
Submitted:
18 November 2023
Posted:
20 November 2023
You are already at the latest version
Abstract
In recent years, the emergence of the concept of immunometabolism has shed light on the pivotal role that cellular metabolism plays in both the activation of immune cells and the development of immune programs. The antiviral response, a widely distributed defense mechanism used by infected cells, serves to not only control infections but also to attenuates their deleterious effects. The exploration of the role of metabolism in orchestrating the antiviral response represents a burgeoning area of research, especially considering the escalating incidence of viral outbreaks coupled with the increasing prevalence of metabolic diseases. Here, we present a review of current knowledge regarding immunometabolism and the antiviral response during viral infections. Initially, we delve into the concept of immunometabolism by examining its application in the field of cancer—a domain that has long spearheaded inquiries into this fascinating intersection of disciplines. Subsequently, we explore examples of immune cells whose activation is intricately regulated by metabolic processes. Progressing with a systematic and cellular approach, our aim is to unravel the potential role of metabolism in antiviral defense, placing significant emphasis on the innate and canonical interferon response.
Keywords:
antiviral response
; immunometabolism
; viral infections
; metabolic diseases
1. Introduction
Metabolism is generally referred to as the sum of all biochemical reactions within an organism designed to produce or consume energy and, by extension, allow the maintenance of life. Metabolism thus includes all the anabolic or catabolic reactions taking place at the cellular or systemic level. On the one hand, it provides the energy necessary for the cell to ensure its activities, whether in the form of ATP or reduced substrates such as NADH or NADPH. On the other hand, via anabolic pathways, metabolism participates in the synthesis of biomass (nucleic acids, amino acids and proteins, lipids, and carbohydrates) useful for the storage of energy, and information, or for the formation of functional units of the cell (i.e., proteins). The balance between anabolism and catabolism is finely regulated to preserve energy homeostasis. This regulation is dependent on enzymes that are very sensitive to changes in energy status (i.e., glucose deprivation vs. high glucose level). Indeed, the cell seeks to maintain an adequate ATP/ADP ratio with either cellular functions or growth.
As metabolism is the keystone of cellular activity, the mechanisms by which it is governed deserve attention. It should be noted that the consequences of metabolic disorders on cell functions are studied in the specific case of human pathologies, such as diabetes, obesity, or cancer. Immune responses (i.e., innate and adaptive) as well as pathogen sensing are among the issues for which a close link with metabolism has also been established [1]. During viral infections, particular metabolic states, namely the metabolic pathways active at the time of infection, seem to be favorable to the virus multiplication. This may be through a direct action of some metabolites in the early stages of the viral cycle or the establishment of antiviral responses (notably interferon-dependent) [2].
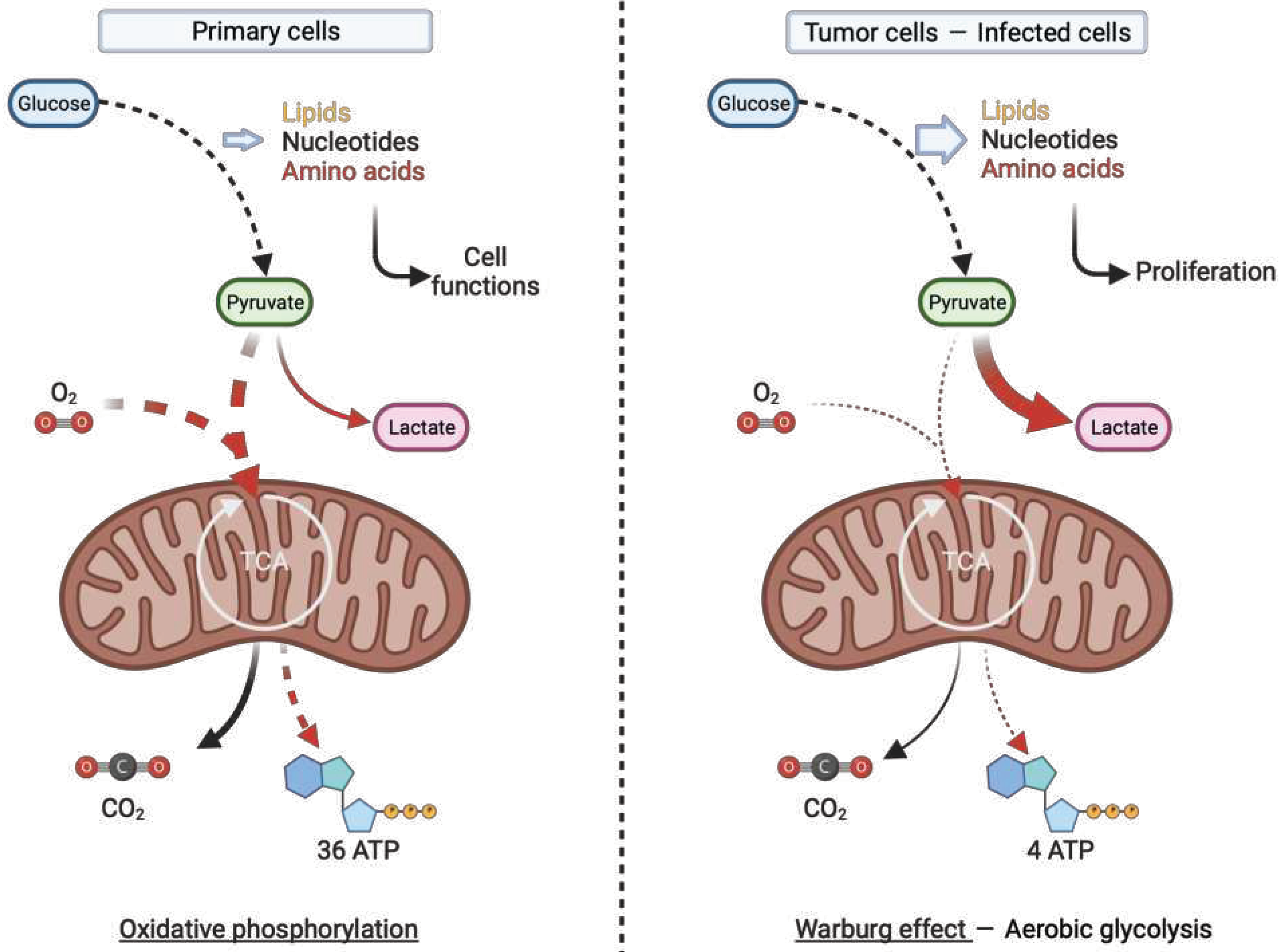
