
Submitted:
17 November 2023
Posted:
20 November 2023
You are already at the latest version
Abstract
Sulfolane, a highly water-soluble industrial solvent, has raised environmental concerns due to its persistence once released into the environment. To assess the extent of contamination effectively, reliable analytical methods are essential. In this review, we delve into the published literature on sulfolane analytical procedures. Existing guidelines for sampling from environmental matrices provide a solid foundation for sul-folane analysis. Notably, there is little variation in the choice of determination method, with GC-MS or GC-FID being favored across studies. However, substantial variability emerges in sample prep-aration methods. Many procedures rely on large quantities of environmentally hazardous solvents, such as dichloromethane, during extraction. Nevertheless, by incorporating extraction enhancement techniques proposed in various studies, it is possible to develop more eco-friendly extraction processes. Overall, this field calls for further re-search to devise efficient and environmentally sustainable analytical methods for sulfolane analysis. Through this review, insights into the challenges at hand and potential solutions can be gained, offering a foundation for the development of novel sulfolane analysis methods applicable to a range of environmental matrices.
Keywords:
Sulfolane
; environmental analysis
; environmental pollution
1. Introduction
Sulfolane, also referred to as tetramethylene sulfone and tetrahydrothiophene 1,1-dioxide, is a highly versatile industrial solvent with the molecular formula C4H8SO2 (Figure 1) [1]. Although first mentioned in the literature in 1916, it was not until the 1960s that it became commercially available. Since then, its usage has steadily increased, and it has found a wide range of applications in various fields [1]. This rise in popularity can be attributed in part to its unique physical and chemical properties (Table 1). These properties include high boiling point, thermal stability, as well as miscibility with a wide range of liquids including water, toluene, acetone, alcohols and aromatic solvents, while being immiscibile with alkanes [2].
Sulfolane has become a desirable solvent for many applications in different fields [1]. In the oil industry, sulfolane is used as an extractive solvent for the separation of benzene, toluene and xylene from aliphatic hydrocarbons, while in the gas treatment processes, it is effective in removing acidic components such as hydrogen sulfide and carbon dioxide from sour natural gas, particularly in the western parts of Canada due to high concentration of hydrogen sulfide in the gas resources [1,7]. Sulfolane is also utilized as a solvent for batteries, a plasticizer, and a solvent in the polymer industry, with an estimated annual production of 18,000 to 36,000 tons, reflecting its variability of function in different fields [1,8].
Despite efforts to recover and reuse sulfolane in industrial processes, a small portion of the solvent is lost during each use cycle. For instance, the loss is estimated to be around 5 parts per million (ppm) in the UOP Sulfolane™ commercial process for extracting aromatics from a mixture of hydrocarbons [9]. This small fraction of sulfolane becomes significant when considering the large amounts of the solvent used annually. In addition, sulfolane can be released into the environment through direct spillage or improper waste management practices, and reports of such incidents are available from different countries, including Canada and the US [8].
Due to its chemical stability, sulfolane persists in the environment for a long time when released, and its high solubility in water can lead to contamination of groundwater resources. Furthermore, sulfolane low adsorption on most soil types means that it can spread over large areas through groundwater after being released into the environment [10]. For example, at a contaminated site in Waterton, Canada, it is estimated that one square mile around the contamination source has been impacted [11].
Numerous studies have indicated that sulfolane exhibits toxicity in rats and mice. However, it is imperative to note that further research is necessary to substantiate these effects in the context of human exposure [12]. The mobility and toxicity of sulfolane have raised concerns over its spread into environmental media, and natural aerobic biodegradation is the major way for sulfolane to be removed when released into the environment. However, the low oxygen levels in groundwater make biodegradation a slow process [13].
To assess the extent and levels of contamination, a sensitive, robust, and accurate analytical method is required. This method must also be versatile and applicable to different environmental matrices, and if possible, should not negatively impact the environment. This review aims at investigating published literature on sulfolane analytical methods and at critically assessing them.
To obtain the relevant literature on sulfolane analysis, the Web of Science database was utilized with appropriate keywords. A total of 49 articles were selected, which provided methods for analyzing sulfolane. Moreover, an exhaustive search for official methods and governmental guidelines for sulfolane analysis was conducted, which yielded two official methods. The reported parameters of the analytical methods were organized into different stages of the analytical process and are summarized in the subsequent sections.
2. Sampling of environmental matrices
Sampling in analytical chemistry is defined as the process of collecting a representative fraction of a material, so that it can be analyzed to determine certain characteristics of the bulk of that material. This step is crucially important in the analytical process because of its immense and immediate effect on the validity of the results [14]. To achieve representativeness, different matrices require different sampling techniques. There are well-established official sampling procedures for semi-volatile compounds such as sulfolane in different matrices published by the US EPA which must be followed to ensure minimizing the error introduced into the analysis by inappropriate sampling [15,16].
3. Sample preparation
Sample preparation is a critical step in sulfolane analysis, as it involves addressing the challenges of low analyte concentrations, and potential co-contaminants that may be present in the sample. Similar to other analytical procedures, sample preparation plays a pivotal role in ensuring accurate and reliable results. The key parameters that vary among different sample preparation procedures include the extraction method, inclusion of any concentration step, and the utilization of internal standards for error correction. These parameters for the reviewed publications are summarized in Table 2. In the subsequent sections, a selection of these papers is discussed which reveals useful insights into different types of extraction, their challenges, and how to deal with them.
3.1. Extraction
Extraction is a widely used process that takes advantage of the differences in chemical distribution between two immiscible phases to separate the constituents of a sample. The main objective of extraction is to partition the analytes of interest into one phase while keeping other sample constituents in the other phase, resulting in a simplified sample composition and reduced interference from other compounds. The extraction itself might lead to analyte concentration if the volume of the extractant is lower than the sample volume and extraction is exhaustive i.e. most of the analyte is removed from the sample into the extraction phase [14].
For sulfolane analysis, two common extraction techniques are liquid-liquid extraction (LLE) and solid-liquid extraction (SLE). In both techniques, a solvent is added to the sample phase, and the analyte partitions into the solvent, which is the extraction phase. The recovery of the extraction process is defined as the fraction of the original analyte that is removed from the sample phase [14]. The goals of the extraction process are to isolate and concentrate the analyte, and to exchange the sample matrix for one that is compatible with the analytical instrument.
There are several factors that can affect the recovery in sulfolane extraction, which will be discussed in the following sections. Understanding these factors is crucial in optimizing the extraction process for sulfolane analysis, ensuring accurate and reliable results.
3.1.1. Extraction solvent
The choice of extraction solvent plays a crucial role in the recovery and sensitivity of the analysis in various ways. The partitioning of the analyte into the extraction phase is influenced by the characteristics and volume of the extraction solvent, which in turn impacts the fraction of the analyte that gets extracted. To increase the extraction efficiency, multiple aliquots of the solvent can be used to extract the sample multiple times. This will result in higher recovery of the analyte and by removing most of the analyte into the extraction phase, exhaustive extraction can be achieved. Additionally, higher sensitivities can be achieved by increasing the amount of analyte extracted.
Blowing down the solvent is an effective way to increase the concentration of the analyte in the extract and improve sensitivity, especially when combined with multiple aliquot extractions that maximizes the amount of extracted analyte. However, this approach may not be practical with high boiling point solvents such as water because of the long evaporation time and high energy requirements. More importantly, when the boiling point of the solvent is high, there is a greater risk of analyte loss due to evaporation, particularly if the boiling point of the analyte is lower than that of the solvent. On the other hand, solvents with low boiling points can be easily evaporated to concentrate the extract or dried to reconstitute the sample in another solvent, resulting in higher concentration and improved sensitivity.
It is important to note that the choice of the extraction solvent also affects gas chromatography (GC) analysis. Typically, the extraction solvent is the same as the final sample solvent. If high boiling point solvents are used for the extraction, it can result in co-elutions with some sample components, leading to the inability to detect early eluting analytes. This highlights the need to carefully consider the boiling point and other characteristics of the extraction solvent in order to optimize the recovery and sensitivity of the analysis. The specific solvents used for the extraction of sulfolane are discussed in the following sections.
3.1.1.1. Dichloromethane
As summarized in Table 2, Dichloromethane (DCM) has been commonly used for the extraction of sulfolane from solid and liquid samples in numerous studies. Despite extensive literature research, no information on the solubility of sulfolane in DCM was found. Nevertheless, reported recovery rates for sulfolane extraction using DCM ranged from 50% to 80%, which falls within the acceptable range based on the only two available official methods for sulfolane determination in environmental samples [45,46]. Despite the acceptable extraction performance demonstrated in various studies, DCM is widely recognized as one of the most harmful solvents to the environment [56]. One of the reasons for this negative reputation is that DCM has been found to contribute to the delay in the recovery of the ozone layer in the Earth's stratosphere [57].
For water samples, most studies utilized a single aliquot of DCM for extraction, with DCM-to-sample ratios ranging from 1:20 [51] to 5:2 [13], with most being around 1:1. Notably, except for Kim et al., 2000, none of the studies exploited the easy evaporation capability of DCM to further concentrate the final samples [51]. The decision to exclude this step could be attributed to achieving adequate sensitivity in their analyses. However, if a single-step extraction is intended, using a lower ratio of extraction solvent to sample could result in a higher concentration of the extract. Despite extracting a lower fraction of analytes, this approach can lead to higher sensitivity. Moreover, by utilizing more efficient extraction procedures, similar sensitivities can be achieved using less solvent, thereby reducing the negative environmental impact of the analysis.
