
Submitted:
15 November 2023
Posted:
16 November 2023
You are already at the latest version
Abstract
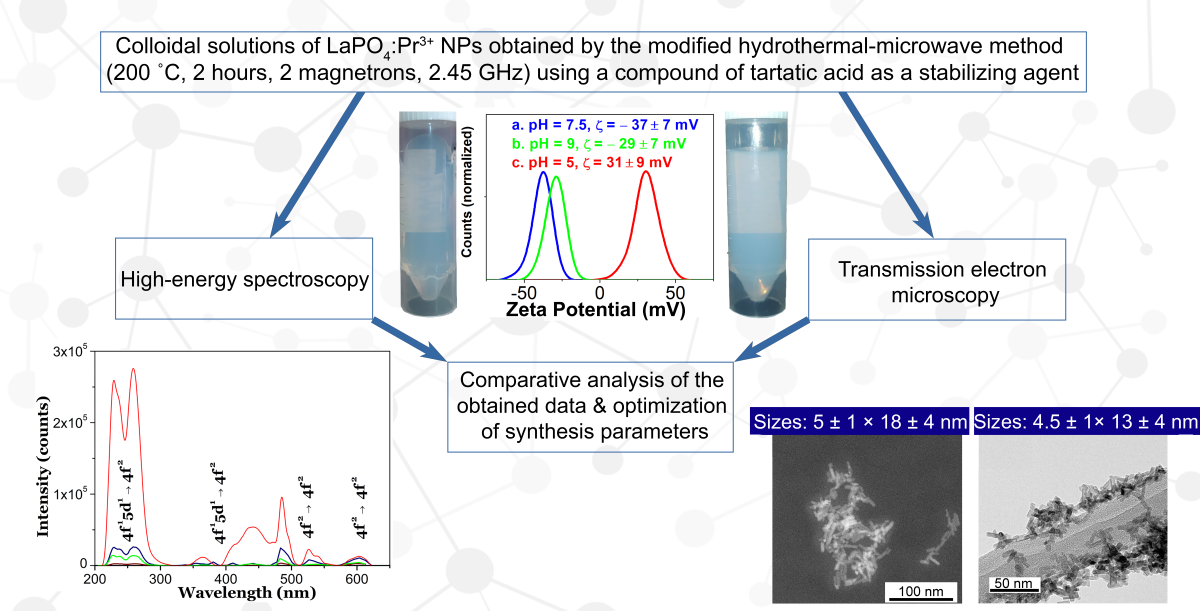
A hydrothermal technique with microwave heating (200 °C, 2 hours, 2 magnetrons, 2.45 GHz) was modified to obtain UV-C luminescent LaPO4: Pr3+ colloidal nanorods for the potential use in X-ray cancer theranostics. For further possible biofunctionalization, a compound of tartaric acid formed under basic conditions was used as a coating and stabilizing agent to provide colloidal properties to the surface of nanoparticles in basic to neutral aqueous media, which was confirmed by zeta potential measurements. In addition, the colloidal properties and clustering processes of synthesized LaPO4:Pr3+ nanorods in aqueous solutions with different pH values were studied in real time using a highly sensitive laser ultramicroscope operating according to the “light-sheet” scheme. A comparative analysis of the transmission electron microscopy, X-ray powder diffraction and high-energy spectroscopy showed the different effects of synthesis parameters on the morphological, crystalline and luminescent properties of the obtained nanorods. Through optimization of the synthesis parameters, stable aqueous solutions of m-LaPO4:Pr3+ nanorods with sizes less than 30 nm and an intensity of UV-C luminescence equal to 5-10% of the unmodified nanofibers were obtained.
Keywords:
UV-C cathodoluminescence
; X-ray excited luminescence
; LaPO4
; Pr3+
; hydrothermal conditions
; microwave heating
; colloidal solution
; ultramicroscopy
1. Introduction
At present, the development of ways to synthesize multifunctional crystalline nanoparticles (NPs) with specified parameters (composition, morphology, structure) is a topical task. To date, the main methods for obtaining nanocrystals is the production of solid NPs via liquid-solid or solid-liquid-solid phase transitions in various dispersion media. For «green» methods of obtaining NPs with specified parameters the various reactor installations (batch or continuous) that operate under relatively mild conditions (temperature up to several hundred degrees, pressure 1–100 atm) are used. Such methods are called chemical; they allow one to obtain reproducible results and high yields of reaction products with minimal energy consumption, which makes them very attractive for many applications.
For medical applications, it is necessary to obtain colloidal solutions of NPs with the following special properties: NP smaller than 50 nm [1], narrow size distribution, low degree of toxicity, high photo- and physico-chemical inertness, low degree of crystalline defects, colloidal stability. The ability to form stable colloids is necessary to prevent NPs aggregation and facilitate their further modification. Materials combining such properties can be used to introduce NPs into a living organism to obtain information about biological tissues (bioimaging) or to deliver drugs or other therapeutic effects to disease centers. Thus, dielectric crystalline NPs and stable colloidal solutions based on them have found applications in theranostics including bioimaging and drug delivery carriers.
One of the promising areas in medicine is the study of the effect of ultraviolet radiation in the UV-C spectral range (200–280 nm) on the deactivation of localized microorganisms and biological tissues resistant to antimicrobial drugs or radiation. UV-C photon is not capable of penetrating into biological tissues; therefore, its delivery by some NPs to a closed area of the disease in a living organism is an urgent task. In particular, UV-C emitting NPs are considered as possible radiosensitizers to enhance cancer radiation therapy due to the strong destructive action of UV-C photons on DNA molecules [2,3].
As is known, some rare earth ions (REI), namely Ce3+, Pr3+, Nd3+, doped into suitable crystalline matrices, are capable of emitting intense radiation in the UV-C spectral range under the high-energy excitation. The high-intense UV-C radiation can be observed due to the presence of high-energy radiative inter-configurational electronic transitions from the lowest-energy state of excited 4fn-15d1 electronic configuration to the ground and some excited states of the 4fn electronic configuration in the above REI. However, it is necessary to choose the best crystal matrix for effective luminescence due to 4fn-15d1 → 4fn transitions. An important condition for the existence of bright 4fn-15d1 → 4fn UV luminescence of REI under high-energy excitation is an efficient energy transfer from the matrix to the REI optical centers. In particular, phosphate matrices ensure efficient energy transfer to REI [4,5,6,7]. Lutetium phosphate NPs doped with Pr3+ ions have been already proposed as efficient UV-C nanoscintillators for biomedical applications [3,8].
Some works [7,9,10,11,12] show that upon high-energy excitation (X-ray, synchrotron radiation, electron beam) of bulk LaPO4 crystals with the monazite structure doped with Pr3+ ions (m-LaPO4, monoclinic system, space group P21/n), it is possible to obtain effective UV-C luminescence (220 – 280 nm) due to the 4f15d1 → 3H4,5,6, 3F2,3,4 transitions in Pr3+ ions. A necessary condition for the existence of 4f15d1 → 4f2 radiative transition of Pr3+ ions in the doped matrix is that the lowest-energy (emitting) 4f15d1 level is below the highest-energy 4f2 level (1S0) [11] Otherwise, luminescence properties of doping Pr3+ ions will be determined by intra-configurational 4f2 → 4f2 transitions from the 1S0 level (two step photon emission process/photon cascade emission process due to 1S0 → 1I6 and 3P0 → 3H4 transitions), which occur mainly in the visible spectral range. Thus, one of the ways to deliver the UV-C radiation to biological tissues can be the creation of drugs based on aqueous colloidal solutions of m-LaPO4: Pr3+ dielectric crystalline NPs with specified morphological parameters.
According to some studies (see, e.g. [13]), nanorods can be more effective drug carriers than nanospheres for anti-cancer therapy because nanorods can penetrate tumors more efficiently than nanospheres of the same effective size due to the enhanced transport through porous media of the tumor. Another factor can be an alignment of nanorods with blood flow, which increases the probability of the delivery to the tumor for nanorods. Accordingly, LaPO4 NPs having a shape of nanorods are of a special interest in our study.
Let us give a small overview of LaPO4: RE3+ NPs synthesis methods. The main method for the synthesis of NPs is their production by various chemical methods using a liquid phase. The use of an intermediate liquid phase makes it possible to reduce the required temperatures for the successful formation of nanocrystals belonging to high-temperature phases without additional annealing of the product. Since the liquid phase separates individual nanocrystals during the synthesis process, the correct choice of synthesis parameters makes it possible to obtain well-faceted crystalline NPs with a low degree of aggregation and stable aqueous colloids based on them.
Recently, Wang et al. [14] investigated a method for synthesizing HTMW to obtain highly crystalline and highly polarized m-LaPO4:Eu3+ nanorod emitters with an average length of about 100 nm or more. It was shown that LaPO4 NPs can be treated with nitric acid, which leads to the formation of a colloidal solution that remains stable for more than a year. It was also shown that m-LaPO4 NPs in an acidic environment (pH = 2) have a modulus twice as large as the zeta potential than in an alkaline environment (pH = 7 – 12), indicating that the sedimentation stability of NPs is much higher in acidic environment.
Based on the report of Espinoza & Jüstel on UV-emitting LaPO4 nanocrystals [15], the X-ray excited optical luminescence (XEOL) spectrum of about 4.5 ± 3 nm m-LaPO4: Pr3+ spherical colloidal NPs obtained by a solvothermal method shows a large shift towards UV-B and UV-A spectral ranges. This circumstance does not allow the effective use of such small m-LaPO4: Pr3+ NPs as UV-C delivery agents and indicates the need to obtain larger colloidal NPs.
Hilario et al. In [16], the hydrothermal synthesis of m-LaPO4 doped with Pr3+ or Pr3+ doped with Gd3+ ions was studied. A series of syntheses carried out by the authors at different pH levels, as well as measurements of the zeta potential of the resulting NPs, showed a higher probability of aggregation of the resulting NPs with increasing pH levels.
On the contrary, H. Meyssamy et al. [17] and H. Song et al. [18] used the hydrothermal method in alkaline aqueous solutions of ammonium hydroxide (pH>11) to obtain faceted NPs of m-LaPO4:Eu3+, Ce3+ with sizes 10 – 50 nm and low aspect ratio of 1:2. As shown in these works, the treatment of freshly prepared suspensions of LaPO4 NPs with an HNO3 solution allows chemical peptization of aggregated NPs with the formation of stable colloidal solutions in acidic conditions.
Riwotzki et al. [19] used a solvothermal synthesis of m-LaPO4: Ce3+, Tb3+ NPs with mean sizes of 5–6 nm. Similarly, N. Niu et al. [20] synthesized m-LaPO4: Ce3+, Tb3+ NPs of irregular shape and rounded morphology with sizes about 15 nm using a solvothermal method. As a result of solvothermal synthesis routes, highly luminescent in UV-A region NPs of m-LaPO4: Ce3+, Tb3+ with a hydrophobic surface were obtained.
To control the processes of nucleation and growth of LaPO4: Ce3+, Tb3+ nanowires, X. Zhu et al. [21] used a combination of an organic dispersion medium, microwave heating and a microfluidic reactor. As a result, non-aggregated hexagonal LaPO4: Ce3+, Tb3+ NPs luminescent in UV-A region with an average length of 60–70 nm and a diameter of about 12 nm were obtained.
The conclusions from this literature review are as follows.
It is possible to obtain a pure monoclinic LaPO4: REI phase of well-crystallized NPs in one step, by using autoclave synthesis methods or synthesis in high-boiling organic solvents.
Depending on the pH values, anion excess ratio, reaction mixture temperature, and the concentration of the initial reagents [14,17,18,22,23], the hydrothermal synthesis allows to obtain m-LaPO4 NPs with characteristic dimensions ranging from 5 to 600 nm with aspect ratio of 2–75. At the same time, the synthesized particles of the LaPO4 via hydrothermal synthesis are most often obtained with big sizes and without a stabilizing charge on the surface in a neutral or basic aqueous media [14,16,24]. Thus, to prevent aggregation of m-LaPO4 NPs in the alkaline region, it is necessary to use a surfactant. To modify the surface of lanthanum containing NPs, anionic surfactants are widely used, in particular, compounds of tartaric and citric acids [25,26].
