against PLK1-PBD and induced apoptosis in the MGC-803 cell line in a dose-dependent manner. Docking studies indicated that this latter compound fits well into the active center of the polo-box domain of polo-like kinase 1, with the binding model showing that 108 is nicely bound to the active site PBD domain of PLK1 by forming three hydrogen bonds with Lys540 and Arg557, one π-cation bond with Lys540, and one π-π bond with Trp414 [134]. The nitroimidazole group has a good affinity with the Lys540-His538 pincer and clinches in the EBR region, while the 6-methoxypyridine group fits perfectly in the HM region. Furthermore, other weak interactions, such as van der Waals and carbon-hydrogen bonds, also contribute to the binding affinity of 108 with PLK1 [134].
Among a series of alkylthio/sulfinyl-8H-thieno[2,3-b]pyrrolizin-8-oximino derivatives synthesized for their antitumor activities by Guo et al. [135], several were evaluated in vitro against Bel-7402, HT-1080, SGC-7901, and A549 cell lines. Among these compounds 111 displayed promising antitumor activity superior to that of Cisplatin [135].
Some pyrimidine-5-carbaldehyde oximes have also been explored for their anticancer activities. In fact, while screening a series of 4-aminopyrimidine-5-carboxaldehyde oxime scaffolds bearing a 4-fluoro-2-methylindol-5-yloxy group at the 6-position and alkyl groups at the oxime side chains, against a panel of 100 kinases, Huang et al. discovered that many of these analogs possess good potency against the VEGFR family of kinases [136]. Compounds 112 (IC50 0.042 μM) and 113 (IC50 0.024 μM) were highly selective for VEGFR-2 compared to other VEGFR family members (VEGFR-1 and VEGFR-3). These compounds also exhibited only marginal inhibition effects on other angiogenesis-related kinases, including FGFR, PDGFR, and Tie2, while demonstrating potent antiproliferative activity, 112 (IC50 0.032, 0.036, and 0.027 μM) and 113 (IC50 0.31, 0.39 and 0.23 μM), against HeLa, HCT116 and A375 cancer cells, respectively [136]. They cause cell accumulation at the G2/M phase of the cell cycle by preventing cells from entering mitosis [136].
Xu et al. also revealed that 4-amino-6-arylaminopyrimidine-5-carbaldehyde oxime derivatives are potent inhibitors of ErbB-2/EGFR kinase and display antiproliferative effects on ErbB-2 overexpressing SK-BR-3 and BT474 cell lines [137]. Among these compounds, those with a 1-(3-fluorobenzyl)indazol-5-amino group as the C-6 side chain produced optimal dual EGFR and ErbB-2 kinase inhibition, with compound 114 being the most potent against both EGFR and ErbB-2, with IC50 values of 8 and 12 nM, respectively [137]. It should be mentioned that a methyl substitution of the C-4 amino group (115) abrogated the activity of these family of compounds against both EGFR and ErbB-2. Such a pronounced drop in activity against theses enzymes suggests that the NH2 group at C-4 is necessary for binding, or that the methyl group prevents the molecule from adopting a proper conformation detrimental to these enzymes’ activities [137]. This observation is supported by the elucidation of the X-ray crystallographic structure of the complex of 116 with EGFR, where the NH2 group forms key hydrogen bonding interactions with the backbone C=O of Met 793, and the oxime side chain is oriented toward the solvent front [137].
Similarly, 4-amino-6-piperazin-1-yl-pyrimidine-5-carbaldehyde oximes have also been shown to display potent inhibitory effects against FLT3 tyrosine kinase while exhibiting strong antiproliferative activity against both FLT3 ITD-mutated human leukemic and wild-type FLT3 BaF3-expressed cell lines [138]. Compound 117 and 118 were among the most potent, with IC50 values of 1.0 and 3.0 nM against FLT3 tyrosine kinase, respectively [138]. Compound 119, however, was the most potent against MV4-11 and FLT3 BaF3-expressed cell lines, with IC50 values of 78 and 8.0 nM, respectively [138].
Considering the synergistic collaboration between c-Met/HGF and VEGFR-2/VEGF, Qiang et al. synthesized a series of 4-aminopyrimidine-5-carbaldehyde oxime analogs as potential dual inhibitors of both HGF/c-Met and VEGF/VEGFR signaling, aiming to develop broad spectrum anticancer agents [139]. Compounds 120 (IC50 0.21 and 0.17 µM), 121 (IC50 0.28 and 0.56 µM), 122 (IC50 0.38 and 0.12 µM), and 123 (IC50 0.42 and 0.22 µM) were among the most potent against both enzymes, respectively. In vitro cell proliferation assays revealed that 120 also possesses potent inhibitory activity[139].
Penta-1,4-diene-3-one derivatives containing quinazoline and oxime ether moieties have also been shown to exhibit strong inhibitory effects against hepatoma SMMC-7721 cells [140,141], with 124 (IC50 0.64 μM) and 125 (IC50 0.63 μM) displaying better activity than that of gemcitabine (IC50 1.40 μM) [141]. Further mechanistic studies indicated that many of these compounds effectively control the migration and inhibit the proliferation of SMMC-7721 cells by impeding DNA replication [141]. Western blot results showed that compounds 124 and 125 induced irreversible apoptosis of SMMC-7721 cells by regulating the expression levels of apoptosis-related proteins [141].
A series of 1,3,8-trisubstituted or 1,8-disubstituted xanthine derivatives were tested in a cell viability assay using the human mammary gland epithelial cell line (MCF-10A). All the compounds exhibited no cytotoxic effects, with more than 90% cell viability at a concentration of 50 μM [142]. Compounds bearing an oxime moiety showed superior antiproliferative activity compared to their non-oxime counterparts against pancreatic (Panc-1), breast (MCF-7), colon (HT-29), and lung (A-549) cancer cell lines. Among them, compounds 126 (IC50 0.91, 0.80, 1.70, and 1.50 μM), 127 (IC50 1.30, 1.00, 1.20, and 1.50 μM), 128 (IC50 1.70, 1.30, 1.70, and 1.80 μM), 129 (IC50 1.91, 1.80, 1.70, and 1.50 μM), and 130 (IC50 1.70, 1.40, 1.80, and 1.89 μM) were the most potent against Panc-1, MCF-7, HT-29, and A-549, respectively [142]. These compounds also increased the level of active caspase-3 in Panc-1 cell lines, with 126, 129, and 130 being the most potent caspase-3 inducers [142]. Compounds 129 and 130 also increased the levels of caspase-8 and 9, activating both intrinsic and extrinsic apoptotic pathways, with a more prominent effect on the intrinsic pathway, and showed potent induction of Bax and cytochrome C while down-regulating Bcl-2 protein levels [142]. Treatment of Panc-1 human pancreatic cancer cells with compound 130 showed cell cycle arrest at the Pre-G1 and G2/M phases, while compounds 126, 128, and 130 also inhibited EGFR, with IC50 values of 0.32, 0.45, and 0.82 μM, respectively [142].
2.3. Anticancer activity of quinone-based oximes and oxime ethers
Quinones are oxidized derivatives of phenolic compounds, with hydroxyl or ether groups increasing the nucleophilicity of the ring and contributing to the large redox potential needed to break aromaticity [143]. Depending on the specific quinone and the site of reduction, they can either re-aromatize or break the conjugation. Consequently, their biological activity is often associated with their redox cycling properties [143,144]. For instance, plastoquinone is well-known for serving as a redox relay in the photosynthetic process, while pyrroloquinoline quinone functions as a biological redox cofactor [145]. Ubiquinones, as implied by their name, are believed to be ubiquitous in the respiratory systems of living creatures [145,146]. Additionally, the auto-oxidation of the neurotransmitter dopamine and its precursor L-Dopa has been shown to generate the comparatively stable dopamine quinone, which inhibits the functioning of the dopamine transporter (DAT) and the tyrosine hydroxylase (TH) enzyme, ultimately leading to low mitochondrial ATP production [147]. This family of compounds is also known to display a wide range of biological and pharmaceutical properties, especially since they constitute a major class of anticancer cytotoxins [144,146]. Consequently, numerous quinone-based oxime derivatives have been investigated for their anticancer activities.
A number of mono-oximes obtained by the oximation of para-benzoquinones were investigated for their cytotoxicity on three tumor cell lines, namely HL60, NCI-H292, and MCF-7. Among them, 5-isopropyl-2-methyl-[1,4]-benzoquinone oxime tosylate (131, IC50 0.3, 3.6, and 1.6 µg/mL), 5-isopropyl-5-methyl-[1,4]-benzoquinone oxime tosylate (132, IC50 0.3, 3.1, and 1.7 µg/mL), and 2,5-dimethyl-[1,4]-benzoquinone oxime tosylate (133, IC50 0.3, 5.9, and 1.6 µg/mL) showed significant activity against the these cell lines, respectively [148].
Alkannin and shikonin are naturally occurring naphthoquinones primarily found in plants, and are key constituents of traditional Chinese medicine known as Zicao, which is derived from the roots of Lithospermum erythrorhizon [149]. These two compounds constitute a pair of enantiomers, differing only by the stereochemistry at carbon 1' of the prenylated side-chain attached at position 2 on the core skeleton (see Figure 8). Alkannin (the S-isomer) and shikonin (the R-isomer) were initially isolated from the roots of Alkanna tinctoria and Lithospermum erythrorhizon, respectively [149,150]. This family of compounds has garnered significant interest due to their intriguing biological activities [150,151]. A large number of alkannin and shikonin derivatives have been either isolated from nature or synthesized and evaluated for their tumor-inhibitory potential [150,151].
For example, a set of alkannin and shikonin oxime derivatives were synthesized and their cytotoxicity investigated against three types of tumor cells (DU145, MCF-7, and K562) as well as a normal cell line (HSF) [152]. As a general trend, alkannin derivatives appear to be more active than their shikonin counterparts bearing the same substituents, with many of the synthesized compounds exhibiting better activity and selectivity towards K562 cells, with no toxicity observed in normal cells [152]. Compound 134 demonstrated the highest potency across all cell lines for both the R and S isomers, while 135 – 139 showed high potency against MCF-7 and K562 cell lines for both isomers.
The same research group expanded their design and synthesis efforts to create a variety of other analogs for a more comprehensive Structure-Activity Relationship (SAR) study by introducing diverse groups or sidechains at the 1’ position, as illustrated in Figure 9 [153,154,155,156]. While the 1’-hydroxy and ether derivatives showed relatively low activity [156], the thio- and benzoylthio-analogs demonstrated significantly superior activity against HCT-15, MGC-803, and K562 cell lines [153,155]. Particularly noteworthy is the fact that the cytotoxic activity of these oxime derivatives against drug-resistant cancer cell lines was much stronger than that against drug-susceptible cell lines. Among the benzoylthio-analogs, 140 (IC50 0.98, 1.21, and 0.85 μM), 141 (IC50 0.50, 0.91, and 0.68 μM), 142 (IC50 0.82, 1.25, and 0.79 μM), and 143 (IC50 0.38, 0.79, and 0.46 μM) displayed significant inhibitory activities against HCT-15, MGC-803, and K562, respectively [153,155]. Similarly, 144 (IC50 0.82, 1.35, and 1.00 μM), 145 (IC50 0.54, 1.09, and 0.76 μM), and 146 (IC50 0.92, 1.54, and 0.98 μM) exhibited the highest potency among the thio-analogs. Compound 143 emerged as the most effective molecule overall, with IC50 values ranging from 0.29 to 1.33 μM against MDR sublines [153,155]. Further mechanistic studies demonstrated that 143 could inhibit colony formation, induce G1 phase arrest, and promote cell apoptosis by augmenting the Bax/Bcl-2 ratio in Bel7402/5-FU cells [153,155].
Subsequent studies revealed that the cytotoxicity of these molecules was not linked to reactive oxygen species (ROS) production or bioreductive alkylation. To identify the molecular target(s) of this molecule, a series of biotinylated derivatives was designed and prepared. Many of these derivatives retained their anticancer activity, including compound 147 (IC50 3.67, 4.76, and 1.26 μM) against K562, MCF-7, and HCT-15, respectively, without exhibiting toxicity against the HSF cell line [154]. Furthermore, cell-based investigations demonstrated that replacing the C4 -linker with a longer chain (C6 or C8) resulted in increased cytotoxicity [154].
The Wnt/β-catenin signaling pathway plays a crucial role in the growth, regulation, development, and differentiation of normal stem cells [157,158,159]. Constitutive activation of the Wnt/β-catenin signaling pathway is found in many human cancer cell lines, making it an appealing target for anticancer therapy. Cell-based screening of a library of compounds, along with computational and structure-based design strategies, led to the development of a series of anthracene-9,10-dione dioximes. When evaluated on a panel of 47 cell lines, including colorectal, lung, ovarian, breast, pancreatic, renal, and hematologic cancers, most of these cell lines showed a sensitivity at concentrations lower than 0.2 μM [160]. The most potent compound (148) exhibited IC50 values of 0.030, 0.09, 0.020, and 0.01 μM against colorectal (HCT-15, HT-29, HCT-116) and adrenal carcinoma (H296R) cell lines, respectively. Compounds 148 (GIC50 0.09 μM), 149 (GIC50 0.03 μM), 150 (GIC50 0.02 μM), 151 (GIC50 0.08 μM) and 152 (GIC50 0.06 μM) also displayed significant growth inhibition (GI) effects against the HT-29 cell line [160]. Most of the cancer cell lines used in this study exhibited high levels of β-catenin expression when treated with these compounds, indicating that these compounds efficiently modulates β-catenin transcriptional activity and β-catenin cellular levels. Fluorescence-activated cell sorting analysis revealed that compound 148 arrested cancer cell lines in the G1 phase of the cell cycle, and induces apoptosis within 48−72 hours of continuous treatment [160]. Consequently, these compounds were recognized as potential agents for treating a broad spectrum of epithelial and hematological malignancies by inhibiting oncogenic Wnt/β-catenin pathway signaling. Furthermore, by blocking the disrupted signaling responsible for early carcinogenesis, metastasis, and the maintenance of the malignant phenotype, compound 148 may prove effective at all stages of a treatment regimen, including metastatic disease, prevention, and the reversal of premalignant, hyperproliferative lesions like colonic polyps [160].
Drug conjugates are chemotherapeutic or cytotoxic agents covalently linked to targeting ligands such as antibodies or peptides via a linker. They have emerged as an important class of therapeutics for cancer treatment, and while antibody-drug conjugates (ADCs) are now clinically established for cancer therapy, peptide-drug conjugates (PDCs) are gaining recognition as a new modality for targeted drug delivery with improved efficacy and reduced side effects in cancer treatment [161,162,163,164]. The linker in a drug conjugate plays a key role in the circulation time of the conjugate and the release of the drug for full activity at the target site. While early technologies relied on random conjugation to either lysine or cysteine residues, resulting in heterogeneous ADCs, recent advancements have led to the preparation of homogeneous ADCs through site-specific conjugation at engineered cysteines, glycosylated amino acids, and bioorthogonal unnatural amino acids [165,166]. As a result, targeted therapy based on polysaccharide- or protein-drug conjugates has attracted significant attention in recent years due to its high efficacy and low side effects [161,162,163,164,165,166].
Doxorubicin is an antibiotic isolated from the Streptomyces peucetius bacterium and belongs to the anthracycline group of chemotherapeutic agents [167,168], which includes daunorubicin, idarubicin, and epirubicin. Doxorubicin, which is marketed under the brand name Adriamycin, among others, has been widely used since the 1960s, and has also been implemented in cancer chemotherapy [169]. This compound is believed to interact with DNA, thereby inhibiting macromolecular biosynthesis [168,169,170] and impeding the progression of topoisomerase II [171]. After disrupting the DNA chain during replication, doxorubicin stabilizes the topoisomerase II complex, preventing the release of the DNA double helix and, as a result, halting the replication process [168].
Schuster et al. [172,173,174,175] reported a series of oxime-linked daunorubicin conjugates, prepared by incorporating a variety of unnatural and D-amino acids into the peptide sequence (see Figure 10). These conjugates were gonadotropin-releasing hormone (GnRH) peptide derivatives, designed to target the overexpressed GnRH receptors found in many cancers. All of these conjugates exhibited noticeable activity against MCF-7 breast cancer cells, with IC50 values ranging from 0.14 to 6.64 μM, as well as against HT-29 colon cancer cells, with IC50 values ranging from 3.31 to 18.00 μM [172,173,174,175]. The lead conjugates 153 and 154 demonstrated stability in 90% human serum for at least 24 hours, and ligand competition assays with GnRH receptors indicated that both compounds had a high binding affinity for these receptors. However, flow cytometry revealed that the cellular uptake of 154 appeared to be higher than that of 153 when used to treat MCF-7 and HT-29 cells for 6 hours [175]. The same research group further demonstrated that conjugates 153 and 154 possessed potent in vivo activity in breast and colorectal cancer-bearing mice using orthotopic models, including 4T1 and MDA-MB-231 breast cancer and HT-29 colorectal cancer in BALB/c and SCID mice, with the results showing a significant reduction in tumor volume and metastasis inhibition, and lower toxicity effects [175].
PDC 155, with a cathepsin B cleavable peptide (GFLG) inserted at the N-terminal of the targeting peptide sequence and two drug molecules conjugated per peptide, was also shown to be highly efficient in uptake by PANC-1 cells, and displayed high cytotoxicity [176]. In vivo evaluation in PANC-1 tumor-bearing SCID male mice showed that tumor volume inhibition was significant for the conjugate 155-treated mice compared to the control group (treated with sterile water) on day 70, with conjugate 155 being able to significantly inhibit tumor growth even at a low dose of 2 mg/kg with minimal side effects [176].
Since integrins are found to be overexpressed on the surface of some tumor cells, making them an appropriate target for cancer drug delivery, Feni et al. [177] used oxime to attach cytotoxic daunorubicin to a linker, employing alkyne−azide click chemistry to construct a consolidated integrin-targeting cyclic diketopiperazine (DKP)-RGD peptide and a cell-penetrating peptide (CPP), to promote effective and targeted cellular uptake. Conjugate 156 exhibited good affinity for tumor-associated integrin receptors, αvβ3 and αvβ5, in an in vitro binding assay. Further studies revealed that the cytotoxicity of 156 (EC50 range 2.7−11.0 μM) towards U87, HT-29, and MCF-7 cell lines was less pronounced than that of free daunorubicin, with the conjugate showing no selectivity toward cells with higher expression of integrin αvβ3 (U87) [177].
Gonadotropin-releasing hormone III (GnRH-III), a decapeptide isolated from the sea lamprey brain, are known to exhibit lower potency than GnRH in stimulating gonadotropin secretion [172]. However, it exerts antiproliferative effects on many tumors expressing the GnRH-R protein [172,173,175]. In their study, Marelli et al. [178] actually demonstrated that oxime bond-linked daunorubicin (Dau)-GnRH-III derived bioconjugates, in which daunorubicin is coupled to the 8Lys in the native form of GnRH-III, and Dau-[4Lys(Ac)]-GnRH-III, in which daunorubicin is attached to the 8Lys of a GnRH-III derivative where 4Ser is replaced by an acetylated lysine possess potent antitumor activity. Their research focused on Castration-Resistant Prostate Cancer (CRPC) cells, and they revealed that both Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III were rapidly internalized into DU145 prostate cancer cells, leading to a significant cytostatic effect [178]. Additionally, both bioconjugates increased the levels of the active form of caspase-3, indicating the involvement of apoptosis in their antitumor activity. The antiproliferative effect of both Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III was counteracted by simultaneous treatment of the cells with Antide, an antagonist of the GnRH-R [178]. Furthermore, after silencing the type I GnRH-R, the antitumor activity of both bioconjugates was completely abolished. These observations suggest that in CRPC cells, daunorubicin-GnRH-III derivative bioconjugates inhibit tumor cell proliferation by triggering the apoptosis process, and exert their antitumor effect through the activation of the type I GnRH-R expressed in these cells [178].
It should be noted that in an earlier study, Orbán et al. [174] also designed and synthesized three oxime bond-linked daunorubicin-GnRH-III bioconjugates and investigated their in vitro antitumor activity on MCF-7 human breast and HT-29 human colon cancer cells. They analyzed the degradation and stability of these bioconjugates in human serum and in rat liver lysosomal homogenate using liquid chromatography in combination with mass spectrometry. It was found that two out of the three prepared bioconjugates remained stable in human serum for at least 24 hours [174]. All three bioconjugates, 157 (IC50 2.2 and 14.2 µM), 158 (IC50 3.9 and 19.4 µM), and 159 (IC50 1.8 and 28.6 µM), exhibited in vitro antitumor effect on both MCF-7 and HT-29 cell lines. However, their antitumor activity was lower than that of free daunorubicin [174]. These results could be explained by the lower cellular uptake of the bioconjugates, the lack of free drug release, and the different DNA-binding properties of the resulting metabolites. Furthermore, the smallest metabolites produced in the lysosomal homogenate were synthesized, and their binding to DNA was assessed by fluorescence spectroscopy. The obtained data indicate that the incorporation of a peptide spacer in the structure of oxime bond-linked daunorubicin-GnRH-III bioconjugates is not required for their antitumor activity [174].
With respect to polysaccharide conjugates, using a versatile green oximation reaction, a dextran-doxorubicin conjugate (160) was synthesized, and the resulting conjugate (see Figure 10) appeared to spontaneously form micelles with a diameter of approximately 100 nm in an aqueous environment [179]. The particles size proved suitable for selective intratumoral accumulation, leveraging the enhanced permeability and retention (EPR) effect, consequently improved the tolerability, the in vivo biodistribution, and the antitumor efficacy of these conjugates [179]. Compound (160) efficiently delivered doxorubicin into HepG2 cells due to its acid-sensitive properties, enabling an 'on-demand' release of doxorubicin in tumor cells in vitro. Furthermore, when administered via intravenous injection, this compound significantly inhibited tumor growth in a H22-xenografted murine model, with the improved effect attributed to its prolonged blood circulation, enhanced drug accumulation, and facilitated intracellular release at the tumor site in comparison to free doxorubicin [179].
2.4. Anticancer activity of flavonoids-based oximes and oxime ethers
Flavonoids constitute a large group of polyphenolic secondary metabolites, characterized by a C6-C3-C6 arrangement as their basic skeleton. They comprise approximately 12 structural classes, each distinguished by the degree of oxidation of the C ring, including flavones, flavonols, flavans, flavanones, flavanols, chalcones, dihydrochalcones, isoflavones, aurones, anthocyanins, anthocyanidins, and catechins [180,181]. These compounds are well-known for their wide range of biological and pharmacological activities, with their cytotoxic and anticancer properties well-documented [181,182,183,184]. Consequently, numerous studies have investigated flavonoid-based oximes for their potential antitumor properties.
Microtubules are highly dynamic structures consisting of α- and β-tubulin heterodimers, and are involved in cell movement, intracellular trafficking, and mitosis [185,186]. In the context of cancer, the tubulin family of proteins is recognized as the target of tubulin-binding chemotherapeutics, which suppress the dynamics of the mitotic spindle to induce mitotic arrest and cell death [185,186]. Furthermore, changes in microtubule stability, the expression of different tubulin isotypes, and altered post-translational modifications have been reported in several types of cancer. Emerging evidence has also suggested that tubulins and microtubule-associated proteins may play a role in a range of cellular stress responses, thus conferring a survival advantage to cancer cells [185,186].
Chiang et al. [187] designed and synthesized a series of flavone-based oximes, and evaluated them for their anti-proliferative effects on a range of cancer cell lines by monitoring proliferation, cell cycle, apoptosis, caspase levels, and mitochondrial membrane potentials. Among these molecules, 161 (see Figure 11) was the most potent, exhibiting IC50 values of 0.45 and 0.43 μM against drug-sensitive NPC-TW01 and HONE-1 cancer cell lines, respectively [187]. The same molecule showed potency against drug-resistant TW01-Taxol-Res and TW01-Met-Res cell lines, with IC50 values of 0.57 and 0.66 μM, respectively, while having little effect on the non-cancer human fibroblast Detroit 551 cell line (IC50 > 20 μM) [187]. This compound also induced cell accumulation in G2/M, and apoptosis in a time- and concentration-dependent manner. Colchicine competition-binding experiments and computer modeling suggested that compound 161 causes microtubule disruption by binding to the colchicine-binding site of tubulin, resulting in mitochondrial membrane damage and cell apoptosis through the activation of caspase-9/-3, without noticeable activation of caspase-8 [187]. In vivo studies demonstrated that at doses of 25 and 50 mg·kg−1, this molecule exhibited good pharmacokinetic properties, and completely inhibited the growth of NPC-TW01 cells in a xenograft nude mouse model [187].
A number of flavanol-based oximes, including E-hesperetin oximes (162 and 163) have also exhibited noticeable cytotoxicity against human cancer cell line SGC-7901 [188]. A naringenin-based oxime (164) was also found to exhibit cytotoxic, genotoxic, and apoptotic effects while inducing and increasing the production of reactive oxygen species in a dose-dependent manner when evaluated against MCF-7, HT-29 and PC-12 cancer [189]. Strong correlations were observed between cell viability, DNA damage, and apoptosis in all cell lines when exposed to these compounds, with the oxime derivative outperforming the parent naringenin in all of these categories [189]. Furthermore, several naringenin oxime derivatives, including 165 (IC50 3.32 µg/mL), 166 (IC50 3.63 µg/mL), and 167 (IC50 4.59 µg/mL), exhibited stronger antiproliferative activity compared to naringenin [190]. In fact, the conversion of the inactive 7,4'-di-O-propylnaringenin (IC50 > 100 µg/mL) into its oxime derivative resulted in approximately 22-fold increase in potency compared to the parent compound [190].
