
Submitted:
14 November 2023
Posted:
16 November 2023
You are already at the latest version
Abstract
Acute myeloid leukemia (AML) is a rare subtype of acute leukemia in the pediatric and adolescent population but causes disproportionate morbidity and mortality in this age group. Standard chemotherapeutic regimens for AML have changed very little in the past 3-4 decades, but addition of targeted agents in recent years have led to improved survival in select subsets of patients as well as a better biologic understanding of the disease. One key paradigm of bench-to-bedside practice in the context of adult AML currently is the focus on leukemia stem cell (LSC)-targeted therapies. Here we review current and emerging immunotherapies and other targeted agents that are in clinical use for pediatric AML, through the lens of what is known (and not known) about their LSC-targeting capability. Based on a growing understanding of pediatric LSC biology, we also briefly discuss potential future agents on the horizon.
Keywords:
pediatric AML
; immunotherapy
; targeted therapy
; leukemia stem cell (LSC)
1. Introduction
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy that affects just over 4 adults per 100,000 and 7 children per 1 million in the United States each year [1,2]. In adults, AML incidence increases with advancing age [1] and the clonal expansion of immature myeloid-lineage blasts is thought to be secondary to sequential accumulation of multiple mutations which collectively confer a growth/proliferation advantage over normal hematopoietic populations [3]. In children, the genetic drivers of AML are fewer in number and tend to be sufficient to generate an acute leukemia phenotype as stand-alone mutations, suggesting biologic differences between adult and pediatric disease [4]. However, in both age groups, outcomes for AML remain suboptimal. Despite intensive treatment regimens including high-dose chemotherapy and at times allogeneic stem cell transplant (SCT), approximately 30-40% of children and young adults and over 80% of elderly adults will relapse [1,2]. Relapsed AML is a scenario where prognosis is particularly poor, and for which additional treatment options including novel agents is a relatively unmet need [5,6]. A better understanding of the pathophysiology and mechanistic drivers of disease will help guide future therapeutic advances.
Leukemia stem cells (LSCs) have been implicated in origination, chemo-resistance, and relapse of both adult and pediatric AML. There has been a general drive among translational researchers and clinicians to better characterize these cells, so as to understand which aspects of their biology might be targeted for therapeutic purposes. The general hypothesis is that LSC targeting agents as an integral part of AML treatment will improve event-free and overall survival for patients. While our knowledge of adult LSCs is substantial and growing constantly, research into pediatric LSCs is relatively nascent. The purpose of this review is to provide an overview of the pediatric LSC literature, within the framework of what is already known about adult LSCs, and to identify which novel agents are thought to have the highest likelihood of successfully eradicating these leukemia-initiating cells.
2. Discovery and Evolving Knowledge of Stem Cells in Adult AML
2.1. Functional Definitions and Immunophenotype
The concept of a myeloid LSC first took shape in the mid-1960’s[7] and was further delineated in the 1980’s, when it was seen in clonogenic assays and murine engraftment models that there was a relatively rare population of AML cells that could recapitulate the phenotypic diversity of the disease[8,9]. Early studies of LSCs in adult AML showed the presence of functionally-defined leukemia-initiating cells exclusively in the CD34+CD38- phenotypic compartment, irrespective of the patient immunophenotype, and correlated frequency of this compartment in patient samples with survival outcomes[10,11]. More recent evaluations of the CD34+CD38- LSC phenotypic assumption have correlated abundance of the CD34+CD38- compartment at diagnosis with post-treatment survival in patients having achieved remission, and demonstrate that quantitation of LSC abundance in this way can supplement more conventional assessment of measurable residual disease (MRD)[12]. CD123 has also been demonstrated to be highly expressed on LSCs but, unlike CD34, generally has minimal or variable expression on hematopoietic stem cells (HSCs), raising the possibility of a therapeutic window for therapeutic targeting[13]. Incidentally, CD123 has also been shown to be a marker of stem cells in high-grade myelodysplastic syndrome (MDS), which carries significant risk of transformation to AML[14].
Additional seminal studies have sought to more precisely quantify LSC frequency through limiting dilution experiments in immunodeficient mice; these studies highlighted the diversity of AML immunophenotypes in primary patient samples and revealed that LSC activity was not restricted to the CD34+CD38- cells, nor even to those lacking lineage markers[15]. Not only has interpatient heterogeneity been shown, but within individual patients LSCs can have multiple immunophenotypes[16,17], and LSC surface marker expression even changes after chemotherapy in many cases[16]. Therefore, while many cell surface proteins have been proposed as LSC markers[13,17,18,19,20,21,22,23], their fidelity across the breadth of AML patients as well as through therapy for a single patient remains questionable.
2.2. LSC Transcriptional Signatures
An alternative approach to identification and study of LSCs has been the development of LSC gene expression signatures. Eppert, et al. published one of the first LSC gene signatures by comparing gene expression microarray data of sorted populations of functionally validated LSCs to populations of non-LSC blasts[24]. This group identified 42 genes whose expression was enriched in LSCs, confirmed that many of these genes were also expressed in normal HSCs, and correlated high expression levels of these genes with poor survival outcomes in 3 independent adult AML cohorts[24]. One of the most frequently cited LSC gene expression signatures is the LSC17 signature developed by Ng and colleagues in 2016[25]. It was developed based on sorted cell populations from 87 adult AML patients that were functionally evaluated for presence or absence of LSC activity. The most upregulated genes in the LSC+ populations compared to the LSC- populations underwent sparse regression analysis in order to identify the minimal gene list that was still prognostic of survival outcomes in a training cohort of adults with AML[25]. These 17 genes were further validated in three additional cohorts of patients, with high LSC17 score associated with poor event-free and overall survival[25].
In 2015 investigators also published a DNA methylation signature based on functionally validated LSC and non-LSC populations from 15 adults with AML[26]. The majority (91%) of the over 3000 differentially methylated regions were hypomethylated in LSCs compared to non-LSCs, representing differential expression of 71 genes[26]. Only 6 of these 71 genes were described in the previous LSC gene expression signatures[26]. It was found that many HOXA cluster genes were upregulated in LSCs, irrespective of genetic driver mutations[26]. Expression of this LSC epigenetic signature was also correlated with outcomes in multiple patient cohorts[26]. Epigenetic dysregulation is a recurring theme in the adult LSC literature[27,28,29,30,31,32,33], with several potential therapeutic targets identified including protein arginine methyltransferase (PRMT6)[28], vitamin D receptor (VDR)[31], and YTH N6-methyladenosine RNA binding protein F2 (YTHDF2)[33], to name a few.
2.3. LSC metabolism, Microenvironment, and Drug Resistance
Additional characterization of LSCs in adult AML has involved evaluation of metabolic and other functional vulnerabilities of these cells. One seemingly unifying feature of functionally validated myeloid LSCs is that they have lower levels of reactive oxygen species (ROS) compared to bulk blasts – a feature which has been used to isolate and study these cells irrespective of their immunophenotype[34,35]. In addition, LSCs in general have a greater dependence on oxidative phosphorylation (OXPHOS), with limited flexibility in transitioning to glycolysis or other energy sources, at least in chemotherapy-naïve cells[34,36]. In chemo-resistant LSCs, which are typically enriched at relapse, both amino acid metabolism[37,38,39] and fatty acid oxidation and transport[28,40,41,42,43] have been shown to play roles in greater metabolic resilience.
LSC interactions with the surrounding niche have also been shown to play a role in their survival and propagation of AML. LSC homing to the bone marrow is facilitated by CXCR4/CXCL12 interactions, which are promoted by AML-associated mutations such as TET2 deletions through epigenetic modifications[27]. LSCs utilizing the CXCR4/CXCL12 axis to hide in the bone marrow have been shown to be more resistant to FLT3 inhibitors such as quizartinib[44]. Similarly, LSCs that had adapted to an adipose niche were shown to be resistant to conventional chemotherapeutic agents[42]. The microenvironment also plays a role in immune evasion of LSCs[45].
Another feature of LSCs is their ability to directly eliminate chemotherapeutic agents through drug efflux pumps[46,47,48] or to upregulate autophagy in response to a variety of insults[49,50,51,52]. Autophagy inhibitors have shown preclinical utility in combination with epigenetic and metabolic agents[49,50] but may have antagonistic activity against certain kinase inhibitors[51].
3. LSC Biology in Pediatric Myeloid Disease
With the upswell of interest and translational research focus on isolating and therapeutically targeting the myeloid LSC in adult medicine, it is not surprising that the literature on pediatric AML stem cells has also seen rapid growth in the past decade. Much of what has been published about pediatric AML stem cell biology borrows heavily from certain foundational tenets of adult AML stem cells, which can at times create limitations in the data. For example, many of the pediatric LSC studies base their investigation of pediatric LSCs on CD34+CD38- sorted populations of cells, which in most cases does enrich for LSCs in the context of adult AML but may miss LSC heterogeneity that we now know is present[57,58,59,60,61,62]. With those caveats in mind, the findings from these studies could still prove therapeutically and prognostically interesting.
3.1. Immunophenotype
Potential immunophenotypic targets for pediatric LSCs have largely been explored in the context of comparing expression levels of published adult LSC markers in pediatric HSCs from healthy controls to CD34+CD38- populations from AML samples. Chavez-Gonzalez et al demonstrated increased expression of both CD123 and CD96, but not CD117 or CD90, in putative LSCs versus normal HSCs[57]. Petersen et al likewise confirmed increased expression of CD123, as well as CLL and IL1RAP, in primitive populations from AML patients versus healthy bone marrow donors, but noted that IL1RAP, CD93, and CD25 expression was not restricted to populations harboring AML-associated genetic mutations[58]. In retrospective analyses both CD123 and CD200 expression have demonstrated correlation with inferior survival and other poor prognostic features (high-risk genetics, persistent MRD)[60,63]. CD200 has been shown to play a role in immune evasion in both adult[64] and pediatric[65] AML, in part through decreasing STAT3-dependent cytokine secretion and reducing OXPHOS in T cells[64]. Another putative LSC marker, CD69, was identified as upregulated in LSCs from pediatric patients who failed to achieve remission with conventional chemotherapy[66].
3.2. LSC Transcriptional Signatures
The bulk of the existing investigation into pediatric LSCs has involved validation of LSC-enriched gene expression signatures, building off of the adult literature. Duployez et al initially evaluated the prognostic relevance of the LSC17 gene signature in two large retrospective pediatric AML data sets, finding that high expression of these 17 genes was strongly associated with inferior event-free and overall survival, including in multivariate analyses[67]. Elsayed et al applied a least absolute shrinkage and selection operator (LASSO) Cox regression model to 32 of the 47 genes originally identified by Ng et al and to survival data from the St. Jude AML02 trial and identified a “pLSC6” gene signature that were most discriminatory of survival outcomes in that patient cohort[68]. They then showed superior association with survival of pLSC6 compared to the LSC17 signature[68]. Most recently, Huang et al reviewed all of the previously published LSC gene signatures in the context of RNA-seq data from 1503 diagnostic samples banked through four large Children’s Oncology Group (COG) trials[69]. This study showed that the larger LSC47 gene signature, from which the LSC17 signature was derived[25], was actually most prognostic of outcomes within genetic risk groups compared to either LSC17 or pLSC6[69]. The prognostic value of LSC47 was then validated in an independent cohort of 212 patients from St. Jude[69]. Together, the transcriptional data from large clinical cohorts provide corroborating evidence of the importance of LSC biology for childhood AML.
3.3. LSC Metabolism, Microenvironment, and Drug Resistance
Although metabolic and functional vulnerabilities of pediatric LSCs are relatively understudied, a growing body of literature is rapidly filling this knowledge gap. Alternative splicing events were identified in a small cohort of pediatric LSC samples that were subjected to single cell proteogenomic analysis in parallel with normal HSCs[59]. Exon skipping was prominent in LSCs but not HSCs, and included alternative splicing of CD47 which led to upregulation of this immune evasive molecule[59]. The exon skipping phenotype was also associated with increased transcript levels of MCL1-L and BCL2-L splice variants, both of which are pro-survival[59]. Rebecsinib, a splicing modulator compound, was shown to reduce viability and colony formation of pediatric LSCs but not cord blood HSCs[59], suggesting that this agent still in preclinical testing could have utility in treating pediatric AML. Rebecsinib has also been proposed as an LSC-targeting therapy in adult AML[70].
Another study compared single cell RNA-seq data in paired pre- and post-chemotherapy bone marrow samples from 13 children with AML, using enrichment of chemo-resistant transcriptional signatures as a surrogate for LSC activity[66]. Traditional LSC signatures were seen in hematopoietic stem cell-like populations, while oxidative phosphorylation (OXPHOS) signatures were upregulated in progenitor populations, particularly from patients with more monocytic AMLs[66]. Gene set enrichment analysis also identified ribosome biology as upregulated in LSCs[66]. Furthermore, compared to LSCs from pre-chemotherapy samples, paired post-chemotherapy LSCs showed activation of genes responsive to reactive oxygen species and heme metabolism, and maintained their LSC or OXPHOS gene signatures[66]. These metabolic features at baseline and in response to chemotherapy may inform selection of adjunctive therapies for pediatric AML in the future. Similarly, analysis of large adult and pediatric transcriptional data sets showed that high RNA levels of the ATP-binding cassette (ABC) transport protein ABCA3 strongly correlated with leukemia-free survival as well as expression of the LSC17 gene signature[71], suggesting that LSCs in particular have enhanced drug resistance compared to bulk AML blasts and warrant careful consideration of their unique vulnerabilities to bypass this resistance.