Since it was previously shown [9,12] that the monoclinic LaPO4 phase doped with Pr3+ ions is an effective UV-C scintillator, we concentrated our efforts on the development of a Hydrothermal Microwave (HTMW) method for the synthesis of m-LaPO4: Pr3+ colloidal NPs with specified physicochemical parameters: optimal NP sizes, high colloidal stability, the ability to emit intense luminescence, and the highest intensity in the spectral region of the UV-C radiation spectrum. A surfactant of alkaline solution of ammonium tartrate was used during syntheses in order to provide the colloidal properties to the surface of LaPO4 NPs for the ability to form the stable aqueous solutions in the neutral or weakly alkaline aqueous media. To study the effect of synthesis parameters on the obtained NPs, several series of syntheses of LaPO4: Pr3+ (1 at.%) NPs were carried out by the HTMW method (200 °C, 2 hours, 2 magnetrons, 2.45 GHz).The influence of various parameters on the morphology and intensity of UV-C luminescence of NPs was studied: the use of solutions of tartaric acid (TA) and ammonium hydroxide (NH4OH) with different amounts of added TA (CTA = 0.027 or 0.08 M) at pH values of 8 or 9, various ratios of excess anions to cations (excess ratio of K2HPO4 = 1 – 10), two concentrations of LaPO4: Pr3+ in the reaction mixture ( = 0.005 or 0.01 M).
Aggregation processes of colloidal NPs synthesized by wet-chemistry methods are a well-known problem in their use for medical and other purposes [27,28,29]. They can occur both during the synthesis of NPs and during the storage of their solutions. Monitoring these processes using an electron microscope is a complex and time-consuming procedure and requires drying the sample, which can distort the information obtained as a result of possible changes in the structure of clusters and their additional aggregation during the drying process. This article presents the results of using a simpler and faster method for monitoring aggregation processes, based on highly sensitive optical microscopy visualization of the Brownian motion of single NPs in solutions and subsequent analysis of their trajectories using the Nanoparticle Tracking Analysis (NTA).
2. Materials and Methods
2.1. Method for the Synthesis of Colloidal Solutions of LaPO4: Pr3+ Nanoparticles
2.1.1. HTMW Synthesis Methodology
The initial chemical reagents used in the synthesis without further purification include: Pr(NO3)3 6H2O (Aldrich, 99.9% purity), La(NO3)3 6H2O (Aldrich, 99.999% purity), K2HPO4 3H2O (RusKhim, analytical grade), tartaric acid import (hps, analytical grade), 25% aqueous solution of NH4OH (SigmaTek, analytical grade). Throughout all syntheses, deionized (DI) type I water (resistance 18.2 MΩ cm) from a water deionizer Crystal EX-1001 (Adrona, Riga, Latvia) was used.
In a typical synthesis the following devices were used: an analytical grade scales HR-100AG (A&D, Japan), syringe pump PSh-10 (Visma Planar, Minsk, Belarus), HTMW device speedwave XPERT (Berghof Products+ Instruments GmbH, Eningen unter Achalm, Germany) with two magnetrons (2.45 GHz, 2 kW maximum output power), centrifuge Z326 (Hermle, Gosheim, Germany), magnetic stirrer C-MAG HS 7 (IKA, Staufen im Breisgau, Germany), ultrasonic bath JP-040S (Skymen, China).
In the general case, the HTMW synthesis of LaPO4: Pr3+ colloidal solutions with surfactant can be divided into 3 stages: coprecipitation at room temperature, HTMW treatment, and the washing stage.
At the coprecipitation stage, three solutions are prepared in DI water, with a total volume of 50 mL or less: a solution of La(NO3)3 6H2O and Pr(NO3)3 6H2O (A), a solution of tartaric acid (C4H6O6) and NH4OH (resulting pH = 8 or 9) (B), a solution of K2HPO4 3H2O (C) and, if necessary, additional DI water to bring the volume of the reaction mixture to 50 mL prior to HTMW treatment. After preparing the solutions, solution B is added dropwise to solution A, resulting in a clear aqueous solution (D), which is then stirred for 15 minutes. Further, depending on the choice of coprecipitation method, solution D is continuously (0.4 mL/min) supplied directly to solution C (or reversed), resulting in the formation of a LaPO4: Pr3+ colloidal gel solutions (E). The solution E is then diluted with DI water to reach 50 mL and transferred into a 100 mL Teflon (PTFE) autoclave with ceramic reinforcing inlay, sealed and placed under microwave irradiation for 2 h at 200 °C using a speedwave XPERT laboratory device.
In the case of surfactant-free synthesis, all steps were the same, except that solution B was replaced by DI water.
2.1.2. Washing Stage Methodology
After the HTMW treatment, the autoclave is allowed to cool naturally to room temperature, and then most of the upper water layer is decanted to collect white precipitates from the bottom of the autoclave. Cloudy and unstable LaPO4: Pr3+ suspension is then transferred to a centrifuge tube with additional DI water. The precipitate is then washed with DI water using a precipitation-redispersion process in which a Hermle Z326 device (11000 rcf, 10 min) was used to precipitate the NPs and an ultrasonic bath was used to redisperse the NPs in a fresh DI water.
After centrifugation, NPs, synthesized with the addition of surfactant solution (C4H6O4 + NH4OH, pH = 8 – 9), could form flocculates, but they were quite easily subjected to the cavitation effect of ultrasound or with the addition of pH-rising agents. As a result, after two cycles of washing of TA-modified NPs with DI water, a homogeneous transparent solution is formed. The third cycle of washing of TA-modified NPs with DI water leads to the loss of the colloidal properties and rapid flocculation upon removal of NPs solution from the ultrasonic bath. To restore the colloidal properties of the TA-modified NPs, 50 μl of 25% ammonium hydroxide solution is added to the centrifuge tube containing 40 ml of modified NPs in suspension. After sonication, the NPs are washed again using the precipitation-redispersion process. At the last stage, the settled transparent particles were redispersed in 5 mL of DI water ( = 10 – 20 mg/mL), forming aqueous colloidal solutions (resulting pH = 7.5 – 8) of LaPO4: Pr3+ NPs, which retain sedimentation stability for several months.
Since tartaric acid is a dibasic carboxylic acid, depending on the pH of the medium, its molecule can carry a negative charge at pH > 7 or remain neutral at pH < 7. Thus, when using the HTMW method modified with the anionic surfactant solution (C4H6O4 + NH4OH), it is necessary to maintain the pH of the synthesis media from neutral to slightly alkaline to prevent NPs aggregation and to maintain colloidal stability in aqueous dispersion media.
Centrifugation of LaPO4: Pr3+ particles synthesized without surfactant solution leads to the formation of dense opaque flakes that cannot be broken with a magnetic stirrer and ultrasound or with addition of alkaline agents. To redisperse non-modified particles into a colloidal solution, as was shown many times in the literature, their surface has to be peptized, which can be achieved by adding a small amount (30– 50 μl) of concentrated HNO3 and sonication, resulting in the formation of a stable colloidal solution of unmodified LaPO4 NPs in an acidic medium.
2.2. Nonoptical Characterization Methods
2.2.1. Transmission Electron Microscopy Analysis
Morphologies of LaPO4: Pr3+ (1 at.%) particles obtained with different synthesis parameters were studied by transmission electron microscopy (TEM ). The TEM images of the as-prepared LaPO4: Pr3+ NPs were taken with Zeiss Libra 200 FT HR microscope at the GPI RAS (Moscow) or with a JEOL JEM-2100 at the National University of Science and Technology «MISIS» (Moscow). The diluted colloids were applied dropwise onto a TEM grid and dried in a vacuum for several hours.
Processing of TEM images for calculation of size distribution was carried out using the ImageJ program. Statistics for each sample, with the exception of NPs obtained by the unmodified HTMW method, included about 500 measurements of individual nanocrystals. For unmodified NPs, the statistics consisted of only 50 measurements, since these particles are not a priority in this study.
2.2.2. X-ray Powder Diffraction
To determine the crystalline phase and assess the degree of crystallinity, X-ray diffraction patterns of LaPO4: Pr3+ (1 at.%) powders were conducted using the D2 Phaser diffractometer (CuKα-radiation). All samples obtained by the HTMW method with a constant temperature parameter of 200 °C and a duration of HTMW treatment for 2 hours correspond to the monoclinic phase.
2.3. Colloidal Properties Control
2.3.1. Zeta Potential Measurements
To assess the sedimentation stability of LaPO4: Pr3+ (1 at.%) particles obtained by modified HTMW method, electrophoretic light scattering (ELS) measurements were performed at different pH levels (pH = 5, 7.5, 9) using a Malvern Zetasizer ULTRA RED LABEL setup (Malvern Panalytical LTD, Worcestershire, UK). Zeta potential measurements at each pH level were made 3 times for 30 seconds each, after which the results were averaged.
2.3.2. Optical Microscopic Diagnostics
To visualize Brownian motion of NPs and to study the processes of association (aggregation and agglomeration) of the synthesized NPs in colloidal solution, we have developed a highly sensitive laser ultramicroscope [30]. It is built according to the “light sheet” scheme and is based on the detection of elastically scattered light at an unshifted frequency. The high sensitivity of the microscope makes it possible to visualize single NPs up to 20 nm in size or less in solution, as well as clusters formed on their basis. The essential advantage of this method is the visualization and analysis of individual trajectories of Brownian motion of single NPs that allows to overcome inherent problems associated with averaging over an ensemble of particles. For example, such averaging is inherent for another method widely used for diagnostics of NPs in solutions: the dynamic light scattering (DLS). The latter is based on measurement the signals of the total elastic light scattering of ensemble of NPs in solution and yields information averaged over an ensemble of particles, which usually heavily weighted towards small value of large, usually contaminant particles. Optical diagnostics of synthesized colloidal solutions using the developed laser ultramicroscope made it possible to eliminate the above ensemble averaging and obtain more detailed information about the colloidal solutions under study.
Numerical analysis of individual trajectories of the Brownian motion (NTA) of the observed NPs made it possible to determine their hydrodynamic radii. In order to avoid the undesirable effect of superimposing images of the observed NPs, the concentration of the solution was reduced to 0.8 µg/mL. The measurements were carried out using radiation from a cw diode laser at 405 nm; the total laser radiation power at the sample did not exceed 50 mW. The studied solutions were placed in a measuring quartz cuvette with internal dimensions of 10 x 10 mm. The laser light was focused into a cuvette in the form of a “light sheet” about 10 × 60 μm in size. Passive temperature stabilization and a special algorithm for the numerical analysis of the recorded images made it possible to eliminate the influence of the thermal convection of the liquid in cuvette on the measurement results.
2.4. Spectral Characterization
Sample preparation for CL and XEOL analyzes is carried out by applying a colloidal solution to a PTFE substrate, followed by mild heat treatment (100°C, 2 hours) in a drying cabinet (ShSU-M, Elektropribor, Russia). After obtaining a dry film on the surface of the PTFE sheet, it is collected with metal spatulas in a plastic Eppendorf cuvette and lightly triturated using plastic spatula. Thus, a fine, well-flowing dry powder of LaPO4:Pr3+ NPs is obtained.
2.4.1. Method for Measuring Cathodoluminescence of the LaPO4: Pr3+ Nanoparticles Powders
In CL experiments, powders of LaPO4: Pr3+ NPs with different morphologies were excited by 10 keV electron beam, with typical 50 nA current. Cathodoluminescence setup (see details in [31]) includes two monochromators for simultaneous recording of emission spectra in the UV-Vis and VUV range. The first one ARC spectraPro 2300i monochromator with different gratings operates in the range of 200–1000 nm using various detectors (Photomultipliers and a CCD camera). The self-made double vacuum monochromator equipped with a solar-blind Hamamatsu photomultiplier R6836 covers the wavelength range 120–270 nm. The electron gun used (Kimball Physics EGG-3101) could be utilized in steady and pulsed (10 ns pulse, repetition rate 5 kHz) operation mode with an excitation spot diameter on the sample of approximately 0.5 mm. Through the window of sample chamber of cryostat, the visible luminescence can be observed for well emitting samples by naked eye during excitation by electron beam.
2.4.2. Method for Measuring X-ray Excited Luminescence of the LaPO4: Pr3+ Nanoparticles Powders
The setup for the measurement of XEOL spectra of LaPO4: Pr3+ powders consists of a water-cooled cathode X-ray tube with Cr anode (30 kV, 30 mA) and a compact mini-spectrometer (FSD-10, «PTC Fiber Optic Devices»), operating in the spectrograph mode, with a fiber-optic radiation input and a photosensitive element–a silicon semiconductor CCD linear image sensor. Spectral sensitivity range of the device: 200–1060 nm, spectral resolution –2 nm. Optical fiber diameter – 200 microns. The end of the fiber light guide is brought directly to the metal cuvette, in which the NPs powder under study is placed.
When measuring luminescence spectra with X-ray excitation, the accumulation time (10 or 50 seconds) is set in the spectrometer control program on a computer depending on the luminescence intensity. The separately measured background is subtracted from the measured luminescence spectra.