Another series of naringenin-based oxime ethers (168 – 171) demonstrated significant antiproliferative activity against human leukemia (HL-60), gynecological isolated cervical (HeLa, Siha), and breast (MCF-7, MDA-MB-231) cancer cell lines [191]. Additional cell cycle analysis suggested that these compounds caused a significant increase in the hypodiploid (subG1) phase and induced apoptosis in HeLa and Siha cells, while leading to cell cycle arrest in the G2/M phase in MCF-7 cells. The proapoptotic potential of some selected compounds was confirmed by the activation of caspase-3 [191]. Antioxidant activities of the prepared molecules were also evaluated using xanthine oxidase, DPPH, and ORAC assays, and the methyl-substituted oxime ether (168) exhibited the most promising activity [191]. In contrast, neither naringenin nor the Z-isomer of compound 164 inhibited the growth of any of the tested cancer cell lines at any of the applied concentrations, while the allyl derivative (170) and the E-isomer of 164 displayed only a limited growth inhibition [191]. The benzyl derivative (171) exerted moderate growth inhibition on MCF-7 cells, similar to that of the methyl derivative (168) on HL-60 cells [191]. Metodiewa et al. [192] also revealed that N,N-diethylaminoethyl flavanone oxime ethers (172 – 175) induce programmed cell death (apoptosis) in Yoshida Sarcoma cells in vivo, with the administration of these compounds in the promotion phase of the disease leading to both growth inhibition (cell cycle perturbation) and apoptosis [192].
A series of flavanone-triazole hybrids were also synthesized and tested for their cytotoxicity against HCT-15, HeLa, NCI-H522, and HEK-293 (normal) cell lines, with compounds 176 – 179 exhibiting significant cytotoxicity across all the cancer cell lines. Compound 178 was more potent against NCI-H522 cell line, while 176 and 177 appeared to be more toxic against the normal cell line HEK-293 [193].
Some chalcone oxime derivatives have also been evaluated as potential tubulin polymerization inhibitors, and for their cytotoxicity against human cancer cell lines, with many of them demonstrating remarkable inhibitory activity against tubulin polymerization by competing for the colchicine-binding site on tubulin [194]. This activity appeared to be associated with G2/M phase cell cycle arrest as well as promising antiproliferative effects [194]. In fact, compound 180 was shown to possess potent tubulin inhibitory activity (IC50 = 1.6 μM) and potent antiproliferative properties against A549, HeLa, and MCF-7 cell lines, with GI50 values of 2.1, 3.5, and 3.6 μM, respectively [194]. Compounds 181 (GI50 2.8, 6.6, and 5.8 μM) and 183 (GI50 3.2, 7.7, and 8.9 μM) also exhibited decent antiproliferative activity against these cancer cell lines [194]. Docking simulations based on the tubulin crystal structure (PDB code: 1SA0) revealed that compound 180 binds tightly to the colchicine domain of tubulin [194].
Furthermore, several chalcone-based oximes containing a ligustrazine moiety were tested for their in vitro antiproliferative inhibitory activity using A-375, MCF-7, A-549, HT-29, and H-460 human cancer cell lines [195]. The effects of these analogs on several cancer markers, including EGFR, BRAFV600E, c-Met, and tubulin polymerization, were also investigated with the objective of identifying their putative target(s).The majority of these compounds exhibited strong anticancer activity, with 183 (IC50 2.43, 0.85, 1.42, 7.83, 1.83 μM), 184 (IC50 1.47, 0.79, 1.32, 3.80, 1.63 μM) and 185 (IC50 0.87, 0.28, 1.25, 2.43, 1.04 μM) being among the most potent against A-375, MCF-7, A-549, HT-29, and H-460 cell lines, respectively, compared to foretinib (IC50 = 1.9, 1.18, 1.15, 3.97, 2.86 μM), used as a positive control [195]. A careful examination of the structure of these molecules revealed that a Z-configuration for the oxime functional group was a requirement for the antiproliferative activity, along with a 2-thiophenyl or a 2-phenylthiophenyl moiety, with or without a bromo group at position-5. Molecular analysis indicates that compound 184 is more active than foretinib in inhibiting EGFR, while compound 185 displayed similar activity to that of foretinib in inhibiting BRAFV600E and c-Met [195]. The inhibition of tubulin polymerization by many of these compounds was reminiscent of that of colchicine [195].
2.5. Anticancer activity of miscellaneous aromatic- and heterocyclic-based oximes and oxime ethers
Other miscellaneous heterocyclic-based oximes and oxime ethers have also been evaluated for their anticancer activity. In fact, while evaluating a series of 4-anilinofuro[2,3-b]quinolines-based oximes in vitro for their anticancer activity against MCF7, NCI-H460, and SF-268 cell lines, Chen et al. [196] revealed that compounds 186 (IC50 0.63 μM, 9.34 μM, and 6.26 μM) and 187 (IC50 0.71 μM, 31.01 μM, and 9.14 μM) possess weak potency against these cancer cell lines, respectively. These compounds also exhibited a high degree of selectivity when compared to their effect on the human lung fibroblast cell line (MRC-5), with IC50 values of 42.85 μM and 47.04 μM, respectively. Mechanistic studies indicated that compound 186 may induce apoptosis in cancer cell lines through a mitotic arrest and mitotic catastrophe process [196]. The pharmacokinetic properties of the hydrochloride salt of 186 demonstrated excellent water solubility (1049 μg/mL) as well as high oral bioavailability (57.1%), with a moderate plasma half-life (3.4 h) [196]. Following an oral dose of 20 mg/kg, the hydrochloride salt of 186 exhibited rapid oral absorption in mice, with a short Tmax of 0.5 h, and a significant second absorption peak appearing at 1.5 h after dosing. Xenograft studies indicated a significant reduction in tumor size in nude mice treated with this salt, with its evaluation in an orthotopic lung cancer model further indicated that the hydrochloride salt of 186 can be readily absorbed through oral administration and distributed throughout lung tissues, while demonstrating significant efficacy in inhibiting the growth of lung cancers [196].
Synthesized derivatives of quinolin-2(1H)-one bearing either an oxime and amide group, were also evaluated for their antiproliferative and antiplatelet activities [197]. Preliminary assays indicated that amide derivatives were either weakly active or completely inactive, while their oxime counterparts exhibited potent inhibitory activities against platelet aggregation induced by collagen, AA (arachidonic acid), and U46619 (the stable thromboxane A(2) receptor agonist) [197]. Among these compounds, (Z)-6-[2-(4-methoxyphenyl)-2-hydroxyiminoethoxy]quinolin-2(1H)-one (188) was the most active against AA-induced platelet aggregation, with an IC50 of 0.58 μM, but was inactive against cell proliferation [197]. Compound 189 (IC50 0.56 μM), 190 (IC50 0.58 μM) and 191 (IC50 0.54 μM) exhibited potent inhibition activities against U46619-induced aggregation [197].
Curcumin-based oxime analogs have also undergone scrutiny for their potential as anticancer agents [198,199,200,201]. Qin et al.[198,199] reported the antiproliferative activity of a series of oxime and oxime ether analogs of cyclohexanone, piperidin-4-one, and tetralone (see Figure 12), which were tested against various cancer cell lines, including HT-29, PC-3, A-549, Panc-1, MCF-7, H-460, and PaCa-2. Among these analogs, 192 (IC50 0.09, 0.05, 0.08, 0.09, 0.07, 0.08, and 0.10 μM), 193 (IC50 0.03, 0.04, 0.04, 0.05, 0.04, 0.03, and 0.04 μM), and 194 (IC50 0.02, 0.03, 0.02, 0.04, 0.02, 0.04, and 0.02 μM) were the most potent [199]. A mechanistic study revealed that compounds 194 and 195 exhibited potent activity against various targets, including BRAFV600E (with IC50 values of 1.3 μM for 194 and 1.5 μM for 195) and EGFR-TK kinases (with IC50 values of 0.04 μM for 194 and 0.05 μM for 195).
Further structure activity relationship study led to other potent analogs, including 196 (IC50 0.2, 0.07, 0.4, 0.3, 0.4, 0.4, and 0.4 μM), 197 (IC50 0.09, 0.07, 0.06, 0.06, 0.07, 0.08, and 0.09 μM), 198 (IC50 0.09, 0.09, 0.07, 0.08, 0.09, 0.08, and 0.02 μM), 199 (IC50 0.02, 0.02, 0.09, 0.02, 0.03, 0.04, and 0.02 μM), and 200 (IC50 0.09, 0.08, 0.09, 0.07, 0.09, 0.07, and 0.08 μM) against the same cancer cell lines, with 199 demonstrating potency comparable to that of the positive control Erlotinib [198,199]. While assessing their mechanistic effects on BRAFV600E, EGFR TK kinases, and tubulin polymerization, as well as for their potential to reverse efflux-mediated resistance developed by cancer cells in vitro, compound 198 displayed the most potent BRAFV600E inhibitory activity, with an IC50 value of 0.9 μM, while analog 197 exhibited the most potent EGFR TK inhibitory activity, with an IC50 of 0.07 μM [199]. Some of the analogs, including 196, 198, and 199, showed a dual role as both anticancer agents and MDR reversal agents [199].
Following a trend of strong anticancer activity observed within this family of compounds, the same research group prepared a number of ligustrazine-containing cyclohexanone- and piperidin-4-one-based oxime analogs, and tested them against A-549, PC-3, MCF-7, PaCa-2 and HT-29 cancer cell lines [200]. Among them, 201 (IC50 0.08, 0.07, 0.04, 0.09 and NA μM), 202 (IC50 0.01, 0.009, 0.01, 0.01 and 0.01 μM), 203 (IC50 0.05, 0.04, 0.03, 0.03 and 0.05 μM), 204 (IC50 0.09, 0.07, 0.08, 0.09 and 0.10 μM), 205 (IC50 0.07, 0.05, 0.08, 0.08 and 0.06 μM) and 206 (IC50 0.02, 0.03, 0.04, 0.03 and 0.04 μM) emerged as the most potent across these cell lines [200]. The evaluation of these compounds for their effect on tubulin polymerization, EGFR TK kinases, KAF and BRAFV600E, as well as for their effect on reversing efflux-based resistance developed by cancer cells, revealed that EGFR was strongly inhibited by 202 and 204, with analogs bearing 1-isopropyl-piperidin-4-one linker showing the best inhibition against KAF [200]. Compounds 201 – 203 exhibited a dual role as anticancer and as multidrug resistance (MDR) reversal agents. Molecular docking studies indicated that 203 fits well inside the ATP binding pocket of the FAK kinase, with the piperidinone core nestled between Ile428, Gly429 and Glu506 residues within the kinase hinge region [200]. Meanwhile, the pyrazine ring bearing three methyl groups generates hydrophobic contacts with Gly429, Glu500, Asp564 and Leu567, located near the gatekeeper residue Met499 and DFG motif of FAK activation loop. This compound also formed an additional H-bond interaction between the hydroxylamine moiety in compound 203 and the Ile428 residue in the hinge region of FAK.
Furthermore, a series of 1-[3-{3-oxopropyl]-4-piperidone oximes, prepared as potential antineoplastic agents, were also evaluated against a panel of leukemic human colon cancer cell lines, including Colo205, HCC-2998, HCT-116, HCT-15, HT29, KM12 and SW-620. Among them, compounds 207 (IC50 1.50, 1.41, 0.22, 0.37, 0.32, 0.23 and 0.20 μM), 208 (IC50 0.35, 0.31, 0.21, 0.27, 0.30, 0.22 and 0.15 μM), 209 (IC50 0.39, 0.36, 0.32, 0.27, 0.32, 0.25 and 0.23 μM), 210 (IC50 0.40, 1.17, 0.49, 0.36, 0.33, 0.29 and 0.20 μM) and 211 (IC50 1.15, 1.58, 0.36, 0.55, 0.35, 0.28 and 0.29 μM) displayed strong cytotoxicity against these cell lines, respectively [201]. As a general trend, quantitative structure–activity relationships revealed that both the cytotoxic potency and selectivity index figures increased as the magnitude of the Hammett sigma values rose. Furthermore, compounds 212 and 213 lowered the mitochondrial membrane potential in CEM cells, while 214 induced transient G2/M accumulation in Ca9-22 cells [201].
The conversion of the 4’-carbonyl of the natural product griseofulvin into an oxime moiety as a strategy to improve its solubility and metabolic stability resulted in analogs 215 (IC50 1.29 and 1.61 μM) and 216 (IC50 10.1 and 14.13 μM), which showed better cytotoxicity than the parent natural product against MDA-MB231 and U2OS cancer cells, respectively, with compound 216 exhibiting about 130-fold increase in PBS solubility compared to the parent compound [202]. Similarly, a series of oxime ester derivatives of the natural product schizandrin, the major compound isolated from Schisandra grandiflora, were evaluated in vitro for their antiproliferative activities against a selected panel of human cancer cell lines (A549, RKO P3, DU145, and HeLa), with normal cells (HEK293) used as control [203]. Several of these derivatives were found to be more potent than the parent compound, notably, 217 and 218, which demonstrated potent activity against DU-145 and RKO P3 cell lines, with IC50 values of 3.42 µM and 3.35 µM, respectively. Cell cycle analysis revealed that both compounds were able to induce apoptosis and cell cycle arrest in the G0/G1 phase [203]. Moreover, the tubulin polymerization assay indicated that 217 and 218 exhibited a potent inhibitory effect on tubulin assembly, with molecular docking studies and competitive binding assays revealing that these two compounds effectively bind to the colchicine binding site of tubulin [203].
Radicicol is a macrocyclic antifungal antibiotic known to bind to the heat shock protein 90 (Hsp90) chaperone, interfering with its function [204,205]. Hsp90 family chaperones have been shown to associate with several signaling molecules and play an essential role in signal transduction, which is important for tumor cell growth [204,205,206].
Due to radicicol’s failure to produce any antitumor activity in vivo in experimental animal models, radicicol 6-oxime and analogs (Figure 13) have been investigated for their effects on human tumor cell growth, both in vitro and in vivo. As a result, compound 219 showed potent antiproliferative activities against various human tumor cell lines in vitro and inhibited v-src- and K-ras-activated signaling [207]. Additionally, this compound depleted Hsp90 family chaperone-associated proteins, such as p185erbB2, Raf-1, cyclin-dependent kinase 4, and mutant p53, at a dose comparable to that required for antiproliferative activity. Compound 219 also competed with geldanamycin for binding to Hsp90, in contrast to 220, the inactive methylether oxime analog of radicicol, which was less potent both in p185erbB2 depletion and Hsp90 binding [207]. More importantly, 219 demonstrated significant growth-inhibitory activity against human breast carcinoma MX-1 cells transplanted into nude mice when administrated at a dose of 100 mg/kg twice daily for five consecutive intravenous injections [207]. The same compound exhibited significant antitumor activity against human breast carcinoma MCF-7, colon carcinoma DLD-1, and vulval carcinoma A431 cell lines in vivo, in an animal model [207]. The depletion of Hsp90-associated signaling molecules (Raf-1 and cyclin-dependent kinase 4) was also confirmed through ex vivo Western blotting analysis using MX-1 xenografts. These findings, in agreement with in vivo antitumor activity, suggests that the antitumor activity of this compound may be mediated, at least in part, by binding to Hsp90 family proteins, leading to the destabilization of Hsp90-associated signaling molecules [207].
The mechanism of action of the E-isomer of another oxime derivative of radicicol (221) was investigated in a comparative analysis to its Z-isomer by examining its inhibitory effects on Hsp90 function and apoptosis induction in erbB2-overexpressing human breast carcinoma KPL-4 cells, in vitro [208]. Direct binding activity to Hsp90 was assessed through Hsp90-binding assays using geldanamycin or radicicol beads. The plasma concentrations of these compounds after intravenous injection in BALB/c mice, and their antitumor activity were studied against KPL-4 cells transplanted into nude mice in an animal model, with the inhibition of Hsp90 function and the induction of apoptosis also evaluated in vivo using tumor specimens from drug-treated animals [208]. It turned out that 221 possesses a strong antiproliferative activity against all breast cancer cell lines tested in vitro and was more potent than its Z-isomer (222) [208]. These results were consistent with the ability of 221 to deplete Hsp90 client proteins and induce apoptosis in KPL-4 cells in vitro, especially since this compound, but not its Z-isomer, showed significant in vivo antitumor activity, accompanied by induction of apoptosis in KPL-4 human breast cancer xenografts [208]. Although the plasma concentrations of these compounds were equivalent, 221, but not 222, depleted Hsp90-related proteins such as erbB2, Raf-1, and Akt in the tumor specimens recovered from nude mice [208]. These results suggest that the inhibition of Hsp90 function, which causes the depletion of Hsp90 client proteins in tumors, contributes to the antitumor activity of 221, and that the stereochemistry of the oxime moiety is important for the biological activity of radicicol oxime derivatives [208].
Two other oxime ethers, 223 (IC50 0.41, 0.44, and 0.58 μM), and 224 (IC50 0.36, 0.54, and 0.57 μM), showed similar potency to that of 219 (IC50 0.24, 0.39, and 0.38 μM) in anti-proliferative assays against MX-1, MCF-7, and A431 cancer cell lines, as well as in tyrosine kinase assays, while exhibiting potent activity in vivo using subcutaneous inoculation in animal tumor models [209]. Guided by the preliminary structure–activity relationships, this research group designed and prepared radicicol-based probes (225 and 226) which could be used to elucidate the role and functions of Hsp90 in cellular processes [209].
Furthermore, a series of O-carbamoylmethyloxime derivatives of radicicol were also evaluated for their in vitro antiproliferative activities against v-src- and K-ras-transformed cells and for their inhibitory activity against v-src tyrosine kinase [210]. O-(piperidinocarbonyl)methyloxime 227, one of the most potent of these derivatives, exhibited superior antiproliferative activity compared to radicicol and its hydroxime 219, with an IC50 value of 25 nM for the inhibition of v-src kinase activity [210]. Compound 227 was also found to decrease the Raf-1 protein level in KNRK5.2 cells and exhibited significant antitumor activity when tested against MX-1 and A431 xenografts in nude mice [210].
2.6. Anticancer activity of small aromatic and phenolic-based oximes and oxime ethers
Some small aromatic and phenolic-based oximes have also been evaluated for their anti-cancer properties, with the structures of all the selected examples provided in Figure 14. While investigating cytotoxicity in a series of thioaryl naphthyl-methanone-based oxime ether analogs towards various cancer cells, 4-(methylthio)phenyl)(naphthalen-1-yl)methanone O-2-(diethylamino)ethyl oxime (228) exhibited the best safety profile [211]. This compound was able to induce apoptosis, impede migration and invasion, while strongly inhibiting the cancer stem cell population and demonstrating potent tumor regression in mouse MCF-7 xenografts after oral administration [211]. Mechanistic studies revealed that this molecule strongly abrogated EGF-induced proliferation, migration, and tyrosine kinase (TK) signaling in breast cancer cells. However, this compound failed to directly inhibit EGFR or other related receptor TKs in a cell-free system [211]. Systematic investigation of a putative target upstream of EGFR revealed that the biological effects of 228 could be abrogated by pertussis toxin [191].
Estrogens are known to play diverse roles in various pathophysiological conditions [212,213,214]. A number of synthetic selective estrogen receptor modulators (SERMs), such as tamoxifen and raloxifene, have been developed and used to treat ER-related diseases, including breast cancer and osteoporosis [212,213,215]. Therefore, a series of bis(4-hydroxy)benzophenone oxime ether derivatives, including 229 (IC50 75 nM), 230 (IC50 46 nM), and 231 (IC50 77 nM), were shown to possess estrogen receptor (ER) agonist properties and complementary anti-proliferative activities arising through an ER-independent mechanism in cancer cells [216]. Furthermore, compounds 231 and its analog 232 were shown to upregulate the activity of an estrogen response element (ERE)-containing luciferase reporter construct (ERE-luciferase) in MCF7 cells [217]. Notably, 231 showed higher activity than E2, the endogenous ligand, at concentrations of 1,000 nM and 100 nM, but displayed weaker activity at 10 nM [217]. The same compound also exhibited a greater agonistic effect than its counterpart (232), inducing a concentration-dependent stimulation of ERE-mediated transcriptional activity with a median effective concentration (EC50) value of 46.3 nM, while suppressing androgen receptor-dependent transcriptional activity [217]. It also induced activation-related phosphorylation of ER and enhanced the transcription of 'growth regulation by estrogen in breast cancer 1' (GREB1), further supporting its ER-stimulating activity [217]. Additionally, compound 231 exerted anti-proliferative effects on various cancer cell lines, including an ER-negative breast cancer cell line, suggesting that it is capable of suppressing the growth of cancer cells independent of its ER-modulating activity [217]. Additionally, a treatment with 231 resulted in a significant apoptotic death of MCF7 and Ishikawa cancer cells, but did not induce apoptosis in non-cancer human umbilical vein endothelial cells [217].
Targeting tubulin polymerization, a series of 5-(furan-2-yl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiol-3-one oximes were prepared and evaluated for their anti-proliferative activities [218]. Among them, 233 (IC50 2.33, 2.72, and 1.96 μM) exhibited potent antiproliferative activity against human gastric adenocarcinoma SGC-7901, human non-small cell lung cancer A549, and human fibrosarcoma HT-1080, respectively [218].
Compound 233 was further assessed against a panel of cancer cell lines, including HeLa (IC50 1.84 μM), MCF-7 (IC50 7.98 μM), A549 (IC50 2.72 μM), HepG-2 (IC50 2.24 μM), and KB (IC50 2.22 μM) [218]. Furthermore, this compound exhibited strong antiproliferative effects against HT-1080 cells in a time- and dose-dependent manner, with the growth inhibitory effect appearing to be related to microtubule depolymerization [218]. Molecular docking studies revealed that 233 interacts and binds efficiently with the colchicine-binding site of tubulin. In addition, a treatment with 233 induced G2/M cell cycle arrest dose-dependently, and subsequently induced cell apoptosis, with the effect associated with both the mitochondrial and death receptor pathways [218]. Western blot studies indicated that upregulation of cyclin B1 and p-cdc2 was related to G2/M arrest [218].
Vascular endothelial growth factor receptors (VEGFRs) are a family of receptor protein tyrosine kinases that play an important role in the regulation of tumor-induced angiogenesis [219]. Consequently, VEGFR inhibitors have been widely used in the treatment of various tumors [219].
Following a hypothesis that a pseudo six-membered ring formed through intramolecular hydrogen bonding could be exploited to mimic planar quinazolines, a series of potential VEGFR-2 inhibitors containing an oxime as a hinge-binding fragment were designed and synthesized [220]. Most of the compounds tested in this study showed potent VEGFR-2 inhibitory activity, especially 234 (IC50 8.7 nM), 235 (IC50 5.3 nM), and 236 (IC50 4.2 nM), which exhibited significant enzymatic inhibitory activity as well as potent antiproliferative activity against cancer cells [220]. Molecular docking suggested that the salicylaldoxime formed two hydrogen bonds with the hinge region [220].
An oxime-bearing derivative of 2-((2,4,5-trifluorobenzyl)oxy)benzaldehyde (237) was also shown to suppress leukemic cell growth by significantly increasing reactive oxygen species (ROS) levels and inducing cell death [221]. The treated cells exhibited apoptotic cell death, as indicated by nuclear condensation, DNA fragmentation, and annexin V staining. This compound also elevated Bax/Bcl2 levels, caspase9, and caspase3/7 activity, while decreasing mitochondrial membrane potential [221]. ROS production was reduced by N-acetyl-L-cysteine (a ROS scavenger) and diphenyleneiodonium chloride (a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor) after exogenous treatment with 238, suggesting that this latter compound promotes apoptosis by modulating ROS levels and regulating NADPH oxidase activity [221]. On the other hand, a series of oxime ether derivatives of phenylpropanoids was shown to display in vitro anti-cancer activities when evaluated against human cancer cell lines, with compounds 238 (IC50: 0.443 and 0.225 µM) and 239 (IC50: 0.513 and 0.407 µM) exhibiting significant potency against lung cancer (A549) and gastric cancer (SMMG-803) cell lines, respectively [222].
While evaluating structurally modified analogs of the antimitotic natural product Curacin A for their antiproliferative activity on breast (MDA-MB231), prostate (PC3), and ovarian (2008) human tumor cell lines, two oxime analogs, 240 (GI50: 0.12, 0.25, and 0.24 µM) and 241 (GI50: 0.98, 1.2, and 9.9 µM), displayed good antiproliferative activity against the three cell lines, respectively [223]. Surprisingly, the less lipophilic and structurally simplified oxime (241), while displaying slightly weaker growth inhibition properties than the parent oxime analog (240), was more potent than Curacin A at inhibiting the assembly of purified tubulin, with the oxime moiety likely acting as a bioisostere of the (Z)-alkene group [223].
Some Suberoyl anilide hydroxamic acid (vorinostat) analogs with the aziridin-1-yl oxime moiety, including compounds 242 (IC50 0.60 and 0.90 mM), 243 (IC50 0.30 and 1.0 mM) and 244 (IC50 0.60 and 0.90 mM) were shown to possess high antiproliferative activity against HT1080 and MG22A human cancer cell lines, while displaying only weak histone deacetylase inhibition activity in HeLa cell line extracts [224].
