
Submitted:
19 November 2023
Posted:
20 November 2023
You are already at the latest version
Abstract
Abstract: The baculovirus expression vector (BEV) system is an efficient, cost effective and scalable method to produce recombinant adeno-associated virus (rAAV) gene therapy vectors. Most BEV designs emulate the wild type AAV transcriptome and translate the AAV capsid proteins, VP1, VP2 and VP3 from a single mRNA transcript with three overlapping open reading frames (ORFs). Non-canonical translation initiation codons for VP1 and VP2 reduce their abundances relative to VP3. Changing capsid ratios to improve rAAV vector efficacy requires theoretical modification of translational context. We have made a Lac repressor inducible system to empirically regulate expression of VP1 and VP2 proteins relative to VP3 in the context of the BEV. We demonstrate the use of this system to tune the abundance, titer and potency of a neurospecific rAAV9 serotype derivative. VP1:VP2:VP3 ratios of 1:1:8 gave optimal potency for this rAAV. It was discovered that ratios of capsid proteins expressed were different than ratios that ultimately were in purified capsids. Over expressed VP1 did not become incorporated into capsids while over expressed VP2 did. Overabundance of VP2 correlated with reduced rAAV titers. This work demonstrates a novel technology for controlling production of rAAV in the BEV system and shows a new perspective on the biology of rAAV capsid assembly.
Keywords:
baculovirus expression vector
; inducible expression
; rAAV
; lac repressor
1. Introduction
Adeno-Associated Viruses (AAVs) are Parvoviruses belonging to the Dependovirus genus and require co-infection of another DNA virus type such as an adenovirus, herpesvirus, or papillomavirus [1,2,3]. AAVs have single stranded DNA genomes that exist as episomes in the nucleus or as proviruses in chromosome 19 of humans [4]. There are 13 characterized primate AAV serotypes with different tissue tropisms and potencies [5]. AAV capsids are icosahedral with T=1 symmetry and 60 viral proteins (VPs) per capsid. These 60 VPs are a mixture of VP1, VP2 and VP3 proteins that share common amino acid sequences. The smallest VP3 protein dominates in abundance, and we and others have observed that VP3 composed capsids can package AAV genomes without the presence of VP1 and VP2 (data not shown). However, the lesser abundant and larger VP1 and VP2 proteins are required for capsid infectivity. In recent years, rAAVs have been harnessed gene therapy vectors with several therapeutics being authorized for clinical use in the United States (RoctavinTM, GlyberaTM, LuxturnaTM, ZolgensmaTM). There have been more than 100 clinical trials of different rAAV drug products [6] and genetically modified capsid serotypes with improved tissue specificities and potencies are on the horizon for a new generation rAAV therapeutics [7,8].
rAAV gene therapeutics involve expressing AAV replicase (Rep) and capsid (Cap) proteins in presence of a therapeutic transgene flanked by AAV inverted terminal repeat sequences (ITRs). The result is a rAAV capsid with a packaged transgene. The rAAV is inoculated into the patient where it transduces targeted cell types. Transgenes are delivered into the cell nucleus where they are converted into a stable double stranded DNA episomes from which the transgene encoded therapeutic gene product is expressed.
Application of the insect cell line-based BEV system to produce rAAV therapeutics was initially described as a triple BEV infection by [9] and later as a dual BEV infection [10]. The BEV system is advantageous for scaled production of rAAV therapeutics with unlimited cell culture production capacity, economically amplified BEV inoculums and absence of endotoxins or potential human pathogens. The dual BEV infection method to produce rAAV in the BEV system involves the co-infection of a RepCap BEV with an ITR transgene BEV. The RepCap expressing BEV is engineered with Rep and Cap genes pointing in opposite directions and expressed from very late baculovirus promoters (Figure 1). Rep78 and Rep52 proteins are translated from a common ORF with Rep52 having its own internal translational initiation codon. The Cap gene encodes for VP1 (81 kDa), VP2 (67 kDa), and VP3 (61 kDa) Cap proteins on a common ORF (Figure 1). The Cap gene, also encodes an out of frame ORF for the 21 kDa assembly-activating protein (AAP). AAP is essential for capsid assembly in multiple AAV serotypes including AAV9 [11,12]. In the BEV system, VP1, VP2 and AAP protein ORFs have non-canonical, non-ATG translational start codons. The abundances of translated VP1, VP2 and AAP proteins are less than VP3 which has a canonical ATG translational start codon.
A challenge of producing potent rAAV capsids using the BEV system has been optimizing VP capsid ratios. The ideal VP1:VP2:VP3 ratios for AAV capsids are imprecise and have been reported as 1:1:20 [13], 1:1:10 [9,14] and 1:1:8 [15,16]. The method of analysis used to determine VP ratios often differs and there may be variations of ideal VP ratios for different AAV serotypes. Furthermore, there are the novel tissue specific rAAV capsid variants [8] which may have different optimal VP ratios. Varying the ratio of VPs has been reported to affect the titer and potency of assembled rAAV capsids [17]. Methods for changing VP ratios produced using BEVs have often involved the modulation of the translational context of VP1 [17,18].
The present work describes a method for expressing rAAV capsid VPs at different ratios for a neurospecific AAV9 capsid variant in the BEV system. Instead of expressing VP1, VP2 and VP3 from a common ORF, we separated VP1, VP2 and VP3 ORFs to be expressed independently in a single BEV construct. The VP1 and VP2 ORFs were placed under the E. coli lac repressor (LacR) inducible regulation in the context of the BEV expression system. We show how controlling the abundances of VP1 and VP2 relative to VP3 affects the potency, yield, and the transgene titer of rAAV capsids. Overabundance of VP1 and VP2 negatively impacts capsids assembly and reduces potency.
2. Materials and Methods
2.1. Insect Cells
Spodoptera frugiperda (Sf9) cells used were a Voyager Therapeutics clonal isolate of the Sf9 cell line [19] which was clonal derivative of the IPLB-SF-21-AE cell line [20]. Sf9 were cultured at 28oC in ESF AF™ Insect Cell Culture Medium (#99-300-01, Expression Systems, Davis, CA). Sf9 cells were grown at 25 mL scale in Mini Bioreactor tubes (#431720, Corning, Tewksbury, MA) and shaken at 250 rpm or grown in 2L shake flasks (#431255, Corning) and shaken at 95 rpm. Human embryonic kidney cells were from the HEK 293T cell line (ATCC CRL-1573). These cells were maintained as adherent cultures in Dulbecco’s Modified Eagle Medium (DMEM) (#10564011, Thermo Fisher Scientific, Waltham MA).
2.2. Virus Stocks
Bacmid DNA was generated in NEB 10-Beta E. coli (#C3020K, New England Biolabs, Ipswich MA) and purified by Qiagen Large Construct Kit (#12462, Qiagen, Germantown, MD) was transfected into Sf9 cells to be converted into replicating BEVs. Bacmid DNA-transfection complexes were prepared by combining 200 ng of bacmid DNA, 5 µL of TransIT-Insect Transfection Reagent (# MIR 6100, Mirus Bio, Madison, WI) and 200 µL of Grace’s Salts (# G8142, Sigma-Aldrich, St. Louis, MO) and transfecting 30 mL of Sf9 cells at 2 x 106 cells/mL. At 5 days post-transfection, BEV inoculum stocks were made as baculovirus-infected insect cells (BIICs). The infected cells were pelleted by centrifugation (5 min, 300 × g), and resuspended at 2 × 107 cells/mL in the original budded virus media. An equal volume of 10% v/v dimethyl sulfoxide, 5 % w/v trehalose in ESF AFTM media was added and resulting BIIC aliquots were stored at -80 oC. BIIC infectious titers were determined using methods described by Nguyen et al., 2023 [21].
2.3. Constructs
2.3.1. Rep and Cap Gene Sources
The replicase (Rep) gene used is based on AAV2 serotype (NCBI:txid10804). The capsid (Cap) gene used in this study was a synthetically engineered neurotropic capsid variant with 98% identity to AAV9 serotype (NCBI:txid235455). It was used as a template for Cap ORFs VP1, VP2 and VP3.
2.3.2. LacR Gene Constructs
The LacR ORF used was engineered to encode an N-terminal SV40 nuclear localization signal as described by Slack et al.,1997 [22]. A polh-LacR cassette (LacRegt) was made which included a 92 bp very late polh promoter corresponding to 4,429 to 4,520 AcMNPV E2 (GenBank KM667940.1) (Figure 1F). An Opgp64-polh-LacR cassette (LacRTn7) was also made (Figure 1G). Its promoter was a hybrid early/late/very late promoter containing a 166 bp early/late promoter region from the OpMNPV gp64 gene [23] upstream of a polh promoter. The OpMPNV gp64 promoter was modified with a G to C mutation to remove its minicistron ATG [24]. The downstream polh promoter was mutated to lack internal ATG start codons by changing ATGs to TTGs such that hybrid early/late/very late promoter had only the ATG translational start codon for LacR.
2.3.3. LacO Promoter Constructs
The capsid genes LacOVP1VC and LacOVP2gta in LacRepCap (Figure 1B) and LacR-LacOVP1 (Figure 2B) had 186 bp AcMNPV p10 promoters corresponding 118,726 to 118,906 of AcMNPV E2 genome (GenBank KM667940.1). These promoters were flanked by the LacO sequences 5’-G A T T G T G A G C G C T C A C A A T T-3’ (Figure 1D). The p10 promoters were modified to have 3 internal ATG start codons changed to TTG.
2.3.4. Making Recombinant VoyBac1.1 Bacmids by T4 Ligase Ligation
All recombinant BEV bacmids were based on the Voyager Therapeutics bacmid VoyBac1.1. This bacmid was derived from bMON14272 [25] which contains a mini-F replicon and the genome for the baculovirus AcMNPV E2 (GenBank KM667940.1). VoyBac1.1 differed from the bMON14272 bacmid by being modified to lack the v-cath (VC) gene [26] as described in Nguyen et al., 2023 [21]. The deleted VC region was replaced by a unique I-Ceul homing endonuclease (HEN) recognition site. This enabled T4 ligase cloning into the VC locus as described previously [27]. Gene cassettes LacRegt, LacOVP1VC, and LacOVP2gta (Figure 1B & 2A) were sequentially cloned by T4 ligase ligation into the I-CeuI HEN site in the VC locus, the unique FseI restriction endonuclease (REN) site in the global transactivator (gta) gene and the unique AvrII REN site in the ecdysosteroid UDP-glycosyltransferase (egt) gene. Each gene cassette for T4 ligation was cut from synthetically made donor plasmids by REN digest and agarose gel purified using the QIAEX II Gel Extraction Kit (#20021, Qiagen, Germantown MD). Purified bacmid DNA was linearized by cutting with unique REN sites in the bacmid genome. Bacmid REN digests were heat inactivated, combined with gel purified gene cassettes, and combined with T4 ligase enzyme. Resulting ligations were transformed into E. coli and bacmid clones were selected on 50 ug/mL kanamycin-LB agar plates. PCR primers flanking the location of insertion in the bacmid were used to screen for single copy insertions and bacmid constructs were sequence verified.
2.3.5. Tn7 Locus Cloning into the VoyBac1.1 Bacmid BEV
Foreign genes were cloned into the attTn7 site (Tn7 locus) of the VoyBac1.1 BEVs, RepCap (Figure 1A), LacRepCap (Figure 1B) and LacR-LacOVP1 (Figure 2A) by transforming NEB 10-beta E. coli with the VoyBac1.1 bacmid, the T7 transposase helper plasmid pMON7124 [25] and a foreign gene cassette containing Tn7L/Tn7R donor plasmid [25]. White colony clones were isolated on LB Agar plates with 7 µg/mL gentamycin, 50 µg/mL kanamycin, 10 µg/mL tetracycline, 100 µg/mL Bluo-gal, and 40 ug/mL Isopropyl β-D-1-thiogalactopyranoside (IPTG) (#L1924, Teknova, Hollister CA). Bacmid clones were cultured in 50 µg/mL kanamycin media, purified of total DNA, retransformed into E. coli, colony isolated on 50 ug/mL kanamycin LB agar plates and screened for colonies lacking helper plasmid and donor plasmid. Recombinant Tn7 locus bacmids were confirmed using PCR.
