
Submitted:
13 November 2023
Posted:
14 November 2023
You are already at the latest version
Abstract
X-linked myotubular myopathy (XLMTM) is a rare congenital myopathy resulting from dysfunction of the protein myotubularin encoded by the MTM1 gene. XLMTM has a high neonatal and infantile mortality rate due to a severe myopathic phenotype and respiratory failure. However, in a minority of XLMTM cases, patients present with milder phenotypes and achieve ambulation and adulthood. Regarding wide phenotypic variability, we investigated the genotype-phenotype correlations in newly diagnosed XLMTM patients in a patients' cohort (previously published data plus three novel variants, n=414). We found a significant association between severe phenotype and truncating variants (p < 0.001), frameshift variants (p < 0.001), nonsense variants (p = 0.006), and in/del variants (p = 0.036). On the contrary, missense variants were significantly associated with the mild and moderate phenotype (p < 0.001). We found no significant association between phenotype and the MTM1-specific functional domain, contrasting with previously published data. Based on the significant facial gestalt difference between XLMTM patients and unaffected controls (p = 0.001), we consider the Face2Gene application as an effective diagnostic tool in XLMTM when using the correct algorithm. The Face2Gene application seems to be a practical, non-invasive diagnostic approach in XLMTM.
Keywords:
MTM1 gene
; myotubularin
; X-linked myotubular myopathy
; centronuclear myopathy
; genotypephenotype correlations
; DeepGestalt technology
; Face2Gene application
1. Introduction
Myotubularin (MTM1), a protein of 603 amino acids, is a member of the myotubularin superfamily. It functions as an endosomal lipid phosphatase, which acts e.g. on phosphatidylinositol 3-phosphate, a crucial lipid in the intracellular signaling pathway. It regulates intracellular membrane trafficking and vesicular transport processes, particularly in myocytes of skeletal muscles [1,2,3]. The myotubularin is constituted by four conserved functional domains: 1) PH-GRAM (Pleckstrin Homology-Glucosyltransferase, Rab-like GTPase Activator, and Myotubularin) from amino acid 29 to 160 of MTM1, which plays a crucial role in targeting the various myotubularins to a specific intracellular compartment; 2) RID (Rac1-Induced recruitment Domain) from position 161 to 272, which is inecessary in the recruitment of the MTM1 protein to the plasma membrane; 3) PTP/DSP (Protein Tyrosine Phosphatase/Dual-Specificity Phosphatase) from position 273 to 471, which dephosphorylates phosphatidylinositol 3-phosphate (PtdIns3P) and phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2) into phosphatidylinositol (PtdIns) and phosphatidylinositol 5-phosphate (PtdIns5P), respectively; and 4) SID (SET-protein Interaction Domain) from position 435 to 486, where interaction between the SID domain of myotubularins and proteins with a SET domain could regulate the expression of some genes. These four functional domains of the MTM1 protein are followed by a C-terminal coiled-coil motif and a PDZ binding domain [3]. Myotubularin is encoded by the MTM1 gene (MIM * 300415, gene locus Xq28), composed of 15 exons [4], while the start codon is located in exon 2. Today, more than 590 variants (LOVD database https://www.lovd.nl/, from which more than 530 are classified as pathogenic and likely pathogenic) have been found in the MTM1 gene, distributed throughout the MTM1 gene with some recurrent variants but no unambiguous hot spots in the gene.
Mutations in the MTM1 gene lead to the congenital myopathy called X-linked myotubular myopathy (XLMTM, MIM # 310400, ORPHA: 596) as a subtype of centronuclear myopathies group, first described in 1966 by Spiro et al. [5]. XLMTM is inherited in an X-linked recessive manner, and its prevalence is 1:50,000 in males [6]. Three clinical forms of XLMTM have been classified as severe, moderate, and mild according to the severity of the disease course, achieving the milestones, ambulation, and ventilatory support needed [7,8,9]. In most cases (more than 80 %), the XLMTM presents in the neonatal period, typically immediately after birth, by severe global hypotonia (floppy infant), hypo- to areflexia, and different degrees of respiratory insufficiency. Weak fetal movements and polyhydramnios can be present even prenatally. Miscarriages or stillbirths also occur. In early infancy (in the first year of life), 25 % of patients die due to respiratory failure [10], and 48 % of patients by 18 months of age [8]. However, some XLMTM patients achieve respiratory and ambulatory independence and adulthood. Gene therapy in XLMTM is under investigation, and symptomatic treatment remains the standard [11], early diagnosis is therefore essential.
The majority of XLMTM patients have characteristic facial features (myopathic facies, uni- or bilateral ptosis, dolichocephaly with a high forehead and long face, midface hypoplasia, and a narrow, high-arched palate with malocclusion) becoming more pronounced with age, a birth length above the 97th percentile, long hands, feet, and fingers, as well as a rare condition called peliosis hepatis in up to 5 % of individuals [7,10,11,12]. Thanks to the typical histopathological picture, relatively low costs, and quick results, invasive muscle biopsy has for a long time been considered a standard diagnostic method. However, the innovative non-invasive approach of DeepGestalt technology to facial features in the diagnosis of XLMTM has not yet been investigated.
As mentioned, some XLMTM patients achieve respiratory and ambulatory independence and adulthood, contrasting with high neonatal and infantile mortality rates. This phenotype variability logically led to questions about genotype and resulting phenotype. In the past, several studies focusing on evaluating genotype-phenotype correlations have been published [8,9,13,14], but these correlations remain unclear.
Our study aimed to introduce a new perspective on genotype-phenotype correlations and reconsider their predictive value in the XLMTM disease course based on data obtained from databases published for more than 20 years, supported by statistical processing, as well as expansion of the MTM1 mutational spectrum by three novel disease-causing variants identified in our patients. Considering the typical facial features, by using DeepGestalt technology [15] via the Face2Gene application (FDNA, Inc., Boston, MA, USA), we tried to determine its relevance in a non-invasive diagnostic approach in XLMTM patients, and whether it has a potential to replace the invasive muscle biopsy as the investigation supplementary to genetic testing in the future.
2. Subjects and Methods
2.1. Subjects involved in the statistical analysis
For the selection of subjects involved in the present analysis we used already published data obtained retrospectively from the PubMed® database, searched by using keywords: myotubularin, MTM1 gene, X-linked myotubular myopathy, XLMTM, X-linked centronuclear myopathy (XLCNM), and genotype-phenotype correlations in XLMTM/XLCNM. In our study, we included only index male subjects (from unrelated families), with disease-causing variants identified in the MTM1 gene and specified XLMTM phenotype (see 2.2. Phenotype severity below). Data were expanded by three novel MTM1 variants detected in patients P1-P4 from our in-house database.
