
Submitted:
05 November 2023
Posted:
17 November 2023
You are already at the latest version
Abstract
Gastrointestinal cancers are a specific group of oncological processes in which the location and nature of growth are of key importance for clinical symptoms and prognosis. At the same time, as research shows, they can pose a very serious threat to the patient's life, especially at an advanced stage of development. The type of therapy used depends on the anatomical location of the cancer, its type and degree of progression. One of the modern forms of therapy used to treat gastrointestinal cancers is photodynamic therapy, which, as already mentioned, has been approved for the treatment of esophageal cancer in the United States. Despite the increasingly rapidly developing clinical use of this treatment method, the exact immunological mechanism it induces on cancer cells has not yet been known. This article presents the current findings on the mode of action of dynamic phototherapy on cells of various gastrointestinal cancers. There are most of them about the impact on colorectal cancer. The types of cell death induced by PDT include apoptosis, necrosis, and pyroptosis. The anticancer effect also results from the induction of destruction of tumor vessels and activation of the immune system. Most reports exist on the mechanism of apoptosis induction, in which the mitochondrial pathway is most often emphasized. Photodynamic therapy may also have a beneficial effect on such aspects of cancer as the ability to develop metastases, or contribute to reducing resistance to known pharmacological agents.
Keywords:
gastrointestinal cancers
; PDT
; Anticancer effect
1. Introduction
Cancer is one of the most important health problems worldwide [1]. One of the more diverse groups of cancers are those originating in the gastrointestinal tract [2,3,4]. There is a steady increase in the number of patients with the condition [5,6]. One of the aggressive cancers in this group with a poor prognosis is esophageal cancer [7,8]. Globally, it is a significant cause of cancer deaths, and the five-year survival rate for patients diagnosed with cancer is only 20% [7,9]. Nonspecific symptoms can delay a patient’s presentation to a physician [10]. This implies a delayed diagnosis, resulting in inoperable tumor stage and the presence of metastases (in more than 50% of patients), which makes this cancer have a poor prognosis [10,11]. This cancer is more often diagnosed in men. More than 95% of new cases of this cancer are adenocarcinoma, more common in developed countries, and squamous cell carcinoma, prevalent in non-industrialized countries. There are many risk factors for developing esophageal cancer. Among them, smoking and obesity predispose both to the development of adenocarcinoma and squamous cell carcinoma, while reflux disease predisposes more to adenocarcinoma and alcohol consumption and esophageal achalasia to squamous cell carcinoma [11]. The third most common cause of cancer deaths worldwide is stomach cancer, with a five-year survival rate of about 20%. Risk factors for the development of this disease are socially prevalent (including high salt intake, a diet low in fruits and vegetables, and H. pylori infection) making it the fifth most common cancer worldwide, with about 1.1 million new cases of the disease in 2020 alone [12,13]. Gastric cancer should be treated in a multidisciplinary manner. Surgical resection is the primary treatment method with demonstrated treatment efficacy. This method is being expanded with adjuvant and neoadjuvant therapies for the treatment of locally advanced lesions. In patients found to have metastases, therapy has unsatisfactory results, with a median survival of about 1 year [14,15]. Analyzing the issue of cancers of the gastrointestinal tract, attention should be paid to cancers of its distal part. According to estimates, colorectal cancer, being the third most common cancer globally, occurs in more patients compared to upper gastrointestinal cancers. Its incidence is increasing year by year, mainly in the developed countries of the Western world, accounting for the fourth most common cause of death among cancer patients, with a 5-year survival rate of 57% [16,17]. Factors such as an unhealthy lifestyle (including unhealthy diet, smoking, and excessive alcohol consumption), the presence of chronic diseases, advanced age, and exposure to adverse environmental factors predispose to the development of this cancer [16,18]. It has been established that the development of this neoplasm is the result of multistage progression of adenoma to carcinoma, caused by mutation of the APC gene, and progression of serrated adenoma to carcinoma, the genetic basis of which remains unknown [19]. The worsening prognosis is due to the fact that both the early stages and precancerous adenomatous polyps and advanced cancer can progress asymptomatically [20]. This strongly hinders earlier diagnosis, resulting in the need for screening in people over the age of 50 [21]. In addition, a major factor contributing to the poor prognosis is the fact that up to 20% of patients may present with metastases, most commonly in the liver [19].
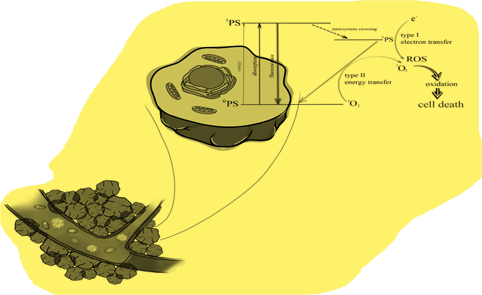
The significant number and growth of new cancer cases worldwide creates the need to discover a new, effective therapeutic method. The search for a modern form of therapy is ongoing, and photodynamic therapy (PDT) may be one of them. This method uses an interaction between a photosensitizer, light of a specific wavelength and oxygen [22]. Numerous studies have demonstrated its effectiveness not only in cancer therapy, but also in the treatment of many non-oncological diseases (e.g., dermatoses) [23,24,25,26]. In the presence of oxygen, a series of reactions of selectively accumulating photosensitizer in tumor tissue, pre-irradiated with light of the appropriate wavelength, results not only in direct cell death through various pathways, but also to the induction of the inflammatory response and the destruction of blood vessels supplying the tumor [27,28]. The phenomenon of photon absorption is key to the excitation of photosensitizers (PS) to a higher energy level and the formation of a triplet state, which in turn is responsible for the production of other reactive oxygen species or singlet oxygen [29]. The delivery of singlet oxygen site-specifically in focused concentrations for hypoxic tumor destruction is a major technological challenge. This review project is, therefore, significant because it presents an integrative approach towards selective generation of singlet oxygen by optical excitation of sensitizer molecules. Figure 1 presents the mechanism of singlet oxygen generation.