Psammaplin A (245) is a marine metabolite previously reported to be a potent inhibitor of two classes of epigenetic enzymes: histone deacetylases and DNA methyltransferases. A library of psammaplin A-based core analogs and moieties was synthesized, and their cytotoxicity was evaluated in A549, MCF7, and W138 cell lines [225]. Many of the compounds, including 246 (IC50 0.16, 0.61, and 1.13 µM) and 247 (IC50 1.59, 1.13, and 7.86 µM), showed significant cytotoxicity in A549, MCF7, and W138 cells respectively, with the cytotoxicity SAR correlating with HDAC inhibition [225]. Compound 245 (IC50 7.5, 1.27, and 3.44 µM) was demonstrated to function as a natural prodrug, with the reduced form being highly potent against HDAC1 in vitro (IC50 0.9 nM) [225]. Furthermore, this compound exhibited high isoform selectivity, with 360-fold potency for HDAC1 over HDAC6 and more than 1000-fold less potent against HDAC7 and HDAC8 [225]. Moreover, treatment with this compound caused upregulation of histone acetylation, but had only a marginal effect on tubulin functions, with no evidence for 245 acting as a DNA methyltransferase (DNMT) inhibitor [225].
2.7. Anticancer activity of steroidal oximes and oxime ethers
Steroids are an important class of natural products, characterized by a basic molecular structure typically made up of 17 carbon atoms arranged in a consecutively fused four-ring system [226,227]. Three of these rings are six-membered cyclohexane rings (rings A, B, and C), while one is a five-membered cyclopentane ring (the D ring), as illustrated in Figure 15. Additional carbons are often added to the structure through the incorporation of methyl, prenyl, or isoprenyl groups at different positions, ranging from estrane (18 carbons), androstane (19 carbons), pregnane (21 carbons), all the way to cholestane (27 carbons) [226,227]. They are known to regulate several key processes in various biological systems, and considering their compelling biochemical properties, including their ability to easily penetrate cell membranes and bind to nuclear and membrane receptors, as well as their suitability for structural modifications, steroids represent an extremely attractive motif for designing potential new treatments for a range of diseases. In fact, since their discovery in 1935, steroids have been widely used in the treatment of several conditions, including autoimmune and inflammatory diseases and cancers [228,229]. Because even minor alterations to their core skeleton can result in significant variations in biological activity, a large number of steroidal derivatives with a wide range of therapeutic activities have been reported [43,229]. Exploiting these attributes, the hydrophobic steroid core has been used as a framework for a variety of oxime-bearing derivatives and analogs, which have exhibited remarkable antiproliferative and antitumor properties in many cases [228]. Interestingly, the introduction of this functional group frequently improves the bioactivity when compared to non-oxime analogous compounds. These improvements can be attributed to either the enhancement of the overall biochemical properties of the resulting molecules or to additional interactions with biological targets [51,230]. Furthermore, it has been observed that varying the position of the hydroximino group on the parent skeleton leads to remarkable changes in the relevant activity.
While screening a small library of steroidal oxime ethers with either an androstene or estrane core skeleton on a panel of cancer cell lines, including NCI-H23, NCI-H522, MDA-MB-231, MDA-MB-435, SW-620, COLO 205, LOX, UACC-62, OVCAR-3, OVCAR-5, U251, and SF-295, Jindal et al. [231] revealed that 17α-ethynylandrostene derivatives 248 – 250, and estrane derivatives 251 and 252 displayed noticeable antiproliferative activities.
(6E)-Hydroximinocholest-4-en-3-one (253), a steroidal oxime previously reported from Cinachyrella alloclada and C. apion [232], was also shown to exhibit selective cytotoxic activity against several types of cancer cells, such as P-388, A-549, HT-29 (IC50: 1.25 g/mL), and MEL-28 tumor cells (IC50: 2.5 g/mL) [233]. Unfortunately, this compound and its analog (254) exhibited only a marginal activity against sk-Hep-1, H-292, PC-3, and Hey-1B [234].
Estrone-16-oxime ethers also shown cytotoxicity toward human cancer cell lines HeLa, MCF7, A2780, and A431. Among them, 255 (IC50 3.52, 4.13, and 4.61 µM) was primarily active against HeLa, MCF7, and A2780, respectively, while 256 (IC50 5.63 and 13.25 µM) exerted more effects on HeLa and A431, respectively [235]. These estrone-16-oxime analogs were able to activate caspase-3 and change the mRNA level expression of endogenous factors regulating the G1-S phase transition (retinoblastoma protein, CDK4, and p16) [235]. Furthermore, several 6E-hydroximino-4-ene steroid oxime derivatives and their oxalate salts were evaluated for their in vitro antineoplastic activity against a panel of cancer cell lines. Many of these compounds showed moderate to good antiproliferative activity against leukemia, colon, melanoma, and renal cancer cell lines, with compounds 257 and 258 demonstrating the highest activity across all cell lines [236].
The anticancer activity of benzylidene pregnenolone oxime derivatives was also screened against HT-29, HTC-15, SF-295, HOP-62, A-545, and MCF-7 cancer cell lines. Compounds 259 (IC50 2.44, 3.53, 3.79, 1.50, 1.42, and 1.90 μM), 260 (IC50 2.43, 1.46, 8.86, 4.18, 1.97, and 1.34 μM), 261 (IC50 2.35, 0.31, 1.67, 3.57, 3.56, and 0.60 μM), and 262 (IC50 2.37, 0.65, 5.44, 0.81, 7.17, and 1.91 μM) exhibited significant cytotoxic activities against these cell lines.
Yanmin et al. [236] also evaluated a group of structurally diverse steroidal oxime ethers bearing either a 3-, 6-, or 22-O-methyloxime or O-benzyloxime for their antiproliferative activities. Some of these compounds exhibited a distinct cytotoxicity against SGC-7901 (263: IC50 17.3 μmol/L; 264: IC50 15.2 μmol/L), Bel-7404 (265: IC50 14 μmol/L; 264: IC50 13.3 μmol/L), and CNE-2 (265: IC50 14.1 μmol/L) cancer cell lines, with the study indicating that derivatives bearing 22-O-methyloxime and 22-O-benzyloxime exhibited better antiproliferative activities [236].
D-Secooxime derivatives of 13β- and 13α-estrone were shown to possess potent antiproliferative activity against a panel of human adherent cell lines (HeLa, MCF-7, A2780, and A431), with compounds 266 (IC50: 1.2 μM, 0.9 μM, 0.8 μM, and 2.6 μM), 267 (IC50: 1.7 μM, 0.7 μM, 0.9 μM, and 2.1 μM), and 268 (IC50: 0.8 μM, 0.4 μM, 0.9 μM, and 0.6 μM) exhibiting the best activity [237]. The E isomer of 266 (the Z isomer), which was also the most selective of the series, induced a cell cycle disturbance in the S phase [237]. Some 4-azasteroidal-20-oxime derivatives of progesterone also displayed anticancer activity in vitro against T24 cells, with compounds 269 (IC50: 1.90 μM), 270 (IC50: 1.94 μM), and 271 (IC50: 1.99 μM) being the most potent of the series [238]. This study indicated that methyl and ethyl oxime-ethers exhibited higher activity compared to the aryl-substituted oxime-esters [238].
Canário et al. [230] also studied several steroidal oximes of the estrane family, many of which exhibited interesting effects against the proliferation of various tumor cell lines when compared to the parent ketone compounds. Among these oximes, compound 272 displayed the highest activity against LNCaP cancer cells, along with a significant selectivity index. Furthermore, this compound induced cell cycle arrest in G2/M prophase and caused condensed and fragmented nuclei, characteristic of apoptosis [230]. In an e-screening assay, compound 272 also promoted the proliferation of T47-D cells, and docking studies indicated that this compound has relevant affinities for ERα and β-tubulin, providing a basis for its mechanism of action and its estrogenic effect. Interestingly, oximes bearing halogens in the A-ring, such as 2,4-diiodoestrone oxime 273 and 2,4-dibromoestrone oxime 274, exhibited selectivity for HepaRG cancer cells [230]. Another A-ring functionalized derivative, 2-nitroestrone oxime (275) (but not its 2,4-dinitro analog), showed higher cytotoxicity against HepaRG and MCF-7 cancer cells [230].
While evaluating O-alkylated oxyimino androst-4-ene derivatives synthesized from the corresponding isomerically pure 3E-oximes in vitro for their cytotoxic activity against eight human cancer cell lines, as well as normal fetal lung (MRC-5) and human foreskin (BJ) fibroblasts for selectivity, most compounds demonstrated strong activity against malignant melanoma (G-361), lung adenocarcinoma (A549), and colon adenocarcinoma (HT-29) cell lines [239]. Compound 276 emerged as the most promising candidate. These compounds also induce significant apoptosis in these cells, and further in vitro investigations against cytochrome P450 enzymes revealed their selective binding to the active sites of human steroid hydroxylases, including CYP7, CYP17A1, CYP19A1, or CYP21A2 [239].
Kolsi et al. [240] reported a series of dehydroabietic oxime derivatives with potential as antitumor agents, targeting pancreatic cancer and cancer-related inflammation. Compounds 277 and 278 were the most potent against human pancreatic cancer Aspc-1 cells, both exhibiting an IC50 value of 8.6 μM. Additionally, they demonstrated anti-inflammatory activity by inhibiting nitric oxide production, a crucial inflammatory mediator in the tumor microenvironment [240]. Further studies revealed that these compounds induced cancer cell differentiation and simultaneously downregulated cyclin D1 expression while upregulating p27 levels, consistent with cell cycle arrest in the G1 phase. Moreover, a kinase profiling study showed that some of these compounds exhibited isoform-selective inhibitory activity on RSK2 and AGC kinase, which are known to be implicated in cellular invasion and metastasis [240].
Both the androgen receptor (AR) and Cytochrome P450 17A1 (CYP17) are important targets for blocking androgen signaling. A steroidal imidazole oxime derivative (279) has demonstrated favorable biological profiles in both enzymatic and cellular assays [241]. This compound exhibited AR inhibition with an IC50 of 0.5 μM, which was 27-fold higher than that of the non-oxime parent compound, and a comparable CYP17 inhibition, with an IC50 of 11 μM [241]. Structure-activity relationship (SAR) studies revealed that introducing an oximino group at the C-3 position of the steroidal scaffold enhances AR antagonistic activity [241]. Additionally, 279 demonstrated promising anti-proliferative effects on LNCap cell lines, with an IC50 value of 23 μM, superior to the positive control Flutamide (IC50 = 28 μM) [241].
Furthermore, molecular docking was employed to investigate the binding mode of compound 279 with both AR (PDB: 4oea) and CYP17 proteins (PDB: 3ruk). This compound precisely fit into the ligand binding domain (LBD) of AR [241]. Further analysis revealed that the hydroxyimino group at the C-3 position established hydrogen bonds with Gln-711, Met745, and Arg752, while the hydroxy group formed hydrogen bonds with Asn705 and Thr877 [241]. Additionally, the docking study with CYP17 provided evidence of its promising CYP17 inhibitory activity, as it positioned well within the active site of CYP17 as illustrated in Figure 16. The nitrogen atom of N-methylimidazole formed a hydrogen bond with the Arg239 amino acid residue, and the oxygen atom at the C-3 position interacted with the heme of CYP17, which is crucial for inhibiting CYP17 activity [241].
A series of oleanolic acid oxime derivatives and their conjugates with aspirin have also been investigated for their effects on the expression and activation of NF-κB in human hepatoma HepG2 cells [242]. The oleanolic acid oxime derivatives exhibited a stronger cytotoxic effect against HepG2 cells compared to their conjugates with aspirin. However, conjugation of oleanolic acid oxime with aspirin resulted in enhanced downregulation of NF-κB expression and activation [242]. Among the aspirin hybrids, compounds 280 (3-(2-acetoxy)benzoyloxyiminoolean-12-en-28-oic acid morpholide) and 281 (3-(2-acetoxy)benzoyloxyiminoolean-12-en-28-oic acid methyl ester), differing by the presence of morpholide and methyl ester groups at the C-17 position of the oleanolic acid molecule (Figure 17), were the most efficient [242]. These oleanolic acid oxime derivatives, and particularly their conjugates with aspirin, downregulated the expression of COX-2 in HepG2 cells by modulating the NF-κB signaling pathway, suggesting their potential application in preventing liver inflammation and cancer [242].
Analogs of natural 5Z,9Z-dienoic acids, hybrid molecules based on the oximes of cholesterol, pregnenolone, and androsterone with 1,14-tetradeca-5Z,9Z-dienedicarboxylic acid were synthesized and evaluated, along with their parent oximes, for in vitro antitumor activity against HeLa, Hek293, U937, Jurkat, and K562 cell lines [243]. Compounds 282 (IC50: 0.82, 1.01, 0.74, 0.62, and 0.64 μM), 283 (IC50: 0.67, 0.53, 0.45, 0.74, and 0.71 μM), 284 (IC50: 0.68, 0.27, 0.38, 0.49, and 0.30 μM), 285 (IC50: 0.51, 0.33, 0.49, 0.76, and 0.58 μM), 286 (IC50: 0.32, 0.34, 0.41, 0.13, and 0.29 μM), and 287 (IC50: 0.02, 0.02, 0.04, 0.03, and 0.17 μM) demonstrated strong inhibition of tumor viability in all the tested cell lines, respectively [243]. Flow cytometry assays indicated that these molecules efficiently induced apoptosis in HeLa, Hek293, U937, Jurkat, and K562 cells [243].
While evaluating a series of androstene oxime-nitrogen mustard bioconjugates for their cytotoxicity against a panel of 60 cancer cell lines, 17E-steroidal oxime-benzoic acid mustard ester 3β-acetoxy-17E-[p-(N,N-bis(2-chloroethyl)amino)]benzoyloxy-imino-androst-5-ene (288) emerged as the most potent conjugate [244]. It exhibited significant growth inhibition on the IGROV1 ovarian cancer cell line, with a GI50 of 0.937 µM. Generally, the androstene oxime-nitrogen mustard conjugates derived from the D-ring of the steroid skeleton displayed superior antineoplastic activity compared to those derived from other rings [244].
Bu et al. [245] evaluated a series of designed and synthesized 5α,8α-epidioxyandrost-3β-ol-17-(O-phenylacetamide)oxime derivatives for their antiproliferative activity against human hepatocellular carcinoma cells (HepG2, Sk-Hep1) and human breast cancer cells (MCF-7, MDA-MB231). Compounds 289 (IC50: 12.14, 14.63, 20.36, and 15.82 µM), 290 (IC50: 9.30, 12.12, 22.74, and 17.53 µM), and 291 (IC50: 14.84, 11.08, 19.50, and 14.25 µM) exhibited moderate in vitro anti-proliferative activities. Additionally, fluorescence imaging revealed that the designed coumarin-289 conjugate (292) primarily localized in mitochondria, resulting in enhanced anticancer activities compared to the parent structure [245].
Photodynamic therapy is progressively emerging as an alternative treatment for both malignant and non-malignant cancers, despite its limitations in targeting specificity, which is critical for preventing systemic toxicity. Pavlíčková et al. [246] reported the synthesis and biological evaluation of a bimodal oxime conjugate (293) composed of a photosensitizing drug (red-emitting pheophorbide a) and nandrolone, a steroid that specifically binds to the androgen receptor, commonly overexpressed in various tumors. Since light-triggered therapies have the potential to make significant advancements in the treatment of hormone-sensitive cancers, this potentially targeted photosensitizer was assessed in prostatic cancer cell lines in vitro, using an AR-positive cell line (LNCaP) and an AR-negative/positive cell line (PC-3) [246]. U-2 OS cells, both with and without stable AR expression, were used as a second cell line model. This conjugate not only exhibited photodynamic activity and AR specificity, but also demonstrated a more pronounced phototoxic effect compared to pristine pheophorbide a. Live-cell fluorescence microscopy provided clear evidence that the 293 localizes in the endoplasmic reticulum and mitochondria [246]. A competitive localization study with an excess of nonfluorescent nandrolone was able to displace this fluorescent conjugate (293) from the cell interior, further confirming its binding specificity. Figure 17 summarizes some examples of anticancer oleanolic acid oxime derivatives and steroidal oximes conjugates.
2.8. Anticancer activity of oxime liganded metals and cyanoximates metallic complexes
The discovery of the anticancer activity of cisplatin in the 1960s and its subsequent clinical success generated interest in the use of metal compounds in cancer treatment [247,248]. In fact, cisplatin is known to play a role in the cure of over 90% of testicular cancer cases, and is vital in the treatment of other cancers, including melanoma, lymphomas, as well as ovarian, bladder, and cervical cancers [247]. To overcome the two main problems associated with cisplatin treatment, namely severe side effects and resistance, several strategies for the design and application of platinum-based drugs that can be activated in the tumor tissue have been developed. For example, classes of kinetically inert platinum-based anticancer prodrugs, which can be converted to more reactive platinum(II) species, taking advantage of the reducing environment in solid tumors, or activated on site through phototherapy, have been developed [249,250].
Furthermore, trans-configured platinum complexes with oxime ligands have emerged as a new class with features distinct from those of established anticancer agents, including different DNA binding behavior, increased cellular accumulation, and a different pattern of protein interaction [248,249,251]. As such, a number of platinum(II) oxime complexes that are inert at physiological pH but can be activated under slightly acidic pH values, converting platinum species into their cytotoxic/activated form, have been developed [252,253,254,255,256], and some illustrative examples are provides in Figure 18.
Some of these kinetically inert platinum(II) complexes bearing a chelating oxime ligand were evaluated for their cytotoxic properties on three human tumor cell lines originating from ovarian carcinoma (CH1), colon carcinoma (SW480), and non-small cell lung cancer (A549) [255]. This evaluation included a comparative investigation under standard pH screening conditions (pH 7.4) as well as in an acidified medium (pH 6.0), with the goal of determining their pH-dependent activation properties [255]. Among these compounds, 294 (IC50 3.9 μM), 295 (IC50 16 μM), 296 (IC50 1.0 μM) were primarily active against CH1 cell line, while 297 (IC50 8.3 and 12 μM) showed potency against both CH1 and SW480, respectively [255]. Compound 298 was the most potent of the series across the board, exhibiting IC50 values of 1.1, 0.97 and 5.6 μM against CH1, SW480 and A549, respectively [255].
Bartel et al.[257] reported cellular accumulation experiments and in vitro DNA platination studies involving three pairs of cis- and trans-configured acetone oxime platinum(II) complexes, as well as one pair of pentan-3-one oxime platinum(II) complexes. They found that, overall, trans-platinum(II) oxime complexes, 299(t) (IC50 0.22 µM), 300(t) (IC50 2.5 µM), 301(t) (IC50 2.8 µM), and 302(t) (IC50 1.9 µM), exhibited higher cytotoxicity compared to their corresponding cis congeners: 299(c) (IC50 3.4 µM), 300(c) (IC50 45 µM), 301(c) (IC50 1.8 µM), and 302(c) (IC50 15 µM), on the cisplatin-resistant SW480 cell line [257]. This study also revealed that the concentration of trans-configured complexes within SW480 cells was up to 100 times higher than that of cisplatin and up to 50 times higher than their cis-configured counterparts. Furthermore, the 'r' values (number of platinum atoms per nucleotide) were more than tenfold higher in cells treated with trans complexes compared to those treated with cisplatin [257].
The interaction of these complexes with DNA was also investigated in cell-free experiments using plasmid DNA (pUC19), in capillary zone electrophoresis with the DNA model 2-deoxyguanosine 5'-monophosphate, and through in vitro experiments measuring the extent of DNA damage in the comet assay. While incubation with cis complexes did not induce any DNA degradation, the trans complexes resulted in pronounced strand cleavage [257]. The most likely explanation for the difference in behavior between these complexes and the influence of their configuration is probably the formation of platinum–DNA mono-adducts in the case of the trans complexes, which potentially contributed to their cytotoxic effect [257].
The cytotoxicity of some trans platinum(II) oxime complexes and their head-to-tail-oriented oximato-bridged dimers featuring a six-membered diplatinacycle and representing a unique class of oxime ligands coordinated to platinum via an oxygen atom, was also evaluated on three human cancer cell lines, including the cisplatin-sensitive CH1 cells as well as cisplatin-resistant SW480 and A549 cells [256]. All these compounds, including 303 (IC50 6.6, 3.5 and 14 μM), 304 (IC50 3.3, 3.3 and 13 μM), 305 (IC50 6.2, 3.6 and 21 μM), 306 (IC50 3.4, 3.3 and 13 μM), 307 (IC50 0.95, 1.2 and 12 μM), 308 (IC50 1.8, 1.9 and 12 μM), 309 (IC50 0.94, 1.0 and 5.1 μM), 310 (IC50 0.96, 1.3 and 5.7 μM) and 311 (IC50 2.2, 2.5 and 9.5 μM), exhibited strong potency against all the three cell lines [256]. The dimerization resulted in a substantial (up to 7-fold) improvement of IC50 values of (aldoxime)Pt(II) compounds, whereas for the analogous complexes featuring ketoxime ligands the reverse trend was observed. Remarkably, the dimers yielded no cross-resistance with cisplatin in SW480 cells, exhibiting up to 2-fold enhanced cytotoxicity in comparison with the CH1 cell line [256]. Furthermore, one monomer 305 as well as one dimer 309 proved to be strong apoptotic agents capable of inducing cell death even in the cisplatin-resistant SW480 cell line [256]. Increasing the alkyl chain length of the oxime ligand using propionaldehyde oxime (305, 306, 309, and 310) instead of acetaldehyde oxime (303, 304, 307, and 308) did not result in a significant increase in cytotoxicity. However, some influence of the E- and Z- configuration of the coordinated aldoximes on the IC50 values was observed, as monomeric complexes 304 and 306 featuring both E-and Z-aldoximes proved to be by a factor of 2-fold or more cytotoxic in CH1 cells compared to their isomers 303 and 305, containing two Z ligands [256].
Pt(II) complexes bearing triphenylphosphine and chelating oximes were found to efficiently penetrate resistant cells and induce significant antiproliferative effects, despite minimal interaction with DNA [258]. Among them, compounds 312 with IC50 values of 3.87, 5.18, 3.83, 14.1, 0.62, 1.43, and 3.15 μM, and 313 with IC50 values of 2.47, 2.50, 2.63, 3.61, 0.31, 1.08, and 2.60 μM, exhibited potency against a panel of human tumor and mesothelial cells, including HeLa, A549, HT-29, MSTO-211H, A2780, A2780cis, and Met-5A, respectively [258]. Notably, compound 313 showed a remarkable cytotoxicity in ovarian carcinoma cells (A2780) and, interestingly, a significant antiproliferative effect on cisplatin-resistant cells (A2780cis). Investigation into the intracellular mechanism of action demonstrated that 313 had a lower ability to bind to DNA compared to cisplatin, used as a reference, but it had notably higher uptake in resistant cells [258]. A significant accumulation in mitochondria, along with the ability to induce concentration-dependent mitochondrial membrane depolarization and intracellular reactive oxygen species production, suggest a mitochondrion-mediated pathway as responsible for the intriguing cytotoxic profile of complex 313 [258].
Eddings et al. [259] reported the synthesis of bivalent palladium and platinum cyanoximates and evaluated their in vitro antiproliferative activity against human cervical cancer HeLa cell lines, using cisplatin as a positive control. Among these compounds, 314 and 315 were found to be active, causing the death of 16% and 28% of the cells, respectively, compared to a 55% reduction observed for cisplatin under the same conditions [259].
With worldwide annual sales of platinum-based anticancer drugs currently reaching around two billion U.S. dollars [251], several other metal compounds have been prepared and evaluated as potential chemotherapeutic agents as illustrated in Figure 19, although only a few have entered clinical trials [260,261].
Oxime-based palladacycles were also found to exhibit significant cytotoxic activity against the HT29, A549, and HeLa cancer cell lines [262,263]. Among them, the dinuclear complex 316 displayed greater anticancer activity in comparison to the mononuclear complexes 317 and 318. All these compounds acted in a time- and dose-dependent manner, resulting in a significant decrease in cell viability when compared to the control group in all three cell lines for each of the complexes [263].
Half-sandwich ruthenium, rhodium, and iridium complexes synthesized from aldoxime, ketoxime, and amidoxime ligands also exhibited in vitro anti-tumor activity against HT-29, BE and MIA PaCa-2 cell lines, especially for complexes 319 and 320 [264]. Complex 319 demonstrated comparable activity but greater selectivity for MIA PaCa-2 cells compared to cisplatin. Further studies revealed that complexes 321, 322, 323, and 324 induced significant apoptosis in Dalton's ascites lymphoma (DL) cells, with 319 showing statistically significant in vivo activity in DL-bearing mice.
While investigating the ability of water-soluble chiral arene Ru(II) complexes bearing amino-oxime ligands to interact with plasmid (pBR322) DNA and to exhibit cytotoxic and antimetastatic effects in vitro against androgen-unresponsive human prostate cancer PC3 cell lines, Benabdelouahab et al. [265] discovered that these compounds significantly reduced cell viability in PC3 cells compared to control, with an inhibitory activity of 75–82% at concentrations of 50 μM. Compounds 325 (IC50 24.4 μM), 326 (IC50 14.8 μM), and 327 (IC50 21.5 μM) showed considerably higher cytotoxicity than cisplatin (IC50 510 μM) under the same experimental conditions [265].
Furthermore, Saad El-Tabl et al. [266,267] and Fatma and Magdy [268] reported a panel of metal complexes derived from 2-hydroxy-3-(hydroxyimino)-4-oxopentan-2-ylidene)benzohydrazide and (E)-3-(2-(5, 6- diphenyl-1,2,4- triazin-3- yl)hydrazono)butan-2- one oxime, respectively. These compounds included iron (328), silver (329), zirconium (330), uranium (331), cobalt (332) and copper (333, 334, 335, and 336) complexes, all of which were evaluated for their anticancer effects. Among them, copper complexes, including 334 (IC50 6.49 μM), 335 (IC50 2.67 μM), and 336 (IC50 2.24 μM), as well as the cobalt complex 332 (IC50 36.8 μM), exhibited noticeable antiproliferative potency against the human liver cancer HepG2 cell lines [266,267].