With respect to microenvironment, mouse studies suggest that inherent differences in niche crosstalk and stromal composition contribute to differences in self-renewal capacity and fitness between HSCs and LSCs, with neonatal marrow favoring normal HSC development and adult marrow favoring LSC propagation[72]. Introducing cells to the marrow of differently aged mice completely reversed the differences seen in phenotype, suggesting that interactions with the bone marrow niche have significant implications for leukemogenesis[72]. Less is known about the contribution of the adipose niche in pediatric AML development and chemo-resistance, with conflicting data as to the relationship between body mass index (BMI) and outcomes in children. One meta-analysis of 11 articles including 2922 patients showed a significant correlation between higher BMI (≥85%) and both event-free and overall survival[73]. However, another multi-national study of 867 pediatric patients with newly diagnosed AML found no relationship between BMI and relapse risk, treatment-related mortality, or overall survival[74]. Additional preclinical and clinical studies are needed to better understand the contributions of different microenvironmental interactions toward pediatric LSC survival.
4. Immunotherapies
As preclinical and clinical knowledge around LSC immunophenotypes has grown, so has the interest in immune-based therapies. Immunotherapies differ significantly from chemotherapy in that they aim to stimulate a patient’s pre-existing immune system to preferentially target and kill the malignant cell population while minimizing harm to healthy cells. Cell membrane surface targets have been the main interest for the development of leukemia-directed immunotherapies, with a particular focus on antibody-based and cell-based therapies. While the landscape of acute lymphoblastic leukemia (ALL) therapy has changed completely as a result of immunotherapies, unfortunately immunotherapy advances have not been as successful to date in the treatment of pediatric AML. When it comes to specifically eradicating the AML LSC population, an ideal target for an immunotherapeutic approach is highly and universally expressed on LSCs but absent in other normal cells and HSCs.
As previously discussed, research efforts to differentiate between the immunophenotypic profile of LSCs compared to HSCs are immense in order to identify and develop new immunotherapies that have an optimal therapeutic window. For AML, few targets have been identified that are exclusive to LSCs; many surface markers that are enriched on LSCs are also found on HSCs, thus the therapeutic window is relatively narrow. CD33 and CD123 are two of the most commonly pursued surface antigens as clinical targets but unfortunately, many clinical trials for these antigens have seen unfavorable toxicity profiles. The off-target effects and prolonged myelosuppression that have been observed further emphasize the ongoing hurdles that need to be overcome in order to develop AML-selective and especially myeloid LSC- immunotherapies. What follows is a brief review of the immunotherapy agents that are recently or currently under investigation for pediatric AML, and what is known about their LSC-targeting capabilities. Currently active clinical trials that include pediatric patients are summarized in Table 1.
4.1. CD33
CD33 is a transmembrane receptor that is expressed highly on the majority of AML cells. It was identified early as a potential cell-surface therapeutic target and its expression on AML LSCs exceeds that of HSCs[75,76]. Gemtuzumab ozogamicin (GO) is a CD33-targeting antibody-drug conjugate (ADC) consisting of a fully humanized CD33 antibody with a calicheamicin payload[77,78]. GO received Food and Drug Administration (FDA) approval in 2000 on the basis of early adult studies that demonstrated that GO could induce remissions in relapsed/refractory (R/R) AML[79]. However, due to toxicities including prolonged cytopenias and increased rates of veno-occlusive disease of the liver, as well as efficacy concerns in which the benefit of GO in R/R AML could not be confirmed in subsequent studies, GO was withdrawn from the market in 2010[80]. These concerning toxicity profiles were likely due to higher GO dosing than what is currently used, as successive larger clinical trials confirmed demonstrable clinical benefit and more manageable toxicities with lower doses of GO[81,82,83,84,85,86,87]. Therefore, GO was reapproved by the FDA in 2016 for adults with relapsed and newly diagnosed AML and for children with relapsed AML[88].
The COG AAML0531 phase 3 clinical trial demonstrated that GO in combination with standard chemotherapy resulted in improved outcomes for certain subsets of pediatric patients with AML[82]. High CD33 surface expression was associated with inferior outcomes in children with de novo AML treated on AAML0531 and several studies have suggested that the benefit of GO is directly correlated with higher CD33 surface expression[82,89].
Recently, significant efforts have been made to optimize the potency of ADCs targeting CD33. Vadastuximab talirine is one such ADC that was found to be more effective at killing AML cells compared to GO but with significant hematologic toxicities described due to its small therapeutic window[90,91,92]. The phase 3 CASCADE study investigating this ADC in older patients with de novo AML was discontinued early due to substantial adverse events suggesting that normal HSCs as well as AML LSCs are being affected.
Lintuzumab-Ac225 is an alternative approach to CD33 targeting and consists of a radiolabeled anti-CD33 antibody. It delivers high-energy radiation over a short radius to CD33-expressing cells. Lintuzumab has been studied as monotherapy and in combination with chemotherapy in adults with AML and has demonstrated significant antileukemic activity with high response rates, even in patients with high-risk and heavily pretreated disease[93]. More clinical trials of Lintuzumab are underway for adults but it has not yet been tested in the pediatric population.
Bi-specific antibodies consist of two distinct antigen-binding domains to interact with two disparate cell surface proteins. Typically, these are designed to be able to recruit lymphoid cells to interact and kill malignant cells. Bi-specific T cell-engagers (BiTEs) have shown promising efficacy for the treatment of R/R ALL and as a result of this success, there has been an increase in focused efforts to develop BiTEs for AML[94,95,96,97,98,99]. Specifically, these bi-specific antibodies for AML are constructed to employ T lymphocytes to recognize a cell surface target on the AML cells, ideally a target with high expression on LSCs including CD33, CD123, and CLL-1[94,95,96,97,98,99]. One such BiTE, AMG 330, binds CD33 and CD3 on T cells causing T-cell mediated destruction of CD33+ cells and preclinically has demonstrated effective LSC killing[96]. Several studies of AMG 330 have demonstrated safety and efficacy in the treatment of adult patients with AML but this agent has not yet been investigated in children[96,97,98].
Chimeric antigen receptor (CAR) T cells have been extensively investigated with promising response rates in the context of R/R lymphoid malignancies. They are now being progressively pursued to treat several other types of malignancies. Unfortunately, in the setting of AML, CAR T cell therapies have not progressed as rapidly as for lymphoid malignancies, again due to difficulty in identifying target antigens that are restricted to the leukemia cells and LSCs.
CD33 has been one of the most studied antigen targets for AML CAR T cells. Preclinical research has demonstrated potent leukemia killing of CD33 specific CAR T cells and several clinical trials are ongoing investigating CD33 CAR T cells in both the adult and pediatric populations (NCT03927261, NCT03971799). To date, there are case reports of patients who have had promising responses to CD33 CAR T cells and have been successfully bridged to SCT[100]. Additionally, dual targeting CD33/CLL-1 CAR T cells are in clinical trials in China (NCT03795779, NCT05248685) in an effort to optimize efficacy and AML specificity of cytotoxic killing. One significant barrier to the success of CAR T cells in this context is that CAR T persistence is often associated with prolonged cytopenias without full hematopoietic recovery. Given the propensity for patients to become aplastic following these therapies, CD33 CAR T cells are largely being investigated as a bridge to SCT. This is in contrast to ADCs and BiTEs which may be able to induce sustained remissions with less myelotoxicity. To combat the off-target effects seen with CD33-directed CAR T cells, research is ongoing into the possibility of genetically inactivating CD33 in HSCs to permit CD33 CAR T cell mediated cytotoxicity with increased specificity for leukemic blasts and LSCs[101].
4.2. CD123
CD123 (the interleukin-3 [IL3] receptor alpha) was the first antigen that was identified to be specific to LSCs. Studies of mouse AML xenograft models established that the CD34+/CD38-/CD123+ population of AML cells held all of the detectable engraftment potential[13]. However, while CD123 is highly expressed on myeloid leukemic blasts in a majority of patients, it is also present on HSCs as well as more differentiated myeloid and lymphoid cells[102]. While the therapeutic window for CD123-directed therapies is narrow for this reason, interest in CD123 as a potential cell-surface therapeutic target persists[63]. Overall, clinical investigations of anti-CD123 immunotherapies have generally shown limited efficacy and unfavorable safety profiles due to the off-leukemia effects of CD123 targeting. As more trials have developed and more patients have been treated with different CD123 directed therapies, it has been observed that patients with high CD123 benefit most from these immunotherapies[63,103].
While the IL3 signaling pathway is important for normal hematopoietic differentiation from HSCs, it has also been shown to be essential for LSCs[103]. In the efforts to develop agents that will be effective in killing AML LSCs, one approach has been to conjugate a cytotoxic drug to the IL3 ligand for the IL3 receptor CD123. Tagraxofusp is an IL3/diphtheria toxin fusion protein that is FDA-approved for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN), a rare malignancy which has high levels of CD123 expression[104]. Tagraxofusp has shown potent cytotoxicity against AML LSCs in preclinical studies, in addition to normal HSCs[105]. In initial trials in adults, clinically meaningful responses were observed after treatment with Tagraxofusp in a small subset of patients with R/R AML that received multiple lines of prior therapy, suggesting promising LSC targeting[106]. Trials in both adult and pediatric patients with R/R AML are currently underway or in development to further evaluate the efficacy and toxicity profile of Tagraxofusp (NCT05476770, NCT05233618, NCT04342962, NCT05720988, NCT05716009).
IMGN632 is an ADC consisting of a CD123-targeting antibody and a DNA-alkylating payload which in preclinical studies has demonstrated compelling antileukemic activity[107]. IMGN632 has been studied in early phase clinical trials both as a single agent and in combination with chemotherapy in adults with AML with promising activity against AML with tolerable safety profiles (NCT04086264)[108,109]. Studies in adults are ongoing but trials involving IMGN632 have not yet expanded to children.
Bi-specific engagers have also been investigated to target AML LSCs through CD123. These strategies include the CD123/CD3 dual-affinity retargeting protein (DART) flotetuzumab, the CD123-targeting bi-specific antibody XmAb14045 and the CD123/CD3 bi-specific IgG1 antibody JNJ-63709178. In early-phase clinical trials in adults with R/R AML, 30% of patients treated with flotetuzumab achieved responses, with the toxicity profile mostly including cytokine release syndrome (CRS) and infusion reactions[110,111]. Based on this data, a clinical trial is ongoing investigating flotetuzumab for the treatment of children with R/R AML (NCT04158739).
Given that significant CRS has been observed with CD123-targeted T cell engagers, the focus for cytotoxic antibody-directed therapies has shifted towards natural killer (NK) cells. SAR443579 is a trifunctional NK cell engager that targets CD123 while co-engaging NKp46 and CD16a on NK cells. In preclinical mouse and primate models, SAR443579 had potent antitumor effects through NK cell activation in the presence of AML blasts with a tolerable toxicity profile and minimal cytokine secretion[112]. In a first-in-human Phase I/II trial of SAR443579 for adult patients with R/R AML, 3/23 total patients (13%) achieved a complete remission (CR), with the highest response rates at higher dose levels (3/8 patients, 37.5%)[113]. The most common adverse events were nausea and infusion reactions, with only 2 cases of low-grade CRS[113]. This first-in-human study of SAR443579 recently opened a pediatric arm enrolling patients greater than 1 year of age with R/R AML (NCT05086315).
Lastly, CAR T cells targeting CD123 are also in development with active CD123 CAR T cell trials enrolling adult and pediatric patients with R/R AML (NCT04265963, NCT04272125, NCT04318678, NCT04678336). Early reports from some of the pivotal CD123 CAR T cell trials have identified few significant off-leukemia effects, including minimal myeloablative toxicities, but with some myelosuppression and limited CAR persistence[114]. For these reasons, and similar to the approach for CD33 CAR T cells, CD123 CAR T cells are primarily being investigated as a bridge to SCT.
4.3. Checkpoint Inhibitors
Immune checkpoint antigen expression has been described in both bulk AML blasts and LSCs, and treatment concepts have been developed with the goal to block checkpoint molecules and thereby overcome immune evasion[115,116]. PD-L1 expression in AML LSCs is hypothesized to be due to 1) oncogenic mutations in key proteins that are part of the JAK-STAT and MYC pathway signaling cascades in LSCs and 2) cytokine production from AML cells leading to cytokine-induced expression of PD-L1[117,118,119]. Additionally, certain drugs used in the treatment of AML, like hypomethylating agents, can promote increased expression of PD-L1 in AML blasts and LSCs[120]. PD-L1 targeting antibodies in combination with these chemotherapy medications have been evaluated in clinical trials for adult patients with AML. In a Phase II study of azacitidine and nivolumab for R/R AML, 70 patients were treated with an overall response rate of 33% and overall survival of 19%[121]. A phase I/II study of the azacitidine/nivolumab combination is currently underway to evaluate safety and efficacy in children with R/R AML (NCT03825367). In a phase II clinical trial assessing the efficacy of nivolumab as maintenance therapy for adults with high-risk AML in remission who were not being considered for SCT, 15 patients were enrolled, of whom 9 patients (60%) had detectable minimal residual disease (MRD) at time of enrollment[122]. The recurrence-free survival was 57.1% and 2 of the 9 patients with detectable MRD at time of enrollment cleared their MRD while on treatment with nivolumab. This study showed only a modest effect on MRD eradication and survival when nivolumab was used as a single agent but has provided the basis for subsequent clinical trials investigating immune checkpoint blockade in combination with chemotherapy as maintenance therapy in adults with high-risk AML.