On the other hand, in a study conducted by Doucette et al., 2005, water samples were extracted with three aliquots of DCM at a DCM-to-sample ratio of 2:5, and the combined extract was reserved for analysis [18]. The reported recovery ranged from 60% to 70%, which barely meets the acceptance criteria set by the two official guidelines for water sample analysis of sulfolane [45,46]. The use of three aliquots of solvent for extraction should result in higher extraction efficiency. However, when the reported recovery by Doucette et al., 2005, is compared to similar studies conducted using single aliquot extraction with lower cumulative solvent volumes, such as the studies by Izadifard et al., 2017, and Khan et al., 2019, the achieved recovery is 10% to 20% lower [18,20,25]. This is unexpected, as the absolute recovery should be directly proportional to the fraction of the analyte partitioned from the sample into the extraction phase, and theoretically, more aliquots of extraction and higher cumulative volumes should result in higher recoveries.
Since the samples used in the study by Doucette et al. were water samples that did not contain particles for the sulfolane to adsorb onto, and the concentrations of sulfolane were not high enough to saturate DCM and decrease sensitivity, one explanation for the lower recovery could be the possible difference in extraction temperature, as can be inferred from the study by Li et al., 2022 [18,55]. In this study, they aimed to inspect the suitability of 1,2-dichloroethane for the extraction of sulfolane from an aqueous phase, and they created a ternary system consisting of water, 1,2-dichloroethane, and sulfolane in varying proportions. They measured the distribution of sulfolane between the organic and aqueous phases and found that at low concentrations of sulfolane, sulfolane tends to be enriched in the organic phase up to 120 times. Additionally, there is an inverse relationship between sulfolane enrichment in the organic phase and temperature in the range of 15 °C to 35 °C. The effect is significant, as the enrichment decreased from 120:1 at 15 °C to 32:1 at 35 °C [55]. Although there is no such study done for DCM extraction of sulfolane, it can be expected that DCM and 1,2-dichloroethane behave similarly due to their similar structure. This relationship between temperature and sulfolane distribution could be the reason for the significant difference in recoveries observed when using seemingly similar extraction procedures. Furthermore, it would be interesting to explore if and to what extent the distribution of sulfolane in other water-immiscible solvents, such as toluene and ethyl acetate, is affected by temperature.
In another study conducted by Greene et al., 1998, DCM was used for soil sample extraction [7]. The extraction was carried out using a Soxhlet apparatus for 6 hours. Soxhlet extraction is an example of a solid-liquid extraction technique where the analyte is continuously removed by the solvent at its boiling point. This method allows higher recoveries compared to room temperature solid-liquid extraction, especially for challenging samples, due to the higher temperature and circulation of distilled solvent over the sample [58]. Following the extraction, the solvent was concentrated using a rotary evaporator, as described by Greene et al., 1999 [49]. Notably, this extraction method is similar to the official soil extraction method used by the government of Alaska, which will be discussed later in this section.
DCM is also the solvent recommended by the only two official guidelines available for sulfolane analysis. In the lab manual for sulfolane analysis by the government of British Columbia, a 100 mL water sample is adjusted to a pH below 2 and then extracted with two aliquots of 100 mL of DCM. The extracted samples are then treated with sodium sulfate to remove water, followed by complete evaporation of the solvent and reconstitution of the sample in 1 mL of isooctane [45]. Although this procedure results in a concentration of sulfolane in the sample by around a factor of 200, the environmental impact of using 200 mL of DCM for every sample is significant. Scaling down the extraction procedure and reevaluating the need for such a high analyte concentration rate may be strategies to reduce solvent usage, especially considering that reasonably low limits of detection have been achieved with simpler and less environmentally hazardous procedures [39]. However, if these methods are to be replaced, extensive validation procedures must be undertaken to ensure their reliability. In contrast, the government of Alaska has not specified any particular method of extraction for water samples, but has designated the required performance parameters for the extraction [46].
For soil samples, the extraction procedure described in the BC lab manual involves adding 1 mL of water to 10 to 12 g of field-moist sample, followed by extraction with 10 mL of DCM. Sodium sulfate and sodium chloride are added to remove water, and after centrifugation, 1 mL of the extraction phase is reserved for analysis [45]. Although this method uses much less DCM compared to the official water method, it does not include a concentration step. It may be possible to increase sensitivity by taking a portion of the DCM extract and concentrating it through evaporation.
On the other hand, the government of Alaska suggests using EPA method 3550C or preferably 3540C for soil samples [46]. The EPA method 3550C involves mixing 40 g of soil sample with 60 g of sodium sulfate to remove water, followed by three 100 mL aliquots of DCM extraction while being sonicated for 3 minutes. The extracts are then filtered and centrifuged to remove any particles [59]. The EPA method 3540C for soil samples involves mixing 10 g of soil sample with sodium sulfate to remove water, followed by Soxhlet extraction with 300 mL of DCM for 16 to 24 hours. The solvent is then evaporated and reconstituted in 100 mL to 125 mL of DCM [60]. Both methods use large amounts of solvents and achieve a subtle increase in the concentration of the analyte in the final extracts. Furthermore, Soxhlet extraction is time-consuming and energy-intensive, and is prone to analyte loss for volatile co-contaminants.
For plant tissues, the Alaskan government requires the sample to be ground, frozen using liquid nitrogen, pulverized, and then extracted with water, followed by extraction with DCM [46]. This method is based on the method by Headley et al., 2002, discussed later in Section 3.1.1.3, with the difference being that toluene has been replaced by DCM as the extraction phase [39].
3.1.1.2. Water
In order to optimize water extraction conditions for soil samples, Brandao et al., 2019, investigated the effects of extraction time, number of extraction steps, and soil-to-water ratio on recovery [44]. They concluded that increasing the extraction time from 30 to 90 minutes did not significantly affect the recovery. However, increasing the water-to-soil ratio from 1:1 to 3:1 resulted in an increase in recovery from 82% to 93%. Additionally, three cycles of extraction with a water-to-soil ratio of 2:1 resulted in increased recovery from 85.2% for the first cycle to 97.2% for the second cycle, and 98.9% for the third cycle. To confirm the remaining sulfolane content in the soil after water extraction, Soxhlet extraction with DCM was used [44]. Although using two or three aliquots of water for the extraction resulted in better recoveries, it may negatively impact sensitivity due to the impracticality of evaporating water and concentrating the final extract. In cases where higher sensitivities are required, a single aliquot of water can be used. However, using only one aliquot of water may result in incomplete extraction, as the recovery is dependent on the partitioning equilibrium, which is influenced by the distribution constant [58]. The distribution constant can be affected by various factors, such as temperature, the composition of the soil sample, the presence of ionic species in the soil sample, and co-contaminants, all of which add uncontrollable parameters to the analysis.
An alternative solution to address this issue is to use a water-immiscible solvent to extract sulfolane from the aqueous extract, as demonstrated by Fedorak, 1996 [47]. While this approach may be beneficial in certain scenarios, such as when there are numerous hydrophobic compounds that require clean-up, the limitation of water as an extraction phase in terms of its inability to extract hydrophobic co-contaminants remains unresolved. Moreover, the addition of an additional extraction step introduces another potential source of errors, which can adversely impact the analytical reliability.
Water samples can be directly injected into analytical instruments for analysis without any pretreatment if the analyte concentration is high enough for detection. This approach, known as direct injection of aqueous samples, eliminates the sample extraction step, which minimizes errors introduced during extraction. However, the samples must be clean and free from excessive constituents, as this method is susceptible to contamination of the analytical instrument. Therefore, this method may not be suitable for dirty samples such as wastewater. To extend the applicability of this technique, centrifugation and sample filtration can be employed to separate the aqueous phase for samples that may contain particles, such as soil slurries [7]. In cases where filtration is used, it is important to confirm that the analyte does not adsorb onto the filter, as demonstrated by Yu et al., 2016, in their study for nylon filters [33].
In an uncommon and novel extraction method, water was used in an ordinary espresso machine to extract sulfolane from biochar. The authors concluded that ground biochar extraction with water in an espresso machine is more efficient than ethyl acetate extraction with Soxhlet for six solvent cycles or dispersion of biochar in ethyl acetate for 48 hours with initial ultrasonication for 45 minutes. They also reported a loss of sulfolane when they tried to evaporate water at 60 °C or 104 °C in the presence of biochar due to unknown reasons [36]. This method of extraction is a simple and inexpensive setup for pressurized liquid extraction. Considering the 10 to 20 bars of pressure offered by these machines, they can be considered low-pressure liquid extractors. However, since the extraction is performed with water just below its boiling point, it cannot be used for the simultaneous extraction of volatile co-contaminants. Additionally, although the authors achieved reproducible recoveries based on their limited set of experiments, it is interesting to see how reproducibly this machine performs over time, as most of these machines lose pressure as they age.