2.2. Phenotype severity
All involved subjects had specified XLMTM phenotype, classified according to [7,8,9] into three groups, presented by: 1) "S" – severe (classic) phenotype with characteristic facial features, long-term ventilator dependence more than 12 hours per day, delayed gross motor milestones, no independent ambulation, and high mortality rate in infancy, 2) "Mo" – moderate (intermediate) phenotype with less severe motor delay, prolonged periods of decreased ventilation support, at least 6 to 8 hours per day without ventilatory support [7], or more than 12 hours without ventilator support per day [9], and 3) "Mi" – mild phenotype with minimal motor delay, and the lack of a need for mechanical ventilation, independent spontaneous respiratory function beyond the newborn period, and no or limited impact of the life span. Cases collected by Laport et al. 2000 were classified only into two groups: 1) "S" – severe subjects with classic phenotype and 2) "M" – mild or moderate subjects who are long-term survivors without heavy ventilator assistance [16]. This fact was taken into account during statistical processing.
2.3. Variant evaluation
Variants reported in already published XLMTM patients were considered as disease-causing and missense variants were tested in silico by the PredictSNP tool (combine MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, SNAP tools) [17] to prove their pathogenicity. The classification of the three novel MTM1 variants (from our in-house database) followed the consensus recommendations of the American College of Medical Genetics [18]. All collected variants were grouped according to their location in the MTM1 gene (in exon or intron and MTM1-specific functional domain: PH-GRAM, RID, PTP/DSP, and SID), variant type, and their impact on the resulting protein (truncating or non-truncating) and predicted to escape or undergo nonsense-mediated mRNA decay.
2.4. Facial gestalt analysis
We used DeepGestalt technology [15] via the Face2Gene application (FDNA, Inc., Boston, MA, USA), for facial dysmorphology comparison. In the facial gestalt analysis, we enrolled only patients with appropriate photo quality. Each cohort had to comprise at least ten photos. We selected four patient cohorts: 1) cohort of XLMTM patients (14 patients), 2) cohort of Myotonic dystrophy type 1 (MD1) patients, whose facial features, in our opinion, are most similar to XLMTM (10 patients), 3) cohort of overall neuromuscular disorders (NMDs patients: other congenital myopathies and muscle dystrophies, except XLMTM patients, together 21 patients), and 4) cohort of unaffected controls (11 patients). Healthy controls showed no obvious dysmorphia and were never suspected to have any genetic syndrome. We compared all cohorts to each other and the cohort of 11 healthy individuals in a binary comparison and as composite photos. A mean area under the curve (AUC) value was generated in the binary comparison, representing the degree of discrimination between the cohorts. Mean AUC ranged between 0 and 1, where 0 means incorrectly classified cohorts, 0.5 random classifications, and 1 means perfect separation between the cohorts. The p-value describes the accuracy of the binary comparison, and p < 0.05 is considered statistically significant, indicating that the Face2Gene application can distinguish between the two cohorts.
2.5. Statistical analysis in genotype-phenotype correlations
For statistical evaluation, we used IBM SPSS 28 Statistics software. For genotype-phenotype correlation: phenotype vs. protein truncation, phenotype vs. mutation type, phenotype vs. MTM1-specific functional domain, phenotype vs. nonsense-mediated mRNA decay, i.e., categorical variables, we used Fisher’s exact test.
3. Results
3.1. Novel variants in the MTM1gene
We have identified three novel MTM1 variants in our patients P1-P3 (Figure 1A). All patients presented a severe (classic) XLMTM phenotype. The hemizygous frameshift variant c.438_439delCA (p.His146Glnfs*10) in exon 6, found in P1, was evaluated as pathogenic. The hemizygous variant c.(342+1_343-1)_(444+1_445-1)del; p.(Asp115_Leu148del), which represents an in-frame exon 6 deletion, detected in P2, and the hemizygous variant c.(1053+1_1054-1)_(1467+1_1468-1)del; p.(Leu352_Gln489del), which represents an in-frame deletion of exons 11-13, found in P3 and his brother P4, were classified as likely pathogenic (Table 1). In P2 and P3, the pathogenicity of these variants was proved by histopathologic and immunohistochemical evaluation of the vastus lateralis muscle biopsy sample (for more details, see Supplementary Materials).
3.2. XLMTM cohort: variants type and their distribution
We collected 414 index cases (411 published cases so far and 3 novel variants from our in-house database); 192 variants (78.4 %) in the MTM1 gene occurred only once, 53 variants (21.6 %) occurred twice or more times. All collected variants are summarized in the Supplementary Table 1.
3.2.1. Exonic and intronic variants
From 385 variants (except large deletions n=29), 307 variants (79.7 %) were located in exons and 78 (20.3 %) in introns. More than half (53.7 %) are located in exon 8, exon 4, exon 9, exon 11, and intron 11 (counted together): 55 variants (14.3 %) were located in exon 8 and 48 (12.5 %) in exon 4 and 41 (10.6 %) in exon 9 and 36 (9.3 %) in exon 11 and 27 (7 %) in intron 11 and 22 (5.7 %) in exon 12 and 21 (5.4 %) in exon 3 and 13, 17 (4.4 %) in exon 14 and 11 and less in the rest (Figure 1B).
3.2.2. Variants in the MTM1-specific functional domain
Most variants were located in the MTM1-specific functional domain (n=276, 66.7 %), 100 variants (24.1 %) were located in RID domain, 85 variants (20.5 %) in PH-GRAM domain, 64 variants (15.5 %) in PTP/DSP domain, 15 variants (3.4 %) in SID domain and 12 variants (2.9 %) in both domains (PTP/DSP and SID). 138 variants (33,3 %) were located outside of the specific domain.
3.2.3. Truncating effect and non-sense mediated mRNA decay
188 variants (45.4 %) were identified as truncating and 133 (32.1 %) as non-truncating. For 93 variants it was not possible to define final protein truncation. Non-sense mediated mRNA decay was associated with 129 variants (31.2 %), in 24 not (5.8 %).