4.4. CD47
CD47 is another immune checkpoint antigen that plays a role in how leukemia evades the immune system and has been found to be amply and invariably expressed on both AML LSCs and normal HSCs[123]. The CD47 antigen is also known as the ‘don’t eat me’ signal that mediates the escape of AML LSCs from phagocytosis by macrophages; blocking this signal can induce an immune response resulting in killing of leukemic cells[124,125]. Magrolimab is an anti-CD47 antibody that through CD47 blockade induces tumor phagocytosis and eliminates LSCs in AML preclinical models. Magrolimab has also shown promising results in early-phase clinical trials in adults with newly diagnosed AML when combined with azacitidine, which has been shown to enhance CD47 expression[126]. In 16 evaluable AML patients treated with magrolimab and azacitidine on a phase Ib clinical trial, 11/16 (69%) had an objective response[127]. Additionally, CD34+CD38- identifiable LSC populations were measured by flow cytometry and complete LSC elimination was observed in 10/16 (63%) of patients with myelodysplastic syndrome or AML that had a clinical response. This LSC eradication suggests the potential for durable responses in these patients and clinical trials further investigating magrolimab efficacy are ongoing. Trials investigating magrolimab for the treatment of AML in the pediatric population have not yet been initiated.
4.5. Folate Receptor 1/Folate Receptor-Alpha
CBFA2T3::GLIS2 translocations are associated with a particularly high-risk subtype of pediatric AML that is associated with megakaryoblastic phenotype in young non-Down syndrome patients[128]. This subtype of AML has proven to be highly chemo-refractory, and survival rates with traditional chemotherapeutic regimens and SCT are quite poor[128]. Transcriptional analysis of cells from patients with CBFA2T3::GLIS2 AML has revealed high expression of the folate receptor-alpha gene (FOLR1), which correlates with prominent surface expression of the FOLR1 protein in the majority of patients[129]. Although no formal validation of FOLR1 as an LSC marker has been undertaken, preclinical modeling of STRO-002, an antibody-drug conjugate (ADC) against FOLR1[129], and separately a FOLR1-specific chimeric antigen receptor T-cell (CAR-T)[130], showed promising anti-tumor efficacy. Limited clinical experience with use of STRO-002 for pediatric AML has been presented in abstract form, in which 16 total patients were treated with this ADC alone or in combination with other chemotherapeutic agents, with 7 (44%) achieving complete remission (CR)[131]. Updated clinical data is expected to be shared at the American Society of Hematology 2023 annual meeting.
An alternative FOLR1-targeting agent, ELU001, has also demonstrated preclinical efficacy in MV4-11 cells engineered to overexpress FOLR1[132]. ELU001 is a C’Dot drug conjugate (CDC), a nanoparticle-based drug delivery system with 21 exatecan molecules per nanoparticle as payload. A Phase 1 clinical trial of ELU001 for children with R/R CBFA2T3::GLIS2 AML is expected to open to enrollment in 2024 (NCT05622591).
5. Other Novel Agents
As has been noted above, immunophenotype is not the only or even the most consistent feature of myeloid LSCs in the pediatric or adult literature. Stem cell transcriptional, signaling, and metabolic programs have proven to be targetable features of LSCs as well. Figure 1 and Figure 2 highlight a few of the relevant aspects of LSC biology that have been explored for therapeutic intent. Below and in Table 2 we summarize non-immunotherapeutic agents that have been or will soon be in clinical trials for pediatric AML, and their relevance to the concept of LSC eradication.
5.1. FLT3 Inhibitors
Fms-like tyrosine kinase 3 (FLT3), also known as CD135, is a receptor tyrosine kinase that is expressed on HSCs and has functions in normal hematopoiesis[133]. Activating mutations in FLT3 such as the internal tandem duplication (ITD) or tyrosine kinase domain (TKD) mutations are common in both adult and pediatric AML[4,134]; FLT3 ITD mutations, in particular, confer a poor prognosis[134]. Tyrosine kinase inhibitors (TKIs) that target FLT3 have generally improved survival both alone and in combination with conventional chemotherapy in adult clinical trials[135,136,137,138,139,140,141]. Safety and preliminary efficacy of FLT3 inhibitors quizartinib and sorafenib have also been demonstrated in pediatric early-phase trials[142,143]. In the COG trial AAML1031, addition of sorafenib for FLT3-mutant AML prolonged event-free and disease-free survival and lowered relapse risk compared to historical cohorts of patients with FLT3-mutant AML (COG AAML0531)[144]. An ongoing phase 3 trial is evaluating the addition of gilteritinib to chemotherapy for all patients with FLT3 ITD or TKD mutations, including a maintenance phase of gilteritinib monotherapy for patients proceeding to SCT. Whether FLT3 is an important survival mechanism for LSCs is an open question. While multiple studies (adult and pediatric) have demonstrated the presence of FLT3 mutations in the phenotypic LSC compartment[145,146,147], it is notable that FLT3 mutations generally are considered a “late” stage modification in the evolution of adult AML[148], and approximately 50% of patients who relapse or progress on FLT3 inhibitor therapy lose their ITD mutations[149]. In one preclinical study, the FLT3 inhibitor sorafenib alone was insufficient to block LSC functionality in serial transplantation experiments of leukemia into mice, but the addition of all-trans-retinoic acid (ATRA) to sorafenib abrogated transmission of leukemia to secondary recipients[150], suggesting that there are combination therapies that can effectively eradicate LSCs. In addition to ongoing pediatric trials evaluating established FLT3 inhibitors, there are two Phase 1 trials of newer agents pexidartinib[151] and MRX-2843[152], both of which are active against common TKI resistance mutations such as the “gatekeeper” F691 mutation.
5.2. Menin Inhibitors
Translocations in the KMT2A gene, a master hematopoietic regulator, are associated with both acute lymphoblastic and myeloid leukemias and generally confer poor prognosis[4,134,153]. The LSC in KMT2A-rearranged leukemias is transformed by the KMT2A translocation in isolation, and generally arises from a granulocyte-macrophage progenitor (GMP) rather than from the HSC[154]. KMT2A rearrangements activate a transcriptional program characterized by upregulation of HOXA genes and MEIS1, which are associated with lineage-inappropriate expression of stemness markers[153,154]. The N-terminus of KMT2A, which is conserved in leukemogenic fusion proteins, recruits transcription elongation complexes to target genes in a manner that is dependent on its interacting protein, menin[155,156]. VTP50469 was the first small-molecule inhibitor of the menin-KMT2A interaction; treatment of KMT2A-rearranged leukemia cell lines and primary samples with this agent led to downregulation of the classic KMT2A gene signature and prolonged survival (even cures) in mice[157]. Similar gene expression signatures and similar benefit from menin inhibition has been shown in preclinical studies for NPM1-mutant AML[158,159], NUP98-rearranged AML[160], and UBTF mutated AML[161,162]. Revumenib has been the first menin inhibitor in clinical trials for AML; the first-in-human Phase 1 monotherapy results were recently published. Of 60 evaluable patients with R/R KMT2A-rearranged or NPM1-mutant AML, 18 (30%) achieved a composite complete remission (CRc), with complete cytogenetic remission in 64% of patients with KMT2A-rearranged AML who had clearance of bone marrow blasts[163]. Preclinical support for the utility of another menin inhibitor, ziftomenib, has been published for both adult and pediatric AML[164,165]. In the adult study, percentages of phenotypically defined stem/progenitor cells were reduced after ziftomenib treatment[164]. Preliminary data from KOMET-001, the Phase 1/2 trial of ziftomenib in adult R/R AML, was presented in abstract form in 2022. At Phase 1b dosing of 600 mg, CRc was 33% with 75% of these patients achieving MRD negativity; overall response rate (ORR) was 42% for the entire cohort and 75% in patients experiencing differentiation syndrome[166]. Additional combination therapy trials of ziftomenib and revumenib are ongoing in the adult age group. For pediatric patients, ziftomenib is currently available only through expanded access, but revumenib and another compound, JNJ-75276617, are open or soon-to-be-open in combination therapy trials (Table 2).
5.3. Venetoclax
Venetoclax is a BCL-2 inhibitor that was designed as a BH3 mimetic compound to block interaction between BCL-2 and pro-apoptotic proteins[167]. While initially showing clinical promise in chronic lymphocytic leukemia (CLL)[168], venetoclax in combination with low-intensity therapies has revolutionized the care of elderly patients with AML, many of whom do not have intensive therapy options and are not medically fit for SCT[169,170]. It has also shown promise in younger adults in combination with both high- and low-intensity chemotherapeutic regimens[171,172]. BCL-2 has been shown to be expressed at high levels in canonical myeloid LSCs, and to be associated with a dependence on OXPHOS[34]. Although there is literature to suggest that the canonical pro-apoptotic role of venetoclax is its primary mechanism[173], there is also a wealth of metabolic data to suggest that mitochondrial respiration in LSCs is affected and plays at least some role in the efficacy of venetoclax[36]. Both pathophysiologic hypotheses assume that venetoclax acts as a priming agent for AML cell death, and so is most effective when combined with other agents. Resistance to venetoclax has been associated with metabolic switching of LSCs to alternative energy sources such as amino acid or fatty acid metabolism[39,40,43]. Phenotypically this has been associated in adult AML with presence of non-canonical monocytic LSC biology[35,174].
Multiple retrospective single-center studies[175,176,177,178] and one multi-center study[179] have evaluated the efficacy of venetoclax combinations in R/R pediatric AML, with ORR ranging from 12.5% to 75% and a significant number of patients reported as able to bridge to SCT. Venetoclax in combination with daratumumab and myeloablative preparatory regimens led to an 85% MRD-negative CR rate in pediatric patients proceeding to transplant with active disease, and resulted in a 2-year EFS of 44% and OS of 65%[180]. A Phase 1 trial combining venetoclax with high-dose cytarabine and idarubicin identified the RP2D as 360 mg/m2 and reported the total cohort ORR as 69% with 70% CRc at the RP2D[181]. Although little is known about LSC-targeting capability or metabolic effects of venetoclax in pediatric AML, a recent single cell analysis including 5 pediatric primary specimens confirmed that 4 out of 5 pediatric AML specimens had venetoclax resistant subpopulations with unique metabolic features[182]. This suggests that at least some of the trends seen in adult AML may hold true in pediatric disease as well. Based on accumulating data about venetoclax resistance mechanisms, new rational combination therapies are being explored in preclinical[183] as well as adult clinical trial settings. Multiple combination regimens are currently in early phase pediatric trials, as well (Table 2).
5.4. PARP Inhibitors
Poly (ADP-ribose) polymerases (PARPs) are enzymes that catalyze addition of polymerized ADP-ribose onto substrates, thereby modifying their function or stability[184,185]. They are primarily implicated in DNA damage response pathways[184,185]. PARP inhibitors such as olaparib or talazoparib combined with DNA damaging agents such as anthracyclines or topoisomerase inhibitors tend to show synergy because of impaired DNA repair in tumor cells[185]. This is the premise of the current Phase 1 pediatric trial of talazoparib combined with topotecan and gemcitabine (POE22-01), which is not known to be LSC-targeting. However, in one adult study of LSCs, lack of cell surface expression of natural killer group 2, member D (NKG2D) ligands was noted to be a consistent feature of these cells and mediated escape of immune surveillance by NK cells[20]. PARP inhibitors were found to induce NKG2D ligand expression on LSCs and enhance immune clearance[20]. Therefore, future trials may explore this mechanism in pediatric AML to ascertain the potential synergy between PARP inhibitors and immunotherapies, for example.
5.5. Epigenetic Modifiers
Given that altered methylation is a key feature of adult LSCs[26], it is logical to anticipate that epigenetic modifiers such as DNA methyltransferase inhibitors (e.g., decitabine), hypomethylating agents (decitabine, azacitidine), or histone deacetylase inhibitors (e.g., vorinostat) might add benefit in combination regimens against pediatric AML. To that end, a Phase 1 feasibility study of epigenetic priming with decitabine followed by standard induction chemotherapy in newly diagnosed pediatric patients with AML was conducted. It enrolled 25 patients and showed no difference in CR rates, MRD negativity rates, or DFS between decitabine priming and standard-of-care arms, although it was not powered for efficacy endpoints[186]. Methylation changes were seen in the decitabine arm, consistent with pharmacodynamic goals[186]. A larger Phase 1 dose expansion trial of decitabine and vorinostat in combination with FLAG chemotherapy was conducted through the TACL (Therapeutic Advances in Childhood Leukemia & Lymphoma) consortium. This trial enrolled 37 patients with a median age of 8.4 years who had R/R AML[187]. The regimen was shown to be safe with no dose-limiting toxicities at the decitabine RP2D of 10 mg/m2[187]. ORR in 35 evaluable patients was 54% of which 90% were MRD negative and 84% were able to be bridged to SCT[187]. Two-year OS was 75.6% for patients who became MRD negative with therapy versus 17.9% for those who remained MRD positive after treatment[187]. Real-world data from 6 centers, including 28 patients who were treated off-study but with the same regimen, showed similar safety and efficacy with ORR of 63% and 93% of responders able to proceed to SCT[188]. Given the relative success of this regimen in a heavily pretreated population, and the promise of venetoclax combination regimens, there is now an open trial adding venetoclax to azacitidine, vorinostat, and FLAG (NCT05317403) in hopes of demonstrating safety and enhanced efficacy.
