
Submitted:
13 November 2023
Posted:
14 November 2023
You are already at the latest version
Abstract
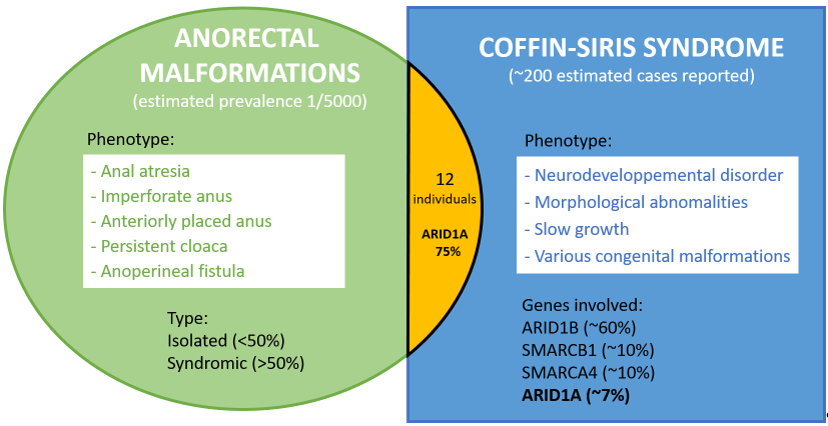
Anorectal malformations (ARMs) represent a wide spectrum of congenital anomalies of the anus and rectum, of which more than half are syndromic. Their etiology is highly heterogeneous and still poorly understood. We report a 4-year-old girl who initially presented with an isolated ARM, and subsequently developed a global developmental delay as part of an ARID1B-related Coffin-Siris syndrome (CSS). The co-occurrence of ARMs and CSS in an individual is unexpected since both diseases are very rare. A review of the literature enabled us to identify 12 individuals with both CSS and ARMs. Among these 12 individuals, 9 had a variant in ARID1A, 2 in ARID1B, and 1 in SMARCA4. This more frequent than expected association between CSS and ARM indicates that some ARMs are likely a rare consequence of CSS, especially for ARID1A-related CSS.
Keywords:
Coffin-Siris syndrome
; ARID1A
; anorectal malformation
; anal atresia
; anteriorly placed anus
; imperforate anus
1. Introduction
Anorectal malformations (ARMs) represent a wide spectrum of congenital anus and rectum anomalies that arise in early embryonic development[1].Their incidence is estimated at 1 in 5,000 live births[2]. The most common malformations include anal atresia (Human Phenotype Ontology HP:0002023), imperforate anus (Monarch Initiative MONDO:0001046), anteriorly placed anus (HP:0001545), persistent cloaca (HP:0012621), and anoperineal fistula (HP:0005218)[3]. More than 50% of ARMs are associated to other malformations (syndromic ARMs)[4,5]. The majority of non-syndromic ARMs are identified after birth, while syndromic ARMs are more often detected by foetal ultrasound during pregnancy[6]. The causes of ARMs are heterogeneous and still poorly known. Both genetic and environmental factors are thought to be implicated[1,2]. Notably, ARMs is a common feature of the VACTERL malformative sequence that accounts for about 15% of ARMs cases, and is typically of unknown etiology[5]. Chromosomal causes of ARMs represent 5 to 15% of the cases. They include aneuploidies (in particular trisomy 21 found approximately in 2% of ARM) and Copy Number Variations (CNV, approximately found in 3% of ARM)[5,7]. Monogenic variants explain up to 1/3 of syndromic ARMs, and are very heterogeneous[8]. The genes involved in syndromic ARMs include but are not limited to: ADNP (Helsmoortel-van der Aa syndrome), BBS1 (Bardet-Biedl syndrome 1), CHD7 (CHARGE syndrome), CREBBP (Rubinstein-Taybi syndrome 1), EP300 (Rubinstein-Taybi syndrome 2), FANCC (Fanconi anemia group C), GLI3 (Pallister–Hall syndrome), KDM6A ( Kabuki syndrome 2), MID1 (Opitz G/BBB syndrome), MNX1 (Currarino syndrome), SALL1 (Townes–Brocks syndrome), SALL4 (Duane-radial ray syndrome ), and SETD2 (SETD2-related disorders)[1,9,10]
Coffin-Siris syndrome (CSS) is a well-known genetic disorder characterized by developmental delay and/or intellectual disability, hypoplastic to absent fifth fingernails and fifth distal phalanges, coarse facial appearance, and other variable features including hypotonia, slow growth, hypertrichosis, sparse scalp hair, feeding difficulties, and organ malformations[11]. CSS is caused by heterozygous variants in genes involved in chromatin remodeling, especially those coding for subunits of the BAF (BRG1-associated factor) complex. Of the 12 genes responsible for CSS, ARID1B is by far the most prevalent (~60% of molecularly diagnosed cases). SMARCB1, SMARCA4 are among the next most common (~10%), following with ARID1A (~7%)[12,13].