To maintain its homeostasis in a fluctuating environment, the cell must have a consistent metabolic status so that a shift in the dynamics of cell metabolism is possible depending on the cell's fate. Schematically, a quiescent cell will tend to favor the use of carbohydrate substrates, degraded to pyruvate to fuel the tricarboxylic acid cycle (TCA) and provide large amounts of energy in the form of ATP. This mechanism is driven via oxidative phosphorylation – OXPHOS, which enables cells to maintain their activities (e.g., enzymatic activities, ATP-dependent pump, ATP/ADP ratio-dependent signaling). On the contrary, a proliferating cell will favor the rapid production of energy through ‘aerobic glycolysis’, leading to the utilization of pyruvate to produce lactate even under aerobic conditions (Figure 1). This mechanism, known as the Warburg effect, was initially observed in cancer cells and tends to facilitate the abundant synthesis of biomass at the expense of efficient energy production [3]. More recently, the ability of some viruses to modulate the metabolism in this way has been established, reminiscent of the Warburg effect [4,5]. During infection, the virus exploits the cellular machinery to its advantage, aiming to rapidly produce and release an important quantity of virions. This intense viral replication within the infected cell necessitates a significant amount of nucleotides, amino acids, and lipids, all supplied in a manner similar to that in a cancer cell, through metabolic reprogramming [4]. This ability to reprogram or redirect the host's metabolism seems to be shared by many viruses, spanning different families and groups, such as the Human Immunodeficiency Virus (HIV), Dengue virus (DENV), Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), or Herpes Simplex Virus (HSV) [4,6]. Moreover, it is plausible that the tendency of viruses to redirect metabolism could elucidate the preferential targeting of cells engaged in aerobic glycolysis, such as tumor cells. These cells, with an existing metabolism suitable for infection and viral replication, emerge as a prime target. This preference may confer oncolytic potential to the infecting virus, capable of reinstating immune responses within tumors.
Another putative reason for modulating the host's metabolism during infection is to impede the establishment of the antiviral cellular response, thereby promoting viral expansion. The hypothesis that certain transient or specific metabolic states due to chronic diseases favor viral infection takes on full significance in this context, partly explaining the differences in systemic reactivity to the same virus.
Figure 1.
Schematic representation of the Warburg effect. According to this concept created by Otto Warburg, proliferating cells, including tumor cells, supply their biomass requirements by an adaptating of their metabolism. This adaptation involves the production of lactate from pyruvate even under aerobic conditions and has therefore been established as aerobic glycolysis. The intermediates of glycolysis are then utilized to provide lipids, nucleotides, and amino acids essential for high-rate cellular proliferation.
Figure 1.
Schematic representation of the Warburg effect. According to this concept created by Otto Warburg, proliferating cells, including tumor cells, supply their biomass requirements by an adaptating of their metabolism. This adaptation involves the production of lactate from pyruvate even under aerobic conditions and has therefore been established as aerobic glycolysis. The intermediates of glycolysis are then utilized to provide lipids, nucleotides, and amino acids essential for high-rate cellular proliferation.