5.6. Selinexor
Selective inhibitors of nuclear export (SINEs) are compounds that inhibit the protein exportin-1 (XPO1)[189], which has numerous ramifications in the context of cancer. Tumor suppressors are expected to be retained in the nucleus and thereby re-activated[189]. Mutant NPM1, a commonly mutated protein in adult AML, requires cytoplasmic localization for its leukemogenic activity, which is inhibited by treatment with SINE compounds such as selinexor or its more potent second generation relative, eltanexor[190]. Selinexor has been shown to potentiate DNA damage induced by both cytotoxic chemotherapy[191] and venetoclax[192]. Furthermore, through reduction of MCL1 protein levels in treated cells, eltanexor further potentiates the anti-leukemic effects of venetoclax[193]. Preclinical studies have shown that both selinexor and to a greater degree eltanexor target LSCs, as demonstrated by limiting dilution engraftment experiments in mice[194,195]. While selinexor monotherapy led to only a 14% response rate in R/R adult AML[196], combination trials with chemotherapy have shown more favorable response rates of 42-50%[189,197,198], aside from one study in elderly AML that showed worse outcomes with addition of selinexor to standard chemotherapy[199]. A Phase 1 trial combining selinexor with fludarabine and cytarabine in 18 pediatric patients with R/R AML demonstrated XPO1 target inhibition and led to an ORR of 47%[200]. Another Phase 1 trial of these agents plus the addition of venetoclax is ongoing at multiple U.S. sites (Table 2).
5.7. Niclosamide
Niclosamide is an anti-parasitic agent that shows antineoplastic activity through inhibition of NFkB and cAMP-response element binding protein (CREB) as well as through upregulation of reactive oxygen species (ROS)[201,202]. A cell line CRISPR/Cas9 library screen identified mitochondrial metabolic pathways as significantly altered after exposure of AML cells to niclosamide – in particular OXPHOS, glycolysis, and mitochondrial membrane potential – suggesting effects on pathways essential to LSCs[203]. Indeed, preclinical studies showed decreased viability of CD34+CD38- cells and decreased colony-forming potential in AML primary samples, both of which suggest a selective reduction in the LSC compartment after niclosamide treatment[201]. There is minimal to no clinical data on niclosamide in AML, however. A Phase 1 monotherapy trial is actively recruiting for pediatric patients with R/R AML (Table 2).
5.8. Uproleselan
Interaction of leukemia cells, including LSCs, with the microenvironment is thought to be protective against chemotherapy toxicity by a variety of mechanisms. LSCs in adults have been shown to express E-selectin ligands, which enhance binding to the bone marrow niche[204]. The E-selectin antagonist uproleselan showed preclinical efficacy to sensitize AML cells to standard chemotherapeutic agents[204], and a recent Phase I/II study in adults with R/R AML and elderly patients with newly diagnosed (ND) AML (Phase 2 only) was recently published[205]. A total of 91 patients were enrolled, approximately 2/3 of which were R/R to prior therapies. Overall response rates were 41% in the R/R cohort and 72% in the ND cohort with over half of responders achieving MRD negativity. Responses were correlated with E-selectin ligand expression on bulk blasts and on immunophenotypically defined LSCs (CD34+CD38-CD123+)[205]. Based on these data, the COG Cellular Therapies for AML Task Force is conducting a Phase I/II trial of uproleselan combined with myeloablative preparatory regimens for allogeneic SCT in pediatric patients with AML (NCT05569512). The primary endpoint is to identify the RP2D of uproleselan with standard transplant conditioning regimens, and secondary endpoints include 12-month leukemia-free survival (LFS) and 2-year CIR and OS.
5.9. Enasidenib
Mutations in the isocitrate dehydrogenase-2 (IDH2) gene occur in 10-15% of adult AML[206], but only 2% of pediatric AML[207]. Mutations in IDH2 affect cytoplasmic and mitochondrial metabolism, and have also been shown to have epigenetic effects leading to differentiation arrest[208]. A selective small-molecule inhibitor of mutant IDH2, enasidenib, has been developed and tested in adult AML in both R/R and newly diagnosed patients. In the R/R setting, response rates for enasidenib monotherapy average 40% but with a response duration of only 5.8 months[209,210]. In propensity matched cohorts, a survival benefit of ~33% was cited over conventional care (low-dose chemotherapy or supportive management)[211]. Response rates in the up-front setting in combination with azacitidine[212] or intensive chemotherapy[213] were higher – 74% for enasidenib + azacitidine versus 36% for azacitidine alone, and 63% for enasidenib + conventional chemotherapy. The drug appears to induce differentiation of leukemic blasts into neutrophils in responding patients[206]. The role of LSC targeting in the mechanism of action of enasidenib remains inadequately characterized.
5.10. Pevonedistat
NEDD8-activating enzyme regulates degradation of proteins involved in essential processes such as cell cycle progression and DNA damage repair[214]. A novel NEDD8-activating enzyme inhibitor, MLN4924 or pevonedistat, was developed to target cancer cells which rely heavily on protein turnover for these cellular processes. In vitro analysis of pevonedistat demonstrated inhibition of NFkB, increased ROS generation[214], and depletion of intracellular nucleotide pools contributing to increased DNA damage[215]. Co-treatment with pevonedistat and cytarabine in vitro led to increased incorporation of cytarabine into DNA synthesis and decreased colony formation of AML cell lines and primary samples[215]. LSC-targeting capability of pevonedistat has not been well-characterized. However, clinical trials involving pevonedistat have not had the hoped-for success. Dose-limiting toxicities, particularly hepatotoxicity and multi-organ failure, are common[216]. Phase 2 and 3 studies randomizing between pevonedistat and azacitidine versus azacitidine alone have not demonstrated significant benefit to the addition of pevonedistat[217,218]. A phase 1/2 study of pevonedistat in combination with venetoclax and azacitidine showed similar CRc rates to venetoclax + azacitidine alone[219]. Early data from the phase 1 pediatric trial of pevonedistat, azacitidine, fludarabine, and cytarabine again suggest high rates of dose-limiting toxicities (25%) with only 3 of 12 patients achieving CR with incomplete count recovery[220]. Data maturation will be needed to determine whether this agent moves forward in later phase pediatric trials.
6. Future Targets
6.1. Immunotherapies
Although not a comprehensive list, below are some potential immunotherapy targets for pediatric myeloid LSCs that may see future clinical development:
- CD70 is a tumor necrosis factor receptor ligand that is not normally expressed in normal tissues or on HSCs during hematopoiesis. It is upregulated on immune cells upon activation but not on resting B or T lymphocytes[221]. It has been demonstrated that CD34+ AML cells and LSCs express CD70 and its receptor CD27, that CD70/CD27 signaling in AML cells activates stem cell expression programs, and that the promoter for CD70 is sensitive to methylation[222,223]. For these reasons, blocking CD70/CD27 signaling in conjunction with hypomethylating agents is being considered as a potential treatment concept for AML. Currently, a CD70-targeting antibody, cusatuzumab, in combination with azaciditine or venetoclax, remains under clinical investigation with promising initial responses but short follow-up of treated patients to date[224].
- Surface expression of CD69 was enriched on LSCs from patients whose disease proved chemoresistant in one study, and CD69 expression in transcriptional data from large retrospective cohorts of pediatric patients correlated with poor outcomes[66]. Therefore, CD69 could represent a future LSC targeting strategy for pediatric AML, although CD69 expression on regulatory T cells and other specialized T-cell subsets may indicate unwanted side effects of immune dysregulation with CD69 targeting[225].
6.2. Other Novel Agents
Although not a comprehensive list, below are some potential novel non-immunotherapy targets for pediatric myeloid LSCs that may see future clinical development:
A 2021 study evaluated AML transcriptional data from 284 pediatric patients, and found that high expression of calcitonin receptor-like (CALCRL), a G-protein coupled receptor with roles in proliferation, apoptosis, and inflammation, was associated with inferior 5-year EFS and OS compared to those with low expression of CALCRL[226]. Antibody- or small molecule-based targeting of the CALCRL ligand calcitonin gene-related peptide (CGRP) are currently being investigated for migraine (NCT03432286, NCT05217927), and may warrant investigation as a novel agent for both adult and pediatric AML. As noted above, given the association of LSCs with alternative splicing and exon skipping[59], a splicing modulator such as rebecsinib might be of future clinical interest in pediatric AML, particularly combined with anti-CD47 agents and/or pro-apoptotic agents such as venetoclax.
Telomerase activity has been postulated to be a dependency in adult LSCs, and genetic deletion of an RNA template subunit (TERC) in mouse models of leukemia significantly impaired LSC functionality[227]. While children with AML tend to have lower telomerase activity on average than adults, those with higher telomerase activity had worse outcomes in a retrospective analysis[228]. Pre-treatment of 6 pediatric patient-derived xenograft (PDX) samples with the telomerase inhibitor imetelstat, alone or in combination with azacitidine or cytarabine-based chemotherapy, reduced LSC viability, prolonged survival in primary murine recipients, and reduced engraftment into secondary recipients[229]. These data suggest that imetelstat could be an LSC-targeting therapy that should be prioritized for combination regimens. There is currently an ongoing clinical trial for adult MDS and AML that is enrolling at multiple Australian sites (NCT05583552).
There is substantial evidence in adult AML and mounting evidence in pediatric AML that these diseases arise as a result of a leukemia-initiating population of LSCs. Identification of phenotypic, metabolic, and other vulnerabilities of these cells is the most direct path forward toward development of more effective and less toxic therapies for patients of all ages with AML. Prioritization of immunotherapeutic and other novel agents for which there is strong preclinical and/or adult clinical data for LSC targeting should be standard practice for pediatric clinical trial design.
Author Contributions
L.A.M. and A.C.W. contributed equally to the writing of this review article. A.C.W. conceptualized the topic and outline of the article; L.A.M. and A.C.W. both contributed to literature review, writing, review, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
L.A.M. and A.C.W. would like to acknowledge all of the patients who have taught them so much about AML. A.C.W. would also like to acknowledge her mentors Drs. Craig Jordan and Dan Pollyea, who brought her into the LSC research community.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.; Antoniou, E.; Waack, K. Pediatric Acute Myeloid Leukemia-Past, Present, and Future. J Clin Med 2022, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Duncavage, E.J.; Chang, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Heath, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017, 31, 872–881. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- DeWolf, S.; Tallman, M.S. How I treat relapsed or refractory AML. Blood 2020, 136, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Thol, F. What to use to treat AML: The role of emerging therapies. Hematology Am Soc Hematol Educ Program 2021, 2021, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, P.J.; Gartler, S.M.; Yoshida, A. Clonal origin of chronic myelocytic leukemia in man. Proceedings of the National Academy of Sciences 1967, 58, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, E. Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982). 1983.
- Griffin, J.D.; Lowenberg, B. Clonogenic cells in acute myeloblastic leukemia. 1986.