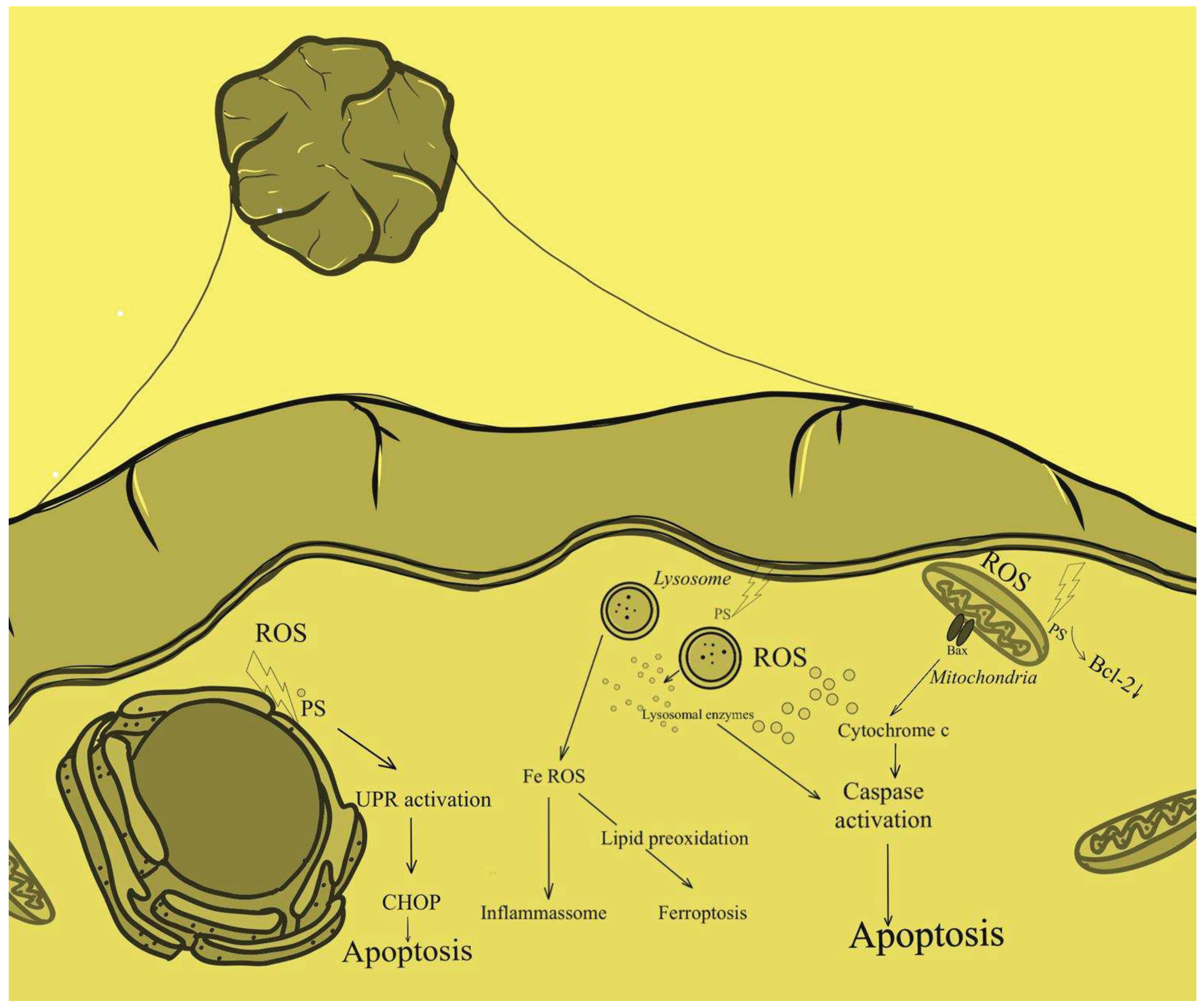
At the same time, PDT-induced damage heals mainly through regeneration rather than scarring [30,31]. Due to the organ-preserving principle of PDT, important structures are preserved with good functional and cosmetic results [30]. In the treatment of gastrointestinal cancers, photodynamic therapy has found application mainly in the treatment of lesions located in the esophagus. In addition, studies have shown that PDT is indicated not only for treating cancer that has already developed, but also for Barett’s esophagus, its precursor state. The origins of photodynamic therapy for esophageal cancer include the palliative treatment of patients with obstructive esophageal cancer. In addition, PDT is being used to treat superficial esophageal cancers characterized by difficulties with endoscopic treatment, and this indication has already been approved for treatment in Japan. In patients in whom local radiotherapy has not achieved the intended therapeutic goals and in whom treatment with other methods may not be sufficiently effective, PDT with second-generation photosensitizers is indicated [32]. The strength of photodynamic therapy is that with its help it is possible to cure early mucosal disease after a single endoscopic procedure [33]. Photodynamic therapy with the photosensitizer porfimer sodium was approved in the United States in 1995 for use in patients with advanced esophageal cancer [34]. Most clinical experiences with gastrointestinal PDT involve patients who are considered to be at low surgical risk, and reported posttreatment follow-up is not only limited but also variable [35]. Despite the demonstrated efficacy of PDT and knowledge of its underlying mechanism of action, reports on the exact mechanisms by which it leads to cell death are still scarce (Figure 2).
2. Materials and methods
The search, which focused on the immunological mechanisms induced by dynamic phototherapy in the treatment of gastrointestinal cancers, was conducted using articles from PubMed, ScienceDirect, Web of Science, and Google Scholar from 1990 to September 2023. The authors of this review worked according to an agreed framework, selecting articles based on their title, language, abstract and access. Duplicate works have been removed. The review included papers describing the immunological view of dynamic phototherapy in the treatment of gastrointestinal cancers, such as esophageal, stomach and colon cancer.
3. Literature review
3.1. Esophageal cancer
The antitumor effect of photodynamic therapy in esophageal cancer is due to a combination of direct cell damage, destruction of tumor blood vessels, and activation of the immune response [36]. However, the exact mechanism of action of PDT has not yet been precisely researched and established. The mechanism of photosensitizer accumulation in cancer cells is also insufficiently understood. One study suggests that in the case of photofrin-II, the mechanism responsible for the accumulation of the photosensitizer in cancer cells is the direct uptake of this compound by the cells, while others negate these conclusions [37,38]. It has been shown that after administration of 5-alpha-aminolevulinic acid, porphyrins accumulate in greater amounts in Barrett’s epithelium and esophageal adenocarcinoma, which results from an imbalance between the activity of porphobilinogen deaminase and ferrochelatase enzymes [39]. PDT causes cell death by apoptosis and necrosis, and induces autophagy and pyroptosis of esophageal cancer cells [40,41,42,43,44]. In the study by Shi Y. et al. PDT using sinoporphyrin sodium (DVDMs-PDT) has been shown to induce apoptosis and autophagy of Eca-109 cells of this tumor. By inducing the formation of reactive oxygen species in Eca-109 esophageal cancer cells, photodynamic therapy leads to the activation of p38MAPK and JNK kinases, and HO-1 heme oxygenase, i.e. proteins responsible, among others, for these types of cellular response to stress [40,45,46,47]. In Eca-109 cells, apoptosis is also induced by ALA-PDT, stopping the cell cycle in the G0/G1 phase and increasing the level of the pro-apoptotic Bax protein, while decreasing the anti-apoptotic Bcl-2 [41,48,49]. Despite the observed increased levels of apoptosis and caspase-3 activity in esophageal adenocarcinoma cells, PDT using Photofrin-II has not been shown to be responsible for these differences [42]. Necrosis of Eca-109 esophageal cancer cells was induced by dynamic phototherapy with hematoporphyrin, while significantly increasing the level of malondialdehyde (a product of peroxidation of omega-6 fatty acids), and not leading to an increase in the expression of caspase-3, a key proenzyme in the apoptosis process [43,50,51]. By inhibiting the last enzyme involved in glycolysis, pyruvate kinase PKM-2, and consequently activating caspase-8 and caspase-9, ultimately leading to the release of gasdermin E (GSDME), PDT can induce pyroptosis of esophageal squamous cell carcinoma cells [44,52]. A reduction in PKM-2 activity was also observed when examining the effect of ALA-PDT on the Warburg effect. It was shown that in esophageal cancer cells, glucose uptake was inhibited within 4 hours after ALA-PDT, however, after 24 hours, a significant increase in the expression of this enzyme and glucose uptake was observed [53]. ALA-PDT enhances the effect of the EGFR inhibitor AG1478 and the PI34 inhibitor LY294002, significantly reducing the expression of EGFR/PI3K and PI3K/AKT proteins, leading to a synergistic reduction in the growth and migration ability of Eca-109 esophageal cancer cells in vitro [54]. PDT increased NF-κB activity and HIF-1α and VEGF gene expression in vitro and in vivo, which may maintain their proliferation, protect against apoptosis, and promote tumor development. Dihydroartemisinin (DHA) may enhance the effect of PDT on esophageal cancer cells [55,56,57]. Yan Jing Li et al. examined the mechanism of action of DHA and showed that it was at least partially due to the deactivation of NF-kB [58].