Indeed, the interactions between metabolism and immunity are increasingly discussed with the emergence of the term ‘immunometabolism’. This facet of immunology has been the subject of numerous advances in the context of cancer (see section 2.a. Immunometabolism and cancer) and has already been extensively presented elsewhere. The aim of this review is rather to discuss, in the light of the latest available literature, how metabolic disorders (diabetes, obesity, and others) might affect the outcome of viral infections. Then, we will discuss the advances made regarding the interplays between metabolism and immunity in the context of viral infections, with an emphasis on the antiviral innate immunity of the infected host cell.
2. Immunometabolism
The immune system is an intricate network of cells and molecules that safeguard the body against threats. Recently, increasing attention has been directed towards the regulation of this network by metabolism, giving rise to the concept of immunometabolism. The study of immunometabolism could provide insight into how obesity fosters inflammation in conditions such as arthritis [7]. It also offers a way of understanding how drugs acts on metabolism or metabolites, such as methotrexate or 25-hydroxycholesterol, to find utility in the treatment of infectious diseases [8,9].
a. Immunometabolism and cancer
A proper introduction to immunometabolism is its contribution to the understanding and treatment of pathologies of cancer origin. Indeed, cancer has been one of the most prolific subjects of study for immunometabolism. Cancer is a group of pathologies that can occur in virtually all organs and have in common the anarchic division of cells, possibly leading in the case of malignant tumors to the invasion of distant tissues. The metabolism of cancer cells is of crucial importance for the development of these pathologies, as cancer cells have a particular metabolism allowing them to support their development. In addition, the tumor metabolic environment also influences the immune system by participating in immune tolerance and immune system failures in the clearance of tumor cells [10].
Indeed, tumors have been shown to modulate the concentration of nutrients critical to immune cell function. Infiltration of immune cells into the tumor tissue is generally associated with a better prognosis [11]. In particular, T cell infiltration is well documented, and the promotion of their activity is even the subject of new therapeutic approaches [12]. The tumor cells microenvironment is specific and results from the metabolism of tumor cells. The latter is based primarily on high-rate of glycolysis to allow the large production of biomass (e.g., amino acids, nucleotides and lipids) required for their sustained division. This leads to a depletion of the available intratumoral glucose pool [13]. Of note, glucose depletion has been associated with decreased infiltration and reactivity of the immune system, including T cells on the tumor lcoation [14]. For example, a deficit in the production of IFN-γ, a key effector secreted by tumor-infiltrating CD8+ T cells, was observed after glucose deprivation [15]. This decrease is induced by the binding of glyceraldehyde phosphate dehydrogenase (GAPDH) to the 3’ untranslated region (3’UTR) of the IFN-γ mRNA when the glycolytic enzyme is not devoted to its metabolic function, namely, at low levels of glucose [15]. Alternatively, phosphoenolpyruvate (PEP), an intermediate glycolysis metabolite, whose level decreases following glucose deprivation, is essential for T cell anti-tumor responses H [16]. It supports T cell receptor (TCR) and nuclear factor of activated T cells (NFAT)-induced signaling, whose activation depends on inositol trisphosphate receptors (IP3R) located on the mitochondrial membrane [16]. Ca2+ then acts as an integrator of TCR signaling and PEP by inhibiting sarco/ER Ca2+−ATPase (SERCA) activity. This increased cytosolic Ca2+ concentration then promotes the anti-tumor activities of TCR-dependent CD4+ and CD8+ T cells [16].
b. A case of metabolic-dependent activation in immune cells: monocyte and macrophage
Macrophages are CD11b+CD68+ leukocytes historically described for their anti-microbial and phagocytic activities. However, many other functions have been associated with them over the years [17]. Indeed, their participation in tissue homeostasis, as well as tissue repair in case of damage, is no longer to be proven [17]. To fulfill these essential but diverse functions, macrophages can adopt a variety of phenotypes. Given the complexity of these phenotypes, it is generally accepted to classify macrophages according to whether they have pro-inflammatory (M1) or anti-inflammatory (M2) activities [17]. Nevertheless, it is important to keep in mind that this classification greatly simplifies the repolarization capabilities of these cells, which rely on transcriptionally dynamic capacities to adopt the phenotype and functions best suited to their tissue microenvironment [17]. Moreover, macrophages are among the only cells to have so many tissue-specific counterparts (i.e., Kupffer cells in the liver, microglia in the brain, and alveolar macrophages in the lung) [17]. All these tissue-specific macrophages have an embryonic origin, unlike the monocyte lineage-macrophages, which derive from the differentiation of monocytes following their tissue infiltration. In the following, we will discuss the metabolic characteristics of macrophages according to whether they are M1 or M2 polarized in vitro and in vivo. Some differences that may occur depending on their tissue of residence, have been reviewed very recently by Wculek et al. [18].