3.2.4. Phenotype severity and variant types
The severe phenotype was present in 330 (79.7 %) subjects, moderate in 19 (4.6 %), mild in 32 (7.7 %), and a combination of mild and moderate phenotype in 33 (8 %) subjects (Figure 1C). From 414 variants the most frequent variants were missense (n=137, 33.1 %), frameshift (n=85, 20.5 %) and nonsense variants (n=70, 16.9 %). From 137 missense variants, 87 (63.5 %) were located in a functional domain as follows: PH-GRAM 15 variants (17.2 %), RID 42 variants (48.3 %), PTP/DSP 25 variants (28.7 %), PTP/DSP/SID 4 variants (4.6 %), and SID 1 variant (1.2 %). Of all 137 missense variants, 75 variants (54.7 %) were associated with severe phenotype, the remaining (n= 62; 45.3 %) with mild to moderate phenotype. Less common were intronic variants (n=45, 10.9 %), splicing variants (n=32, 7.7 %), large deletions (n=29, 7.0 %), in/del variants (n=13, 3.1 %), variants affecting codon initiation (n=2, 0.5 %), and silent variants (n=1, 0.2 %). 209 variants (63.5 %) associated with severe phenotype were located in the specific domain: PH-GRAM 63 variants (30.1 %), RID 71 variants (34 %), PTP/DSP 50 variants (23.9 %), PTP/DSP/SID 11 variants (5.3 %), and SID 14 variants (6.7 %).
3.2.5. Recurrent variants in the MTM1 gene
The most frequent variants were: c.1261-10A>G in intron 11 (n=26), missense variant c.721C>T, p.(Arg241Cys) in exon 9 and the RID domain (n=13), missense variant c.205C>T, p.(Arg69Cys) in exon 4 and the PH-GRAM domain (n=12), frameshift variant c.141_144del, p.(Glu48Leufs*24) in exon 4 and the PH-GRAM domain (n=10); nonsense variant c.109C>T, p.(Arg37*) in exon 3 and the PH-GRAM domain (n=9), missense variant c.614C>T, p.(Pro205Leu) in exon 8 and the RID domain (n=9), and missense variant c.1262G>A, p.(Arg421Gln) in exon 12 and the PTP/DSP domain (n=8).
3.2.6. Inter-individual phenotype variability
In the cohort of 414 unrelated subjects, we found 53 variants (21.6 %) occurring more than once. In 13 variants, we saw inter-individual variability, where phenotype varied from mild to severe in the case of the same mutation (Table 2).
3.3. Genotype-phenotype correlations
For genotype-phenotype correlations, we used Fisher’s exact test. We found that 95.2 % of all truncating variants are associated with severe phenotype; this association is highly significant (p < 0.001). Focusing on variant type vs. phenotype, we found that frameshift variants were significantly associated with the severe phenotype (p < 0.001), as well as nonsense variants (p = 0.006) and deletions/insertions (p = 0.036). On the other hand, missense variants were significantly associated with the mild and moderate phenotype (p < 0.001) compared with the other variants vs. the mild and moderate phenotype. We found no significant association between phenotype and MTM1-specific functional domain, as well as phenotype and nonsense-mediated mRNA decay (Figure 2A-D).
3.4. Facial gestalt analysis
The Face2Gene software found significant differences between XLMTM patients and unaffected controls (p = 0.001) (Figure 3) and between unaffected controls and NMDs patients (p = 0.023). Some differences in facial gestalt could also be seen between XLMTM and MD1 patients and XLMTM patients and other NMDs patients, but these are not statistically significant (see Table 3)
4. Discussion
X-linked myotubular myopathy (XLMTM) is a rare neuromuscular disorder associated with a high rate of neonatal and infantile mortality in patients by 18 months of age (about 46 % of all XLMTM cases) based on severe myopathic phenotype [8]. In our opinion, the percentage of mortality is underestimated because not all cases of abortion, stillbirth, or early death of newborns are genetically investigated in the sense of congenital myopathy. For that reason and the high risk of recurrence in the family, early recognition of XLMTM is essential. On the other side, the possible prediction of the prognosis and further disease course in newly diagnosed XLMTM patients seems to centribute, and genotype-phenotype correlations could be one of these non-invasive prognostic tools.
Our study is the most extensive genotype-phenotype comparison in the XLMTM and expands the spectrum of known variants in the MTM1 gene. Using the PubMed® database plus three novel variants, we selected an XLMTM cohort of 414 index subjects with 245 different disease-causing variants. All subjects involved are males from unrelated families. Female subjects were excluded due to different disease manifestations resulting from X-linked inheritance (asymptomatic or milder phenotype in females, resulting not only from mutation type but also different degrees of skewed X chromosome inactivation) [19,20].
In our study, the majority of variants in the MTM1 gene are associated with the severe (classic) form of XLMTM (79.7%) following already published data [8,10]. According to the previously published studies focusing on phenotype correlations with the type of variant (truncating vs. non-truncating) [8], it seems that truncating variants as nonsense, frameshift, and large deletions almost lead always to severe (classic) XLMTM phenotype, while non-truncating variants as splice site, intronic variants, as well as missense variants are associated in the minority of cases with the milder phenotype [9,13,14]. These studies were based on data from cohorts n=116 males [8] and n=146 males [9]. According to the data from n=414 male subjects involved in our study, we can assume that truncating variants such as frameshift, nonsense, and in/del variants are significantly associated with the severe phenotype. Missense variants are significantly associated with the moderate or mild phenotype. Thanks to this finding, we can suppose better outcomes in individuals affected by missense variants.
In our cohort, missense variants are the most common changes in the MTM1 gene (n=137, 33.1 %). According to Oliveira et al., 2013, who centered their analysis on missense variants using data from the LOVD database, it was suggested that missense variants are not randomly scattered in the protein because 96 % of them are located in myotubularin regions with known function (i.e. functional domains) in contrast with 59 % of nonsense variants [9]. In our study, only 63.5 % (n=87/137) of missense variants and 82.8 % of nonsense variants (n=58/70) were located in the specific functional domains, which in the case of missense variants corresponds with sequence of functional domains, i.e. 71 % of the length of the total MTM1 protein sequence [9]. We conclude a random scatter of the missense variants in the MTM1 protein.
Large deletions are, in almost all cases, associated with severe phenotype. One exception in our data set represents the deletion of the entire exon 15 reported by Tanner et al., 1999, associated with mild XLMTM phenotype. This phenomenon could be explained by the fact that exon 15 is located at the end of the MTM1 gene, representing less vital regions of the gene [21]. This observation could be supported by other mild phenotype frameshift variants in exon 15 [7]. No other pathogenic variants in exon 15 have been found (LOVD database https://www.lovd.nl/). In conclusion, large deletions, except for the isolated deletion of exon 15, are associated with a severe phenotype in all cases.
According to [9,13,14], domain-specific correlations show that mutations in PTP/DSP, SET, and RID domains are associated almost always with severe (classic) phenotypes. However, variants outside these domains are more likely to be associated with a moderate or mild phenotype. Pathogenic variants in PTP/DSP domains should be significantly associated with severe phenotype due to the crucial enzyme activity (dephosphorylation of PtdIns3P and PtdIns(3,5)P2 into PtdIns and PtdIns5P, respectively). However, our study did not find a significant association between phenotype and the MTM1-specific functional domain nor phenotype and nonsense-mediated mRNA decay.