3. Results
3.1. Morphology and Structure of LaPO4: Pr3+ Nanoparticles
The TEM images of dry products (Figure 1) show partly aggregated individual nanocrystals of LaPO4: Pr3+ (1 at.%) with dimensions between 5 and 600 nm with different morphologies of nanofibers, nanorods or rounded particles.
Simple HTMW synthesis ( = 0.01 M, K2HPO4 excess ratio = 1.25, pH = 5) results in nanofibers with dimensions of about 7 – 15 × 30 – 600 nm (diameter × length) (Fig. 1a). This result is close with the literature, although the aspect ratio of particles obtained by HTMW method is quite high. Nanorods with sizes 8 ± 1.5 × 46 ± 12 nm (Fig. 1b) were obtained by modifying the HTMW method with solutions of tartaric acid (TA) and ammonium hydroxide ( = 0.01 M, K2HPO4 excess ratio = 1.25, CTA= 0.08 М, pH = 9). The samples consisting of short nanorods (4.5 ± 1 × 13 ± 4 nm and 5 ± 1 nm × 18 ± 4 nm) were prepared with the same K2HPO4 excess ratio equal to five and the concentration of TA (CTA= 0.027 М), but with different during HTMW conditions: concentrations of equal to 0.01 M and 0.005 M and pH of 9 and 8 (respectively, Fig. 1c, d). Nanorods with diameters of 6.5 ± 1.5 nm and a pronounced double distribution of lengths with maxima at 31 ± 6 and 49 ± 9 nm (Fig. 1e) were prepared with lower cation concentration and K2HPO4 excess ratio ( = 0.005 M, K2HPO4 excess ratio = 2, CTA= 0.027 М, pH = 8). The small faceted nanocrystals with dimensions of 6 ± 2 × 10 ± 4 nm (Figure 1f) were synthesized with = 0.005 M and ammonium tartrate (CTA= 0.027 М, pH = 8) and a tenfold K2HPO4 excess ratio.
Analyzing the TEM results, it was found that increasing the pH level of the reaction mixture leads to a significant decrease in the size of NPs. In turn, the introduction of a large excess of anions to cations into the reaction mixture leads to a further decrease in the size of NPs and a decrease in polydispersity, which is a consequence of a decrease in the solubility of lanthanum phosphate. Moreover, a decrease in the concentration of reagents and an excess of anions leads to the formation of larger individual NPs (Figure 1d,e). This fact may be associated with a decrease in the degree of ion saturation of the medium, due to which the probability of nucleation processes decreases and the process of dissolution-growth of individual NPs becomes more accessible.
It can be seen that the particles obtained in an alkaline medium (pH > 7) are partially aggregated and are in a fractal-like structures, which may be due to the lower degree of sediment stability of LaPO4 NPs in alkaline media [14,16,24]. However, the degree of aggregation of the obtained particles is much lower than in the literature [16,23], where an alkaline medium was also used for synthesis. Thus, the use of tartaric acid allowed to facilitate the growth of individual NPs and weaken their aggregation.
3.2. XRD Analysis
As is known, under appropriate conditions, lanthanum phosphate crystallizes with a monazite structure with space group P21/n.
XRD data (Figure 2a) for a sample of nanofibers obtained by a simple HTMW method without additional substances, revealed that the sample consists of a single crystalline phase of the pure monoclinic phase of m-LaPO4 (space group P21/n). The predominance of the intensity peak with an index of (120) indicates the formation of crystals with strong anisotropic growth in one direction, which is confirmed by TEM data (Figure 1a).
3.3. Colloidal Stability of Modified Nanoparticles
The stabilization of LaPO4: REI NPs in pH-modified aqueous media is explained by the adsorption of H+ or OH− ions on the surface of NPs [14,16,24]. At the same time, in the case of LaPO4 NPs the processes of H+ ions adsorption is more efficient than OH− ions. As a result of the disproportion of the processes of pH-determining ions adsorption, m-LaPO4: Pr3+ NPs without coating agents are more stable in an acidic than in an alkaline media [14,16]. On the other hand, as has been shown in many works, the use of organic compounds with carboxyl groups makes it possible to decrease the aggregation probability of NPs in an alkaline medium [25,26,32].
To control the stability of colloidal solutions of the synthesized TA-modified nanocrystals, measurements of their zeta potentials were carried out, as well as optical studies of their clustering processes using a highly sensitive ultramicroscope.
3.3.1. Zeta Potential Measurements
For zeta potential measurements, a freshly synthesized TA-modified LaPO4: Pr3+ NPs with sizes with sizes 5 ± 1 × 18 ± 4 nm (Figure 1c) were redispersed in DI water with a concentration of 10 mg/mL and a low amount of NH4OH (resulting pH = 8). Next, one part of the colloid was placed into a DTS0012 polystyrene cuvette and diluted with 9 parts of one of the dispersants: DI water (resulting pH = 7.5), aqueous nitric acid solution (resulting pH = 5) or aqueous sodium hydroxide solution (resulting pH = 9). The results of measurements of the zeta potential at different pH (Figure 3) show that when the colloid is diluted with DI water (Figure 3a), the particles have the maximum negative zeta potential (−37 ± 7 mV). An increase in pH to 9 (Figure 3b) leads to a decrease in the zeta potential (−29 ± 7 mV), and when the colloid is diluted with an acidic medium with pH = 5, the zeta potential changes its value to positive (31 ± 9 mV) and small part of NPs become non-charged.
The values of the zeta potential in alkaline media obtained from ELS (Figure 3a,b) significantly exceed the literature data for unmodified m-LaPO4: REI NPs. [14,16,24]. This result indicates the successful formation of stable adsorption layer with the help of tartaric acid compound on the NPs surface. The adsorbed deprotonated hydroxyl groups of tartaric acid form the negative charge near the surface of LaPO4: Pr3+ NPs, leading to higher values of ζ-potential of TA-coated NPs (Figure 3a,b). As a result of synthesis with tartaric acid, almost all particles have a high-energy surface in alkaline media (for most particles |ζ| > 30 mV). Increasing the pH level is equivalent to the process of adding an indifferent electrolyte to a solution, which leads to a decrease in the zeta potential in a more alkaline environment (Figure 3b). Dilution of the colloid with a weak solution of nitric acid leads to recharging of the surface of the particles (Figure 3c), as well as to partial protonation of tartaric acid.
Thus, the use of tartaric acid compound made it possible to expand the pH range of sedimentation stability of the obtained particles into alkaline region, while retaining the ability to form colloidal solutions in acidic conditions.
3.3.2. Colloidal Nanoparticles Cluster Sizes
More information about the processes of association of synthesized NPs in different aqueous media was provided by optical control using laser ultramicroscope and NTA analysis of recorded trajectories of Brownian motion of colloidal solutions under study (Figure 4). For these experiments, a colloidal solution of TA-modified LaPO4: Pr3+ nanocrystals with sizes of 5 ± 1 × 18 ± 4 nm (Figure 1c) was prepared with a concentration of 10 mg/mL in DI water with a low amount of NH4OH (resulting pH = 8). Then it was diluted with DI water in a ratio of 1:250 and subjected to ultrasonic treatment for 30 minutes. Next, the solution was diluted in a ratio of 1:50 in various dispersion media: DI water (resulting pH = 7), weak aqueous sodium hydroxide solution (resulting pH = 9), aqueous solutions of nitric acid (resulting pH = 2 or 5). Then, the prepared solutions were transferred into an optical cuvette for measurements. The registered trajectories of the Brownian motion of NPs were analyzed using NTA method (for more details see [30]).
The results of the analysis are shown in Figure 4. It can be seen that the values of the hydrodynamic radii of NPs under study in highly diluted solutions are scattered over a wide range of sizes and significantly exceed the sizes of primary NPs as measured by TEM (Figure 1e). It is also seen that, all distributions are well fitted by the sum of two different Gaussians: one with the distribution maximum and half-width, which little dependent on the studied solution (red) and the other, which parameters differ greatly in the cases of different dispersion media (green). The results of the analysis are shown in Figure 4. It can be seen that the values of the hydrodynamic radii of NPs under study in highly diluted solutions are scattered over a wide range of sizes and significantly exceed the sizes of primary NPs as measured by TEM (Figure 1e). It is also seen that, all distributions are well fitted by the sum of two different Gaussians: one with the distribution maximum and half-width, which little dependent on the studied solution (red) and the other, which parameters differ greatly in the cases of different dispersion media (green).
In a DI aqueous medium, the NPs form clusters with a wide size distribution and large hydrodynamic radii of more than 100 nm (Figure 4a). The size distribution became the smallest and narrowest when the particles were diluted in a slightly alkaline medium (Figure 4b). In an acidic medium (Figure 4c,d), the values of hydrodynamic radii also became smaller, but the resulting sizes and distributions are larger than in an alkaline medium.
The results obtained can be explained based on the following considerations. In the case of pH = 7 (Figure 4a), the NP surface becomes close to the isoelectric point, which leads to additional sticking of clusters formed during synthesis into loose agglomerates up to several hundred nanometers in size (Figure 4a, green line). The interactions between the constituent particles in these agglomerates are likely caused by van der Waals forces overcoming a weak electrical repulsion near the isoelectric point of m-LaPO4. When the pH of the environment changes, the surface of the NPs is populated with positive (H+) or negative (OH-) ions, which leads to an increase in the surface charge. As a result, large agglomerates break up into particles with a narrower size distribution (Figure 4b,c, green lines), which, apparently, represent smaller aggregates with strong interactions between individual nanocrystals (van der Waals forces and chemical bonds). The smaller size of NPs clusters with narrow size distributions in pH-modified aqueous media (Figure 4b,c, green lines) may explain their ability to form loose agglomerates in DI water (Figure 4a, green line). Another fraction (Figure 4, red lines) may consist of larger and denser clusters of NPs, which have a higher degree of aggregation and are formed during synthesis. Due to the large size and high density of NPs in this fraction, they are not able to form agglomerates with NPs among this fraction, therefore, their distribution remains practically unchanged in all dispersion media.
3.4. High-Energy Excitation Spectroscopy Results
3.4.1. Cathodoluminescence Spectroscopy
In the CL spectra of Pr3+ doped LaPO4 (1 at.%) NPs (Figure 5) three emission bands can be recognized in the UV-C region which correspond to electronic transitions 4f15d1 → 3H4 (230 nm), 4f15d1 → 3H5 (shoulder at ~236 nm) and 4f15d1 → 3H6, 3F2 (258 nm). The observed features are in general agreement with data reported in [7,9]. At 485 nm there is an intense line which is clearly identified as Pr3+ 4f2 → 4f2 transition 3P0 → 3H4. The narrow bands in the spectral range of 510–620 nm can also be associated with some 4f2 → 4f2 transitions (see notations in Fig. 5). Between these narrow emission lines and emission bands in UV, there are always several broad emission bands in the range from 350 to 475 nm which have a correlation with known transitions from 4f15d1 state to excited 4f2 levels of Pr3+ ion.
3.4.2. XEOL Spectroscopy
Due to high speed and simplicity, XEOL measurements were performed for all LaPO4: Pr3+ (1 at.%) powders (Figure 6 and Figure 7) to monitor the development of LaPO4: Pr3+ NPs synthesis. To improve the legibility of XEOL data, the graphs of the spectra were smoothed by averaging the data with a step of 2 nm.
The obtained XEOL spectra are dominated by a broad UV band peaking at 257 nm, which is due to Pr3+ 4f15d1 – 3H6, 3F2 inter-configurational transitions. The observed strong decrease of luminescence intensity of the UV band towards shorter wavelengths can be related to the low sensitivity of the silicon detector in high-energy ultraviolet spectral range. For the largest-size NPs synthesized by standard HTMW method, in addition to the intense UV band, some other 4f15d1 – 4f2 and 4f2 – 4f2 transitions can be clearly observed in the visible range as the structured broad and narrow bands (see notations in Figure 6). For the smaller NP samples prepared by the modified HTMW method (Figure 6b–f), the narrow lines overlapping the spectra do not correlate with possible 4f2 – 4f2 transitions in Pr3+ and are due to the background noise signal (Figure 7d). In any case, the low signal to noise ratio of the measured XEOL spectra of NPs prepared with modified HTMW method indicates a rather low UV luminescence intensity in comparison with unmodified nanofibers.
