
Submitted:
14 November 2023
Posted:
15 November 2023
You are already at the latest version
Abstract
In this work we developed two new polyfunctional hybrid systems, in which the presence of Fc redox "antennas" on peptide scaffolds allows a modulation of their electronic properties. Specifically, we synthesized two helical hexapeptides containing four Aib (α-amionoisobutyric acid) and two L-Dap (2,3-diamino propionic acid) residues. L-Dap side chains were then functionalized with Fc moieties. The structures of the two 310 helical peptides, namely Z-Aib-L-Dap(Fc)-Aib-Aib- L-Dap(Fc)-Aib-NH-iPr and Z-Aib-L-Dap(Fc)-Aib- L-Dap(Fc)-Aib-Aib-NH-iPr, were investigated by X-ray diffraction, 2D-NMR, CD and IR spectroscopies. Due to the helical conformation, in Z-Aib-L-Dap(Fc)-Aib-Aib- L-Dap(Fc)-Aib-NH-iPr the Fc groups are located on the same face of the helix, in Z-Aib-L-Dap(Fc)-Aib- L-Dap(Fc)-Aib-Aib-NH-iPr on opposite faces. Surprisingly, two bands were found in DPV for Z-Aib- L-Dap(Fc)-Aib- L-Dap(Fc)-Aib-Aib-NH-iPr, indicating electrostatic interaction between the Fc groups despite their longer reciprocal distance with respect to that in Z-Aib- L-Dap(Fc)-Aib-Aib- L-Dap(Fc)-Aib-NH-iPr. CD experiments at different concentrations evidenced aggregation for Z-Aib- L-Dap(Fc)-Aib-L-Dap(Fc)-Aib-Aib-NH-iPr even at high dilution, thus suggesting that the Fc-Fc electrostatic interaction could be of intermolecular nature.
Keywords:
ferrocene
; peptide
; cyclic voltammetry
; X-ray diffraction
1. Introduction
Systems containing both ferrocene (Fc) and biomolecules, such as amino acids, peptides, and nucleic acids, arise in conjugated organometallic compounds possessing novel properties. For instance, Fc-peptide conjugates were synthetized to exploit the intramolecular H-bonding network of foldamers and the Fc redox properties with the aim at studying the electronic interactions in peptide chains.
Bio-organometallics containing pendant ferrocenyl peptide conjugates were widely investigated [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22] allowing the design of systems with new redox activated properties among which potential bio-inspired electronic materials [9], changes in the self-assembly [12] and sensory applications [13,14,19].
In particular, we have functionalized with one Fc group, acting as a probe, peptides displaying different secondary structures, such as 310-helices [23,24] with α-amino isobutyric acid (Aib) [25] or L-2,3-diamino propionic (L-Dap) acid [26], and the 2.05-helix [24,27] based on Cα,β-didehydroalanine (ΔAla) [28]. Conversely, there are only few reports of bis-ferrocenyl peptides. To our knowledge, the unique examples of end-capped homopeptides were reported by us, namely Fc-CO-(Aib)n-NH−Fc (n = 1−5) [29], Fc-CO-(ΔAla)n-NH−Fc (n = 1−4) [30], Fc-CO-[L-Dap(Boc)]n-NH-Fc [31]. Besides, two dinuclear ferrocene-based conjugates containing a sequence of two amino acids (Fc−CO−L-Pro−L-Ala−NH−Fc and Fc−CO−D-Pro−D-Ala−NH−Fc) were synthetized by Nuskol et al. [32]. An intramolecular Fc-N-H⋅⋅⋅O=C-Fc hydrogen bond (β-turn) [33] is responsible for dipeptide folding.
By exploiting the terminal, redox-active Fc groups, utilizing electrochemical techniques, we were able to investigate the end-to-end effects of the positive charges generated by single and double oxidations. We demonstrated that charge transfer along the peptide chain is hugely affected by nature and length of the peptide 3D-structure.
The aim of the present work is the synthesis of new polyfunctional hybrid systems behaving as “electron reservoirs”, in which the simultaneous presence of Fc redox "antennas" inserted on peptide scaffolds can allow a realistic modulation of their electronic properties as a function of the applied electric potential. In particular, we designed and synthesized two helical hexapeptides containing different Aib/ L-Dap sequences but each with two Fc moieties covalently bound. The L-Dap side chain allowed the introduction of the Fc functionality. This is the first example of a helical peptide containing two Fc moieties appended laterally and not at the peptide ends, as so far reported by us and others. Aib, a highly helicogenic amino acid, was used exactly with the purpose to promote and stabilize the peptide helical structure, and in particular a 310-helix.
The two hexapeptides have the following sequence: Z-Aib-L-Dap(Fc)-Aib-Aib-L-Dap(Fc)-Aib-NH-iPr (1) and Z-Aib-L-Dap(Fc)-Aib-L-Dap(Fc)-Aib-Aib-NH-iPr (2) (Scheme 1).
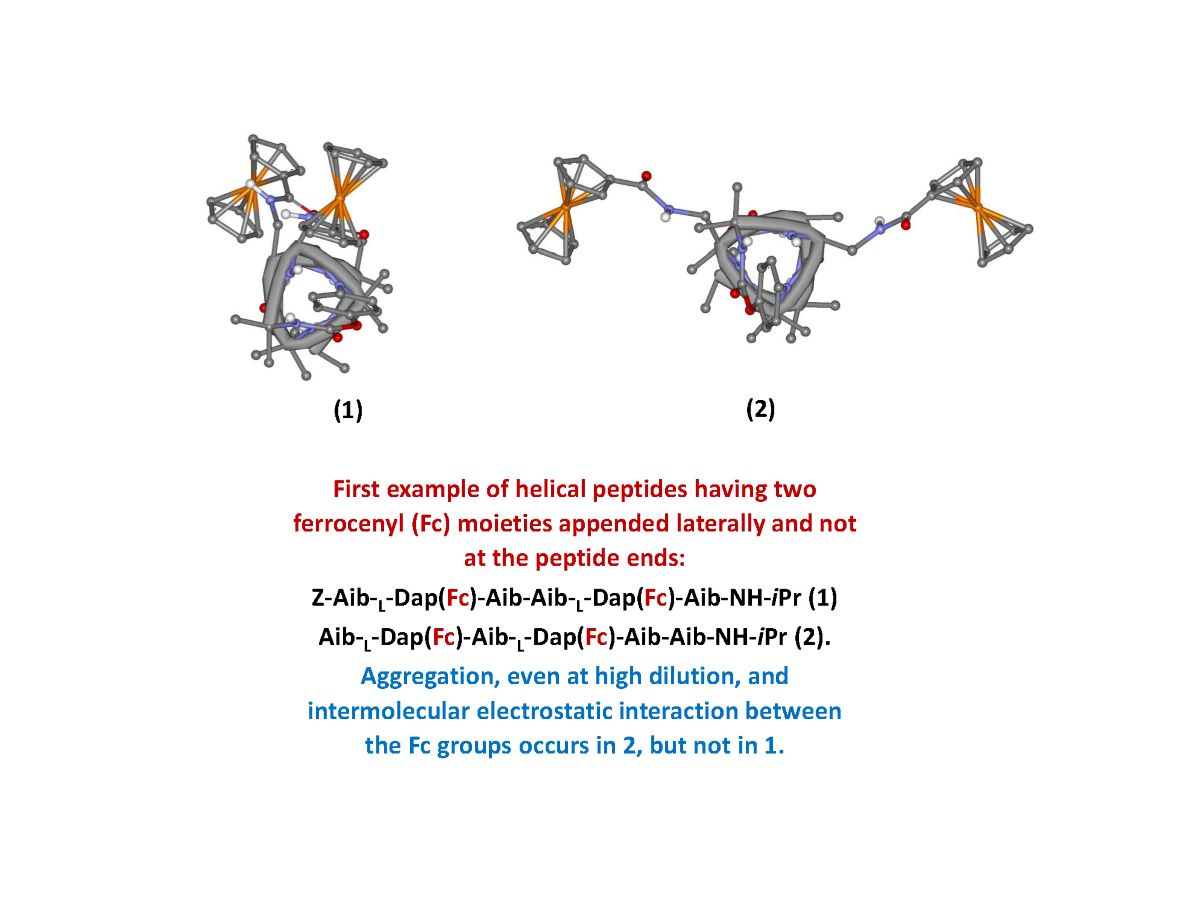
The two ferrocenes are linked to the side chain of L-Dap at reciprocal position 1,4 and 1,3. Due to the peptide conformation, a 310-helix, the Fc groups are expected to be spatially placed on the same side of the 310-helix in 1 and on opposite sides in 2, as clearly shown in Figure 1.
Information on peptide secondary structure in solution was obtained by means of NMR, IR and circular dichroism measurements. We were also able to determine the solid-state conformation of an intermediate peptide, namely Z-Aib-L-Dap(Boc)-Aib-NHiPr, by means of single-crystal X-ray diffraction analysis.
The metal-metal electronic interaction was assessed by electrochemical studies (CV and DPV).
2. Results and Discussion
2.1. Peptide Synthesis
The peptides were synthesized by solution methods, step-by-step, starting from the C-terminal residue. The formation of all amide bonds was achieved by activating the carboxylic function with EDC [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide] in the presence of HOBt (1-hydroxybenzotriazole). HOBt is a racemization suppressant, but it has also a catalytic effect towards the amide bond formation [34]. Therefore, it was used also in the coupling steps involving the achiral Aib residue. Yields were in general above 70%. This is a satisfactory result in view of the presence of four, sterically-hindered Aib residues in each hexapeptide. Only in the last step of the two hexapeptide syntheses, i.e., the one-shot incorporation of two bulky Fc moieties, we obtained moderate yields (35% and 34%). In both cases a silicagel flash chromatography was required to isolate the pure, doubly Fc-labelled hexapeptides. Experimental details of the synthetic procedures and characterizations of the peptides synthesized are given in the Supporting Information.
2.2. Crystal-State Conformational Analysis
The molecular structure of Z-Aib-L-Dap(Boc)-Aib-NHiPr, as determined by single-crystal X-ray diffraction analysis, is illustrated in Figure 2 with atom numbering. Relevant crystal data and structure refinement parameters are listed in Table S1.
Relevant backbone and side-chain torsion angles are listed in Table S2. Intra- and intermolecular H-bond parameters are reported in Table S3.