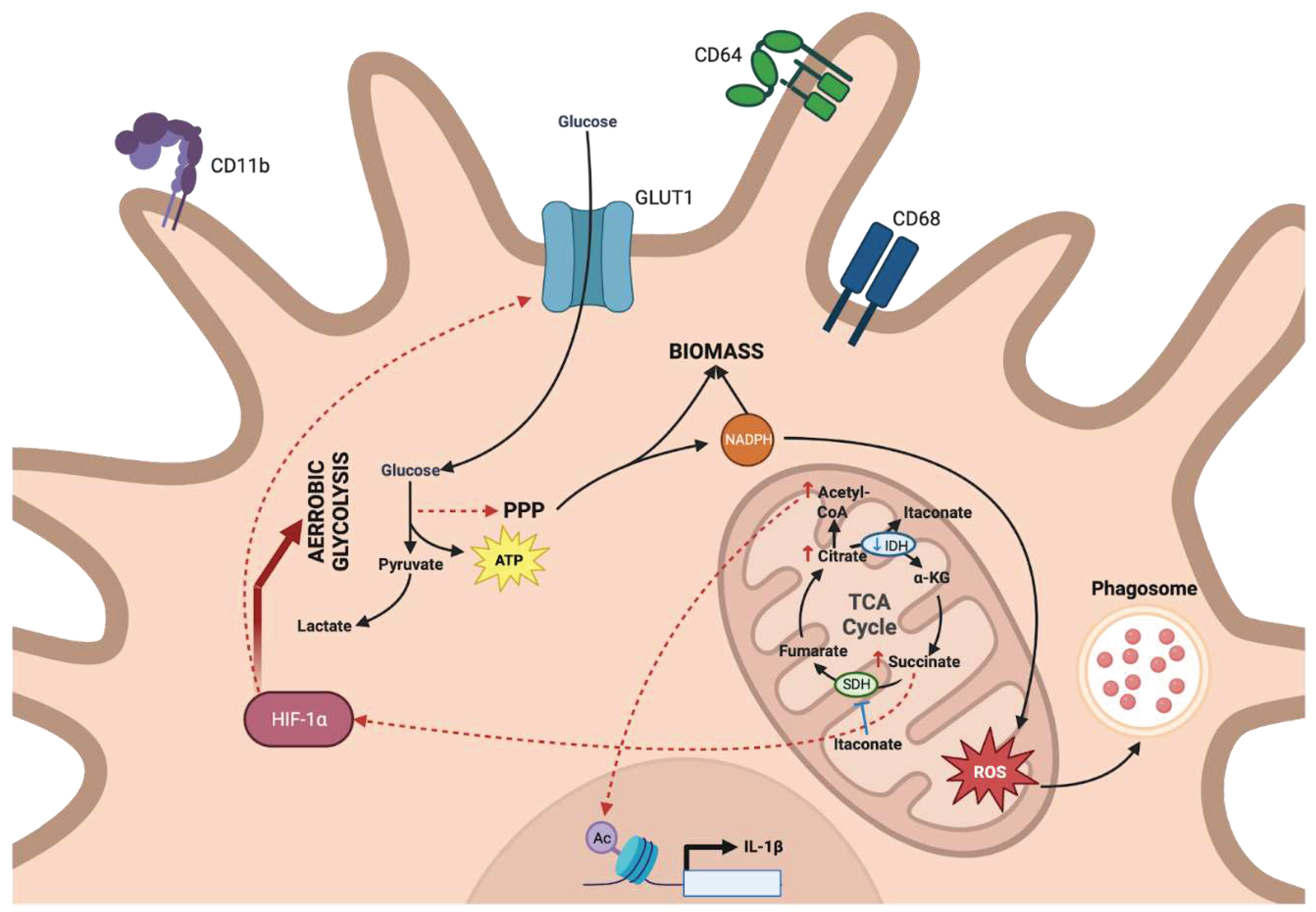
M1 macrophages (CD11b+CD68+CD64+) have a predominantly pro-inflammatory activity whose activation in vitro occurs either through TLR stimulation by LPS or by cytokine stimulation with TNF or IFNγ. This activation reprograms cell metabolism in a way that supports the establishment of an oxidative burst by the production of reactive oxygen species (ROS) and nitrogen species (RNS), as well as the production of antimicrobial effectors. M1 macrophages are characterized by predominant glycolytic metabolism (Figure 2) as evidenced by their increased consumption of glucose, enabled in particular by an increase in the Glucose Type 1 receptor (GLUT1) [19,20]. Hypoxia Inducible Factor-1α (HIF-1α) was shown to be a transcription factor essential for the onset of this metabolic reprogramming [21,22]. Stabilization of HIF-1α is mediated in M1 macrophages by blocking TCA at two distinct levels. First, the inhibition of isocitrate dehydrogenase (IDH-1) leads to citrate accumulation, as well as itaconate synthesis [23]. On the one hand, the accumulated citrate is converted to acetyl-CoA, which is used for histone acetylation and inflammatory gene transcription [24]. On the other hand, itaconate inhibits the activity of the succinate dehydrogenase complex (SDH), leading to the accumulation of succinate [25]. Ultimately, the succinate generated in this way stabilizes HIF-1α [22]. Notably, this metabolic reprogramming has been shown to be essential for IL-1β production in response to LPS stimulation [22]. Mitochondrial activity in M1 macrophages is then diverted from ATP production to mitochondrial ROS production. The latter will serve within phagosomes, participating in antigen processing and presentation [26]. At this stage, ATP production is therefore mainly dependent on aerobic glycolysis, similarly to the effect described by Warburg for cancer cells [5,22]. This also supplies the necessary intermediates for the pentose phosphate pathway (PPP) leading in the end to an increase in the NADPH pool, as well as to the production of biomass (here as nucleotides) and ROS [27].