3.2. Stomach cancer
There is little information available regarding the mechanism of action of dynamic phototherapy on gastric cancer cells. One study showed that the mechanism involved in the specific accumulation of porphyrins in gastric cancer cells is related to the presence of nitric oxide and heme carrier protein-1. In addition, intracellular levels of porphyrins in cells were found to increase after administration of the NO donor following 5-aminolevulinic acid treatment, and heme carrier protein-1was also found to be capable of transporting other porphyrins [59]. It has been shown that through the intrinsic production of free radicals and an increase in the internal level of Ca2+ ions, photodynamic therapy using the photosensitizer DH-II-24 at a low dose induces apoptosis. In the case of high-dose PDT, a massive and prolonged increase in cellular Ca2+ ion levels was observed, resulting in the induction of necrosis process. Moreover, low dose activated caspase-3 [60]. It was observed that ALA-PDT applied to human gastric cancer xenografts in vivo caused apoptosis and necrosis of its cells, and in histological examination, most of the tumor blood vessels were hyperemic [61]. It has been shown that photodynamic therapy via the photosensitizer photofrin in the MKN45 gastric cancer cell line within 15 minutes leads to an increase in the activity of caspase-3 and caspase-9, and chromatin condensation, the reduction of rhodamine 123 uptake begins after 30 minutes and induces after 60 minutes mitochondrial damage and apoptosis [62]. Moreover, due to its ability to activate the immune system, PDT has a specific effect on metastatic lesions [63]. We investigated the effects of PDT on gastric adenocarcinoma cells in patients receiving immune checkpoint inhibitors. Immune cell infiltration increased in tumors after PDT, which is associated with upregulation of the B2M gene, which is lost in tumor cells. TCR analysis revealed specific clonal expansion after PDT in cytotoxic T cells but constriction in Treg cells [64,65].
3.3. Colon cancer
The largest number of reports on the effects of dynamic phototherapy on gastrointestinal cancer are related to colorectal cancer. However, the exact sequence of reactions occurring after PDT has not yet been precisely explained [66]. It has been established that PDT leads to direct killing of cancer cells by singlet oxygen species, and indirect killing of cells through damage to blood vessels and the induced immune response [67]. The effectiveness of PDT itself depends on the concentration of the photosensitizer in the cell, but it has been shown that precise intracellular localization has an additional impact on the way in which the therapy causes damage. Moreover, the degree of differentiation of cancer cells also affects the effectiveness of therapy. It was shown that well-differentiated tumor cells had a better response to PDT using PpIX than less differentiated ones [68]. Research results indicate that the internalization of a photosensitizer may be the result of partitioning, pinocytosis and endocytosis, and the target place of its accumulation in the cell is different for different photosensitizers [69,70]. In the case of PPIX, it was found that the tumor-preferential accumulation of this compound is influenced by the difference in activity between porphobilinogen deaminase and ferrochelatase [71]. Photodynamic therapy causes the death of colorectal cancer cells by apoptosis and necrosis [66,72,73,75,76,77,78,79,80,81,82,83,84,85,86,88,89,90,91]. One of the most important mechanisms of apoptosis triggered by PDT appears to be the mitochondrial pathway. PDT using hexaminolevulinaine as a photosensitizer leads to the loss of mitochondrial membrane potential, the release of cytochrome c from mitochondria into the cytosol and rapid activation of caspase-9 and caspase-3, and consequently to apoptosis of 320 DM colon cancer cells [83]. Identical observations were made in the case of PDT with silicon (IV) phthalocyanine conjugated [77]. It has been shown that the calcium signal plays an important role in the apoptosis of SW480 cells induced by PDT with the photosensitizer ALA [73]. However, the role of this signal may also contribute to the failure of PDT, as it induces the activation of the ERK pathway, which plays a key role in the survival and development of cancer cells, and calcium ions released from the endoplasmic reticulum resulted in an increase in the expression level of the chaperone protein GRP78, which in many cancer models, both in both in vitro and in vivo, it confers a growth advantage and drug resistance to solid tumors [73,74,77,78]. Another study highlighting the involvement of the mitochondrial apoptosis pathway is the study by Guoqing Ouyang et al. showing that PDT with PpIX led to an increase in the expression of the pro-apoptotic protein bax and caspase-3, while the expression of the anti-apoptotic bcl-2 was decreased [82]. It was shown that cell lines with cytosolic or mitochondrial localization of PpIX were characterized by a loss of mitochondrial transmembrane potential, which led to growth arrest [68]. In turn, in the case of dynamic phototherapy with pyropheophorbide methylester (PPME), accumulating in the endoplasmic reticulum/Golgi apparatus and lysosomes, it was not demonstrated that transmitters such as calcium ions, Bid proteins, Bap31, phosphorylated Bcl-2 and caspase-12 were involved. in triggering the release of cytochrome c from mitochondria when provoking apoptosis [61]. Loss of mitochondrial functionality and therefore apoptosis was also induced by PDT using [Ir-b]Cl and [Rh-b]Cl complexes, and PpIX attached to triphenylphosphonium (TPP), which has the ability to target mitochondria [76,86]. Moreover, it is assumed that the leakage of lysosomal protease into the cytosol may also be involved in the induction of apoptosis [85]. The effect of PDT on gene expression, which can contribute to resistance, also appears to be important. A study by H. Abrahamse et al. showed that the level of tumorigenic features of PDT-treated cells affects the nature of proapoptotic and anti-apoptotic gene expression. It was found that apoptosis and up-regulation of 3 genes and 20 genes down-regulation were observed in cells showing increased tumorigenic features. In addition, it was noted that these cells showed an increased risk of resistance to therapy. In contrast, cells showing reduced neoplastic features responded more favorably to photodynamic therapy, as manifested by the observed up-regulation of 16 genes and 22 genes down-regulation [80]. As mentioned earlier, PDT can also cause necrosis of colorectal cancer cells, however, there are no precise reports on the specific mechanism by which this cell death occurs. A study conducted on colon adenocarcinoma cells showed that the predominant mechanism of cell death induced by photodynamic therapy with Foscan was necrosis, and apoptosis was observed, only at the lowest and moderate fluence levels. The use of higher fluences (at identical levels of photocytotoxicity) did not affect caspase-3 activation [92]. It is known that protoporphyrin IX (PpIX) bound to the cell membrane leads to loss of membrane integrity and induces cell necrosis, while 21-selenoporphyrin probably induces necrosis in the endothelial cells of newly formed tumor vessels [68,90]. Necrosis may also be induced by other photosensitizers [89].
So far, it has been established that cellular interactions in the tumor microenvironment also participate in the induction of cancer cell death. It has been shown that due to their plasticity, macrophages residing in or recruited from the tumor can enhance tumor development by promoting tumor cell migration and endothelial stimulation. The increased cytotoxicity of PDT mediated by the production of nitric oxide, interleukin-6 (IL-6) and tumor necrosis factor alpha was in turn achieved in the presence of non-resident macrophages with a strong anti-tumor phenotype [93]. On the other hand, in a study conducted by A. Jalili et al, it was found that inactivation of colon cancer cells after PDT was followed by necrosis and apoptosis and an increase in the expression of some heat shock proteins [88]. It was observed that after photodynamic therapy, immature dendritic cells co-cultured with tumor cells showed an effective ability to phagocytose killed tumor cells. In addition, they acquired features of functional maturity and generated significant amounts of interleukin-12, resulting in increased activity of macrophages, NK cells and monocytes. Moreover, it was observed that these dendritic cells also stimulated the cytotoxic activity of NK cells and T cells, and promoted their migration to lymph nodes [87,88]. Vascular PDT of colon adenocarcinomas expressing the β-galactosidase antigen was shown to completely cure metastasis in mice with antigen-positive tumors, and their T cells were able to recognize the epitope derived from this antigen and specifically destroy the antigen-positive tumor cells. When the antigen was lost, the metastatic lesions were not cured [94]. The effect of PDT on the ability of cells to migrate and metastasize seems to be significant. It is known that PDT using low concentrations (5 μM) of hyperforin and aristophorin not only inhibits cell cycle progression and induces apoptosis, but also reduces the expression of metalloproteinases-2/-9 and cell adhesion potential [75]. Similar observations were found in the case of PDT therapy using m-THPC, which also reduces the colony formation and migration ability of SW480 and SW620 colorectal cancer cells [81]. The possible mechanism of this effect was investigated during PDT involving the photosensitizer chlorin-e6 (Ce6-PDT). It was shown that the therapy led to inhibition of proliferation, almost complete disappearance of pseudopodia, a decrease in the migration ability of SW480 cells and an increase in the expression of F-actin, α-tubulin, β-tubulin, vimentin and E-cadherin. It is assumed that the possible inhibition of cancer cell migration was due to the increased expression of E-cadherin, the loss of which is often observed during metastases, causing the disappearance of pseudopodia and destruction of the cytoskeleton [95,96]. In another study, it was shown that under the influence of Ce6-PDT, the healing and migration rate of SW620 cells was significantly reduced, the pseudopodia of the cells were reduced or disappeared, the original microfilament structure was destroyed and the expression of F-actin was significantly reduced. The Rac1/PAK1/LIMK1/cofilin signaling pathway, which is one of the main pathways through which Rho GTPases regulate microfilaments, was downregulated by Ce6-PDT [97]. Another aspect of PDT’s action is the ability to reduce the resistance of colorectal cancer cells. As shown by M. Luo et al. photodynamic therapy with the photosensitizer chlorin-e6 can inhibit oxaliplatin (L-OHP)-induced autophagy while promoting apoptosis and increasing the expression of procaspase-3 protein, while the combination of Ce-6PDT with L-OHP led to the same effects and an increase in the expression of proapoptotic Bcl -2 and reduced the migration capacity of SW620 colorectal cancer cells [91].