The peptide backbone is folded and stabilized by two intramolecular N−H⋅⋅⋅O=C H-bonds, one between the N3−H group and the O0 urethane carbonyl oxygen, and the other between the C-terminal isopropylamido NT−H group and the Aib(1) carbonyl oxygen (Figure 2 and Table S3).
Two consecutive β-turns [33] are thus formed, which give rise to an incipent 310-helix [23]. Indeed, the sets of φ,ψ backbone torsion angles adopted by Aib(1) [φ1,ψ1 = −51.5(2)°, −37.7(2)°], L-Dap(2) [φ2,ψ2 = −64.3(2)°, −26.2(2)°], and Aib(3) [φ3,ψ3 = −77.9(2)°, −15.9(3)°], are not far from those of a regular, right-handed 310-helix (φ,ψ = −57°, −30°) [24]. The largest distortion is shown by Aib(3), with φ,ψ values in between those typical for the i+2 corner position of type-III β-turn (the building unit of the 310-helix) and type-I (φ,ψ = −90°, 0°) [33].
As for the Nγ Boc-protected side chain of the L-Dap residue, the χ1 torsion angle is found in the g+ disposition, and the value of the torsion angle about the Cβ−Nγ bond is 107.0(2)° (Table S2). As a result, the potential H-bond donor and acceptor of the protected L-Dap side chain point away from the peptide backbone. In conclusion, the crystal state structure of this tripeptide amide supports the scope of our design, i.e., building stable 310-helical structures to the side of which append the Fc groups.
2.3. Solution Conformational Analysis
We investigated the preferred conformation of 1 and 2 in a solvent of low polarity (CDC13) by means of FT-IR absorption and 1HNMR. We also carried out circular dichroism measurements, but in this case we used MeOH as solvent because chloroform does not allow to examine the amide absorption region (190-250 nm), not being transparent at those wavelengths.
Figure 3 shows the FT-IR absorption spectra in the N–H stretching region (amide A) of Fc-hexapeptides 1 and 2 and of their shorter intermediates (from dimer to hexamer) at 1 mM concentration in CDCl3. In most cases two bands are present above 3400 cm−1 (free, solvated NH groups) and one major band in the range 3375–3310 cm−1 (H-bonded NH groups). The intensity of the low-frequency band increases and, concomitantly, the absorption maximum shifts to lower wavenumbers as the main-chain length increases. At 0.1 mM concentrations only minor changes occur in the spectra (Figure S2). Therefore, the observed H-bonding bands below 3375 cm−1 should be interpreted as arising almost exclusively from intramolecular NH···O=C interactions. This conclusion clearly supports the view that Fc-hexapeptides 1 and 2 adopt a helical conformation, most likely of the 310 type. Indeed, at the level of the dipeptide amides, Z-L-Dap-Aib-NHiPr and Z-(Aib)2-NHiPr (“di” in Figure 3, left and right panel, respectively), a β-turn is clearly formed. Obviously, it is more stable for Z-(Aib)2-NHiPr (“di” in the right panel) because of the presence of two, turn inducers, Aib residues. The absorptions above 3400 cm−1 are ascribable to the N-terminal amide NHs, not involved in a β-turn or a 310-helix, and to the Dap side-chain amide or urethane NHs.
Interestingly, while the H-bonding band at about 3320 cm−1 is relatively sharp for 1, in the case of 2 it is wide and displays a shoulder at about 3350 cm−1. Taking into account that the solution of 2 was cloudy, very likely the shape of the curve is due to aggregation. When this occurs through NH···O=C intermolecular H-bonds, a tenfold dilution dissolves the aggregates and the curves change dramatically. However, this is not the case for 2: at 0.1 mM peptide concentration the broad curve is still there (Figure S2). Therefore, it is possible that 2 forms aggregates through π···π stacking of the Fc moieties appended to the peptide side chains. Indeed, it is reasonable to assume that 2 behaves differently from 1 if we consider the spatial arrangements shown in Figure 1.
We were able to assign the 1H resonances of all Fc-peptide conjugates by 2D-NMR analysis. In particular, we evaluated the conformational preferences of 1 in CDCl3 solution. A helical structure is clearly present. Indeed, we observed sequential cross-peaks α/βCHi→NHi+1 and NHi→NHi+1 (Figure 4) and two long range α/βCHi→NHi+2 connectivity. These cross-peaks, involving Dap2 and Aib4 (data not shown), and Aib1 and Aib3 (Figure 5) are typical of the 310-helix. This finding is in agreement with the conformational properties observed in our FT-IR absorption analysis and also detected by CD spectroscopy.
Under the same conditions, peptide 2 exhibited high overlapping of signals in the amide region, thus preventing a complete assignment of all resonances. This evidence may be ascribed to aggregation phenomena, in agreement with the conclusions of our FT-IR and CD analyses.
The far-UV CD spectrum of 1 in methanol solution (Figure 6) displays two negative maxima at 218 and 205 nm, assigned to the n → π* transition and to the parallel component of the π → π* transition. The positive maximum at 190 nm is due to the antiparallel component of the π → π* transition. The presence of these three bands denotes that a peptide is folded into a helix, of type α or 310.
At 0.5 mM concentrations, both peptides display the typical curve of a helical peptide (Figure 6 left). However, for 2 the intensity of the maximum at 220 nm is more pronounced than that at 205 nm. The θ220/θ205 ratio is much higher than 1. This behavior is usually attributed to peptide aggregation [35,36]. Indeed, upon dilution the θ220/θ205 ratio drops below 1, thus confirming the hypothesis of aggregation for 2 at 0.5 mM concentration.
The CD spectra in the 300-600 nm region display a similar behavior for both compounds with two positive maxima at 344 nm and 407 nm together with two negative maxima at 307 nm and 468 nm, arising from the achiral Fc chromophore transitions (Figure 6, right panel). These induced CD bands are brought about by the ordered conformation of the host peptide. At 322 nm and 420 nm, corresponding to the UV absorptions of the ferrocene moiety, the CD signal for both compounds cancel out due to the Cotton effect.
2.4. CV and DPV Analysis
Ferrocene is known to be very stable in solution and air, easy to functionalize and displays a reversible electrochemical behavior, making it suitable in labeling biomolecules [37]. Moreover, the electrochemistry of Fc-peptides gives insights on effect of folding and length of the peptide chain [25,26,27,28,29,30,31,32].
Cyclic voltammetry (CV) of the bis-Fc hexapeptides 1 and 2, recorded under argon in CH2Cl2/0.1 M nBu4NPF6 at potential scan rates in the range 0.1−5 Vs−1, shows reversible oxidation waves with oxidation potential Ep = 0.54 and 0.62 V vs SCE, respectively. (Figure 7a), consistent with the chemical reversibility criteria in the range of scan rates of 0.1−5 V s−1 as they all showed cathodic/anodic peak current ratios of ia/ic = 1.
However, at first glance, a comparison of the two curves suggests the presence of an incipient splitting in 2, thus implying that the ferrocenyl groups are electrochemically distinguishable in this peptide. However, the 2:1 (1.97) ratio of the two peaks areas (0.67 and 0.58 V), evidenced by Gaussian deconvolution of the differential pulse voltammetry (DPV) analysis reported in Figure 7b, indicates the two different species are present.
The FT-IR and CD analyses indicate that 2 undergoes to intermolecular aggregation. Therefore, the splitting of the DPV peak may arise from different, intermolecular electrostatic interactions between Fc groups belonging to different peptide molecules.
A possible interpretation of the DPV peaks is sketched in Figure 8, which could explain the 2:1 ratio. In fact, the aggregation of three peptides 2 in an antiparallel orientation (Figure 8a), would result in two different relative position of Fc, the one having two couples of Fc in close proximity (red) interacting through stacking and responsible of a 1:1 splitting of the DPV peaks at Ep = 0.54 and 0.62 V due to electrostatic interaction (two one electron waves), the other possessing two far-away disposed Fc (blue), causing a single peak superposed at 0.54 V (one two-electron wave). Conversely, aggregation of only two peptides (Figure 8b) should arise a 3:1 ratio.
2.5. Vis-MIR-IR Chemical Oxidation
The oxidized species production was examined in the visible (Figure 8) and IR (Figure 9) regions. Stable solutions of 12+ and 22+ in CH2Cl2/0.1 M nBu4NPF6 were obtained by succesive addition of increasing amounts of acetylferrocenium[BF4] (up to 2 equivalent) to the the neutral peptides. In the visible region, the absorption band appearing at 620 nm is due to the Fc+-CO- group [26]. For example, the visible spectra acquired by the stepwise oxidation of 1 (bold black line, Figure 9) clearly indicates the formation of 12+ dication (bold red line) due to the simultaneous oxidation of both Fc groups.
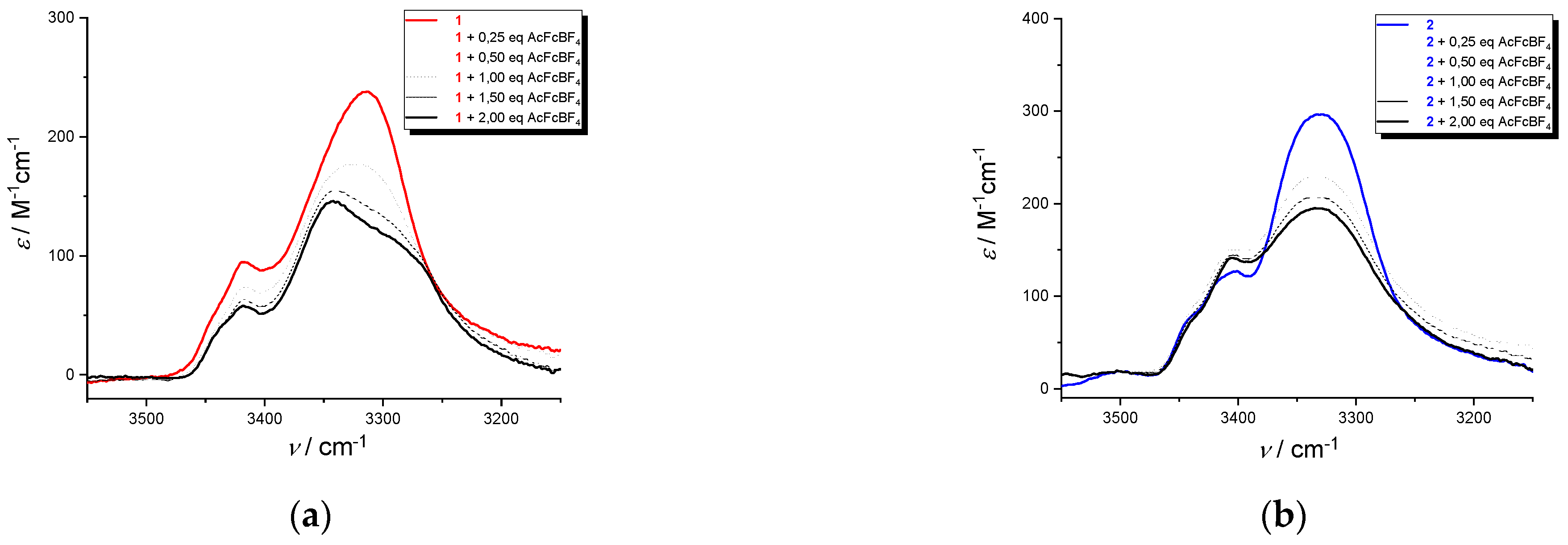
Upon oxidation with acetylferricinum of 1 and 2, the bands in the range 3440-3400 cm−1, due to a non H-bonded amide N−H (Dap side-chain NH and the two N-terminal amide NH) are insensitive to oxidation, (Figure 10a,b). As for the amide A of 1 at 3315 cm−1 (NHpept/H-bond), arising from the intramolecular H-bonds stabilizing the peptide helix,, a split in two components is observed. It suggests a release (3343 cm−1) and a reinforcement (3285 cm−1) of H-bond due to the presence of the positive charges on Fc groups, with a fast equilibrium between bonded and non-bonded conformations.