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- van Rhenen, A.; Feller, N.; Kelder, A.; Westra, A.H.; Rombouts, E.; Zweegman, S.; van der Pol, M.A.; Waisfisz, Q.; Ossenkoppele, G.J.; Schuurhuis, G.J. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res 2005, 11, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Khan, N.; D'Alessandro, A.; Nemkov, T.; Winters, A.; Jones, C.L.; Zhang, W.; Pollyea, D.A.; Jordan, C.T. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat Commun 2018, 9, 3694. [Google Scholar] [CrossRef] [PubMed]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest 2011, 121, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O'Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Sachs, K.; Sarver, A.L.; Noble-Orcutt, K.E.; LaRue, R.S.; Antony, M.L.; Chang, D.; Lee, Y.; Navis, C.M.; Hillesheim, A.L.; Nykaza, I.R.; et al. Single-Cell Gene Expression Analyses Reveal Distinct Self-Renewing and Proliferating Subsets in the Leukemia Stem Cell Compartment in Acute Myeloid Leukemia. Cancer Res 2020, 80, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Gonen, M.; Sun, Z.; Figueroa, M.E.; Patel, J.P.; Abdel-Wahab, O.; Racevskis, J.; Ketterling, R.P.; Fernandez, H.; Rowe, J.M.; Tallman, M.S.; et al. CD25 expression status improves prognostic risk classification in AML independent of established biomarkers: ECOG phase 3 trial, E1900. Blood 2012, 120, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.H.; Roy, N.; Chakraborty, S.; Desrichard, A.; Chung, S.S.; Woolthuis, C.M.; Hu, W.; Berezniuk, I.; Garrett-Bakelman, F.E.; Hamann, J.; et al. CD97 is a critical regulator of acute myeloid leukemia stem cell function. J Exp Med 2019, 216, 2362–2377. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Kersten, B.; Valkering, M.; Wouters, R.; van Amerongen, R.; Hanekamp, D.; Kwidama, Z.; Valk, P.; Ossenkoppele, G.; Zeijlemaker, W.; Kaspers, G.; et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br J Haematol 2016, 173, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol 2013, 98, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Askmyr, M.; Agerstam, H.; Hansen, N.; Gordon, S.; Arvanitakis, A.; Rissler, M.; Juliusson, G.; Richter, J.; Jaras, M.; Fioretos, T. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 2013, 121, 3709–3713. [Google Scholar] [CrossRef] [PubMed]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Dai, B.; Gentles, A.J.; Majeti, R.; Feinberg, A.P. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 2015, 6, 8489. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xue, M.; Deng, X.; Dong, L.; Nguyen, L.X.T.; Ren, L.; Han, L.; Li, C.; Xue, J.; Zhao, Z.; et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell 2023, 30, 1072–1090. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gao, Z.; Zhang, T.; Wang, Y.; Xie, X.; Han, G.; Li, Y.; Yin, R.; Chen, Y.; Wang, P.; et al. Decoding m(6)A RNA methylome identifies PRMT6-regulated lipid transport promoting AML stem cell maintenance. Cell Stem Cell 2023, 30, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Wang, P.; Han, G.; Zhang, T.; Chang, J.; Yin, R.; Shan, Y.; Wen, J.; Xie, X.; et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 2020, 27, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Hierro, L.; van Galen, P.; Prado, M.A.; Higby, K.J.; Togami, K.; Mowery, C.T.; Paulo, J.A.; Xie, Y.; Cejas, P.; Furusawa, T.; et al. Chromatin accessibility promotes hematopoietic and leukemia stem cell activity. Nat Commun 2020, 11, 1406. [Google Scholar] [CrossRef] [PubMed]
- Paubelle, E.; Zylbersztejn, F.; Maciel, T.T.; Carvalho, C.; Mupo, A.; Cheok, M.; Lieben, L.; Sujobert, P.; Decroocq, J.; Yokoyama, A.; et al. Vitamin D Receptor Controls Cell Stemness in Acute Myeloid Leukemia and in Normal Bone Marrow. Cell Rep 2020, 30, 739–754. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.; Anokye, J.; Yeung, M.M.; Lam, E.Y.N.; Chan, Y.C.; Weng, C.F.; Yeh, P.; Knezevic, K.; Butler, M.S.; Hoegl, A.; et al. HBO1 is required for the maintenance of leukaemia stem cells. Nature 2020, 577, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O'Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D'Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Velasco-Hernandez, T.; Mess, J.; Lalioti, M.E.; Romero-Mulero, M.C.; Obier, N.; Karantzelis, N.; Rettkowski, J.; Schonberger, K.; Karabacz, N.; et al. GPRC5C Drives Branched-Chain Amino Acid Metabolism in Leukemogenesis. Blood Adv 2023. [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D'Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019. [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D'Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Culp-Hill, R.; Stevens, B.M.; Jones, C.L.; Pei, S.; Dzieciatkowska, M.; Minhajuddin, M.; Jordan, C.T.; D'Alessandro, A. Therapy-Resistant Acute Myeloid Leukemia Stem Cells Are Resensitized to Venetoclax + Azacitidine by Targeting Fatty Acid Desaturases 1 and 2. Metabolites 2023, 13, 467. [Google Scholar] [CrossRef] [PubMed]
- Subedi, A.; Liu, Q.; Ayyathan, D.M.; Sharon, D.; Cathelin, S.; Hosseini, M.; Xu, C.; Voisin, V.; Bader, G.D.; D'Alessandro, A.; et al. Nicotinamide phosphoribosyltransferase inhibitors selectively induce apoptosis of AML stem cells by disrupting lipid homeostasis. Cell Stem Cell 2021, 28, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D'Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.R.; Sheth, V.; Li, H.; Harris, M.W.; Qiu, S.; Crossman, D.K.; Kumar, H.; Agarwal, P.; Nagasawa, T.; Paterson, A.J.; et al. Microenvironmental CXCL12 deletion enhances Flt3-ITD acute myeloid leukemia stem cell response to therapy by reducing p38 MAPK signaling. Leukemia 2023, 37, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ma, R.; Ma, S.; Tian, L.; Lu, T.; Zhang, J.; Mundy-Bosse, B.L.; Zhang, B.; Marcucci, G.; Caligiuri, M.A.; et al. ILC1s control leukemia stem cell fate and limit development of AML. Nat Immunol 2022, 23, 718–730. [Google Scholar] [CrossRef] [PubMed]
- de Grouw, E.P.; Raaijmakers, M.H.; Boezeman, J.B.; van der Reijden, B.A.; van de Locht, L.T.; de Witte, T.J.; Jansen, J.H.; Raymakers, R.A. Preferential expression of a high number of ATP binding cassette transporters in both normal and leukemic CD34+CD38- cells. Leukemia 2006, 20, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.M.; Hogge, D.E.; Ling, V. MDR1 and BCRP1 expression in leukemic progenitors correlates with chemotherapy response in acute myeloid leukemia. Exp Hematol 2008, 36, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.G.; Wang, R.Y.; Kuehnle, I.; Weidner, D.; Marini, F.; Brenner, M.K.; Andreeff, M.; Goodell, M.A. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001, 98, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK-ULK1-Mediated Autophagy Confers Resistance to BET Inhibitor JQ1 in Acute Myeloid Leukemia Stem Cells. Clin Cancer Res 2017, 23, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Chung, H.; Kim, Y.R.; Kim, J.S.; Cheong, J.W.; Min, Y.H. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J Exp Clin Cancer Res 2020, 39, 66. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Kumar, H.; Yan, C.; Li, H.; Paterson, A.J.; Anderson, N.R.; He, J.; Yang, J.; Xie, M.; Crossman, D.K.; et al. Autophagy inhibition impairs leukemia stem cell function in FLT3-ITD AML but has antagonistic interactions with tyrosine kinase inhibition. Leukemia 2022, 36, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv 2021, 5, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Pabon, C.M.; Abbas, H.A.; Konopleva, M. Acute myeloid leukemia: Therapeutic targeting of stem cells. Expert Opin Ther Targets 2022, 26, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Stelmach, P.; Trumpp, A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica 2023, 108, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Gonzalez, A.; Dorantes-Acosta, E.; Moreno-Lorenzana, D.; Alvarado-Moreno, A.; Arriaga-Pizano, L.; Mayani, H. Expression of CD90, CD96, CD117, and CD123 on different hematopoietic cell populations from pediatric patients with acute myeloid leukemia. Arch Med Res 2014, 45, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.A.; Rosenberg, C.A.; Brondum, R.F.; Aggerholm, A.; Kjeldsen, E.; Rahbek, O.; Ludvigsen, M.; Hasle, H.; Roug, A.S.; Bill, M. Immunophenotypically defined stem cell subsets in paediatric AML are highly heterogeneous and demonstrate differences in BCL-2 expression by cytogenetic subgroups. Br J Haematol 2022, 197, 452–466. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, I.; Mondala, P.K.; Steel, S.K.; Balaian, L.; Ladel, L.; Mason, C.N.; Diep, R.H.; Pham, J.; Cloos, J.; Kaspers, G.J.L.; et al. Detection and targeting of splicing deregulation in pediatric acute myeloid leukemia stem cells. Cell Rep Med 2023, 4, 100962. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, E.Z.; Madney, Y.; Eldin, D.N.; Shafik, N.F. Overexpression of CD200 and CD123 is a major influential factor in the clinical course of pediatric acute myeloid leukemia. Exp Mol Pathol 2021, 118, 104597. [Google Scholar] [CrossRef] [PubMed]
- Thakral, D.; Singh, V.K.; Gupta, R.; Jha, N.; Khan, A.; Kaur, G.; Rai, S.; Kumar, V.; Supriya, M.; Bakhshi, S.; et al. Integrated single-cell transcriptome analysis of CD34 + enriched leukemic stem cells revealed intra- and inter-patient transcriptional heterogeneity in pediatric acute myeloid leukemia. Ann Hematol 2023, 102, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Depreter, B.; De Moerloose, B.; Vandepoele, K.; Uyttebroeck, A.; Van Damme, A.; Terras, E.; Denys, B.; Dedeken, L.; Dresse, M.F.; Van der Werff Ten Bosch, J.; et al. Deciphering molecular heterogeneity in pediatric AML using a cancer vs. normal transcriptomic approach. Pediatr Res 2021, 89, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.J.; Eidenschink Brodersen, L.; Alonzo, T.A.; Wang, J.; Pardo, L.; Sung, L.; Cooper, T.M.; Kolb, E.A.; Aplenc, R.; Tasian, S.K.; et al. CD123 Expression Is Associated With High-Risk Disease Characteristics in Childhood Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 2022, 40, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Herbrich, S.; Baran, N.; Cai, T.; Weng, C.; Aitken, M.J.L.; Post, S.M.; Henderson, J.; Shi, C.; Richard-Carpentier, G.; Sauvageau, G.; et al. Overexpression of CD200 is a Stem Cell-Specific Mechanism of Immune Evasion in AML. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Cieniewicz, B.; Uyeda, M.J.; Chen, P.P.; Sayitoglu, E.C.; Liu, J.M.; Andolfi, G.; Greenthal, K.; Bertaina, A.; Gregori, S.; Bacchetta, R.; et al. Engineered type 1 regulatory T cells designed for clinical use kill primary pediatric acute myeloid leukemia cells. Haematologica 2021, 106, 2588–2597. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S.; He, F.; Tian, Y.; Hu, H.; Gao, L.; Zhang, L.; Chen, A.; Hu, Y.; Fan, L.; et al. Single-cell transcriptomics reveals multiple chemoresistant properties in leukemic stem and progenitor cells in pediatric AML. Genome Biol 2023, 24, 199. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Villenet, C.; Petit, A.; Rousseau, A.; Ng, S.W.K.; Paquet, A.; Gonzales, F.; Barthelemy, A.; Lepretre, F.; et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia 2018. [CrossRef] [PubMed]
- Elsayed, A.H.; Rafiee, R.; Cao, X.; Raimondi, S.; Downing, J.R.; Ribeiro, R.; Fan, Y.; Gruber, T.A.; Baker, S.; Klco, J.; et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia 2020, 34, 735–745. [Google Scholar] [CrossRef]
- Huang, B.J.; Smith, J.L.; Farrar, J.E.; Wang, Y.C.; Umeda, M.; Ries, R.E.; Leonti, A.R.; Crowgey, E.; Furlan, S.N.; Tarlock, K.; et al. Integrated stem cell signature and cytomolecular risk determination in pediatric acute myeloid leukemia. Nat Commun 2022, 13, 5487. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.A.; Ma, W.; Ladel, L.; Pham, J.; Balaian, L.; Steel, S.K.; Mondala, P.K.; Diep, R.H.; Wu, C.N.; Mason, C.N.; et al. Reversal of malignant ADAR1 splice isoform switching with Rebecsinib. Cell Stem Cell 2023, 30, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Ceraulo, A.; Lapillonne, H.; Cheok, M.H.; Preudhomme, C.; Dombret, H.; Terre, C.; Lambert, J.; Leverger, G.; Bertrand, Y.; Mortreux, F.; et al. Prognostic impact of ABCA3 expression in adult and pediatric acute myeloid leukemia: An ALFA-ELAM02 joint study. Blood Adv 2022, 6, 2773–2777. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Jeong, S.Y.; Lee, H.R.; Oh, I.H. Age-related differences in the bone marrow stem cell niche generate specialized microenvironments for the distinct regulation of normal hematopoietic and leukemia stem cells. Sci Rep 2019, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Orgel, E.; Genkinger, J.M.; Aggarwal, D.; Sung, L.; Nieder, M.; Ladas, E.J. Association of body mass index and survival in pediatric leukemia: A meta-analysis. Am J Clin Nutr 2016, 103, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, D.J.A.; Asdahl, P.H.; Abrahamsson, J.; Ha, S.Y.; Jonsson, O.G.; Kaspers, G.J.L.; Koskenvuo, M.; Lausen, B.; De Moerloose, B.; Palle, J.; et al. Associations between pretherapeutic body mass index, outcome, and cytogenetic abnormalities in pediatric acute myeloid leukemia. Cancer Med 2019, 8, 6634–6643. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.D. CD33 as a target for selective ablation of acute myeloid leukemia. Clin Lymphoma 2002, 2 Suppl 1, S9–11. [Google Scholar] [CrossRef]
- Hauswirth, A.W.; Florian, S.; Printz, D.; Sotlar, K.; Krauth, M.T.; Fritsch, G.; Schernthaner, G.H.; Wacheck, V.; Selzer, E.; Sperr, W.R.; et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest 2007, 37, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.D. Monoclonal antibodies to the myeloid stem cells: Therapeutic implications of CMA-676, a humanized anti-CD33 antibody calicheamicin conjugate. Leukemia 2000, 14, 474–475. [Google Scholar] [CrossRef]
- Majeti, R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 2011, 30, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 2016, 34, 747–755. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J Clin Oncol 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 2014, 15, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terre, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terre, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Sung, L.; Pollard, J.A.; Aplenc, R.; Loken, M.R.; Gamis, A.S.; et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Clin Cancer Res 2016, 22, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin Cancer Res 2018, 24, 3242–3246. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012, 119, 3705–3711. [Google Scholar] [CrossRef] [PubMed]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.S.; DeAngelo, D.J.; Kovacsovics, T.J.; et al. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood 2018, 132, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.; et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.; Guru Murthy, G.S.; Szabo, A.; Hamadani, M.; Michaelis, L.C.; Carlson, K.-S.; Runaas, L.; Gauger, K.; Desai, A.G.; Chen, M.M. Lintuzumab-Ac225 with combination with intensive chemotherapy yields high response rate and MRD negativity in R/R AML with adverse features. Blood 2022, 140, 157–158. [Google Scholar] [CrossRef]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood, The Journal of the American Society of Hematology 2017, 129, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.; Cheung, N. Acute myeloid leukemia targets for bispecific antibodies. Blood cancer journal 2017, 7, e522. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M. Preclinical characterization of AMG 330, a CD3/CD33-bispecific t-cell–engaging antibody with potential for treatment of acute myelogenous leukemia. Molecular cancer therapeutics 2014, 13, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.H.; Gudgeon, C.J.; Laszlo, G.S.; Newhall, K.J.; Sinclair, A.M.; Frankel, S.R.; Kischel, R.; Chen, G.; Walter, R.B. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS ONE 2015, 10, e0135945. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood, The Journal of the American Society of Hematology 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R. Targeting CD123 in acute myeloid leukemia using a T-cell–directed dual-affinity retargeting platform. Blood, The Journal of the American Society of Hematology 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-s.; Wang, Y.; Lv, H.-y.; Han, Q.-w.; Fan, H.; Guo, B.; Wang, L.-l.; Han, W.-d. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Molecular therapy 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453 e1419. [Google Scholar] [CrossRef]
- Ehninger, A.; Kramer, M.; Rollig, C.; Thiede, C.; Bornhauser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J 2014, 4, e218. [Google Scholar] [CrossRef]
- Mitchell, K.; Steidl, U. Targeting Immunophenotypic Markers on Leukemic Stem Cells: How Lessons from Current Approaches and Advances in the Leukemia Stem Cell (LSC) Model Can Inform Better Strategies for Treating Acute Myeloid Leukemia (AML). Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Konopleva, M. Approval of tagraxofusp-erzs for blastic plasmacytoid dendritic cell neoplasm. Blood Adv 2020, 4, 4020–4027. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Goswami, S.; Gopalakrishnan, B.; Ramaswamy, R.; Wasmuth, R.; Tran, M.; Mo, X.; Gordon, A.; Bucci, D.; Lucas, D.M.; et al. The interleukin-3 receptor CD123 targeted SL-401 mediates potent cytotoxic activity against CD34(+)CD123(+) cells from acute myeloid leukemia/myelodysplastic syndrome patients and healthy donors. Haematologica 2018, 103, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv 2018, 2, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Wang, E.S.; Papadantonakis, N.; Deconinck, E.; Erba, H.P.; Pemmaraju, N.; Lane, A.A.; Rizzieri, D.A. Clinical profile of IMGN632, a novel CD123-targeting antibody-drug conjugate (ADC), in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML) or blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 2019, 134, 734. [Google Scholar] [CrossRef]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Wang, E.S.; Todisco, E.; Tarella, C.; Martinelli, G.; Erba, H.P.; Deconinck, E.; Sweet, K.L. A phase I/II study of IMGN632, a novel CD123-targeting antibody-drug conjugate, in patients with relapsed/refractory acute myeloid leukemia, blastic plasmacytoid dendritic cell neoplasm, and other CD123-positive hematologic malignancies. 2020.