2.3.6. Tn7L-ITR-SEAP-ITR -Tn7R Donor Plasmid and ITR-SEAP Bacmid BEV
To make the ITR-SEAP BEV, a pUC57 based Tn7L-ITR-SEAP-ITR-Tn7R donor plasmid was synthesized (GenScript, Piscataway, NJ) for Tn7 transposition [25] into VoyBac1.1. The inverted terminal repeat (ITR) sequences flanking the secreted embryonic alkaline phosphatase (SEAP) expression cassette were based the AAV2 serotype. The promoter region included a human cytomegalovirus (CMV) gene promoter (GenBank X03922.1) and additional proprietary enhancer elements and was 872 bp in length. The SEAP ORF was based on Homo sapiens (GenBank NM_001632.5) region 53-1568. The 3’ UTR region was 996 bp and included a poly adenylation signal as well as proprietary stuffer sequences such that the final size of ITR-SEAP-ITR transgene cassette was 3,723 bp.
2.4. Sucrose Cushion rAAV Purification
Twenty-five mL volumes of BEV infected Sf9 cells (6.0 x 106 cells/ml) were lysed in culture media by adding 1.25 mL of 10 % w/v triton X-100, 1.25 mL of 2 M arginine and 1.2 ul of 250U/ul benzonase (#E1014, Millipore, Burlington MA). The 50 mL mini bioreactor tubes were placed back at 28 oC and shaken for 2 h at 260 rpm in an orbital shaker. A 300 ul aliquot was set aside for crude lysate rAAV titer determination and the remaining was centrifuged for 7 min at 7,000 x g. Soluble lysate supernatants were transferred to open top, 38.5 ml ultracentrifuge tubes (#344058, Beckman Coulter, Indianapolis IN). Lysates were underlaid with 6 mL of 20% w/w sucrose in PBS (#P0200, Teknova, Mansfield MA). Tubes were placed in an SW32 Ti rotor (#369650, Beckman Coulter) and centrifuged at 30,000 rpm (67,214 x g min to 153,445 x g max) for 1 h at room temperature in a Beckman Coulter Optima LE80 Ultracentrifuge. The supernatant lysate upper layer and sucrose solution were aspirated leaving only the sucrose cushion pellet at the bottom of the tube. 500 ul of PBS was added to the pellets and pellets were suspended in the PBS, transferred to 1.5 mL cryotubes, and suspended. After suspension, cryotubes were centrifuged for 7 min at 20,000 x g in a microcentrifuge. The supernatants were collected and transferred to fresh 2.0 mL cryotubes. These supernatants were stored at 4 oC while experiments were conducted and at -20 oC if longer term storage was needed.
2.5. Affinity Purification, CE-SDS Analysis, and Empty/Full Ratios
Eight hundred mL cultures of BEV infected Sf9 cells were lysed in culture media supplemented with 0.5 % v/v triton X-100, 200 uM arginine and benzonase as described earlier. Cell lysate rAAV capsids were affinity captured using POROSTM Capture SelectTM AAV9 affinity resin (#A27354, Thermo Fisher Scientific). Resin captured rAAV were washed with 20 mM Tris, 1M NaCl (pH 8.0), bridge washed with 50 mM Na2PO4, 350 mM NaCl (pH 5.5), and then eluted with 200 mM glycine, 50 mM NaCl (pH 3.0). Eluted rAAV were neutralized by adding Tris-base to 150 mM final concentration. At all steps in purification contained 0.001% v/v pluronic F-68 (#24040032, Thermo Fisher Scientific). Capillary electrophoresis sodium dodecyl sulfate (CE-SDS) was done using a Sciex PA-800 capillary electrophoresis system. Affinity chromatography purified rAAV capsid samples were diluted in water to a protein concentration of 100 ug/ml. Quantification of separated proteins was done by measuring peak area absorbances at 220 nm and was normalized to the molecular weight to accurately calculate ratios of VP1, VP2 and VP3. Empty-full ratios of capsids were determined using size exclusion chromatography multiangle light scattering (SEC-MALS) as described [28].
2.6. Western Blots
For total cell lysate Western blots, Sf9 cells were collected by centrifugation for 2 min, 20,000 x g and then cell pellets were resuspended at 2.0 x 103 cells/ul in PBS, pH 7.4 (#0010072, Thermo Fisher Scientific). 4X NuPAGE™ LDS Sample Buffer (#NP0007, Thermo Fisher Scientific) and 10X NuPAGE™ Sample Reducing Agent (#NP0004, Thermo Fisher Scientific) were combined to 2X concentrations in PBS, pH 7.4. Resulting 2X LDS Sample Buffer/Reducing Agent was combined with equal volumes of PBS diluted cells. For sucrose cushion sample Western blots, 40 ul of PBS, pH 7.4 solubilized sucrose cushion pellets were diluted with 15 ul of 4X LDS Sample Buffer and 6 ul of 10X Sample Reducing agent. The final sample represented material generated from 2.0 x 105 Sf9 cells/ul. All SDS-PAGE samples were suspended and then heat denatured for 10 min at 80 oC. Volumes of 10 ul were loaded onto NuPAGE™ 4-12% Bis-Tris 1.0 mm X 17 well SDS-PAGE gels (#NP0329BOX, Thermo Fisher Scientific). Proteins were fractionated by electrophoresis in 1X MOPS buffer (#BP-178, Boston BioProducts, Milford MA) for 90 min at 120 V and then transferred to nitrocellulose membranes (#1704159, Bio-Rad, Hercules CA). Blots were blocked 1 h with 5% w/v Blotting-Grade Blocker (#1706404, Bio-Rad) diluted in 1X TBS-T (#IBB-180X, Boston BioProducts). Primary mouse monoclonal antibodies were diluted to 1:2000 in TBS-T and incubated with blocked blots for 90 min. Anti-Lac Repressor antibody used was mouse monoclonal antibody Anti-LacI [9A5] (#ab33832, Abcam, Cambridge MA). Anti-Capsid antibody used was the mouse monoclonal antibody, anti-AAV VP1/VP2/VP3 antibody (B1) (#03-61058, American Research Products, Waltham MA). Secondary goat anti-mouse horse radish peroxidase (HRP) conjugated antibodies were diluted 1:10,000 in TBS-T and incubated for 90 min with primary antibody probed blots after they had been washed with 2X TBS-T three times. The secondary antibody used for all Enhanced Chemiluminescence (ECL) Western blots was a HRP conjugated goat anti-mouse IgG H&L antibody (Abcam, Goat Anti-Mouse IgG H&L (#ab6789, Abcam). Western blot signals were detected using Clarity Western ECL Substrate (#170-5060, Bio-Rad) and an Azure Imager c300 (Azure Biosystems, Dublin CA). All images were collected as Tiff files and analyzed using ImageJ software [29].
2.7. rAAV Transduction Assays.
HEK 293T cells were seeded into 96-well plates at 5.0 x 104 cells/well in 80 ul of Opti-MEM™ Media (#51985091, Thermo Fisher Scientific) supplemented with 5% v/v fetal calf serum. After 2 days of incubation at 37 oC in 5% v/v CO2, cells were transduced with rAAV containing sucrose cushion samples. In a U-bottom 96-well plate, 20 ul volumes of 1/10, 1/20, 1/40 and 1/80 diluted AAV samples in PBS were combined with 200 ul of Opti-MEM media and 8 ul of 100X Antibiotic-Antimycotic (#15240112, Thermo Fisher Scientific). Thirty-five ul volumes of these rAAV dilutions were transferred to wells of the HEK 293T cell plates. This was done in triplicate for each rAAV sample.
2.8. Determination of rAAV Titer by Q-PCR.
Packaged genome copies of transgenes were determined by Q-PCR as described previously [21].
2.9. SEAP Activity Assay Assays.
AAV samples were added to 96-well plates of freshly seeded HEK 293T cells. At 4 days post transduction with rAAV, the media from HEK 293T cells was transferred from 96-well tissue culture plates to 96-well, PCR plates, that were sealed with aluminum covers and heated at 65 oC for 15 min to inactivate cellular alkaline phosphatases. Fifty ul volumes of heat-treated media were transferred into 96-well microtiter plates and combined with 50 ul volumes of p-nitrophenylphosphate BioFX AP-Yellow One (#NC9444916, Thermo Fisher Scientific). Samples were incubated for 24h at room temperature in the dark to endpoint. The optical density at 405 nm (OD405) was read using a Synergy HTX microplate reader (BioTek, Winooski VT). Every assay included a recombinant shrimp alkaline phosphatase (#M0371S, New England Biolabs) standard which had been serially diluted in Opti-MEM media and not heat treated. Shrimp alkaline phosphatase was confirmed by the same assay to have equal unit phosphatase unit activity as calf intestinal alkaline phosphatase (#18009027, Thermo Fisher Scientific).
2.10. Statistical Calculations.
Standard deviations for Q-PCR derived rAAV genome titers or SEAP activity assays were calculated from the average of three repeated samples. Standard deviations for potency (SEAP activity/ rAAV genome) were calculated from the square route of the sum of the squared coefficients of variation of Q-PCR titer and SEAP activity.
3. Results
3.1. Multiple loci BEV cloning of Rep, VP1, VP2, VP3 gene cassettes.
VP1, VP2, LacR and LacR-Rep-VP3 gene cassettes were cloned into VoyBac1.1 in separated locations to ensure genetic stability (See Materials and Methods and Figure 1B). In the 141 kbp LacRepCap BEV these cassettes were separated by 31 kbp, 72 kbp, 23 kbp and 15 kbp respectively. Expression of VP1, VP2, and VP3 was driven by the very late baculovirus p10 promoters. LacR and Rep expression was driven by the polh promoter. A second cloned LacR gene was expressed from a hybrid Opgp64/very late polh promoter (See Materials and Methods and Figure 1G). The LacR protein is a tetramer that binds to two LacO sequences simultaneously [30]. We placed LacO sequences on either side of p10 promoters driving expression of VP1 and VP2 (Figure 1D).
3.2. LacR was confirmed to be able to regulate expression of a LacOVP1 construct in the context of baculovirus infection.
To our knowledge, LacR has not be reported regulating very late baculovirus promoters in the context of baculovirus infection. In Slack & Blissard, 1997 [22], LacR was only used to regulate transcription of plasmids transiently transfected into Sf9 cells. An intermediate recombinant BEV construct called LacR-LacOVP1 (Figure 2A) was used to confirm the ability of LacR to regulate the very late baculovirus p10 promoter. (See Methods & Figure 2A). The LacR-LacOVP1 BEV was evaluated under different IPTG inducer concentrations (Figure 2B and D). We observed a gradient of VP1 expression proportional to IPTG concentration. VP1 expression was maximal at 50 uM IPTG and minimal at 0 uM IPTG and the range repression on this BEV construct was about 50% based on ECL Western blot signal. To improve repression, we later included the second LacR gene with Opgp64/polh hybrid promoter in our ultimate design (Figure 1).
In Western blots, it was noticed that there was a 60 kDa protein in addition to the expected 82.1 kDa VP1 protein (Figure 2B). This corresponded to the 60.6 kDa VP3 protein and could be from leaky translational scanning of the VP1 mRNA transcript. The 60 kDa protein abundance had the same response to IPTG as did VP1. An unexpected observation was to see LacR protein abundance correlating inversely to IPTG concentration. This could be explained by competition for translation as IPTG induction released LacR transcriptional repression of the LacO-p10-LacO promoter controlling VP1 expression.
3.3. Time course of baculovirus infection in Sf9 cells reveals shifting VP ratios in inducible system.
After assembling the complete LacRepCap baculovirus with dual copies of LacR, a time course experiment was done to evaluate LacR and capsid VP protein production (Figure 3). LacR expression at 14 hpi preceded the expression of VP1, VP2 and VP3 capsid. However, between 14 hpi and 24 hpi, VP1 was more abundant than VP2 and similar in abundance to VP3 (Figure 3C). The LacO-p10-LacO very late promoter of VP1 should have had the same temporal expression profile as VP2. We speculate that more abundant early VP1 translation may be from leaky scanning of upstream baculovirus gp64 gene mRNA transcripts which would not be affected by LacR. The LacOVP2 gene was cloned downstream of the baculovirus gta gene promoter which is 50-fold less transcribed than the gp64 gene [31]. We chose to continue working with the current construct design with the acknowledgment that the cumulative capsid ratios seen in Western blots had imperfect homogeneity at the beginning of the baculovirus infection cycle. The vast majority of VP1 and VP2 transcripts would come at very late times from the p10 promoters which are expressed at much higher levels than gp64.