This splitting is absent the amide A band of 2 at 3330 cm−1 (NHpept/H-bond), indicating that the H-bonds stabilizing the helical conformation are not affected by the presence of the positive charges.
3. Materials and Methods
3.1. Synthesis and Analyses
The synthesis of the peptides were performed by solution methods in an oxygen and moisture-free atmosphere. Solvents were dried by reflux over the appropriate drying agent and distilled under stream of argon. Ferrocenecarboxylic acid was Alfa Aesar product, 1-hydroxy-benzotriazole hydrate (HOBt), diisopropylethylamine (DIEA) and 2-aminopropane were Sigma Aldrich products, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), and Nα-benzyloxycarbonyl-Nβ-(t-butyloxycarbonyl)-L-2,3-diaminopropionic acid [Z-L-Dap(Boc)-OH] were Iris Biotech products. Z-Aib-OH and Z-Aib-NHiPr were prepared by reported procedure. Microanalyses were performed at the Department of Chemical Sciences, University of Padova. MS spectra were obtained using an Agilent 6130 ESI-TOF mass spectrometer coupled to a HPLC system, collecting data in the positive mode. All compounds were analyzed by analytical HPLC and were >95% pure.
3.2. X-Ray Diffraction
Crystals of Z-Aib-L-Dap(Boc)-Aib-NHiPr were grown by slow evaporation from an ethyl acetate / hexane solvent mixture. X-Ray diffraction data were collected with a Gemini E four-circle kappa diffractometer (Agilent Technologies) equipped with a 92 mm EOS CCD detector, using graphite monochromated Cu Kα radiation (λ = 1.54184 Å). Data collection and reduction were performed with the CrysAlisPro software system (version 1.171.40.60a, Rigaku Corporation, Wroclaw, Poland). A semi-empirical absorption correction based on the multi-scan technique using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm, was applied. The structure was solved by ab initio procedures of the SIR 2014 program [38]. The trial solution with the best combined figure of merit allowed location of all non-H atoms. Refinement was carried out by full-matrix least-squares on F2, using all data, by application of the SHELXL-2014 program [39], with anisotropic displacement parameters for all of the non-H atoms. H-Atoms were calculated at idealized positions and refined using a riding model. Relevant crystal data and structure refinement parameters are listed in Table S1. CCDC 2306243 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
3.3. Nuclear Magnetic Resonance
1H spectra were obtained on a Bruker Avance NEO-600 spectrometer operating at 600 MHz equipped with a Prodigy cryoprobe and on a Bruker Avance III HD spectrometer operating at 400.13 MHz (T=298K). The peptide concentration in solution was 1 mM in spectrograde CHCl3-d1 (99.8 % d1 containing 0.5 %wt. of silver foils as stabilizers and 0.03 % (v/v) tetramethylsilane - Sigma Aldrich) and DMSO-d6 (99.96% D-Eurisotop). Processing and evaluation of the experimental data were carried out using the TOPSPIN software packages. All homonuclear spectra were acquired by collecting 400 experiments, each consisting of 32 scans and 2K data points. The spin systems of the coded amino acid residues were identified using standard double-quantum filtered-COSY [40] and clean TOCSY spectra [41]. In the latter case, the spin-lock pulse sequence was 70 ms long. NOESY experiments were utilized for sequence-specific assignments [42,43].
3.4. Cyclic Voltammetry
The experiments were performed in an air-tight three-electrode cell connected to a vacuum/argon Schlenk line. Dichloromethane solvent was pre-dried with anhydrous calcium chloride, refluxed over calcium hydride and distilled under stream of Argon. Solvent and nBu4NPF6 were degassed in a Schlenk flask by manifold freeze-pump-thaw cycles and transferred by cannula in the cell. The reference electrode was a SCE (Tacussel ECS C10) separated from the solution by a bridge compartment filled with the same solvent/supporting electrolyte solution used in the cell. The counter electrode was a platinum spiral with around 1 cm2 apparent surface area. The working electrode was a disk obtained from cross section of a gold wire with 0.5 and 0.125 mm diameter sealed in glass. Between successive scans, the working electrode was polished on alumina according to standard procedures and sonicated before use. An EG&G PAR-175 signal generator was used. The currents and potentials were recorded on a Lecroy 9310L oscilloscope. The potentiostat was home-built with a positive feedback loop for compensation of the ohmic drop [43].
3.5. Differential Pulse Voltammetry
Autolab PGSTAT 30 potentiostat/galvanostat (EcoChemie, The Netherlands) run by a PC with GPES software was used for the DPV experiments. The measurements were conducted an air-tight three electrode cell, the same used for the CV experiments. Peak amplitude of 50 mV, pulse width of 0.05 s, 2 mV increment per cycle, and pulse period of 0.1 s.
3.6. UV-Vis Analysis
Solution absorption spectra in the UV-vis region were recorded at 293 K with a JASCO V770 double beam spectrophotometer using quartz cells with 1 mm optical path. A spectrum of CH2Cl2 (baseline) was recorded under the same conditions. Oxidation was achieved in an air-tight container connected to a vacuum/argon line by incremental addition of oxidizing agent solution (acetylferrocenium[BF4]/CH2Cl2 from 0.1 to 2.0 equivalents). HPLC grade CH2Cl2 ≥ 99% was purchased from Carlo Erba and distilled on CaH2 (≥ 97% powder Sigma Aldrich). Cells with path lengths of 1 mm (with quartz windows) were used.
3.7. FT-IR Analysis
Solution FT-IR absorption spectra were recorded at 293 K using a FT-IR Nicolet Nexus 670 spectrophotometer, nitrogen flushed, equipped with a sample-shuttle device, at 2 cm−1 nominal resolution, averaging 25 scans. Solvent (baseline) spectra were recorded under the same conditions. Spectrograde CHCl3-d1 (99.8%, d). HPLC grade CH2Cl2 ≥ 99% was purchased from Carlo Erba and distilled on CaH2 (≥ 97% powder Sigma Aldrich). For spectral elaboration, the software SpectraCalc provided by Galactic (Salem, MA) was employed. Cells with path lengths of 1 mm (with CaF2 windows) were used.
3.8. Circular Dichroism
The CD spectra were obtained on a Jasco (Tokyo, Japan) model J-1500 spectropolarimeter. Cylindrical fused quartz cells (Hellma, Müllheim, Germany) of 0.02 cm and rectangular cells of 0.1 cm path length were used. The values are expressed in terms of [θ]T (deg x cm2 x dmol−1). Spectrograde CH3OH (Merck) was used as solvent.
4. Conclusions
Surprisingly, two bands were found in the DPV for 2, indicating electrostatic interaction between the Fc groups despite their longer reciprocal distance, whereas a single and narrow peak was observed for 1. Circular Dichroism (CD) and FT-IR absorption experiments at different concentrations confirmed the tendence of 2 to aggregate even at high dilution, a behavior compatible with the presence of intermolecular, Fc-Fc electrostatic interactions.
To conclude, we reported for the first time the synthesis and spectroscopic characterizations of helical peptides having two Fc moieties appended to their sides, not to their ends. We observed quite different behaviors, primarily depending on the spatial localization of the two Fc groups (same or opposite sides of the peptide helix). In particular, we hypothesized the possible formation of aggregates through stacking interactions of side-chain Fc.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Synthetic details, Figure S1: Packing mode of Z-Aib-L-Dap(Boc)-Aib-NHiPr as viewed down the b axis. Intermolecular N-H…O=C H-bonds are indicated by dashed lines, Figure S2, FT-IR spectra in the amide A region of the Fc-hexapeptides 1 (left) and 2 (right) and of their shorter intermediates, from the dipeptide to the hexapeptide. Peptide concentration: 0.1 mM in CDCl3; Table S1: Crystal data and structure refinement for Z-Aib-L-Dap(Boc)-Aib-NHiPr; Video S1: title, Table S2: Selected torsion angles [°] for Z-Aib-L-Dap(Boc)-Aib-NHiPr, Table S3: Hydrogen bonds for Z-Aib-L-Dap(Boc)-Aib-NHiPr [Å and °].
Author Contributions
Conceptualization, S.S. and F.F.; X-Ray diffraction analysis, M.C.; solution conformational analyses, B.B., A.B., R.C. and R.S.; electrochemical studies, S.S., R.C.; Circular Dichroism F.F. and R.S.: data curation, S.S., B.B.; A.B., R.C. R.S. and M.C.; writing—original draft preparation, S.S.; writing—review and editing, S.S., B.B., A.B., R.C., R.S., M.C. and F.F.; funding acquisition, S.S. and F.F. All authors have read and agreed to the published version of the manuscript.