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Uy, G.L.; Godwin, J.; Rettig, M.P.; Vey, N.; Foster, M.; Arellano, M.L.; Rizzieri, D.A.; Topp, M.S.; Huls, G.; Lowenberg, B. Preliminary results of a phase 1 study of flotetuzumab, a CD123 x CD3 bispecific Dart® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood 2017, 130, 637. [Google Scholar] [CrossRef]
- Gauthier, L.; Virone-Oddos, A.; Beninga, J.; Rossi, B.; Nicolazzi, C.; Amara, C.; Blanchard-Alvarez, A.; Gourdin, N.; Courta, J.; Basset, A. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nature Biotechnology 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.S.; Jongen-Lavrencic, M.; Garciaz, S.; Huls, G.A.; Maiti, A.; Boissel, N.; De Botton, S.; Fleming, S.; Zwaan, C.M.; C. de Leeuw, D. A first-in-human study of CD123 NK cell engager SAR443579 in relapsed or refractory acute myeloid leukemia, B-cell acute lymphoblastic leukemia, or high-risk myelodysplasia. 2023.
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: A first-in-human clinical trial. Blood 2017, 130, 811. [Google Scholar] [CrossRef]
- Masarova, L.; Kantarjian, H.; Ravandi, F.; Sharma, P.; Garcia-Manero, G.; Daver, N. Update on Immunotherapy in AML and MDS: Monoclonal Antibodies and Checkpoint Inhibitors Paving the Road for Clinical Practice. Adv Exp Med Biol 2018, 995, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Kantarjian, H.; Garcia-Mannero, G.; Ravandi, F.; Sharma, P.; Daver, N. Harnessing the Immune System Against Leukemia: Monoclonal Antibodies and Checkpoint Strategies for AML. Adv Exp Med Biol 2017, 995, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O'Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Driss, V.; Liu, J.; Kuranda, K.; Leleu, X.; Jouy, N.; Hetuin, D.; Quesnel, B. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol Immunother 2010, 59, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Kronig, H.; Kremmler, L.; Haller, B.; Englert, C.; Peschel, C.; Andreesen, R.; Blank, C.U. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol 2014, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer discovery 2019, 9, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Reville, P.K.; Kantarjian, H.M.; Ravandi, F.; Jabbour, E.; DiNardo, C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients: A single-arm, open-label, phase II study. Blood Cancer J 2021, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front Oncol 2019, 9, 1380. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Gip, P.; McKenna, K.M.; Zhao, F.; Mata, O.; Choi, T.S.; Duan, J.; Sompalli, K.; Majeti, R.; Weissman, I.L. Combination treatment with 5F9 and azacitidine enhances phagocytic elimination of acute myeloid leukemia. Blood 2018, 132, 2729. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S. The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: Ongoing phase 1b results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Masetti, R.; Pigazzi, M.; Togni, M.; Astolfi, A.; Indio, V.; Manara, E.; Casadio, R.; Pession, A.; Basso, G.; Locatelli, F. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 2013, 121, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Le, Q.; Castro, S.; Pardo, L.; McKay, C.N.; Perkins, L.; Smith, J.; Kirkey, D.; Abrahams, C.; Bedard, K.; et al. Targeting FOLR1 in high-risk CBF2AT3-GLIS2 pediatric AML with STRO-002 FOLR1-antibody-drug conjugate. Blood Adv 2022, 6, 5933–5937. [Google Scholar] [CrossRef]
- Le, Q.; Hadland, B.; Smith, J.L.; Leonti, A.; Huang, B.J.; Ries, R.; Hylkema, T.A.; Castro, S.; Tang, T.T.; McKay, C.N.; et al. CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Miller, L.; Massaro, S.; Williams, R.; Krieger, E.; Pauly, M.; Nelson, C.; Bhojwani, D.; Johnson, R.; Horton, T.M. Anti-Leukemic Activity of STRO-002 a Novel Folate Receptor-α (FR-α)-Targeting ADC in Relapsed/Refractory CBF2AT3-GLIS2 AML. Blood 2022, 140, 159–161. [Google Scholar] [CrossRef]
- Le, Q.; Chen, F.; Tang, T.; McKay, C.N.; Wu, F.; Turker, M.; Gardinier, T.; Patel, V.; Venkatesan, A.; Khor, T. Therapeutic targeting of CBFA2T3-GLIS2 infant AML with ELU001-folate receptor alpha-directed C’Dot-drug-conjugate. Cancer Research 2022, 82, 1075. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Weber, D.; Fiedler, W.; Salih, H.R.; Wulf, G.; Salwender, H.; Schroeder, T.; Kindler, T.; Lubbert, M.; Wolf, D.; et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 2019, 133, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Rollig, C.; Wollmer, E.; Wasch, R.; Bornhauser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3-Internal Tandem Duplication Mutation (SORMAIN). J Clin Oncol 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Serve, H.; Krug, U.; Wagner, R.; Sauerland, M.C.; Heinecke, A.; Brunnberg, U.; Schaich, M.; Ottmann, O.; Duyster, J.; Wandt, H.; et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: Results from a randomized, placebo-controlled trial. J Clin Oncol 2013, 31, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol 2015, 16, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Perl, A.E.; Dohner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol 2018, 19, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A Phase I Study of Quizartinib Combined with Chemotherapy in Relapsed Childhood Leukemia: A Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Clin Cancer Res 2016, 22, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Chang, B.; Cooper, T.; Gross, T.; Gupta, S.; Neudorf, S.; Adlard, K.; Ho, P.A.; McGoldrick, S.; Watt, T.; et al. Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer 2015, 62, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Fisher, B.; Hirsch, B.; Kahwash, S.; Getz, K.; et al. Sorafenib in Combination With Standard Chemotherapy for Children With High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report From the Children's Oncology Group Protocol AAML1031. J Clin Oncol 2022, 40, 2023–2035. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Murphy, K.M.; Pham, R.; Kim, K.T.; Stine, A.; Li, L.; McNiece, I.; Smith, B.D.; Small, D. Internal tandem duplications of the FLT3 gene are present in leukemia stem cells. Blood 2005, 106, 673–680. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.B.; Woods, W.G.; Lange, B.J.; Sweetser, D.A.; Radich, J.P.; Bernstein, I.D.; Meshinchi, S. FLT3 internal tandem duplication in CD34+/CD33- precursors predicts poor outcome in acute myeloid leukemia. Blood 2006, 108, 2764–2769. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J Hematol Oncol 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Strang, E.; Theis, F.; Jahn, N.; Panina, E.; Blatte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Greenblatt, S.M.; Shirley, C.M.; Duffield, A.S.; Bruner, J.K.; Li, L.; Nguyen, B.; Jung, E.; Aplan, P.D.; Ghiaur, G.; et al. All-trans retinoic acid synergizes with FLT3 inhibition to eliminate FLT3/ITD+ leukemia stem cells in vitro and in vivo. Blood 2016, 127, 2867–2878. [Google Scholar] [CrossRef]
- Smith, C.C.; Levis, M.J.; Frankfurt, O.; Pagel, J.M.; Roboz, G.J.; Stone, R.M.; Wang, E.S.; Severson, P.L.; West, B.L.; Le, M.H.; et al. A phase 1/2 study of the oral FLT3 inhibitor pexidartinib in relapsed/refractory FLT3-ITD-mutant acute myeloid leukemia. Blood Adv 2020, 4, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front Pediatr 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rucker, F.G.; Dohner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, M.; Blair, H.; Troester, S.; Szoltysek, K.; Cameron, R.; Ashtiani, M.; Krippner-Heidenreich, A.; Grebien, F.; McGeehan, G.; Zwaan, C.M.; et al. The MLL-Menin Interaction is a Therapeutic Vulnerability in NUP98-rearranged AML. Hemasphere 2023, 7, e935. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Ma, J.; Huang, B.J.; Hagiwara, K.; Westover, T.; Abdelhamed, S.; Barajas, J.M.; Thomas, M.E.; Walsh, M.P.; Song, G.; et al. Integrated Genomic Analysis Identifies UBTF Tandem Duplications as a Recurrent Lesion in Pediatric Acute Myeloid Leukemia. Blood Cancer Discov 2022, 3, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.M.; Rasouli, M.; Umeda, M.; Hiltenbrand, R.L.; Abdelhamed, S.; Mohnani, R.; Arthur, B.; Westover, T.; Thomas, M.E., 3rd; Ashtiani, M.; et al. Acute myeloid leukemias with UBTF tandem duplications are sensitive to Menin inhibitors. Blood 2023. [CrossRef] [PubMed]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef]
- Fiskus, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Das, K.; Takahashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor ziftomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, C.D.; Niswander, L.M.; Kessler, L.; Tomkinson, B.; Burrows, F.; Tasian, S.K. Preclinical In Vivo Activity of the Menin Inhibitor Ziftomenib (KO-539) in Pediatric KMT2A-Rearranged Acute Lymphoblastic Leukemia. Blood 2022, 140, 3491–3492. [Google Scholar] [CrossRef]
- Erba, H.P.; Fathi, A.T.; Issa, G.C.; Altman, J.K.; Montesinos, P.; Patnaik, M.M.; Foran, J.M.; De Botton, S.; Baer, M.R.; Schiller, G.J. Update on a phase 1/2 first-in-human study of the menin-KMT2A (MLL) inhibitor ziftomenib (KO-539) in patients with relapsed or refractory acute myeloid leukemia. Blood 2022, 140, 153–156. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol 2018, 19, 216–228. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J Clin Oncol 2021, 39, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Shelton, I.T.; Gillen, A.E.; Stevens, B.M.; Gasparetto, M.; Wang, Y.; Liu, L.; Liu, J.; Brunetti, T.M.; Engel, K.; et al. A Novel Type of Monocytic Leukemia Stem Cell Revealed by the Clinical Use of Venetoclax-Based Therapy. Cancer Discov 2023, 13, 2032–2049. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Maloney, K.W.; Treece, A.L.; Gore, L.; Franklin, A.K. Single-center pediatric experience with venetoclax and azacitidine as treatment for myelodysplastic syndrome and acute myeloid leukemia. Pediatr Blood Cancer 2020, 67, e28398. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Li, Y.; Ashcraft, E.; Karol, S.E.; Rubnitz, J.E.; Epperly, R.; Madden, R.; Mamcarz, E.; Obeng, E.; Qudeimat, A. Venetoclax-based therapy as a bridge to allogeneic hematopoietic cell transplantation in children with relapsed/refractory AML. Bone Marrow Transplantation 2023, 58, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Marinoff, A.E.; Aaronson, K.; Agrawal, A.K.; Braun, B.S.; Golden, C.; Huang, B.J.; Michlitsch, J.; Southworth, E.; Thrall, A.; Vo, K.T.; et al. Venetoclax in combination with chemotherapy as treatment for pediatric advanced hematologic malignancies. Pediatr Blood Cancer 2023, 70, e30335. [Google Scholar] [CrossRef] [PubMed]
- Trabal, A.; Gibson, A.; He, J.; McCall, D.; Roth, M.; Nunez, C.; Garcia, M.; Buzbee, M.; Toepfer, L.; Bidikian, A.; et al. Venetoclax for Acute Myeloid Leukemia in Pediatric Patients: A Texas Medical Center Experience. Cancers (Basel) 2023, 15, 1983. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Baccelli, F.; Leardini, D.; Gottardi, F.; Vendemini, F.; Di Gangi, A.; Becilli, M.; Lodi, M.; Tumino, M.; Vinci, L.; et al. Venetoclax-based therapies in pediatric advanced MDS and relapsed/refractory AML: A multicenter retrospective analysis. Blood Adv 2023, 7, 4366–4370. [Google Scholar] [CrossRef] [PubMed]