An important aspect of PDT is the possibility of developing tumor resistance to this type of therapy, resulting from the cells’ response to stress, hypoxia, or heterogeneity of photosensitivity uptake by individual tumor cells [98,99,100]. It is known that in response to hypoxia, cells can induce HIF-1α-mediated autophagy, leading to increased colon cancer cell survival and reduced cell death after PDT. By binding to hypoxia-responsive elements in the VMP1 promoter, stabilization of HIF-1α has been shown to significantly increase the VMP1-related autophagy process [101]. An important factor involved in tumorigenesis is hypoxia-inducible factor-1 alpha (HIF-1α), which may also contribute to the development of PDT resistance [102,103]. The resistance phenotype of human colon cancer spheroids to photodynamic therapy using methyl-5-aminoleuvlinic acid may be the result of highly upregulated transcriptional activity of hypoxia-inducible factor-1α. The degree of resistance can be reduced by RNA interference of hypoxia-inducible factor-1 alpha, while inhibition of the MEK/ERK signaling pathway and removal of ROS is responsible for the abrogation of hypoxia-inducible factor-1 alpha regulation [103]. The ineffectiveness of PDT may also be influenced by elevated levels of the heat shock protein Hsp27, since phosphorylation of this protein plays an important role in cytoprotective processes, which consequently increases the resistance of cells to photooxidative damage [104]. By examining the effect of YM155, a small molecule inhibitor of survivin expression, on HT-29 colorectal adenocarcinoma cells resistant to dynamic phototherapy with hypericin, it was shown that a protein belonging to proteins that inhibit apoptosis plays a key role in cancer progression and therapeutic resistance [105]. Further, the interaction of hypericin with the mechanisms of elimination of anticancer drugs by cancer cells is unclear. Moreover, the interaction of hypericin with the mechanisms of elimination of anticancer drugs by cancer cells is unclear. It is known that they are complex. Activities of multidrug resistance-related protein 1 and breast cancer resistance protein were increased in colon cancer cells exposed to hypericin, and cytochrome P450 inhibition led to increased levels of this photosensitizer [106]. On the other hand, examining the contribution of the mechanism of export by p-glycoprotein, it was shown that the use of verapamil, a p-glycoprotein antagonist, can reverse the resistance of HRT-18 colorectal cancer cells to dynamic phototherapy with hematoporphyrin, which suggests a significant role of p-glycoprotein in reducing sensitivity to treatment [107]. One study examined the effect of histone deacetylase (HDAC) inhibitors on the development of resistance to PDT with hypericin by colorectal cancer cells. The combination of histone deacetylase inhibitors with photodynamic therapy using hypericin showed a significant reduction in cancer cell resistance to treatment. The nature, specificity and potency of histone deacetylase inhibition were dependent on the specific inhibitor, suggesting that histone deacetylase may initiate cellular resistance to photodynamic therapy [108]. In colon cancer cells undergoing photodynamic therapy, 1096 long non-coding RNAs were identified, and an interaction between them was found to be responsible for the onset of resistance to PDT [109]. Epigenetic changes may also account for colorectal cancer cell resistance [110]. Stem cells are considered resistant to photodynamic therapy, a potential factor limiting the therapeutic efficacy of P5DT. Due to their ability to self-renew in long-term cycles, they show increased resistance to therapies, which increases the risk of relapse [111]. Photodynamic therapy has demonstrated the ability to increase the sensitivity of colorectal cancer cells to drug treatment, particularly L-oxaliplatin. A complex mechanism for this phenomenon has been identified, including reduced efflux of L-oxaliplatin, inhibition of glutathione S-transferase activity and reduced intracellular glutathione levels [112]. Photodynamic therapy shows synergism with drugs that block programmed death ligand 1, potentially increasing the effectiveness of treatment. Analysis by Z. Yuan and colleagues showed that this combination is able to effectively inhibit the growth of both the primary tumor and distant metastases. In addition, it contributes to the maintenance of long-term host immune memory, which counteracts cancer recurrence. The mechanism of this synergy has been shown to include induction of cell apoptosis and activation of the systemic immune response, which can be further supported by blockade with drugs that block programmed death ligand 1 [113]. It was found that the efficacy of photodynamic therapy against colon cancer cells can be enhanced by stimulating the apoptosis process using the specific 5-lipoxygenase inhibitor MK-886. Further analyzes of individual ROS groups revealed the effect of increasing MK-886 concentration on peroxide accumulation, which was accompanied by a decrease in the level of hydrogen peroxide in cells. A clonogenicity test revealed impaired colony formation when both agents were combined compared to MK-886 or PDT alone [114].
4. Conclusions
Cancer treatment using dynamic phototherapy poses many challenges. One of them is the possibility of cancer cells becoming resistant to this type of therapy. This article presents evidence that mechanisms such as removal of photosensitizer from cancer cells, induction of autophagy in response to damage, natural increased resistance of tumor stem cells and, finally, increased presence of various cytoprotective proteins are involved in this process. Interactions between tumor cells and other cells are also an important aspect, as they may contribute to weakening the effect of PDT and even to accelerating tumor development. Further research is necessary to determine the exact mechanisms of action of dynamic phototherapy on gastrointestinal cancer cells, taking into account the type of photosensitizers, classification of cancers and their stage of advancement. Understanding the precise impact of PDT on the treatment of this disease may help discover new photosensitizers, their transport mechanism, or determine the appropriate, most effective therapeutic regimens. It also seems extremely promising to investigate the mechanisms by which photodynamic therapy can lead to the activation of the immune system and, as a result, to the treatment of metastases. Moreover, based on this review, it can be concluded that a thorough examination of the mechanisms responsible for cellular resistance to dynamic phototherapy may contribute to the discovery of new therapeutic agents that can inhibit this resistance. To sum up, the immunological mechanisms of the action of dynamic phototherapy on gastrointestinal cancer cells are still insufficiently understood, and their detailed examination may contribute to increasing the effectiveness of this therapy.
