
Submitted:
16 November 2023
Posted:
17 November 2023
You are already at the latest version
Abstract
In the modern era of genomic research, the scientific community is witnessing an explosive growth in the volume of published findings. While this abundance of data offers invaluable insights, it also places a pressing responsibility on genetic professionals and researchers to stay informed about the latest findings and their clinical significance. Genomic variant interpretation is currently facing a challenge in identifying the most up-to-date and relevant scientific papers, while also extracting meaningful information to accelerate the process from clinical assessment to reporting. Computer-aided literature search and summarization can play a pivotal role in this context. By synthesizing complex genomic findings into concise, interpretable summaries, this approach facilitates the translation of extensive genomic datasets into clinically relevant insights. To bridge this gap, we present VarChat (varchat.engenome.com), an innovative tool based on generative AI, developed to find and summarize the fragmented scientific literature associated with genomic variants into brief yet informative texts.VarChat provides users with a concise description of specific genetic variants, detailing their impact on related proteins and possible effects on human health. Additionally, VarChat offers direct links to related scientific trustable sources, and encourages deeper research. Availability: VarChat is freely available at varchat.engenome.com
Keywords:
Genomics
; Variant Interpretation
; Generative AI
; Genome analysis
; Rare Diseases
; Bioinformatics
1. Introduction
The rise of genomics and personalized medicine is generating a tremendous amount of data, with genomic variants as a primary research focus. These variants can be linked to disease susceptibility, drug responses, and other phenotypic outcomes1, and the vast majority is well-documented in scientific papers.
Not just the identification of these variants from sequencing data, but also the effective curation and interpretation of this large amount of information may be challenging, and several methods have been proposed to automate this process2,3. Many efforts have been made to condense this knowledge in dedicated databases and publicly available resources, like ClinVar, gnomAD4,5, and OMIM6.
The scientific literature, with its rich repository of knowledge, offers a wealth of insights into these genomic variants. However, the enormous volume of publications, coupled with the not yet perfect standardization in applying a nomenclature describing genomic variants, makes manual curation challenging7. Furthermore, the enhancement of genomic variant discovery relies on the curation process, which is significantly improved by accessing not only the abstracts but also the full texts and supplementary data of scientific articles8–10.
Acknowledging this challenge, several tools have been developed to support genomic variant research in the scientific literature.
Among these, LitVar11,12 is a semantic search engine designed specifically for linking genomic variant data in PubMed and PMC. By employing advanced text mining techniques, LitVar not only retrieves standardized variant information but also visualizes the relationships between variants and other associated entities, such as diseases and chemicals/drugs.
Variomes13 is another tool designed as a high-recall search engine, focusing on aiding the curation of genomic variants. Different parameters allow for personalizing the search by specifying the timeline and adding keywords for papers re-ranking.
Finally, SynVar14 has been developed to ensure effective retrieval of variant-containing documents, providing descriptions in both standard and non-standard formats found in the literature.
A significant limitation of these approaches is their inability to synthesize variants’ information into concise, human-readable texts that are suitable for clinical reports. Many systems prioritize data aggregation and categorization but fall short in generating comprehensive yet succinct textual interpretations.
Conversely, Large Language Models (LLM) based on generative AI, such as the widely recognized chatGPT (www.openai.com), Bard (google AI), Falcon (www.tii.ae) and Claude 2 (Anthropic, www.claude.ai), have the innate capability of comprehension and summarization of complex texts. These models have become integral to solutions widely used in our daily life and have demonstrated exceptional performance across multiple Natural Language Processing tasks, showcasing strong comprehension and reasoning abilities15,16.
The balance between providing detailed and accurate genomic insights and ensuring readability for a diverse audience, including those without deep genomic and computational expertise, is a challenge yet to be fully addressed.
For this purpose, we introduce VarChat, the first generative AI based tool designed to search and summarize scientific literature about a human genomic variant and provide a concise text explaining the variant, insights from existing research, and associated references.
VarChat is freely available at varchat.engenome.com.
2. Methods
VarChat requires as input genomic variants coordinates according to HGVS nomenclature17 together with gene symbols. For every queried variant, VarChat produces concise and coherent summaries through an LLM model, enabling researchers and clinicians to capture the core insights of articles associated with these variants. In addition to textual summarization, the system provides the user with the 15 most relevant references when available.
VarChat graphical user interface is implemented in ReactJS and optimized for desktop and mobile, while the Restful API is built upon a serverless and scalable infrastructure leveraging on Amazon AWS Lambda functions, FastAPI and Python 3.