3.4. There is competition among very late promoters as IPTG induces LacR repression of LacO regulated very late promoters.
After the time course expression experiment, the RepLacCap BEV was titrated for IPTG induction of LacOVP1 and LacOVP2 expression (Figure 4). As with the LacR-LacOVP1 BEV, the responsive concentrations of IPTG were between 0 uM and 50 uM. Cummulative VP1 and VP2 abundances increased proportionally in response to IPTG induction of LacR repression of LacO-p10-LacO promoters. Estimated VP1:VP2:VP3 capsid ratios in infected Sf9 cells ranged of ranged from 15:17:68 at 0 uM IPTG to 30:32:38 at 50 uM and 100 uM IPTG. Capsid VP ratios produced in RepCap BEV infected Sf9 cells were unaffected by IPTG and VP1:VP2:VP3 ratios were 8:3:89 and 8:4:87 at 0 uM and 100 uM IPTG respectively. LacR was not able to repress VP1 and VP2 expression enough to obtain a 1:1:10 ratio. When VP1 and VP2 became more abundant, there was an unexpected reciprocal drop in VP3 abundance. This can be explained by the LacO-p10-LacO promoters for VP1 and VP2 drawing away very late transcription factors from the p10 promoter driving expression of VP3.
3.5. Increasing abundances of VP1 and VP2 led to reduced capsid titers at three ITR-SEAP:LacRepCap BEV co-infection ratios.
After determining IPTG concentrations for the regulation of capsid ratios, we then optimized co-infection ratios for the LacRepCap BEV with a SEAP transgene carrying BEV called ITR-SEAP. Sf9 cells were co-infected with ITR-SEAP and LacRepCap BEVs at co-infection ratios ranging from 9:1 to 1:12. In these 25 mL scale mini bioreactor shake flask experiments, initial infection MOIs ranged from 0.01 to 0.12 TCID50 units per cell with for example the 9:1 ratio co-infection receiving 0.09 TCID50 units ITR-SEAP BEV and 0.01 TCID50 units of LacRepCap BEV per cell. Low MOI infections were done to better emulate larger scale production of rAAV in the BEV Sf9 system. For the non-Lac regulated ITR-SEAP:RepCap BEV the optimal co-infection ratio was 3:1. It was surprising to find that the 3:1 co-infection ratio of ITR-SEAP:LacRepCap produced a low titer rAAV compared to ratios as high as 1:12 (Figure 5B). It is possible that the LacRepCap BEV was replicating more slowly but it had a similar BV growth curve as the RepCap BEV (data not shown). Another explanation for needing more of the LacRepCap BEV could be a reduced abundance of Rep protein in the LacRepCap BEV due LacR and capsid VP expression drawing away very late baculovirus transcription factors from the polh promoter driving Rep expression. Western blot analysis for Rep expression did not indicate deficiency of Rep expression in the LacRepCap BEV compared to Rep expression from the RepCap BEV (data not shown). Higher ITR-SEAP:RepCap co-infection ratios of 9:1, 6:1 and 3:1 also produced more abundant VP1 expression relative to VP2 expression. This resembled the overabundance of VP1 that was observed in the time course experiment of LacRepCap at 18 hpi and 22 hpi (Figure 3). In this case, the excessive ITR-SEAP BEVs co-infecting Sf9 cells with LacRepCap BEVs were providing more early and late transcription factors to express the baculovirus gp64 gene upstream of LacOVP1 which we have speculated has translatable transcripts of VP1. Despite the imbalance in VP1 and VP2 at high co-infection ratios, there was still some Lac regulated control of VP ratios. Under all co-infection conditions, rAAV titers were higher when VP1 and VP2 expression was repressed by LacR.
3.6. A high throughput method was developed to purify rAAV from BEV infected Sf9 cells
This inducible system required optimizing both IPTG concentrations and ITR-SEAP:LacRepCap BEV co-infection ratios for rAAV potency on HEK 293T cells. Screening of IPTG concentrations and co-infection conditions often required 25 to 30 samples per experiment. A simple, faster, and reproducible method of rAAV purification was employed based on a single step sucrose cushion ultracentrifugation method [32]. We achieved rapid and consistent recovery of pelleted rAAV particles through 20% sucrose after only 1 h of ultracentrifugation. The recovered soluble rAAV in PBS resuspended sucrose cushion pellets were found to be suitable for HEK 293T cell transduction assays and did not require buffer exchange. This method allowed 30 samples to be processed in one day on one centrifuge. Control empty capsids were found to purify through 20% sucrose cushions at similar abundance to DNA containing capsids produced in presence of ITR-SEAP transgenes. Thus, there was no discrimination of empty and full capsids in this purification method. A disadvantage of this method of AAV capsid purification was that only about 1% of total capsids in cell lysates were being recovered from the pellet after 1 h of ultracentrifugation. Despite this inefficiency, there was more than sufficient material to carry out several 96-well plate HEK 293T transduction assays.
One additional issue we had with purifying rAAV capsids only by sucrose cushion was that baculovirus capsids co-purified. When we used these samples to transduce HEK 293T cells, there was a SEAP background from the ITR-SEAP BEV alone control group which was not detected in the RepCapLac BEV alone control group (data not shown). Initially, we thought the background SEAP activity was coming from baculovirus virion transduction of HEK 239T cells. However, we could not detect the baculovirus envelope fusion protein GP64 by Western blot and the background SEAP activity did not diminish after lysate treatments with higher concentrations of detergents. We discovered that there was SEAP expression by the ITR-SEAP BEV in Sf9 cells from the ITR-SEAP transgene despite the transgene having a mammalian CMV promoter. Resulting SEAP enzyme may be associated with baculovirus capsids co-purifying with AAV capsids through sucrose cushions. The baculovirus capsid protein ORF1629 has an affinity for phosphatase enzymes [33]. The ITR-SEAP BEV alone control preparation SEAP activities were subtracted as residual SEAP background from all ITR-SEAP:RepCapLac co-infection groups. The background residual SEAP activity was not present when rAAV were purified by immunoaffinity purification and baculovirus capsids were eliminated.
3.7. The yield of assembled capsids decreased as VP1 and VP2 expression was increased relative to VP3.
The ITR-SEAP:LacRepCap co-infection ratios of 1:1, 1:3 and 1:6 were selected for further optimization with IPTG concentration. Twenty-five mL scale ITR-SEAP:LacRepCap co-infections of Sf9 cells were done at eight different IPTG concentrations ranging from 0 uM to 200 uM and the rAAV capsids were purified by 20% sucrose cushion from the cell lysates of baculovirus infected insect cells. Western blots were used to detect total expressed VP1, VP2 and VP3 capsid proteins in cell lysates and what was recovered as assembled capsids in sucrose cushion pellets (Figure 6). Total capsid proteins present in cell lysates was only changed marginally as IPTG concentrations went up. The 1:1 co-infection ratio cell lysate had lower total capsid protein yield relative to the 1:3 and 1:6 co-infection ratios. Total capsid protein abundance in sucrose cushion purified samples was more affected over varying IPTG concentrations. For all three co-infection groups increased expression of VP1 and VP2 relative to VP3 reduced the yield of capsid proteins passing through the sucrose cushion. We interpret this as assembled capsids passing through the sucrose cushion and soluble proteins remaining in solution above the sucrose cushion. The capsid ratios in cell lysates differed from the capsid ratios in corresponding sucrose cushion samples. (Figure 6E and 6F). Interestingly, the abundance of VP1 in sucrose cushion samples relative to VP2 and VP3 was more constant over the 0 to 200 uM IPTG compared to the cell lysate samples. There appears to be a limit to the amount of VP1 that can be incorporated into assembled rAAV capsids and less of a limit for VP2 incorporation into capsids.
3.8. The potency of rAAV capsids was tunable using the Lac inducible system for small scale production.
Potency was determined by transducing HEK 293T cells with rAAV capsids containing the ITR-SEAP reporter transgene and measuring the SEAP enzymatic activity relative to rAAV dosage on cells. Before doing potency transduction assays, we measured the rAAV titers from sucrose cushion purified rAAV (Figure 7A). There was a drop in the rAAV titer of sucrose cushion purified capsids with increased IPTG concentrations. This likely was due to the lower abundance of capsid proteins in sucrose cushion samples as IPTG concentrations increased (Figure 6D). The potency of recovered capsids did not follow capsid abundance and capsid titer which both peaked at 0 uM IPTG. Instead, potency was highest between 1 uM and 2 uM IPTG for all three co-infection groups (Figure 7B). Potency dropped by six-fold at 0 uM IPTG and at 5 uM IPTG. Western blots did not show significant differences in VP ratios between 0 uM and 5 uM IPTG. It is possible that there were different ratios over the baculovirus infection cycle which were not observable at the cumulative final harvest time point.
3.9. Larger scale production of rAAV in Sf9 cells confirmed that there was limited incorporation of VP1 into capsids.
Data presented thus far was generated from sucrose cushion purified rAAV samples generated at small 25 mL scale. Production of rAAV for drug therapeutics involves larger scale Sf9 cultures and affinity chromatography-based purification [ 34]. We scaled up to 800 mL Sf9 cultures and co-infected these cells with ITR-SEAP and LacRepCap BEVs at 1:6 ratio (0.1:0.6 moi). These co-infections were done at four IPTG concentrations; 0 uM, 2 uM, 10 uM and 50 uM IPTG. Sf9 cell lysates made at this time were subjected to affinity chromatography to purify rAAV capsids. A portion of those affinity purified capsids were further purified by sucrose cushion ultracentrifugation. Expressed capsid VP ratios in cell lysates were determined by Western blot (Figure 8A) or by capillary electrophoresis sodium dodecyl sulfate (CE-SDS) for both affinity purified capsids (Figure 8B) and affinity/sucrose cushion purified capsids (Figure 8C). The most noticeable trend was the nearly constant proportion of VP1 in both affinity and affinity/sucrose cushion purified rAAV capsids regardless of the IPTG concentration. In contrast, the relative abundance of VP2 in purified capsids increased with IPTG concentration just as it did in cell lysates. These data corroborate with 25 mL scale data and show that capsid assembly is not stochastic with there being a limit to the amount of VP1 that can be incorporated into capsids.