3. Conclusions
This report represents one of the most comprehensive assessments of the anticancer potential of structurally diverse molecular frameworks bearing oxime and oxime ether moieties. Specific examples have been carefully selected to illustrate the distinct contributions of these functional groups to general strategies for molecular design, modulation of biological activities, computational modeling, and structure-activity relationship studies. The primary focus is on the role of this functional group in enhancing the physicochemical and anticancer properties of the resulting molecules. Additional mechanistic investigations, molecular dynamics, and docking studies indicate that the mechanism and the potential drug targets of the resulting compounds also depend on the chemical framework to which the functional group is attached. In some cases, the oxime or oxime ether groups enhanced the already available anticancer properties, while in others, these groups were fully responsible for the observed properties, as the scaffold without this functional group was completely inactive. The pharmacokinetic and pharmacodynamic profiles, as well as drug-likeness, also appeared to be impacted to a greater extent by the addition of the oxime or oxime ether functional group. Regardless of the situation, these compounds appear to inhibit, interact with, or interfere with a number of key markers in cancer pathogenesis and malignancy. As a result, it is not unlikely that some of these oxime and/or oxime ether-bearing molecular frameworks could soon advance to clinical trials or even make it to the market.
Author Contributions
J.F prepared the first draft, reviewed and edited the manuscript, J.B.M, C.M.M, J.E.W, K.J., L.G.B and A.A.P performed the literature search, helped prepared the first draft and proof-read the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: J.F. acknowledges the financial support from NSF CHE-1954734 and from the Edward G. Schlieder Foundation Professorships.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All needed information is contained within the manuscript, no supplemental data available.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shi Y-B, Chen S-Y, Liu R-B. The new insights into autophagy in thyroid cancer progression. J Transl Med. 2023, 21, 413. [Google Scholar] [CrossRef]
- Cao J, Wan S, Chen S, Yang L. ANXA6: a key molecular player in cancer progression and drug resistance. Discover Oncol. 2023, 14, 53. [Google Scholar] [CrossRef]
- Zhou M, Zheng M, Zhou X, Tian S, Yang X, Ning Y, et al. The roles of connexins and gap junctions in the progression of cancer. Cell Commun Signaling. 2023, 21, 8. [Google Scholar] [CrossRef]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Deo SVS, Sharma J, Kumar S. GLOBOCAN 2020 report on global cancer burden: challenges and opportunities for surgical oncologists. Ann Surg Oncol. 2022, 29, 6497–6500. [Google Scholar] [CrossRef]
- La Vecchia C, Negri E, Carioli G. Progress in cancer epidemiology: avoided deaths in Europe over the last three decades. Eur J Cancer Prev. 2022, 31, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Mazidimoradi A, Momenimovahed Z, Allahqoli L, Tiznobaik A, Hajinasab N, Salehiniya H, et al. The global, regional and national epidemiology, incidence, mortality, and burden of ovarian cancer. Health Sci Rep. 2022, 5, e936. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi C, Lippi G. Current cancer epidemiology. J Epidemiol Glob Health. 2019, 9, 217–222. [Google Scholar] [CrossRef]
- Ferlay J EM, Lam F, Colombet M, Mery L, Piñeros M, et al. Global cancer observatory: Cancer Today. Lyon: International Agency for Research on Cancer. In: Organization WH. https://gco.iarc.fr/today2020.
- Li C, He W-Q. Global prediction of primary liver cancer incidences and mortality in 2040. J Hepatol. 2023, 78, e144–e146. [Google Scholar] [CrossRef]
- Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M, et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Yu N, Hwang M, Lee Y, Song BR, Kang EH, Sim H, et al. Patient-derived cell-based pharmacogenomic assessment to unveil underlying resistance mechanisms and novel therapeutics for advanced lung cancer. J Exp Clin Cancer Res. 2023, 42, 37. [Google Scholar] [CrossRef]
- LaFargue CJ, Amero P, Noh K, Mangala LS, Wen Y, Bayraktar E, et al. Overcoming adaptive resistance to anti-VEGF therapy by targeting CD5L. Nat Commun. 2023, 14, 2407. [Google Scholar] [CrossRef] [PubMed]
- Liu Z-L, Chen H-H, Zheng L-L, Sun L-P, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduction Targeted Ther. 2023, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Amaresan R, Gopal U. Cell surface GRP78: a potential mechanism of therapeutic resistant tumors. Cancer Cell Int. 2023, 23, 100. [Google Scholar] [CrossRef] [PubMed]
- Zhuang H, Yu B, Tao D, Xu X, Xu Y, Wang J, et al. The role of m6A methylation in therapy resistance in cancer. Mol Cancer. 2023, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Naldini MM, Casirati G, Barcella M, Rancoita PMV, Cosentino A, Caserta C, et al. Longitudinal single-cell profiling of chemotherapy response in acute myeloid leukemia. Nat Commun. 2023, 14, 1285. [Google Scholar] [CrossRef] [PubMed]
- Chen Z, Zhang Y, Li W, Gao C, Huang F, Cheng L, et al. Single cell profiling of female breast fibroadenoma reveals distinct epithelial cell compositions and therapeutic targets. Nat Commun. 2023, 14, 3469. [Google Scholar] [CrossRef]
- Cortes-Selva D, Dasgupta B, Singh S, Grewal IS. Innate and innate-like cells: the future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol Sci. 2021, 42, 45–59. [Google Scholar] [CrossRef]
- Bohineust A, Tourret M, Derivry L, Caillat-Zucman S. Mucosal-associated invariant T (MAIT) cells, a new source of universal immune cells for chimeric antigen receptor (CAR)-cell therapy. Bull Cancer. 2021, 108, S92–S95. [Google Scholar] [CrossRef]
- Gao Z, Bai Y, Lin A, Jiang A, Zhou C, Cheng Q, et al. Gamma delta T-cell-based immune checkpoint therapy: attractive candidate for antitumor treatment. Mol Cancer. 2023, 22, 31. [Google Scholar] [CrossRef]
- Hong L, Aminu M, Li S, Lu X, Petranovic M, Saad MB, et al. Efficacy and clinicogenomic correlates of response to immune checkpoint inhibitors alone or with chemotherapy in non-small cell lung cancer. Nat Commun. 2023, 14, 695. [Google Scholar] [CrossRef] [PubMed]
- Xiang Y, Gong M, Deng Y, Wang H, Ye D. T cell effects and mechanisms in immunotherapy of head and neck tumors. Cell Commun Signaling. 2023, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- 25. Villarreal-González RV, González-Díaz S, Vidal-Gutiérrez O, Cruz-de la Cruz Cdl, Pérez-Ibave DC, Garza-Rodríguez ML. Hypersensitivity reactions to anticancer chemotherapy and monoclonal antibodies: safety and efficacy of desensitization. J Oncol Pharm Pract. 2023, 10781552231189461. [CrossRef] [PubMed]
- Suzuki H, Asano T, Ohishi T, Yoshikawa T, Suzuki H, Mizuno T, et al. Antitumor activities in mouse xenograft models of canine fibroblastic tumor by defucosylated mouse-dog chimeric anti-HER2 monoclonal antibody (H77Bf). Monoclonal Antibodies Immunodiagn Immunother. 2023, 42, 34–40. [Google Scholar] [CrossRef]
- Tanaka M, Ito A, Shiozawa S, Hara-Chikuma M. Anti-tumor effect of aquaporin 3 monoclonal antibody on syngeneic mouse tumor model. Transl Oncol. 2022, 24, 101498. [Google Scholar] [CrossRef] [PubMed]
- Yong T, Wei Z, Gan L, Yang X. Extracellular-vesicle-based drug delivery systems for enhanced antitumor therapies through modulating the cancer-immunity cycle. Adv Mater (Weinheim, Ger). 2022, 34, 2201054. [Google Scholar] [CrossRef] [PubMed]
- Moslehi M, Moazamiyanfar R, Dakkali MS, Rezaei S, Rastegar-Pouyani N, Jafarzadeh E, et al. Modulation of the immune system by melatonin; implications for cancer therapy. Int Immunopharmacol. 2022, 108, 108890. [Google Scholar] [CrossRef]
- Castelli C, Rivoltini L, Rodolfo M, Tazzari M, Belgiovine C, Allavena P. Modulation of the myeloid compartment of the immune system by angiogenic- and kinase inhibitor-targeted anti-cancer therapies. Cancer Immunol Immunother. 2015, 64, 83–89. [Google Scholar] [CrossRef]
- Mahadevan, S. Role of oximes in nitrogen metabolism in plants. Annu Rev Plant Physiol. 1973, 24, 69–88. [Google Scholar] [CrossRef]
- Soerensen M, Neilson EHJ, Moeller BL. Oximes: unrecognized chameleons in general and specialized plant metabolism. Mol Plant. 2018, 11, 95–117. [Google Scholar] [CrossRef]
- Meyer V, Janny A. Nitrogenous acetone derivatives. Ber. Dtsch. Chem. Ges 1882, 15, 1164–1167.
- Tapper BA, Conn EE, Butler GW. Conversion of α-ketoisovaleric acid oxime and isobutyraldoxime to linamarin in flax seedlings. Arch Biochem Biophys. 1967, 119, 593. [Google Scholar] [CrossRef]
- Underhill, EW. Biosynthesis of mustard oil glucosides. Conversion of phenylacetaldehyde oxime and 3-phenylpropionaldehyde oxime to glucotropaeolin and gluconasturtiin. Eur J Biochem. 1967, 2, 61. [Google Scholar] [CrossRef]
- Kindl, H. Oxidases and oxygenases in higher plants. I. Occurrence of indole-3-acetaldehyde oxime and its formation from L-tryptophan. Hoppe-Seyler's Z Physiol Chem. 1968, 349, 519. [Google Scholar]
- Halkier BA, Olsen CE, Moeller BL. The biosynthesis of cyanogenic glucosides in higher plants. The (E)- and (Z)-isomers of p-hydroxyphenylacetaldehyde oxime as intermediates in the biosynthesis of dhurrin in Sorghum bicolor (L.) Moench. J Biol Chem. 1989, 264, 19487. [Google Scholar]
- Andrawes NR, Bagley WP, Herrett RA. Metabolism of 2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime (Temik aldicarb pesticide) in potato plants. J Agr Food Chem. 1971, 19, 731. [Google Scholar] [CrossRef]
- Bartley WJ, Andrawes NR, Chancey EL, Bagley WP, Spurr HW. Metabolism of temik aldicarb pesticide [2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime] in the cotton plant. J Agr Food Chem. 1970, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Ridgway RL, Jones SL, Coppedge JR, Lindquist DA. Systemic activity of 2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime (UC-21149) in the cotton plant with special reference to the boll weevil. J Econ Entomol. 1968, 61, 1705. [Google Scholar] [CrossRef]
- Massolini G, Carmellino ML, Baruffini A. Activity of some benzophenone oximes on plant-pathogenic fungi. Farmaco, Ed Sci. 1987, 42, 117. [Google Scholar]
- Kula K, Nagatsky R, Sadowski M, Siumka Y, Demchuk OM. Arylcyanomethylenequinone oximes: an overview of synthesis, chemical transformations, and biological activity. Molecules. 2023, 28, 5229. [Google Scholar] [CrossRef]
- Gomes AR, Pires AS, Roleira FMF, Tavares-da-Silva EJ. The structural diversity and biological activity of steroid oximes. Molecules. 2023, 28, 1690. [Google Scholar] [CrossRef]
- Omidi S, Kakanejadifard A. A review on biological activities of Schiff base, hydrazone, and oxime derivatives of curcumin. RSC Adv. 2020, 10, 30186–30202. [Google Scholar] [CrossRef]
- Abele, E. Synthesis, reactions and biological activity of derivatives of oximes of three-membered heterocycles. Heterocycl Lett. 2013, 3, 229. [Google Scholar]
- Abele E, Abele R. Oximes of nucleosides and related compounds: synthesis, reactions and biological activity. Curr Org Synth. 2018, 15, 650–665. [Google Scholar] [CrossRef]
- Dai H, Li G, Chen J, Shi Y, Ge S, Fan C, et al. Synthesis and biological activities of novel 1,3,4-thiadiazole-containing pyrazole oxime derivatives. Bioorg Med Chem Lett. 2016, 26, 3818–3821. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru J, Zviagin E, Skouta R. FDA-Approved oximes and their significance in medicinal chemistry. Pharmaceuticals. 2022, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, GA. L-Canaline: a potent antimetabolite and anti-cancer agent from leguminous plants. Life Sci. 1997, 60, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Fylaktakidou KC, Hadjipavlou-Litina DJ, Litinas KE, Varella EA, Nicolaides DN. Recent developments in the chemistry and in the biological applications of amidoximes. Curr Pharm Des. 2008, 14, 1001–1047. [Google Scholar] [CrossRef]
- Canario C, Silvestre S, Falcao A, Alves G. Steroidal oximes: useful compounds with antitumor activities. Curr Med Chem. 2018, 25, 660–686. [Google Scholar] [CrossRef]
- Schepetkin IA, Plotnikov MB, Khlebnikov AI, Plotnikova TM, Quinn MT. Oximes: novel therapeutics with anticancer and anti-inflammatory potential. Biomolecules. 2021, 11, 777. [Google Scholar] [CrossRef]
- Dhuguru J, Skouta R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules. 2020, 25, 1615. [Google Scholar] [CrossRef]
- Ma MZ, Yao BY. Progress in indirubin treatment of chronic myelocytic leukemia. J Tradit Chin Med. 1983, 3, 245–248. [Google Scholar]
- Wang H, Wang Z, Wei C, Wang J, Xu Y, Bai G, et al. Anticancer potential of indirubins in medicinal chemistry: biological activity, structural modification, and structure-activity relationship. Eur J Med Chem. 2021, 223, 113652. [Google Scholar] [CrossRef]
- Xiao Z, Hao Y, Liu B, Qian L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leuk Lymphoma. 2002, 43, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Wang L, Zhou G-B, Liu P, Song J-H, Liang Y, Yan X-J, et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc Natl Acad Sci USA. 2008, 105, 4826–4831. [Google Scholar] [CrossRef] [PubMed]
- Fogaca MV, Candido-Bacani PdM, Benicio LM, Zapata LM, Cardoso PdF, Tempesta de Oliveira M, et al. Effects of indirubin and isatin on cell viability, mutagenicity, genotoxicity and BAX/ERCC1 gene expression. Pharm Biol (Abingdon, U K). 2017, 55, 2005–2014. [Google Scholar] [CrossRef]
- Alex D, Lam IK, Lin ZX, Lee SMY. Indirubin shows anti-angiogenic activity in an in vivo zebrafish model and an in vitro HUVEC model. J Ethnopharmacol. 2010, 131, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hoessel R, Leclerc S, Endicott JA, Nobel MEM, Lawrie A, Tunnah P, et al. Indirubin, the active constituent of a Chinese antileukemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef]
- Kumagai T, Aratsu Y, Sugawara R, Sasaki T, Miyairi S, Nagata K. Indirubin, a component of Ban-Lan-Gen, activates CYP3A4 gene transcription through the human pregnane X receptor. Drug Metab Pharmacokinet. 2016, 31, 139–145. [Google Scholar] [CrossRef]
- Eisenbrand G, Hippe F, Jakobs S, Muehlbeyer S. Molecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicine. J Cancer Res Clin Oncol. 2004, 130, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Morgan, DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Campa VM, Baltziskueta E, Bengoa-Vergniory N, Gorroño-Etxebarria I, Wesołowski R, Waxman J, et al. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget. 2014, 5, 8173–8187. [Google Scholar] [CrossRef] [PubMed]
- Marchand B, Arsenault D, Raymond-Fleury A, Boisvert F-M, Boucher M-J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J Biol Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef] [PubMed]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3β by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Hur E-M, Zhou F-Q. GSK3 signaling in neural development. Nat Rev Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud-Kolar N, Vougogiannopoulou K, Skaltsounis A-L. Indirubin derivatives: a patent review (2010 - present). Expert Opin Ther Pat. 2015, 25, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Ribas J, Bettayeb K, Ferandin Y, Knockaert M, Garrofe-Ochoa X, Totzke F, et al. 7-Bromoindirubin-3'-oxime induces caspase-independent cell death. Oncogene. 2006, 25, 6304–6318. [Google Scholar] [CrossRef]
- Cheng X, Merz K-H, Vatter S, Zeller J, Muehlbeyer S, Thommet A, et al. Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. J Med Chem. 2017, 60, 4949–4962. [Google Scholar] [CrossRef]
- Wang Z, Wang Y, Feng M, Tan X, Cheng J, Hua W, et al. Facile synthesis, X-ray structure and anticancer activity of N1-alkylindirubin-3'-oxime. Youji Huaxue. 2009, 29, 1606–1610. [Google Scholar]
- Lee J-W, Moon MJ, Min H-Y, Chung H-J, Park E-J, Park HJ, et al. Induction of apoptosis by a novel indirubin-5-nitro-3'-monoxime, a CDK inhibitor, in human lung cancer cells. Bioorg Med Chem Lett. 2005, 15, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
- Choi SJ, Moon MJ, Lee SD, Choi S-U, Han S-Y, Kim Y-C. Indirubin derivatives as potent FLT3 inhibitors with anti-proliferative activity of acute myeloid leukemic cells. Bioorg Med Chem Lett. 2010, 20, 2033–2037. [Google Scholar] [CrossRef]
- Sano M, Ichimaru Y, Kurita M, Hayashi E, Homma T, Saito H, et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 3'-oxime and 5-methoxyindirubin 3'-oxime in vitro and in vivo. Cancer Lett (N Y, NY, U S). 2017, 397, 72–82. [Google Scholar] [CrossRef]
- Song JH, Lee J-E, Cho K-M, Park S-H, Kim H-J, Kim Y-C, et al. 5-diphenylacetamido-indirubin-3'-oxime as a novel mitochondria-targeting agent with anti-leukemic activities. Mol Carcinog. 2016, 55, 611–621. [Google Scholar] [CrossRef]
- Zheng Y, Li X, Kuang L, Wang Y. New insights into the characteristics of DRAK2 and its role in apoptosis: from molecular mechanisms to clinically applied potential. Front Pharmacol. 2022, 13, 1014508. [Google Scholar] [CrossRef] [PubMed]
- Jung ME, Byun BJ, Kim H-M, Lee JY, Park J-H, Lee N, et al. Discovery of indirubin derivatives as new class of DRAK2 inhibitors from high throughput screening. Bioorg Med Chem Lett. 2016, 26, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Yoon KB, Lee HJ, Chung HJ, Lee J, Choi J, Heo JD, et al. Discovery of LDD-1075 as a potent FLT3 inhibitor. Oncol Lett. 2019, 17, 4735–4741. [Google Scholar] [CrossRef]
- Cheng X, Rasque P, Vatter S, Merz K-H, Eisenbrand G. Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg Med Chem. 2010, 18, 4509–4515. [Google Scholar] [CrossRef]
- Jautelat R, Brumby T, Schaefer M, Briem H, Eisenbrand G, Schwahn S, et al. From the insoluble dye indirubin towards highly active, soluble CDK2-inhibitors. ChemBioChem. 2005, 6, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Yu A-S, Zhao L. Effects of the GSK-3β inhibitor (2Z,3E)-6-bromoindirubin-3'-oxime upon ovarian cancer cells. Tumor Biol. 2016, 37, 4857–4864. [Google Scholar] [CrossRef] [PubMed]
- Ribas J, Yuste VJ, Garrofe-Ochoa X, Meijer L, Esquerda JE, Boix J. 7-Bromoindirubin-3'-oxime uncovers a serine protease-mediated paradigm of necrotic cell death. Biochem Pharmacol. 2008, 76, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Broecker-Preuss M, Becher-Boveleth N, Gall S, Rehmann K, Schenke S, Mann K. Induction of atypical cell death in thyroid carcinoma cells by the indirubin derivative 7-bromoindirubin-3'-oxime (7BIO). Cancer Cell Int. 2015, 15, 97–1. [Google Scholar] [CrossRef]
- Ichimaru Y, Fujii T, Saito H, Sano M, Uchiyama T, Miyairi S. 5-Bromoindirubin 3'-(O-oxiran-2-ylmethyl)oxime: a long-acting anticancer agent and a suicide inhibitor for epoxide hydrolase. Bioorg Med Chem. 2017, 25, 4665–4676. [Google Scholar] [CrossRef] [PubMed]
- Ichimaru Y, Saito H, Uchiyama T, Metori K, Tabata K, Suzuki T, et al. Indirubin 3'-(O-oxiran-2-ylmethyl)oxime: A novel anticancer agent. Bioorg Med Chem Lett. 2015, 25, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Choi S-J, Lee J-E, Jeong S-Y, Im I, Lee S-D, Lee E-J, et al. 5,5'-Substituted Indirubin-3'-oxime derivatives as potent cyclin-dependent kinase inhibitors with anticancer activity. J Med Chem. 2010, 53, 3696–3706. [Google Scholar] [CrossRef]
- Kim J-K, Shin EK, Kang Y-H, Park JHY. Indirubin-3'-monoxime, a derivative of a chinese antileukemia medicine, inhibits angiogenesis. J Cell Biochem. 2011, 112, 1384–1391. [Google Scholar] [CrossRef]
- Kim W-S, Lee M-J, Kim D-H, Lee J-E, Kim J-I, Kim Y-C, et al. 5'-OH-5-nitro-Indirubin oxime (AGM130), an Indirubin derivative, induces apoptosis of Imatinib-resistant chronic myeloid leukemia cells. Leuk Res. 2013, 37, 427–433. [Google Scholar] [CrossRef]
- Ahn M-Y, Kim T-H, Kwon S-M, Yoon H-E, Kim H-S, Kim J-I, et al. 5-Nitro-5'-hydroxy-indirubin-3'-oxime (AGM130), an indirubin-3'-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur J Pharm Sci. 2015, 79, 122–131. [Google Scholar] [CrossRef]
- Yan L, Lai F, Chen X, Xiao Z. Discovery of novel indirubin-3'-monoxime derivatives as potent inhibitors against CDK2 and CDK9. Bioorg Med Chem Lett. 2015, 25, 2447–2451. [Google Scholar] [CrossRef]
- Lindberg MF, Meijer L. Dual-specificity, tyrosine phosphorylation-regulated kinases (DYRKs) and cdc2-like kinases (CLKs) in human disease, an overview. Int J Mol Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Myrianthopoulos V, Kritsanida M, Gaboriaud-Kolar N, Magiatis P, Ferandin Y, Durieu E, et al. Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. ACS Med Chem Lett. 2013, 4, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Meijer L, Skaltsounis A-L, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, et al. GSK-3-selective inhibitors derived from tyrian purple indirubins. Chem Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Nam S, Buettner R, Turkson J, Kim D, Cheng JQ, Muehlbeyer S, et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc Natl Acad Sci USA. 2005, 102, 5998–6003. [Google Scholar] [CrossRef] [PubMed]
- Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci USA. 2004, 101, 16789–16794. [Google Scholar] [CrossRef]
- Kurita M, Hanada S, Ichimaru Y, Saito H, Tabata K, Asami S, et al. Indirubin 3'-epoxide induces caspase-independent cell death in human neuroblastoma. Biol Pharm Bull. 2016, 39, 993–999. [Google Scholar] [CrossRef]
- Shin E-K, Kim J-K. Indirubin derivative E804 inhibits angiogenesis. BMC Cancer. 2012, 12, 164. [Google Scholar] [CrossRef]
- Chan Y-K, Kwok H-H, Chan L-S, Leung KS-Y, Shi J, Mak N-K, et al. An indirubin derivative, E804, exhibits potent angiosuppressive activity. Biochem Pharmacol. 2012, 83, 598–607. [Google Scholar] [CrossRef]
- Nam S, Scuto A, Yang F, Chen WY, Park S, Yoo H-S, et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol Oncol. 2012, 6, 276–283. [Google Scholar] [CrossRef]
- Cheng X, Alborzinia H, Merz K-H, Steinbeisser H, Mrowka R, Scholl C, et al. Indirubin derivatives modulate TGFβ/BMP signaling at different levels and trigger ubiquitin-mediated depletion of nonactivated R-smads. Chem Biol (Oxford, U K). 2012, 19, 1423–1436. [Google Scholar] [CrossRef]
- Nam S, Wen W, Schroeder A, Herrmann A, Yu H, Cheng X, et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol. 2013, 7, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Heshmati N, Wagner B, Cheng X, Scholz T, Kansy M, Eisenbrand G, et al. Physicochemical characterization and in vitro permeation of an indirubin derivative. Eur J Pharm Sci. 2013, 50, 467–475. [CrossRef] [PubMed]
- Jakobs S, Merz KH, Vatter S, Eisenbrand G. Molecular targets of indirubins. Int J Clin Pharmacol Ther. 2005, 43, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Ginzinger W, Egger A, Muehlgassner G, Arion VB, Jakupec MA, Galanski M, et al. Water-soluble cationic derivatives of indirubin, the active anticancer component from Indigo naturalis. Chem Biodiversity. 2012, 9, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Kim W-H, Jeong P, Kim S-W, Cho H, Lee J-m, Seo S, et al. hA novel indirubin derivative that increases somatic cell plasticity and inhibits tumorigenicity. Bioorg Med Chem. 2019, 27, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Jeong P, Moon Y, Lee J-H, Lee S-D, Park J, Lee J, et al. Discovery of orally active indirubin-3'-oxime derivatives as potent type 1 FLT3 inhibitors for acute myeloid leukemia. Eur J Med Chem. 2020, 195, 112205. [Google Scholar] [CrossRef]
- Lee HJ, Jeong P, Moon Y, Choi J, Heo JD, Kim Y-C, et al. Characterization of LDD-2633 as a novel RET kinase inhibitor with anti-tumor effects in thyroid cancer. Pharmaceuticals. 2021, 14, 38. [Google Scholar] [CrossRef]
- Lee HJ, Lee J, Jeong P, Choi J, Baek J, Ahn SJ, et al. Discovery of a FLT3 inhibitor LDD1937 as an anti-leukemic agent for acute myeloid leukemia. Oncotarget. 2018, 9, 924–936. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar N, Myrianthopoulos V, Vougogiannopoulou K, Gerolymatos P, Horne DA, Jove R, et al. Natural-based indirubins display potent cytotoxicity toward wild-type and T315I-resistant leukemia cell lines. J Nat Prod. 2016, 79, 2464–2471. [Google Scholar] [CrossRef]
- Ferandin Y, Bettayeb K, Kritsanida M, Lozach O, Polychronopoulos P, Magiatis P, et al. 3'-Substituted 7-halogenoindirubins, a new class of cell death inducing agents. J Med Chem. 2006, 49, 4638–4649. [Google Scholar] [CrossRef] [PubMed]
- Liu L, Kritsanida M, Magiatis P, Gaboriaud N, Wang Y, Wu J, et al. A novel 7-bromoindirubin with potent anticancer activity suppresses survival of human melanoma cells associated with inhibition of STAT3 and Akt signaling. Cancer Biol Ther. 2012, 13, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Ndolo KM, An SJ, Park KR, Lee HJ, Yoon KB, Kim Y-C, et al. Discovery of an indirubin derivative as a novel c-Met kinase inhibitor with in vitro anti-tumor effects. Biomol Ther. 2019, 27, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Vougogiannopoulou K, Ferandin Y, Bettayeb K, Myrianthopoulos V, Lozach O, Fan Y, et al. Soluble 3',6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J Med Chem. 2008, 51, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Nguyen DT, Truong GN, Van Vuong T, Van TN, Manh CN, Dao CT, et al. Synthesis of new indirubin derivatives and their in vitro anticancer activity. Chem Pap. 2019, 73, 1083–1092. [Google Scholar] [CrossRef]
- Anh DT, Hai P-T, Dung DTM, Dung PTP, Huong L-T-T, Park EJ, et al. Design, synthesis and evaluation of novel indirubin-based N-hydroxybenzamides, N-hydroxypropenamides and N-hydroxyheptanamides as histone deacetylase inhibitors and antitumor agents. Bioorg Med Chem Lett. 2020, 30, 127537. [Google Scholar] [CrossRef]
- Dan NT, Quang HD, Van Truong V, Huu Nghi D, Cuong NM, Cuong TD, et al. Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3β of new indirubin-3'-oxime derivatives. Sci Rep. 2020, 10, 11429. [Google Scholar] [CrossRef]
- Qu H-E, Huang R-Z, Yao G-Y, Li J-L, Ye M-Y, Wang H-S, et al. Synthesis and pharmacological evaluation of novel bisindole derivatives bearing oximes moiety: Identification of novel proapoptotic agents. Eur J Med Chem. 2015, 95, 400–415. [Google Scholar] [CrossRef]
- Grosso C, Cardoso AL, Lemos A, Varela J, Rodrigues MJ, Custodio L, et al. Novel approach to bis(indolyl)methanes: de novo synthesis of 1-hydroxyiminomethyl derivatives with anti-cancer properties. Eur J Med Chem. 2015, 93, 9–15. [Google Scholar] [CrossRef]
- Dandu R, Zulli AL, Bacon ER, Underiner T, Robinson C, Chang H, et al. Design and synthesis of dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole oximes as potent dual inhibitors of TIE-2 and VEGF-R2 receptor tyrosine kinases. Bioorg Med Chem Lett. 2008, 18, 1916–1921. [Google Scholar] [CrossRef]
- Tseng C-H, Chen Y-R, Tzeng C-C, Liu W, Chou C-K, Chiu C-C, et al. Discovery of indeno[1,2-b]quinoxaline derivatives as potential anticancer agents. Eur J Med Chem. 2016, 108, 258–273. [Google Scholar] [CrossRef]
- Alvarez C, Alvarez R, Corchete P, Lopez JL, Perez-Melero C, Pelaez R, et al. Diarylmethyloxime and hydrazone derivatives with 5-indolyl moieties as potent inhibitors of tubulin polymerization. Bioorg Med Chem. 2008, 16, 5952–5961. [Google Scholar] [CrossRef]
- Pereira R, Benedetti R, Perez-Rodriguez S, Nebbioso A, Garcia-Rodriguez J, Carafa V, et al. Indole-Derived Psammaplin A analogues as epigenetic modulators with multiple inhibitory activities. J Med Chem. 2012, 55, 9467–9491. [Google Scholar] [CrossRef]
- Sharma A, Talimarada D, Yadav UP, Singh N, Reddy AS, Bag D, et al. Design and synthesis of new tubulin polymerization inhibitors inspired from combretastatin A-4: an anticancer agent. ChemistrySelect. 2020, 5, 11560–11572. [Google Scholar] [CrossRef]
- El-Wahab HAAA, Ali AM, Abdel-Rahman HM, Qayed WS. Synthesis, biological evaluation, and molecular modeling studies of acetophenones-tethered 1,2,4-triazoles and their oximes as epidermal growth factor receptor inhibitors. Chem Biol Drug Des. 2022, 100, 981–993. [Google Scholar] [CrossRef]
- Fadaly WAA, Elshaier YAMM, Hassanein EHM, Abdellatif KRA. New 1,2,4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: synthesis, cyclooxygenase inhibition, anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg Chem. 2020, 98, 103752. [Google Scholar] [CrossRef]
- Abdel-Aziz AAM, El-Azab AS, Al Saif NA, Alanazi MM, El-Gendy MA, Obaidullah AJ, et al. Synthesis, anti-inflammatory, cytotoxic, and COX-1/2 inhibitory activities of cyclic imides bearing 3-benzenesulfonamide, oxime, and β-phenylalanine scaffolds: a molecular docking study. J Enzyme Inhib Med Chem. 2020, 35, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz M, Abuo-Rahma GE-DAA, Beshr EAM, Ali TFS. New nitric oxide donating 1,2,4-triazole/oxime hybrids: Synthesis, investigation of anti-inflammatory, ulcerogenic liability and antiproliferative activities. Bioorg Med Chem. 2013, 21, 3839–3849. [Google Scholar] [CrossRef] [PubMed]
- Abuo-Rahma GE-DAA, Abdel-Aziz M, Beshr EAM, Ali TFS. 1,2,4-Triazole/oxime hybrids as new strategy for nitric oxide donors: synthesis, anti-inflammatory, ulcerogenicity and antiproliferative activities. Eur J Med Chem. 2014, 71, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Park H-J, Lee K, Park S-J, Ahn B, Lee J-C, Cho H, et al. Identification of antitumor activity of pyrazole oxime ethers. Bioorg Med Chem Lett. 2005, 15, 3307–3312. [Google Scholar] [CrossRef] [PubMed]
- Xiong B, Chen S, Zhu P, Huang M, Gao W, Zhu R, et al. Design, synthesis, and biological evaluation of novel thiazolyl substituted bis-pyrazole oxime derivatives with potent antitumor activities by selectively inducing apoptosis and ROS in cancer cells. Med Chem (Sharjah, United Arab Emirates). 2019, 15, 743–754. [Google Scholar] [CrossRef]
- Dai H, Ge S, Guo J, Chen S, Huang M, Yang J, et al. Development of novel bis-pyrazole derivatives as antitumor agents with potent apoptosis induction effects and DNA damage. Eur J Med Chem. 2018, 143, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Sun J, Liu H-Y, Xu R-F, Zhu H-L. Identification of nitroimidazole-oxime derivatives targeting the polo-box domain of polo-like kinase 1. Bioorg Med Chem. 2017, 25, 6581–6588. [Google Scholar] [CrossRef] [PubMed]
- Guo S-c, Zhao Y-f, Li R-d, Xie L-j, Yang Y-b, Gong P. Synthesis and biological evaluation of novel tricyclic oximino derivatives as antitumor agents. Chem Res Chin Univ. 2008, 24, 47–53. [Google Scholar] [CrossRef]
- Huang S, Li R, Connolly PJ, Xu G, Gaul MD, Emanuel SL, et al. Synthesis and biological study of 4-aminopyrimidine-5-carboxaldehyde oximes as antiproliferative VEGFR-2 inhibitors. Bioorg Med Chem Lett. 2006, 16, 6063–6066. [Google Scholar] [CrossRef] [PubMed]
- Xu G, Searle LL, Hughes TV, Beck AK, Connolly PJ, Abad MC, et al. Discovery of novel 4-amino-6-arylaminopyrimidine-5-carbaldehyde oximes as dual inhibitors of EGFR and ErbB-2 protein tyrosine kinases. Bioorg Med Chem Lett. 2008, 18, 3495–3499. [Google Scholar] [CrossRef] [PubMed]
- Gaul MD, Xu G, Kirkpatrick J, Ott H, Baumann CA. 4-Amino-6-piperazin-1-yl-pyrimidine-5-carbaldehyde oximes as potent FLT-3 inhibitors. Bioorg Med Chem Lett. 2007, 17, 4861–4865. [Google Scholar] [CrossRef] [PubMed]
- Qiang H, Gu W, Huang DD, Shi W, Qiu Q, Dai Y, et al. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-carbaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorg Med Chem. 2016, 24, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Xue W, Li Q, Fu Y, Liu F, Huai Z, Xia R, et al., inventors; Guizhou University, assignee. Process for preparation of quinazoline-containing 1,4-pentadiene-3-one oxime ether derivative, and application to treatment of human liver cancer patent. CN108530427. 2018.
- Su S, Chen M, Li Q, Wang Y, Chen S, Sun N, et al. Novel penta-1,4-diene-3-one derivatives containing quinazoline and oxime ether fragments: Design, synthesis and bioactivity. Bioorg Med Chem. 2021, 32, 115999. [Google Scholar] [CrossRef]
- Hisham M, Youssif BGM, Osman EEA, Hayallah AM, Abdel-Aziz M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur J Med Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef]
- Huynh MT, Anson CW, Cavell AC, Stahl SS, Hammes-Schiffer S. Quinone 1 e- and 2 e-/2 H+ reduction potentials: identification and analysis of deviations from systematic scaling relationships. J Am Chem Soc. 2016, 138, 15903–15910. [Google Scholar] [CrossRef]
- El-Najjar N, Gali-Muhtasib H, Ketola RA, Vuorela P, Urtti A, Vuorela H. The chemical and biological activities of quinones: overview and implications in analytical detection. Phytochem Rev. 2011, 10, 353–370. [Google Scholar] [CrossRef]
- O'Brien, PJ. Molecular mechanisms of quinone cytotoxicity. Chem-Biol Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Bolton JL, Dunlap T. Formation and biological targets of quinones: cytotoxic versus cytoprotective effects. Chem Res Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska J, Prendecki M, Lianeri M, Kozubski W. Molecular effects of L-dopa therapy in Parkinson disease. Curr Genomics. 2014, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Feitosa CM, de Sousa Silva S, Militao GGC, Pergentino de Sousa D, Rashed K, Lima LKF. Benzoquinone mono oximes derivatives with anticancer activity. Int J Pharmacogn (Panchkula, India). 2020, 7, 369–375. [Google Scholar] [CrossRef]
- Kaur K, Sharma R, Singh A, Attri S, Arora S, Kaur S, et al. Pharmacological and analytical aspects of alkannin/shikonin and their derivatives: An update from 2008 to 2022. Chin Herb Med. 2022, 14, 511–527. [Google Scholar] [CrossRef]
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Wang R, Zhou S, Li S. Cancer therapeutic agents targeting hypoxia-inducible factor-1. Curr Med Chem. 2011, 18, 3168–3189. [Google Scholar] [CrossRef]
- Wang R, Zhang X, Song H, Zhou S, Li S. Synthesis and evaluation of novel alkannin and shikonin oxime derivatives as potent antitumor agents. Bioorg Med Chem Lett. 2014, 24, 4304–4307. [Google Scholar] [CrossRef]
- Huang G, Dong J-Y, Zhang Q-J, Meng Q-Q, Zhao H-R, Zhu B-Q, et al. Discovery and synthesis of sulfur-containing 6-substituted 5,8-dimethoxy-1,4-naphthoquinone oxime derivatives as new and potential anti-MDR cancer agents. Eur J Med Chem. 2019, 165, 160–171. [Google Scholar] [CrossRef]
- Huang G, Meng Q-Q, Zhou W, Zhang Q-J, Dong J-Y, Li S-S. Design and synthesis of biotinylated dimethylation of alkannin oxime derivatives. Chin Chem Lett. 2017, 28, 453–457. [Google Scholar] [CrossRef]
- Huang G, Zhao H-R, Meng Q-Q, Zhang Q-J, Dong J-Y, Zhu B-q, et al. Synthesis and biological evaluation of sulfur-containing shikonin oxime derivatives as potential antineoplastic agents. Eur J Med Chem. 2018, 143, 166–181. [Google Scholar] [CrossRef]
- Huang G, Zhao H-r, Zhou W, Dong J-y, Zhang Q-j, Meng Q-q, et al. 6-Substituted 1,4-naphthoquinone oxime derivatives (I): synthesis and evaluation of their cytotoxic activity. Monatsh Chem. 2017, 148, 1011–1023. [Google Scholar] [CrossRef]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Klaus A, Saga Y, Taketo MM, Tzahor E, Birchmeier W. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci USA. 2007, 104, 18531. [Google Scholar] [CrossRef]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Soldi R, Horrigan SK, Cholody MW, Padia J, Sorna V, Bearss J, et al. Design, synthesis, and biological evaluation of a series of anthracene-9,10-dione dioxime β-catenin pathway inhibitors. J Med Chem. 2015, 58, 5854–5862. [Google Scholar] [CrossRef]
- Gogia P, Ashraf H, Bhasin S, Xu Y. Antibody-drug conjugates: a review of approved drugs and their clinical level of evidence. Cancers. 2023, 15, 3886. [Google Scholar] [CrossRef] [PubMed]
- Sun T, Niu X, He Q, Liu M, Qiao S, Qi R-Q. Development, efficacy and side effects of antibody-drug conjugates for cancer therapy (Review). Mol Clin Oncol. 2023, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Samantasinghar A, Sunildutt NP, Ahmed F, Soomro AM, Salih ARC, Parihar P, et al. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. Biomed Pharmacother. 2023, 161, 114408. [Google Scholar] [CrossRef]
- Suzuki Y, Zhou S, Ota Y, Harrington M, Miyagi E, Takagi H, et al. Toxicity profiles of antibody drug conjugates for anticancer treatment: a systematic review and meta-analysis. JNCI Cancer Spectr. 2023. [CrossRef]
- Xiao T, Ali S, Mata DGMM, Lohmann AE, Blanchette PS. Antibody-drug conjugates in breast cancer: ascent to destiny and beyond-A 2023 review. Curr Oncol. 2023, 30, 6447–6461. [Google Scholar] [CrossRef]
- Alas M, Saghaeidehkordi A, Kaur K. Peptide-drug conjugates with different linkers for cancer therapy. J Med Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Nakano I, Soe CZ, Codd R. Isolation of doxorubicin from a bacterial culture using immobilised metal ion affinity chromatography. RSC Adv. 2015, 5, 46437–46442. [Google Scholar] [CrossRef]
- Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Momparier RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976, 36, 2891. [Google Scholar]
- Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994, 45, 649. [Google Scholar]
- Pommier Y, Leo E, Zhang H-L, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol (Cambridge, MA, U S). 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Schuster S, Biri-Kovacs B, Szeder B, Buday L, Gardi J, Szabo Z, et al. Enhanced in vitro antitumor activity of GnRH-III-daunorubicin bioconjugates influenced by sequence modification. Pharmaceutics. 2018, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Schuster S, Biri-Kovacs B, Szeder B, Farkas V, Buday L, Szabo Z, et al. Synthesis and in vitro biochemical evaluation of oxime bond-linked daunorubicin-GnRH-III conjugates developed for targeted drug delivery. Beilstein J Org Chem. 2018, 14, 756–771. [Google Scholar] [CrossRef]
- Orban E, Mezo G, Schlage P, Csik G, Kulic Z, Ansorge P, et al. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin-GnRH-III bioconjugates and DNA-binding properties of daunorubicin-amino acid metabolites. Amino Acids. 2011, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Randelovic I, Schuster S, Kapuvari B, Fossati G, Steinkuhler C, Mezo G, et al. Improved in vivo anti-tumor and anti-metastatic effect of GnRH-III-daunorubicin analogs on colorectal and breast carcinoma bearing mice. Int J Mol Sci. 2019, 20, 4763. [Google Scholar] [CrossRef]
- Dokus LE, Lajko E, Randelovic I, Mezo D, Schlosser G, Kohidai L, et al. Phage display-based homing peptide-daunomycin conjugates for selective drug targeting to PANC-1 pancreatic cancer. Pharmaceutics. 2020, 12, 576. [Google Scholar] [CrossRef]
- Feni L, Parente S, Robert C, Gazzola S, Arosio D, Piarulli U, et al. Kiss and run: promoting effective and targeted cellular uptake of a drug delivery vehicle composed of an integrin-targeting diketopiperazine peptidomimetic and a cell-penetrating peptide. Bioconjugate Chem. 2019, 30, 2011–2022. [Google Scholar] [CrossRef]
- Marelli MM, Manea M, Moretti RM, Marzagalli M, Limonta P. Oxime bond-linked daunorubicin-GnRH-III bioconjugates exert antitumor activity in castration-resistant prostate cancer cells via the type I GnRH receptor. Int J Oncol. 2015, 46, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Xu W, Ding J, Xiao C, Li L, Zhuang X, Chen X. Versatile preparation of intracellular-acidity-sensitive oxime-linked polysaccharide-doxorubicin conjugate for malignancy therapeutic. Biomaterials. 2015, 54, 72–86. [Google Scholar] [CrossRef]
- Fotie, J. The antiprotozoan potential of flavonoids. Pharmacogn Rev. 2008, 2, 6–19. [Google Scholar]
- Chen S, Wang X, Cheng Y, Gao H, Chen X. A Review of classification, biosynthesis, biological activities and potential applications of flavonoids. Molecules. 2023, 28, 4982. [Google Scholar] [CrossRef]
- Shi S, Li J, Zhao X, Liu Q, Song S-J. A comprehensive review: biological activity, modification and synthetic methodologies of prenylated flavonoids. Phytochemistry (Elsevier). 2021, 191, 112895. [Google Scholar] [CrossRef]
- Juca MM, Cysne Filho FMS, de Almeida JC, Mesquita DdS, Barriga JRdM, Dias KCF, et al. Flavonoids: biological activities and therapeutic potential. Nat Prod Res. 2020, 34, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Paramas AM, Ayuda-Duran B, Martinez S, Gonzalez-Manzano S, Santos-Buelga C. The mechanisms behind the biological activity of flavonoids. Curr Med Chem. 2019, 26, 6976–6990. [Google Scholar] [CrossRef]
- Guo C, Alfaro-Aco R, Zhang C, Russell RW, Petry S, Polenova T. Structural basis of protein condensation on microtubules underlying branching microtubule nucleation. Nat Commun. 2023, 14, 3682. [Google Scholar] [CrossRef] [PubMed]
- Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Chiang C-Y, Wang T-C, Lee C-H, Chen C-S, Wang S-H, Lin Y-C, et al. WTC-01, a novel synthetic oxime-flavone compound, destabilizes microtubules in human nasopharyngeal carcinoma cells in vitro and in vivo. Br J Pharmacol. 2015, 172, 4671–4683. [Google Scholar] [CrossRef]
- Liu Z, Wei W, Gan C, Huang Y, Liu S, Cui J. Synthesis and cytotoxicity of E-hesperetin oximes against SGC-7901. Youji Huaxue. 2013, 33, 1970–1974. [Google Scholar] [CrossRef]
- Kocyigit A, Koyuncu I, Dikilitas M, Bahadori F, Turkkan B. Cytotoxic, genotoxic and apoptotic effects of naringenin-oxime relative to naringenin on normal and cancer cell lines. Asian Pac J Trop Biomed. 2016, 6, 872–880. [Google Scholar] [CrossRef]
- Kozlowska J, Grela E, Baczynska D, Grabowiecka A, Aniol M. Novel O-alkyl derivatives of naringenin and their oximes with antimicrobial and anticancer activity. Molecules. 2019, 24, 679. [Google Scholar] [CrossRef]
- Latif AD, Gonda T, Vagvolgyi M, Kusz N, Kulmany A, Ocsovszki I, et al. Synthesis and in vitro antitumor activity of naringenin oxime and oxime ether derivatives. Int J Mol Sci. 2019, 20, 2184. [Google Scholar] [CrossRef]
- Metodiewa D, Kochman A, Koceva-Chyla A. Anticancer potential of N,N-diethylaminoethyl ethers of flavanone oximes: a comparison with mitoxantrone action on rat Yoshida sarcoma cells in vivo. Anticancer Res. 1999, 19, 1249–54.