Figure 2.
General metabolism involved in pro-inflammatory M1 macrophages orientation. M1 macrophages develop a glycolytic metabolism enabled by the overexpression of the glucose transporter, GLUT1, in which HIF-1α is involved. Stabilization of this factor is, on the one hand, linked to the inhibition of isocitrate dehydrogenase (IDH-1), leading to citrate accumulation and itaconate synthesis. Accumulated citrate is converted into acetyl-CoA which is used for histone acetylation and thus promotes the transcription of inflammatory genes. On the other hand, itaconate inhibits the succinate dehydrogenase complex (SDH) activity, resulting in succinate accumulation. The succinate thus produced stabilizes HIF-1α. The mitochondrial activity of M1 macrophages is dedicated to the production of mitochondrial reactive oxygen species, to be used within phagosomes. ATP production therefore relies primarily on aerobic glycolysis.
Figure 2.
General metabolism involved in pro-inflammatory M1 macrophages orientation. M1 macrophages develop a glycolytic metabolism enabled by the overexpression of the glucose transporter, GLUT1, in which HIF-1α is involved. Stabilization of this factor is, on the one hand, linked to the inhibition of isocitrate dehydrogenase (IDH-1), leading to citrate accumulation and itaconate synthesis. Accumulated citrate is converted into acetyl-CoA which is used for histone acetylation and thus promotes the transcription of inflammatory genes. On the other hand, itaconate inhibits the succinate dehydrogenase complex (SDH) activity, resulting in succinate accumulation. The succinate thus produced stabilizes HIF-1α. The mitochondrial activity of M1 macrophages is dedicated to the production of mitochondrial reactive oxygen species, to be used within phagosomes. ATP production therefore relies primarily on aerobic glycolysis.
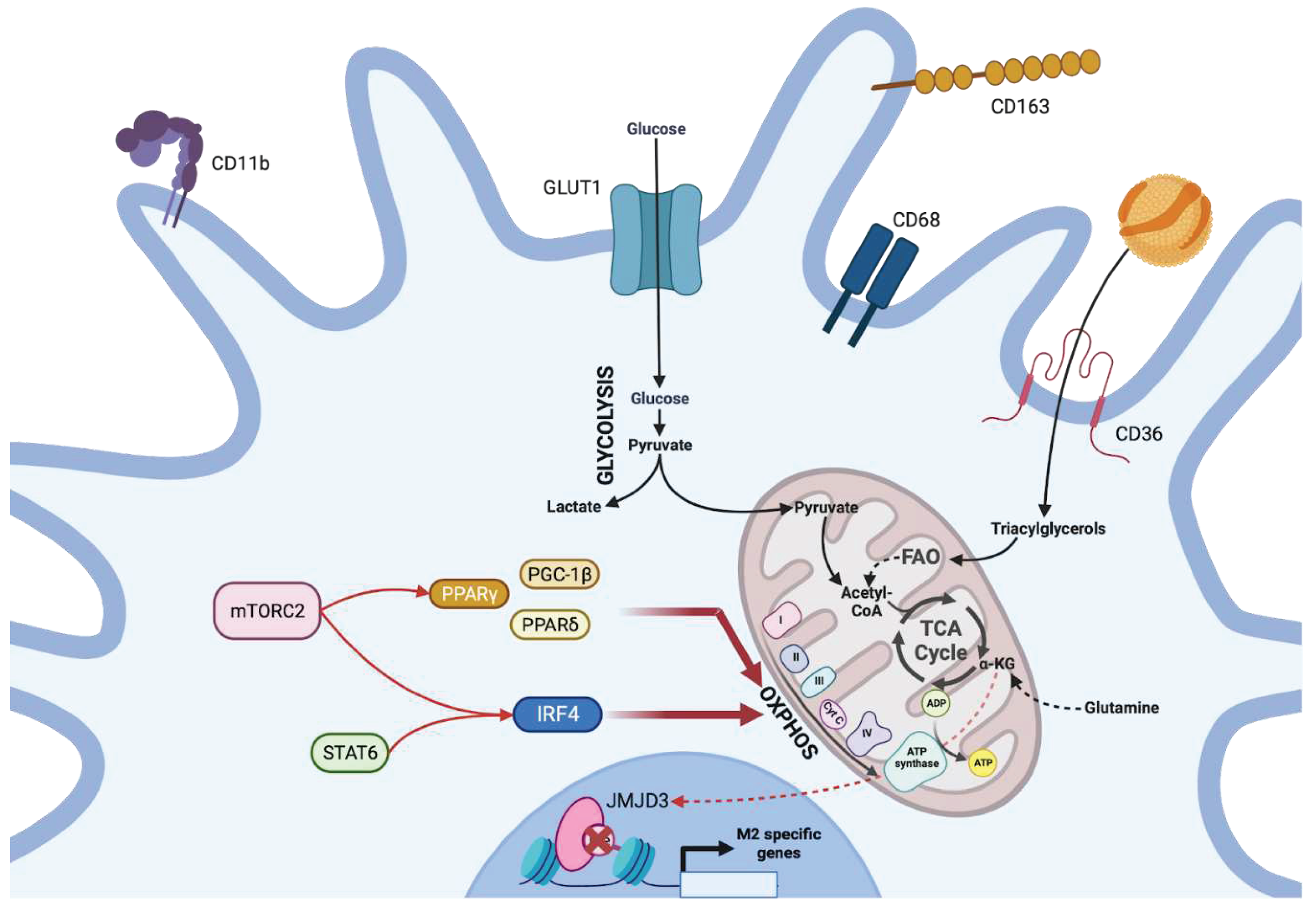
Unlike M1 macrophages, macrophages with a M2 phenotype (CD11b+CD68+CD163+) are obtained in vitro in response to IL-4 or IL-13 and display anti-inflammatory properties as well as involvement in tissue repair [17]. In contrast to M1, M2 macrophages have a cellular metabolism based on increased OXPHOS and mitochondrial respiration (Figure 3), as evidenced by the elevation of OCR in macrophages polarized towards an anti-inflammatory phenotype [28]. Transcriptional shifts leading to this metabolism are controlled in M2 macrophages by activation of several factors including PPARδ, PPARγ, PGC-1β, and IRF4. These factors activation depends on both STAT6 and the mTORC2 complex [28,29]. Some