In this article, we describe anorectal malformation in an individual with ARID1B-related Coffin-Siris syndrome. We then searched the literature for other cases with both ARMs and CSS in an attempt to establish a causal link between these two rare conditions.
2. Results
2.1. Case Report
Our patient is a 4-year-old female with a typical ARID1B-related Coffin-Siris syndrome and anal malformation. She has no family history of ARMs. During pregnancy, ultrasound revealed mild intrauterine growth retardation (HP:0001511) and mega cisterna magna (MONDO:0019953), with no other cerebral abnomalities on fetal MRI. Anteriorly placed anus associated with anoperineal fistula was discovered at birth and was corrected surgically at 2 days of life.The anoplasty was complicated by anal stenosis, followed by anal dialation repair after 1 month. Abdomino-pelvic ultrasound showed a bicornuate uterus (MONDO:0015842). Later, she showed a global developmental delay(HP:0001263) associated with hypotonia (HP:0001252), growth delay (weight < 3rd percentile, HP:0001510), severe feeding difficulties (HP:0008872), and unilateral exotropia (HP:0000577). She sat at 9 months and walked independently at 30 months. Clinical examination showed hypoplasic distal phalanx of the 5th fingers (HP:0008398), slightly coarse facies (HP:0000280), wide-set eyes (HP:0000316), and sparse anterior scalp hair (HP:0004768). Array-CGH was normal. Trio exome sequencing identified a pathogenic de novo variation in ARID1B (Table 1).
2.2. Litterature review
At the time we made the diagnosis of CSS in our patient, there was no established link between Coffin-Siris syndrome and anorectal malformations. We performed a literature review on Pubmed using the terms 'coffin-siris syndrome' or the names of the 12 genes involed in CSS (ARID1A, ARID1B, ARID2, BICRA, DPF2, SMARCD1, SMARCC2, SMARCE1, SMARCA4, SMARCB1, SOX11 and SOX4), and the term 'anorectal malformation' or 'anal malformation' or ‘anteposition of the anus'. We identified 11 individuals with ARM and CSS, for a total of 12 individuals including our case (Table 1). Within this cohort, 9 had a variant in ARID1A, 2 in ARID1B including our patient, and 1 in SMARCA4. The coordinates of the variants were available for 6/12 individuals. All of these variants were hetereozygous and predicted loss of function (pLoF) as expected in CSS. Five were truncating (3 frameshift, 1 gene deletion and 1 intergenic microdeletion) and 1 was a missense. For the remaining 6 individuals, the coordinates of the variants were not availabe because the authors reported separetely the variation and the phenotype[15]. In our cohort of 12 individuals, the types of ARMs were anal atresia (5/12), anteriorly placed anus (5/12), imperforate anus (2/12). Association to anorectal fistula was reported only in our case but we do not know whether it was present or absent in the other cases. We found no clear correlation between the type of CSS-causing genetic variation and type of ARM.