3.1.1.3. Toluene
Toluene has been utilized as a solvent for extracting sulfolane from water samples [39]. Notably, it has been employed for the back-extraction of sulfolane from soil or plant tissue extracts. The reported recoveries for direct water extraction and the extraction of water extract from soil samples were remarkably high at 127% and 126%, respectively [39]. However, no explanation was provided for these high recoveries, but it is possible that the non-symmetric solubility of the toluene-water system formed during extraction [61] and the high extraction efficiency of toluene for sulfolane contributed to these results. It should be noted that the high boiling point of toluene may hinder the analysis of volatile co-contaminants by GC.
The high extraction efficiency of toluene for sulfolane has been effectively utilized in the extraction of sulfolane from plant tissue samples by Headley et al., 2002 [39]. In their procedure, they homogenized plant tissue in Mili-Q water to extract the sulfolane into the aqueous phase. In a slightly different approach, they ground the plant tissue to a fine powder using liquid nitrogen and then suspended it in Mili-Q water. In both cases, the tissue was left in water for 40 minutes with intermittent swirling. Subsequently, they centrifuged and filtered the supernatant to remove any particles. Next, they back-extracted the aqueous extract with three aliquots of toluene. The toluene portions were combined, and the extract was concentrated 15 times using nitrogen blowdown before being reserved for analysis by GC-MS. The average recovery achieved was 80.3% with an RSD of 11%. Remarkably, the method's performance was maintained over a period of 18 months, indicating its ruggedness [39].
The decision to use water as an intermediate extraction solvent for plant tissue is reasonable due to the hydrophilic nature of this tissue. By leaving it in an aqueous solution, water can penetrate and swell the tissue, enhancing partitioning of the analytes into the bulk of water. Aprotic organic solvents, on the other hand, do not swell the tissue and poorly penetrate it, resulting in poor extraction efficiency. Moreover, plant tissue may contain hydrophobic compounds that can interfere with the analysis, and using water as an intermediate step, with its low solubility for these compounds, is a preferable choice over other protic solvents such as methanol. Additionally, the use of water as an intermediate solvent also serves as a clean-up step. However, the lower recovery rate of 80% compared to soil and water samples in the study suggests possible sorption of sulfolane onto the plant tissue, which needs to be monitored when developing a method for plant tissue analysis.
3.1.1.4. Ethyl acetate
Ethyl acetate has been successfully used for plasma sample extraction with good recoveries, as demonstrated in a study by Versace et al., 2012 [40]. Considering that plasma is a complex aqueous solution with many constituents, it is reasonable to explore the use of ethyl acetate for sulfolane extraction from water samples, and potentially environmental samples. Moreover, ethyl acetate is known to have minimal impact on the environment [56].
In the study by Versace et al., 2012, the extraction procedure involved adjusting the pH of 100 microliters of plasma by adding 50 microliters of 1 M sodium hydroxide, resulting in a highly basic pH that caused aggregation of plasma proteins. Subsequently, the plasma was extracted with 500 microliters of ethyl acetate, followed by centrifugation to separate the organic phase. 50 microliters of isopropanol were then added to the ethyl acetate extract, and the ethyl acetate was evaporated using N2 gas to concentrate the extract approximately 5 times, while the remaining isopropanol extract was reserved for analysis. Despite the complexity of the plasma sample and the abundance of constituents in the matrix that could potentially sorb sulfolane, the study achieved a remarkable recovery rate of 93% [40].
A notable feature of the described extraction procedure is the use of small volumes of samples and solvents. Additionally, as mentioned earlier, the environmental impact of ethyl acetate is comparatively low. When compared to the government of Alaska official method for sulfolane extraction from soil samples, the solvent usage in the study by Versace et al., 2012, is three orders of magnitude lower [40,46]. Furthermore, similar studies, such as a study by Silinski et al., 2019, support the possibility of using ethyl acetate for sulfolane analysis [41]. In fact, Heydari et al., 2019, used ethyl acetate for water sample extraction, although no information about the extraction performance was provided in their publication [23]. However, it is worth noting that DCM has been used as a replacement for ethyl acetate in their recent publications [28,62]. This may be due to the inadequate extraction performance of ethyl acetate in their procedure, possibly resulting from the lack of addition of a salt or ionic strength modifier (discussed in section 3.1.2) [23].
3.1.1.5. Other solvents
Acetonitrile has been utilized as an extraction solvent to extract residual sulfolane content from ground polyphenyl sulfone polymers, as reported by Eckardt et al., 2018 [37]. In the same study, chloroform was also used for extracting sulfolane from an ethanol-water (50:50) mixture to analyze the sulfolane leaching extent from bottles using the ethanol-water mixture as a food simulant. Although efficiency parameters were not reported, all three solvents - ethanol, acetonitrile, and chloroform - have the potential to be used as extraction solvents for sulfolane [37]. It's worth noting that while chloroform is as hazardous to the environment as DCM, acetonitrile is less hazardous, and ethanol is considered one of the most environmentally friendly solvents [56].
On the other hand, 1,2-dichloroethane has been investigated as an extraction solvent for sulfolane from water samples. However, it poses environmental hazards similar to DCM and has a higher boiling point, resulting in longer blowout time, greater risk of analyte loss, and increased risk of co-elution with volatile compounds in GC analysis.
3.1.2. Dissolved electrolyte effect
In two-phase solvent systems used for extraction, a fraction of the organic phase can dissolve in the water phase. This fraction can be significant, reaching up to 8% for solvents like ethyl acetate [63], while for highly non-polar solvents like toluene, it is typically below 1% [61]. This dissolved organic phase can alter the distribution coefficient and reduce the partitioning of the solute from water into the organic phase. To mitigate this effect, an electrolyte can be added to the aqueous phase [63], which limits the fraction of the organic phase that dissolves in water and can improve the extraction efficiency. For instance, sodium chloride at 80% of its saturation concentration [19,47] or at its saturation concentration [35,53,54] has been used to enhance the partitioning of sulfolane into DCM, which serves as the organic phase. Similarly, in the study conducted by Versace et al., 2012, adjusting the pH of water by adding sodium hydroxide increased the amount of electrolytes dissolved in the water, which consequently improved the extraction efficiency of ethyl acetate [40].
3.1.3. pH adjustment
If a species dissociates at the pH of the extraction, the undissociated fraction of the analyte would primarily take part in the partitioning equilibrium, while the ionized species resulting from dissociation would mostly stay in the aqueous phase [14]. Hence it is crucial to adjust the pH in a way that most of the analyte stays neutral for the extraction procedure. However, because of the high pKa value (12.9), sulfolane does not dissociate significantly in the pH range typical of environmental samples. Hence, pH adjustment might not be necessary for most samples. It should be pointed out that it is required by the official method suggested by the government of BC official method for water samples [45]. One of the possible reasons for using pH adjustment in sulfolane analysis might be to limit the extraction of matrix co-contaminants. For instance, this principle has been exploited by Versace et al., 2012, Shipkowski et al., 2021, and Silinski et al., 2019, in plasma samples to aggregate and remove proteins present in the matrix [40,41,42].
4. Separation and analysis techniques
In contrast to the various extraction methods discussed in the literature, there has been limited diversity in the separation and analytical techniques utilized for the determination of sulfolane. Almost all researchers have employed GC to analyze sulfolane due to the low sensitivity of common LC detectors for this compound. Sulfolane does not absorb UV light at wavelengths that can be distinguished from the mobile phase, and thus cannot be detected by UV detectors. Furthermore, it is not efficiently ionized by ESI, leading to inadequate sensitivity of LC-MS systems [38]. In contrast, sulfolane is completely compatible with the two most widely used GC detectors, FID and MS. In the following sections, the conditions and configurations reported for the GC analysis of sulfolane are summarized.
4.1. GC inlet
All of the reviewed studies utilized the split/splitless type of inlet. In this inlet system, the temperature must be high enough to causes all sample components to have non-insignificant vapor pressure, so that they can be transferred effectively into the column. As shown in Table 3, temperatures in the range of 165 °C to 300 °C were successfully employed for sulfolane analysis.
With the split/splitless inlet system, different modes of injection can be used for introducing the sample into the GC column. Several studies have utilized split mode injection, as shown in Table 3. In this injection mode, the sample is vaporized, but most of the vapor exits the inlet, and only a small fraction of the vapor is transferred into the capillary column. This injection mode is ideal for injecting high-concentration samples, often eliminating the need for sample dilution. Additionally, thanks to a narrow injection band, narrower peaks can be achieved in this mode [65]. However, since the concentration of sulfolane is typically low in environmental samples, more concentration must typically be performed in the sample preparation step to analyze the samples in this mode.
In most studies, the splitless mode of injection has been used, as indicated in Table 3. In this mode, most of the sample vapor is carried by the carrier gas into the column. Since the transfer of the vapor into the column could take several seconds to minutes depending on the carrier gas flow and inlet volume, the initial temperature of the column oven must be lower than the boiling point of the sample solvent to refocus the injection band on the column head through solvent effect and prevent peak widening due to slow transfer of the vapors into the column [65]. This type of injection results in higher sensitivity since more analyte is transferred into the instrument and is useful when the analyte concentration is low in the sample.