3.10. Larger scale production did not show expected optimal potency at 2 uM IPTG.
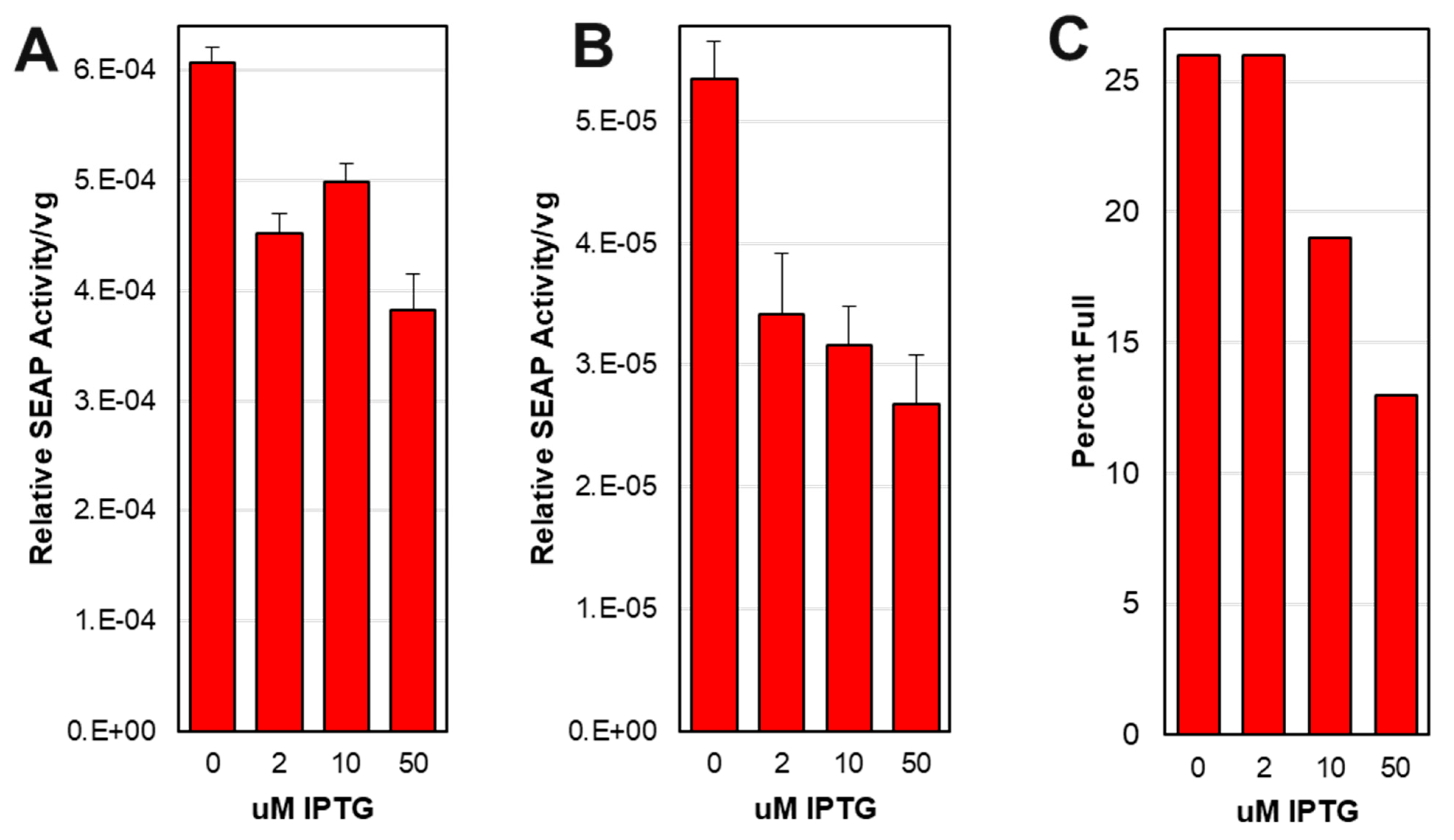
We did not see the expected increased potency when capsids were produced in presence of 2 uM IPTG as was observed in smaller 25 mL scale experiments (Figure 7B). Instead, rAAV potency on HEK 293T cells was highest at 0 uM IPTG (Figure 9A and 9B). The percent of capsids containing ITR-SEAP transgenes in affinity purified samples was measured using SEC-MALS and was found highest at 0 uM IPTG and lowest at 50 uM IPTG (Figure 9C).
4. Discussion
The E. coli Lac repressor (LacR) was selected in this study as a regulator of VP1 and VP2 expression due to its success in other eukaryotic virus platforms such as Vaccinia [35,36,37] and adenoviruses [38]. LacR is a component of the E. coli Lac Operon, that was the first described inducible regulatory system [39] and which has been well characterized (For review see Lewis 2005 [40]). LacR was previously shown to be functional in insect cells and was allosterically regulated (induced) by IPTG [22,41].
In nature, VP1, VP2, and VP3 evolved to be overlapping ORFs due the restricted genome capacity AAV. These overlapping ORFs were emulated when researchers first cloned rAAV contructs into the BEV system [9,10]. The BEV system does not have a restricted for genome size like AAV. Capsid protein VP1, VP2 and VP3 ORFs were separately cloned and stably expressed from three different loci in the BEV genome. We also cloned and expressed the E. coli LacR gene into another unique loci into the BEV genome. This modular recombinant BEV design was stable and allowed for independent modifications of the various elements. Engineering the baculovirus p10 promoters of VP1 and VP2 genes with double LacO’s enabled IPTG inducible LacR regulation. This is the first report of a very late “hyperexpressed” baculovirus promoter being placed under inducible regulation. Due to the strength of p10 transcription, the repression of VP1 and VP2 expression was not complete. Repression was sufficient for the intended purpose of regulating the expression of VP1 and VP2 relative to VP3 for successful AAV capsid assembly.
IPTG induction of LacR repression of VP1 and VP2 expression relative to VP3 was tunable such that the potency of resulting capsids could be optimized in small 25 mL scale Sf9 cultures. This did not translate to larger 800 mL scale production with the non-induced LacR repression producing the most potent rAAV. A possible reason for this was that the ITR-SEAP BEV and RepLacCap BEV co-infection ratios at larger scale needed to be further optimized. As shown in Figure 7, having too little LacRepCap BEV relative to ITR-SEAP BEV reduced the ability to tune potency with IPTG.
From this work, it can be concluded that there was decline in total assembled capsids and rAAV titers as expressed VP1 and VP2 abundances were increased relative to VP3. Our data agree with the findings of Gao et al., 2014 [42] that showed VP1 and VP2 abundances influenced capsid yield. We saw this trend of expressed VP1 and VP2 abundance affecting yield to be consistent with repeated experiments and with other rAAV9 capsid variants (data not shown). Our results contradict the findings of Bosma et al., 2018 [17] and show a decline in the percent full resulting from increased expression of VP1 and VP2 (Figure 9C). A major difference in that study was that it used a rAAV5 serotype which does not require expression of the frame shifted AAP protein found nestled in the common sequences of VP1 and VP2 ORFs.
We did not address in this study the impact of AAP expression as VP1 and VP2 expression was modulated by IPTG induction of VP1 and VP2 expression. Both VP1 and VP2 ORFs have complete copies of the AAP ORF. The canonical ATG start codons for VP1 and VP2 would likely lead to less translational leaky scanning and thus less translation from the downstream non-canonical CTG start codon for AAP. However, as VP1 and VP2 transcription increased with IPTG induction, there would be more AAP produced as are result of leaking translational leaky scanning.
Capsid ratio analysis methods like CE-SDS or Western blot used here show only the mean distribution of VP1, VP2 and VP3 in purified capsids. The VP ratios found in individual capsids is a random normal distribution of VP1, VP2 and VP3 dependent on starting abundances of VP’s during assembly [43]. To complicate this further, the BEV in this Lac inducible system did not offer a steady state relative expression of VP1, VP2 and VP3 due to transcriptional interference from surrounding baculovirus genes in the separated cloning loci.
Our data show that an upper limit to the amount of VP1 that which can be included in assembled capsids. This is best illustrated in Figure 8 where crude cell lysate abundance of VP1 was 20% at 50 uM IPTG and was only 9% in the affinity/sucrose cushion purified capsids. At the same time, VP2 abundance in the cell lysate was 31% and was 27% in the affinity/sucrose cushion purified capsids. It is concluded that assembled capsids tolerate a higher abundance of VP2 compared to VP1. Overabundance of VP1 leads to reduced levels of capsid assembly and reduced potency. We have also previously observed that deficiency of VP1 in capsids does not affect capsid assembly or titer but does reduce capsid potency. Too much or too little VP1 reduces potency.
It has been suggested that IPTG should not be used in processes to make human therapeutics because it is too costly and is toxic to humans [44]. Kaneda et al., 1998 [45] reported IPTG to be nontoxic to Hela cells at 5,000 uM, Cronin et al., 2001 [46] found IPTG toxicity in mice did not occur until concentrations were at 20,000 uM and Figge et al.,1988 [47] reported toxicity of IPTG to monkey cell lines only when concentrations reached 50,000 uM. In the current study, we found IPTG to be well tolerated by Sf9 cells and were able to culture Sf9 cells in IPTG concentrations as high as 18,000 uM IPTG with no effect on cell growth. This concentration of IPTG was 1000-fold higher than the working IPTG concentration range needed in our inducible system. In addition, after rAAV virions are purified from baculovirus infected insect cell lysates by affinity chromatography, IPTG would not be expected to be present at significant concentrations in the final drug product. At time of this study, the cost of the ESF AF insect cell culture media was $62/L. Animal free high purity IPTG (mw 238.3) cost $205 for 5g (CAS 367-93-1 Calbiochem). Even at the highest foreseeable concentration of 100 uM IPTG, we would need 24 mg of IPTG/L costing $0.98/L, a 1.6% increase in media cost. The optimal IPTG concentration for rAAV potency in this study was 2 uM IPTG thus cutting IPTG cost to 0.03% of media cost. IPTG is not too costly or too toxic at the working concentrations for this system.
The purpose of this work was to improve the potency of rAAV9 serotype capsids produced in the Sf9/BEV system. While we demonstrated repression of VP1 and VP2 expression via LacR regulation, further refinement of the transcriptional elements may enable LacR regulation that is more viable and tunable for rAAV. We also desired better understanding of how AAV capsid protein ratios affected rAAV. This was achieved and we gained better understanding of the semi-stochastic nature of AAV assembly.
Author Contributions
Conceptualization J.S.; methodology, J.S. and C.G.; validation, C.G., A.I., and J.S.; investigation, J.S.; writing-original draft preparation, J.S.; writing-review and editing, J.S.; supervision, J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Additional data require confidential disclosure agreement (CDA) with Voyager Therapeutics
Acknowledgments
We acknowledge Zeynep Guillemin, Andrade Hendricks, Peter Slade, Shamik Sharma, and Luis Maranga for their valuable support and facilitation of this work.
Conflicts of Interest
C.N., A.I. and J.S. own stock shares in Voyager Therapeutics.
Disclosures
Authors C.N: A.I., and J.S. were employees of Voyager Therapeutics, Inc. when this work was done. This work pertains to patent applications by Voyager Therapeutics, Inc., in which C.N., A.I., and J.S. are named inventors.