- Gutam M, Mokenapelli S, Yerrabelli JR, Banerjee S, Roy P, Chitneni PR. Synthesis and cytotoxicity of novel (E)-2-phenylchroman-4-one-O-((1-substituted-1H-1,2,3-triazol-4-yl)methyl)oxime derivatives. Synth Commun. 2020, 50, 1883–1891. [Google Scholar] [CrossRef]
- Wang Y-T, Qin Y-J, Zhang Y-L, Li Y-J, Rao B, Zhang Y-Q, et al. Synthesis, biological evaluation, and molecular docking studies of novel chalcone oxime derivatives as potential tubulin polymerization inhibitors. RSC Adv. 2014, 4, 32263–32275. [Google Scholar] [CrossRef]
- Bukhari SNA. Synthesis and evaluation of new chalcones and oximes as anticancer agents. RSC Adv. 2022, 12, 10307–10320. [Google Scholar] [CrossRef] [PubMed]
- Chen Y-W, Chen Y-L, Tseng C-H, Liang C-C, Yang C-N, Yao Y-C, et al. Discovery of 4-anilinofuro[2,3-b]quinoline derivatives as selective and orally active compounds against non-small-cell lung cancers. J Med Chem. 2011, 54, 4446–4461. [Google Scholar] [CrossRef] [PubMed]
- Chen IL, Chang K-M, Miaw C-L, Liao C-H, Chen J-J, Wang T-C. Synthesis, antiproliferative, and antiplatelet activities of oxime- and amide-containing quinolin-2(1H)-one derivatives. Bioorg Med Chem. 2007, 15, 6527–6534. [Google Scholar] [CrossRef] [PubMed]
- Qin H-L, Leng J, Youssif BGM, Amjad MW, Raja MAG, Hussain MA, et al. Synthesis and mechanistic studies of curcumin analog-based oximes as potential anticancer agents. Chem Biol Drug Des. 2017, 90, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Qin H-L, Leng J, Zhang C-P, Jantan I, Amjad MW, Sher M, et al. Synthesis of α,β-unsaturated carbonyl-based compounds, oxime and oxime ether analogs as potential anticancer agents for overcoming cancer multidrug resistance by modulation of efflux pumps in tumor cells. J Med Chem. 2016, 59, 3549–3561. [Google Scholar] [CrossRef]
- Zha G-F, Qin H-L, Youssif BGM, Amjad MW, Raja MAG, Abdelazeem AH, et al. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur J Med Chem. 2017, 135, 34–48. [Google Scholar] [CrossRef]
- Roayapalley PK, Dimmock JR, Contreras L, Balderrama KS, Aguilera RJ, Sakagami H, et al. Design, synthesis and tumour-selective toxicity of novel 1-[3-{3,5-Bis(benzylidene)-4-oxo-1-piperidino}-3-oxopropyl]-4-piperidone oximes and related quaternary ammonium salts. Molecules. 2021, 26, 7132. [Google Scholar] [CrossRef]
- Petersen AB, Konotop G, Hanafiah NHM, Hammershoej P, Raab MS, Kramer A, et al. Strategies for improving the solubility and metabolic stability of griseofulvin analogues. Eur J Med Chem. 2016, 116, 210–215. [Google Scholar] [CrossRef]
- Dileep Kumar G, Siva B, Bharathi K, Devi A, Pavan Kumar P, Anusha K, et al. Synthesis and biological evaluation of Schizandrin derivatives as tubulin polymerization inhibitors. Bioorg Med Chem Lett. 2020, 30, 127354. [Google Scholar] [CrossRef]
- Stancato LF, Silverstein AM, Owens-Grillo JK, Chow YH, Jove R, Pratt WB. The hsp90-binding antibiotic geldanamycin decreases Raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J Biol Chem. 1997, 272, 4013–4020. [Google Scholar] [CrossRef]
- Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998, 16, 2639–2645. [Google Scholar] [CrossRef]
- Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, et al. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones. 1998, 3, 100–108. [Google Scholar] [CrossRef]
- Soga S, Neckers LM, Schulte TW, Shiotsu Y, Akasaka K, Narumi H, et al. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 1999, 59, 2931–2938. [Google Scholar]
- Soga S, Sharma SV, Shiotsu Y, Shimizu M, Tahara H, Yamaguchi K, et al. Stereospecific antitumor activity of radicicol oxime derivatives. Cancer Chemother Pharmacol. 2001, 48, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Agatsuma T, Ogawa H, Akasaka K, Asai A, Yamashita Y, Mizukami T, et al. Halohydrin and oxime derivatives of radicicol: synthesis and antitumor activities. Bioorg Med Chem. 2002, 10, 3445–3454. [Google Scholar] [CrossRef] [PubMed]
- Ikuina Y, Amishiro N, Miyata M, Narumi H, Ogawa H, Akiyama T, et al. Synthesis and antitumor activity of novel O-carbamoylmethyloxime derivatives of radicicol. J Med Chem. 2003, 46, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti B, Akhtar T, Rai B, Yadav M, Akhtar Siddiqui J, Dhar Dwivedi SK, et al. Thioaryl naphthylmethanone oxime ether analogs as novel anticancer agents. J Med Chem. 2014, 57, 8010–8025. [Google Scholar] [CrossRef] [PubMed]
- Martinkovich S, Shah D, Planey SL, Arnott JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interventions Aging. 2014, 9, 1437. [Google Scholar] [CrossRef]
- Sfogliarini C, Pepe G, Cesta CM, Allegretti M, Locati M, Vegeto E. The immune activity of selective estrogen receptor modulators is gene and macrophage subtype-specific yet converges on Il1b downregulation. Biomed Pharmacother. 2023, 165, 115008. [Google Scholar] [CrossRef]
- Tienforti D, Castellini C, Di Giulio F, Totaro M, Dalmazio G, Spagnolo L, et al. Selective modulation of estrogen receptor in obese men with androgen deficiency: A systematic review and meta-analysis. Andrology. 2023, 11, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O. Estrogens and estrogen receptor modulators in cancer research and therapy. Cancers 2023, 15. [Google Scholar] [CrossRef]
- Kim T, Kim H-I, An J-Y, Lee J, Lee N-R, Heo J, et al. Identification of novel estrogen receptor (ER) agonists that have additional and complementary anti-cancer activities via ER-independent mechanism. Bioorg Med Chem Lett. 2016, 26, 1844–1848. [Google Scholar] [CrossRef] [PubMed]
- Kim H-I, Kim T, Kim J-E, Lee J, Heo J, Lee N-R, et al. NJK14013, a novel synthetic estrogen receptor-α agonist, exhibits estrogen receptor-independent, tumor cell-specific cytotoxicity. Int J Oncol. 2015, 47, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Zuo D, Pang L, Shen J, Guan Q, Bai Z, Zhang H, et al. 5-(Furan-2-yl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiol-3- one oxime (6f), a new synthetic compound, causes human fibrosarcoma HT-1080 cell apoptosis by disrupting tubulin polymerisation and inducing G2/M arrest. Int J Oncol. 2017, 50, 2069–2078. [Google Scholar] [CrossRef]
- Liu Y, Li Y, Wang Y, Lin C, Zhang D, Chen J, et al. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J Hematol Oncol. 2022, 15, 89. [Google Scholar] [CrossRef]
- Gao H, Su P, Shi Y, Shen X, Zhang Y, Dong J, et al. Discovery of novel VEGFR-2 inhibitors. Part II: Biphenyl urea incorporated with salicylaldoxime. Eur J Med Chem. 2015, 90, 232–240. [Google Scholar] [CrossRef]
- Jo A, Kwak J-H, Woo S-Y, Kim B-Y, Son Y, Choi H-S, et al. Oxime derivative TFOBO promotes cell death by modulating reactive oxygen species and regulating NADPH oxidase activity in myeloid leukemia. Sci Rep. [CrossRef]
- Liu S, Li Y, Wei W, Wei J. Synthesis and biological evaluation of phenylpropanoid derivatives. Med Chem Res. 2016, 25, 1074–1086. [Google Scholar] [CrossRef]
- Wipf P, Reeves JT, Balachandran R, Day BW. Synthesis and biological evaluation of structurally highly modified analogues of the antimitotic natural product curacin A. J Med Chem. 2002, 45, 1901–1917. [Google Scholar] [CrossRef] [PubMed]
- Nikitjuka A, Shestakova I, Romanchikova N, Jirgensons A. Synthesis and biological evaluation of aziridin-1-yl oxime-based vorinostat analogs as anticancer agents. Chem Heterocycl Compd (N Y, NY, U S). 2015, 51, 647–657. [Google Scholar] [CrossRef]
- Baud MGJ, Leiser T, Haus P, Samlal S, Wong AC, Wood RJ, et al. Defining the mechanism of action and enzymatic selectivity of Psammaplin A against its epigenetic targets. J Med Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Bhatti HN, Khera RA. Biological transformations of steroidal compounds: A review. Steroids. 2012, 77, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Rasheed A, Qasim M. A review of natural steroids and their applications. Int J Pharm Sci Res. 2013, 4, 520–531. [Google Scholar]
- Gupta A, Sathish Kumar B, Negi AS. Current status on development of steroids as anticancer agents. J Steroid Biochem Mol Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Bansal R, Suryan A. A comprehensive review on steroidal bioconjugates as promising leads in drug discovery. ACS Bio Med Chem Au. 2022, 2, 340–369. [Google Scholar] [CrossRef]
- Canario C, Matias M, Brito V, Santos AO, Falcao A, Silvestre S, et al. New estrone oxime derivatives: Synthesis, cytotoxic evaluation and docking studies. Molecules. 2021, 26, 2687. [Google Scholar] [CrossRef]
- Jindal DP, Chattopadhaya R, Guleria S, Gupta R. Synthesis and antineoplastic activity of 2-alkylaminoethyl derivatives of various steroidal oximes. Eur J Med Chem. 2003, 38, 1025–1034. [CrossRef]
- Rodriguez J, Nunez L, Peixinho S, Jimenez C. Isolation and synthesis of the first natural 6-hydroximino-4-en-3-one- steroids from the sponges Cinachyrella spp. Tetrahedron Lett. 1997, 38, 1833–1836. [Google Scholar] [CrossRef]
- Deive N, Rodriguez J, Jimenez C. Synthesis of cytotoxic 6E-hydroximino-4-ene steroids: structure/activity studies. J Med Chem. 2001, 44, 2612–2618. [Google Scholar] [CrossRef]
- Cui J, Huang L, Fan L, Zhou A. A facile and efficient synthesis of some (6E)-hydroximino-4-en-3-one steroids, steroidal oximes from Cinachyrella spp. sponges. Steroids. 2008, 73, 252–256. [Google Scholar] [CrossRef]
- Berenyi A, Minorics R, Ivanyi Z, Ocsovszki I, Ducza E, Thole H, et al. Synthesis and investigation of the anticancer effects of estrone-16-oxime ethers in vitro. Steroids. 2013, 78, 69–78. [Google Scholar] [CrossRef]
- Acharya PC, Bansal R. Synthesis and antiproliferative activity of some androstene oximes and their O-alkylated derivatives. Arch Pharm (Weinheim, Ger). 2014, 347, 193–199. [Google Scholar] [CrossRef]
- Mernyak E, Fiser G, Szabo J, Bodnar B, Schneider G, Kovacs I, et al. Synthesis and in vitro antiproliferative evaluation of D-secooxime derivatives of 13β- and 13α-estrone. Steroids. 2014, 89, 47–55. [Google Scholar] [CrossRef]
- Chen S-R, Shen F-J, Feng G-L, Yuan R-X. Synthesis and anticancer activity of 4-azasteroidal-20-oxime derivatives. J Chem Res. 2015, 39, 527–530. [Google Scholar] [CrossRef]
- Ajdukovic JJ, Jakimov DS, Rarova L, Strnad M, Dzichenka YU, Usanov S, et al. Novel alkylaminoethyl derivatives of androstane 3-oximes as anticancer candidates: synthesis and evaluation of cytotoxic effects. RSC Adv. 2021, 11, 37449–37461. [Google Scholar] [CrossRef] [PubMed]
- Kolsi LE, Leal AS, Yli-Kauhaluoma J, Liby KT, Moreira VM. Dehydroabietic oximes halt pancreatic cancer cell growth in the G1 phase through induction of p27 and downregulation of cyclin D1. Sci Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Hou Q, He C, Lao K, Luo G, You Q, Xiang H. Design and synthesis of novel steroidal imidazoles as dual inhibitors of AR/CYP17 for the treatment of prostate cancer. Steroids. 2019, 150, 108384. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuzniak V, Bednarczyk-Cwynar B, Paluszczak J, Szaefer H, Narozna M, Zaprutko L, et al. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HepG2 hepatoma cells. Bioorg Chem. 2019, 93, 103326. [Google Scholar] [CrossRef]
- D'Yakonov VA, Tuktarova RA, Dzhemileva LU, Ishmukhametova SR, Yunusbaeva MM, Dzhemilev UM. Catalytic cyclometallation in steroid chemistry V: Synthesis of hybrid molecules based on steroid oximes and (5Z,9Z)-tetradeca-5,9-dienedioic acid as potential anticancer agents. Steroids. 2018, 138, 14–20. [Google Scholar] [CrossRef]
- Acharya PC, Bansal R. Synthesis of androstene oxime-nitrogen mustard bioconjugates as potent antineoplastic agents. Steroids. 2017, 123, 73–83. [Google Scholar] [CrossRef]
- Bu M, Cao T, Li H, Guo M, Yang BB, Zeng C, et al. Synthesis and biological evaluation of novel steroidal 5α,8α-epidioxyandrost-6-ene-3β-ol-17-(O-phenylacetamide)oxime derivatives as potential anticancer agents. Bioorg Med Chem Lett. 2017, 27, 3856–3861. [Google Scholar] [CrossRef]
- Pavlickova V, Jurasek M, Rimpelova S, Zaruba K, Sedlak D, Simkova M, et al. Oxime-based 19-nortestosterone-pheophorbide a conjugate: bimodal controlled release concept for PDT. J Mater Chem B. 2019, 7, 5465–5477. [Google Scholar] [CrossRef]
- Alderden RA, Hall MD, Hambley TW. The discovery and development of cisplatin. J Chem Educ. 2006, 83, 728–734. [Google Scholar] [CrossRef]
- 248. Kelland LR, Sharp SY, O'Neill CF, Raynaud FI, Beale PJ, Judson IR. Discovery and development of platinum complexes designed to circumvent cisplatin resistance. J Inorg Biochem. 1999, 77, 111–115. [CrossRef]
- Correa-Morales JE, Giraldo-Moreno S, Mantilla-Manosalva N, Cuellar-Valencia L, Borja-Montes OF, Bedoya-Muñoz LJ, et al. Prevention and treatment of cisplatin-induced ototoxicity in adults: A systematic review. Clin Otolaryngol. 2023. [CrossRef]
- Chattaraj A, Syed MP, Low CA, Owonikoko TK. Cisplatin-Induced Ototoxicity: A concise review of the burden, prevention, and interception strategies. JCO Oncol Pract. 2023, 19, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Sharon Y, Motiei M, Tzror-Azankot C, Sadan T, Popovtzer R, Rosenbaum E. A ′golden′ alternative for prevention of cisplatin nephrotoxicity in bladder cancer. Cancer Nanotechnol. 2023, 14, 72. [Google Scholar] [CrossRef]
- Kukushkin VY, Belsky VK, Tudela D. Unusual reactivity mode of coordinated oximes: platinum(IV)-assisted ring closure by reaction with acetone. Inorg Chem. 1996, 35, 510. [Google Scholar] [CrossRef] [PubMed]
- Kukushkin VY, Izotova YA, Tudela D, Guedes da Silva MFC, Pombeiro AJL. Organometallic and coordination complexes. Platinum(II) complexes of propanone oxime. Inorg Synth. 2004, 34, 81–85. [Google Scholar]
- Kukushkin VY, Nishioka T, Tudela D, Isobe K, Kinoshita I. Hydrogen-bonding patterns in oxime/oximato platinum(II) species providing the formation of one-dimensional chains, two-dimensional networks, and cages. Inorg Chem. 1997, 36, 6157–6165. [Google Scholar] [CrossRef]
- Scaffidi-Domianello YY, Legin AA, Jakupec MA, Arion VB, Kukushkin VY, Galanski MS, et al. Synthesis, characterization, and cytotoxic activity of novel potentially pH-Sensitive nonclassical platinum(II) complexes featuring 1,3-dihydroxyacetone oxime ligands. Inorg Chem. 2011, 50, 10673–10681. [Google Scholar] [CrossRef]
- Scaffidi-Domianello YY, Legin AA, Jakupec MA, Roller A, Kukushkin VY, Galanski MS, et al. Novel oximato-bridged platinum(II) di- and trimer(s): synthetic, structural, and in vitro anticancer activity studies. Inorg Chem. 2012, 51, 7153–7163. [Google Scholar] [CrossRef]
- Bartel C, Bytzek AK, Scaffidi-Domianello YY, Grabmann G, Jakupec MA, Hartinger CG, et al. Cellular accumulation and DNA interaction studies of cytotoxic trans-platinum anticancer compounds. J Biol Inorg Chem. 2012, 17, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Hyeraci M, Colalillo M, Labella L, Marchetti F, Samaritani S, Scalcon V, et al. Platinum(II) complexes bearing triphenylphosphine and chelating oximes: antiproliferative effect and biological profile in resistant cells. ChemMedChem. 2020, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Eddings D, Barnes C, Gerasimchuk N, Durham P, Domasevich K. First bivalent palladium and platinum cyanoximates: synthesis, characterization, and biological activity. Inorg Chem. 2004, 43, 3894–3909. [Google Scholar] [CrossRef]
- Sharma S, Chamola G. Systematic review of organometallic half-sandwich complexes: medicinal application and biological evaluation of anticancer activity. AIP Conf Proc. 2023, 2735, 030014. [Google Scholar] [CrossRef]
- Zhang J-J, Xu Q-J, Schmidt C, Maaty MAAe, Song J, Yu C, et al. Elucidating the multimodal anticancer mechanism of an organometallic terpyridine platinum(II) N-heterocyclic carbene complex against triple-negative breast cancer in vitro and in vivo. J Med Chem. 2023, 66, 3995–4008. [Google Scholar] [CrossRef] [PubMed]
- Samiee S, Shiralinia A, Hoveizi E, Gable RW. A new family of oxime palladacycles mixed with unsymmetrical phosphorus ylides; synthesis, structural, cytotoxicity and catalytic activity studies. J Organomet Chem. 2019, 900, 120927. [Google Scholar] [CrossRef]
- Samiee S, Shiralinia A, Hoveizi E, Gable RW. Mono- and dinuclear oxime palladacycles bearing diphosphine ligands: An unusual coordination mode for dppe. Appl Organomet Chem. 2019, 33, n. [Google Scholar] [CrossRef]
- Palepu NR, Adhikari S, J RP, Verma AK, Shepherd SL, Phillips RM, et al. Half-sandwich ruthenium, rhodium and iridium complexes featuring oxime ligands: Structural studies and preliminary investigation of in vitro and in vivo anti-tumour activities. Appl Organomet Chem. 2017, 31, n/a. [Google Scholar] [CrossRef]
- Benabdelouahab Y, Munoz-Moreno L, Frik M, de la Cueva-Alique I, El Amrani MA, Contel M, et al. Hydrogen bonding and anticancer properties of water-soluble chiral p-cymene RuII compounds with amino-oxime ligands. Eur J Inorg Chem. 2015, 2015, 2295–2307. [Google Scholar] [CrossRef]
- El-Tabl AS, Abd El-Waheed MM, Shakdofa MME, Abou El-Fadl NA. Synthesis, spectroscopic characterization and antitumor activity of new metal complexes of isonicotinoylhydrazide oxime. Main Group Chem. 2013, 12, 153–168. [Google Scholar] [CrossRef]
- El-Tabl AS, Abd El-Waheed MM, Wahba MA, Abd El-Halim Abou El-Fadl N. Synthesis, characterization, and anticancer activity of new metal complexes derived from 2-hydroxy-3-((hydroxyimino)-4-oxopentan-2-ylidene)benzohydrazide. Bioinorg Chem Appl. 2015, 1–14. [CrossRef]
- Samy F, Shebl M. Synthesis, spectroscopic, biological, and theoretical studies of new complexes from (E)-3-(2-(5, 6-diphenyl-1,2,4-triazin-3-yl)hydrazono)butan-2-one oxime. Appl Organomet Chem. 2020, 34, e5502. [Google Scholar] [CrossRef]
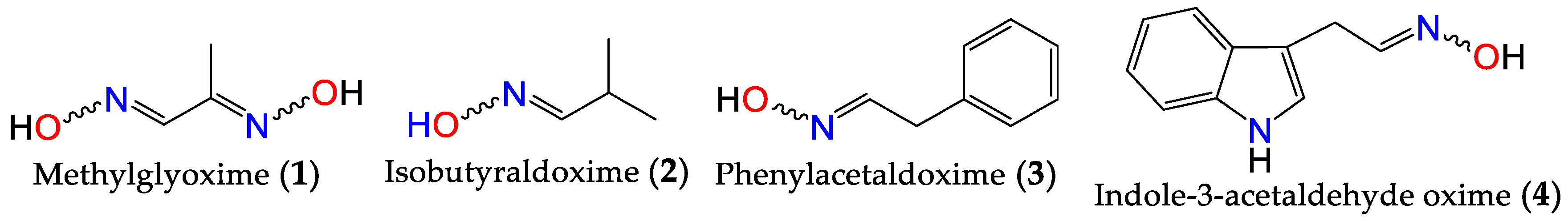
Figure 2.
Illustrative examples of anticancer indirubin-3’-oxime derivatives, bearing a diversity of substitution patterns on the core bis-indole scaffold.
Figure 2.
Illustrative examples of anticancer indirubin-3’-oxime derivatives, bearing a diversity of substitution patterns on the core bis-indole scaffold.
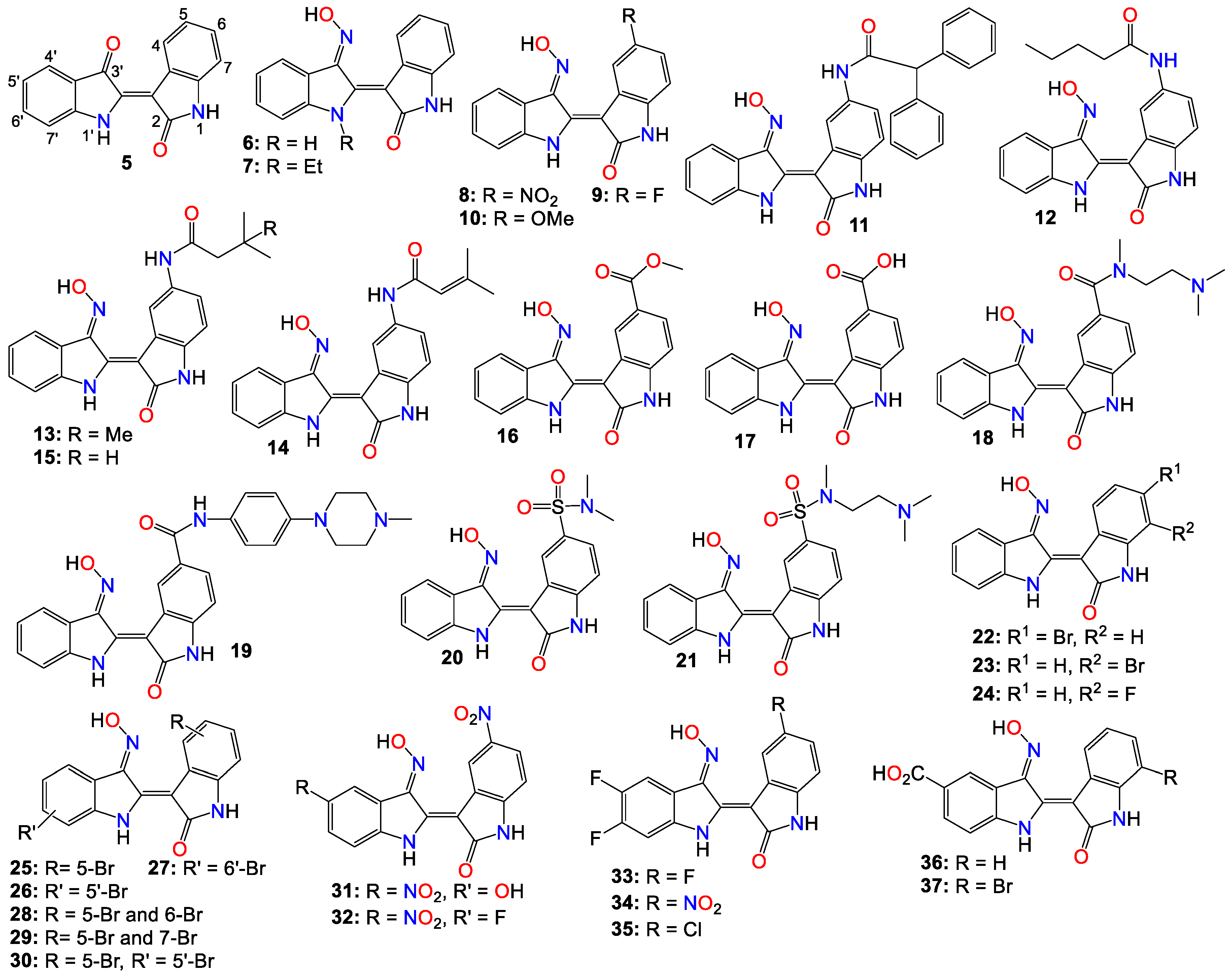
Figure 3.
Crystal structure of the 37-DYRK2 complex revealing a nontypical binding mode, in which the orientation of the indirubin core is in good accordance with the predicted binding mode [94] (https://pubs.acs.org/doi/10.1021/ml300207a). This image is reused with the expressed permission from ACS, and any further permissions related to this material should be directed to ACS.
Figure 3.
Crystal structure of the 37-DYRK2 complex revealing a nontypical binding mode, in which the orientation of the indirubin core is in good accordance with the predicted binding mode [94] (https://pubs.acs.org/doi/10.1021/ml300207a). This image is reused with the expressed permission from ACS, and any further permissions related to this material should be directed to ACS.
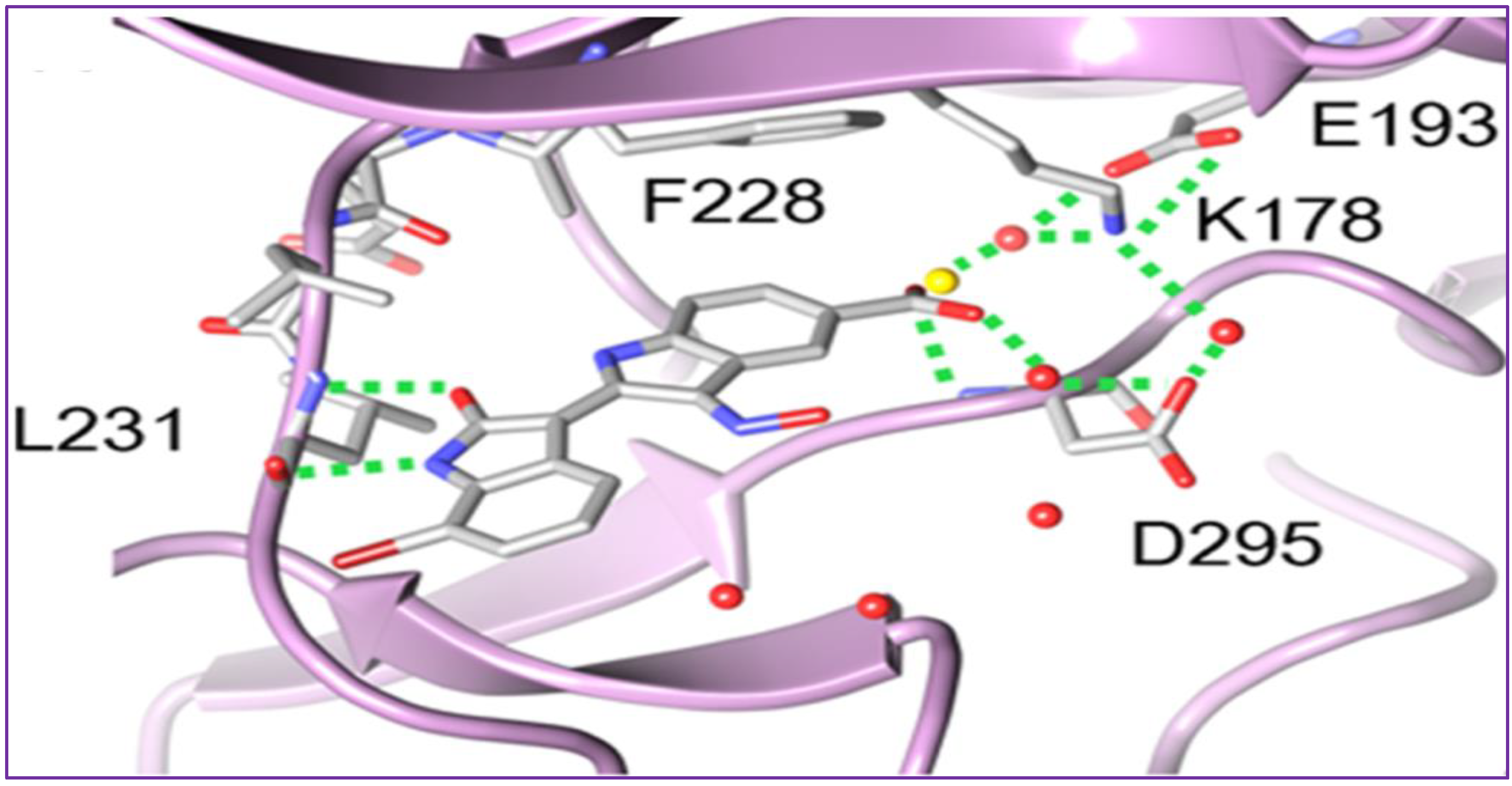
Figure 4.