Author Contributions
D.A.; P.W.; K.D. and D.B.- A.; Conceptualization, D.A.; P.W.; K.D. and D.B.- A.; methodology, D.A.; P.W.; K.D. and D.B.- A.; software, D.A.; P.W.; K.D. and D.B.- A.; validation, D.A.; P.W.; K.D. and D.B.- A.; formal analysis, D.A.; P.W.; K.D. and D.B.- A.; investigation, D.A.; P.W.; K.D. and D.B.- A.; resources, D.A.; P.W.; K.D. and D.B.- A.; data curation, D.A.; P.W.; K.D. and D.B.- A.; writing—original draft preparation, D.A.; P.W.; K.D. and D.B.- A.; writing—review and editing, D.A.; P.W.; K.D. and D.B.- A.; visualization, D.A.; P.W.; K.D. and D.B.- A.; supervision, D.A.; project administration, D.A.; funding acquisition, D.A.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
MDPI Research Data Policies at https://www.mdpi.com/ethics.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mattiuzzi C, Lippi G. Current Cancer Epidemiology. J Epidemiol Glob Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev 2014, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Lander S, Lander E, Gibson MK. Esophageal Cancer: Overview, Risk Factors, and Reasons for the Rise. Curr Gastroenterol Rep. 9 October 2023.
- Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ, Watanabe T. Colorectal cancer. Nat Rev Dis Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Tong Y: Gao H, Qi Q, Liu X, Li J, Gao J, Li P, Wang Y, Du L, Wang C. High fat diet, gut microbiome and gastrointestinal cancer. Theranostics 2021, 11, 5889–5910. [Google Scholar] [CrossRef] [PubMed]
- Yumo Xie, Lishuo Shi, Xiaosheng He, Yanxin Luo, Gastrointestinal cancers in China, the USA, and Europe. Gastroenterology Report 2021, 9, 91–104. [CrossRef] [PubMed]
- Abbas G, Krasna M. Overview of esophageal cancer. Ann Cardiothorac Surg 2017, 6, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Huang FL, Yu SJ. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J Surg 2018, 41, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Li J, Xu J, Zheng Y, Gao Y, He S, Li H, Zou K, Li N, Tian J, Chen W, He J. Esophageal cancer: Epidemiology, risk factors and screening. Chin J Cancer Res 2021, 33, 535–547. [Google Scholar] [CrossRef]
- Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med 2003, 349, 2241–2252. [Google Scholar] [CrossRef]
- Short MW, Burgers KG, Fry VT. Esophageal Cancer. Am Fam Physician 2017, 95, 22–28. [Google Scholar]
- Gastric cancer Elizabeth C Smyth, MD Magnus Nilsson, PhD Heike I Grabsch, PhD Nicole CT van Grieken, PhD Florian Lordick, MD.
- Ilic M, Ilic I. Epidemiology of stomach cancer. World J Gastroenterol 2022, 28, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Vincenzo Catalano, Roberto Labianca, Giordano D. Beretta, Gemma Gatta, Filippo de Braud, Eric Van Cutsem, Gastric cancer. Critical Reviews in Oncology/Hematology 2009, 71, 127–164. [Google Scholar]
- Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int J Mol Sci 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht HA, Hassanipour S, Shojaie L, Vali M, Ghaffari-Fam S, Ghelichi-Ghojogh M, Maleki Z, Arab-Zozani M, Abdzadeh E, Delam H, Salehiniya H, Shafiee M, Mohammadi S. Survival Rate of Colorectal Cancer in Eastern Mediterranean Region Countries: A Systematic Review and Meta-Analysis. Cancer Control 2020, 27, 1073274820964146. [Google Scholar]
- Haraldsdottir S, Einarsdottir HM, Smaradottir A, Gunnlaugsson A, Halfdanarson TR. Krabbamein í ristli og endaþarmi [Colorectal cancer - review]. Laeknabladid 2014, 100, 75–82. [Google Scholar]
- Cappell, M.S. Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin North Am 2008, 37, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, A., Levin, T.R. Current and future colorectal cancer screening strategies. Nat Rev Gastroenterol Hepatol 2022, 19, 521–531.
- Rkein AM, Ozog DM. Photodynamic therapy. Dermatol Clin 2014, 32, 415–425. [Google Scholar] [CrossRef]
- Chiba, K.; Aihara, Y.; Oda, Y.; Fukui, A.; Tsuzuk, S.; Saito, T.; Nitta, M.; Muragaki, Y.; Kawamata, T. Photodynamic therapy for malignant brain tumors in children and young adolescents. Front. Oncol. 2022, 12, 957267. [Google Scholar] [CrossRef]
- Aldosari, L.I.N.; Hassan, S.A.B.; Alshadidi, A.A.F.; Rangaiah, G.C.; Divakar, D.D. Short-term influence of antimicrobial photodynamic therapy as an adjuvant to mechanical debridement in reducing soft-tissue inflammation and subgingival yeasts colonization in patients with peri-implant mucositis. Photodiagn. Photodyn. Ther. 2023, 42, 103320. [Google Scholar] [CrossRef] [PubMed]
- Gil-Pallares, P.; Navarro-Bielsa, A.; Almenara-Blasco, M.; Gracia-Cazaña, T.; Gilaberte, Y. Photodynamic Therapy, a successful treatment for granular parakeratosis. Photodiagn. Photodyn. Ther. 2023, 42, 103562. [Google Scholar] [CrossRef]
- Alexiades-Armenakas, M. Laser-mediated photodynamic therapy. Clin. Dermatol. 2006, 24, 16–25. [Google Scholar] [CrossRef]
- Kwiatkowski S, Knap B, Przystupski D, Saczko J, Kędzierska E, Knap-Czop K, Kotlińska J, Michel O, Kotowski K, Kulbacka J. Photodynamic therapy - mechanisms, photosensitizers and combinations. Biomed Pharmacother 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J. Photodynamic therapy of cancer: an update. CA Cancer J Clin 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Juarranz A, Jaén P, Sanz-Rodríguez F, Cuevas J, González S. Photodynamic therapy of cancer. Basic principles and applications. Clin Transl Oncol 2008, 10, 148–154. [Google Scholar]
- Alexander, C. Kübler, Photodynamic therapy. Medical Laser Application 2005, 20, 37–45. [Google Scholar]
- Triesscheijn M, Baas P, Schellens JH, Stewart FA. Photodynamic therapy in oncology. Oncologist 2006, 11, 1034–1044. [Google Scholar] [CrossRef]
- Yano T, Wang KK. Photodynamic Therapy for Gastrointestinal Cancer. Photochem Photobiol 2020, 96, 517–523. [Google Scholar] [CrossRef]
- Barr, H. Photodynamic therapy in gastrointestinal cancer: a realistic option? Drugs Aging 2000, 16, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Qumseya BJ, David W, Wolfsen HC. Photodynamic Therapy for Barrett’s Esophagus and Esophageal Carcinoma. Clin Endosc 2013, 46, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Webber J, Herman M, Kessel D, Fromm D. Photodynamic treatment of neoplastic lesions of the gastrointestinal tract. Recent advances in techniques and results. Langenbecks Arch Surg 2000, 385, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Filip AG, Clichici S, Daicoviciu D, Olteanu D, Mureşan A, Dreve S. Photodynamic therapy--indications and limits in malignant tumors treatment. Rom J Intern Med. 2008, 46, 285–293. [Google Scholar]
- Gao S, Liang S, Ding K, Qu Z, Wang Y, Feng X. Specific cellular accumulation of photofrin-II in EC cells promotes photodynamic treatment efficacy in esophageal cancer. Photodiagnosis Photodyn Ther 2016, 14, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Gao SG, Wang LD, Feng XS, Qu ZF, Shan TY, Xie XH. [Absorption and elimination of photofrin-II in human immortalization esophageal epithelial cell line SHEE and its malignant transformation cell line SHEEC]. Ai Zheng 2009, 28, 1248–1254. (In Chinese) [Google Scholar]
- Hinnen P, de Rooij FW, van Velthuysen ML, Edixhoven A, van Hillegersberg R, Tilanus HW, Wilson JH, Siersema PD. Biochemical basis of 5-aminolaevulinic acid-induced protoporphyrin IX accumulation: a study in patients with (pre)malignant lesions of the oesophagus. Br J Cancer 1998, 78, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Shi Y, Zhang B, Feng X, Qu F, Wang S, Wu L, Wang X, Liu Q, Wang P, Zhang K. Apoptosis and autophagy induced by DVDMs-PDT on human esophageal cancer Eca-109 cells. Photodiagnosis Photodyn Ther 2018, 24, 198–205. [Google Scholar] [CrossRef]
- Chen X, Zhao P, Chen F, Li L, Luo R. Effect and mechanism of 5-aminolevulinic acid-mediated photodynamic therapy in esophageal cancer. Lasers Med Sci 2011, 26, 69–78. [Google Scholar] [CrossRef]
- Apoptosis associated with esophageal adenocarcinoma: influence of photodynamic therapy.