In a study by Kasanke et al., 2017 [34], pulsed splitless mode of injection was utilized. In this mode, the pressure, and consequently the carrier gas flow rate is raised temporarily during ijection. The vaporized sample is transferred into the column faster and more effectively by the increased flow rate, and the injection band is narrower. Additionally, since the pressure in the inlet is higher at the time of injection, solvent expansion volume is lower, so higher volumes of sample can be injected. Both the higher injection volume and narrower band have the potential to increase sensitivity [66].
4.2. GC column
In earlier publications, there are reports of using packed columns for GC analysis of sulfolane. However, recent studies have exclusively employed capillary columns due to their higher separation efficiency. In the following sections, the properties of these capillary columns are outlined. For complete specifications of the columns used in each study refer to Table 3.
4.2.1. Stationary phase
The majority of studies on sulfolane have utilized non-polar columns coated with (5%-phenyl)-methylpolysiloxane as the stationary phase. This type of stationary phase results in weak retention of sulfolane, and consequently, the elution time is not long. However, if other polar compounds or volatile non-polar compounds are present in the sample, they may co-elute with sulfolane. On the other hand, several researchers employed columns with more polar stationary phases, specifically polar polyethylene glycol [19,36], and mid-polar trifluoropropyl methyl polysiloxane [34].
Due to the high polarity of sulfolane, polyethylene glycol columns retain it strongly, and the analysis time is typically longer due to the lower maximum temperature limit of these columns. Therefore, the use of polyethylene glycol columns is only justified when there are polar components in the sample that need to be separated from sulfolane for reliable analysis.
In the study by Kasanke et al., 2017 [34], mid-polar trifluoropropyl methyl polysiloxane columns were used to exclude hydrocarbon co-contaminants that might have been present in the samples.
4.2.2. Column dimensions
The dimensions of the columns used for sulfolane analysis vary in length from 15 m to 30 m, in inner diameter (ID) from 0.25 mm to 0.53 mm, and in film thickness from 0.25 µm to 1 µm (Table 3). While longer columns may provide better separation, they result in longer analysis time, which can lead to band broadening and reduced sensitivity. Therefore, it is recommended to choose the shortest column that can separate the sample components [65].
Columns with narrower diameters and thinner films result in higher chromatographic resolution, but have lower sample capacity. This is not a major issue for detection with MS, which is characterized by high sensitivity. In the case of FID detectors, thicker stationary phases may be preferred to achieve higher sensitivity through the use of larger injection volumes [65].
4.3. GC detectors
As previously mentioned, both MS and flame ionization detectors are commonly used for sulfolane analysis. However, MS provides superior sensitivity and selectivity compared to FID. In fact, reported limits of detection (LODs) for MS range from 0.516 µg/L to 20 µg/L, while FID LODs range from 500 µg/L to 6000 µg/L in aqueous samples. Additionally, MS allows the use of isotopically labeled internal standards, which results in more reliable and reproducible results.
Electron impact ionization (EI) was the preferred mode for all mass spectrometric techniques. The mass spectrum shown in Figure 2 indicates that the molecular ion for sulfolane appears at 120 m/z, with two abundant fragment ions at m/z 41 and 56, corresponding to the loss of SO2CH3 and SO2, respectively. Different publications have used either the ion at m/z 120 or 41 for quantification, but Headley et al., 1999, reported 5 times higher sensitivity when using m/z 41 as the quantifier ion [38].
In the official method by the government of Alaska, the molecular ion of sulfolane which appears at m/z 120 in the mass spectrum is required to be used as the quantifier ion for sulfolane detection. Additionally, the ions at m/z 41, 55, and 56 are specified as qualifier ions, corresponding to the loss of CH3SO2, C2H4SO2, and C2H5SO2 fragments, respectively [46]. On the other hand, the official method by the government of BC does not specify any particular ion for the detection of sulfolane using MS [45].
4.4. Internal standard
An internal standard is a compound added at a known level to samples, standards, and blanks to correct for analysis errors. Two approaches are employed for adding internal standards. In the first approach, internal standards are added before sample processing (surrogate standards). This method allows the standards to correct for errors at all stages of the analysis, including sample preparation, separation, and final determination. However, it is crucial to choose an internal standard that is not present in the samples, and mimics the partitioning, injection, separation, and detector response profile of the original compound. This can be challenging when using a non-selective detector, such as FID [14].
In contrast, when using mass spectrometry (MS), isotopically labeled compounds can serve as internal standards. These compounds have the same structure as the analyte, but some of their atoms have been replaced by their stable isotopes. This method provides a close match between the chemical and physical properties of the internal standard and the original compound, but MS can separate its signal from that of the analyte due to its different molecular weight [14]. A common isotopically labeled internal standard for sulfolane analysis is sulfolane-d8, which has been employed in several studies listed in Table 2, and is required by the official methods for sulfolane analysis [45,46].
The second approach involves adding the internal standard directly to the final sample before injection and analysis. This type of internal standard addition is mostly used to correct errors introduced by the analytical instrumentation, such as uncertainty in injection volume or differences in analysis conditions at different times. In this case, any compound that would not be present in the samples can serve as an internal standard. Naphtalene-d8 is required as this type of internal standard in the Alaskan government's official method for sulfolane analysis [46].
5. Conclusions
The release of sulfolane into the environment and its subsequent spread through groundwater contamination raise significant health and environmental concerns. To effectively mitigate these risks, it is essential to evaluate the extent of contamination at different sites, which necessitates the use of a reliable analytical method. By examining the existing literature on this subject, valuable insights can be gained into the development of such methods and the challenges it may entail. In this review, various analytical methods for the analysis of sulfolane were thoroughly examined, summarized, and critically assessed.
One area where the analytical procedures for sulfolane analysis greatly differ is in the sample preparation step. Not all procedures yielded sufficiently sensitive methods, and many of the acceptable ones required large amounts of solvents, which can be hazardous to the environment. However, it might be possible to modify or downscale these procedures to reduce the required solvent volumes for extraction.
Moreover, one can exploit the physical and chemical properties of sulfolane to enhance the efficiency of extraction. For example, the Alaskan government's official method utilizes Soxhlet extraction and takes advantage of the thermal stability of sulfolane to improve extraction efficiency from soils. However, as previously mentioned, this method demands a significant amount of solvent and a long extraction time. While Soxhlet extraction cannot be easily downscaled, this thermal stability can be leveraged by other high-temperature extraction techniques to enhance efficiency and speed, potentially reducing solvent consumption. One such technique is microwave-assisted extraction (MAE), which utilizes microwave radiation to rapidly heat the sample and extraction solvent. Additionally, since the extraction is carried out at higher than atmospheric pressure, the temperature can be increased above the atmospheric boiling point of the solvent, thereby shortening the extraction time.
The capability of the extraction method to extract sulfolane co-contaminants is another important and often overlooked aspect for evaluating the sample preparation method. Examining co-contaminants alongside sulfolane may provide a better understanding of the origin and fate of the contamination. For instance, due to the extensive usage of sulfolane in the petrochemical industry, environmental samples might contain hydrocarbons as co-contaminants along with sulfolane. Thus, it is crucial to evaluate the extraction method's capability to extract these compounds simultaneously.
When it comes to determining sulfolane, GC-MS and GC-FID are the exclusive techniques employed for the separation and analysis in the analytical process. While GC-FID might not always offer sufficient sensitivity for detecting the low concentrations of sulfolane found in environmental samples, GC-MS has proven to be a highly sensitive and selective technique for quantifying sulfolane. Nevertheless, other analytical techniques, such as LC-MS/MS with ionization modes other than ESI, should be considered if co-contaminants are not suitable for GC analysis.
In summary, based on the literature reviewed in this study, there is a clear need for a more efficient and environmentally friendly sample preparation procedure for analyzing sulfolane in environmental samples. This method should accommodate the specific requirements of different environmental matrices, including soil or plant samples, and should be adaptable to different analytical instruments. Furthermore, it should be optimized to simultaneously analyze potential co-contaminants.
Acknowledgments
Funding for this research was provided by Shell (through Morwick G360 Group) and NSERC. Restek is gratefully acknowledged for their continuous support of our research.
References
- Clark, E. Sulfolane and Sulfones. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; p. 1921120603120118.a01 ISBN 978-0-471-23896-6.
- Bak, A.; Kozik, V.; Dybal, P.; Kus, S.; Swietlicka, A.; Jampilek, J. Sulfolane: Magic Extractor or Bad Actor? Pilot-Scale Study on Solvent Corrosion Potential. Sustainability 2018, 10, 3677. [CrossRef]
- Travis, C.C.; Arms, A.D. Bioconcentration of Organics in Beef, Milk, and Vegetation. Environ. Sci. Technol. 1988, 22, 271–274. [CrossRef]
- The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; O’Neil, M.J., Ed.; 13th ed.; Merck: Whitehouse Station, N.J, 2001; ISBN 978-0-911910-13-1.
- Coetzee, J.F. Sulpholane: Purification, Tests for Purity and Properties. Pure Appl. Chem. 1977, 49, 211–216. [CrossRef]
- Willer, R. Sulfoxides. In Kirk-Othmer Encyclopedia of Chemical Technology; Kirk-Othmer, Ed.; Wiley, 2000 ISBN 978-0-471-48494-3.