Today, more than 589 variants (LOVD database https://www.lovd.nl/, from which more than 530 are classified as pathogenic and likely pathogenic) have been found in the MTM1 gene. Considering that the variants detected in the MTM1 gene are in a high percentage pathogenic or likely pathogenic (more than 90 %), it can be assumed that every patient in whom a variant in the MTM1 gene is detected, deserves special attention, and detailed clinical evaluation in the context of XLMTM in males as well as in female probands is appropriate.
We found that from 385 point disease-causing variants in the MTM1 gene, more than half (53.7 %) are located in exon 8, exon 4, exon 9, exon 11, and intron 11. For now, this high number of pathogenic variants found in these exons could be explained by their length without other known explanation or correlation. Some variants in the MTM1 gene occur more than once. Tsai et al., 2005 described a potential hot spot, the recurrent pathogenic intronic variant c.1261-10A>G in a Japanese XLMTM cohort [22]. In the present cohort, we can see this variant 26 times, and according to our findings, it is the most frequently occurring variant in the MTM1 gene, in all cases associated with the severe (classic) XLMTM phenotype.
Inter-individual variability (phenotype from classic to mild form) as well as between members of the same family have been previously described: two families diagnosed according to clinical examination and the histopathological findings in the biopsied muscle of affected individuals [23] in the three-generation family with variant c.540T>G, p.(Asn180Lys) [24] and very mild phenotype in grandfather with pathogenic variant c.1210G>A, p.(Glu404Lys) with more severe phenotype in his grandson [25]. Bertazzi et al., 2014 described patients’ disease-causing variants affecting the PH-GRAM domain and their association with XLMTM phenotype severity [3]. While the variant p.(Val49Phe) was associated with a two-fold decrease in phosphatase activity, p.(Arg69Cys) displays wild-type phosphatase activity [26]. Our data show that the first patient is associated with the severe phenotype [27], but we collected 12 subjects with variant p.(Arg69Cys) with phenotypes varying from mild to severe. What causes inter-individual and intra-familial phenotype differences in XLMTM has yet to be discovered. We can only presume environmental factors, epigenetic factors, gene interactions, gene expression, DNA methylation, or some unknown mechanisms.
In the era of massive development and the availability of non-invasive genetic methods, the question of using invasive procedures such as muscle biopsy to diagnose neuromuscular diseases remains controversial. Based on muscle biopsy findings, some authors suggested non-genetic starting points for determining the phenotype. Other studies, e.g., McEntagart et al., 2002, try to find if the level of myotubularin expression correlates with XLMTM phenotype [8]. Pierson et al., 2007 described that myofiber size correlated with MTM1 mutation type and patient outcome [28]. According to Bryen et al., 2021, all procedures considering clinical scoring/phenotype, genetics, and muscle biopsy are equally crucial in the diagnostics of XLMTM [29]. Despite the constantly improving harmless genetic diagnostic methods, we consider muscle biopsy, regardless of its invasiveness, to be an essential diagnostic tool in diagnosing neuromuscular diseases, especially in cases with rapid progression. Furthermore, muscle tissue can be used for possible functional studies and variant pathogenicity proof.
Considering the typical facial features in XLMTM, by using DeepGestalt technology [15] via the Face2Gene application (FDNA, Inc., Boston, MA, USA), we tried to determine its relevance in a non-invasive diagnostic approach in XLMTM patients. The Face2Gene software found significant differences between XLMTM patients and unaffected controls and between unaffected controls and NMD patients. Some differences in facial gestalt could also be seen between XLMTM and MD1 patients and XLMTM patients and other NMD patients, but these are not statistically significant. Currently, XLMTM does not appear as one of the syndromes in the RARE tab with a composite image, meaning that the DeepGestalt algorithm (vs. DG 22.3.0) cannot discern it yet from other possible syndromes. Therefore, the second algorithm, GestaltMatcher, based on facial gestalt comparison, available in the ultra-rare tab [30], will need to be used for these cases. Based on our findings, the Face2Gene application is a potential diagnostic tool in XLMTM.
5. Conclusion
The presented genotype-phenotype correlations show that truncating variants such as frameshift, nonsense and in/del variants are significantly associated with a severe phenotype. In contrast, missense variants lead significantly in nearly half of the patients to mild and moderate phenotypes. No significant association was found between phenotype and MTM1-specific functional domains, as well as phenotype and nonsense-mediated mRNA decay. Other associations are suggestive but are statistically not significant. Interindividual phenotypic variability, even between members of the same family, is a known phenomenon in XLMTM, which needs further investigation. Due to this fact, the genotype-phenotype correlations are helpful but still need to be clarified and are, as a single approach, insufficient for predicting the disease’s course in XLMTM patients. Despite the increasing availability and effectiveness of molecular-genetic methods, muscle biopsy remains an irreplaceable, albeit invasive, method in diagnosing congenital myopathies, including XLMTM. Based on our findings, the Face2Gene represents a promising non-invasive diagnostic tool for XLMTM when using the correct algorithm. However, the idea that this innovative non-invasive approach including the Face2Gene application and the result of molecular-genetic testing will prospectively supplement an invasive muscle biopsy, is now closer to reality.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Materials (Case Reports including Figure 1-4 and additional Methods and Results) and Supplementary Table 1 (Excel table, Sheet 1-2).
Author Contributions
Conceptualization and Methodology: K.K., D.W. and S.W.; Software: K.K., S.W., A.S., and A.F., Validation: A.S., A.F., R.G.F., J.A.M., M.Š., D.G., M.K., D.W. and S.W.; Investigation (clinical and molecular-genetic): K.K., A.S., A.F., R.G.F., J.A.M., M.Š., D.G., M.K., and D.W.; Neuropathologic evaluation: O.K., K.O. and S.W., Writing – Original Draft Preparation: K.K.; Writing – Review and Editing: D.W. and S.W.; Data Curation and Visualization: K.K., A.F., and A.S.; Supervision: D.W., S.W. and J.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
Grant: APVV-17-0296, APVV-22-257.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the Johannes Kepler University Linz (Approval No: 1253/2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent was obtained from all patients’ legal representatives for the publication.
Data Availability Statement
All data are available in the article or in the Supplementary Materials. Genetic variants (3 novel variants) reported in this study have been submitted to ClinVar, and they can be accessed at: https://www.ncbi.nlm.nih.gov/clinvar/.
Acknowledgments
The authors would like to thank the patients’ families.
Conflicts of Interest
The author as well as the co-authors have no conflict of interest to declare.