of these factors are associated with lipid metabolism and lead, in particular here, to the upregulation of the scavenger membrane protein CD36 that allows internalization of lipids used for fatty acid oxidation (FAO) [29,30]. While CD36 has been shown to be critical for polarization in M2 [30], the role of FAO remains ambiguous. Indeed, the use of etoxomir to inhibit FAO or genetic models that prevent it, do not appear to affect M2 polarization [31,32], but rather the ability of M2 macrophages to produce ATP. Thus, one might conclude that FAO does not appear to be essential for M2 macrophages to perform their inflammatory functions, with fatty acids serving instead as a substrate for energy production through OXPHOS [31]. In contrast, glutaminergic metabolism appears to be of paramount importance with glutamine providing one-third of the carbon derivatives of TCA [33]. Glutamine leads to the accumulation of α-ketoglutarate, which orchestrates M2 polarization by activating Jumonji domain-containing protein D3 (JMJD3) demethylases [34]. They mediate histone demethylation of M2-specific gene promoters [34]. As a result, these epigenetic modifications are partly responsible for M2 polarization.
Figure 3.
General metabolism involved in anti-inflammatory M2 macrophages orientation. In M2 macrophages, activation of the PPARδ, PPARγ, PGC-1β and IRF4 factors enables transcriptional changes directing the cellular metabolism towards OXPHOS. The activation of these factors depends on both STAT6 and mTORC2. In particular, overexpression of the scavenger CD36 provides lipidic substrates required for fatty acid oxidation. Fatty acid oxidation is essential for ATP production via OXPHOS, rather than for M2 macrophage functions. On the other hand, glutamine, which supplies the Krebs cycle, is essential since it orchestrates M2 polarization through the activation of JMJD3 demethylases.
Figure 3.
General metabolism involved in anti-inflammatory M2 macrophages orientation. In M2 macrophages, activation of the PPARδ, PPARγ, PGC-1β and IRF4 factors enables transcriptional changes directing the cellular metabolism towards OXPHOS. The activation of these factors depends on both STAT6 and mTORC2. In particular, overexpression of the scavenger CD36 provides lipidic substrates required for fatty acid oxidation. Fatty acid oxidation is essential for ATP production via OXPHOS, rather than for M2 macrophage functions. On the other hand, glutamine, which supplies the Krebs cycle, is essential since it orchestrates M2 polarization through the activation of JMJD3 demethylases.