- Greene, E.; Gieg, L.; Coy, D.; Fedorak, P. Sulfolane Biodegradation Potential in Aquifer Sediments at Sour Natural Gas Plant Sites. Water Res. 1998, 32, 3680–3688. [CrossRef]
- Canadian Environmental Quality Guidelines for Sulfolane: Water and Soil : Scientific Supporting Document; Canadian Council of Ministers of the Environment: Winnipeg, Man., 2006; ISBN 978-1-896997-59-9.
- UOP SULFOLANE PROCESS. In Handbook of Petroleum Refining Processes; Meyers, R.A., Ed.; McGraw-Hill Education: New York, 2016 ISBN 978-0-07-185049-0.
- Kim, C.; Clarke, W.; Lockington, D. Competitive Adsorption of Sulfolane and Thiolane on Clay Materials. Korean J. Chem. Eng. 1999, 16, 215–220. [CrossRef]
- Stewart, O. Sulfolane Technical Assistance and Evaluation Report; 2010;
- Testing Status of Sulfolane 11054 Available online: https://ntp.niehs.nih.gov/go/ts-11054 (accessed on 17 September 2023).
- Mulder, D.; Yu, L.; Achari, G. A Laboratory and Field Investigation of Aerobic Biodegradation of Sulfolane in Groundwater. J. Chem. Technol. Biotechnol. 2021, 96, 2865–2871. [CrossRef]
- Skoog, D.A.; West, D.M.; Holler, F.J.; Crouch, S.R. Fundamentals of Analytical Chemistry; Tenth edition.; Cengage: Boston, MA, 2022; ISBN 978-0-357-45043-7.
- Groundwater Sampling Operating Procedure Available online: https://www.epa.gov/sites/default/files/2015-06/documents/Groundwater-Sampling.pdf (accessed on 16 April 2023).
- Soil Sampling Operating Procedure Available online: https://www.epa.gov/sites/default/files/2015-06/documents/Soil-Sampling.pdf (accessed on 17 September 2023).
- Saint-Fort, R. Sulfolane Attenuation by Surface and Subsurface Soil Matrices. J. Environ. Sci. Health Part A Tox. Hazard. Subst. Environ. Eng. 2006, 41, 1211–1231. [CrossRef]
- Doucette, W.; Chard, J.; Moore, B.; Staudt, W.; Headley, J. Uptake of Sulfolane and Diisopropanolamine (DIPA) by Cattails (Typha Latifolia). Michrochemical J. 2005, 81, 41–49. [CrossRef]
- Lee, J.K.; Lee, W.J.; Cho, Y.-J.; Park, D.H.; Lee, Y.-W.; Chung, J. Variation of Bacterial Community Immobilized in Polyethylene Glycol Carrier during Mineralization of Xenobiotics Analyzed by TGGE Technique. Korean J. Chem. Eng. 2010, 27, 1816–1821. [CrossRef]
- Izadifard, M.; Achari, G.; Langford, C. Degradation of Sulfolane Using Activated Persulfate with UV and UV-Ozone. Water Res. 2017, 125, 325–331. [CrossRef]
- Dominic, J.A.; Somathilake, P.; Achari, G.; Langford, C.H.; Tay, J.-H. Sunlight Mediated Passive Wastewater Treatment Technology Using Photochemical Reduction of Ferric Iron for Decontamination of Various Aqueous Contaminants. Sol. Energy 2018, 173, 470–477. [CrossRef]
- Izadifard, M.; Achari, G.; Langford, C. Mineralization of Sulfolane in Aqueous Solutions by Ozone/CaO2 and Ozone/CaO with Potential for Field Application. Chemosphere 2018, 197, 535–540. [CrossRef]
- Heydari, G.; Langford, C.; Achari, G. Passive Solar Photocatalytic Treatment of Emerging Contaminants in Water: A Field Study. Catalysts 2019, 9. [CrossRef]
- Khan, M.; Yu, L.; Achari, G.; Tay, J. Degradation of Sulfolane in Aqueous Media by Integrating Activated Sludge and Advanced Oxidation Process. Chemosphere 2019, 222, 1–8. [CrossRef]
- Khan, M.; Yu, L.; Tay, J.; Achari, G. Coaggregation of Bacterial Communities in Aerobic Granulation and Its Application on the Biodegradation of Sulfolane. J. Hazard. Mater. 2019, 377, 206–214. [CrossRef]
- Dinh, M.; Hakimabadi, S.G.; Pham, A.L.-T. Treatment of Sulfolane in Groundwater: A Critical Review. J. Environ. Manage. 2020, 263, 110385. [CrossRef]
- Khan, M.; Yu, L.; Achari, G. Field Evaluation of a Pressurized Ozone Treatment System to Degrade Sulfolane in Contaminated Groundwaters. J. Environ. Chem. Eng. 2020, 8. [CrossRef]
- Khan, M.; Yu, L.; Hollman, J.; Tay, J.; Achari, G. Integration of Aerobic Granulation and UV/H2O2 Processes in a Continuous Flow System for the Degradation of Sulfolane in Contaminated Water. Environ. Sci. Water Res. Technol. 2020, 6, 1711–1722. [CrossRef]
- Yang, Y.; Yu, L.; Gopal, A. Aerobic Biodegradation of Sulfolane Using Archaea and Pseudomonas Strains. J. Chem. Technol. Biotechnol. 2022, 97, 1763–1770. [CrossRef]
- Yu, L.; Iranmanesh, S.; Keir, I.; Achari, G. A Field Pilot Study on Treating Groundwater Contaminated with Sulfolane Using UV/H2O2. Water 2020, 12. [CrossRef]
- Dharwadkar, S.; Yu, L.; Achari, G. Enhancement of LED Based Photocatalytic Degradation of Sulfolane by Integration with Oxidants and Nanomaterials. Chemosphere 2021, 263. [CrossRef]
- Dharwadkar, S.; Yu, L.; Achari, G. Photocatalytic Degradation of Sulfolane Using a LED-Based Photocatalytic Treatment System. Catalysts 2021, 11. [CrossRef]
- Yu, L.; Mehrabani-Zeinabad, M.; Achari, G.; Langford, C. Application of UV Based Advanced Oxidation to Treat Sulfolane in an Aqueous Medium. Chemosphere 2016, 160, 155–161. [CrossRef]
- Kasanke, C.; Leigh, M. Factors Limiting Sulfolane Biodegradation in Contaminated Subarctic Aquifer Substrate. PLOS ONE 2017, 12. [CrossRef]
- Chang, S.; Wu, C.; Yang, C.; Lin, C. Evaluation Use of Bioaugmentation and Biostimulation to Improve Degradation of Sulfolane in Artificial Groundwater. Chemosphere 2021, 263. [CrossRef]
- Stevens, S.; Richardson, D. Analysis of Sulfolane in Biochar. Appita 2018, 71, 349–353.
- Eckardt, M.; Greb, A.; Simat, T.J. Polyphenylsulfone (PPSU) for Baby Bottles: A Comprehensive Assessment on Polymer-Related Non-Intentionally Added Substances (NIAS). Food Addit. Contam. Part Chem. Anal. Control Expo. Risk Assess. 2018, 35, 1421–1437. [CrossRef]
- Headley, J.V.; Peru, K.M.; Dickson, L.C. Gas Chromatographic–Mass Spectrometric Determination of Sulfolane in Wetland Vegetation Exposed to Sour Gas-Contaminated Groundwater. J. Chromatogr. A 1999, 859, 69–75. [CrossRef]
- Headley, J.; Dickson, L.; Peru, K. Comparison of Levels of Sulfolane and Diisopropanolamine in Natural Wetland Vegetation Exposed to Gas-Condensate Contaminated Ground Water. Commun. Soil Sci. Plant Anal. 2002, 33, 3531–3544. [CrossRef]
- Versace, F.; Uppugunduri, C.R.S.; Krajinovic, M.; Théorêt, Y.; Gumy-Pause, F.; Mangin, P.; Staub, C.; Ansari, M. A Novel Method for Quantification of Sulfolane (a Metabolite of Busulfan) in Plasma by Gas Chromatography–Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2012, 404, 1831–1838. [CrossRef]
- Silinski, M.A.R.; Uenoyama, T.; Cooper, S.D.; Fernando, R.A.; Robinson, V.G.; Waidyanatha, S. Development and Validation of an Analytical Method for Quantitation of Sulfolane in Rat and Mouse Plasma by GC-MS. J. Anal. Toxicol. 2019, 43, 477–481. [CrossRef]
- Shipkowski, K.; Cora, M.; Cesta, M.; Robinson, V.; Waidyanatha, S.; Witt, K.; Vallant, M.; Fallacara, D.; Hejtmancik, M.; Masten, S.; et al. Comparison of Sulfolane Effects in Sprague Dawley Rats, B6C3F1/N Mice, and Hartley Guinea Pigs after 28 Days of Exposure via Oral Gavage. Toxicol. Rep. 2021, 8, 581–591. [CrossRef]
- Greene, E.; Fedorak, P. Nutrient Stimulation of Sulfolane Biodegradation in a Contaminated Soil from a Sour Natural Gas Plant and in a Pristine Soil. Environ. Technol. 2001, 22, 619–629. [CrossRef]
- Brandao, M.; Yu, L.; Garcia, C.; Achari, G. Advanced Oxidation Based Treatment of Soil Wash Water Contaminated with Sulfolane. Water 2019, 11. [CrossRef]
- BC Lab Manual for Sulfolane Analysis 2017.