Funding
S.S. acknowledges the University of Padova that funded this research through Grant P-DiSC-2018. S.S., B.B. and F.F. are grateful to the Italian Ministry of Research for the financial support through PRIN 2020 N. 2020833Y75.
Data Availability Statement
CCDC 2306243 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
References
- Moriuchi, T. Helical Chirality of Ferrocene Moieties in Cyclic Ferrocene-Peptide Conjugates. Eur. J. Inorg.Chem. 2022, e202100. [Google Scholar] [CrossRef]
- Tassinari, F.; Jayarathna, D.R.; Kantor-Uriel, N.; Davis, K.L.; Varade, V.; Achim, C.; Naaman, R. Chirality Dependent Charge Transfer Rate in Oligopeptides. Adv. Mat. 2018, 30, 1706423. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Tong, F.; Zhang, W.; Zhou, Y.; He, S.Q.; Xie, R.; Lei, T.; Wang, Y.S.; Peng, S.J.; Li, Z.F.; Leong, J.; Gao, H.L.; Lu, L.G. Self-Delivered Supramolecular Nanomedicine with Transformable Shape for Ferrocene-Amplified Photodynamic Therapy of Breast Cancer and Bone Metastases. Adv. Funct. Mater. 2021, 31, 2104645. [Google Scholar] [CrossRef]
- Gómez, J.; Sierra, D.; Ojeda, C.; Thavalingam, S.; Miller, R.; Guzmán, F.; Metzler-Nolte, N. Solid-phase synthesis and evaluation of linear and cyclic ferrocenoyl/ruthenocenoyl water-soluble hexapeptides as potential antibacterial compounds. J. Biol. Inorg. Chem. 2021, 26, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Gurunarayanan, V.; Ramapanicker, R. Amphiphilic conjugates of ferrocene with amino acids and peptides: Design, synthesis, and studies on their aggregation behavior. J. Pep. Sci. 2021, 27, e3332. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.C.S.; Piccoli, J.P.; Santos-Filho, N.A.; Clementino, L.C.; Fusco-Almeida, A.M.; De Annunzio, S.R.; Fontana, C.R.; Verga, J.B.; Eto, S.F.; Pizauro-Junior, J.M.; Graminha, M.A.S. Cilli; E.M. Antimicrobial Activity of RP-1 Peptide Conjugate with Ferrocene Group. PLoS ONE 2020, 15, e0228740. [Google Scholar] [CrossRef]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef]
- Falcone, N.; Kraatz, H.-B. Ferrocene Peptide-based Supramolecular Gels: Current Trends and Applications. In Advances in Bioorganometallic Chemistry; Hirao, T.; Moriuchi, T., Eds.; Elsevier, 2019; Chapter 3, pp 57−74. [CrossRef]
- Yu, J.; Horsley, J.R.; Abell, A.D. Peptides as Bio-Inspired Electronic Materials: An Electrochemical and First-Principles Perspective. Acc. Chem. Res. 2018, 51, 2237–2246. [Google Scholar] [CrossRef]
- Albada, B.; Metzler-Nolte, N. Highly Potent Antibacterial Organometallic Peptide Conjugates. Acc. Chem. Res. 2017, 50, 2510–2518. [Google Scholar] [CrossRef]
- Hirao, T.; Moriuchi, M.; Groß, A. Functionalized Redox Systems. Synthetic Reactions and Design of π- and Bio-Conjugates. In Functionalized Redox Systems Chp. 4 Bioconjugates to Induce Chirality Organization. Editors: Hirao, T., Ed. Springer, 2015; Chapter 4, pp 111–150.
- Adhikariab, B.; Kraatz, H.-B. Redox-triggered changes in the self-assembly of a ferrocene–peptide conjugated. Chem. Commun. 2014, 50, 5551–5553. [Google Scholar]
- Takahashi, S.; Anzai, J.-I. Recent Progress in Ferrocene-Modified Thin Films and Nanoparticles for Biosensors. Materials 2013, 6, 5742–5762. [Google Scholar] [CrossRef] [PubMed]
- Zuccarello, L.; Barbosa, C.; Todorovic, S.; Silveira, C.M. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts 2021, 11, 218. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, P.; Jin, X.; Wang, Y.; Yuan, H.; Zhu, X. Enzymatic biofuel cells based on protein engineering: Recent advances and future prospects. Biomater. Sci. 2020, 8, 5230–5524. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Myung, N.V. Recent Advances in the Direct Electron Transfer-Enabled Enzymatic Fuel Cells. Front. Chem. 2021, 8, 620153. [Google Scholar] [CrossRef]
- Zhang, L.; Yagnik, G.; Peng, J.; Wang, J.; Xu, H.H.; Hao, Y.; Liu, Y.-N.; Zhou, F. Kinetic Studies of Inhibition of the Aβ(1–42) Aggregation Using a Ferrocene-tagged β-Sheet Breaker Peptide. Anal Biochem. 2013, 434, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, R.; Kraatz, H.B. Stimuli-Responsive Supramolecular Gelation in Ferrocene–Peptide Conjugates. Chem. Eur. J. 2013, 19, 17296–17300. [Google Scholar] [CrossRef] [PubMed]
- Martić, S.; Labib, M.; Shipman, P.-O.; Kraatz, H.-B. Ferrocene-peptido conjugates: From synthesis to sensory applications. Dalton Trans. 2011, 40, 7264–7290. [Google Scholar] [CrossRef] [PubMed]
- Moriuchi, T.; Kikushima-Honda, N.; Ohmura, S.D.; Hirao, T. Design and characterization of ferrocene–peptide–oligoaniline conjugates. Tetrahedron Letters 2010, 4530–4533. [Google Scholar] [CrossRef]
- Okamoto, S.; Morita, T.; Kimura, S. Electron Transfer through a Self-Assembled Monolayer of a Double-Helix Peptide with Linking the Terminals by Ferrocene. Langmuir 2009, 25, 3297–3304. [Google Scholar] [CrossRef]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef]
- Toniolo, C.; Benedetti, E. The polypeptide 310-helix. Trends Biochem. Sci. 1991, 16, 350–353. [Google Scholar] [CrossRef]
- Aravinda, S.; Datta, S.; Shamala, N.; Balaram, P. Hydrogen-Bond Lengths in Polypeptide Helices: No Evidence for Short Hydrogen Bonds. Angew. Chem. Int. Ed. 2004, 43, 6728–6731. [Google Scholar] [CrossRef]
- Donoli, A.; Marcuzzo, V.; Moretto, A.; Toniolo, C.; Cardena, R.; Bisello, A.; Santi, S. Charge Mapping in 310-Helical Peptide Chains by Oxidation of the Terminal Ferrocenyl Group. Org. Lett. 2011, 13, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Biondi, B.; Bisello, A.; Cardena, R.; Schiesari, R.; Facci, M.; Cerveson, L.; Rancan, M.; Formaggio, F.; Santi, S. Conformational Analysis and Through-Chain Charge Propagation in Ferrocenyl-Conjugated Homopeptides of 2,3-Diaminopropionic acid (Dap). Eur. J. Inorg. Chem. 2022, e202100. [Google Scholar] [CrossRef]
- Crisma, M.; Formaggio, F.; Alemán, C.; Torras, J.; Ramakrishnan, C.; Kalmankar, N.; Balaram, P.; Toniolo, C. The fully-extended conformation in peptides and proteins. Pept. Sci. 2018, 110, e23100. [Google Scholar] [CrossRef]
- Biondi, B.; Bisello, A.; Cardena, R.; Schiesari, R.; Cerveson, L.; Facci, M.; Rancan, M.; Formaggio, F.; Santi, S. Flat, Ferrocenyl-Conjugated Peptides: A Combined Electrochemical and Spectroscopic Study. ChemElectroChem 2021, 8, 2693–2700. [Google Scholar] [CrossRef]
- Donoli, A.; Marcuzzo, V.; Moretto, M.; Crisma, M.; Toniolo, C.; Cardena, R.; Bisello, A.; Santi, S. New bis-Ferrocenyl End-Capped Peptides: Synthesis and Charge Transfer Properties. Pept. Sci. 2012, 100, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Santi, S.; Bisello, A.; Cardena, R.; Tomelleri, S.; Schiesari, R.; Biondi, B.; Crisma, M.; Formaggio, F. Flat, Cα,β-Didehydroalanine Foldamers with Ferrocene Pendants: Assessing the Role of α-Peptide Dipolar Moments. ChemPlusChem 2021, 86, 723–730. [Google Scholar] [CrossRef]
- Santi, S.; Biondi, B.; Cardena, R.; Bisello, A.; Schiesari, R.; Tomelleri, S.; Facci, M.; Crisma, F.; Formaggio, F. Helical versus Flat Bis-Ferrocenyl End-Capped Peptides: The Influence of the Molecular Skeleton on Redox Properties. Molecules 2022, 27, 6128. [Google Scholar] [CrossRef]
- Nuskol, M.; Studen, M.; Meden, A.; Kodrin, I.; Cakic Semencic, M. Tight turn in dipeptide bridged ferrocenes: Synthesis, X-ray structural, theoretical and spectroscopic studies. Polyhedron 2019, 161, 137–144. [Google Scholar] [CrossRef]
- Venkatachalam, C.M. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 1968, 6, 1425–1436. [Google Scholar] [CrossRef]
- König, W.; Geiger, R. A new method for synthesis of peptides: Activation of the carboxyl group with dicyclohexylcarbodiimide using 1-hydroxybenzotriazoles as additives. Chem. Ber. 1970, 103, 788–798. [Google Scholar] [CrossRef]
- Zhou, N.E.; Kay, C.M.; Hodges, R.S. Synthetic model proteins: The relative contribution of leucine residues at the nonequivalent positions of the 3-4 hydrophobic repeat to the stability of the two-stranded α-helical coiled-coil. Biochemistry 1992, 31, 5739–5746. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, P.; Kennedy, R.J.; Kemp, D.S. Large Circular Dichroism Ellipticities for N-Templated Helical Polypeptides Are Inconsistent with Currently Accepted Helicity Algorithms. Angew. Chem. Int. Ed. 1999, 38, 1290–1292. [Google Scholar] [CrossRef]
- Metzler-Nolte, N.; Salmain, M. Ferrocenes: Ligands, Materials and Biomolecules; Štepnicka, P., Ed.; Wiley: Chichester, UK, 2008; pp. 499–639. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Rance, M.; Sørensen, O.W.; Bodenhausen, G.; Wagner, G.; Ernst, R.R.; Wüthrich, K. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983, 117, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, C.; Otting, G.; Wüthrich, K.; Ernst, R.R. Clean TOCSY for proton spin system identification in macromolecules. J. Am. Chem. Soc. 1988, 110, 7870–7872. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids, Wiley, 1991.