As with other cells of the immune system, deficits in macrophage metabolism can occur pathologically, as in the case of atheromatous intraplaque foamy macrophages or in obesity [18].
c. The metabolic-dependent activation of tissue sentinels: dendritic cells
Dendritic cells (DCs, CD11c+) are antigen-presenting cells (APCs), which like macrophages, have variable origins [35]. In fact, several subtypes of DCs can be distinguished according to their origin. Conventional DCs (cDC1 and cDC2) and plasmacytoid DCs (pDCs) derived from specific progenitors, and others derived from monocytes (mo-DCs) [35]. As APCs, DCs are found in a resting state in many tissues (e.g., Langerhans cells in the dermis) where their role is to sense their environment, and then activate in case of danger. Activation occurs in response to pathogen associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs) and results in initiating the immune response by the secretion of cytokines, as well as by the presentation of antigen in lymphoid tissues [35].
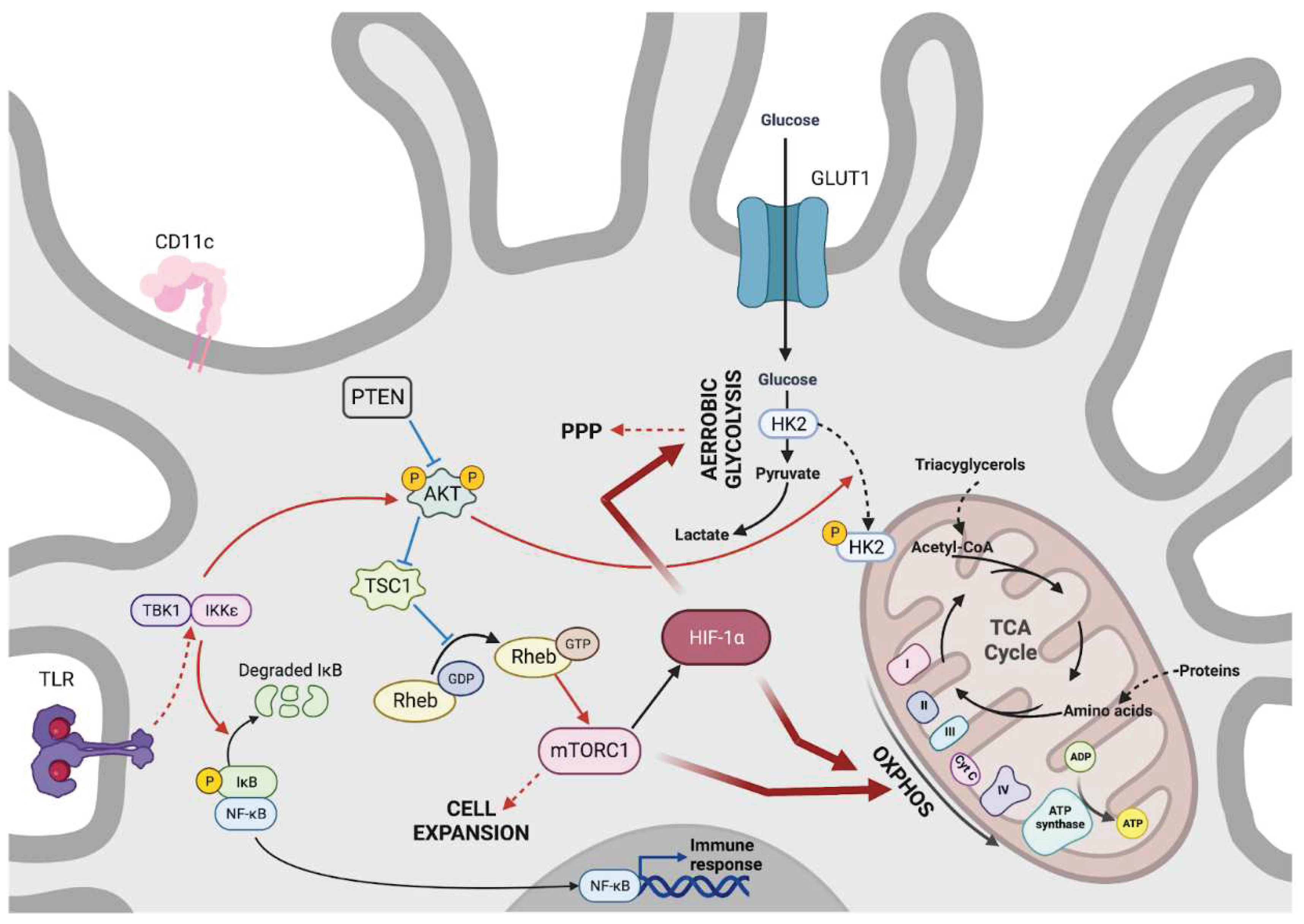
The metabolism of resting DCs has already been studied both in vitro and in vivo. Thus, it is known that the mTORC1 complex has a preponderant activity here again. Indeed, in mice, the use of rapamycin leads to the inhibition of the ex vivo growth of pDCs and cDCs from the bone marrow (BM); in vivo the pool of DCs is considerably reduced [36]. Confirming these results obtained in mouse models, the use of rapamycin on human mo-DCs results in impaired survival [37]. Conditional expression models were used to further explore the importance of mTORC1 in DCs survival. Phosphatase and TENsin homolog (Pten) deletion using a Cd11c-Cre system leads to expansion of DCs in mice. This result is consistent with the previous one, PTEN being a negative regulator of mTOR [36]. Deletion of Tsc1, another negative regulator of mTOR, using a similar system, led to an increased growth of cultured BM-DCs [38]. As one of the main factors that regulate cellular metabolism, mTORC1 directs quiescent DCs to use OXPHOS to satisfy their low energy requirement [39].