References
- Taylor, G. S., Maehama, T., & Dixon, J. E. (2000). Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proceedings of the National Academy of Sciences of the United States of America, 97(16), 8910–8915. [CrossRef]
- Robinson, F. L., & Dixon, J. E. (2006). Myotubularin phosphatases: policing 3-phosphoinositides. Trends in cell biology, 16(8), 403–412. [CrossRef]
- Bertazzi, Dimitri & De Craene, Johan-Owen & Friant, Sylvie. (2014). Myotubularin MTM1 Involved in Centronuclear Myopathy and its Roles in Human and Yeast Cells. Journal of molecular and genetic medicine: an international journal of biomedical research. [CrossRef]
- Laporte, J., Guiraud-Chaumeil, C., Tanner, S. M., Blondeau, F., Hu, L. J., Vicaire, S., Liechti-Gallati, S., & Mandel, J. L. (1998). Genomic organization of the MTM1 gene implicated in X-linked myotubular myopathy. European journal of human genetics : EJHG, 6(4), 325–330. [CrossRef]
- Spiro, A. J., Shy, G. M., & Gonatas, N. K. (1966). Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Archives of neurology, 14(1), 1–14. [CrossRef]
- Laporte, J., Blondeau, F., Buj-Bello, A., & Mandel, J. L. (2001). The myotubularin family: from genetic disease to phosphoinositide metabolism. Trends in genetics : TIG, 17(4), 221–228. [CrossRef]
- Herman, G. E., Finegold, M., Zhao, W., de Gouyon, B., & Metzenberg, A. (1999). Medical complications in long-term survivors with X-linked myotubular myopathy. The Journal of Pediatrics, 134(2), 206–214. [CrossRef]
- McEntagart, M., Parsons, G., Buj-Bello, A., Biancalana, V., Fenton, I., Little, M., Krawczak, M., Thomas, N., Herman, G., Clarke, A., & Wallgren-Pettersson, C. (2002). Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscular disorders : NMD, 12(10), 939–946. [CrossRef]
- Oliveira, J., Oliveira, M. E., Kress, W., Taipa, R., Pires, M. M., Hilbert, P., Baxter, P., Santos, M., Buermans, H., den Dunnen, J. T., & Santos, R. (2013). Expanding the MTM1 mutational spectrum: novel variants including the first multi-exonic duplication and development of a locus-specific database. European journal of human genetics : EJHG, 21(5), 540–549. [CrossRef]
- Dowling JJ, Lawlor MW, Das S. X-Linked Myotubular Myopathy. 2002 Feb 25 [Updated 2018 Aug 23]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1432/.
- Lawlor, M. W., & Dowling, J. J. (2021). X-linked myotubular myopathy. Neuromuscular disorders : NMD, 31(10), 1004–1012. [CrossRef]
- Motoki, T., Fukuda, M., Nakano, T., Matsukage, S., Fukui, A., Akiyoshi, S., Hayashi, Y. K., Ishii, E., & Nishino, I. (2013). Fatal hepatic hemorrhage by peliosis hepatis in X-linked myotubular myopathy: a case report. Neuromuscular disorders : NMD, 23(11), 917–921. [CrossRef]
- Amburgey, K., Tsuchiya, E., de Chastonay, S., Glueck, M., Alverez, R., Nguyen, C. T., Rutkowski, A., Hornyak, J., Beggs, A. H., & Dowling, J. J. (2017). A natural history study of X-linked myotubular myopathy. Neurology, 89(13), 1355–1364. [CrossRef]
- Beggs, A. H., Byrne, B. J., De Chastonay, S., Haselkorn, T., Hughes, I., James, E. S., Kuntz, N. L., Simon, J., Swanson, L. C., Yang, M. L., Yu, Z. F., Yum, S. W., & Prasad, S. (2018). A multicenter, retrospective medical record review of X-linked myotubular myopathy: The recensus study. Muscle & nerve, 57(4), 550–560. [CrossRef]
- Gurovich, Y., Hanani, Y., Bar, O. et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med 25, 60–64 (2019). [CrossRef]
- Laporte, J., Biancalana, V., Tanner, S. M., Kress, W., Schneider, V., Wallgren-Pettersson, C., Herger, F., Buj-Bello, A., Blondeau, F., Liechti-Gallati, S., & Mandel, J. L. (2000). MTM1 mutations in X-linked myotubular myopathy. Human mutation, 15(5), 393–409. [CrossRef]
- Bendl, J., Stourac, J., Salanda, O., Pavelka, A., Wieben, E. D., Zendulka, J., Brezovsky, J., & Damborsky, J. (2014). PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS computational biology, 10(1), e1003440. [CrossRef]
- Richards, S., Aziz, N., Bale, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–423 (2015). [CrossRef]
- Savarese, M., Musumeci, O., Giugliano, T., Rubegni, A., Fiorillo, C., Fattori, F., Torella, A., Battini, R., Rodolico, C., Pugliese, A., Piluso, G., Maggi, L., D’Amico, A., Bruno, C., Bertini, E., Santorelli, F. M., Mora, M., Toscano, A., Minetti, C., & Nigro, V. (2016). Novel findings associated with MTM1 suggest a higher number of female symptomatic carriers. Neuromuscular disorders : NMD, 26(4-5), 292–299. [CrossRef]
- Biancalana, V., Scheidecker, S., Miguet, M., Laquerrière, A., Romero, N. B., Stojkovic, T., Abath Neto, O., Mercier, S., Voermans, N., Tanner, L., Rogers, C., Ollagnon-Roman, E., Roper, H., Boutte, C., Ben-Shachar, S., Lornage, X., Vasli, N., Schaefer, E., Laforet, P., Pouget, J., … Laporte, J. (2017). Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues. Acta neuropathologica, 134(6), 889–904. [CrossRef]
- Tanner, S. M., Schneider, V., Thomas, N. S., Clarke, A., Lazarou, L., & Liechti-Gallati, S. (1999). Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscular disorders : NMD, 9(1), 41–49. [CrossRef]
- Tsai, T. C., Horinouchi, H., Noguchi, S., Minami, N., Murayama, K., Hayashi, Y. K., Nonaka, I., & Nishino, I. (2005). Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240 kb deletion in Xq28 without male hypogenitalism. Neuromuscular disorders : NMD, 15(3), 245–252. [CrossRef]
- Barth, P. G., & Dubowitz, V. (1998). X-linked myotubular myopathy--a long-term follow-up study. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society, 2(1), 49–56. [CrossRef]