Similar to CL measurements, the highest XEOL intensity was obtained for LaPO4: Pr3+ (1 at.%) particles with the sizes of ~ 7–15 × 30–600 nm synthesized by standard HTMW method (Figure 6a). For this particular sample, the XEOL spectrum was recorded only for 10 seconds, as the detector was saturated at higher measurement times. Nevertheless, the recorded spectrum of these nanofibers (Figure 6a) is approximately 5 times more intense than that of NPs prepared by the modified HTMW method and recorded for 50 seconds (Figure 6b–f). For the NPs, prepared with modified HTMW method, the highest-intensity of UV-C XEOL was detected from the samples prepared with = 0.005 M, CTA = 0.027 M, and pH = 8, with an excess ratio of K2HPO4 equal to 2 or 5 (respectively, Figure 6b,c). A further increase in the excess K2HPO4 leads to the virtual absence of the UV-C XEOL (Figure 6f). A decrease in the concentration of cations during HTMW treatment from 0.01 M to 0.005 M lead to a noticeable increase of the UV-C luminescence intensity (Figure 6d,e).
At the same time, XEOL spectra for NPs obtained by different coprecipitation methods (Figure 7) show that the final spectra are affected by cation concentrations at the coprecipitation stage, while the direction of coprecipitation does not have a strong effect on the final spectra. Thus, using higher cation concentrations during the coprecipitation step and subsequent dilution of fresh gel prior to HTMW treatment results in enhanced UV-C XEOL.
Thus, it has been established that the amount of added NH4OH and excess K2HPO4, as well as concentration of LaPO4: Pr3+ in the reaction mixture have the greatest influence on the XEOL spectra of LaPO4: Pr3+ NPs.
4. Discussion
As shown earlier [14,16,24], the LaPO4: REI NPs have the isoelectric point near pH-neutral region, while stabilization of LaPO4: REI NPs in aqueous colloids in pH-modified media is explained by the chemical adsorption of H+ or OH− ions on the surface nanocrystals. At the same time, the adsorption of H+ ions on the surface of LaPO4: REI NPs occurs more efficiently than of OH−. As a result of the disproportion in the adsorption of pH-determining ions, LaPO4: Pr3+ NPs without coating agents are more stable in acidic than in alkaline environment [14,24]. On the other hand, as has been shown in many works, the use of surfactants with carboxyl groups makes it possible to decrease the aggregation probability of NPs in an alkaline medium [25,26,32].
The reaction of chemical surface peptization of non-modified LaPO4 NPs as a result of the adsorption of pH-determining ions in an acidic or alkaline environment can be represented by the following expressions:
{[LaPO4]m}0 + nH+ ↔ {[LaPO4]m·nH+}n+
{[LaPO4]m}0 + nOH− ↔ {[LaPO4]m·nOH−}n−
When TA-modified particles are dispersed in an acidic or alkaline medium, the hydroxyl end of the organic molecule can also undergo reversible chemical transformations:
R-COOH0 ↔ R-COO−
Thus, for TA-modified NPs placed to an alkaline medium, the total expression of chemical adsorption and neutralization of the hydroxyl groups with OH- ions can be written as
{[LaPO4]m·yR-COOH}0 ↔ {[LaPO4]m·nOH−·yR—COO−}−(n+y)
After the adsorption of pH-determining ions, the charged surface of the NP is surrounded by a solvation aqueous shell, which leads to a decrease in the excess energy of the system. For this case, the surface of the LaPO4 NPs is surrounded by the polar ends of water molecules, opposite in sign to the surface:
{[LaPO4]m}0 + nH+ ↔ {[LaPO4]m·nH+}n+ xH2O
{[LaPO4]m·yR-COOH}0 ↔ {[LaPO4]m·nOH−·yR—COO−}−(n+y)·xH2O
The results of these reactions are shown in Scheme 1.
As can be seen (Scheme 1), the solvate aqueous shell, as well as the adsorbed tartaric acid molecules, lead to a decrease in the aggregation probability. However, optical microscopy studies have shown, that despite the use of a tartaric acid component as capping agent, the product of modified HTMW synthesis was not individual nanocrystals, but clusters with hydrodynamic radii up to 100 nm and more. It was shown that the synthesized TA-modified NPs have the narrowest hydrodynamic radii distributions in a weakly alkaline aqueous medium, which is a consequence of the adsorption of the tartaric acid compound on the NPs surface. In addition, the resulting particles also have good stability in an acidic environment due to the high efficiency of adsorption of H+ ions on the surface of LaPO4: Pr3+ NPs.
The dense aggregates of TA-modified LaPO4: Pr3+ nanocrystals are formed during the synthesis (coprecipitation, HTMW treatment and cooling of the reaction mixture) and not during storage of the colloidal solution. The source of the observed NPs clusters with narrow distributions in modified aqueous media is probably the washing stage (Section 2.1.2). As mentioned earlier, after HTMW synthesis, a rapidly settling precipitate of crystalline particles is formed, while the colloidal properties appear after several washing cycles. As a result of the washing process, loose aggregates break up into smaller aggregates, which leads to transparency and stabilization of the dispersion of NPs. The double distributions of hydrodynamic radii in different pH are likely due to the different properties of hard clusters: size, shape, and density; however, this assumption requires further research.
The comparison of obtained data on UV-C luminescence intensities for NPs prepared under various synthesis conditions and having different sizes and aspect ratios between width and length of individual nanocrystals are presented in Table 1. It should be mentioned that we cannot make reliable quantitative analysis of the dependence of NP luminescence intensities on synthesis conditions (NP morphology) because the procedure of preparing NP powder samples for each measurement of XEOL or CL spectrum can differ from time to time. For example, the temperature and duration of the drying procedure can be slightly different, and the time between synthesis and the measurement also varied in the wide range (from several days to few months) which can induce some aging effects, e.g., NP aggregation. Thus, our qualitative analysis can indicate only some tendencies in changing the NP luminescence intensity depending on synthesis conditions.
An obvious result, which can be seen in the Table 1 is that in general the UV-C luminescence intensity is higher for larger size NPs. The highest intensity was obtained for NPs with the size of ~ 7–15 × 30–600 nm synthesized without surfactants (natural pH = 5), while the intensity is much weaker for NPs prepared at basic medium (pH = 8 – 9). As it is commonly accepted the luminescence intensity of NPs depends on the degree of luminescence quenching in the near-surface defect layer as well as on the concentration of defects or impurities in the volume of NPs. Thus, an acidic environment provides a better quality of LaPO4 NPs due to a more accessible dissolution-growth mechanism during the HTMW treatment, while a basic aqueous medium can inhibit the dissolution of particles and lead to particles with smaller sizes and a larger amount of crystal structural defects (for example, quenching OH− groups).
Most of performed synthesis experiments resulted in the production of fiber-like morphology of LaPO4: Pr3+ NPs (nanorods, nanowires) having different sizes and aspect ratios between width and length. It is obvious that nanorod have a larger surface-to-volume ratio than rounded NPs with the same volume, i.e., it is expected that near-surface luminescence quenching is stronger for nanofibers than for nanospheres of the same particle volume. However, NPs with a size of 6 × 10 nm, i.e., having a shape close to round, demonstrate a very low intensity of XEOL UV-C luminescence (Figure 6f), as compared to particles of the similar sizes 4.5 × 13 nm (Figure 6e). Besides that, the results of CL (Figure 5b,c) and XEOL (Figure 6b,d) analysis for samples with comparable lengths but different diameters (Figure 1b, e) indicate that powders of particles with smaller diameters show better UV-C luminescence intensities. The different intensities of UV-C radiation from samples with similar size distributions can be explained by the lower crystallinity of NPs obtained in a more alkaline medium or with a higher excess of anions.
As a common conclusion from this consideration, it can be assumed that there is no simple direct dependence of Pr3+ UV-C luminescence intensity only on the size of particles but there is some dependence of luminescence intensity on the synthesis conditions, the optimization of which allows to increase the intensity of Pr3+ UV-C luminescence.
5. Conclusions
The hydrothermal method with microwave (HTMW) heating (200 °C, 2 hours, 2 magnetrons, 2.45 GHz) has been successfully applied for synthesis of colloidal solutions of nanorods of monoclinic LaPO4 doped with Pr3+. The influence of synthesis conditions on the intensity of UV-C luminescence of Pr3+ ions at the inter-configurational 4f15d1 – 4f2 transitions in the resulting NPs was assessed using electron-beam and X-ray excitation. Measurements of zeta potential showed that the TA-modified colloids have better stability in basic water media, which can facilitate biofunctionalization of such NPs in the alkaline pH range. The processes of aggregation of the resulting NPs into clusters under conditions of strong dilution in solutions of deionized water, or weakly alkaline and acidic aqueous solutions were studied on single cluster level using the developed highly sensitive laser ultramicroscope to visualize and to analyze the individual trajectories of the Brownian motion of the observed clusters. It was shown that the synthesized nanorods form clusters with narrow distribution and minimal hydrodynamic radii in slightly alkaline or acidic aqueous solutions. The qualitative analysis of the relationship between the morphology of LaPO4:Pr3+ NPs and the intensity of their UV-C luminescence has revealed that there is no direct dependence of Pr3+ UV-C intensity on the sizes of NPs alone, but there is a strong dependence of luminescence intensity on the synthesis conditions. In particular, the pH level and anion excess ratio must be maintained optimal during synthesis for the prevention of strong quenching of luminescence. In addition, an increase in scintillation intensity can be achieved by increasing the cation concentration at the coprecipitation stage and decreasing it (diluting) at the hydrothermal stage. These synthesis parameters can affect the NP growth mechanism, resulting in NPs with different sizes (TEM results) and degree of crystallinity (XRD and high-energy spectroscopy results). Thus, optimization of the HTMW synthesis conditions makes it possible to influence the conditions of nucleation and crystallization, which in turn significantly increase the intensity of UV-C luminescence of monoclinic TA-modified LaPO4 nanorods doped with Pr3+ ions under high-energy excitation.
Author Contributions
Conceptualization, Y.O., Y.V., V.M. and E.O.; methodology, A.S. E.O. and Y.V.; software, G.S.; validation, Y.O., Y.V. and V.M.; formal analysis, A.S., G.S.; investigation, A.S., A.P., S.B., L.I., O.U., U.Q. S.M. and G.S.; resources, A.S. and E.O.; data curation, A.S. and G.S.; writing—original draft preparation, A.S., Y.V.; writing—review and editing, Y.O., V.M. and Y.V.; visualization, A.S. and G.S.; supervision, Y.O., V.M. and Y.V.; project administration Y.V.; funding acquisition, Y.V. All authors have read and agreed to the published version of the manuscript.
Funding
“The research was funded by a grant of the Russian Science Foundation, grant number 22-22-00998”. U.Q. and S.M. are grateful for the support of the Estonian Research Council (project PUT PRG629).
Data Availability Statement
Not applicable.