3. Discussion
In this study, we discuss the phenotype-genotype correlation between anorectal malformations (ARMs) and Coffin-Siris syndrome (CSS). There have been more than 300 molecularly confirmed cases of Coffin-Siris syndrome reported in the literature[12,13,15].If we estimate at 500 the number of CSS in the literature, our cohort of 12 cases with ARMs would indicate a 1/40 prevalence of ARMs in CSS patients, which is 125 times higher than the 1/5000 estimated prevalence of ARMs in the general population. Of course, these calculations are only rough estimates, since we know neither the exact number of published CSS cases nor the number of cases in which clinical signs of ARMs have been sought and reported, and nor the exact prevalence of ARMs in the general population. In any case, the prevalence of ARMs in patients with CSS appears to be much higher than expected by chance. In particular, there is a significant enrichment of ARID1A variants (9/12) in our cohort compared with the usual low prevalence of ARID1A variants (~15%) in CSS. Thus, ARID1A could be particularly at risk of anorectal malformation among the genes responsible for Coffin-Siris syndrome.
4. Conclusions
In this study, we observed a higher than expected incidence of anorectal malformations among Coffin-Siris syndrome patients, especially in those carrying an ARID1A variant. The true prevalence of ARMs in CSS patients is difficult to estimate and may be close to 1/40, corresponding to more than 100 times the estimated prevalence in the general population. We conclude that anorectal malformations are a feature of Coffin-Siris syndrome. Given that other genetic syndromes affecting chromatin remodeling pathways are also known to cause anorectal malformations, it is tempting to think that chromatin remodeling defects, of either genetic or environmental origin, could be a common mechanism leading to anorectal malformations.
Author Contributions
Conceptualization, Francis Ramond; Data curation, Inès Harzallah; Funding acquisition, Renaud Touraine; Investigation, Ralah Alharbi and Francis Ramond; Supervision, Renaud Touraine; Validation, Anna Suchet-Dechaud and Francis Ramond; Writing – original draft, Ralah Alharbi; Writing – review & editing, Anna Suchet-Dechaud, Inès Harzallah and Francis Ramond.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of CHU Saint-Etienne (reference IORG0007394).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moore, S.W. Associations of Anorectal Malformations and Related Syndromes. Pediatr Surg Int 2013, 29, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Wijers, C.H.W.; de Blaauw, I.; Marcelis, C.L.M.; Wijnen, R.M.H.; Brunner, H.; Midrio, P.; Gamba, P.; Clementi, M.; Jenetzky, E.; Zwink, N.; et al. Research Perspectives in the Etiology of Congenital Anorectal Malformations Using Data of the International Consortium on Anorectal Malformations: Evidence for Risk Factors across Different Populations. Pediatr Surg Int 2010, 26, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.J.; Levitt, M.A. Anorectal Malformations. Clin Colon Rectal Surg 2018, 31, 061–070. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, A. EUROCAT Working Group Descriptive Epidemiology of Isolated Anal Anomalies: A Survey of 4.6 Million Births in Europe. Am J Med Genet 2001, 103, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cuschieri, A. EUROCAT Working Group Anorectal Anomalies Associated with or as Part of Other Anomalies. Am J Med Genet 2002, 110, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, A.N.; Pandey, V. Anorectal Malformations. J Indian Assoc Pediatr Surg 2015, 20, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.; Dworschak, G.C.; Waffenschmidt, L.; Schierbaum, L.; Bendixen, C.; Heilmann-Heimbach, S.; Sivalingam, S.; Buness, A.; Schwarzer, N.; Boemers, T.M.; et al. Genome-Wide Identification of Disease-Causing Copy Number Variations in 450 Individuals with Anorectal Malformations. Eur J Hum Genet 2023, 31, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Belanger Deloge, R.; Zhao, X.; Luna, P.N.; Shaw, C.A.; Rosenfeld, J.A.; Scott, D.A. High Molecular Diagnostic Yields and Novel Phenotypic Expansions Involving Syndromic Anorectal Malformations. Eur J Hum Genet 2023, 31, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Dworschak, G.C.; van Rooij, I.A.L.M.; Reutter, H.M. The Role of De Novo Variants in Formation of Human Anorectal Malformations. Genes 2021, 12, 1298. [Google Scholar] [CrossRef] [PubMed]
- Exome Chip Association Study Excluded the Involvement of Rare Coding Variants with Large Effect Sizes in the Etiology of Anorectal Malformations | PLOS ONE Available online:. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0217477 (accessed on 27 October 2023).