- Chen XH, Luo RC, Li LB, Ding XM, Lv CW, Zhou XP, Yan X. [Mechanism of photodynamic therapy against human esophageal carcinoma xenografts in nude mice]. Nan Fang Yi Ke Da Xue Xue Bao 2009, 29, 2222–2224. (In Chinese) [Google Scholar]
- Li L, Song D, Qi L, Jiang M, Wu Y, Gan J, Cao K, Li Y, Bai Y, Zheng T. Photodynamic therapy induces human esophageal carcinoma cell pyroptosis by targeting the PKM2/caspase-8/caspase-3/GSDME axis. Cancer Lett 2021, 520, 143–159, Erratum in: Cancer Lett 2022, 525, 203–205. [Google Scholar] [CrossRef]
- Xue Q, Wang P, Wang X, Zhang K, Liu Q. Targeted inhibition of p38MAPK-enhanced autophagy in SW620 cells resistant to photodynamic therapy-induced apoptosis. Lasers Med Sci 2015, 30, 1967–1975. [Google Scholar] [CrossRef]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Puente Y, Zapata-Benavides P, Tari AM, López-Berestein G. Bcl-2-related antisense therapy. Semin Oncol 2002, 29 (Suppl. 11), 71–76.
- Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells". J. Biol. Chem. 2005, 280, 10491–10500. [Google Scholar] [CrossRef]
- Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014, 2014, 360438. [Google Scholar]
- Asadi M, Taghizadeh S, Kaviani E, Vakili O, Taheri-Anganeh M, Tahamtan M, Savardashtaki A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol Appl Biochem 2022, 69, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. The Oncologist 2004, 9 (Suppl. 5), 4–9. [Google Scholar] [CrossRef]
- Gan J, Li S, Meng Y, Liao Y, Jiang M, Qi L, Li Y, Bai Y. The influence of photodynamic therapy on the Warburg effect in esophageal cancer cells. Lasers Med Sci 2020, 35, 1741–1750. [Google Scholar] [CrossRef]
- Zhang X, Cai L, He J, Li X, Li L, Chen X, Lan P. Influence and mechanism of 5-aminolevulinic acid-photodynamic therapy on the metastasis of esophageal carcinoma. Photodiagnosis Photodyn Ther 2017, 20, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Li Y, Sui H, Jiang C, Li S, Han Y, Huang P, Du X, Du J, Bai Y. Dihydroartemisinin Increases the Sensitivity of Photodynamic Therapy Via NF-κB/HIF-1α/VEGF Pathway in Esophageal Cancer Cell in vitro and in vivo. Cell Physiol Biochem. 2018, 48, 2035–2045. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A. Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode. Cancer Biology & Medicine 2017, 14, 254–270. [Google Scholar]
- Zhou JH, Du XX, Jia de X, Wu CL, Huang P, Han Y, Sui H, Wei XL, Liu L, Yuan HH, Zhang TT, Zhang WJ, Xie R, Lang XH, Liu T, Jiang CL, Wang LY, Bai YX. Dihydroartemisinin accentuates the anti-tumor effects of photodynamic therapy via inactivation of NF-κB in Eca109 and Ec9706 esophageal cancer cells. Cell Physiol Biochem. 2014, 33, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa H, Ito H, Terasaki M, Matano D, Taninaka A, Shigekawa H, Matsui H. Nitric oxide regulates the expression of heme carrier protein-1 via hypoxia inducible factor-1α stabilization. PLoS One 2019, 14, e0222074. [Google Scholar]
- Yoo JO, Lim YC, Kim YM, Ha KS. Differential cytotoxic responses to low- and high-dose photodynamic therapy in human gastric and bladder cancer cells. J Cell Biochem 2011, 112, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Zhou GJ, Huang ZH, Yu JL, Li Z, Ding LS. [Effect of 5-aminolevulinic acid-mediated photodynamic therapy on human gastric cancer xenografts in nude mice in vivo]. Zhonghua Wei Chang Wai Ke Za Zhi 2008, 11, 580–583. (In Chinese) [Google Scholar]
- Takahira K, Sano M, Arai H, Hanai H. Apoptosis of gastric cancer cell line MKN45 by photodynamic treatment with photofrin. Lasers Med Sci. 2004, 19, 89–94. [Google Scholar] [CrossRef]
- Wang H, Ewetse MP, Ma C, Pu W, Xu B, He P, Wang Y, Zhu J, Chen H. The "Light Knife" for Gastric Cancer: Photodynamic Therapy. Pharmaceutics 2022, 15, 101. [Google Scholar] [CrossRef]
- Yu Y, Xu B, Xiang L, Ding T, Wang N, Yu R, Gu B, Gao L, Maswikiti EP, Wang Y, Li H, Bai Y, Zheng P, Ma C, Wang B, Wang X, Zhang T, Chen H. Photodynamic therapy improves the outcome of immune checkpoint inhibitors via remodelling anti-tumour immunity in patients with gastric cancer. Gastric Cancer 2023, 26, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Zhao Y, Cao Y, Chen Y, Wu L, Hang H, Jiang C, Zhou X. B2M gene expression shapes the immune landscape of lung adenocarcinoma and determines the response to immunotherapy. Immunology 2021, 164, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Xue Q, Wang X, Wang P, Zhang K, Liu Q. Role of p38MAPK in apoptosis and autophagy responses to photodynamic therapy with Chlorin e6. Photodiagnosis Photodyn Ther 2015, 12, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Peng CL, Lin HC, Chiang WL, Shih YH, Chiang PF, Luo TY, Cheng CC, Shieh MJ. Anti-angiogenic treatment (Bevacizumab) improves the responsiveness of photodynamic therapy in colorectal cancer. Photodiagnosis Photodyn Ther 2018, 23, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Krieg RC, Messmann H, Schlottmann K, Endlicher E, Seeger S, Schölmerich J, Knuechel R. Intracellular localization is a cofactor for the phototoxicity of protoporphyrin IX in the gastrointestinal tract: in vitro study. Photochem Photobiol 2003, 78, 393–399. [Google Scholar] [CrossRef]
- Siboni G, Weitman H, Freeman D, Mazur Y, Malik Z, Ehrenberg B. The correlation between hydrophilicity of hypericins and helianthrone: internalization mechanisms, subcellular distribution and photodynamic action in colon carcinoma cells. Photochem Photobiol Sci 2002, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Orenstein A, Kostenich G, Roitman L, Shechtman Y, Kopolovic Y, Ehrenberg B, Malik Z. A comparative study of tissue distribution and photodynamic therapy selectivity of chlorin e6, Photofrin II and ALA-induced protoporphyrin IX in a colon carcinoma model. Br J Cancer 1996, 73, 937–944. [Google Scholar] [CrossRef]
- Krieg RC, Messmann H, Rauch J, Seeger S, Knuechel R. Metabolic characterization of tumor cell-specific protoporphyrin IX accumulation after exposure to 5-aminolevulinic acid in human colonic cells. Photochem Photobiol 2002, 76, 518–525. [Google Scholar] [CrossRef]
- Ibarra LE, Porcal GV, Macor LP, Ponzio RA, Spada RM, Lorente C, Chesta CA, Rivarola VA, Palacios RE. Metallated porphyrin-doped conjugated polymer nanoparticles for efficient photodynamic therapy of brain and colorectal tumor cells. Nanomedicine (Lond) 2018, 13, 605–624. [Google Scholar] [CrossRef]
- Zheng JH, Shi D, Zhao Y, Chen ZL. [Role of calcium signal in apoptosis and protective mechanism of colon cancer cell line SW480 in response to 5-aminolevulinic acid-photodynamic therapy]. Ai Zheng 2006, 25, 683–688. (In Chinese) [Google Scholar]
- Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 2020, 19, 1997–2007. [Google Scholar]
- Šemeláková M, Mikeš J, Jendželovský R, Fedoročko P. The pro-apoptotic and anti-invasive effects of hypericin-mediated photodynamic therapy are enhanced by hyperforin or aristoforin in HT-29 colon adenocarcinoma cells. J Photochem Photobiol B 2012, 117, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Xu Y, Yao Y, Wang L, Chen H, Tan N. Hyaluronic Acid Coated Liposomes Co-Delivery of Natural Cyclic Peptide RA-XII and Mitochondrial Targeted Photosensitizer for Highly Selective Precise Combined Treatment of Colon Cancer. Int J Nanomedicine 2021, 16, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Chan CM, Lo PC, Yeung SL, Ng DK, Fong WP. Photodynamic activity of a glucoconjugated silicon(IV) phthalocyanine on human colon adenocarcinoma. Cancer Biol Ther 2010, 10, 126–134. [Google Scholar] [CrossRef]
- Risheng Ye, Yi Zhang, Amy S. Lee ; The ER Chaperone GRP78 and Cancer, Protein Misfolding Disorders. A Trip into the ER 2009, 1, 47.