Acknowledgments
U.Q. and S.M. are grateful for the personal assistance of Eduard Feldbach in the CL measurements. The authors are grateful to the Center for Collective Use of the GPI RAS for the equipment provided. Y.V., G.S., E.O. and A.S. are grateful for a support of the Russian Science Foundation, grant number 22-22-00998 for small scientific groups. All the work except the CL measurements was done under support of the RSF.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- Gai, S.; Li, C.; Yang, P.; Lin, J. Recent Progress in Rare Earth Micro/Nanocrystals: Soft Chemical Synthesis, Luminescent Properties, and Biomedical Applications. Chem. Rev. 2014, 114(4), 2343–2389. [Google Scholar] [CrossRef]
- Miwa, S.; Yano, S.; Hiroshima, Y.; Tome, Y.; Uehara, F.; Mii, S.; Efimova, E.V.; Kimura, H.; Hayashi, K.; Tsuchiya, H.; Hoffman, R.M. Imaging UVC-induced DNA damage response in models of minimal cancer. J. Cell. Biochem. 2013, 114(11), 2493–2499. [Google Scholar] [CrossRef]
- Müller, M.; Espinoza, S.; Jüstel, T.; Held, K.D.; Anderson, R.R.; Purschke, M. UVC-Emitting LuPO4:Pr3+ Nanoparticles Decrease Radiation Resistance of Hypoxic Cancer Cells. Radiat. Res. 2020, 193(1), 82–87. [Google Scholar] [CrossRef]
- Wisniewski, D.; Tavernier, S.; Wojtowicz, A. J.; Wisniewska, M.; Bruyndonckx, P.; Dorenbos, P.; Van Loef, E.; Van Eijk, C.W.E.; Boatner, L.A. LuPO4:Nd and YPO4:Nd — new promising VUV scintillation materials. Nucl. Instrum. Methods Phys. Res., Sect. A. 2002, 486(1-2), 239–243. [Google Scholar] [CrossRef]
- Makhov, V.N.; Kirikova, N.Y.; Kirm, M.; Krupa, J.C.; Liblik, P.; Lushchik, A.; Zimmerer, G. Luminescence properties of YPO4:Nd3+: a promising VUV scintillator material. Nucl. Instrum. Methods Phys. Res., Sect. A. 2002, 486(1-2), 437–442. [Google Scholar] [CrossRef]
- Mikhailin, V.V.; Spassky, D.A.; Kolobanov, V.N.; Meotishvili, A.A.; Permenov, D.G.; Zadneprovski, B.I. Luminescence study of the LuBO3 and LuPO4 doped with RE3+. Radiat. Meas. 2010, 45(3-6), 307–310. [Google Scholar] [CrossRef]
- Malyy, T.S.; Vistovskyy, V.V.; Khapko, Z.A.; Pushak, A.S.; Mitina, N.E.; Zaichenko, A.S.; Gektin, A.V.; Voloshinovskii, A. S. Recombination luminescence of LaPO4-Eu and LaPO4-Pr nanoparticles. J. Appl. Phys. 2013, 113(22), 224305. [Google Scholar] [CrossRef]
- Espinoza, S.; Volhard, M.-F.; Kätker, H.; Jenneboer, H.; Uckelmann, A.; Haase, M.; Müller, M.; Purschke, M.; Jüstel, T. Deep Ultraviolet Emitting Scintillators for Biomedical Applications: The Hard Way of Downsizing LuPO4:Pr3+. Part. Part. Syst. Charact. 2018, 35(12), 1800282. [Google Scholar] [CrossRef]
- Srivastava, A.M.; Setlur, A.A.; Comanzo, H.A.; Beers, W.W.; Happek, U.; Schmidt, P. The influence of the Pr3+ 4f15d1 configuration on the 1S0 emission efficiency and lifetime in LaPO4. Opt. Mater. 2011, 33(3), 292–298. [Google Scholar] [CrossRef]
- Okamoto, S.; Uchino, R.; Kobayashi, K.; Yamamoto, H. Luminescent properties of Pr3+-sensitized LaPO4:Gd3+ ultraviolet-B phosphor under vacuum-ultraviolet light excitation. J. Appl. Phys. 2009, 106(1), 013522. [Google Scholar] [CrossRef]
- Srivastava, A.M. Aspects of Pr3+ luminescence in solids. J. Lumin. 2016, 169(B), 445–449. [Google Scholar] [CrossRef]
- Bagatur’yants, A.A.; Iskandarova, I.M.; Knizhnik, A.A.; Mironov, V.S.; Potapkin, B.V.; Srivastava, A.M.; Sommerer, T.J. Energy level structure of 4f5d states and the Stokes shift In LaPO4:Pr3+: A theoretical study. Phys. Rev. B. 2008, 78(16), 165125. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Popović, Z.; Chen, O.; Cui, J.; Fukumura, D.; Bawendi, M.G.; Jain, R.K. Fluorescent Nanorods and Nanospheres for Real-Time In Vivo Probing of Nanoparticle Shape-Dependent Tumor Penetration, Angew. Chem., Int. Ed. 2011, 50(48), 11417–11420. [CrossRef]
- Wang, Z.; Kim, J.; Magermans, L.; Corbella, F.; Florea, I.; Larquet, E.; Kim, J.; Gacoin, T. Monazite LaPO4:Eu3+ nanorods as strongly polarized nano-emitters, Nanoscale 2021, 13 (40), 16968-16976. [CrossRef]
- Espinoza, S.; Jüstel, T. UV Emitting LaPO4 Nanocrystals. Poster. In Proceedings of the 22nd Meeting of the Deutsche Akademie for Photobiologie und Phototechnologie, Hotel Mercure, Freiburg, Germany, May 2017 . Available online: https://www.researchgate.net/publication/318913422_UV_Emitting_LaPO4_Nanocrystal (accessed on 15 July 2023).
- Hilario, E.G.; Rodrigues, L.C.V.; Caiut, J.M.A. Spectroscopic study of the 4f5d transitions of LaPO4 doped with Pr3+ or co-doped with Pr3+ and Gd3+ in the vacuum ultra violet region. Nanotechnology. 2022, 33(30), 305703. [Google Scholar] [CrossRef]
- Meyssamy, H.; Riwotzki, K.; Kornowski, A.; Naused, S.; Haase, M. Wet-Chemical Synthesis of Doped Colloidal Nanomaterials: Particles and Fibers of LaPO4: Eu, LaPO4: Ce, and LaPO4: Ce,Tb. Adv. Mater. 1999, 11(10), 840–844. [Google Scholar] [CrossRef]
- Song, H.; Yu, L.; Lu, S.; Wang, T.; Liu, Z.; Yang, L. Remarkable differences in photoluminescent properties between LaPO4: Eu one-dimensional nanowires and zero-dimensional nanoparticles. Appl. Phys. Lett. 2004, 85(3), 470–472. [Google Scholar] [CrossRef]
- Riwotzki, K.; Meyssamy, H.; Schnablegger, H.; Kornowski, A.; Haase, M. Liquid-Phase Synthesis of Colloids and Redispersible Powders of Strongly Luminescing LaPO4:Ce,Tb Nanocrystals. Angew. Chem., Int. Ed. 2001, 40(3), 573–576. [Google Scholar] [CrossRef]
- Niu, N.; Yang, P.; Wang, Y.; Wang, W.; He, F.; Gai, S.; Wang, D. LaPO4:Eu3+, LaPO4:Ce3+, and LaPO4: Ce3+, Tb3+ nanocrystals: Oleic acid assisted solvothermal synthesis, characterization, and luminescent properties. J. Alloys Compd. 2011, 509(6), 3096–3102. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, Q.; Li, Y.; Wang, H. Facile crystallization control of LaF3/LaPO4: Ce, Tb nanocrystals in a microfluidic reactor using microwave irradiation. J. Mater. Chem. 2010, 20(9), 1766–1771. [Google Scholar] [CrossRef]
- Enikeeva, M.O.; Kenges, K.M.; Proskurina, O.V.; Danilovich, D.P.; Gusarov, V.V. ; Influence of Hydrothermal Treatment Conditions on the Formation of Lanthanum Orthophosphate Nanoparticles of Monazite Structure, Russ. J. Appl. Chem. 2020, 93(4), 529–539. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Zhang, Z.; Wang, X. Effects of pH value on growth morphology of LaPO4 nanocrystals: investigated from experiment and theoretical calculations. Appl. Phys. A. 2016, 112(5), 508. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.A.; Chini, S.F.; Sadeghi, A. Size and shape tailored sol-gel synthesis and characterization of lanthanum phosphate (LaPO4) nanoparticles. Mater. Des. 2019, 181, 108058. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.-G.; Zhu, Q.; Kim, B.-N.; Sun, X. Tartrate promoted hydrothermal growth of highly [001] oriented (La0.95−xBixEu0.05)PO4 (x = 0–0.01) nanowires with enhanced photoluminescence. Mater. Des. 2017, 126, 115–122. [Google Scholar] [CrossRef]
- Safronikhin, A.V.; Ehrlich, G.V.; Lisichkin, G.V. Synthesis of lanthanum fluoride nanocrystals and modification of their surface. Russ. J. Gen. Chem. 2011, 81(2), 277–281. [Google Scholar] [CrossRef]
- Majolino, D.; Mallamace, F.; Migliardo, P.; Micali, N.; Vasi, C. Elastic and quasielastic light-scattering studies of the aggregation phenomena in water solutions of polystyrene particles. Phys. Rev. A. 1989, 40(8), 4655–4674. [Google Scholar] [CrossRef]
- Lazzari, S.; Nicoud, L.; Jaquet, B.; Lattuada, M.; Morbidelli, M. Fractal-like structures in colloid science. Adv. Colloid Interface Sci. 2016, 235, 1–13. [Google Scholar] [CrossRef]
- Timofeeva, E.; Orlovskaya, E.; Popov, A.; Shaidulin, A.; Kuznetsov, S.; Alexandrov, A.; Uvarov, O.; Vainer, Yu.; Silaev, G.; Rähn, M.; Tamm, A.; Fedorenko, S.; Orlovskii, Yu. The Influence of Medium on Fluorescence Quenching of Colloidal Solutions of the Nd3+: LaF3 Nanoparticles Prepared with HTMW Treatment. Nanomaterials. 2022, 12(21), 3749. [Google Scholar] [CrossRef]
- Silaev, G. O.; Krasheninnikov, V. N.; Shaidulin, A. T.; Uvarov, O. V.; Orlovskaya, E. O.; Orlovskii, Yu. V.; Vainer, Yu. G. Optical and Electron Microscopy of Clusters of Nd3+:LaF3 Nanoparticles Synthesized by the HTMW Method. Phys. Wave Phenom. 2023, 31(3), 160–170. [Google Scholar] [CrossRef]
- Feldbach, E.; Tõldsepp, E.; Kirm, M.; Lushchik, A.; Mizohata, K.; Räisänen, J. Radiation resistance diagnostics of wide-gap optical materials. Opt. Mater. 2016, 55, 164–167. [Google Scholar] [CrossRef]
- Li, C.; Hassan, A.; Palmai, M.; Snee, P.T.; Baveye, P.C. Colloidal stability and aggregation kinetics of nanocrystal CdSe/ZnS quantum dots in aqueous systems: effects of pH and organic ligands. J. Nanopart. Res. 2020, 22, 349. [Google Scholar] [CrossRef]
Figure 1.
Figure 1.TEM images and size distributions (diameter × length) of m-LaPO4: Pr3+ (1 at.%) NPs prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.08 M, pH=9; c) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH=9; d) nanorods (5 ± 1 × 18 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8; e) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 2, CTA = 0.027 M, pH= 8; f) small faceted NPs (6 ± 2 × 10 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 10, CTA = 0.027 M, pH=8.
Figure 1.
Figure 1.TEM images and size distributions (diameter × length) of m-LaPO4: Pr3+ (1 at.%) NPs prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.08 M, pH=9; c) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH=9; d) nanorods (5 ± 1 × 18 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8; e) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 2, CTA = 0.027 M, pH= 8; f) small faceted NPs (6 ± 2 × 10 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 10, CTA = 0.027 M, pH=8.
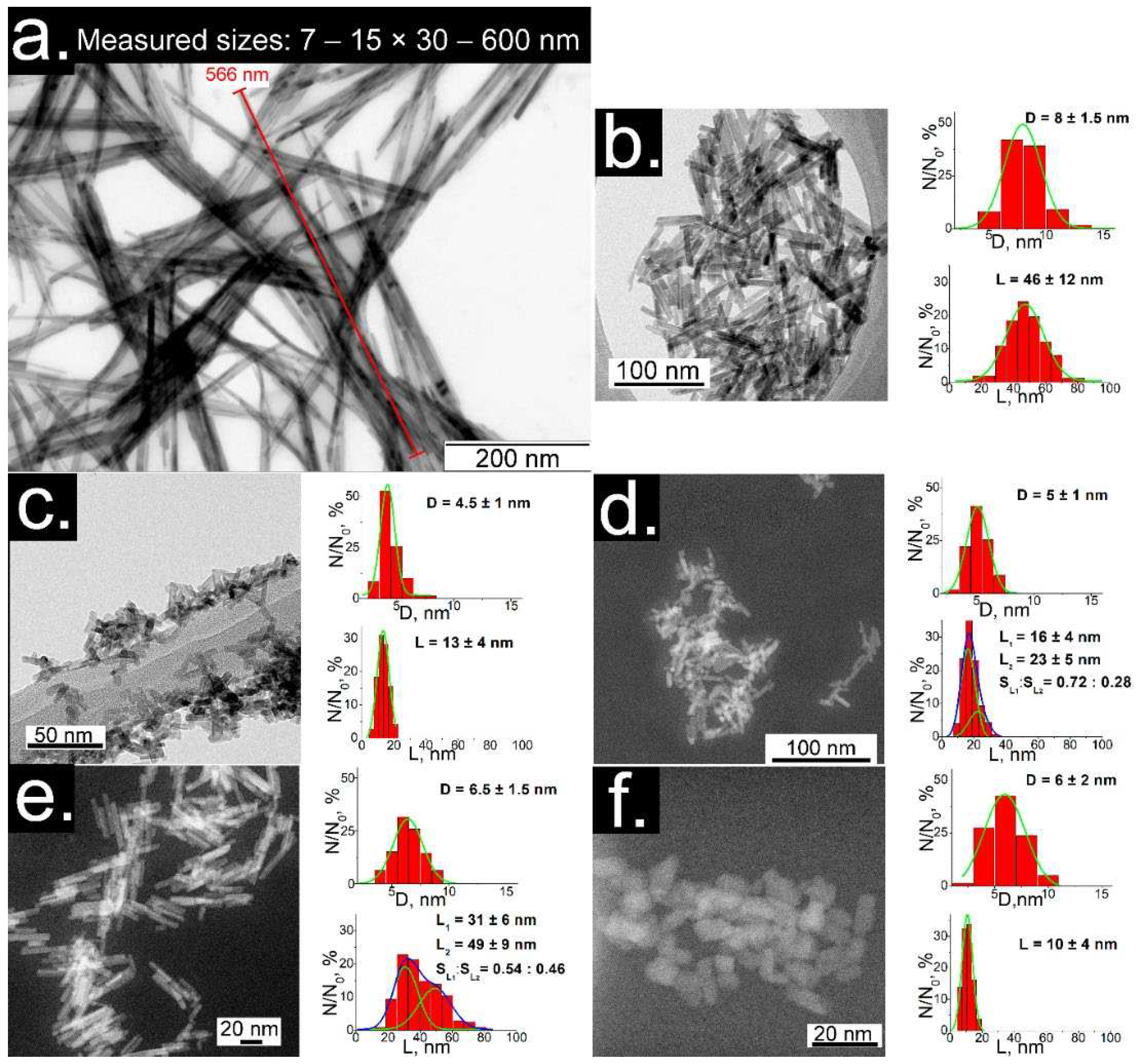
Figure 2.