- Klimentova, M.; Shelikhova, L.; Ilushina, M.; Kozlovskaya, S.; Blagov, S.; Popov, A.; Kashpor, S.; Fadeeva, M.; Olshanskaya, J.; Glushkova, S.; et al. Targeted Therapy With Venetoclax and Daratumumab as Part of HSCT Preparative Regimen in Children With Chemorefractory Acute Myeloid Leukemia. Transplant Cell Ther 2023, 29, 127 e121–127 e129. [Google Scholar] [CrossRef] [PubMed]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Das Gupta, S.; et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: A phase 1, dose-escalation study. Lancet Oncol 2020, 21, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Stuani, L.; Jager, A.; Sahal, A.; Koladiya, A.; Sarno, J.; Domizi, P.; Liu, Y.; De Mas, V.; Recher, C.; Vergez, F. Single-Cell Proteomic Analysis Defines Metabolic Heterogeneity in Response to Venetoclax in AML. Blood 2022, 140, 1028–1029. [Google Scholar] [CrossRef]
- Hege, K.; Kalpage, H.; Hüttemann, M.; Pitman, H.; Taub, J.W.; Polin, L.; Li, J.; Ge, Y. Enhancing the antileukemic activity of venetoclax against leukemia stem cells by targeting oxidative phosphorylation through dual inhibition of PI3K and HDAC. Cancer Research 2021, 81, 2463. [Google Scholar] [CrossRef]
- Kuhlen, M.; Klusmann, J.H.; Hoell, J.I. Molecular Approaches to Treating Pediatric Leukemias. Front Pediatr 2019, 7, 368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; So, C.W. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Exp Hematol 2016, 44, 902–907. [Google Scholar] [CrossRef]
- Gore, L.; Triche, T.J., Jr.; Farrar, J.E.; Wai, D.; Legendre, C.; Gooden, G.C.; Liang, W.S.; Carpten, J.; Lee, D.; Alvaro, F.; et al. A multicenter, randomized study of decitabine as epigenetic priming with induction chemotherapy in children with AML. Clin Epigenetics 2017, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Pommert, L.; Schafer, E.S.; Malvar, J.; Gossai, N.; Florendo, E.; Pulakanti, K.; Heimbruch, K.; Stelloh, C.; Chi, Y.Y.; Sposto, R.; et al. Decitabine and vorinostat with FLAG chemotherapy in pediatric relapsed/refractory AML: Report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. Am J Hematol 2022, 97, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Chao, K.; Stevens, A.M.; Jo, E.; Hilsenbeck, S.G.; Gossai, N.P.; Doan, A.; Colace, S.I.; Guinipero, T.; Otterson, D.; et al. Real-world experience in treating pediatric relapsed/refractory or therapy-related myeloid malignancies with decitabine, vorinostat, and FLAG therapy based on a phase 1 study run by the TACL consortium. Pediatr Blood Cancer 2022, 69, e29812. [Google Scholar] [CrossRef]
- Bhatnagar, B.; Zhao, Q.; Mims, A.S.; Vasu, S.; Behbehani, G.K.; Larkin, K.; Blachly, J.S.; Badawi, M.A.; Hill, K.L.; Dzwigalski, K.R.; et al. Phase 1 study of selinexor in combination with salvage chemotherapy in Adults with relapsed or refractory Acute myeloid leukemia. Leuk Lymphoma 2023, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pianigiani, G.; Gagliardi, A.; Mezzasoma, F.; Rocchio, F.; Tini, V.; Bigerna, B.; Sportoletti, P.; Caruso, S.; Marra, A.; Peruzzi, S.; et al. Prolonged XPO1 inhibition is essential for optimal antileukemic activity in NPM1-mutated AML. Blood Adv 2022, 6, 5938–5949. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Kashyap, T.; Yu, X.; Meng, X.; Lai, T.H.; McNeil, B.; Bhatnagar, B.; Shacham, S.; Kauffman, M.; Dorrance, A.M.; et al. XPO1 Inhibition using Selinexor Synergizes with Chemotherapy in Acute Myeloid Leukemia by Targeting DNA Repair and Restoring Topoisomerase IIalpha to the Nucleus. Clin Cancer Res 2016, 22, 6142–6152. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wu, S.; Liu, S.; Li, X.; Gai, Y.; Lin, H.; Wang, Y.; Edwards, H.; Ge, Y.; Wang, G. Venetoclax enhances DNA damage induced by XPO1 inhibitors: A novel mechanism underlying the synergistic antileukaemic effect in acute myeloid leukaemia. J Cell Mol Med 2022, 26, 2646–2657. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.A.; Friedlander, S.Y.; Arrate, M.P.; Chang, H.; Gorska, A.E.; Fuller, L.D.; Ramsey, H.E.; Kashyap, T.; Argueta, C.; Debler, S.; et al. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv 2020, 4, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Montero, J.; Berezovskaya, A.; Le, B.T.; Kentsis, A.; Christie, A.L.; Conway, A.S.; Chen, W.C.; Reed, C.; Mansour, M.R.; et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia 2016, 30, 190–199. [Google Scholar] [CrossRef]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Savona, M.; Baz, R.; Andreeff, M.; Gabrail, N.; Gutierrez, M.; Savoie, L.; Mau-Sorensen, P.M.; Wagner-Johnston, N.; Yee, K.; et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 2017, 129, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Klement, P.; Fiedler, W.; Gabdoulline, R.; Dallmann, L.K.; Wienecke, C.P.; Schiller, J.; Kandziora, C.; Teich, K.; Heida, B.; Buttner, K.; et al. Molecular response patterns in relapsed/refractory AML patients treated with selinexor and chemotherapy. Ann Hematol 2023, 102, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Martinez Sanchez, M.P.; Megias-Vericat, J.E.; Rodriguez-Veiga, R.; Vives, S.; Bergua, J.M.; Torrent, A.; Suarez-Varela, S.; Boluda, B.; Martinez-Lopez, J.; Cano-Ferri, I.; et al. A phase I trial of selinexor plus FLAG-Ida for the treatment of refractory/relapsed adult acute myeloid leukemia patients. Ann Hematol 2021, 100, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.; Lowenberg, B.; Manz, M.; Biemond, B.J.; Westerweel, P.E.; Klein, S.K.; Fehr, M.; Sinnige, H.A.M.; Efthymiou, A.; Legdeur, M.; et al. Addition of the nuclear export inhibitor selinexor to standard intensive treatment for elderly patients with acute myeloid leukemia and high risk myelodysplastic syndrome. Leukemia 2022, 36, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Alexander, T.B.; Lacayo, N.J.; Choi, J.K.; Ribeiro, R.C.; Pui, C.H.; Rubnitz, J.E. Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination With Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. J Clin Oncol 2016, 34, 4094–4101. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.D.; Cox, N.; Dahl, G.V.; Lacayo, N.J.; Davis, K.L.; Capolicchio, S.; Smith, M.; Sakamoto, K.M. Niclosamide suppresses acute myeloid leukemia cell proliferation through inhibition of CREB-dependent signaling pathways. Oncotarget 2018, 9, 4301–4317. [Google Scholar] [CrossRef] [PubMed]
- Wojcicki, A.; Chae, H.-D.; Han, K.; Youn, M.; Wilkes, M.C.; Lacayo, N.J.; Bassik, M.; Sakamoto, K.M. Genetic Modulators of Niclosamide Sensitivity and Resistance in Acute Myeloid Leukemia. Blood 2020, 136, 29. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun 2020, 11, 2042. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; Magnani, J.L.; Thackray, H.M.; Feldman, E.J.; O'Dwyer, M.E.; et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood 2022, 139, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Miller, D.W.; Lynch, J.A.; Lemoff, A.S.; Cai, Z.; Pounds, S.B.; Radtke, I.; Yan, B.; Schuetz, J.D.; Rubnitz, J.E.; et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 2011, 25, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- de Botton, S.; Montesinos, P.; Schuh, A.C.; Papayannidis, C.; Vyas, P.; Wei, A.H.; Ommen, H.; Semochkin, S.; Kim, H.J.; Larson, R.A.; et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: A randomized phase 3 trial. Blood 2023, 141, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- de Botton, S.; Brandwein, J.M.; Wei, A.H.; Pigneux, A.; Quesnel, B.; Thomas, X.; Legrand, O.; Recher, C.; Chantepie, S.; Hunault-Berger, M.; et al. Improved survival with enasidenib versus standard of care in relapsed/refractory acute myeloid leukemia associated with IDH2 mutations using historical data and propensity score matching analysis. Cancer Med 2021, 10, 6336–6343. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Zeidan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): A single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O'Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Keaton, M.; Carraway, H.; Sekeres, M.A.; Maciejewski, J.P.; Carew, J.S. The NEDD8-activating enzyme inhibitor MLN4924 disrupts nucleotide metabolism and augments the efficacy of cytarabine. Clinical cancer research 2015, 21, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Erba, H.P.; DeAngelo, D.J.; Bixby, D.L.; Altman, J.K.; Maris, M.; Hua, Z.; Blakemore, S.J.; Faessel, H.; Sedarati, F.; et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: A phase 1 study. Br J Haematol 2015, 169, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Watts, J.; Radinoff, A.; Sangerman, M.A.; Cerrano, M.; Lopez, P.F.; Zeidner, J.F.; Campelo, M.D.; Graux, C.; Liesveld, J.; et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia 2021, 35, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Ades, L.; Girshova, L.; Doronin, V.A.; Diez-Campelo, M.; Valcarcel, D.; Kambhampati, S.; Viniou, N.A.; Woszczyk, D.; De Paz Arias, R.; Symeonidis, A.; et al. Pevonedistat plus azacitidine vs azacitidine alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast-percentage AML. Blood Adv 2022, 6, 5132–5145. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Muftuoglu, M.; Ong, F.; Nasr, L.; Macaron, W.; Montalban-Bravo, G.; Alvarado, Y.; Basyal, M.; Daver, N.; Dinardo, C.D.; et al. A phase 1/2 study of azacitidine, venetoclax and pevonedistat in newly diagnosed secondary AML and in MDS or CMML after failure of hypomethylating agents. J Hematol Oncol 2023, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Liu, X.; Minard, C.G.; Isikwei, E.A.; Reid, J.M.; Horton, T.M.; Fox, E.; Weigel, B.J.; Cooper, T. Feasibility of pevonedistat combined with azacitidine, fludarabine, cytarabine in pediatric relapsed/refractory AML: Results from COG ADVL1712. Pediatr Blood Cancer 2023, 70, e30672. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The cloning of CD70 and its identification as the ligand for CD27. J Immunol 1994, 152, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Schurch, C.M.; Buhrer, E.D.; Hinterbrandner, M.; Huguenin, A.L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med 2017, 214, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Flieswasser, T.; Van den Eynde, A.; Van Audenaerde, J.; De Waele, J.; Lardon, F.; Riether, C.; de Haard, H.; Smits, E.; Pauwels, P.; Jacobs, J. The CD70-CD27 axis in oncology: The new kids on the block. J Exp Clin Cancer Res 2022, 41, 12. [Google Scholar] [CrossRef]
- Riether, C.; Pabst, T.; Hopner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Muller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med 2020, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Hajighasemi, S.; Kiaie, N.; Gheibi Hayat, S.M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. The pivotal role of CD69 in autoimmunity. J Autoimmun 2020, 111, 102453. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Woste, M.; Mikesch, J.H.; Arteaga, M.F.; Angenendt, A.; Sandmann, S.; Berdel, W.E.; Lenz, G.; Dugas, M.; Meshinchi, S.; et al. Calcitonin receptor-like (CALCRL) is a marker of stemness and an independent predictor of outcome in pediatric AML. Blood Adv 2021, 5, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Bruedigam, C.; Bagger, F.O.; Heidel, F.H.; Paine Kuhn, C.; Guignes, S.; Song, A.; Austin, R.; Vu, T.; Lee, E.; Riyat, S.; et al. Telomerase inhibition effectively targets mouse and human AML stem cells and delays relapse following chemotherapy. Cell Stem Cell 2014, 15, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Manshouri, T.; Smith, F.O.; Giles, F.J.; Cortes, J.; Estey, E.; Kantarjian, H.; Keating, M.; Jeha, S.; Albitar, M. Telomerase activity is prognostic in pediatric patients with acute myeloid leukemia: Comparison with adult acute myeloid leukemia. Cancer 2003, 97, 2212–2217. [Google Scholar] [CrossRef]
- Barwe, S.P.; Huang, F.; Kolb, E.A.; Gopalakrishnapillai, A. Imetelstat Induces Leukemia Stem Cell Death in Pediatric Acute Myeloid Leukemia Patient-Derived Xenografts. J Clin Med 2022, 11, 1923. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of LSC biology in adult AML. Created with BioRender.com.