- Biancalana, V., Caron, O., Gallati, S., Baas, F., Kress, W., Novelli, G., D’Apice, M. R., Lagier-Tourenne, C., Buj-Bello, A., Romero, N. B., & Mandel, J. L. (2003). Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Human genetics, 112(2), 135–142. [CrossRef]
- Hoffjan, S., Thiels, C., Vorgerd, M., Neuen-Jacob, E., Epplen, J. T., & Kress, W. (2006). Extreme phenotypic variability in a German family with X-linked myotubular myopathy associated with E404K mutation in MTM1. Neuromuscular disorders : NMD, 16(11), 749–753. [CrossRef]
- Amoasii, L., Bertazzi, D. L., Tronchère, H., Hnia, K., Chicanne, G., Rinaldi, B., Cowling, B. S., Ferry, A., Klaholz, B., Payrastre, B., Laporte, J., & Friant, S. (2012). Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS genetics, 8(10), e1002965. [CrossRef]
- Herman, G. E., Kopacz, K., Zhao, W., Mills, P. L., Metzenberg, A., & Das, S. (2002). Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Human mutation, 19(2), 114–121. [CrossRef]
- Pierson, C. R., Agrawal, P. B., Blasko, J., & Beggs, A. H. (2007). Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscular disorders : NMD, 17(7), 562–568. [CrossRef]
- Bryen, S. J., Oates, E. C., Evesson, F. J., Lu, J. K., Waddell, L. B., Joshi, H., Ryan, M. M., Cummings, B. B., McLean, C. A., MacArthur, D. G., Kornberg, A. J., & Cooper, S. T. (2021). Pathogenic deep intronic MTM1 variant activates a pseudo-exon encoding a nonsense codon resulting in severe X-linked myotubular myopathy. European journal of human genetics : EJHG, 29(1), 61–66. [CrossRef]
- Hsieh, T. C., Bar-Haim, A., Moosa, S., Ehmke, N., Gripp, K. W., Pantel, J. T., Danyel, M., Mensah, M. A., Horn, D., Rosnev, S., Fleischer, N., Bonini, G., Hustinx, A., Schmid, A., Knaus, A., Javanmardi, B., Klinkhammer, H., Lesmann, H., Sivalingam, S., Kamphans, T., … Krawitz, P. M. (2022). GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nature genetics, 54(3), 349–357. [CrossRef]
- Annoussamy, M., Lilien, C., Gidaro, T., Gargaun, E., Chê, V., Schara, U., Gangfuß, A., D’Amico, A., Dowling, J. J., Darras, B. T., Daron, A., Hernandez, A., de Lattre, C., Arnal, J. M., Mayer, M., Cuisset, J. M., Vuillerot, C., Fontaine, S., Bellance, R., Biancalana, V., … Servais, L. (2019). X-linked myotubular myopathy: A prospective international natural history study. Neurology, 92(16), e1852–e1867. [CrossRef]
- Fattori, F., Maggi, L., Bruno, C., Cassandrini, D., Codemo, V., Catteruccia, M., Tasca, G., Berardinelli, A., Magri, F., Pane, M., Rubegni, A., Santoro, L., Ruggiero, L., Fiorini, P., Pini, A., Mongini, T., Messina, S., Brisca, G., Colombo, I., Astrea, G., … D’Amico, A. (2015). Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. Journal of neurology, 262(7), 1728–1740. [CrossRef]
- Laporte, J., Guiraud-Chaumeil, C., Vincent, M. C., Mandel, J. L., Tanner, S. M., Liechti-Gallati, S., Wallgren-Pettersson, C., Dahl, N., Kress, W., Bolhuis, P. A., Fardeau, M., Samson, F., & Bertini, E. (1997). Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Human molecular genetics, 6(9), 1505–1511. [CrossRef]
- Buj-Bello, A., Biancalana, V., Moutou, C., Laporte, J., & Mandel, J. L. (1999). Identification of novel mutations in the MTM1 gene causing severe and mild forms of X-linked myotubular myopathy. Human mutation, 14(4), 320–325. [CrossRef]
- Liechti-Gallati, S., Müller, B., Grimm, T., Kress, W., Müller, C., Boltshauser, E., Moser, H., & Braga, S. (1991). X-linked centronuclear myopathy: mapping the gene to Xq28. Neuromuscular disorders : NMD, 1(4), 239–245. [CrossRef]
- de Gouyon, B. M., Zhao, W., Laporte, J., Mandel, J. L., Metzenberg, A., & Herman, G. E. (1997). Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Human molecular genetics, 6(9), 1499–1504. [CrossRef]
- Al-Hashim, A., Gonorazky, H. D., Amburgey, K., Das, S., & Dowling, J. J. (2017). A novel intronic mutation in MTM1 detected by RNA analysis in a case of X-linked myotubular myopathy. Neurology. Genetics, 3(5), e182. [CrossRef]
- Paila, U., Chapman, B. A., Kirchner, R., & Quinlan, A. R. (2013). GEMINI: integrative exploration of genetic variation and genome annotations. PLoS computational biology, 9(7), e1003153. [CrossRef]
- Bonne, G., Rivier, F., & Hamroun, D. (2017). The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscular disorders : NMD, 27(12), 1152–1183. [CrossRef]
- Plon, S. E., Eccles, D. M., Easton, D., Foulkes, W. D., Genuardi, M., Greenblatt, M. S., Hogervorst, F. B., Hoogerbrugge, N., Spurdle, A. B., Tavtigian, S. V., & IARC Unclassified Genetic Variants Working Group (2008). Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Human mutation, 29(11), 1282–1291. [CrossRef]
- den Dunnen, J. T., & Antonarakis, S. E. (2000). Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Human mutation, 15(1), 7–12. [CrossRef]
- Dubowitz V, Sewry C.A, Oldfors A: Muscle Biopsy. A practical approach, 2021, 5th edition, Elsevier.
Figure 1.
Gene structure, variants distribution, and phenotype severity proportion. (A):MTM1 gene: Gene structure and three novel variants identified in Patients 1-4 (P1-P4); (B): Distribution of the disease-causing variants: n=385 variants (except large deletions, n=29) within the MTM1 gene; (C): Phenotype severity proportion: (n=414). Abbreviations: In – intron, Ex – exon.
Figure 1.
Gene structure, variants distribution, and phenotype severity proportion. (A):MTM1 gene: Gene structure and three novel variants identified in Patients 1-4 (P1-P4); (B): Distribution of the disease-causing variants: n=385 variants (except large deletions, n=29) within the MTM1 gene; (C): Phenotype severity proportion: (n=414). Abbreviations: In – intron, Ex – exon.