XRD pattern of LaPO4: Pr3+ (1 at.%) powders prepared with HTMW (200 °C, 2 hours): a) nanofibers obtained with = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b,c) NPs obtained with = 0.005 M, CTA = 0.027 M, pH = 8, but with K2HPO4 excess ratio equal to five (b) and ten (c).
Figure 2.
XRD pattern of LaPO4: Pr3+ (1 at.%) powders prepared with HTMW (200 °C, 2 hours): a) nanofibers obtained with = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b,c) NPs obtained with = 0.005 M, CTA = 0.027 M, pH = 8, but with K2HPO4 excess ratio equal to five (b) and ten (c).
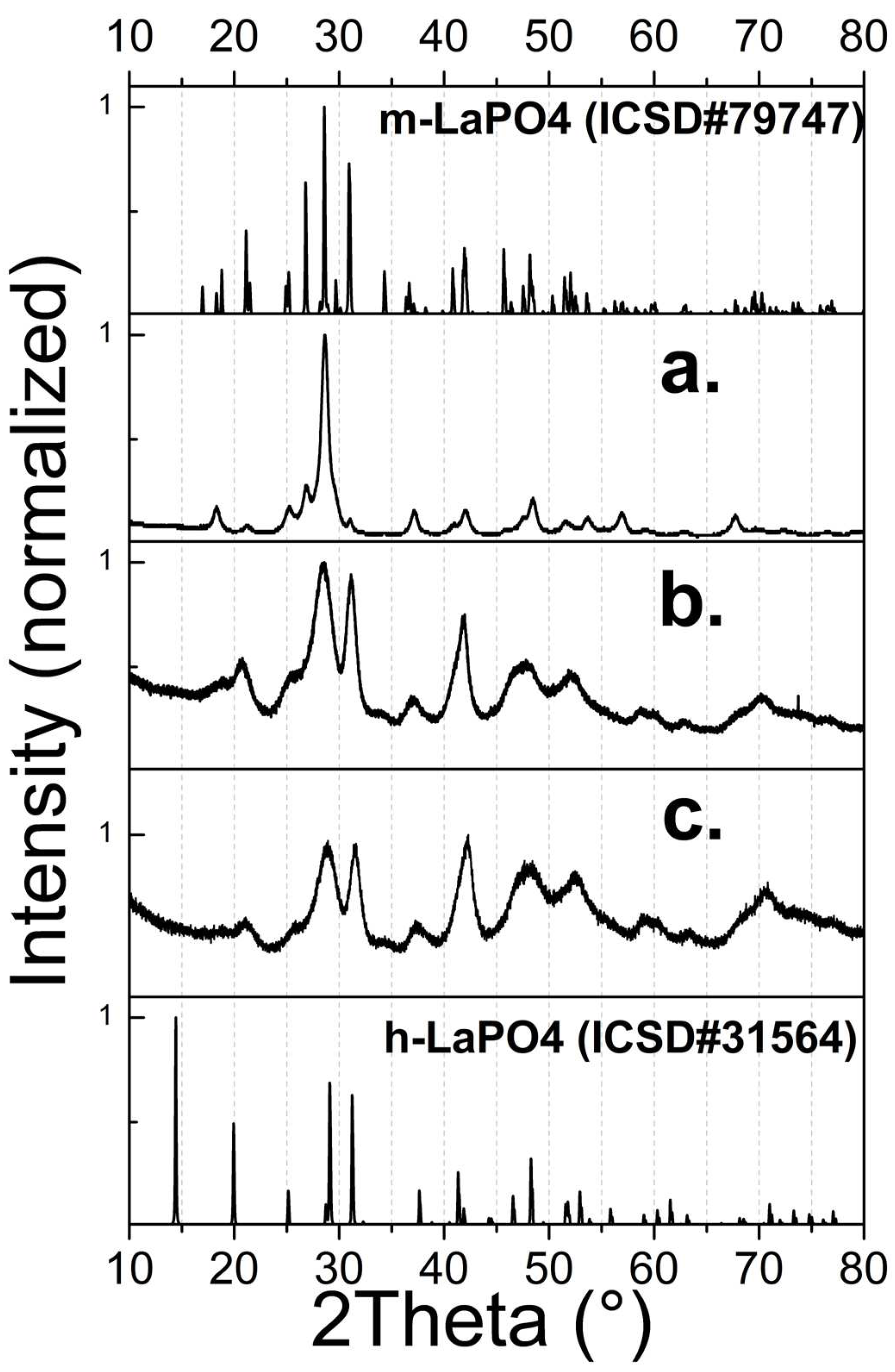
Figure 3.
Zeta potential measurements of the TA-modified LaPO4: Pr3+ (1 at.%) NPs with sizes 5 ± 1 × 18 ± 4 nm (Fig. 1c) obtained by HTMW method (200 , 2 hours) with = 0.005 M, K2HPO4 excess ratio = 5, pH = 8, CTA = 0.027 M, dispersed in different aqueous media by diluting 0.1 mL of colloid with concentration of 10 mg/mL with 0.9 mL of dispersants: a) blue line - DI water, resulting pH = 7.5; b) green line - weak NaOH solution, resulting pH = 9; c) red line - weak HNO3 solution, resulting pH = 5.
Figure 3.
Zeta potential measurements of the TA-modified LaPO4: Pr3+ (1 at.%) NPs with sizes 5 ± 1 × 18 ± 4 nm (Fig. 1c) obtained by HTMW method (200 , 2 hours) with = 0.005 M, K2HPO4 excess ratio = 5, pH = 8, CTA = 0.027 M, dispersed in different aqueous media by diluting 0.1 mL of colloid with concentration of 10 mg/mL with 0.9 mL of dispersants: a) blue line - DI water, resulting pH = 7.5; b) green line - weak NaOH solution, resulting pH = 9; c) red line - weak HNO3 solution, resulting pH = 5.
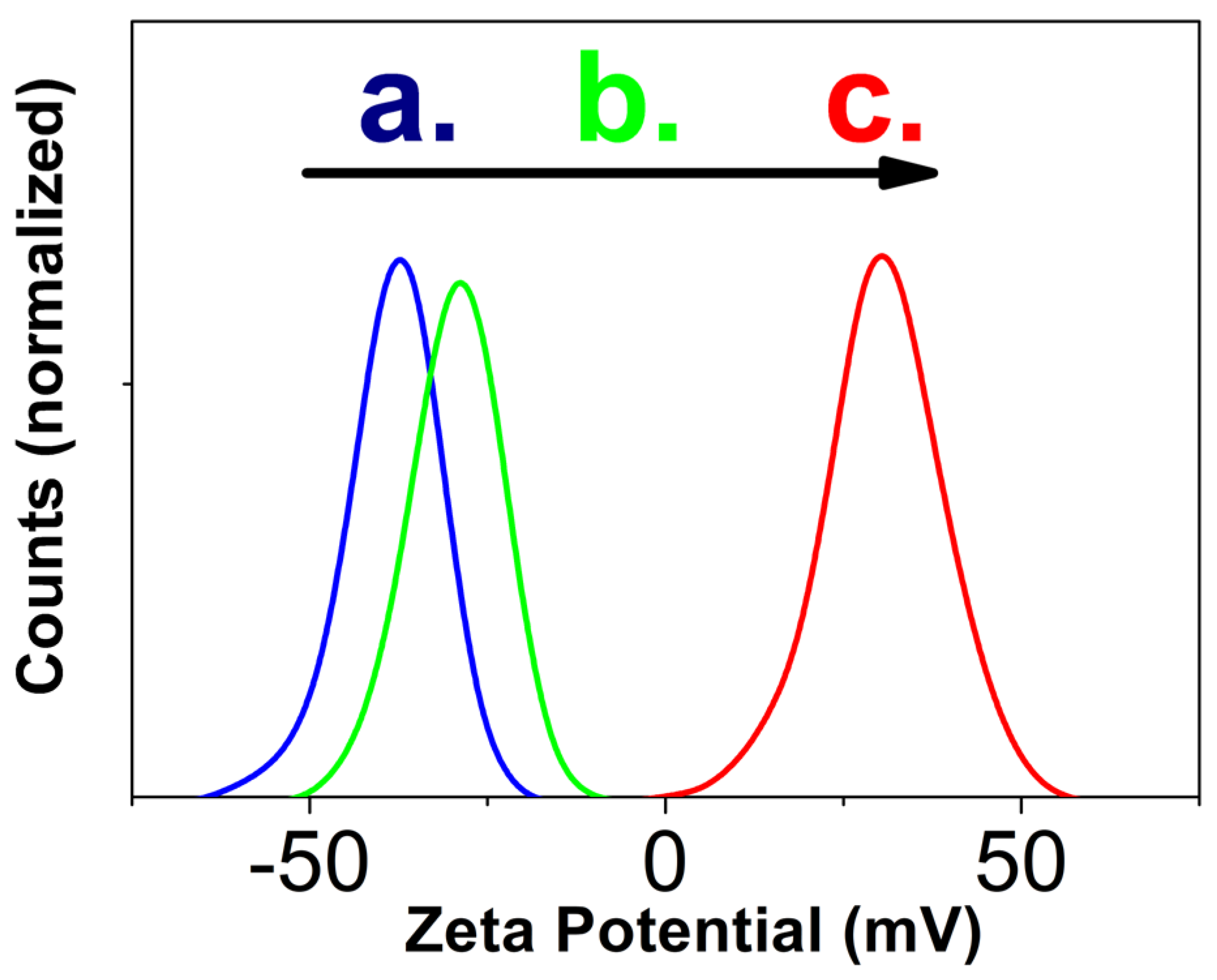
Figure 4.
The distributions of hydrodynamic radii of the synthesized colloidal LaPO4: Pr3+ (1 at.%) NPs with primary sizes of 5 ± 1 × 18 ± 4 nm (fig. 1c) and the clusters formed by them, measured in various aqueous solutions using the developed high-sensitivity laser ultramicroscope: a) - in a purified deionized water with pH=7; b) - in a weak NaOH solution with pH=9; c,d) in weak HNO3 solutions with pH=5 (c) and pH=2 (d). All distributions are well fitted by the sum of two different Gaussians (R2 > 0.98).
Figure 4.
The distributions of hydrodynamic radii of the synthesized colloidal LaPO4: Pr3+ (1 at.%) NPs with primary sizes of 5 ± 1 × 18 ± 4 nm (fig. 1c) and the clusters formed by them, measured in various aqueous solutions using the developed high-sensitivity laser ultramicroscope: a) - in a purified deionized water with pH=7; b) - in a weak NaOH solution with pH=9; c,d) in weak HNO3 solutions with pH=5 (c) and pH=2 (d). All distributions are well fitted by the sum of two different Gaussians (R2 > 0.98).