- Gariboldi MB, Ravizza R, Baranyai P, Caruso E, Banfi S, Meschini S, Monti E. Photodynamic effects of novel 5,15-diaryl-tetrapyrrole derivatives on human colon carcinoma cells. Bioorg Med Chem 2009, 17, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse H, Houreld NN. Genetic Aberrations Associated with Photodynamic Therapy in Colorectal Cancer Cells. Int J Mol Sci 2019, 20, 3254. [Google Scholar] [CrossRef] [PubMed]
- Abdulrehman G, Xv K, Li Y, Kang L. Effects of meta-tetrahydroxyphenylchlorin photodynamic therapy on isogenic colorectal cancer SW480 and SW620 cells with different metastatic potentials. Lasers Med Sci 2018, 33, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Ouyang G, Liu Z, Xiong L, Chen X, Li Q, Huang H, Lin L, Miao X, Ma L, Chen W, Wen Y. [Role of PpIX-based photodynamic therapy in promoting the damage and apoptosis of colorectal cancer cell and its mechanisms]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2017, 42, 874–881. (In Chinese) [Google Scholar]
- Shahzidi S, Stokke T, Soltani H, Nesland JM, Peng Q. Induction of apoptosis by hexaminolevulinate-mediated photodynamic therapy in human colon carcinoma cell line 320DM. J Environ Pathol Toxicol Oncol. 2006, 25, 159–171. [Google Scholar] [CrossRef]
- Ali SM, Olivo M. Bio-distribution and subcellular localization of Hypericin and its role in PDT induced apoptosis in cancer cells. Int J Oncol 2002, 21, 531–540. [Google Scholar]
- Matroule JY, Carthy CM, Granville DJ, Jolois O, Hunt DW, Piette J. Mechanism of colon cancer cell apoptosis mediated by pyropheophorbide-a methylester photosensitization. Oncogene 2001, 20, 4070–4084. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Arnaiz C, Acuña MI, Busto N, Echevarría I, Martínez-Alonso M, Espino G, García B, Domínguez F. Thiabendazole-based Rh(III) and Ir(III) biscyclometallated complexes with mitochondria-targeted anticancer activity and metal-sensitive photodynamic activity. Eur J Med Chem 2018, 157, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Wang X, Zhang A, Qiu X, Yang K, Zhou H. The IL-12 family cytokines in fish: Molecular structure, expression profile and function. Dev Comp Immunol 2023, 141, 104643. [Google Scholar] [CrossRef] [PubMed]
- Jalili A, Makowski M, Switaj T, Nowis D, Wilczynski GM, Wilczek E, Chorazy-Massalska M, Radzikowska A, Maslinski W, Biały L, Sienko J, Sieron A, Adamek M, Basak G, Mróz P, Krasnodebski IW, Jakóbisiak M, Gołab J. Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin Cancer Res 2004, 10, 4498–4508. [Google Scholar] [CrossRef]
- Cogno IS, Gilardi P, Comini L, Núñez-Montoya SC, Cabrera JL, Rivarola VA. Natural photosensitizers in photodynamic therapy: In vitro activity against monolayers and spheroids of human colorectal adenocarcinoma SW480 cells. Photodiagnosis Photodyn Ther 2020, 31, 101852. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska E, Ziółkowski P, Pacholska E, Latos-Grazyński L, Chmielewski P, Radzikowski C. The new sensitizing agents for photodynamic therapy: 21-selenaporphyrin and 21-thiaporphyrin. Anticancer Res 1997, 17, 3313–3319. [Google Scholar]
- TLuo M, Ji J, Yang K, Li H, Kang L. The role of autophagy in the treatment of colon cancer by chlorin e6 photodynamic therapy combined with oxaliplatin. Photodiagnosis Photodyn Ther 2022, 40, 103082. [Google Scholar] [CrossRef]
- Necrotic and apoptotic features of cell death in response to Foscan photosensitization of HT29 monolayer and multicell spheroids.