- Boros, S.; Gáspári, Z.; Batta, G. Accurate NMR Determinations of Proton-Proton Distances. Annu. Rep. NMR Spectro. 2018, 94, 1–39. [Google Scholar]
- Amatore, C.; Lefrou, C.; Pflüger, F. On-line compensation of ohmic drop in submicrosecond time resolved cyclic voltammetry at ultramicroelectrodes. J. Electroanal. Chem. 1989, 270, 43–59. [Google Scholar] [CrossRef]
Scheme 1.
Molecular structures of the two Fc-hexapeptides investigated.
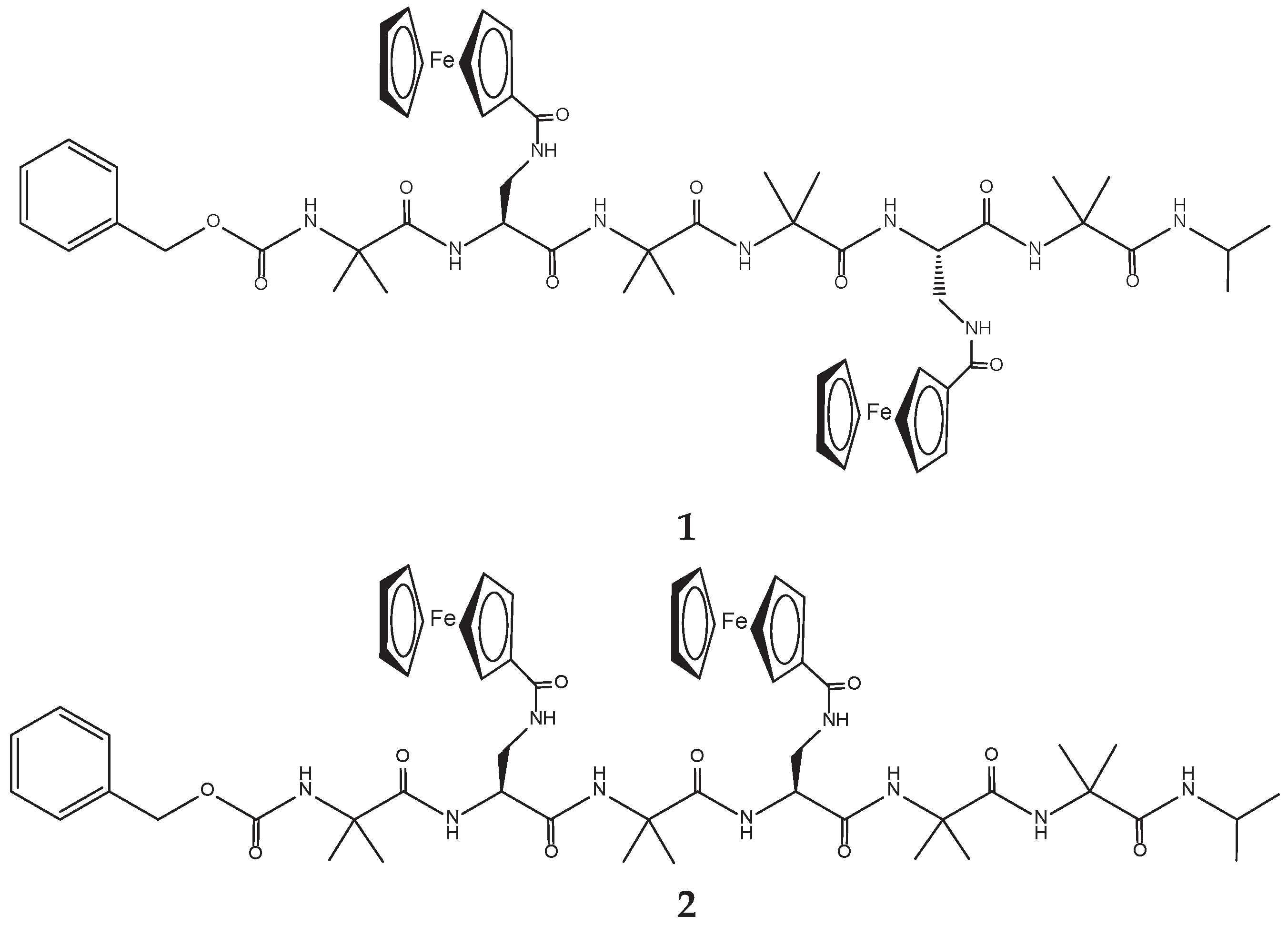
Figure 1.
Molecular models of 1 and 2 folded into a 310-helix.
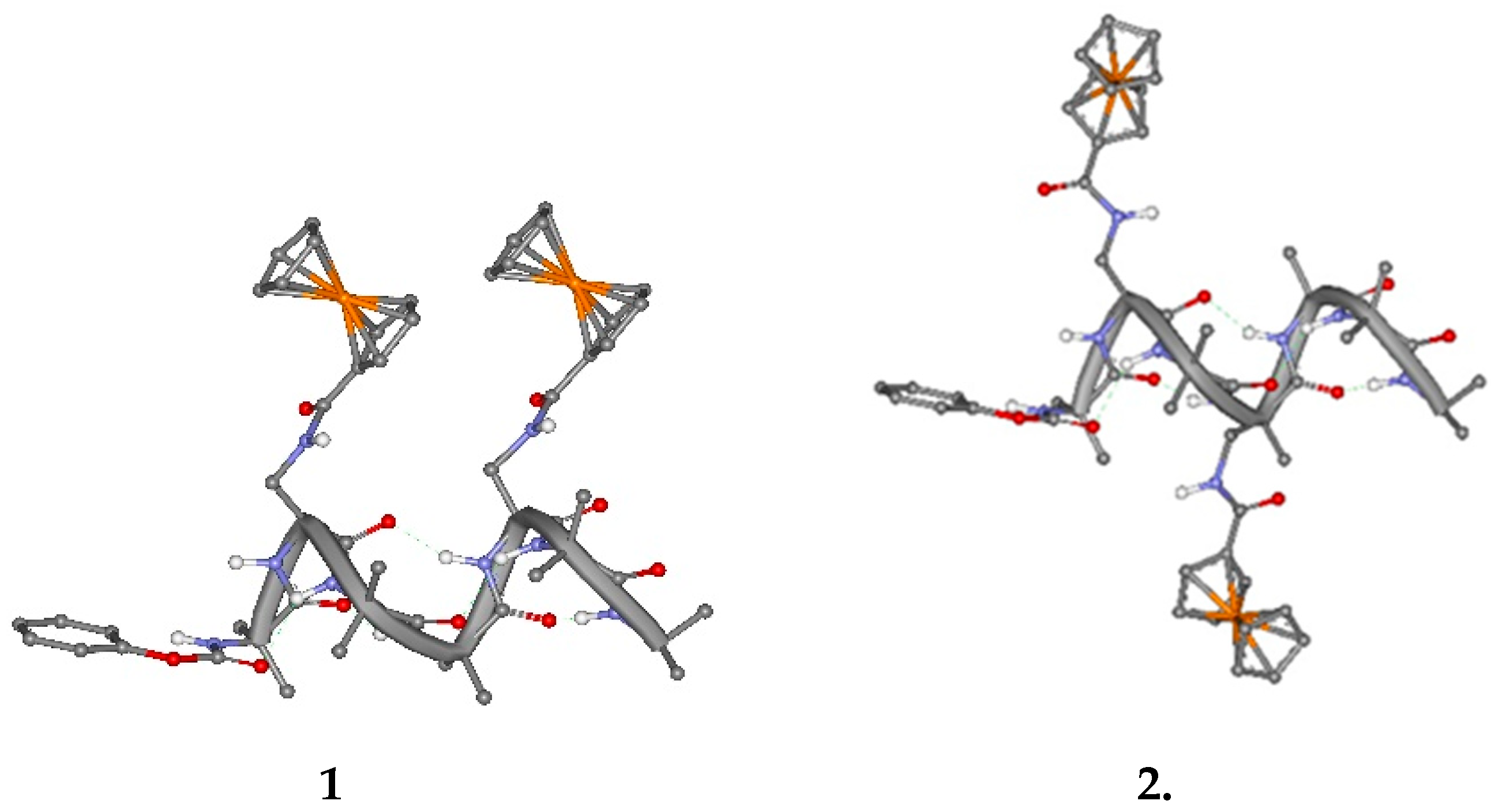
Figure 2.
X-Ray diffraction structure of Z-Aib-L-Dap(Boc)-Aib-NHiPr. Most of the H-atoms are omitted for clarity. Anisotropic displacement ellipsoids for the non-H atoms are drawn at the 30% probability level. The two intramolecular N-H⋅⋅⋅O=C H-bonds are indicated by turquoise lines.
Figure 2.
X-Ray diffraction structure of Z-Aib-L-Dap(Boc)-Aib-NHiPr. Most of the H-atoms are omitted for clarity. Anisotropic displacement ellipsoids for the non-H atoms are drawn at the 30% probability level. The two intramolecular N-H⋅⋅⋅O=C H-bonds are indicated by turquoise lines.