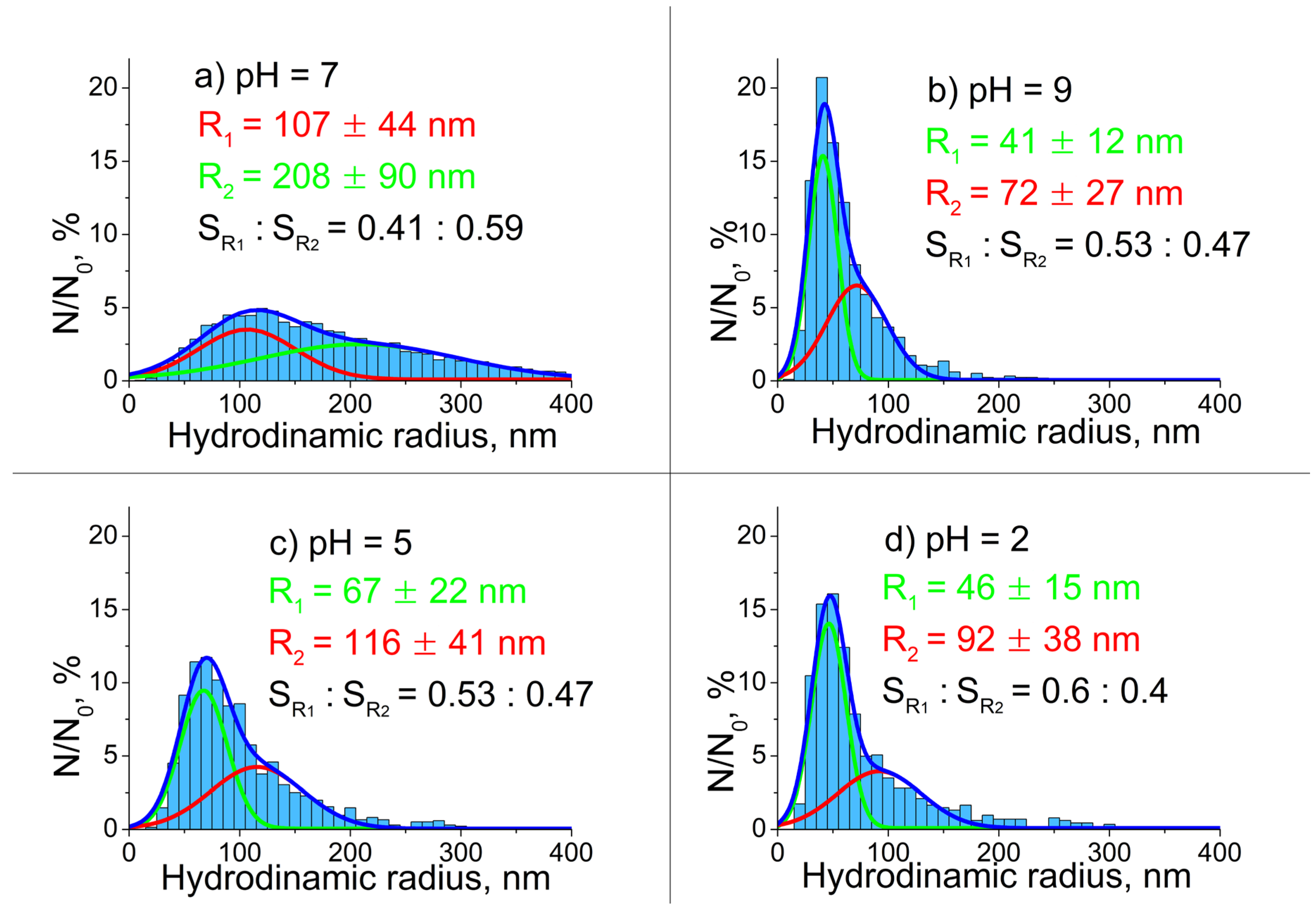
Figure 5.
CL spectra of m-LaPO4: Pr3+ (1 at.%) NPs prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH= 8; c) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.027 M, pH= 9; d) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH = 9.
Figure 5.
CL spectra of m-LaPO4: Pr3+ (1 at.%) NPs prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no additional components (natural pH = 5); b) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH= 8; c) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.027 M, pH= 9; d) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH = 9.
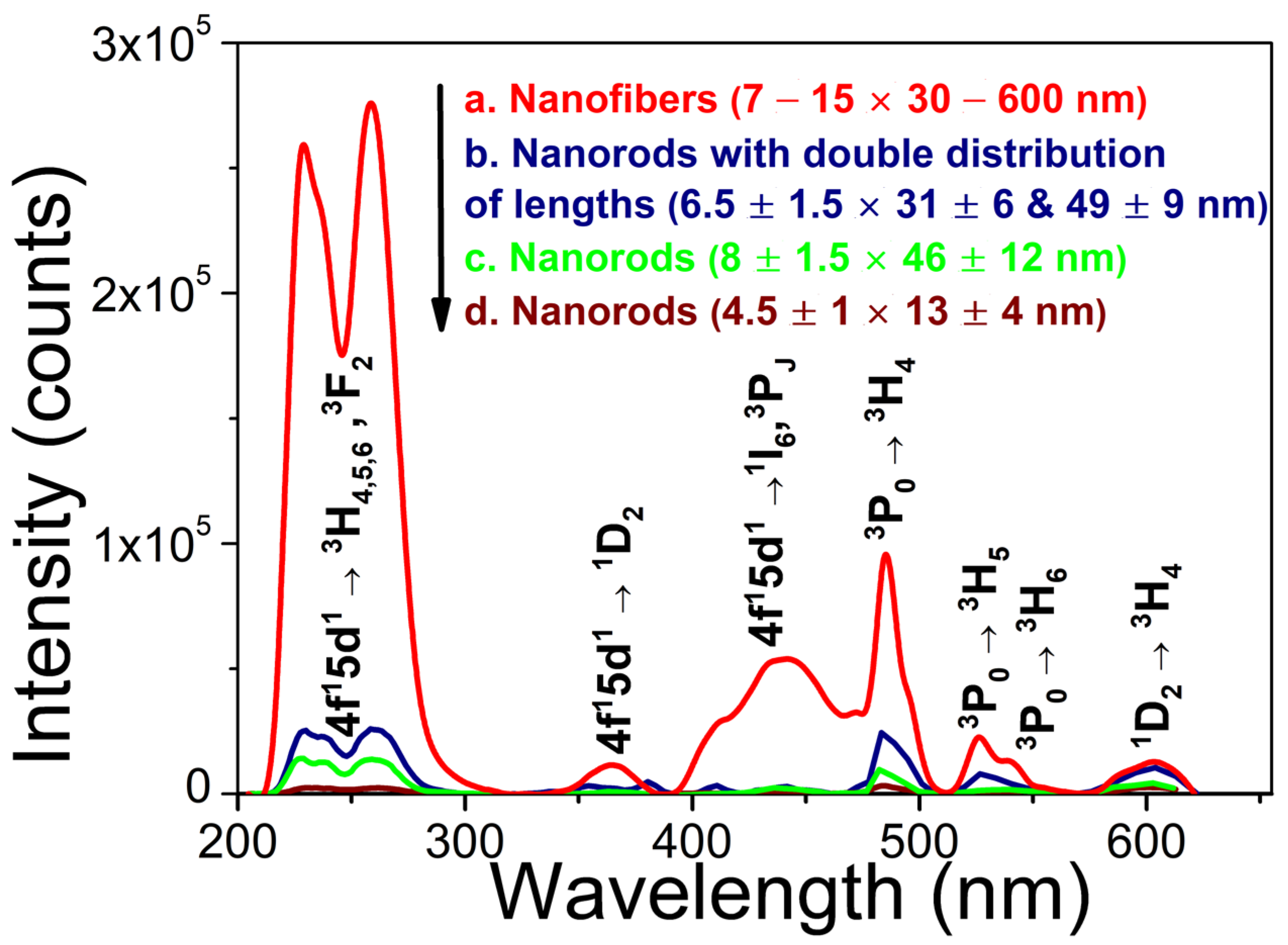
Figure 6.
XEOL spectra of m-LaPO4: Pr3+ (1 at.%) powders prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no surfactant (natural pH = 5); b) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 2, CTA = 0.027 M, pH = 8; c) nanorods (5 ± 1 × 18 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8; d) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.08 M, pH = 9; e) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH = 9; f) Small faceted nanoparticles (6 ± 2 × 10 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 10, CTA = 0.027 M, pH = 8.
Figure 6.
XEOL spectra of m-LaPO4: Pr3+ (1 at.%) powders prepared with HTMW method (200 °C, 2 hours) obtained with various synthesis conditions: a) nanofibers (7 – 15 × 30 – 600 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, no surfactant (natural pH = 5); b) nanorods with double distribution of lengths (6.5 ± 1.5 × 31 ± 6 & 49 ± 9 nm), = 0.005 M, K2HPO4 excess ratio = 2, CTA = 0.027 M, pH = 8; c) nanorods (5 ± 1 × 18 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8; d) nanorods (8 ± 1.5 × 46 ± 12 nm), = 0.01 M, K2HPO4 excess ratio = 1.25, CTA = 0.08 M, pH = 9; e) nanorods (4.5 ± 1 × 13 ± 4 nm), = 0.01 M, K2HPO4 excess ratio = 5, CTA = 0.08 M, pH = 9; f) Small faceted nanoparticles (6 ± 2 × 10 ± 4 nm), = 0.005 M, K2HPO4 excess ratio = 10, CTA = 0.027 M, pH = 8.
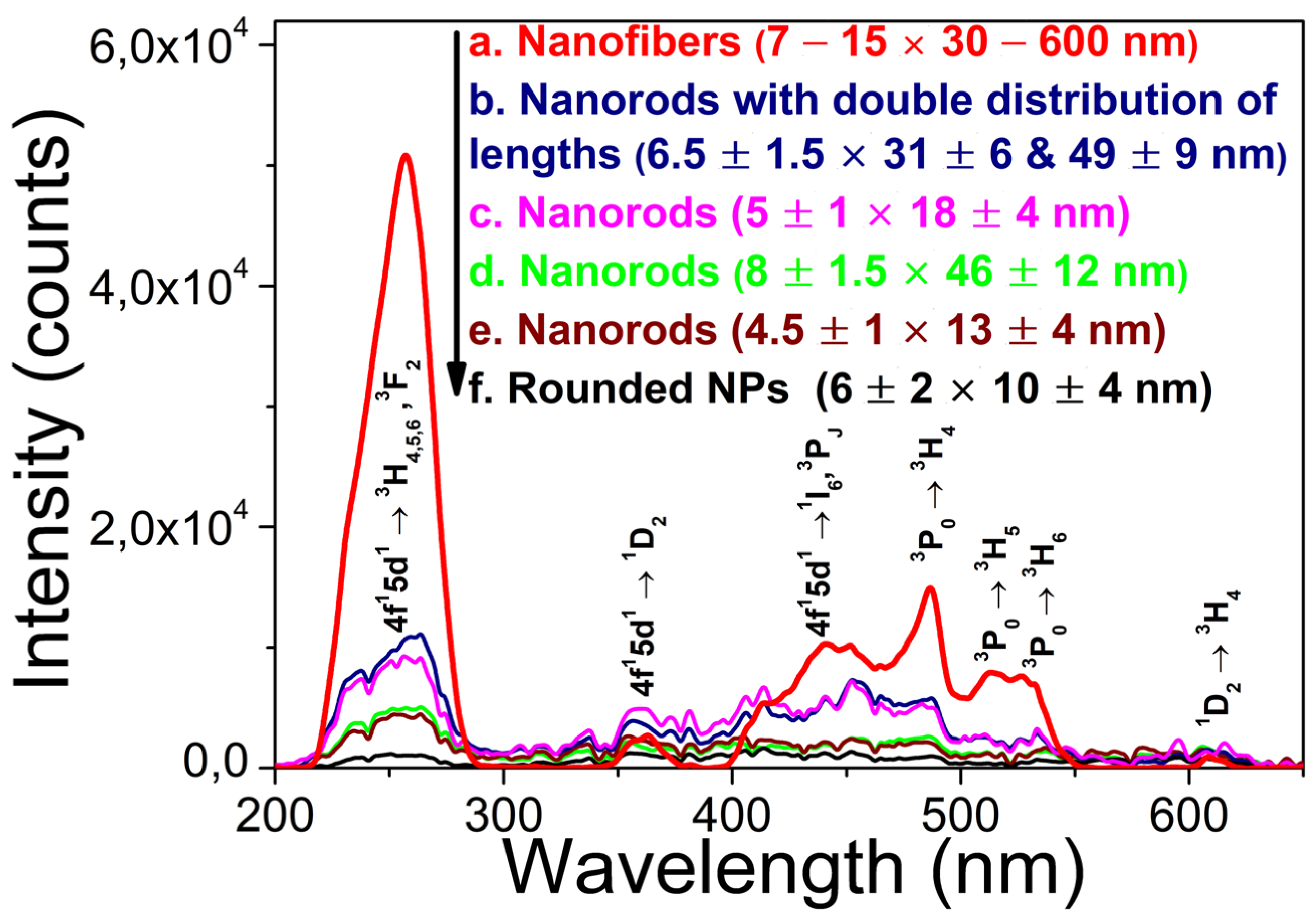
Figure 7.