3. Results and Discussion
VarChat workflow is described in Figure 1. Users can search genomic variants by HGVS nomenclature17 (choosing between coding DNA reference sequence, protein reference sequence, mitochondrial DNA reference sequence or even both coding DNA reference sequence and protein reference sequence) together with the gene symbol. Examples of valid queries are: “BRAF p.V600E”, “GJB2 c.35delG”, and “PINK1 c.926G>A p.G309D”. We empirically observed that the chances of retrieving references from the scientific literature increase with the use of both coding and protein coordinates in a single query. For mitochondrial variants, we recommend using mitochondrial coordinates, such as MT-ND4 m.11778G>A, as an example. All the scientific papers mentioning the variant in the abstract, in the full text or in the supplementary information are retrieved. The 15 most relevant ones are shown to the user and the direct links to Pubmed full text papers are provided. The abstracts related to the searched variant are then exploited by VarChat for summarization purposes.
Based on the information at hand, VarChat generates a summary derived from the relevant publications and the insights of its LLM model. Specifically, if the variant is referenced in the abstract of PubMed publications, that text serves as an additional content for the summarization. If not, the response is entirely produced by the VarChat LLM. Regardless of the scenario, if there's a variant match with the scientific literature, the list of the corresponding PubMed references for the variant is displayed.
The system is designed to be trustworthy for users. Being an LLM-based system, VarChat can be prone to producing 'hallucinations’, a phenomenon where these models generate information that is not supported by the input data or is factually incorrect18. This aspect can be particularly challenging when LLMs are used for tasks that require high levels of accuracy and reliability.
To enhance the transparency of the process, VarChat clearly informs users about the source of its responses, indicating whether the answer was derived from PubMed references or generated solely from the knowledge of VarChat LLM.
After receiving a response, users have the option to provide feedback using a 5-star rating system and can also add a comment. This information will be exploited to fine-tune the system and identify key areas for improvement.
To the best of our knowledge, no similar tools currently exist.
5. Conclusion
VarChat is the first generative AI-based tool specifically designed to support genomic variant interpretation by efficiently finding and summarizing relevant scientific literature, thus acting as a genetic assistant.
We plan to improve VarChat by expanding its search capabilities and integrating different sources of information such as ClinVar and other variant-centric databases and systems. Users will be able to use chat functions to engage with the tool, extend discussions about variants, pose further questions, and delve into areas of particular interest.
VarChat holds the potential to serve the community of genetic professionals as a valuable aid in assessing human genetic variations through generative AI thus enhancing understanding of variants’ impact and their implications.
Acknowledgments
We extend our gratitude to the enGenome team for their insightful discussions and invaluable support in the development of VarChat.
Competing Interests
All the authors collaborate with enGenome srl. FDP, IL and SZ are full employees of enGenome srl. ER, IL and SZ have shares of enGenome.
Availability
VarChat is freely available at varchat.engenome.com.
References
- Karchin, R. & Nussinov, R. Genome Landscapes of Disease: Strategies to Predict the Phenotypic Consequences of Human Germline and Somatic Variation. PLoS Comput. Biol. 12, e1005043 (2016).
- Nicora, G., Zucca, S., Limongelli, I., Bellazzi, R. & Magni, P. A machine learning approach based on ACMG/AMP guidelines for genomic variant classification and prioritization. Sci. Rep. 12, 2517 (2022).
- Sarah L. Stenton et al. Critical assessment of variant prioritization methods for rare disease diagnosis within the Rare Genomes Project. medRxiv 2023.08.02.23293212 (2023). [CrossRef]
- Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
- Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
- Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514-517 (2005).
- Lee, K., Wei, C.-H. & Lu, Z. Recent advances of automated methods for searching and extracting genomic variant information from biomedical literature. Brief. Bioinform. 22, bbaa142 (2021).
- Wei, C.-H., Kao, H.-Y. & Lu, Z. PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 41, W518–W522 (2013).
- Khare, R., Leaman, R. & Lu, Z. Accessing Biomedical Literature in the Current Information Landscape. in Biomedical Literature Mining (eds. Kumar, V. D. & Tipney, H. J.) vol. 1159 11–31 (Springer New York, 2014).
- Pasche, E. et al. Assessing the use of supplementary materials to improve genomic variant discovery. Database 2023, baad017 (2023).
- Allot, A. et al. LitVar: a semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res. 46, W530–W536 (2018).
- Allot, A. et al. Tracking genetic variants in the biomedical literature using LitVar 2.0. Nat. Genet. 55, 901–903 (2023).
- Pasche, E. et al. Variomes: a high recall search engine to support the curation of genomic variants. Bioinformatics 38, 2595–2601 (2022).
- Mottaz, A. et al. Designing an Optimal Expansion Method to Improve the Recall of a Genomic Variant Curation-Support Service. in Studies in Health Technology and Informatics (eds. Séroussi, B. et al.) (IOS Press, 2022). [CrossRef]
- Borji, A. & Mohammadian, M. Battle of the Wordsmiths: Comparing ChatGPT, GPT-4, Claude, and Bard. GPT-4 Claude Bard June 12 2023 (2023).
- Ye, J. et al. A Comprehensive Capability Analysis of GPT-3 and GPT-3.5 Series Models. (2023). [CrossRef]
- Den Dunnen, J. T. et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37, 564–569 (2016).
- Zhang, Y. et al. Siren’s Song in the AI Ocean: A Survey on Hallucination in Large Language Models. ArXiv Prepr. ArXiv230901219 (2023).