- Kosho, T.; Okamoto, N. ; Coffin-Siris Syndrome International Collaborators Genotype-Phenotype Correlation of Coffin-Siris Syndrome Caused by Mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet 2014, 166C, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, F.; Tsurusaki, Y.; Okamoto, N.; Teik, K.W.; Mizuno, S.; Suzumura, H.; Isidor, B.; Ong, W.P.; Haniffa, M.; White, S.M.; et al. Genetic Abnormalities in a Large Cohort of Coffin–Siris Syndrome Patients. J Hum Genet 2019, 64, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Vasko, A.; Drivas, T.G.; Schrier Vergano, S.A. Genotype-Phenotype Correlations in 208 Individuals with Coffin-Siris Syndrome. Genes 2021, 12, 937. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.; Lefebvre, M.; Brehin, A.-C.; Thauvin, C.; Patrier, S.; Sparks, T.N.; Norton, M.; Yu, J.; Huang, E. Prenatal Presentation of Multiple Anomalies Associated with Haploinsufficiency for ARID1A. Eur J Med Genet 2022, 65, 104407. [Google Scholar] [CrossRef] [PubMed]
- van der Sluijs, P.J.; Joosten, M.; Alby, C.; Attié-Bitach, T.; Gilmore, K.; Dubourg, C.; Fradin, M.; Wang, T.; Kurtz-Nelson, E.C.; Ahlers, K.P.; et al. Discovering a New Part of the Phenotypic Spectrum of Coffin-Siris Syndrome in a Fetal Cohort. Genetics in Medicine 2022, 24, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Halgren, C.; Kjaergaard, S.; Bak, M.; Hansen, C.; El-Schich, Z.; Anderson, C.; Henriksen, K.; Hjalgrim, H.; Kirchhoff, M.; Bijlsma, E.; et al. Corpus Callosum Abnormalities, Intellectual Disability, Speech Impairment, and Autism in Patients with Haploinsufficiency of ARID1B. Clinical Genetics 2012, 82, 248–255. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Molecular and clinical data of individuals with Coffin-Siris syndrome and anorectal malformation.
Table 1.
Molecular and clinical data of individuals with Coffin-Siris syndrome and anorectal malformation.
| Gene | Variant | ARM Type | Reference |
|---|---|---|---|
| ARID1A | NM_006015:c.6134_6138del p.(K2045Rfs*52) | IA, APA | [8] |
| NM_006015:c.4886dup p.(V1630Cfs*18) | APA | [14] | |
| arr[hg19] 1p36.11(26,797,508_27,052,080)×1 | IA | [14] | |
| n.a* | AA | [15] | |
| n.a* | AA | [15] | |
| n.a* | AA | [15] | |
| n.a* | AA | [15] | |
| n.a* | APA | [15] | |
| n.a* | APA | [15] | |
| ARID1B | arr[hg19] 6q25.3(157,079,676-157,806,675)x1 | AA | [16] |
| NM_017519.3:c.5227_5228insCA p.(L1743Pfs*12) | APA, AF | This study | |
| SMARCA4 | NM_001128849:c.2656A>G p.(M886V) | Not specified | [8] |
ARM = anorectal malformation. IA = imperforate anus. APA = anteriorly placed anus. AA = anal atresia. AF = anoperineal fistula. n.a = not available. *The authors reported separately the variation and the phenotype in a cohort, not allowing to link a specific ARID1A genotype to a CSS phenotype individually.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