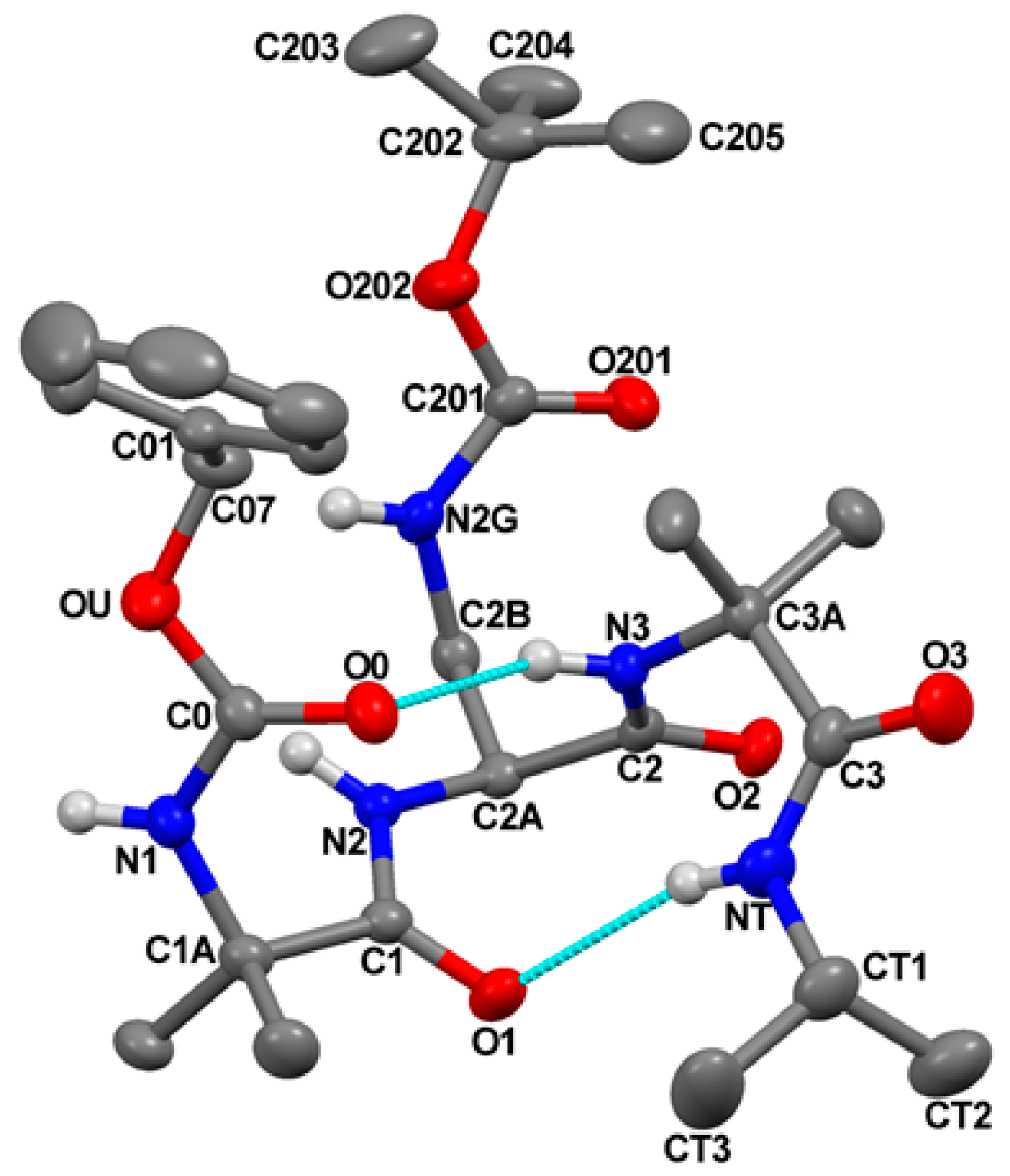
Figure 3.
FT-IR spectra in the amide A region of the Fc-hexapeptides 1 (left) and 2 (right) and of their shorter intermediates, from the dipeptide to the hexapeptide. Peptide concentration: 1 mM in CDCl3.
Figure 3.
FT-IR spectra in the amide A region of the Fc-hexapeptides 1 (left) and 2 (right) and of their shorter intermediates, from the dipeptide to the hexapeptide. Peptide concentration: 1 mM in CDCl3.
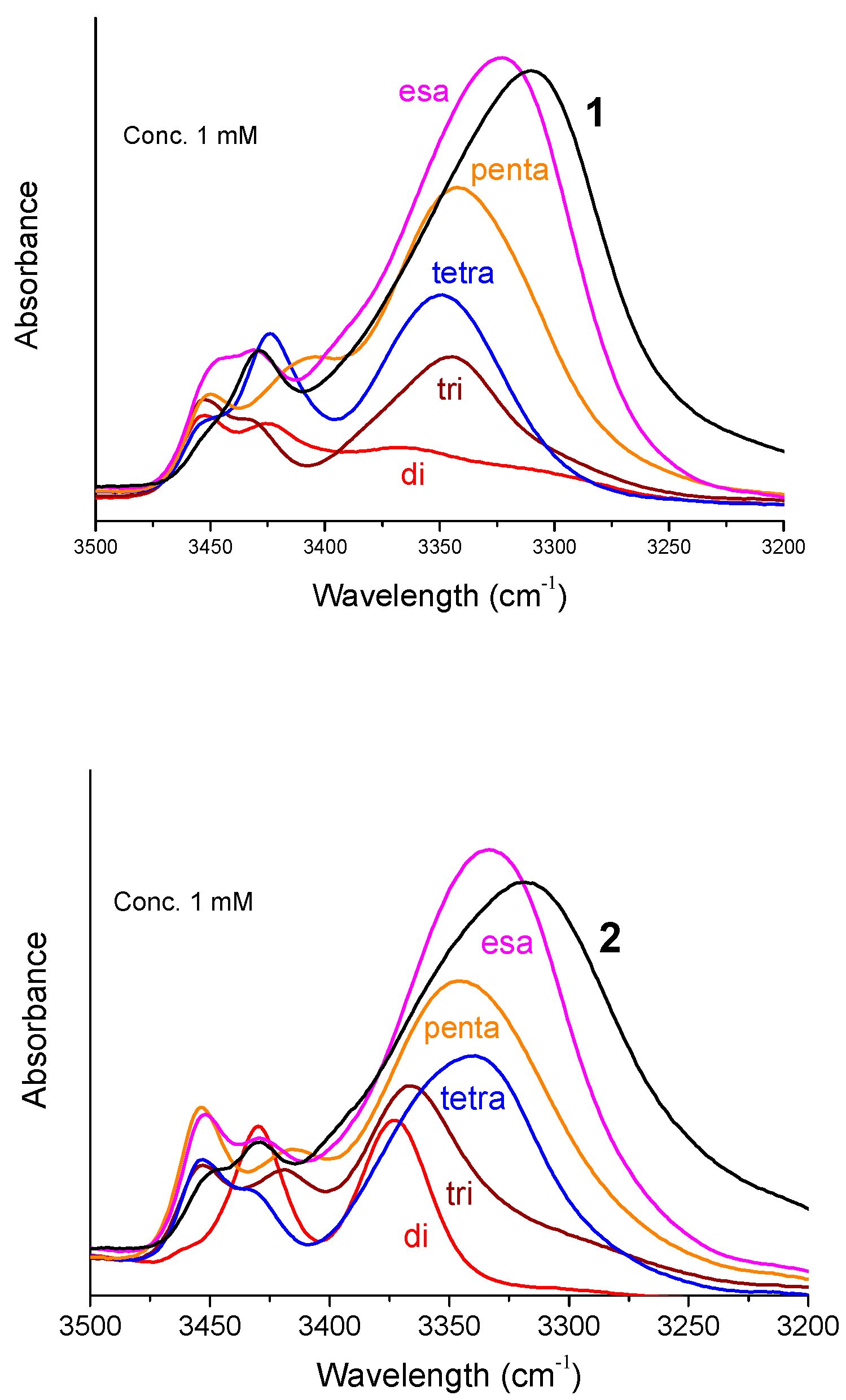
Figure 4.
Amide protons region of the NOESY spectrum of 1 in CDCl3 solution, T=25°C.
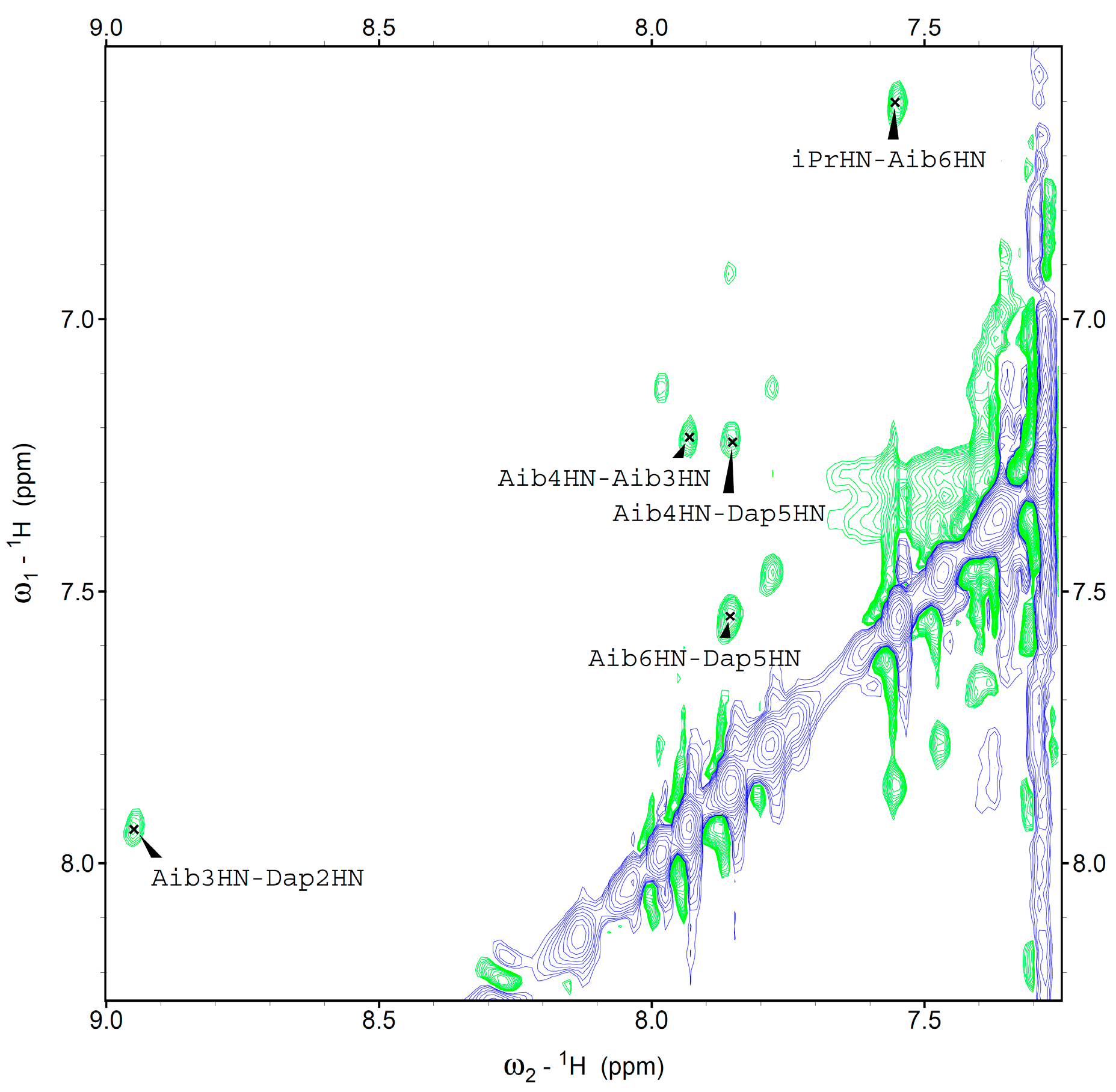
Figure 5.