XEOL spectra of m-LaPO4: Pr3+ (1 at.%) powders prepared by HTMW method (200 °C, 2 hours) obtained with same synthesis conditions = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8), but with different coprecipitation techniques: a)coprecipitation by adding 0.25 mmol of cations (10 mL solution) to 1.25 mmol anions (15 mL solution) and then diluted with 25 mL of deionized water; b) coprecipitation by adding 1.25 mmol of anions (10 mL solution) to 0.25 mmol cations (15 mL solution) and then diluted with 25 mL of deionized water; c) coprecipitation by adding 0.2 mmol of cations (5 mL solution) to 1.25 mmol cations (45 mL solution);d) background spectrum.
Figure 7.
XEOL spectra of m-LaPO4: Pr3+ (1 at.%) powders prepared by HTMW method (200 °C, 2 hours) obtained with same synthesis conditions = 0.005 M, K2HPO4 excess ratio = 5, CTA = 0.027 M, pH = 8), but with different coprecipitation techniques: a)coprecipitation by adding 0.25 mmol of cations (10 mL solution) to 1.25 mmol anions (15 mL solution) and then diluted with 25 mL of deionized water; b) coprecipitation by adding 1.25 mmol of anions (10 mL solution) to 0.25 mmol cations (15 mL solution) and then diluted with 25 mL of deionized water; c) coprecipitation by adding 0.2 mmol of cations (5 mL solution) to 1.25 mmol cations (45 mL solution);d) background spectrum.
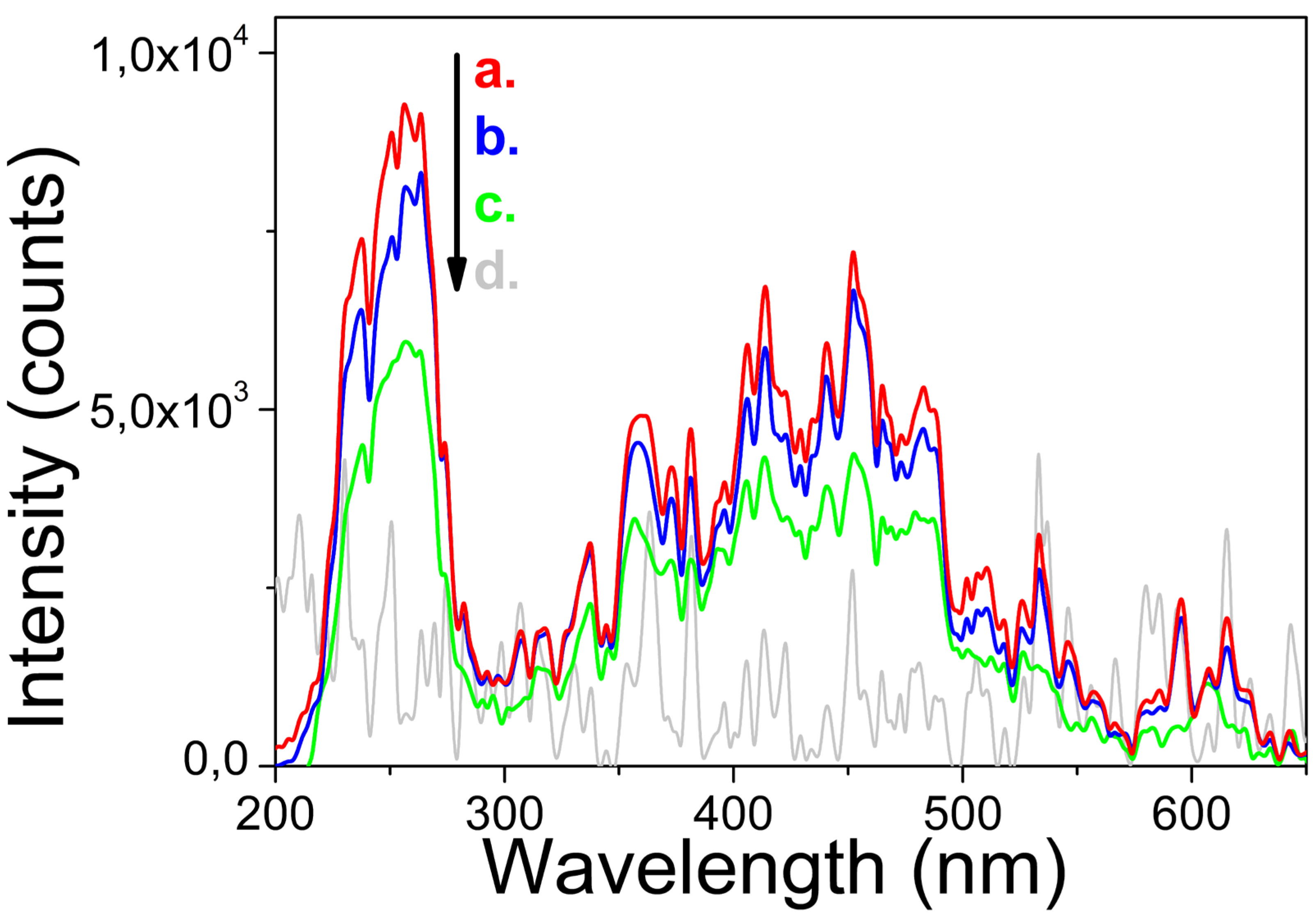
Scheme 1.
Representation of processes on the surface of LaPO4 NP in different aqueous media: (a) pH = 7 is near the isoelectric point of m-LaPO4, no charge in the surface, the near polar H2O molecules (shown by blue circle and red circles) are randomly positioned around NP; (b) pH < 5 is an acidic environment in which H+ ions are adsorbed on the surface of the LaPO4 NP, leading to a positive charge on the surface and polarization of H2O molecules with negative fields around the NP; (c) pH > 9 is a basic environment in which OH− ions are adsorbed on the surface of the LaPO4 NP, which leads to a negative surface charge and polarization of H2O molecules with positive fields around the NP; (d) pH > 9 with TA-modified LaPO4 NP causes the adsorption of OH− ions, as well as deprotonation of the carboxyl groups of tartaric acid, which leads to a higher total negative charge of the modified NPs and to a more stable solvate shell due to stronger interactions between NPs surface and aqueous medium.
Scheme 1.
Representation of processes on the surface of LaPO4 NP in different aqueous media: (a) pH = 7 is near the isoelectric point of m-LaPO4, no charge in the surface, the near polar H2O molecules (shown by blue circle and red circles) are randomly positioned around NP; (b) pH < 5 is an acidic environment in which H+ ions are adsorbed on the surface of the LaPO4 NP, leading to a positive charge on the surface and polarization of H2O molecules with negative fields around the NP; (c) pH > 9 is a basic environment in which OH− ions are adsorbed on the surface of the LaPO4 NP, which leads to a negative surface charge and polarization of H2O molecules with positive fields around the NP; (d) pH > 9 with TA-modified LaPO4 NP causes the adsorption of OH− ions, as well as deprotonation of the carboxyl groups of tartaric acid, which leads to a higher total negative charge of the modified NPs and to a more stable solvate shell due to stronger interactions between NPs surface and aqueous medium.
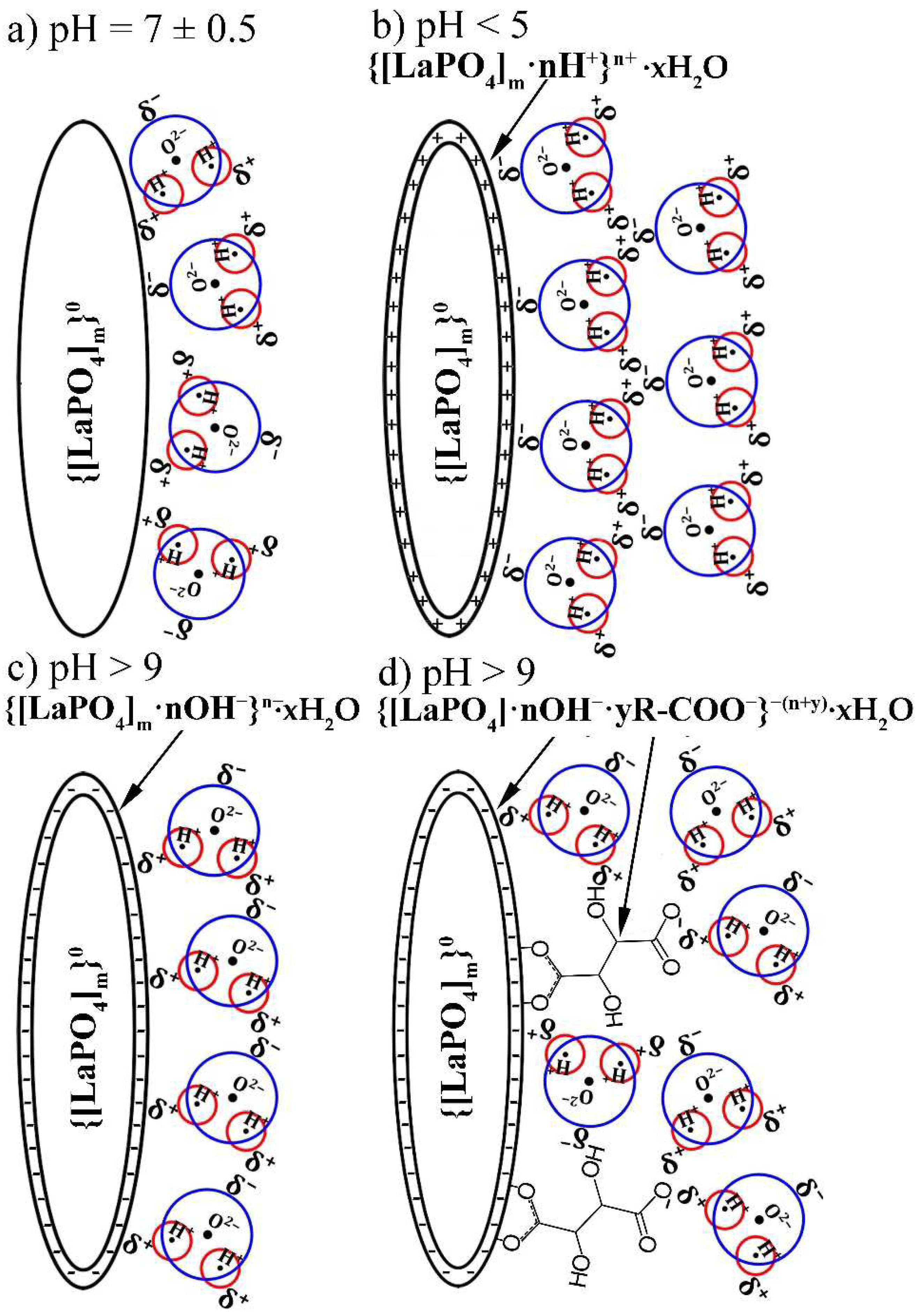

Table 1.
The comparison of morphology parameters with XEOL and CL UV-C intensities for LaPO4: Pr3+ (1 at.%) NPs at room temperature obtained under various synthesis conditions.
Table 1.
The comparison of morphology parameters with XEOL and CL UV-C intensities for LaPO4: Pr3+ (1 at.%) NPs at room temperature obtained under various synthesis conditions.
| Synthesis conditions | TEM | CL | XEOL | |||
| Cat:An | , M | CTA, M | pH | Size, nm | Relative Intensity, arb. units | Relative Intensity, arb. units |
| 1:1.25 | 0.01 | - | 5 | 7 – 15 × 30 – 600 | 1 | 1 |
| 1:2 | 0.005 | 0.027 | 8 | 6.5 ± 1.5 × 31 ± 6 & 49 ± 9 | 0.095 | 0.057 |
| 1:5 | 0.005 | 0.027 | 8 | 5 ± 1 × 18 ± 4 | - | 0.049 |
| 1:1.25 | 0.01 | 0.08 | 9 | 8 ± 1.5 × 46 ± 12 | 0.052 | 0.028 |
| 1:5 | 0.01 | 0.08 | 9 | 4.5 ± 1× 13 ± 4 | 0.009 | 0.024 |
| 1:10 | 0.005 | 0.027 | 8 | 6 ± 2 × 10 ± 4 | - | 5.8 10-3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