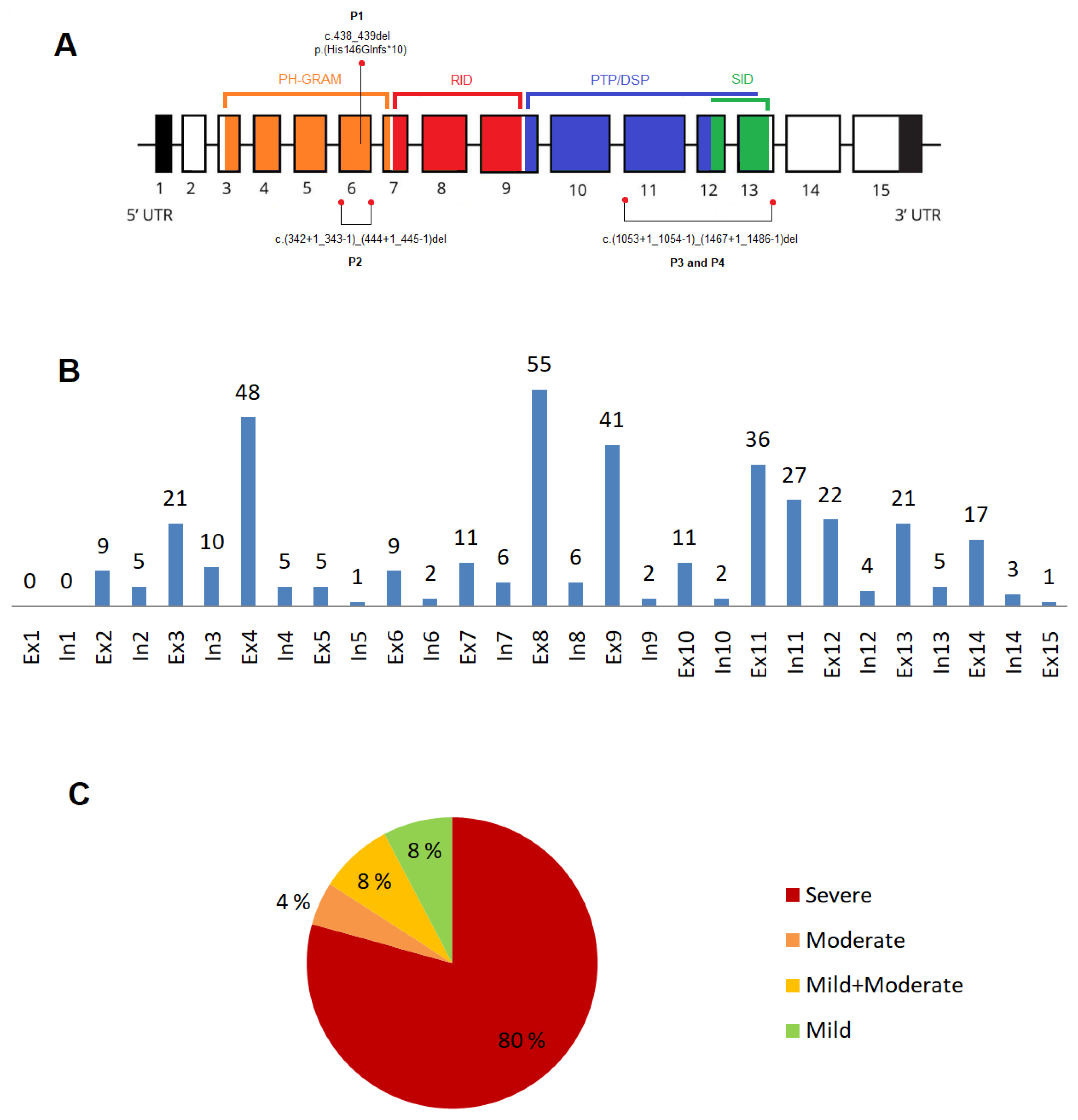
Figure 2.
Genotype-phenotype correlations in XLMTM. (A): Type of disease-causing variant in the MTM1 gene vs. phenotype: frameshift, nonsense, and in/del variants are significantly associated with a severe phenotype, as well as missense variants significantly correlate with mild or moderate phenotype, (B): truncation vs. phenotype: truncating variants are significantly associated with severe phenotype, (C): MTM1-specific domain vs. phenotype: there is no significant difference between specific domain and phenotype severity, and (D): nonsense-mediated mRNA decay (NMD) vs. phenotype: there is no significant difference between nonsense-mediated mRNA decay and phenotype severity.
Figure 2.
Genotype-phenotype correlations in XLMTM. (A): Type of disease-causing variant in the MTM1 gene vs. phenotype: frameshift, nonsense, and in/del variants are significantly associated with a severe phenotype, as well as missense variants significantly correlate with mild or moderate phenotype, (B): truncation vs. phenotype: truncating variants are significantly associated with severe phenotype, (C): MTM1-specific domain vs. phenotype: there is no significant difference between specific domain and phenotype severity, and (D): nonsense-mediated mRNA decay (NMD) vs. phenotype: there is no significant difference between nonsense-mediated mRNA decay and phenotype severity.
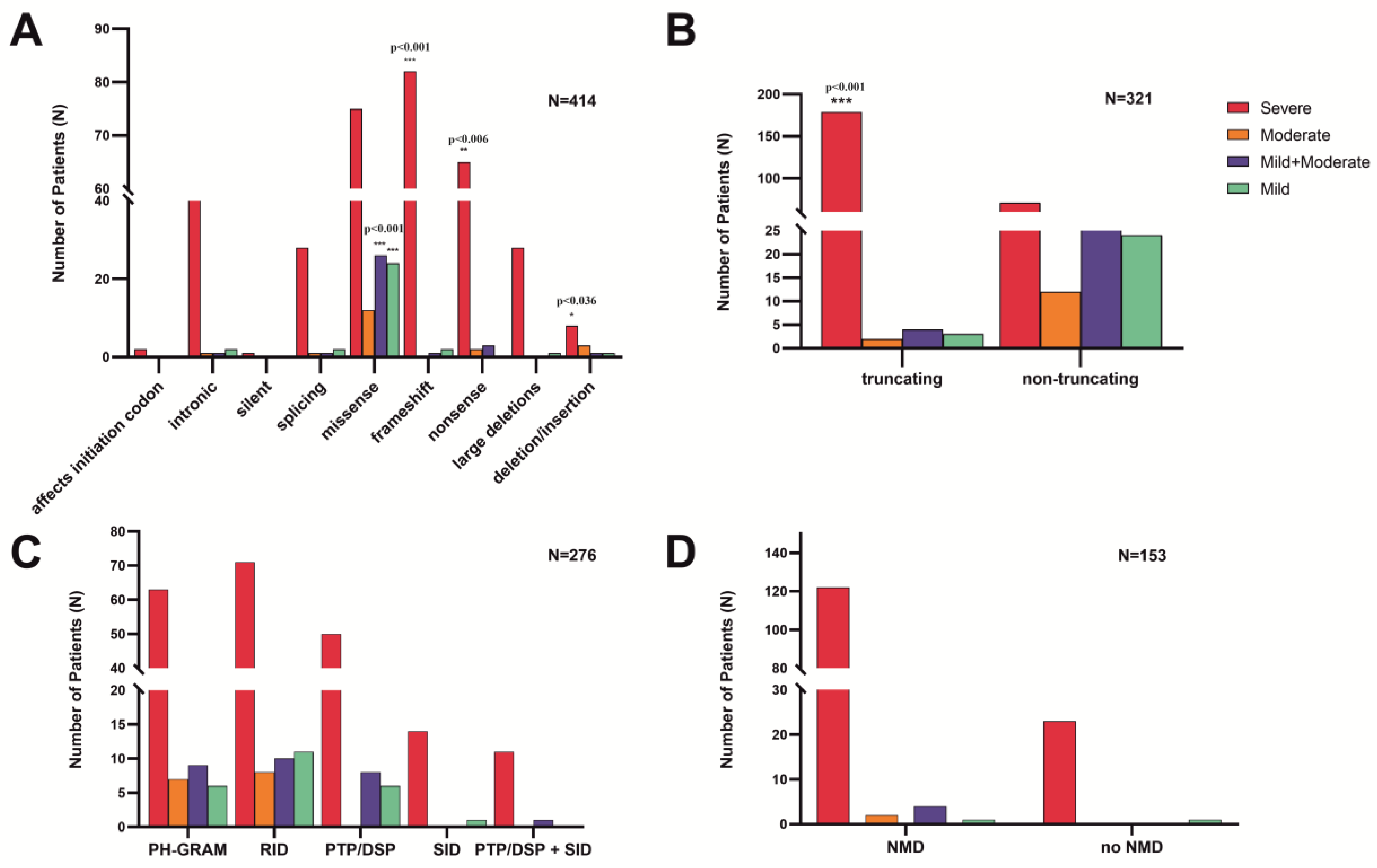
Figure 3.