References
- Blacklow, N.R.; Hoggan, M.D.; Rowe, W.P. Immunofluorescent studies of the potentiation of an adenovirus-associated virus by adenovirus 7. J. Exp. Med. 1967, 125, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ogston, P.; Raj, K.; Beard, P. Productive replication of adeno-associated virus can occur in human papillomavirus type 16 (HPV-16) episome-containing keratinocytes and is augmented by the HPV-16 E2 protein. J. Virol. 2000, 74, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Kotin, R.M.; Menninger, J.C.; Ward, D.C.; Berns, K.I. Mapping and direct visualization of a region-specific viral DNA integration site on chromosome 19q13-qter. Genomics 1991, 10, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V; et al. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, M.; Wang, W.; Child, M.A.; Ren, X.Q.; Huang, C.; Ren, A.Z.; Tocci, J.; Chen, Q.; Bittner, K.; Tyson, K.; Pande, N.; Chung, C.H.; Paul, S.M.; Hou, J. Rapid evolution of blood-brain-barrier-penetrating AAV capsids by RNA-driven biopanning. Mol. Ther. Methods Clin. Dev. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Urabe, M.; Ding, C.; Kotin, R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002, 13, 1935–1943. [Google Scholar] [CrossRef]
- Smith, R.H.; Levy, J.R.; Kotin, R.M. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer AAV stocks from insect cells. Mol. Ther. 2009, 17, 1888–1896. [Google Scholar] [CrossRef]
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.C.; Pacouret, S.; Cepeda Diaz, A.K.; Blake, J.; Andres-Mateos, E.; et al. The assembly-activating protein promotes stability and interactions between AAV’s viral proteins to nucleate capsid assembly. Cell Rep. 2018, 23, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Maizel JVJr Inman, J.K.; Shatkin, A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971, 8, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, B.; Yarbrough, J.; Domsic, J.; Bennett, A.; Bothner, B.; et al. Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 2013, 87, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.B.; Ozer, H.L.; Hoggan, M.D. Structural proteins of adenovirus-associated virus type 3. J. Virol. 1971, 8, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, S.; Kleinschmidt, J.A.; Böttcher, B. Electron cryo-microscopy and image reconstruction of adeno-associated virus type 2 empty capsids. EMBO Rep. 2001, 2, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Bosma, B.; du Plessis, F.; Ehlert, E.; Nijmeijer, B.; de Haan, M.; et al. Optimization of viral protein ratios for production of AAV serotype 5 in the baculovirus system. Gene Ther. 2018, 25, 415–424. [Google Scholar] [CrossRef]
- Kondratov, O.; Zolotukhin, S. Exploring the Comprehensive Kozak Sequence Landscape for AAV Production in Sf9 System. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Summers, M.D.; Smith, G.E. A manual of methods for baculovirus vectors and insect cell culture procedures. Tex. Agric. Exp. Stn. Bull. No. 1555 1987. [Google Scholar]
- Vaughn, J.L.; Goodwin, R.H.; Tompkins, G.J.; McCawley, P. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 1977, 13, 213–217. [Google Scholar] [CrossRef]
- Nguyen, C.; Ibe-Enwo, A.; Slack, J. A Baculovirus Expression Vector Derived Entirely from Non-Templated, Chemically Synthesized DNA Parts. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.M.; Blissard, G.W. Identification of two independent transcriptional activation domains in the Autographa californica multicapsid nuclear polyhedrosis virus IE1 protein. J. Virol. 1997, 71, 9579–9587. [Google Scholar] [CrossRef]
- Blissard, G.W.; Rohrmann, G.F. Baculovirus gp64 gene expression: analysis of sequences modulating early transcription and transactivation by IE1. J. Virol. 1991, 65, 5820–5827. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.J.; Blissard, G.W. Baculovirus gp64 gene expression: negative regulation by a minicistron. J. Virol. 1997, 71, 7448–7460. [Google Scholar] [CrossRef] [PubMed]
- Luckow, V.A.; Lee, S.C.; Barry, G.F.; Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 1993, 67, 4566–4579. [Google Scholar] [CrossRef]
- Slack, J.M.; Kuzio, J.; Faulkner, P. Characterization of v-cath, a cathepsin L-like proteinase expressed by the baculovirus Autographa californica multiple nuclear polyhedrosis virus. J. Gen. Virol. 1995, 76 Pt 5, 1091–1098. [Google Scholar] [CrossRef]
- Lihoradova, O.A.; Ogay, I.D.; Abdukarimov, A.A.; Azimova, S.h.S.; Lynn, D.E.; et al. The Homingbac baculovirus cloning system: An alternative way to introduce foreign DNA into baculovirus genomes. J. Virol. Methods 2007, 140, 59–65. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, N.L.; Berguig, G.Y.; Karim, O.A.; Cortesio, C.L.; De Angelis, R.; Khan, A.A.; Gold, D.; Maga, J.A.; Bhat, V.S. Comprehensive characterization and quantification of adeno associated vectors by size exclusion chromatography and multi angle light scattering. Sci. Rep. 2021, 11, 3012. [Google Scholar] [CrossRef]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Oehler, S.; Eismann, E.R.; Krämer, H.; Müller-Hill, B. The three operators of the lac operon cooperate in repression. EMBO J. 1990, 9, 973–979. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zhong, S.; Fei, Z.; Hashimoto, Y.; Xiang, J.Z.; et al. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Papaneri, A.; Walker, M.; Scappini, E.; Keys, R.D.; et al. A simple, two-step, small-scale purification of recombinant adeno-associated viruses. J. Virol. Methods 2020, 281, 113863. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Koyano, Y.; Kang, W.; Kokusho, R.; Kamita, S.G.; et al. The baculovirus uses a captured host phosphatase to induce enhanced locomotory activity in host caterpillars. PLoS Pathog. 2012, 8, e1002644. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Smith, J.K.; Yu, J.C.; Banala, V.; Emmanuel, S.N.; et al. Characterization of AAV-Specific Affinity Ligands: Consequences for Vector Purification and Development Strategies. Mol. Ther. Methods Clin. Dev. 2020, 19, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, T.R.; Fernandez, M.P.; Moss, B. Transfer of the inducible lac repressor/operator system from Escherichia coli to a vaccinia virus expression vector. Proc. Natl. Acad. Sci. USA 1989, 86, 2549–2553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Crews, C.J.; Derdeyn, C.A.; Blackwell, J.L. Lac-regulated system for generating adenovirus 5 vaccine vectors expressing cytolytic human immunodeficiency virus 1 genes. J. Virol. Methods 2009, 160, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Xiao, W.; Americo, J.L.; Earl, P.L.; Moss, B. Novel nonreplicating vaccinia virus vector enhances expression of heterologous genes and suppresses synthesis of endogenous viral proteins. mBio 2017, 8, e00790-17. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Cummings, D.; Evelegh, C.; Graham, F.L.; Prevec, L. Development and use of a 293 cell line expressing lac repressor for the rescue of recombinant adenoviruses expressing high levels of rabies virus glycoprotein. J. Gen. Virol. 1999, 80 Pt 2, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Lewis, M. The lac repressor. C R Biol. 2005, 328, 521–548. [Google Scholar] [CrossRef]
- Slack, J.M.; Blissard, G.W. Measurement of membrane fusion activity from viral membrane fusion proteins based on a fusion-dependent promoter induction system in insect cells. J. Gen. Virol. 2001, 82 Pt 10, 2519–2529. [Google Scholar] [CrossRef]
- Gao, K.; Li, M.; Zhong, L.; Su, Q.; Li, J.; et al. Empty virions in AAV8 vector preparations reduce transduction efficiency and may cause total viral particle dose-limiting side-effects. Mol. Ther. Methods Clin. Dev. 2014, 1, 20139. [Google Scholar] [CrossRef]
- Wörner, T.P.; Snijder, J.; Bennett, A.; Agbandje-McKenna, M.; Makarov, A.A.; et al. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 2020, 17, 395–398. [Google Scholar] [CrossRef]
- Briand, L.; Marcion, G.; Kriznik, A.; Heydel, J.M.; Artur, Y.; et al. A self-inducible heterologous protein expression system in Escherichia coli. Sci. Rep. 2016, 6, 33037. [Google Scholar] [CrossRef]
- Kaneda, Y.; Kinoshita, K.; Sato, M.; Saeki, Y.; Yamada, R.; et al. The induction of apoptosis in HeLa cells by the loss of LBP-p40. Cell Death Differ. 1998, 5, 20–28. [Google Scholar] [CrossRef]
- Cronin, C.A.; Gluba, W.; Scrable, H. The lac operator-repressor system is functional in the mouse. Genes Dev. 2001, 15, 1506–1517. [Google Scholar] [CrossRef]
- Figge, J.; Wright, C.; Collins, C.J.; Roberts, T.M.; Livingston, D.M. Stringent regulation of stably integrated chloramphenicol acetyl transferase genes by E. coli lac repressor in monkey cells. Cell 1988, 52, 713–722. [Google Scholar] [CrossRef]
Figure 1.
BEV Constructs and Promoter designs used. The RepCap BEV (A) and LacRepCap BEV (B) designs are shown. The RepCap baculovirus had foreign genes transposed into a mini-AttTn7 locus while the LacRepCap Baculovirus also had foreign genes also T4 ligase ligated into the VC locus, the gta locus and the egt locus. (C) The RepCap BEV had overlapping VP1, VP2, VP3 ORFs expressed by very late (VL) p10 promoter. VP1 and VP2 had non-canonical translational initiation codons and VP3 had an ATG start codon. The LacRepCap BEV had VP1, VP2 and VP3 ORFs were separately cloned with ATG translational start codons and p10 promoters. (D) The p10 promoters of VP1 and VP2 were flanked by LacO spaced 188 bp apart. (E) Both BEVs had overlapping Rep78/Rep52 ORFs co-expressed by a VL polyhedrin (polh) promoter (E) with Rep78 having a non-canonical translational initiation codon and Rep52 having an ATG start codon. The LacRepCap BEV had two LacR genes in the virus. (F) The EGT locus LacR had a VL polh promoter. (G) The copy of LacR in the the Tn7 locus also had an early (E) and late (L) OpMNPV gp64 promoter upstream of the VL polh promoter.
Figure 1.
BEV Constructs and Promoter designs used. The RepCap BEV (A) and LacRepCap BEV (B) designs are shown. The RepCap baculovirus had foreign genes transposed into a mini-AttTn7 locus while the LacRepCap Baculovirus also had foreign genes also T4 ligase ligated into the VC locus, the gta locus and the egt locus. (C) The RepCap BEV had overlapping VP1, VP2, VP3 ORFs expressed by very late (VL) p10 promoter. VP1 and VP2 had non-canonical translational initiation codons and VP3 had an ATG start codon. The LacRepCap BEV had VP1, VP2 and VP3 ORFs were separately cloned with ATG translational start codons and p10 promoters. (D) The p10 promoters of VP1 and VP2 were flanked by LacO spaced 188 bp apart. (E) Both BEVs had overlapping Rep78/Rep52 ORFs co-expressed by a VL polyhedrin (polh) promoter (E) with Rep78 having a non-canonical translational initiation codon and Rep52 having an ATG start codon. The LacRepCap BEV had two LacR genes in the virus. (F) The EGT locus LacR had a VL polh promoter. (G) The copy of LacR in the the Tn7 locus also had an early (E) and late (L) OpMNPV gp64 promoter upstream of the VL polh promoter.
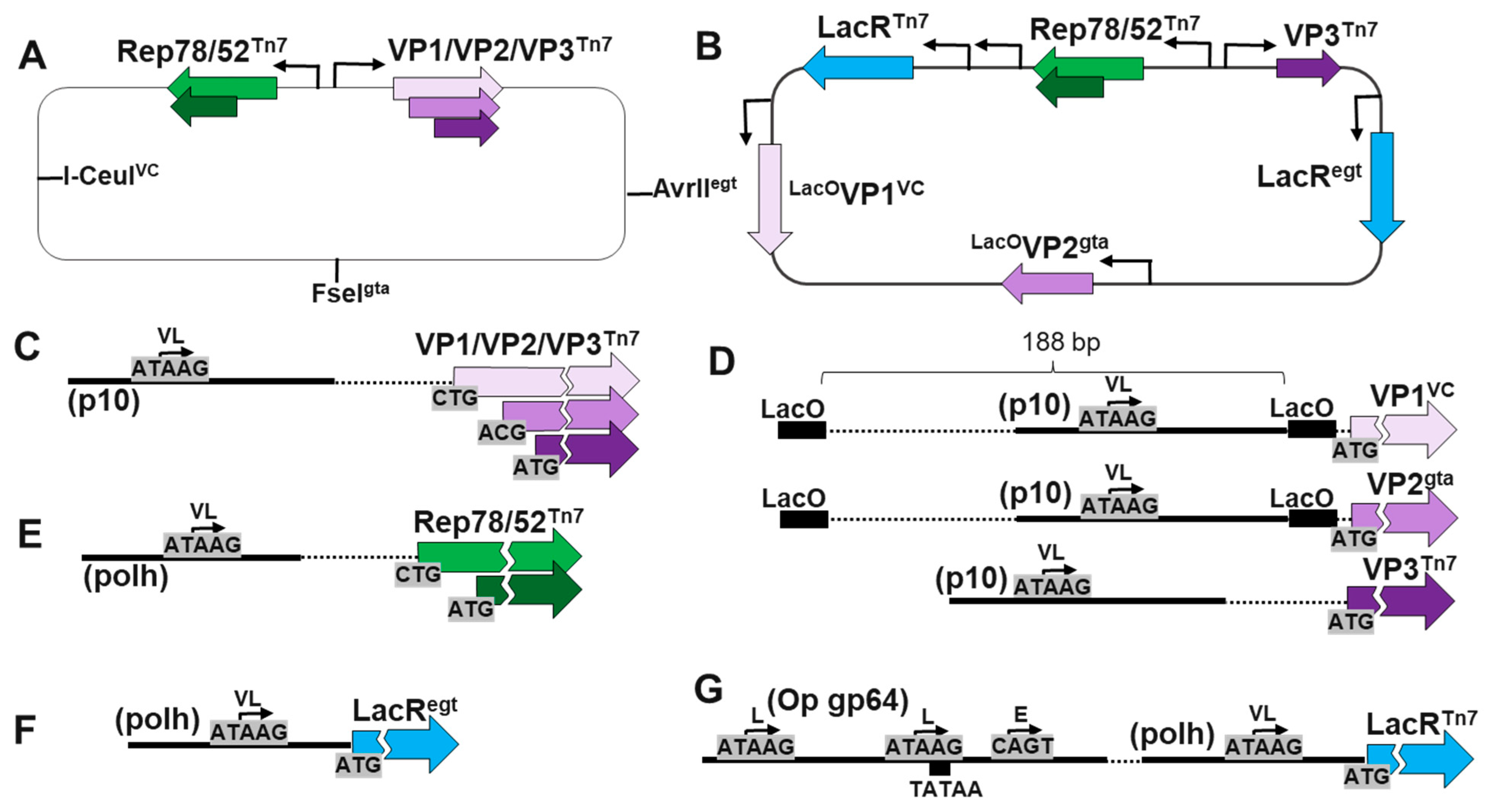
Figure 2.