- Sulfolane Key Elements Document, Version 4 Available online: https://dec.alaska.gov/media/6165/key-element-all-media-2013-7-22.pdf.
- Fedorak, P.; Coy, D. Biodegradation of Sulfolane in Soil and Groundwater Samples from a Sour Gas Plant Site. Environ. Technol. 1996, 17, 1093–1102. [CrossRef]
- Luther, S.; Dudas, M.; Fedorak, P. Sorption of Sulfolane and Diisopropanolamine by Soils, Clays and Aquifer Materials. J. Contam. Hydrol. 1998, 32, 159–176. [CrossRef]
- Greene, E.A.; Coy, D.L.; Fedorak, P.M. Laboratory Evaluations of Factors Affecting Biodegradation of Sulfolane and Diisopropanolamine. Bioremediation J. 1999, 3, 299–313. [CrossRef]
- Kim, C.; Clarke, W.; Lockington, D. Determination of Retardation Coefficients of Sulfolane and Thiolane on Soils by K-Ow-K-Oc and Solubility Parameter, Batch and Column Experiments. Environ. Geol. 2000, 39, 741–749. [CrossRef]
- Kim, C.; Lockington, D.; Clarke, W.; Kim, C. Preliminary Determination of Pollutants Plume in Groundwater at Hazardous Solid Waste Disposal Site by Employing CPT and Rig. Environ. Technol. 2000, 21, 17–30. [CrossRef]
- Kim, C.; Chon, B. Oxygen Uptake Characteristics of Soil Inoculum Amended with Thiophene Derivatives. Korean J. Chem. Eng. 2002, 19, 773–779. [CrossRef]
- Yang, C.; Liu, S.; Su, Y.; Chen, Y.; Lin, C.; Lin, K. Bioremediation Capability Evaluation of Benzene and Sulfolane Contaminated Groundwater: Determination of Bioremediation Parameters. Sci. Total Environ. 2019, 648, 811–818. [CrossRef]
- Lin, C.-W.; Liu, S.-H.; Wu, C.-F.; Chang, S.-H. Critical Factors for Enhancing the Bioremediation of a Toxic Pollutant at High Concentrations in Groundwater: Toxicity Evaluation, Degrader Tolerance, and Microbial Community. J. Environ. Manage. 2021, 277, 111487. [CrossRef]
- Li, Z.; Song, J.; Zhang, S.; Wang, J.; Jian, X. Measurement and Correlation of Liquid-Liquid Equilibrium for the Ternary System (Water+1,2-Dichloroethane plus Sulfolane) at 288.15, 298.15, and 308.15 K. Chin. J. Chem. Eng. 2022, 51, 109–114. [CrossRef]
- Tobiszewski, M.; Namieśnik, J.; Pena-Pereira, F. Environmental Risk-Based Ranking of Solvents Using the Combination of a Multimedia Model and Multi-Criteria Decision Analysis. Green Chem. 2017, 19, 1034–1042. [CrossRef]
- Hossaini, R.; Chipperfield, M.P.; Montzka, S.A.; Leeson, A.A.; Dhomse, S.S.; Pyle, J.A. The Increasing Threat to Stratospheric Ozone from Dichloromethane. Nat. Commun. 2017, 8, 15962. [CrossRef]
- Handbook of Sample Preparation; Pawliszyn, J., Lord, H.L., Eds.; Wiley: Hoboken, N.J, 2010; ISBN 978-0-470-09934-6.
- EPA Method 3550C Available online: https://www.epa.gov/sites/default/files/2015-12/documents/3550c.pdf (accessed on 17 April 2023).
- EPA Method 3540C Available online: https://www.epa.gov/hw-sw846/sw-846-test-method-3540c-soxhlet-extraction.
- Neely, B.J.; Wagner, J.; Robinson, R.L.Jr.; Gasem, K.A.M. Mutual Solubility Measurements of Hydrocarbon–Water Systems Containing Benzene, Toluene, and 3-Methylpentane. J. Chem. Eng. Data 2008, 53, 165–174. [CrossRef]
- Khan, M.; Yu, L.; Achari, G. Sulfolane in Contaminated Sites: Environmental Toxicity and Bioremediation Technologies. Environ. Rev. 2022, 30, 217–227. [CrossRef]
- Altshuller, A.P.; Everson, H.E. The Solubility of Ethyl Acetate in Aqueous Electrolyte Solutions. J. Am. Chem. Soc. 1953, 75, 4823–4827. [CrossRef]
- Yang, Y.; Yu, L.; Iranmanesh, S.; Keir, I.; Achari, G. Laboratory and Field Investigation of Sulfolane Removal from Water Using Activated Carbon. J. Environ. Eng. 2020, 146. [CrossRef]
- Skoog, D.A.; Holler, F.J.; Crouch, S.R. Principles of Instrumental Analysis; Seventh edition.; Cengage Learning: Australia, 2018; ISBN 978-1-305-57721-3.
- Kovacevik, B.; Zdravkovski, Z.; Mitrev, S. PESTICIDE ANALYSIS IN WATER SAMPLES USING GC-MS PULSED SPLITLESS INJECTION. 2016.
- Thiophene, Tetrahydro-, 1,1-Dioxide Available online: https://webbook.nist.gov/cgi/inchi?ID=C126330&Mask=200 (accessed on 22 September 2023).
Figure 1.
Structure of sulfolane.
Figure 2.
Mass spectrum of sulfolane from NIST website (Thiophene, Tetrahydro-, 1,1-Dioxide, 2014). The most abundant fragment ions appear at m/z 41, 56, and 55, and the molecular ion appears at m/z 120 [67].
Figure 2.
Mass spectrum of sulfolane from NIST website (Thiophene, Tetrahydro-, 1,1-Dioxide, 2014). The most abundant fragment ions appear at m/z 41, 56, and 55, and the molecular ion appears at m/z 120 [67].
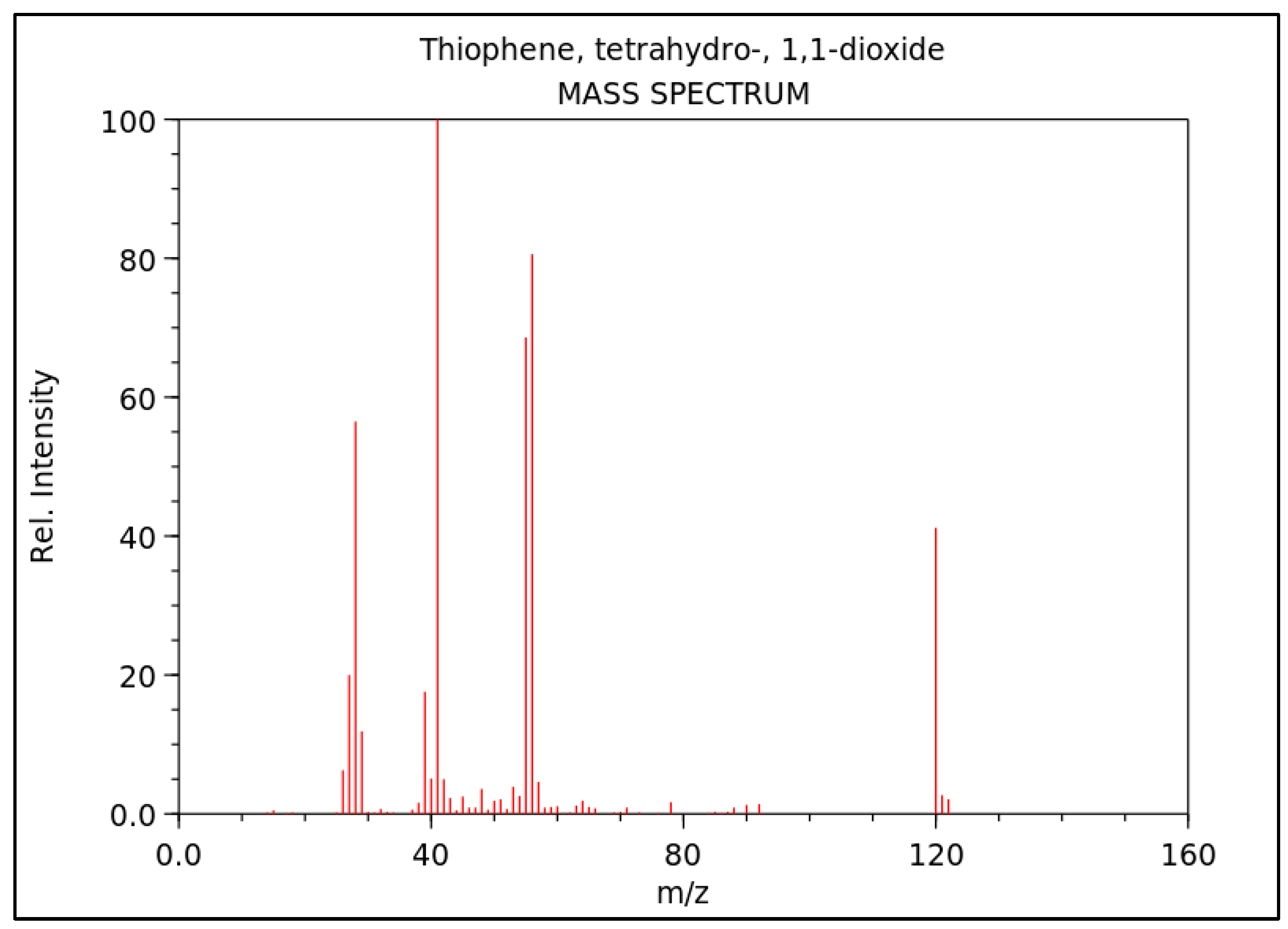

Table 1.