Cβ/NH region of the NOESY spectrum of 1 in CDCl3 solution, T=25 °C.
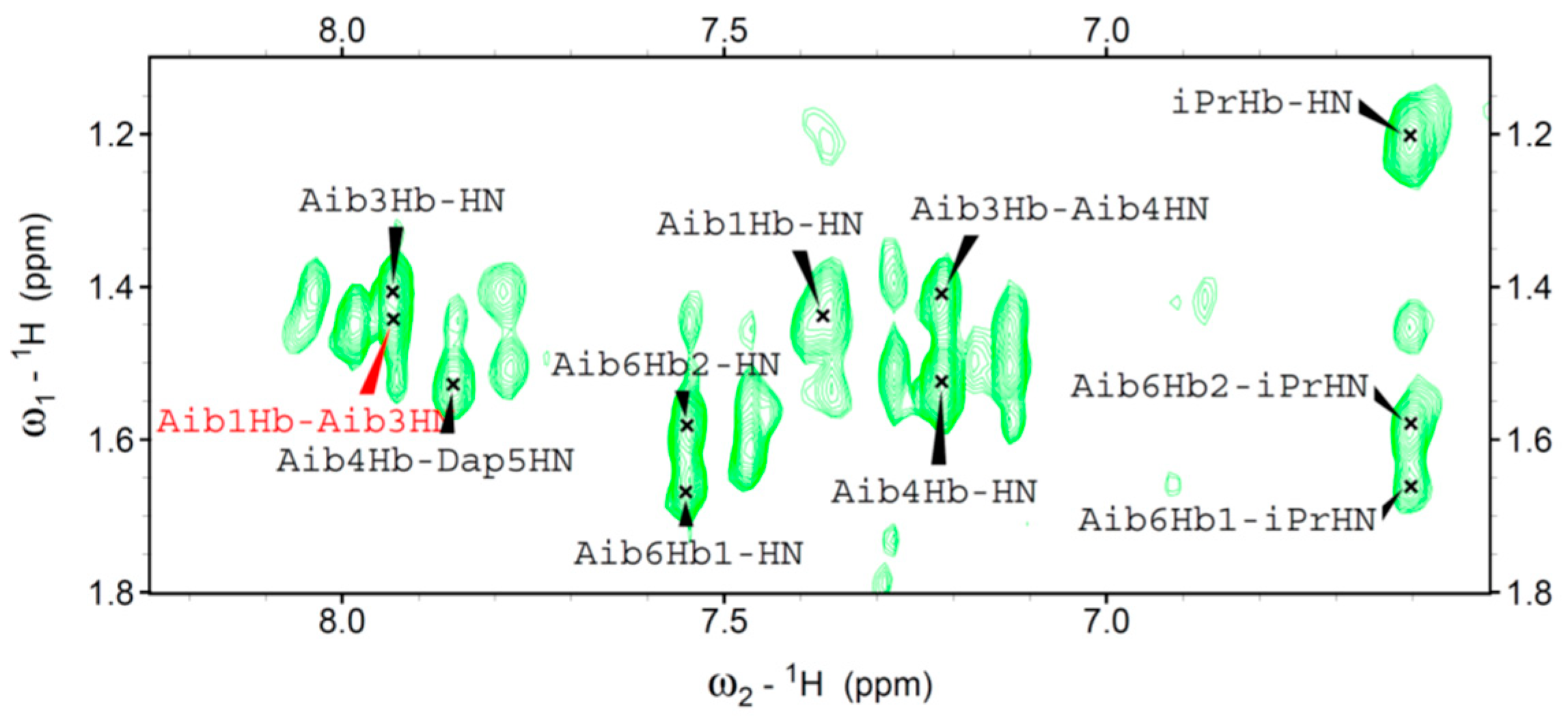
Figure 6.
Left: far-UV CD spectra of 1 (red) and 2 (blue) at 0.5 mM concentration and of 2 at 0,1 mM concentration in MeOH. Right: near UV and visible CD spectra of 1 and 2 at 0.5 mM concentration in CH3OH.
Figure 6.
Left: far-UV CD spectra of 1 (red) and 2 (blue) at 0.5 mM concentration and of 2 at 0,1 mM concentration in MeOH. Right: near UV and visible CD spectra of 1 and 2 at 0.5 mM concentration in CH3OH.
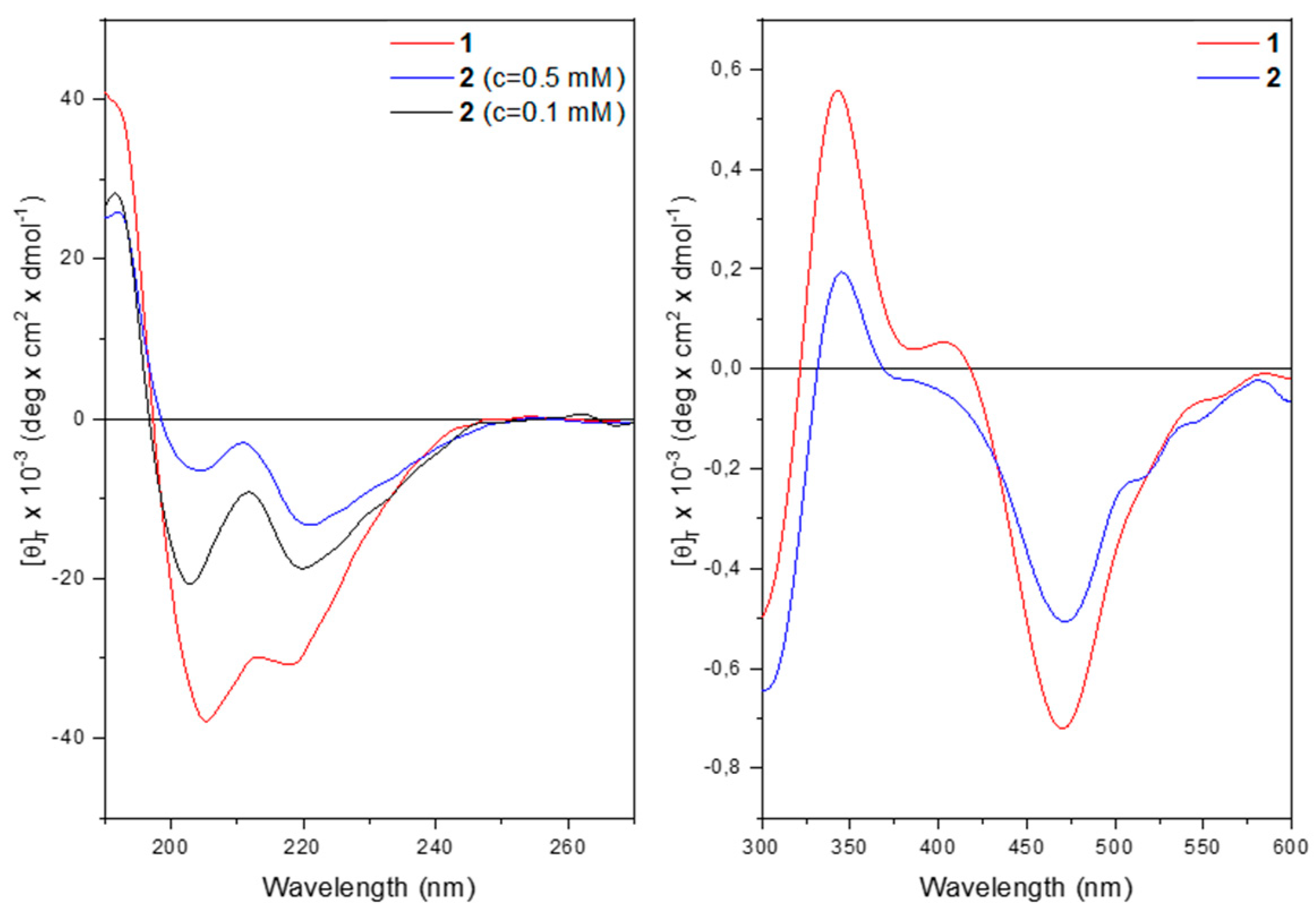
Figure 7.
(a) Cyclic voltammetry, scan rate 0.1 Vs−1, and (b) DPVof 1 (bold red line) and 2 (bold blue line) in CH2Cl2 containing 0.1 M nBu4NPF6 as the supporting electrolyte; Au disk electrode (d = 0.5 mm); T = 20 °C. The current i is normalized according to the equation: iv-1/2c−1, as for to a diffusion-controlled current variation, with concentration c = 0.4 and 1.7 mM.
Figure 7.
(a) Cyclic voltammetry, scan rate 0.1 Vs−1, and (b) DPVof 1 (bold red line) and 2 (bold blue line) in CH2Cl2 containing 0.1 M nBu4NPF6 as the supporting electrolyte; Au disk electrode (d = 0.5 mm); T = 20 °C. The current i is normalized according to the equation: iv-1/2c−1, as for to a diffusion-controlled current variation, with concentration c = 0.4 and 1.7 mM.