LacR Regulation of LacO-p10-VP1 Expression. Sf9 cells were infected at 10 moi with a recombinant LacR-LacOVP1 BEV (A) with a polh promoter LacR in locus A and lacO-p10 promoter VP1 in locus B. At 72 hpi cells were collected, fractionated by SDS-PAGE and then Western blot probed with (B) anti-capsid monoclonal antibody and (C) anti-lac repressor antibody. Relative abundances of ECL proteins were quantified using ImageJ software and plotted (D).
Figure 2.
LacR Regulation of LacO-p10-VP1 Expression. Sf9 cells were infected at 10 moi with a recombinant LacR-LacOVP1 BEV (A) with a polh promoter LacR in locus A and lacO-p10 promoter VP1 in locus B. At 72 hpi cells were collected, fractionated by SDS-PAGE and then Western blot probed with (B) anti-capsid monoclonal antibody and (C) anti-lac repressor antibody. Relative abundances of ECL proteins were quantified using ImageJ software and plotted (D).
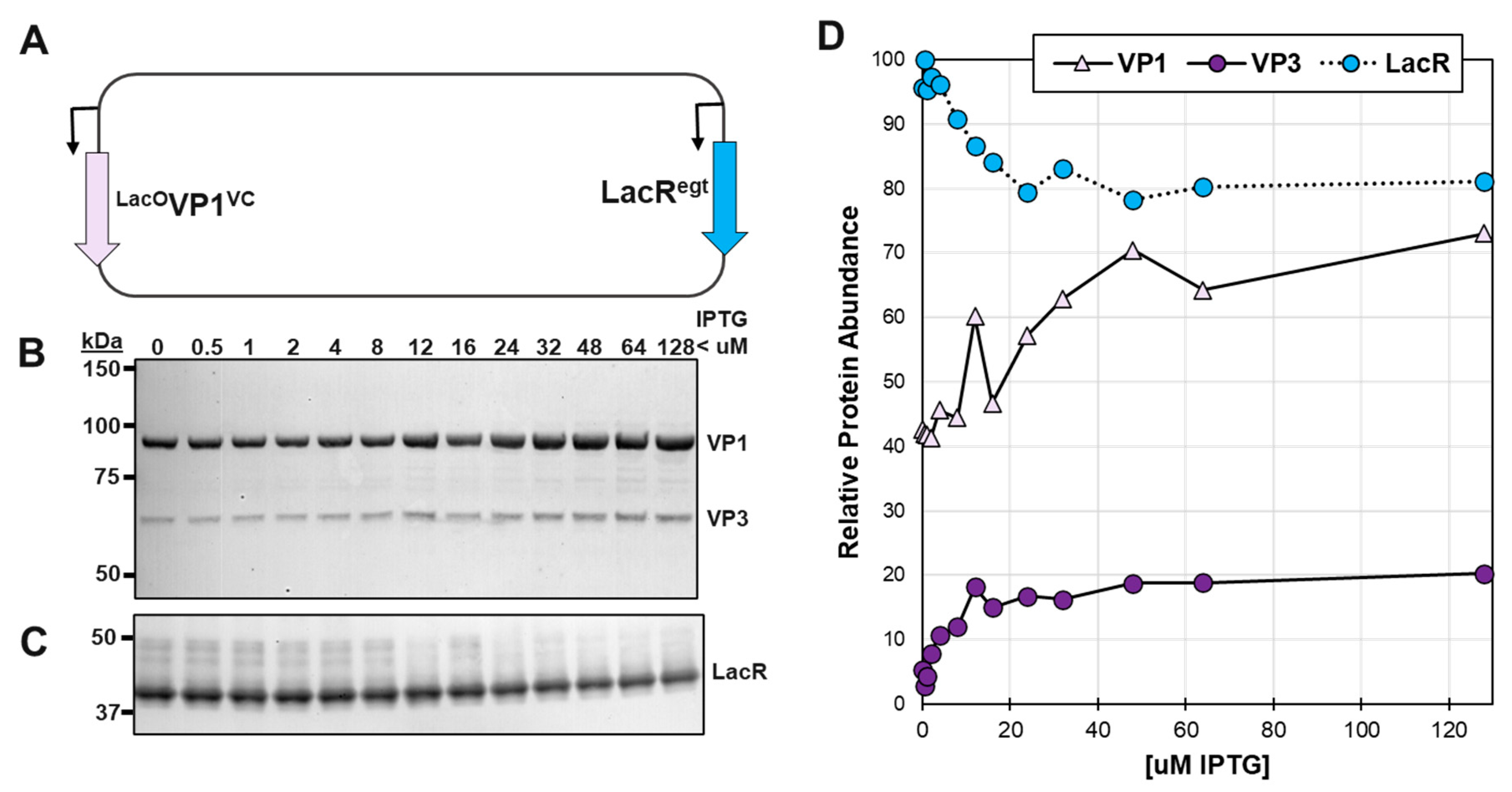
Figure 3.
Time Course of Cap and Lac Expression in Baculovirus Infected Cells. Sf9 cells were infected at 10 MOI with LacRepCap baculovirus. Cells were collected at different hours post infection from 0 hpi to 50 hpi. (A) Cell proteins were fractionated by SDS-PAGE and then Western blot probed with anti-capsid monoclonal antibody and anti-lac repressor antibody. (B) ECL signals of Western detected proteins were quantified using ImageJ software. (C) Relative capsid VP ratios are expressed in bar graph format.
Figure 3.
Time Course of Cap and Lac Expression in Baculovirus Infected Cells. Sf9 cells were infected at 10 MOI with LacRepCap baculovirus. Cells were collected at different hours post infection from 0 hpi to 50 hpi. (A) Cell proteins were fractionated by SDS-PAGE and then Western blot probed with anti-capsid monoclonal antibody and anti-lac repressor antibody. (B) ECL signals of Western detected proteins were quantified using ImageJ software. (C) Relative capsid VP ratios are expressed in bar graph format.
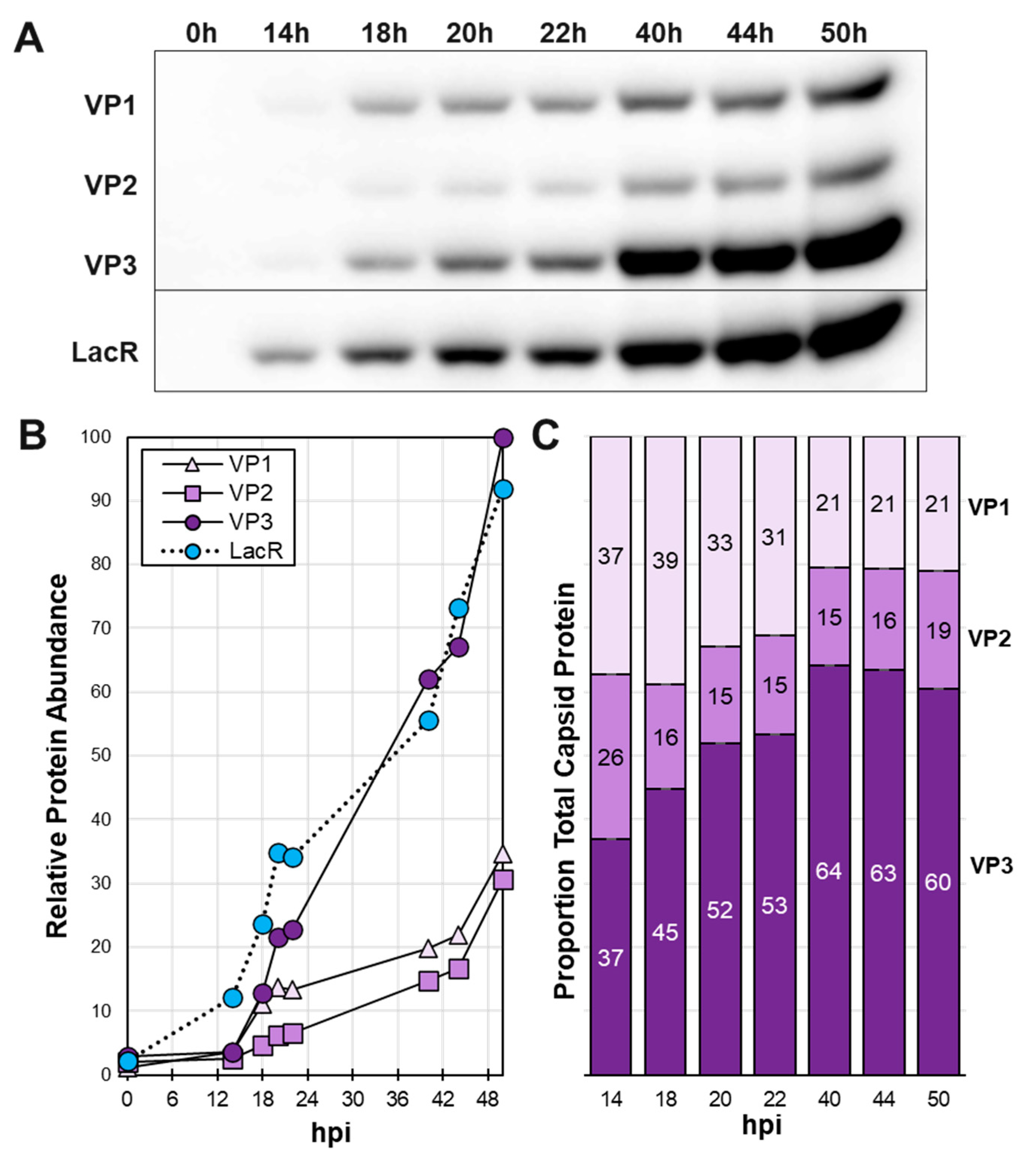
Figure 4.
Titration of IPTG Induction of VP1 and VP2 expression. Sf9 cells were infected with LacRepCap baculovirus or RepCap baculovirus in presence of different concentrations of IPTG. Cells were collected at 72 hpi and their proteins were fractionated by SDS-PAGE and Western blot probed with anti-Capsid monoclonal antibody. Relative capsid abundances in LacRepCap (A) and RepCap (B) infected cells were observed by Western ECL signals which were quantified using ImageJ software. (C) Capsid abundances for LacRepCap baculovirus infected cells graphed relative to IPTG concentration (D). Calculated capsid ratios are expressed in bar graph format for the LacRepCap baculovirus and the RepCap baculovirus.
Figure 4.
Titration of IPTG Induction of VP1 and VP2 expression. Sf9 cells were infected with LacRepCap baculovirus or RepCap baculovirus in presence of different concentrations of IPTG. Cells were collected at 72 hpi and their proteins were fractionated by SDS-PAGE and Western blot probed with anti-Capsid monoclonal antibody. Relative capsid abundances in LacRepCap (A) and RepCap (B) infected cells were observed by Western ECL signals which were quantified using ImageJ software. (C) Capsid abundances for LacRepCap baculovirus infected cells graphed relative to IPTG concentration (D). Calculated capsid ratios are expressed in bar graph format for the LacRepCap baculovirus and the RepCap baculovirus.