Chemical and physical properties of sulfolane.
| property | Value | Ref |
| Molecular formula | C4H8SO2 | [1] |
| Molecular weight | 120.17 g/mol | [1] |
| Melting point | 28.5 °C | [1] |
| Boiling point | 287.3 °C | [1] |
| Log P | -0.4 | [3] |
| Solubility in water | Miscible at 30 °C | [4] |
| pKa | 12.9 * | [5] |
* Despite extensive research, there is only one reported pKa value for sulfolane. However, this value seems questionable given that dimethylsulfoxide has a pKa value of 35.1, and shares structural similarities with sulfolane [6].
Table 2.
Summary of sample preparation methods used in the articles reviewed for the paper.
| Sample type | Method of extraction | Extraction phase | NO. of extraction aliquotes | Solvent to sample ratio (mL:mL or mL:g) | Centrifugation | Filtration | Salting out agent (saturation %) | Water removal of extract | Solvent evaporation (concentration factor) | pH adjustment | Internal standard | Recovery (%) | Ref. |
| Aqueous solution | AQ1 direct injection | N/A | N/A | N/A | Yes | Filter paper | No | No | No | No | No | N/A | [17] |
| Aqueous solution | LLE2 | DCM4 | 3 | 2:5 | No | No | No | No | No | No | No | 60 to 70 | [18] |
| Aqueous solution | LLE2 | DCM4 | 1 | 2:5 | No | Yes | NaCl (80%) | No | No | No | Ethylene glycol butyl ether | N/A | [19] |
| Aqueous solution | LLE2 | DCM4 | 1 | 1:1 | No | No | No | No | No | No | No | 80 | [20] |
| Aqueous solution | LLE2 | DCM4 | 1 | 2:5 | No | N/A | No | No | No | No | No | N/A | [21] |
| Aqueous solution | LLE2 | DCM4 | 1 | 2:1 | No | No | No | No | No | No | No | 80 | [22] |
| Aqueous solution | LLE2 | Ethyl acetate | 1 | 2:1 | No | No | No | No | No | No | No | N/A | [23] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | 80±5 | [24] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | 80±5 | [25] |
| Aqueous solution | LLE2 | Toluene | 1 | 2:1 | No | No | No | No | No | No | Sulfolane-d8 | N/A | [26] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | 80±5 | [27] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | 80±5 | [28] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | N/A | [29] |
| Aqueous solution | LLE2 | DCM4 | 1 | 3:5 | No | Yes | No | No | No | No | No | 80±5 | [30] |
| Aqueous solution | LLE2 | DCM4 | 1 | 1:1 | No | 0.2 µm PTFE filter | No | No | No | No | No | N/A | [31] |
| Aqueous solution | LLE2 | DCM4 | 1 | 1:1 | No | 0.2 µm PTFE filter | No | No | No | No | No | N/A | [32] |
| Aqueous suspension | AQ1 direct injection | N/A | N/A | N/A | Yes | 0.22 µm nylon filter | No | No | No | No | No | N/A | [33] |
| Aqueous suspension | LLE2 | DCM4 | 3 | N/A | N/A | N/A | No | No | No | No | Sulfolane-d8 and nitrobenzene-d8 | N/A | [34] |
| Aqueous suspension | LLE2 | DCM4 | 1 | 1:10 | No | 0.22 µm Teflon filter | NaCl (100%) | No | No | No | No | N/A | [35] |
| Aqueous suspension | LLE2 | DCM4 | 1 | 3:5 | No | 0.45 µm syringe filter | No | No | No | No | No | N/A | [29] |
| Biochar | Soxhlet extraction | Ethyl acetate | 6 | N/A | No | No | No | No | N/A | No | N-butanol | 43 to 50 | [36] |
| Ethanol/water (1:1) mix | LLE2 | Chloroform | 1 | 1:5 | No | No | No | By Na2SO4 | No | No | Dicyclohexylmethanol | 45.5 | [37] |
| Homogenized water plant tissue mixture | LLE2 | Toluene | 3 | 5:1 | Yes | 0.2 µm cellulose acetate membrane filter | No | No | Yes (15) | No | No | 80±12 | [38] |
| Homogenized water plant tissue mixture | LLE2 | Toluene | 3 | 5:1 | Yes | 0.2 µm cellulose acetate membrane filter | No | No | Yes (15) | No | No | 80±12 | [39] |
| Homogenized water plant tissue mixture | LLE2 | DCM4 | 3 | 2:5 | No | 0.2 µm membrane filter | No | No | No | No | No | 50 to 60 | [18] |
| Plasma | LLE2 | Ethyl acetate | 1 | 5:1 | Yes | No | Yes | No | Yes (8) | by NaOH | Sulfolane-d8 | 93 | [40] |
| Plasma | LLE2 | Ethyl acetate | 1 | 5:1 | Yes | No | Yes | No | Yes (8) | by NaOH | Sulfolane-d8 | 74.4 to 88.7 | [41] |
| Plasma | LLE2 | Ethyl acetate | 1 | 5:1 | Yes | No | Yes | No | No | by NaOH | Sulfolane-d8 | >85 | [42] |
| PPSF polymer | SLE3 | Acetonitrile | 1 | 20:1 | No | No | No | No | No | No | Dicyclohexylmethanol | N/A | [37] |
| Soil | SLE3 | Water | 2 | 5:1 | Yes | No | No | No | No | No | No | 65 to 102 | [43] |
| Soil | SLE3 | Water | 1 to 3 | 1:1 to 3:1 | Yes | 0.45 µm filter | No | No | No | No | No | 82 to 99 | [44] |
| Soil | SLE3 | DCM4 | 1 | 1:1 | Yes | No | No | By Na2SO4 and NaCl | No | No | Sulfolane-d8 | 80-120 | [45] |
| Soil | SLE3 | DCM4 | 3 | 5:2 | Yes | Yes | No | By Na2SO4 | If required | No | Sulfolane-d8 | 70-120 | [46] |
| Soil | Soxhlet extraction | DCM4 | 1 | 30:1 | No | No | No | By Na2SO4 | Yes (3) | No | Sulfolane-d8 | 70-120 | [46] |
| Soil Slurry | AQ1 direct injection | Water | N/A | N/A | Yes | No | No | No | No | No | No | N/A | [7] |
| Soil Slurry/soil water mix | LLE2 | DCM4 | 3 | 1:2 | Yes | No | NaCl (80%) | By Na2SO4 | Yes (N/A) | No | Dibenzothiophene | N/A | [47] |
| Soil water mix | LLE2 | Toluene | 3 | 5:1 | Yes | 0.2 µm cellulose acetate membrane filter | No | No | Yes (15) | No | No | 126 | [39] |
| Water | AQ1 direct injection | N/A | 0 | N/A | Yes | No | No | No | No | No | No | N/A | [48] |
| Water | AQ1 direct injection | Water | N/A | N/A | Yes | No | No | No | No | No | No | N/A | [49] |
| Water | LLE2 | DCM4 | 1 | 1:1 | Yes | 0.2 µm membrane filter | No | By Na2SO4 | No | No | No | N/A | [10] |
| Water | LLE2 | DCM4 | 1 | 1:1 | Yes | 0.2 µm embrane filter | No | No | No | No | No | N/A | [50] |
| Water | LLE2 | DCM4 | 3 | 1:20 | No | Glass wool | No | By Na2SO4 | Yes (15) | By NaOH to pH 10 | No | N/A | [51] |
| Water | LLE2 | Toluene | 3 | 5:1 | Yes | 0.2 µm cellulose acetate membrane filter | No | No | Yes (15) | No | No | 127 | [39] |
| Water | LLE2 | DCM4 | 1 | 1:1 | No | 0.2 µm membrane filter | No | By Na2SO4 | No | No | No | N/A | [52] |
| Water | LLE2 | DCM4 | 1 | 1:10 | No | No | NaCl (100%) | No | No | By Na2CO3 to pH 5-8 | No | N/A | [53] |
| Water | LLE2 | DCM4 | 1 | 1:10 | No | No | NaCl (100%) | No | No | By Na2CO3 to pH 5-8 | No | N/A | [54] |
| Water | LLE2 | DCM4 | 1 | 5:2 | No | 0.45 µm PTFE | No | No | No | No | No | N/A | [13] |
| Water | LLE1 | 1,2-dichloroethane | 1 | 1:1 | No | No | No | No | No | No | No | N/A | [55] |
| Water | LLE2 | DCM4 | 2 | 1:1 | No | No | Yes | By Na2SO4 | Yes (200) | Yes (pH<2) | Sulfolane-d8 | 80-120 | [45] |
1 Aqueous extract. 2 Liquid-liquid extraction. 3 Solid-liquid extraction. 4 Dichloromethane.
Table 3.