Illustrative examples of anticancer indirubin-3’-oxime ether derivatives.
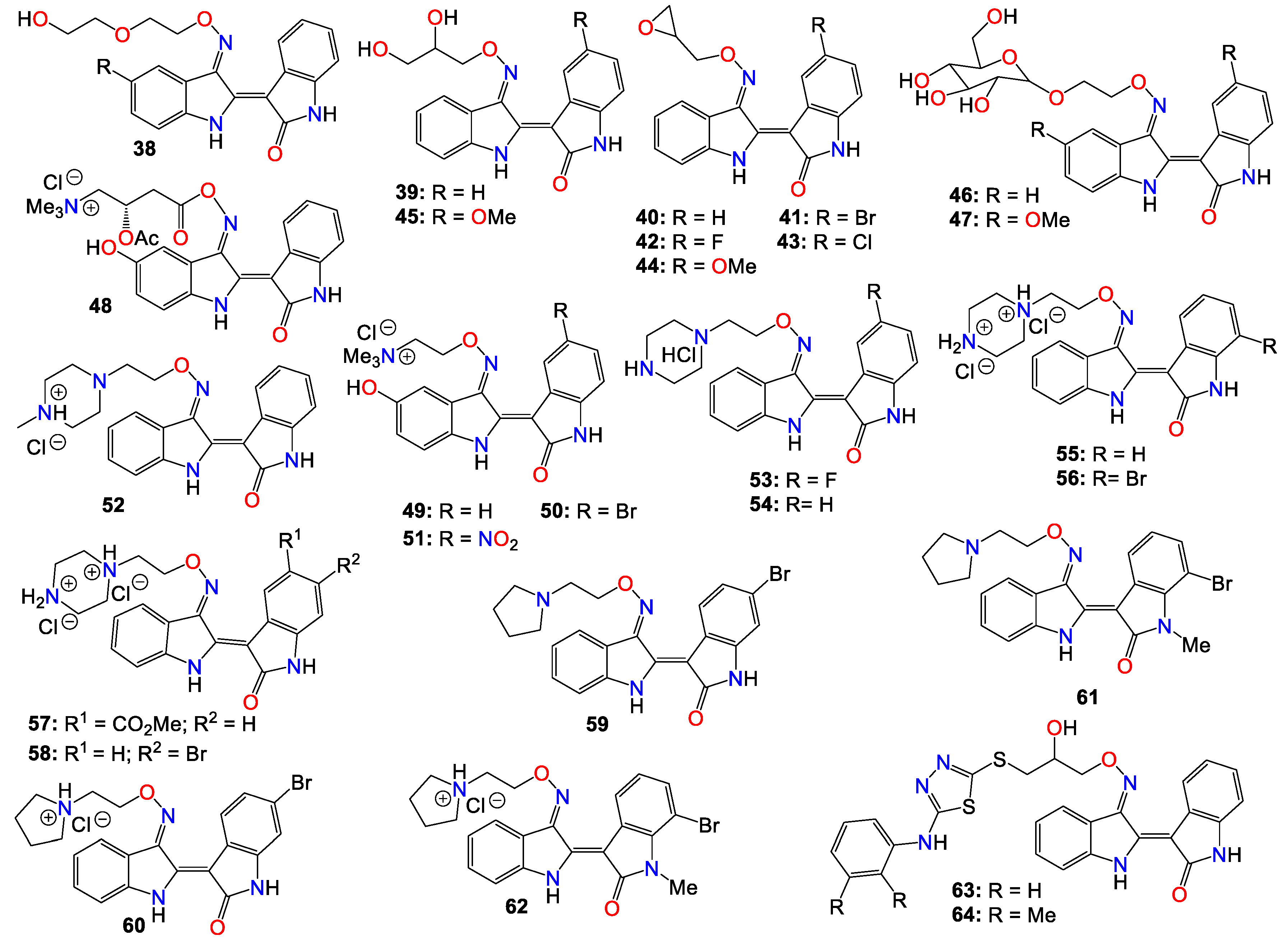
Figure 5.
Illustrative examples of anticancer indirubin-3’-oxime derivative with substituents at N1 of the bisindole core.
Figure 5.
Illustrative examples of anticancer indirubin-3’-oxime derivative with substituents at N1 of the bisindole core.
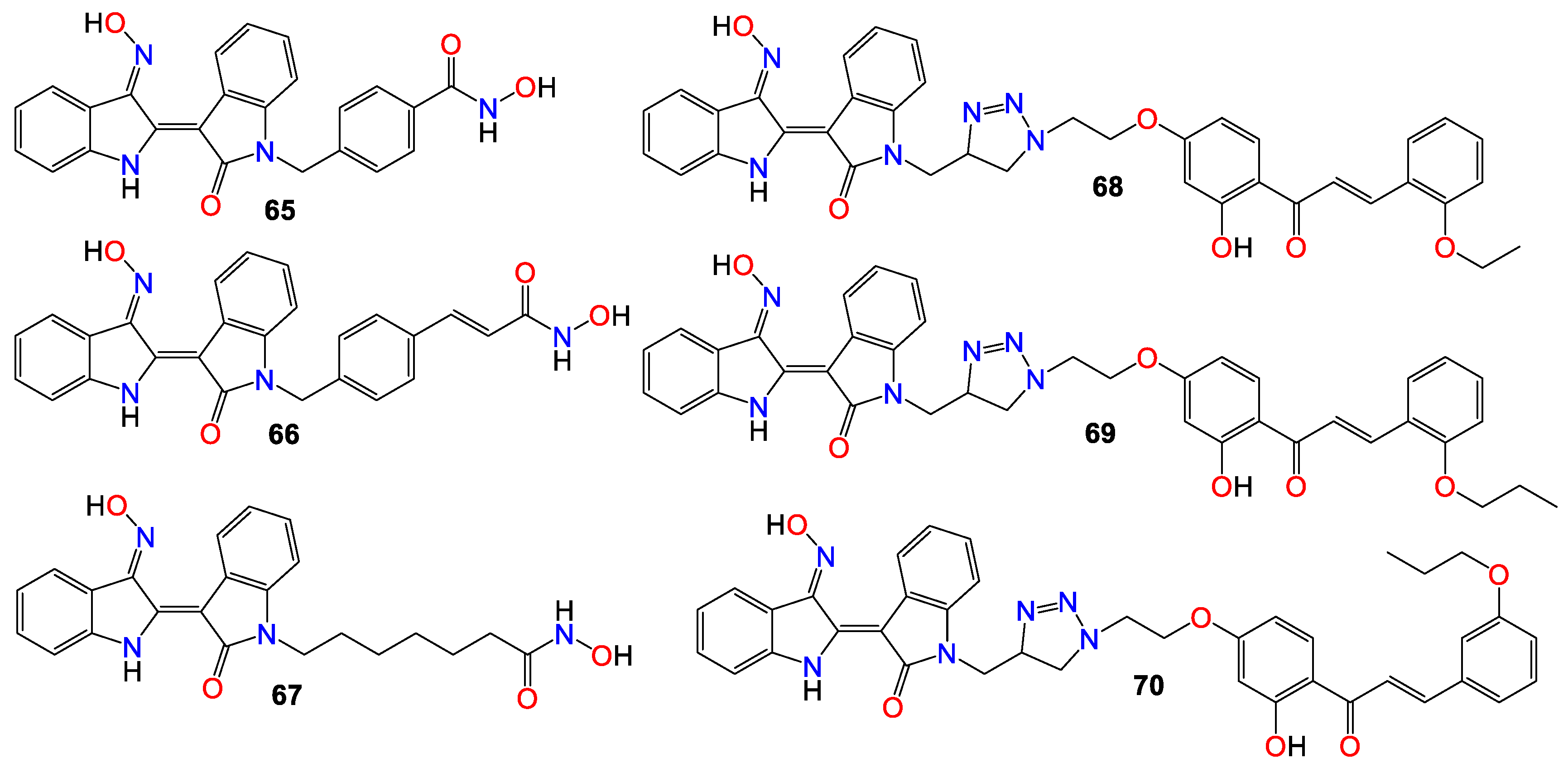
Figure 6.
Illustrative examples of other antitumor indole derivatives bearing an oxime moiety.
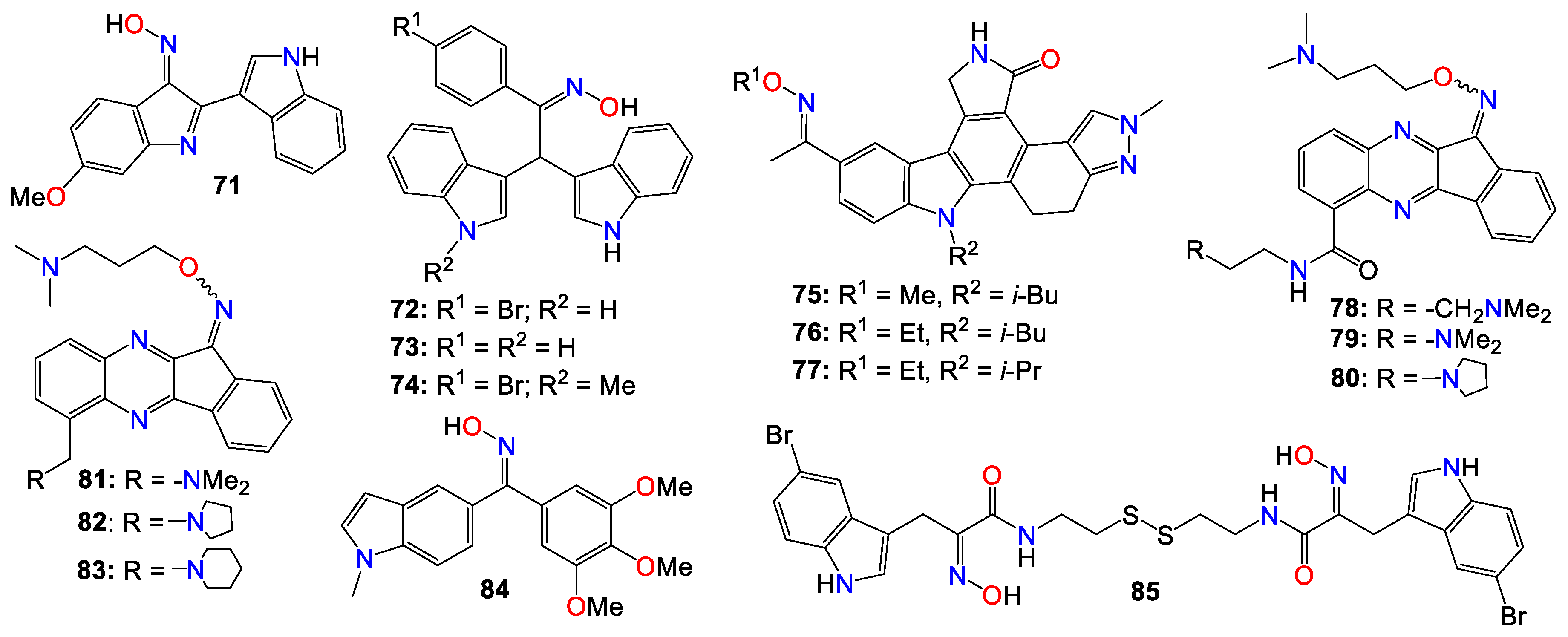
Figure 7.
Illustrative examples of anticancer triazole- and pyrazole-based oximes and oxime ethers.
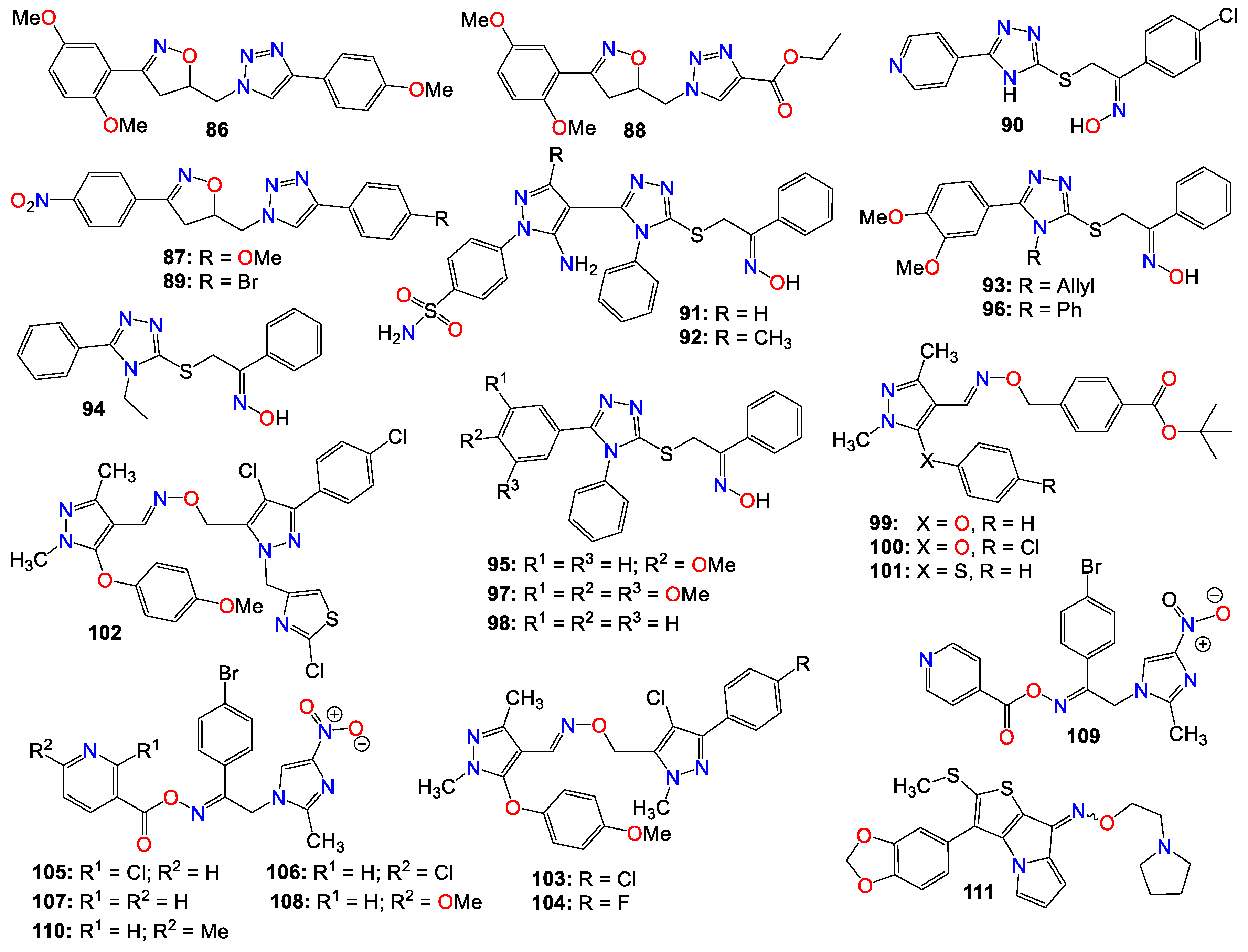
Figure 8.
Illustrative examples of miscellaneous small nitrogen-containing hetorocyclic based oximes with anticancer properties.
Figure 8.
Illustrative examples of miscellaneous small nitrogen-containing hetorocyclic based oximes with anticancer properties.
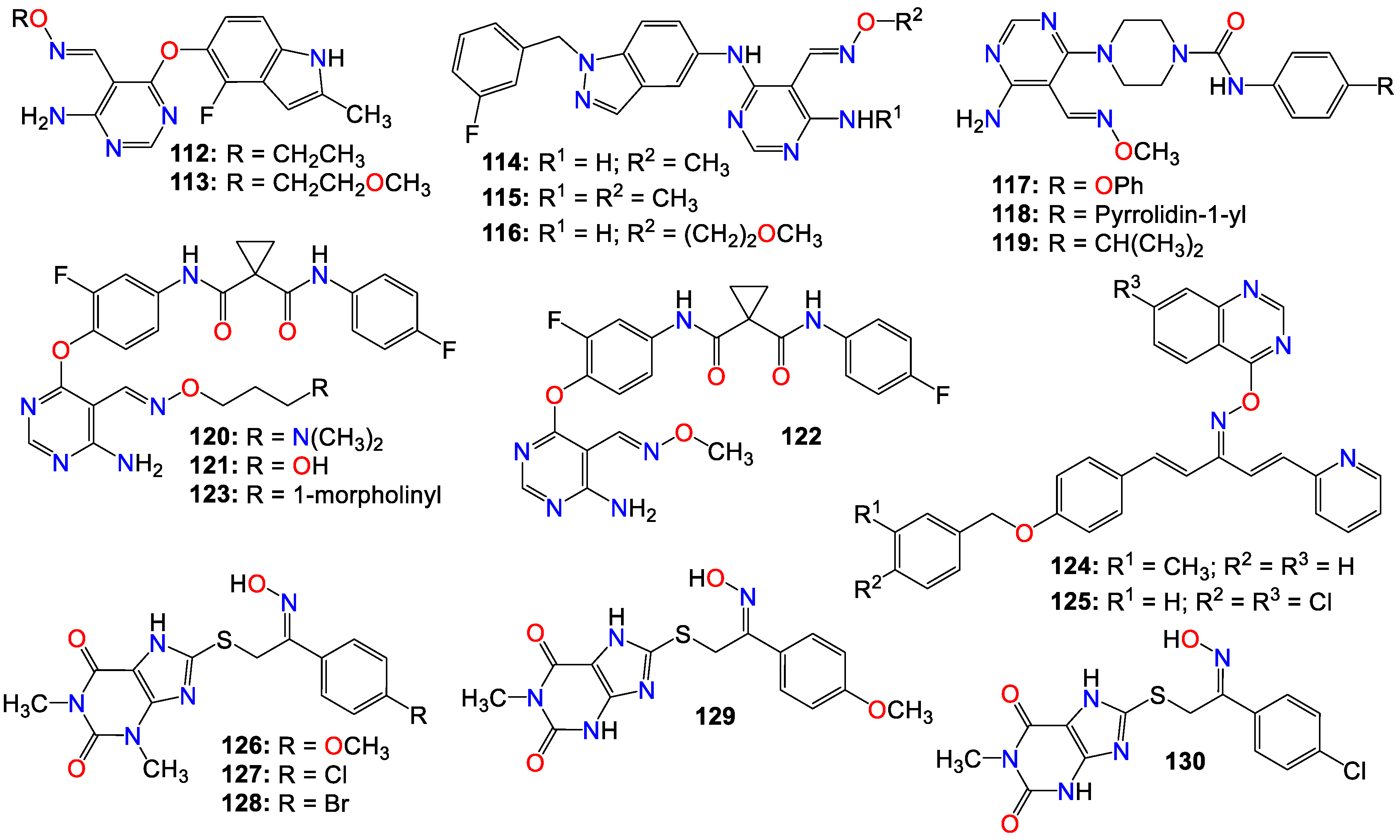
Figure 9.
Illustrative examples of anticancer of quinone-based oximes and oxime ethers.
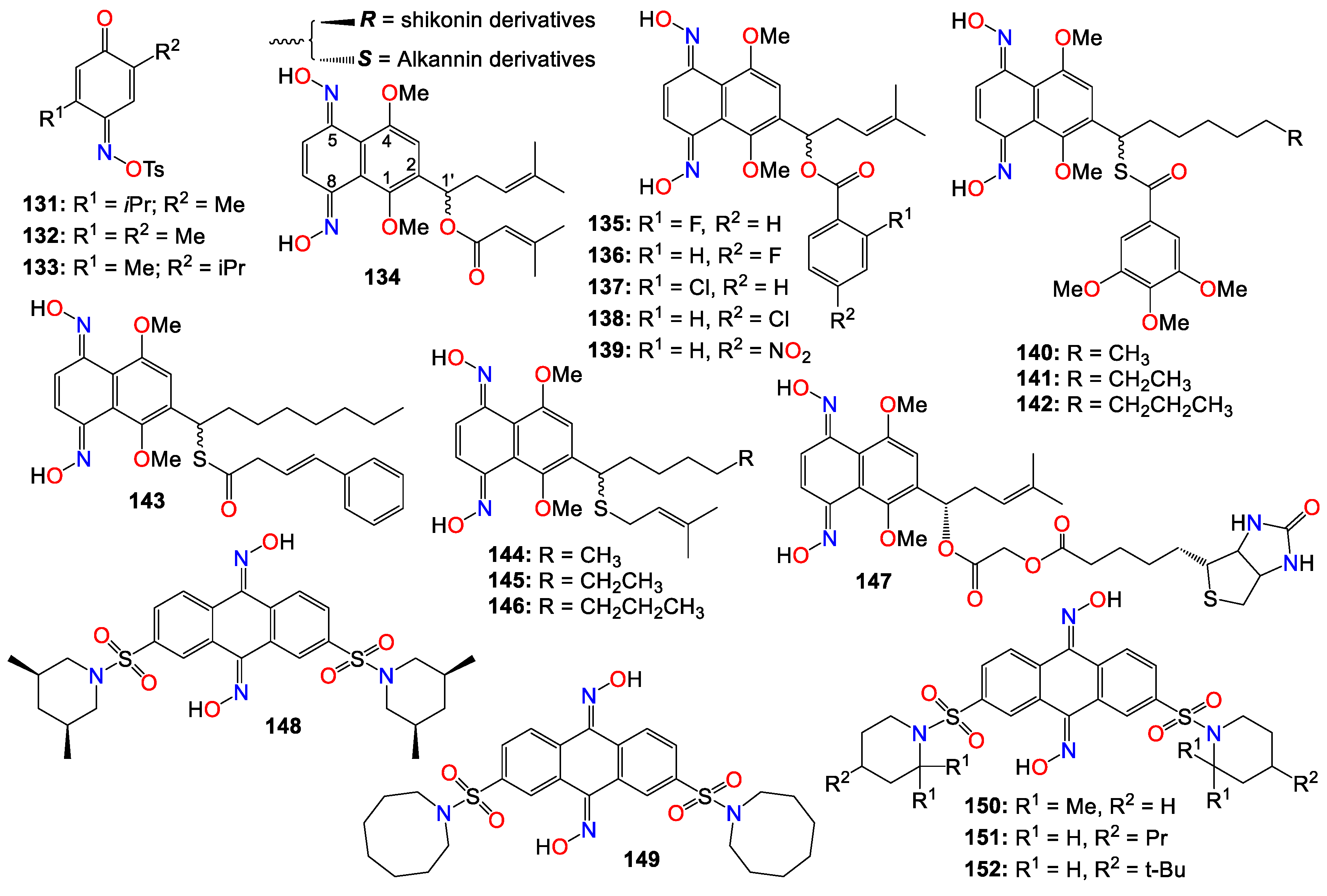
Figure 10.
Illustrative examples of anticancer quinone oxime bond-linked bioconjugates.
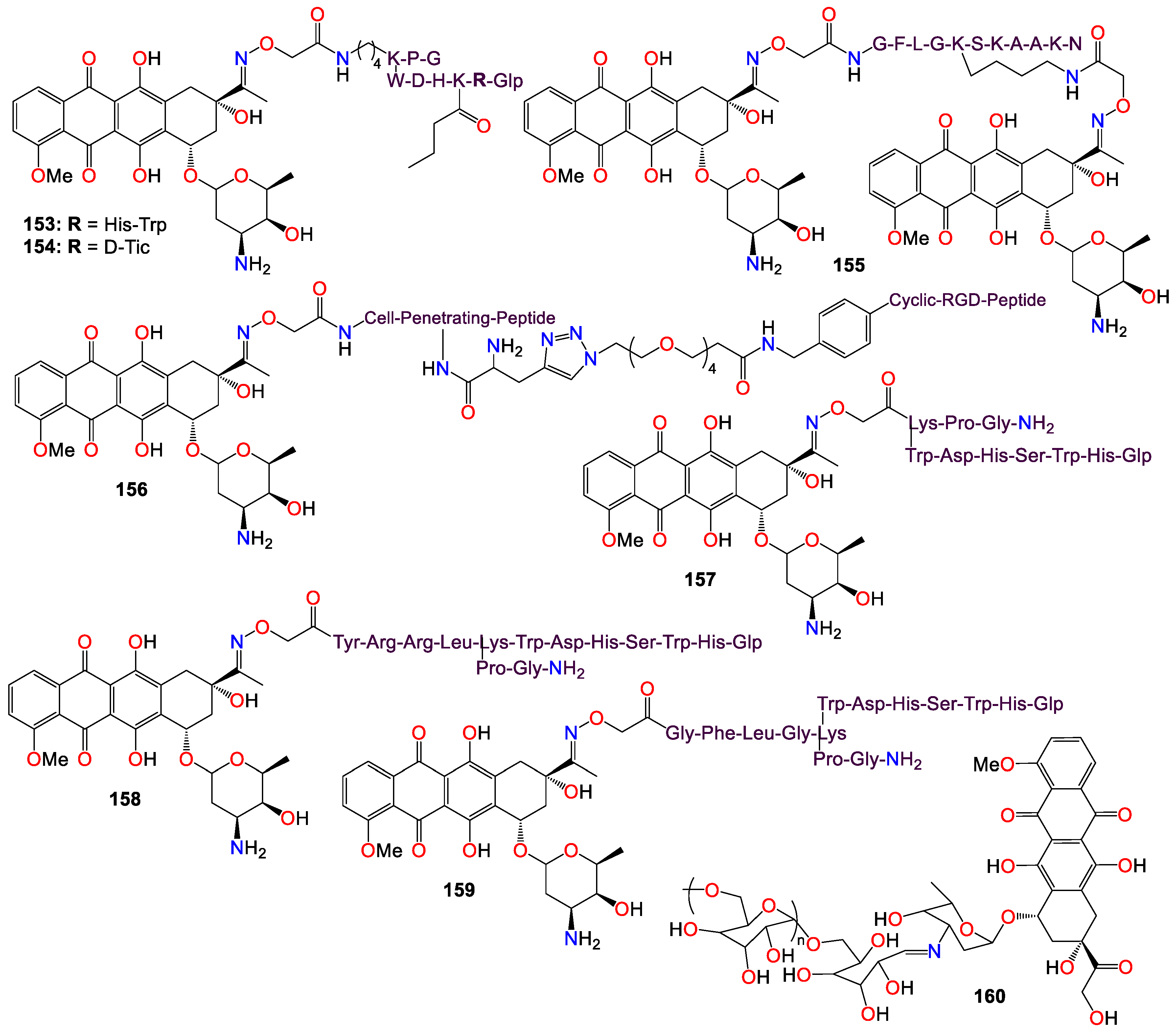
Figure 11.