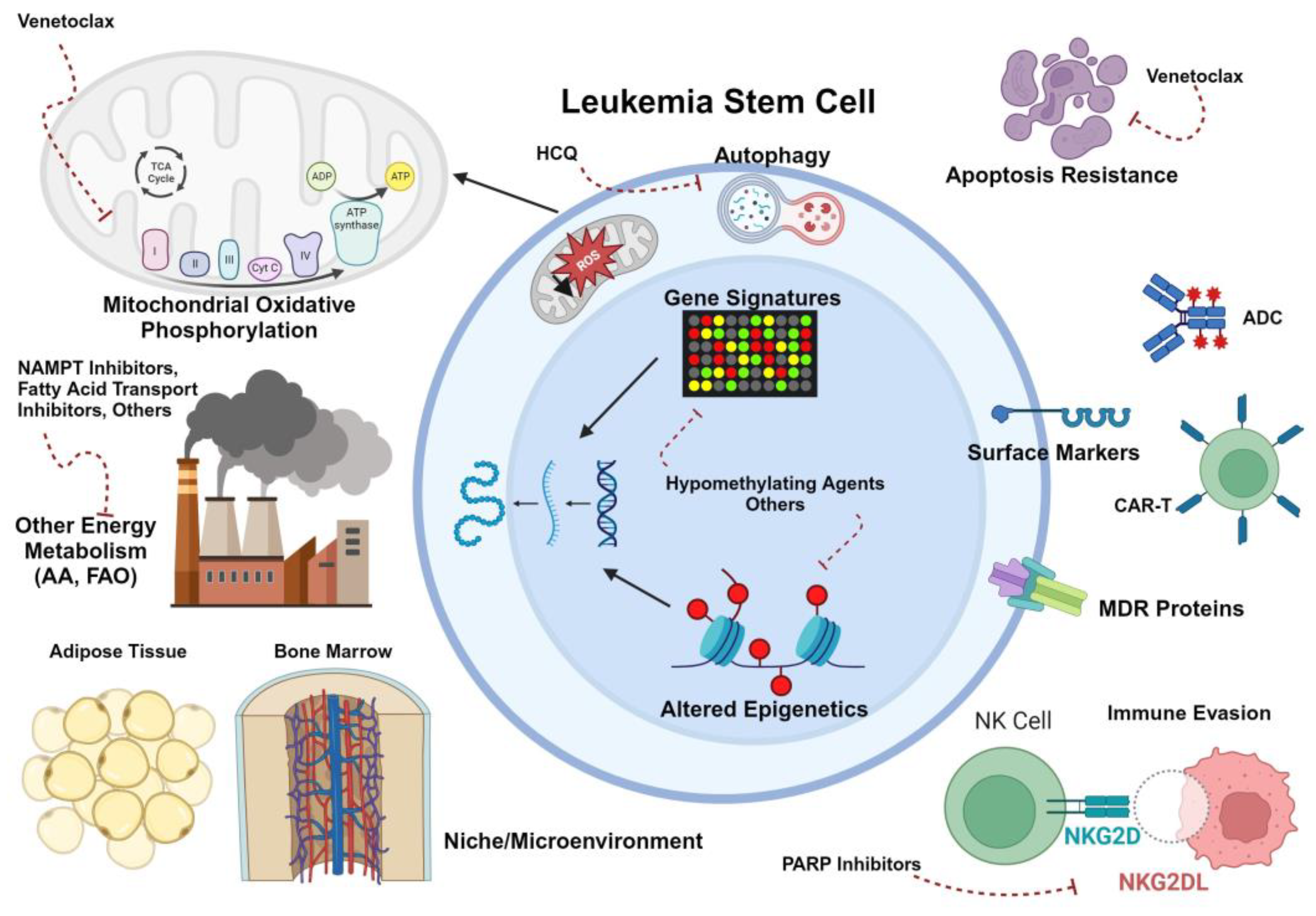
Figure 2.
Schematic diagram of what is known about LSC biology in pediatric AML. Created with BioRender.com.
Figure 2.
Schematic diagram of what is known about LSC biology in pediatric AML. Created with BioRender.com.
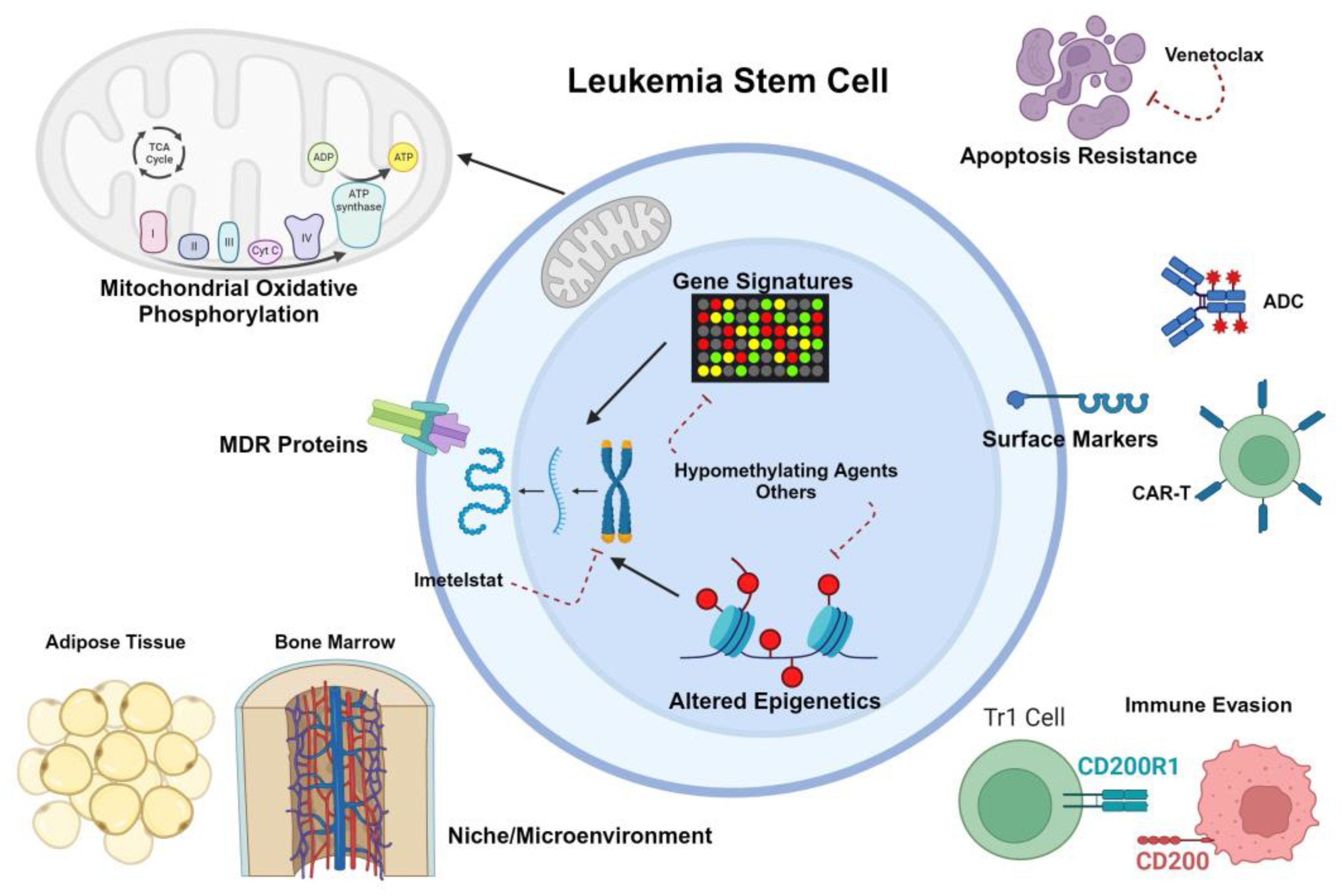

Table 1.
Select immunotherapy clinical trials for pediatric patients with AML.
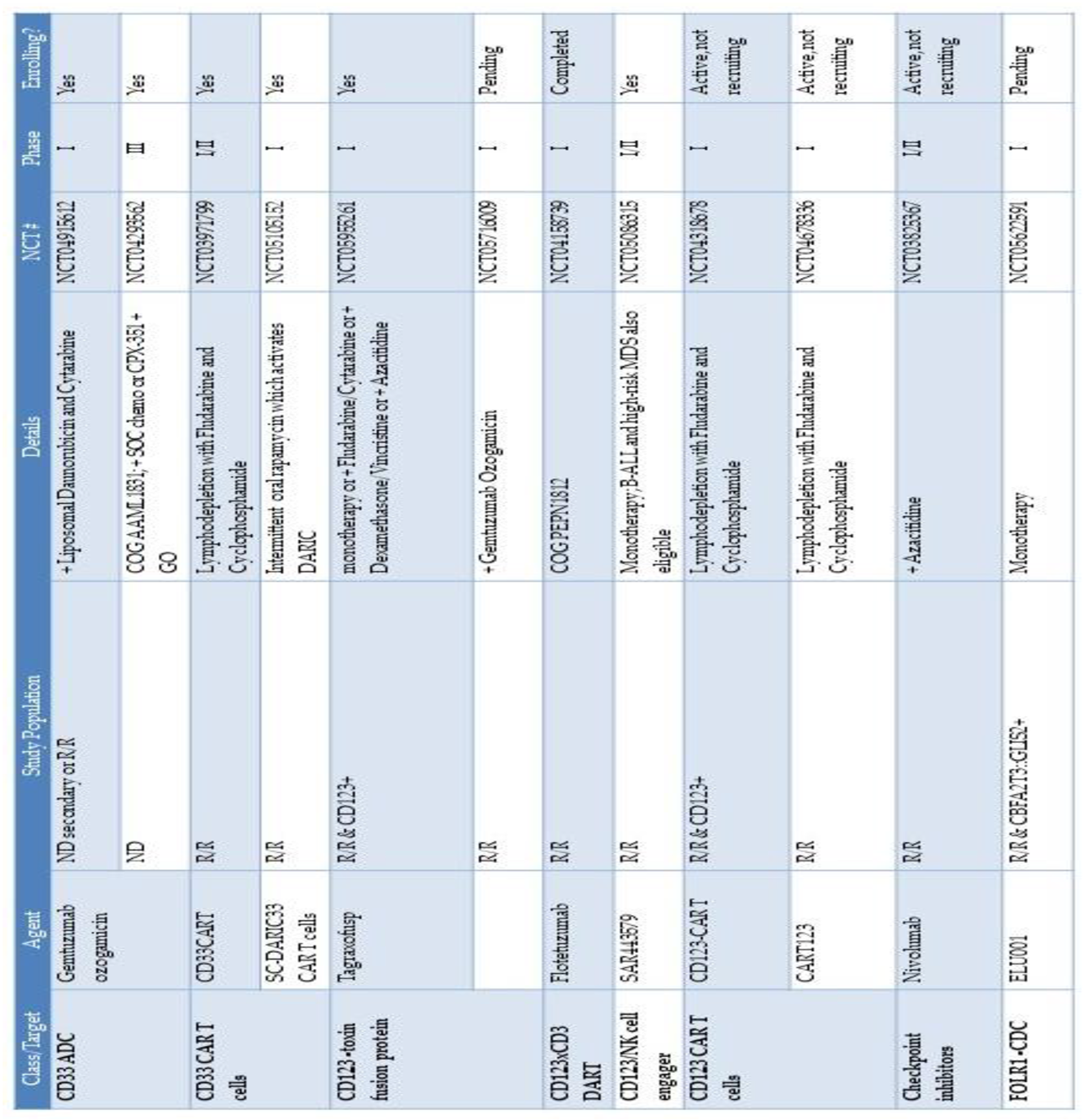 |
ADC = Antibody-drug conjugate; ND = newly diagnosed; R/R = relapsed/refractory; COG = Children’s Oncology Group; SOC = standard of care; CAR = Chimeric Antigen Receptor; DART = dual-affinity retargeting protein; NK = natural killer; B-ALL = B-cell acute lymphoblastic leukemia; MDS = Myelodysplastic Syndrome; FOLR1 = folate receptor alpha; CDC = C’Dot Drug Conjugate.
Table 2.
Non-immunotherapy agents in clinical trials for pediatric patients with AML.
 |
ND = newly diagnosed; R/R = relapsed/refractory; COG = Children’s Oncology Group; SOC = standard of care; GO = gemtuzumab ozogamicin; HSCT = hematopoietic stem cell transplant; FLA(G) = Fludarabine/Cytarabine(/G-CSF); HMA = hypomethylating agent; HDACi = histone deacetylase inhibitor; CREB = cAMP response element binding protein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