Summary of separation and determination methods of sulfolane.
| Sample type | Sample solvent | Carrier gas | Carrier gas flow rate/Pressure | Mode of injection | Injection volume | Inlet temperature (°C) | Type of column | Column stationary phase | Column dimensions | Detector | LLOD4 | LLOQ5 | Detector temperature (°C) | Ref. |
| Aqueous solution | DCM1 | N2 | 10 mL/min | N/A | N/A | N/A | WCOT2 | DB-5 | 30 m x 0.45 mm x 0.25 μm | FID | N/A | N/A | N/A | [18] |
| Aqueous solution | Water | N2 | 360 mL/min | Splitless | N/A | 280 | SCOT3 | OS-138 | 7.5 m x 0.51 mm | FID | 6 mg/L | N/A | N/A | [17] |
| Aqueous solution | DCM1 | He | 1 mL/min | Splitless | 1 | 250 | WCOT2 | DB-WAXETR | 30 m x 0.32 mm x 1 μm | MS | N/A | N/A | N/A | [19] |
| Aqueous solution | DCM1 | He | 250 KPa | Splitless | 2 | 165 | WCOT2 | ZB-5 | N/A | FID | 1 mg/L | N/A | 250 | [20] |
| Aqueous solution | DCM1 | He | 10 mL/min | Splitless | 2 | 250 | WCOT2 | ZB-5 | N/A | FID | N/A | N/A | 320 | [21] |
| Aqueous solution | DCM1 | He | 250 kPa | Splitless | 2 | 165 | WCOT2 | ZB-5 | N/A | FID | 1 mg/L | N/A | 250 | [22] |
| Aqueous solution | Ethyl acetate | N/A | N/A | N/A | N/A | N/A | WCOT2 | ZB-5 | N/A | FID | N/A | N/A | N/A | [23] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | 10 μg/L | N/A | N/A | [24] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | 10 μg/L | N/A | N/A | [25] |
| Aqueous solution | Toluene | He | 1 mL/min | Splitless | 0.5 | 285 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | MS | 20 μg/L | 70 μg/L | Ion source: 285 | [26] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | 10 μg/L | N/A | N/A | [27] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | 10 μg/L | N/A | N/A | [28] |
| Aqueous solution | DCM1 | He | N/A | Splitless | 1 | 165 | WCOT2 | ZB-5 | N/A | FID | 0.3 mg/L | 1 mg/L | 330 | [64] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | 10 μg/L | N/A | N/A | [30] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 165 | WCOT2 | ZB-5 | N/A | FID | N/A | N/A | 330 | [31] |
| Aqueous solution | DCM1 | He | 1.07 mL/min | Splitless | 1 | 165 | WCOT2 | ZB-5 | N/A | FID | N/A | N/A | 330 | [32] |
| Aqueous suspension | Water | He | N/A | Splitless | 1 | 165 | WCOT2 | ZB-5 | N/A | FID | 1 mg/L | N/A | 250 | [33] |
| Aqueous suspension | DCM1 | N/A | N/A | Pulsed-splitless | N/A | N/A | WCOT2 | RTX-200 | 30 m | MS | N/A | 40 μg/L | N/A | [34] |
| Aqueous suspension | DCM1 | N2 | N/A | Splitless | 1 | 200 | WCOT2 | Stabliwax | 30 m x 0.53 x 1 µm | FID | N/A | N/A | 250 | [35] |
| Aqueous suspension | DCM1 | He | N/A | Splitless | 1 | N/A | WCOT2 | 5-MSI | N/A | FID | N/A | N/A | 330 | [29] |
| Biochar | Ethyl acetate | He | N/A | Split (10:1) | 1 | N/A | WCOT2 | ZB-Wax-Plus | 30 m x 0.32 mm x 0.5 μm | FID | 10 mg/L | N/A | N/A | [36] |
| Ethanol/water (1:1) solution | Chloroform | He | 1 mL/min | Splitless | 1 | N/A | WCOT2 | HP-5 | 30 m x 0.25 mm x 0.25 µm | MS | 10 μg/kg of ethanol/water (1:1) | N/A | N/A | [37] |
| Homogenized water plant tissue mixture | Toluene | He | 25 cm/s | Splitless | N/A | 250 | WCOT2 | DB-5 | 25 m x 0.25 mm x 0.25 µm | MS | 90 ng per gram of wet plant tissue | 300 ng per g of wet plant tissue | Ion source: 280 | [38] |
| Homogenized water plant tissue mixture | Toluene | He | 25 cm/s | Splitless | N/A | 250 | WCOT2 | DB-5 | 25 m x 0.25 mm x 0.25 µm | MS | 90 ng per gram of wet plant tissue | 300 ng per g of wet plant tissue | Ion source: 280 | [39] |
| Homogenized water plant tissue mixture | DCM1 | N2 | 10 mL/min | N/A | N/A | N/A | WCOT2 | DB-5 | 30 m x 0.45 mm x 0.25 µm | FID | N/A | N/A | N/A | [18] |
| Plasma | Isopropanol | He | 1 mL/min | Splitless | 1 | 250 | WCOT2 | ZB-5 | 15 m x 0.25 mm x 0.25 µm | MS/MS | N/A | 20 μg/L | Ion source: 200 | [40] |
| Plasma | Isopropanol | He | 1 mL/min | Split (50:1) | 1 | 250 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | MS | 0.516 μg/L | 20 μg/L | Ion source: 230 | [41] |
| Plasma | Ethyl acetate | He | 1 mL/min | Split (50:1) | 1 | 250 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | MS | 1.25 μg/L | N/A | Ion source: 230 | [42] |
| PPSF polymer | Acetonitrile | He | 1 mL/min | Splitless | 1 | N/A | WCOT2 | HP-5 | 30 m x 0.25 mm x 0.25 µm | MS | N/A | N/A | N/A | [37] |
| Soil | Water | He | 25 mL/min | Splitless | 2 | 250 | Packed | Tenax-GC coated with 5% polyphenyl ether | 1.2 m x 0.32 cm | FID | 1 mg/L | N/A | 250 | [43] |
| Soil | DCM1 | He | 250 KPa | Splitless | 1 | 250 | WCOT2 | ZB-5 | N/A | MS | <1 mg/L | <1 mg/L | 330 | [44] |
| Soil Slurry | Water | He | 24 mL/min | Splitless | 2 | 250 | Packed | Tenax-GC coated with 5% polyphenyl ether | 2 m x 0.3cm | FID | 0.5 mg/L | N/A | 250 | [7] |
| Soil Slurry or soil water mixture | DCM1 | N/A | N/A | Split (20:1) | N/A | 250 | WCOT2 | DB-5 | 25 m x 0.25 mm x 0.25 µm | FID | N/A | N/A | 250 | [47] |
| Soil water mixture | Toluene | He | 25 cm/s | Splitless | N/A | 250 | WCOT2 | DB-5 | 25 m x 0.25 mm x 0.25 µm | MS | 90 ng per gram of wet plant tissue | N/A | ion source: 280 | [39] |
| Water | Water | N2 | 30 mL/min | Splitless | 2 | 250 | Packed | Tenax-GC coated with 5% polyphenyl ether | 1.8 m x 0.32 cm | FID | 5 mg/L | N/A | 250 | [48] |
| Water | Water | He | 25 mL/min | Splitless | 2 | 250 | Packed | Tenax-GC coated with 5% polyphenyl ether | 1.2 m x 0.32 cm | FID | 1 mg/L | N/A | 250 | [49] |
| Water | DCM1 | He | 1.7 mL/min | N/A | N/A | 300 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | FID | N/A | N/A | 350 | [10] |
| Water | DCM1 | He | N/A | N/A | N/A | N/A | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | FID | N/A | N/A | N/A | [50] |
| Water | DCM1 | He | 1.7 mL/min | Split (N/A) | 5 | 300 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | FID | N/A | N/A | 350 | [51] |
| Water | Toluene | He | 25 cm/s | Splitless | N/A | 250 | WCOT2 | DB-5 | 25 m x 0.25 mm x 0.25 μm | MS | 1 ng/mL | N/A | ion source: 280 | [39] |
| Water | DCM1 | He | 1.7 mL/min | N/A | N/A | 300 | WCOT2 | DB-5 | 30 m x 0.25 mm x 0.25 µm | FID | N/A | N/A | 350 | [52] |
| Water | DCM1 | He | N/A | N/A | 0.2 | 200 | WCOT | Stabliwax | 30 m x 0.53 mm x 1 µm | FID | N/A | N/A | 250 | [53] |
| Water | DCM1 | He | N/A | N/A | 0.2 | 200 | WCOT2 | Stabliwax | 30 m x 0.53 mm x 1 µm | FID | N/A | 0.44 mg/L | 250 | [54] |
| Water | DCM1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | FID | 1 mg/L | N/A | N/A | [13] |
| Water | 1,2-dichloroethane | N2 | 4.5 mL/min | Split (8:1) | 0.2 | 250 | WCOT2 | DB-FFAP | 30 m x 0.53 mm x 1 µm | FID | N/A | N/A | 250 | [55] |
1 Dichloromethane. 2 Wall coated open tubular. 3 Support coated open tubular. 4 Lower limit of detection. 5 Lower limit of quantification.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