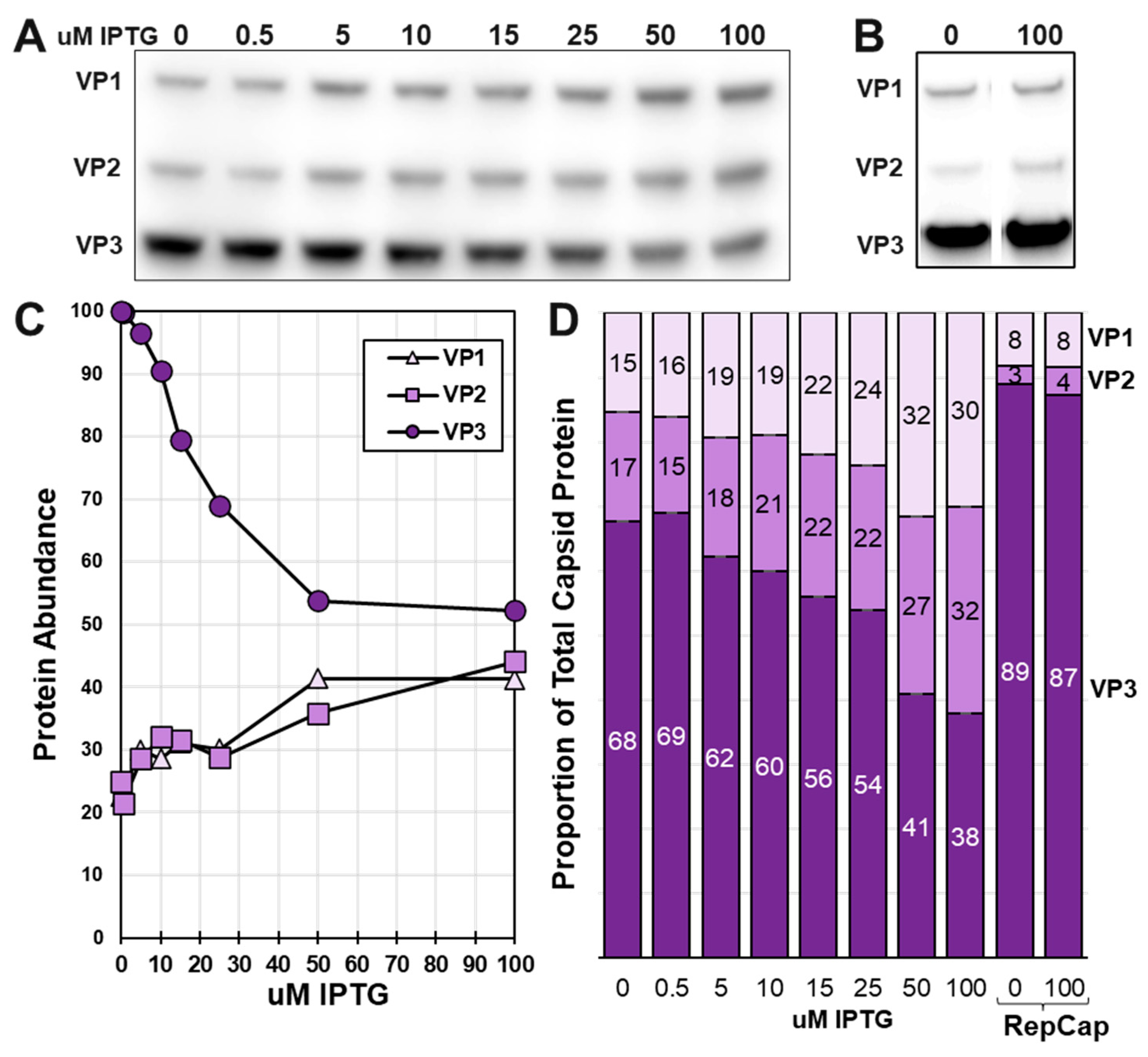
Figure 5.
Optimizing ITR-SEAP and LacRepCap BEV Co-infection Ratios. Sf9 cells were co-infected with an ITR-SEAP and LacRepCap BEVs at different co-infection ratios in the presence of 0 uM and 200 uM IPTG. (A) Cell lysate protein samples were fractionated by SDS-PAGE and Western blot probed with anti-capsid antibody. (B) Crude cell lysate titers for ITR-SEAP-GFP-ITR were determined by Q-PCR. (C) Corresponding estimated capsid ratios are expressed in bar graph format for 0 uM IPTG and 200 uM IPTG.
Figure 5.
Optimizing ITR-SEAP and LacRepCap BEV Co-infection Ratios. Sf9 cells were co-infected with an ITR-SEAP and LacRepCap BEVs at different co-infection ratios in the presence of 0 uM and 200 uM IPTG. (A) Cell lysate protein samples were fractionated by SDS-PAGE and Western blot probed with anti-capsid antibody. (B) Crude cell lysate titers for ITR-SEAP-GFP-ITR were determined by Q-PCR. (C) Corresponding estimated capsid ratios are expressed in bar graph format for 0 uM IPTG and 200 uM IPTG.
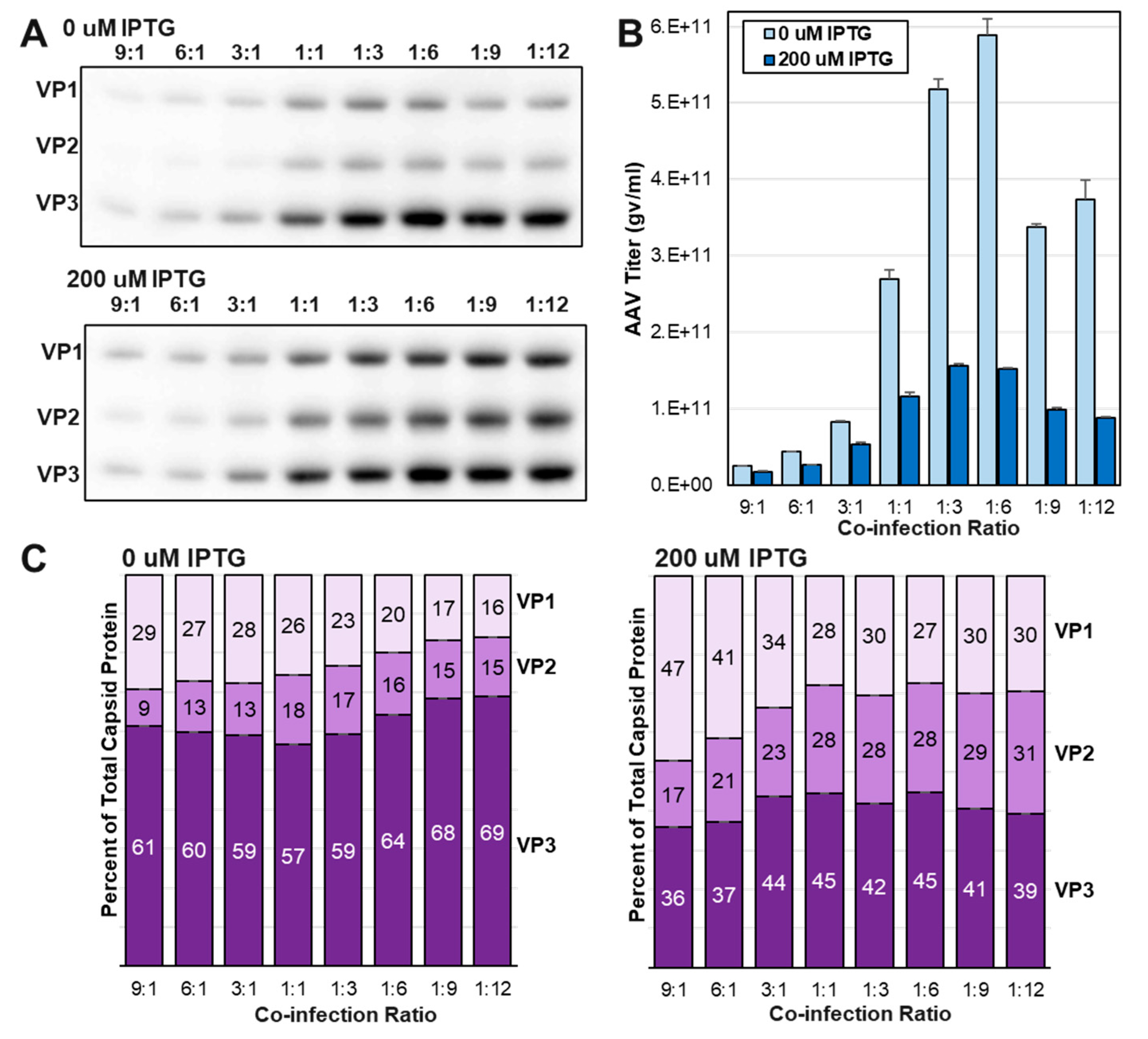
Figure 6.
Effect of IPTG of total AAV Capsid Protein Abundance in Cells and in Purified AAV Capsids. Sf9 cells were co-infected with ITR and LacRepCap baculoviruses at co-infection ratios of 1:1, 1:3 and 1:6 in the presence of different concentrations of IPTG. Protein samples were obtained from cell lysates (A) and from sucrose cushion ultracentrifugation pellet fractions of cell lysates (B). All samples were fractionated by SDS-PAGE and Western blot probed with anti-capsid monoclonal antibody. Immunolabelled capsid proteins were detected by ECL and quantified by using ImageJ software. Total capsid protein abundances in cell lysates (C) and sucrose cushion pellets (D) were plotted relative to IPTG concentration for each co-infection group; 1:1 (open triangle), 1:3 (black diamond) and 1:6 (open circle). Estimated capsid ratios for the 1:6 co-infection is expressed in bar graph format for cell lysate (E) and sucrose cushion samples (F).
Figure 6.
Effect of IPTG of total AAV Capsid Protein Abundance in Cells and in Purified AAV Capsids. Sf9 cells were co-infected with ITR and LacRepCap baculoviruses at co-infection ratios of 1:1, 1:3 and 1:6 in the presence of different concentrations of IPTG. Protein samples were obtained from cell lysates (A) and from sucrose cushion ultracentrifugation pellet fractions of cell lysates (B). All samples were fractionated by SDS-PAGE and Western blot probed with anti-capsid monoclonal antibody. Immunolabelled capsid proteins were detected by ECL and quantified by using ImageJ software. Total capsid protein abundances in cell lysates (C) and sucrose cushion pellets (D) were plotted relative to IPTG concentration for each co-infection group; 1:1 (open triangle), 1:3 (black diamond) and 1:6 (open circle). Estimated capsid ratios for the 1:6 co-infection is expressed in bar graph format for cell lysate (E) and sucrose cushion samples (F).
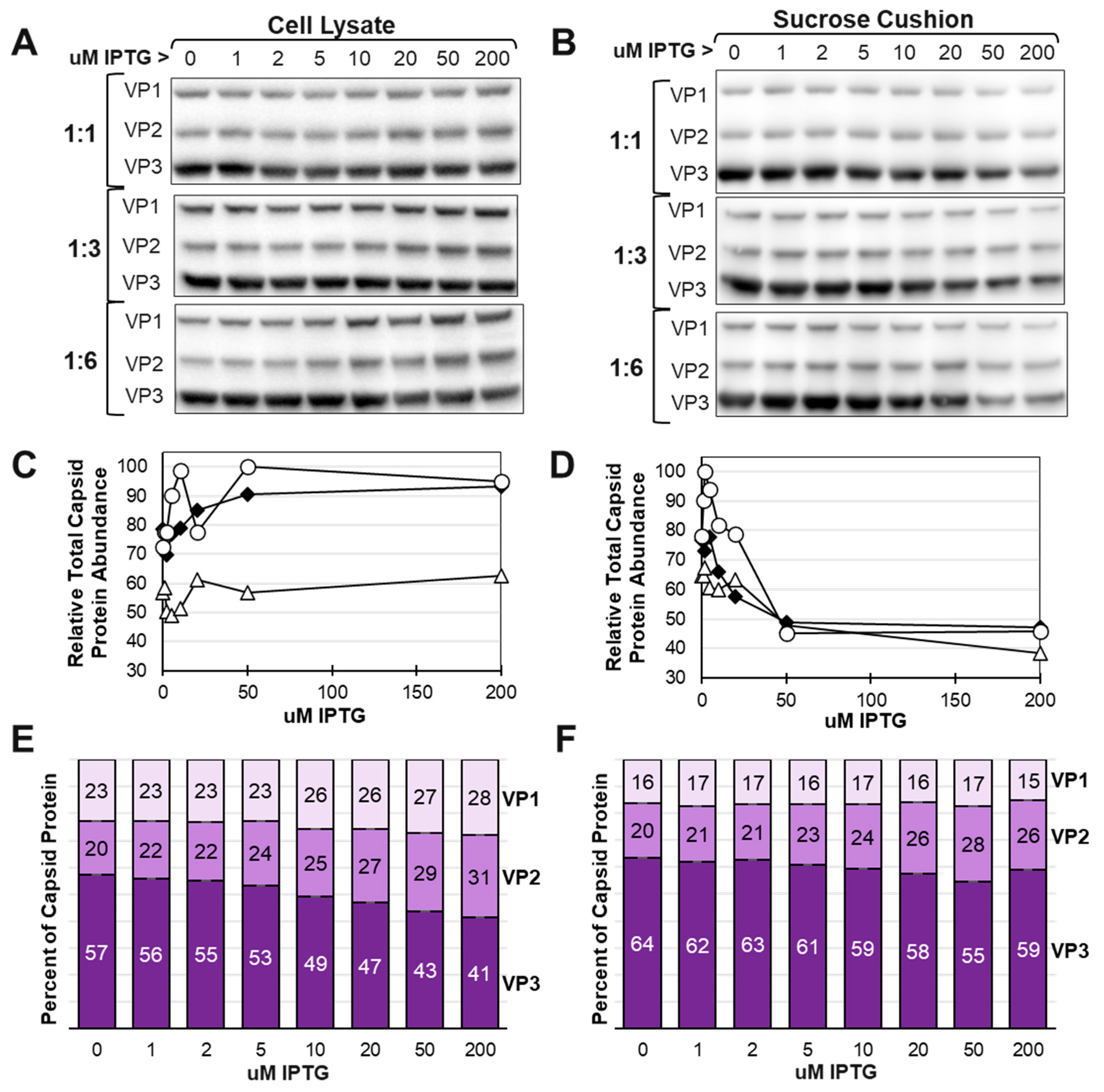
Figure 7.