Illustrative examples of anticancer flavonoids-based oximes and oxime ethers.
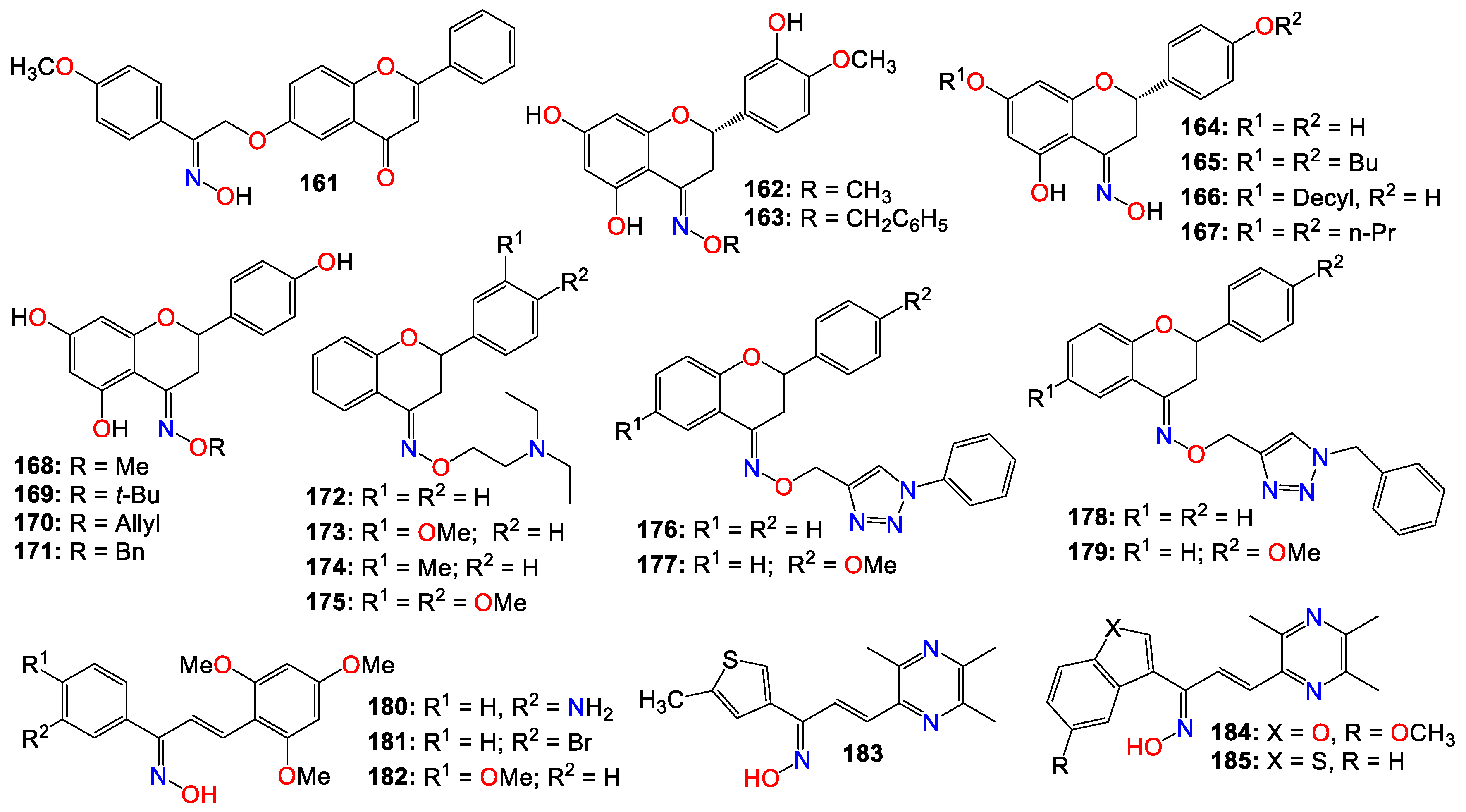
Figure 12.
Illustrative examples of anticancer quinolinone-, cyclohexanone-, piperidin-4-one- and tetralone-based oximes.
Figure 12.
Illustrative examples of anticancer quinolinone-, cyclohexanone-, piperidin-4-one- and tetralone-based oximes.
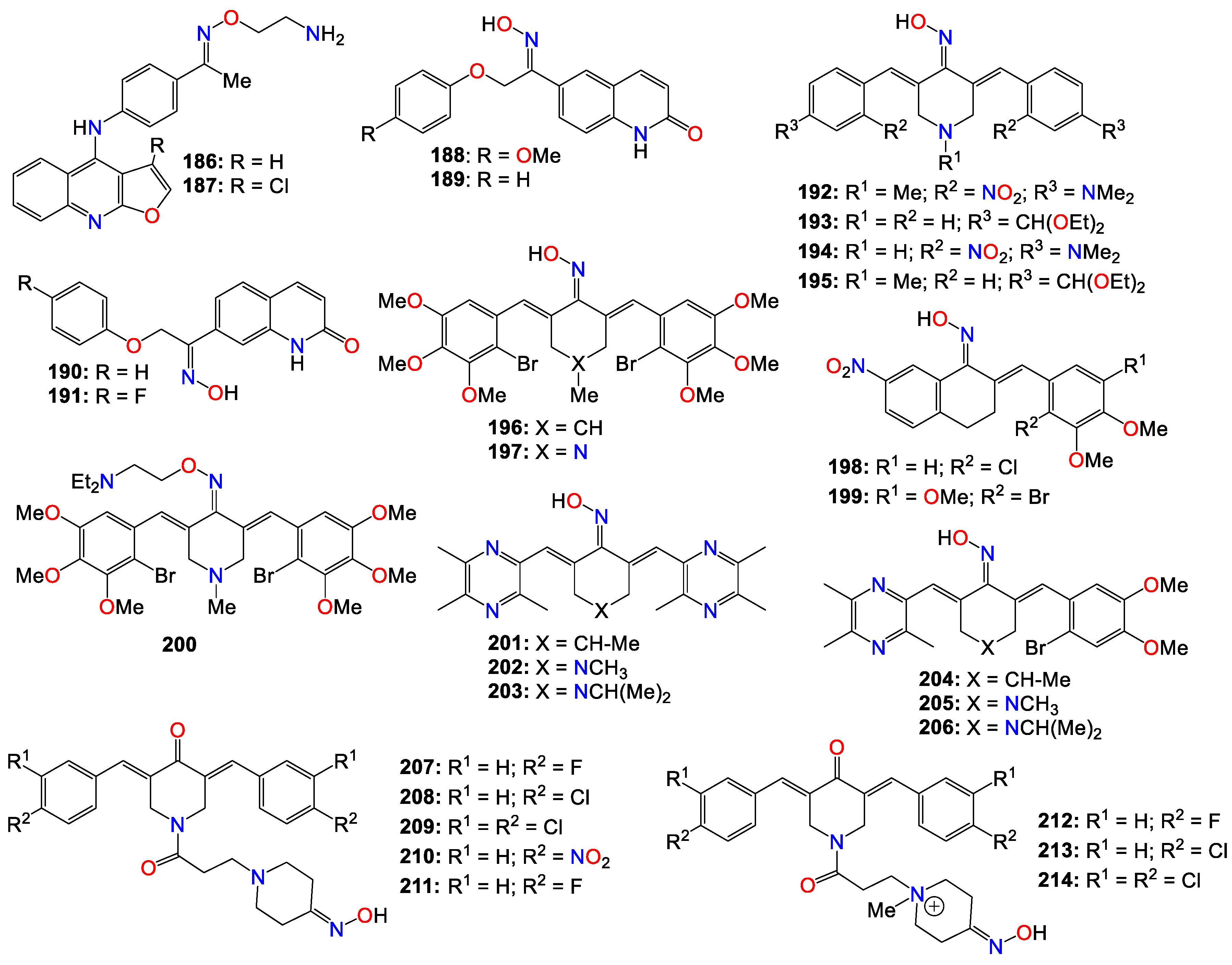
Figure 13.
Illustrative examples of radicicol oxime and oxime ether derivatives with antitumor activities.
Figure 13.
Illustrative examples of radicicol oxime and oxime ether derivatives with antitumor activities.
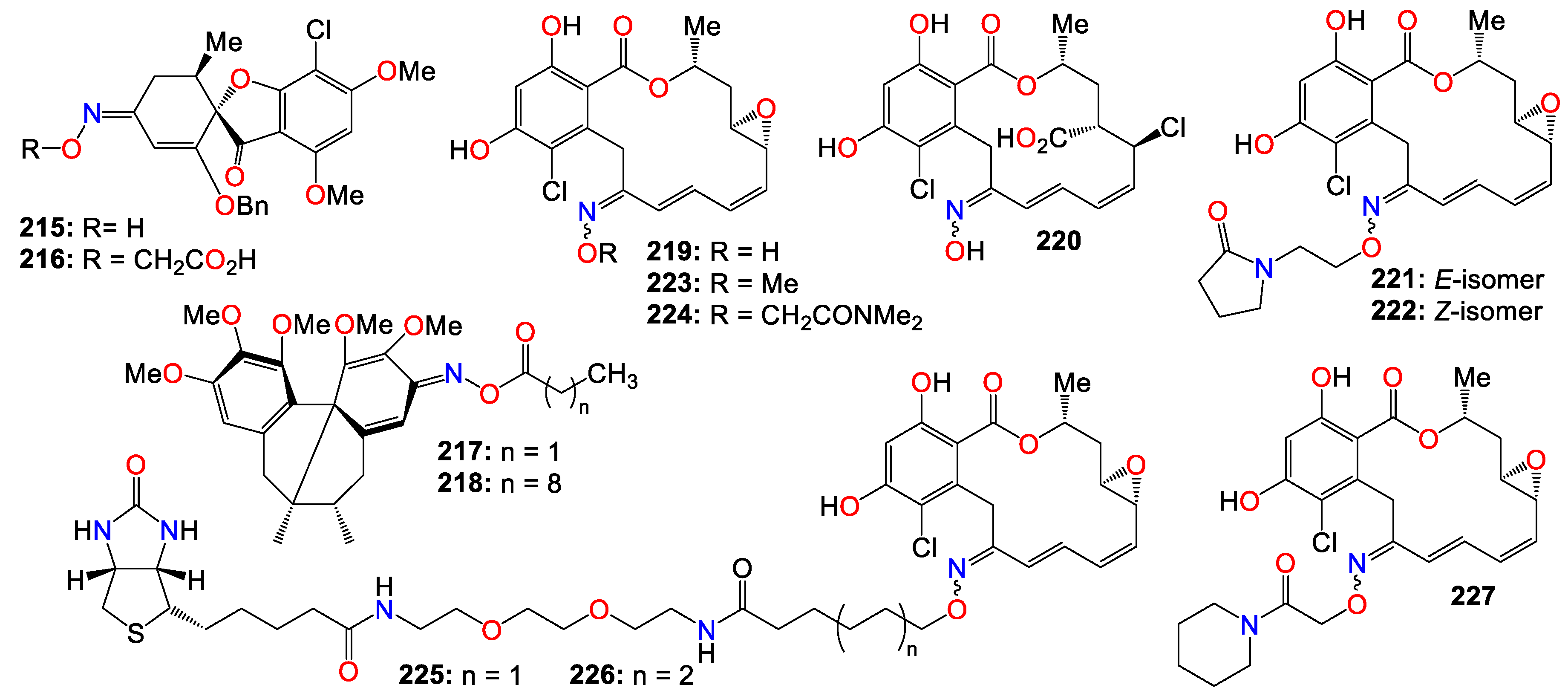
Figure 14.
Illustrative examples of anticancer small aromatic and phenolic-based oximes and oxime ethers.
Figure 14.
Illustrative examples of anticancer small aromatic and phenolic-based oximes and oxime ethers.
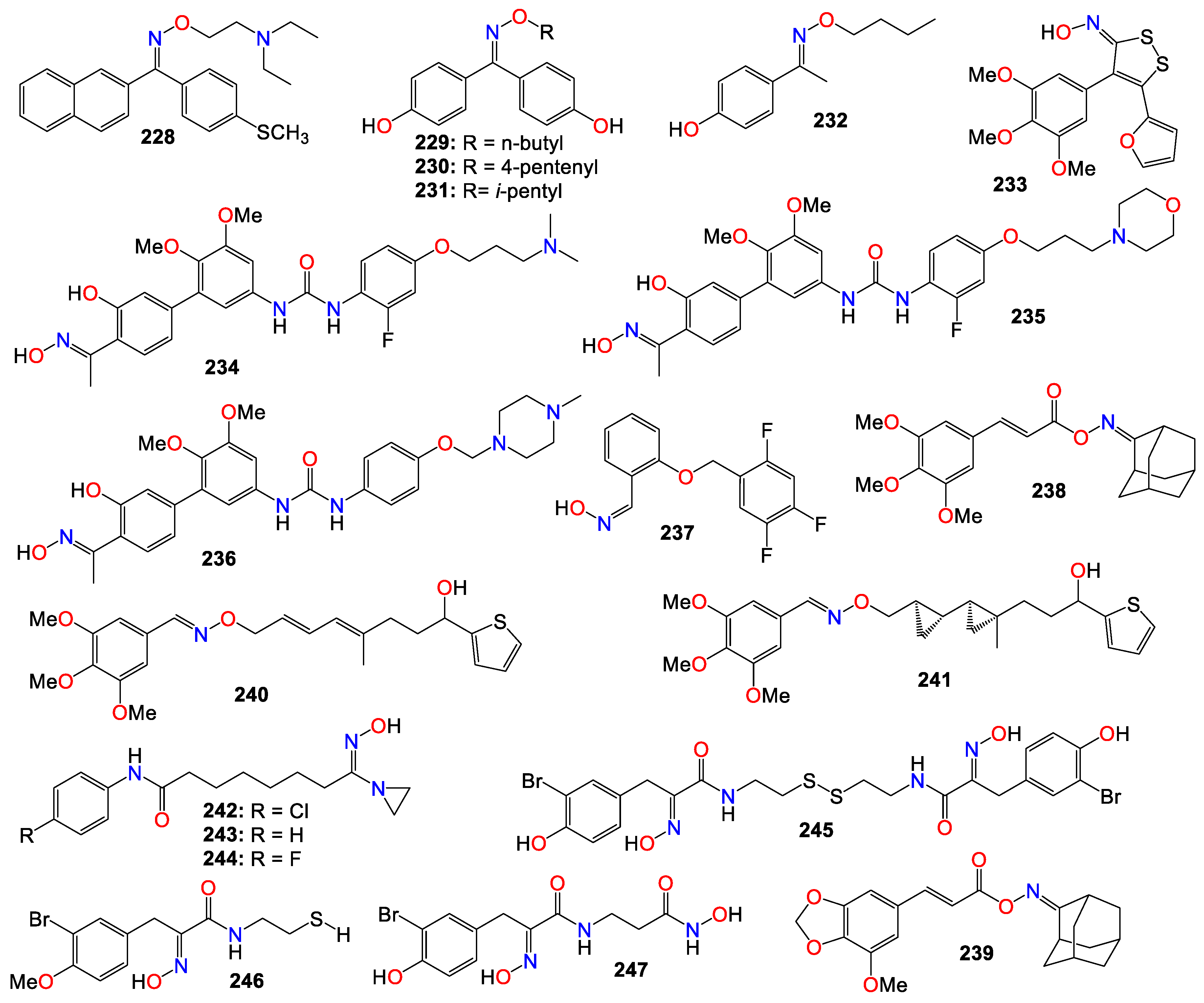
Figure 15.
Illustrative examples of anticancer steroidal oximes and oxime ethers.
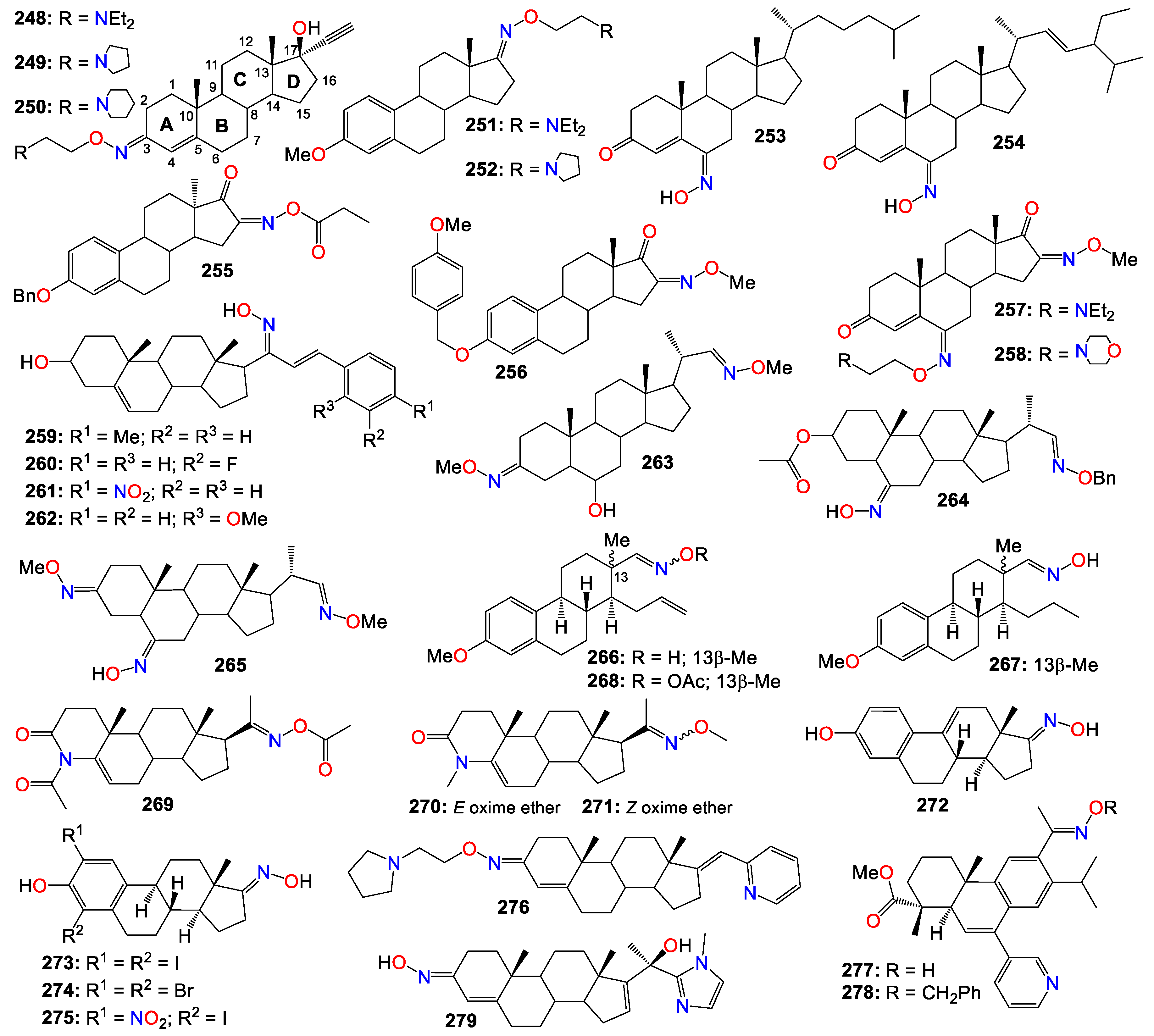
Figure 16.
The binding mode of 279 docked into the active site of CYP17, with the red imaginary line representing hydrogen bond [241] (https://doi.org/10.1016/j.steroids.2019.03.003). This image was reused with the expressed permission from Elsevier, and any further permissions related to this material should be directed to Elsevier.
Figure 16.
The binding mode of 279 docked into the active site of CYP17, with the red imaginary line representing hydrogen bond [241] (https://doi.org/10.1016/j.steroids.2019.03.003). This image was reused with the expressed permission from Elsevier, and any further permissions related to this material should be directed to Elsevier.
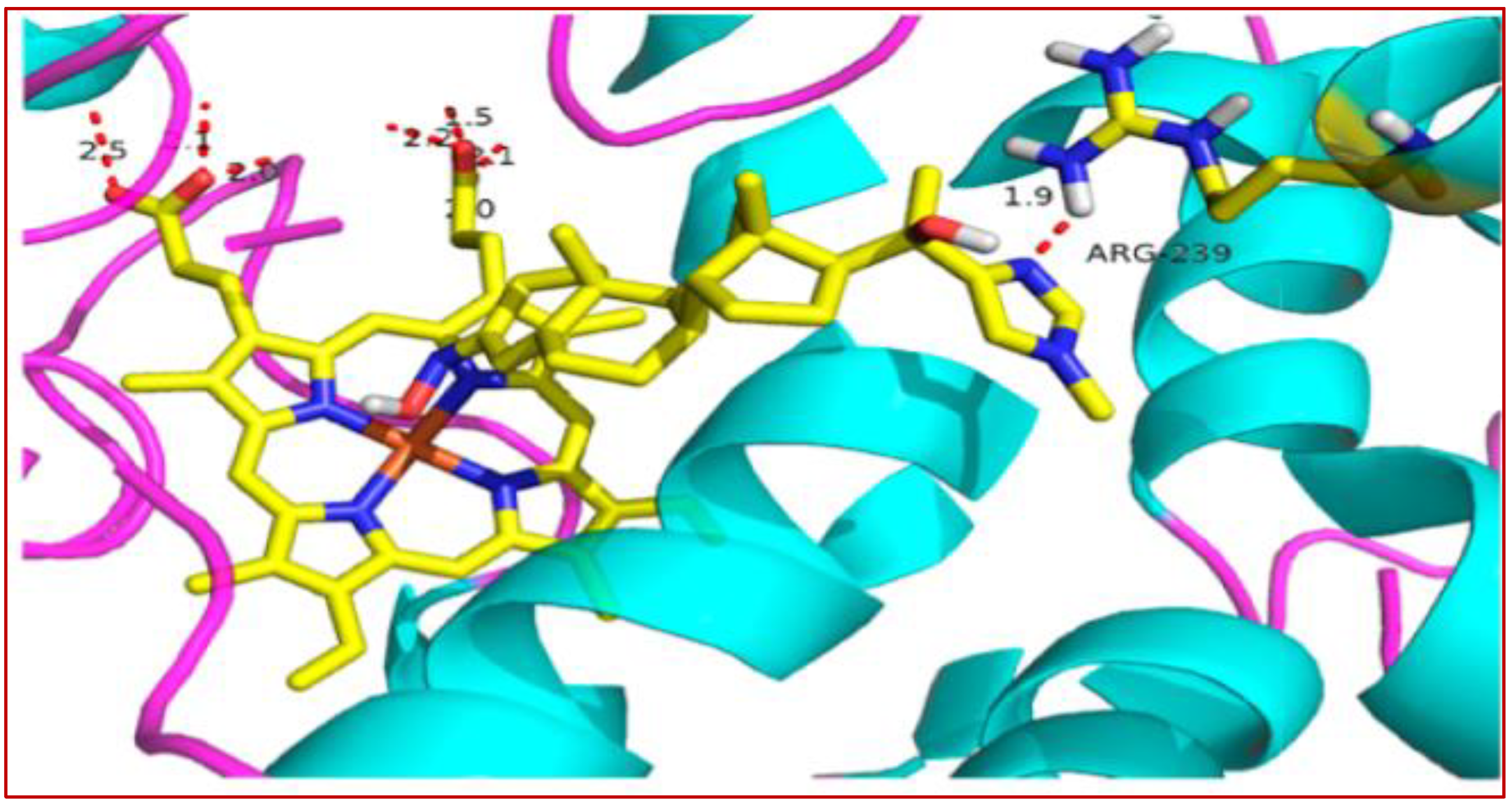
Figure 17.
Illustrative examples of anticancer oleanolic acid oxime derivatives and steroidal oximes conjugates.
Figure 17.
Illustrative examples of anticancer oleanolic acid oxime derivatives and steroidal oximes conjugates.
Figure 18.
Illustrative examples of anticancer platinum complexes bearing chelating oxime ligands.
Figure 19.
Illustrative examples of miscellaneous oxime-chelated metal complexes as potential chemotherapeutic agents.
Figure 19.
Illustrative examples of miscellaneous oxime-chelated metal complexes as potential chemotherapeutic agents.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