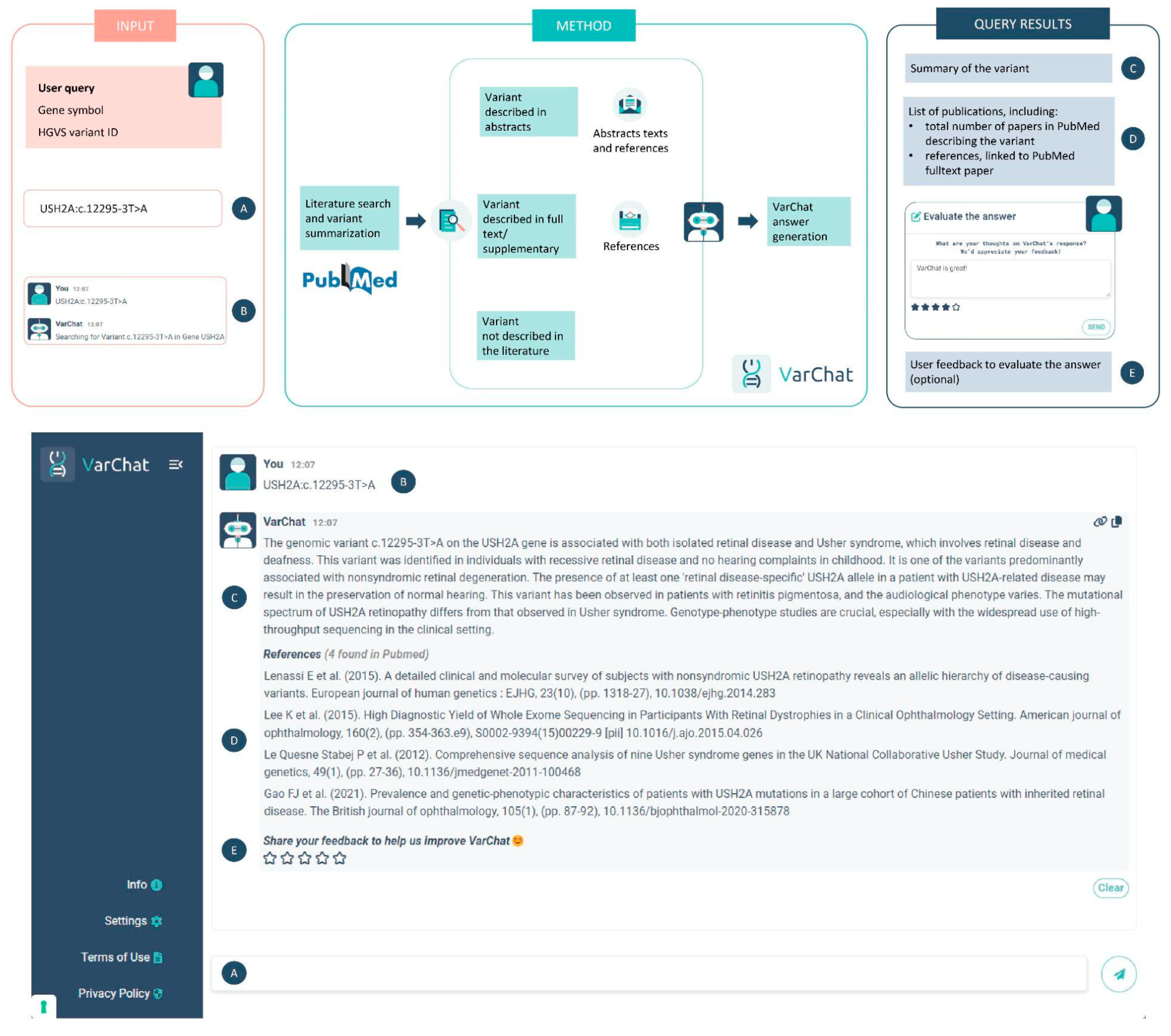
Figure 1.
VarChat workflow and platform preview (A) User prompt enabling variant’s query. Users can search genomic variants with HGVS nomenclature along with the gene symbol. (B) Variant searched by VarChat. (C) VarChat retrieves the literature associated with the searched variant and provides a comprehensive summarization. (D) If available, up to 15 references (sorted by relevance) are displayed, with direct links to Pubmed. The total number of publications found is also shown. (E) Feedback system: users can evaluate the answer through a 5-star scoring system and provide feedback.
Figure 1.
VarChat workflow and platform preview (A) User prompt enabling variant’s query. Users can search genomic variants with HGVS nomenclature along with the gene symbol. (B) Variant searched by VarChat. (C) VarChat retrieves the literature associated with the searched variant and provides a comprehensive summarization. (D) If available, up to 15 references (sorted by relevance) are displayed, with direct links to Pubmed. The total number of publications found is also shown. (E) Feedback system: users can evaluate the answer through a 5-star scoring system and provide feedback.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