- Pansa MF, Lamberti MJ, Cogno IS, Correa SG, Rumie Vittar NB, Rivarola VA. Contribution of resident and recruited macrophages to the photodynamic intervention of colorectal tumor microenvironment. Tumour Biol 2016, 37, 541–552. [Google Scholar] [CrossRef]
- Mroz P, Szokalska A, Wu MX, Hamblin MR. Photodynamic therapy of tumors can lead to development of systemic antigen-specific immune response. PLoS One 2010, 5, e15194. [Google Scholar]
- Na TY, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci U S A 2020, 117, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Ma H, Yang K, Li H, Luo M, Wufuer R, Kang L. Photodynamic effect of chlorin e6 on cytoskeleton protein of human colon cancer SW480 cells. Photodiagnosis Photodyn Ther 2021, 33, 102201. [Google Scholar] [CrossRef] [PubMed]
- Wufuer R, Ma HX, Luo MY, Xu KY, Kang L. Downregulation of Rac1/PAK1/LIMK1/cofilin signaling pathway in colon cancer SW620 cells treated with Chlorin e6 photodynamic therapy. Photodiagnosis Photodyn Ther 2021, 33, 102143. [Google Scholar] [CrossRef] [PubMed]
- Dobre M, Boscencu R, Neagoe IV, Surcel M, Milanesi E, Manda G. Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells. Pharmaceutics 2021, 13, 1032. [Google Scholar] [CrossRef] [PubMed]
- Yuan Z, Liu C, Sun Y, Li Y, Wu H, Ma S, Shang J, Zhan Y, Yin P, Gao F. Bufalin exacerbates Photodynamic therapy of colorectal cancer by targeting SRC-3/HIF-1α pathway. Int J Pharm 2022, 624, 122018. [Google Scholar] [CrossRef] [PubMed]
- West CM, Moore JV. Mechanisms behind the resistance of spheroids to photodynamic treatment: a flow cytometry study. Photochem Photobiol 1992, 55, 425–430. [Google Scholar] [CrossRef]
- Rodríguez ME, Catrinacio C, Ropolo A, Rivarola VA, Vaccaro MI. A novel HIF-1α/VMP1-autophagic pathway induces resistance to photodynamic therapy in colon cancer cells. Photochem Photobiol Sci 2017, 16, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M, Ramezani F. Up-down regulation of HIF-1α in cancer progression. Gene 2021, 798, 145796. [Google Scholar] [CrossRef] [PubMed]
- Lamberti MJ, Pansa MF, Vera RE, Fernández-Zapico ME, Rumie Vittar NB, Rivarola VA. Transcriptional activation of HIF-1 by a ROS-ERK axis underlies the resistance to photodynamic therapy. PLoS One 2017, 12, e0177801. [Google Scholar]
- Wang HP, Hanlon JG, Rainbow AJ, Espiritu M, Singh G. Up-regulation of Hsp27 plays a role in the resistance of human colon carcinoma HT29 cells to photooxidative stress. Photochem Photobiol 2002, 76, 98–104. [Google Scholar] [CrossRef]
- Gyurászová K, Mikeš J, Halaburková A, Jendželovský R, Fedoročko P. YM155, a small molecule inhibitor of survivin expression, sensitizes cancer cells to hypericin-mediated photodynamic therapy. Photochem Photobiol Sci 2016, 15, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Jendzelovský R, Mikes J, Koval’ J, Soucek K, Procházková J, Kello M, Sacková V, Hofmanová J, Kozubík A, Fedorocko P. Drug efflux transporters, MRP1 and BCRP, affect the outcome of hypericin-mediated photodynamic therapy in HT-29 adenocarcinoma cells. Photochem Photobiol Sci 2009, 8, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Purkiss SF, Grahn MF, Williams NS. Haematoporphyrin derivative--photodynamic therapy of colorectal carcinoma, sensitized using verapamil and adriamycin. Surg Oncol 1996, 5, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Halaburková A, Jendželovský R, Kovaľ J, Herceg Z, Fedoročko P, Ghantous A. Histone deacetylase inhibitors potentiate photodynamic therapy in colon cancer cells marked by chromatin-mediated epigenetic regulation of CDKN1A. Clin Epigenetics 2017, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Liu K, Yao H, Wen Y, Zhao H, Zhou N, Lei S, Xiong L. Functional role of a long non-coding RNA LIFR-AS1/miR-29a/TNFAIP3 axis in colorectal cancer resistance to pohotodynamic therapy. Biochim Biophys Acta Mol Basis Dis 2018, 1864 Pt B, 2871–2880. [CrossRef] [PubMed]
- Luo M, Yang X, Chen HN, Nice EC, Huang C. Drug resistance in colorectal cancer: An epigenetic overview. Biochim Biophys Acta Rev Cancer 2021, 1876, 188623. [Google Scholar]
- Hodgkinson N, Kruger CA, Abrahamse H. Targeted photodynamic therapy as potential treatment modality for the eradication of colon cancer and colon cancer stem cells. Tumour Biol 2017, 39, 1010428317734691. [Google Scholar]
- Lin S, Lei K, Du W, Yang L, Shi H, Gao Y, Yin P, Liang X, Liu J. Enhancement of oxaliplatin sensitivity in human colorectal cancer by hypericin mediated photodynamic therapy via ROS-related mechanism. Int J Biochem Cell Biol 2016, 71, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Yuan Z, Fan G, Wu H, Liu C, Zhan Y, Qiu Y, Shou C, Gao F, Zhang J, Yin P, Xu K. Photodynamic therapy synergizes with PD-L1 checkpoint blockade for immunotherapy of CRC by multifunctional nanoparticles. Mol Ther 2021, 29, 2931–2948. [Google Scholar] [CrossRef]
- Kleban J, Mikes J, Horváth V, Sacková V, Hofmanová J, Kozubík A, Fedorocko P. Mechanisms involved in the cell cycle and apoptosis of HT-29 cells pre-treated with MK-886 prior to photodynamic therapy with hypericin. J Photochem Photobiol B 2008, 93, 108–118. [Google Scholar] [CrossRef]
Figure 1.
The mechanism of PDT.
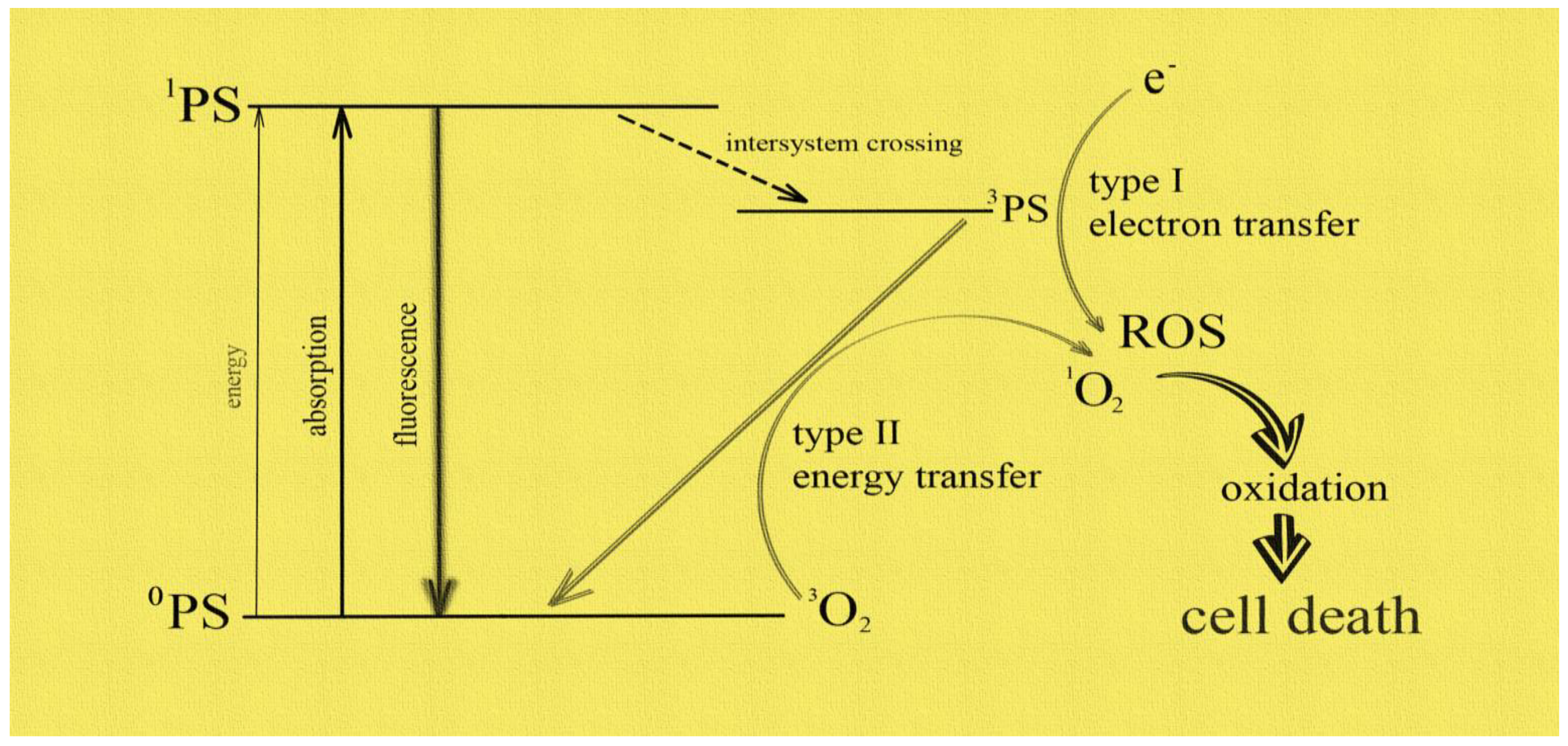
Figure 2.
Cellular mechanism of PDT.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