Composite images from Face2Gene for XLMTM patients and healthy controls.


Table 1.
Three novel variants in the MTM1gene.
| Patient ID | Ancestry (Ethnicity) |
MTM1 variant | Type | Exon | Domain | ACMG classification | Origin | Phenotype |
|---|---|---|---|---|---|---|---|---|
|
P1/F1 M |
Slovakia (Caucasian) |
c.438_439delCA (p.His146Glnfs*10) | frameshift | ex 6 | PH-GRAM | pathogenic | maternal | Severe |
|
P2/F2 M |
Austria (Caucasian) |
c.(342+1_343-1)_(444+1_445-1)del p.(Asp115_Leu148del) |
in-frame deletion | ex 6 | PH-GRAM | likely pathogenic | maternal | Severe |
|
P3/F3 M |
Austria (Caucasian) |
c.(1053+1_1054-1)_(1467+1_1468-1)del; p.(Leu352_Gln489)del | in-frame deletion | ex 11-13 | PTP/DSP-SID | likely pathogenic | maternal | Severe |
|
P4/F3 M |
Austria (Caucasian) |
c.(1053+1_1054-1)_(1467+1_1468-1)del; p.(Leu352_Gln489)del | in-frame deletion | ex 11-13 | PTP/DSP-SID | likely pathogenic | maternal | Severe |
Abbreviations: P – patient, F – family, M- male, ex – exon.
Table 2.
Recurrent disease-causing variants in MTM1 gene with inter-individual phenotype variability in MTM1 cohort (n=414).
Table 2.
Recurrent disease-causing variants in MTM1 gene with inter-individual phenotype variability in MTM1 cohort (n=414).
| Variant | Type | Exon/intron | Domain | Count | Phenotype | Reference |
|---|---|---|---|---|---|---|
| c.98_103del, p.(Glu33_Ala34del) | deletion | ex 3 | PH-GRAM | 2 | 1S/1Mi | [31,32] |
| c.109C>T, p.(Arg37*) | nonsense | ex3 | PH-GRAM | 9 | 8S/1Mo | [20,22,24,27,33,34] |
| c.139_141del, p.(Lys47del) | deletion | ex4 | PH-GRAM | 5 | 4S/1Mo | [24,30,33,35] |
| c.142G>T, p.(Glu48*) | nonsense | ex 4 | PH-GRAM | 2 | 1S/1Mo | [7,21] |
| c.205C>T, p.(Arg69Cys) | missense | ex 4 | PH-GRAM | 12 | 2S/1Mo/6M/3Mi | [16,27,31,32,36] |
| c.232-26_232-23del | intronic | in4 | - | 2 | 1S/1Mo | [31,37] |
| c.590C>T, p.(Thr197Ile) | missense | ex8 | RID | 3 | 1S/1Mo/1Mi | [16,32] |
| c.614C>T, p.(Pro205Leu) | missense | ex8 | RID | 9 | 8S/1Mi | [21,22,24,27,31] |
| c.679G>A, p.(Val227Met) | missense | ex9 | RID | 3 | 1S/1M/1Mi | [16,27,31] |
| c.695A>G, p.(His232Arg) | missense | ex9 | RID | 2 | 1S/1Mo | [21,31] |
| c.721C>T, p.(Arg241Cys) | missense | ex9 | RID | 13 | 3S/1Mo/5M/4Mi | [16,21,31,33,34,35] |
| c.1262G>A, p.(Arg421Gln) | missense | ex12 | PTP/DSP | 8 | 7S/1M | [16,24,27,33,36] |
| c.1558C>T,p.(Arg520*) | nonsense | ex 14 | - | 3 | 1S/2M | [16,18] |
Abbreviations:in – intron, ex – exon, S – severe, Mo – moderate, M – mild and moderate [16], Mi– mild.
Table 3.
Binary comparison of the facial features of XLMTM patients, MD1 patients, NMDs patients, and healthy controls by using the Face2Gene application.
Table 3.
Binary comparison of the facial features of XLMTM patients, MD1 patients, NMDs patients, and healthy controls by using the Face2Gene application.
| Binary comparison | No. of cases | Mean AUC | AUC SD | p value for AUC |
|---|---|---|---|---|
| XLMTM vs. Unaffected | 14 vs. 11 | 1.00 | 0.01 | 0.001 |
| XLMTM vs. MD1 | 14 vs. 10 | 0.88 | 0.07 | 0.074 |
| XLMTM vs. NMDs | 14 vs. 11 | 0.63 | 0.09 | 0.229 |
| Unaffected vs. NMDs | 11 vs. 11 | 0.99 | 0.02 | 0.023 |
Abbreviations: AUC: area under the curve, SD: standard deviation, XLMTM: a cohort of X-linked myotubular myopathy patients, MD1: a cohort of patients with Myotonic dystrophy type 1, NMDs: a cohort of overall patients with neuromuscular disorder except XLMTM. A p-value < 0.05 represents a high degree of discrimination.
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