Titer and Potency of AAV Produced at Different IPTG Concentration. Sf9 cells were co-infected with ITR-SEAP-GFP and LacRepCap baculoviruses. Sucrose cushion purified AAV sample Q-PCR titers are plotted against IPTG concentration (A). Titers generated at ITR-SEAP-GFP:LacRepCap co-infection ratios 1:1 (open triangle), 1:3 (black diamond) and 1:6 (open circle) are shown. Cells were lysed and AAV capsids were purified by sucrose cushion ultracentrifugation. These AAV samples used to transduce HEK 293T cells and the alkaline phosphatase activity was measured after 4 days. Potency was measured as alkaline phosphatase activity relative to genome titer is and is plotted against IPTG concentration (B). The graph is split scaled to emphasize the potency values from capsids produced at IPTG concentrations between 0 uM and 10 uM.
Figure 7.
Titer and Potency of AAV Produced at Different IPTG Concentration. Sf9 cells were co-infected with ITR-SEAP-GFP and LacRepCap baculoviruses. Sucrose cushion purified AAV sample Q-PCR titers are plotted against IPTG concentration (A). Titers generated at ITR-SEAP-GFP:LacRepCap co-infection ratios 1:1 (open triangle), 1:3 (black diamond) and 1:6 (open circle) are shown. Cells were lysed and AAV capsids were purified by sucrose cushion ultracentrifugation. These AAV samples used to transduce HEK 293T cells and the alkaline phosphatase activity was measured after 4 days. Potency was measured as alkaline phosphatase activity relative to genome titer is and is plotted against IPTG concentration (B). The graph is split scaled to emphasize the potency values from capsids produced at IPTG concentrations between 0 uM and 10 uM.
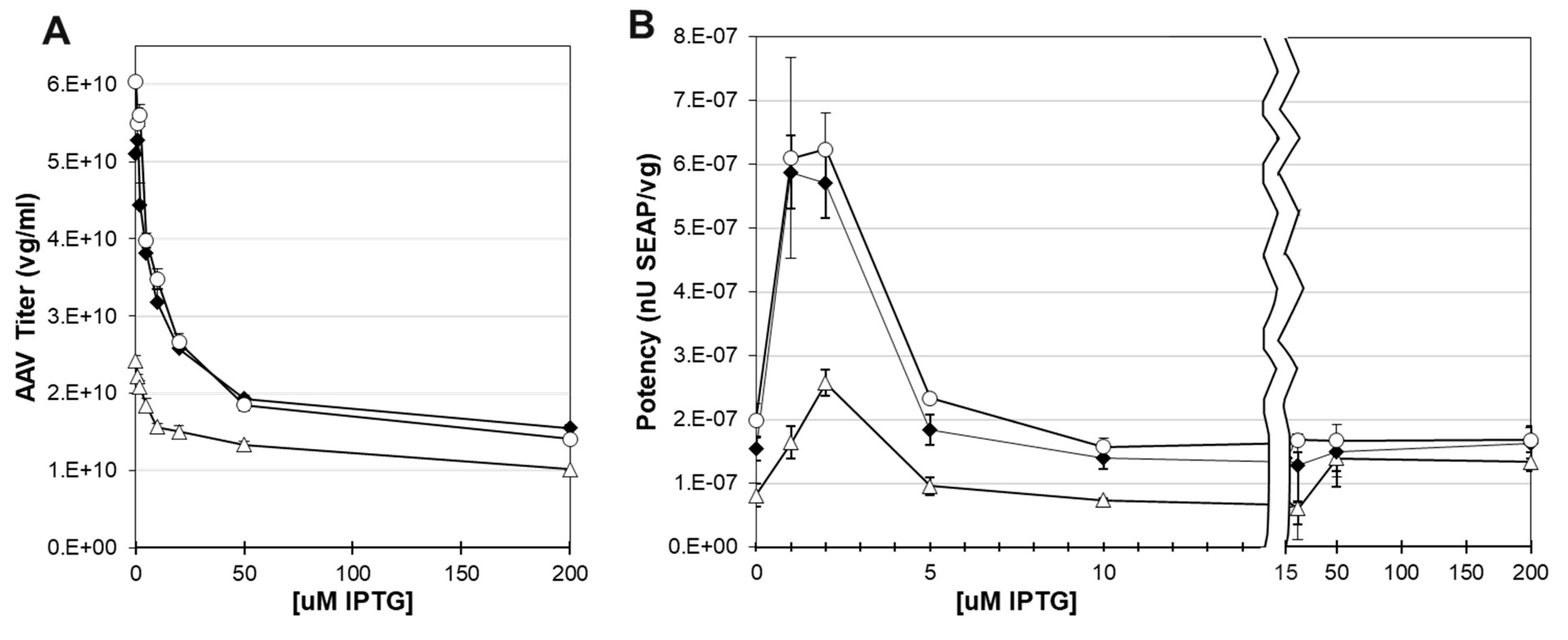
Figure 8.
Comparison of Capsid Ratios Before and After Affinity Purification. Sf9 cells were cultured at 800 ml scale and co-infected with ITR-SEAP-GFP and LacRepCap baculoviruses at a 1:6 co-infection ratio in the presence of 0 uM, 2 uM, 10 uM and 50 uM IPTG. Capsids were affinity purified and then further sucrose cushion purified. Total capsid proteins in cell lysates were analyzed by Western blot and quantified from ECL images using ImageJ software (A). Affinity purified capsids (B) and affinity/sucrose cushion purified capsids (C) were analyzed by CE-SDS.
Figure 8.
Comparison of Capsid Ratios Before and After Affinity Purification. Sf9 cells were cultured at 800 ml scale and co-infected with ITR-SEAP-GFP and LacRepCap baculoviruses at a 1:6 co-infection ratio in the presence of 0 uM, 2 uM, 10 uM and 50 uM IPTG. Capsids were affinity purified and then further sucrose cushion purified. Total capsid proteins in cell lysates were analyzed by Western blot and quantified from ECL images using ImageJ software (A). Affinity purified capsids (B) and affinity/sucrose cushion purified capsids (C) were analyzed by CE-SDS.
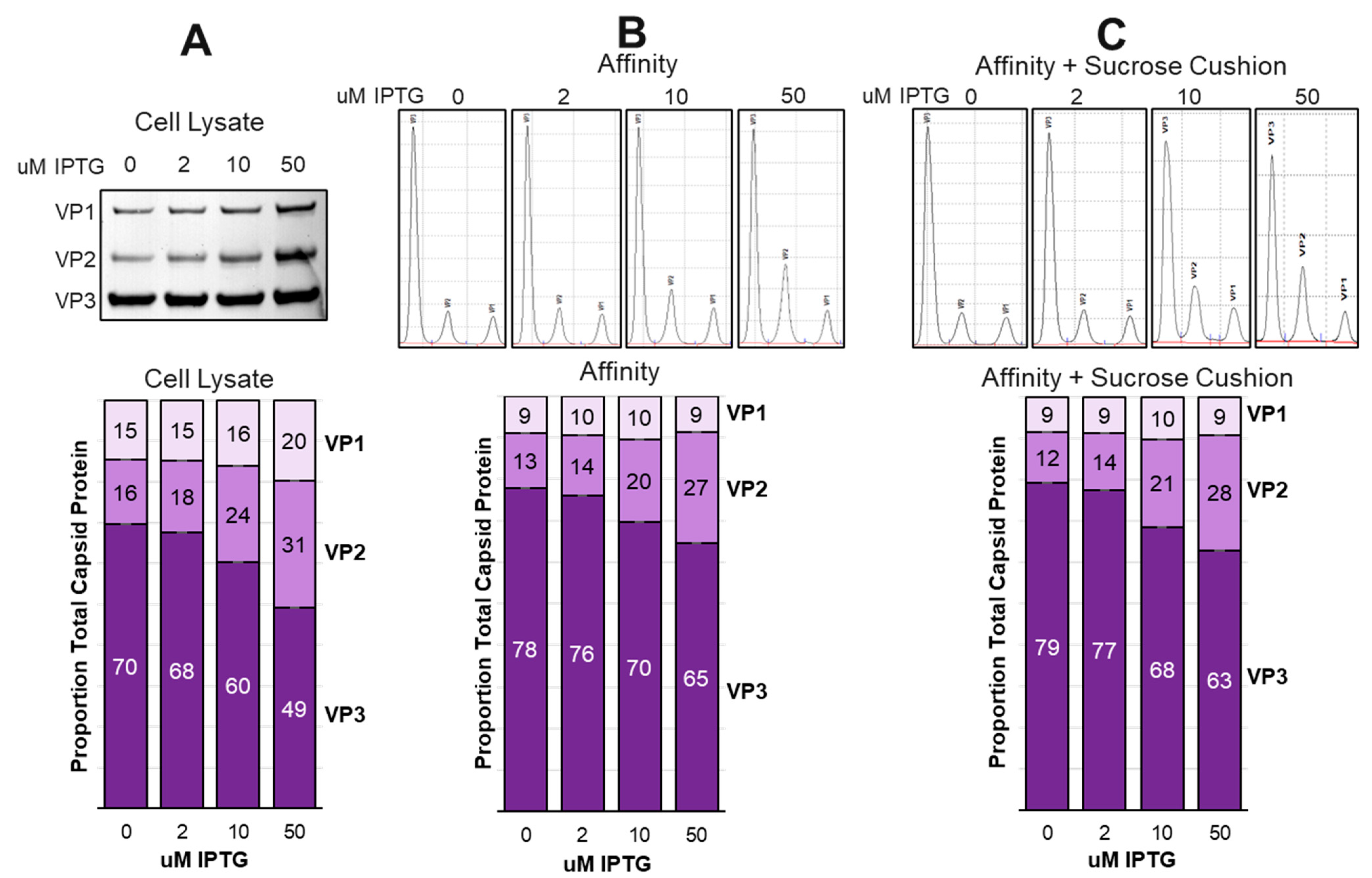
Figure 9.
Affinity purified and affinity/sucrose cushion purified AAV capsids derived from ITR-SEAP-GFP/ LacRepCap baculovirus co-infection of Sf9 cells were used to transduce HEK 293T cells. The alkaline phosphatase activity was measured after 4 days. Potency was measured as alkaline phosphatase activity relative to AAV viral genome (vg) added to HEK 293T cells and is plotted against IPTG concentration for affinity purified AAV (A) and for affinity/sucrose cushion purified AAV (B). Affinity purified capsids were also subjected to SEC-MALS analysis and the percent full ( ITR-SEAP-GFP genomes) in capsids relative to IPTG concentration is shown (C).
Figure 9.
Affinity purified and affinity/sucrose cushion purified AAV capsids derived from ITR-SEAP-GFP/ LacRepCap baculovirus co-infection of Sf9 cells were used to transduce HEK 293T cells. The alkaline phosphatase activity was measured after 4 days. Potency was measured as alkaline phosphatase activity relative to AAV viral genome (vg) added to HEK 293T cells and is plotted against IPTG concentration for affinity purified AAV (A) and for affinity/sucrose cushion purified AAV (B). Affinity purified capsids were also subjected to SEC-MALS analysis and the percent full ( ITR-SEAP-GFP genomes) in capsids relative to IPTG concentration is shown (C).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