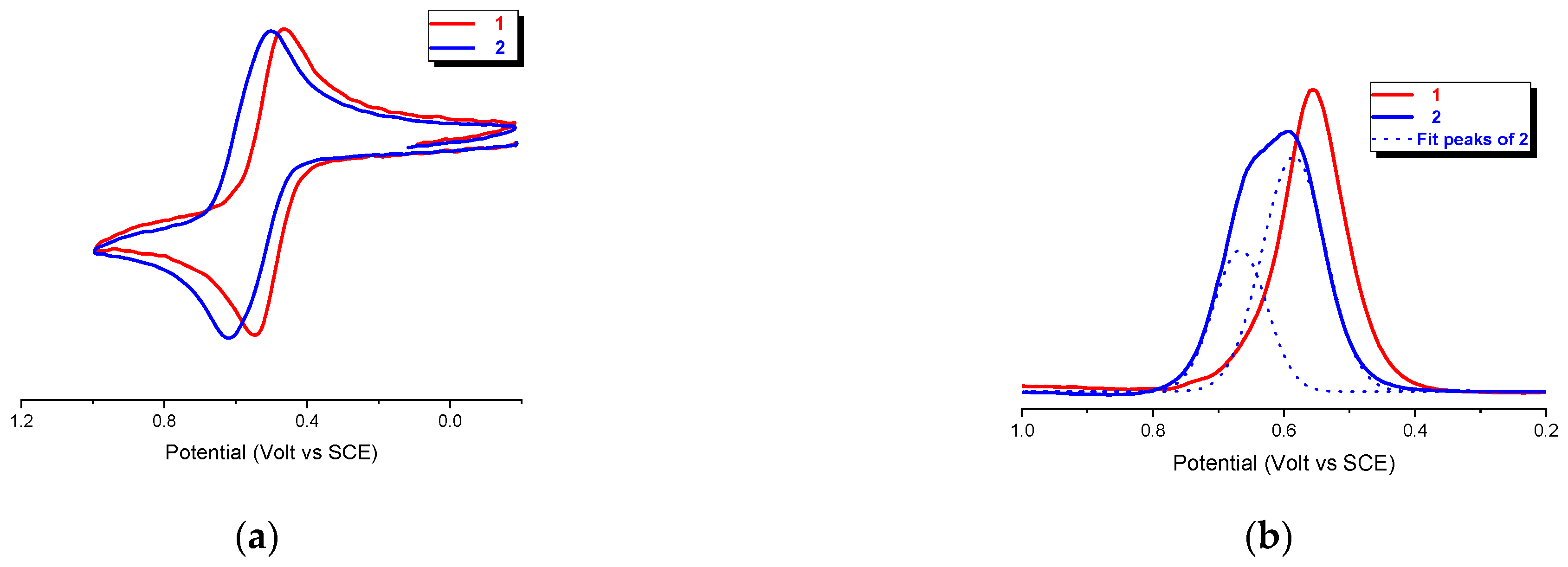
Figure 8.
Schematic representation of possible aggregation modes of 2.
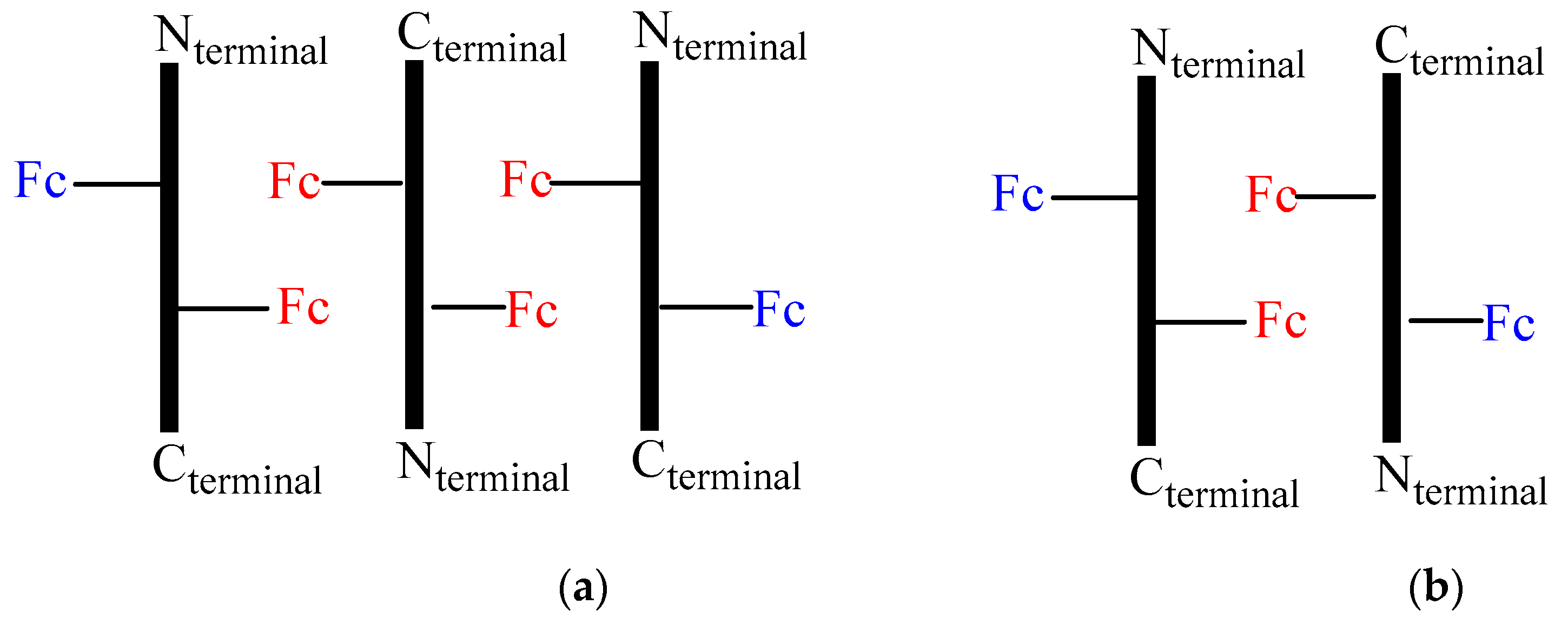
Figure 9.
Visible absorption spectra in CH2Cl2/0.1 M nBu4NPF6 of 1 obtained by oxidation of the neutral parent compound with up to 2 equivalent of acethylferrocenium [BF4]. c = 3.0 mM.
Figure 9.
Visible absorption spectra in CH2Cl2/0.1 M nBu4NPF6 of 1 obtained by oxidation of the neutral parent compound with up to 2 equivalent of acethylferrocenium [BF4]. c = 3.0 mM.
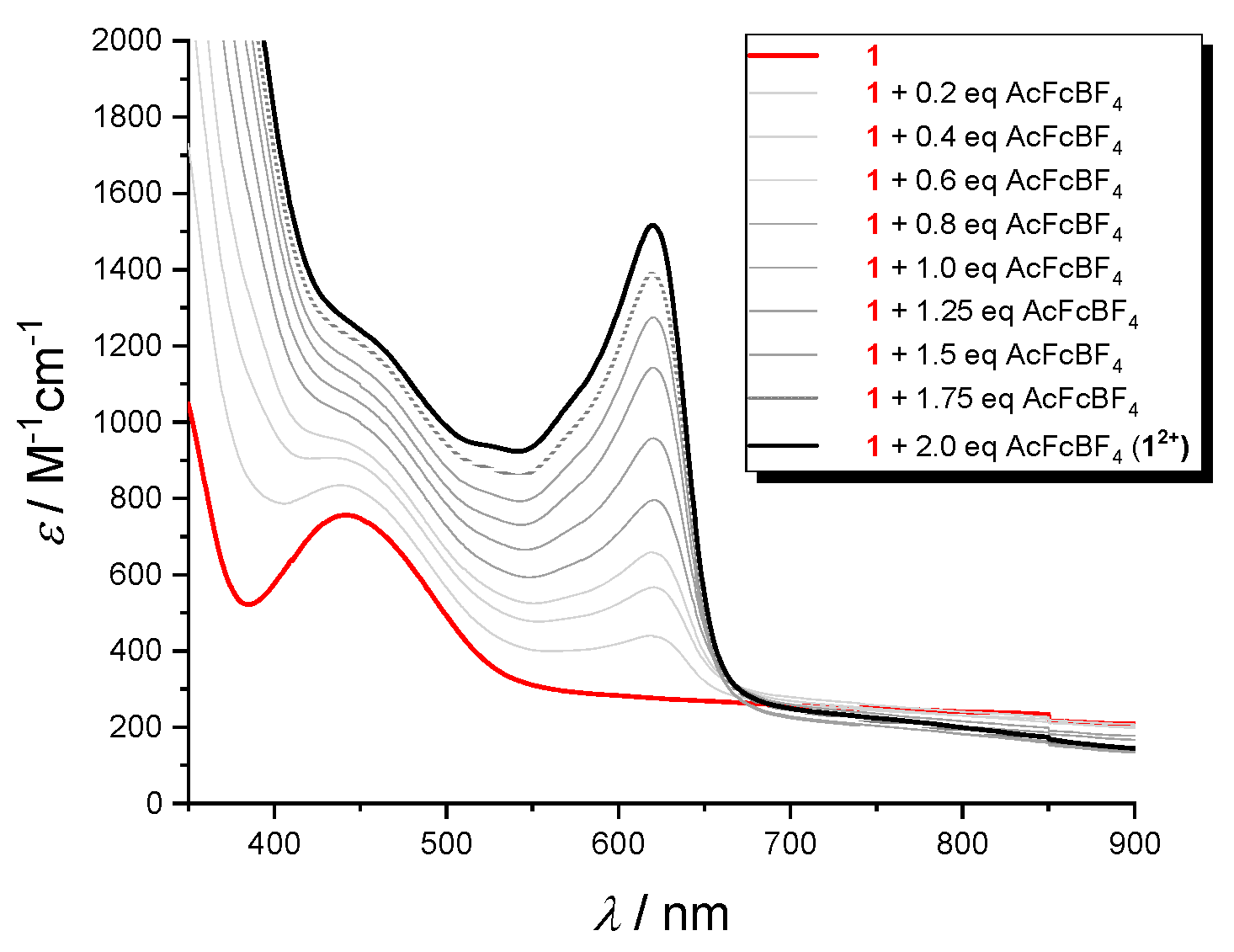
Figure 10.
IR absorption spectra in the amide A region, in CH2Cl2/0.1 M nBu4NPF6 of 12+ (a) and 22+ (b) obtained by addition of 2 equivalent of acethylferrocenium[BF4] to 1 and 2. c = 1.4 mM.
Figure 10.
IR absorption spectra in the amide A region, in CH2Cl2/0.1 M nBu4NPF6 of 12+ (a) and 22+ (b) obtained by addition of 2 equivalent of acethylferrocenium[BF4] to 1 and 2. c = 1.4 mM.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