As for the other immune cells presented so far, the activation of DCs following stimulation of one of their pattern recognition receptors (PRRs) leads to a shift in metabolism with glucose becoming the main fuel for cellular metabolism. This process takes place in two steps. Indeed, shortly after the activation of DCs in a TLR-dependent manner, a rapid increase in glycolytic activity and entry of intermediates into the PPP was observed [40]. These changes are initiated by activation of AKT via TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) [40]. AKT phosphorylates and subsequently activates hexokinase 2 (HK2), the glycolysis-limiting enzyme whose activity increases after association with the mitochondrial surface [40]. Lately, maintenance of glycolysis within DCs relies on activation by the mTOR complex of HIF-1α [41]. Interestingly, if inhibition of HIF-1α or mTOR occurs early after TLRs stimulation, it has no impact on DCs activation, in contrast to early inhibition of AKT. This illustrates the importance of the timing of different signaling pathways in DCs activation [40]. In contrast, activation of the AMP- activated protein kinase (AMPK) pathway appears to control DCs activation, by promoting OXPHOS, in particular, through PGC1α activation, which favors mitochondrial biogenesis. In fact, AMPK knockdown has been associated with a potentiation of DCs activation [42]. Whereas its activation or that of PGC1α, leads to tolerogenic DCs [42].
Figure 4.
Dendritic cell metabolism. Maintenance of dendritic cells quiescence depends on an overriding activity of mTORC1. PTEN and TSC1, negative regulators of mTOR limit dendritic cell growth. mTORC1 orientates quiescent cells towards OXPHOS utilization, with protein catabolism and triacylglycerol providing amino acids and fatty acids to supply ATP production via the Krebs cycle. Activated dendritic cells take up glucose as the main fuel for a metabolism geared towards aerobic glycolysis. Activation of mTORC1and HIF-1α, in this case, is enabled by TBK1 and IKKε activity on AKT downstream of dendritic cells PRR.
Figure 4.
Dendritic cell metabolism. Maintenance of dendritic cells quiescence depends on an overriding activity of mTORC1. PTEN and TSC1, negative regulators of mTOR limit dendritic cell growth. mTORC1 orientates quiescent cells towards OXPHOS utilization, with protein catabolism and triacylglycerol providing amino acids and fatty acids to supply ATP production via the Krebs cycle. Activated dendritic cells take up glucose as the main fuel for a metabolism geared towards aerobic glycolysis. Activation of mTORC1and HIF-1α, in this case, is enabled by TBK1 and IKKε activity on AKT downstream of dendritic cells PRR.
Therefore, we have shown here that essential changes in immune cell metabolism are required during their activation to enable them to support their function, whether as a support or effector. We will now discuss the contributions of immunometabolism in certain pathological situations.
3. At the systemic level: Interplays between host metabolic status and infectious diseases
It is gaining increasing awareness that the resolution of an infection depends on multiple intrinsic physiological factors. In this light, the host metabolic status and the related comorbidities are now taken into account in understanding immune responses, whoch are often more or less efficient during the course of an infectious disease, conditioning resolution or subsequent pathophysiological processes.
a. Diabetes, obesity and other metabolic disorders
Metabolic diseases (e.g., type 2 diabetes, obesity) represent a growing risk for the world’s population, particularly through their cardiovascular (myocardial infarction, stroke) and neurological (diabetic neuropathy) complications. Previously restricted to high-income countries and western lifestyles, these diseases are now tending to develop in low- and middle-income countries. As depicted in this section, these metabolic disorders may present an associated risk of infection. It was also recently demonstrated during the COVID-19 epidemic that one of the complications of SARS-CoV-2 infection is the development of post-infection diabetes.
i. Diabetes
Diabetes is a chronic disease characterized by hyperglycemia and chronic low-grade inflammation. Two main types of diabetes can be distinguished. Type 1 or insulin-dependent diabetes (T1D) is an autoimmune disease targeting the β-cells of the islets of Langerhans in the pancreas which secrete insulin, a key hormone in the regulation of circulating glucose. In contrast, the pathophysiology of type 2 or insulin-resistant diabetes (T2D) involves hyperinsulinemia, due to a progressive resistance of cells to insulin, following the installation of a low-grade inflammation [1]. Diabetes accounts for a total of more than 460 million cases in 2019 with a projection to 700 million cases in 2045, the majority of cases being T2D [43]. Beyond its cardiovascular and neurological complications, diabetes could lead to impaired host defenses and an increased risk of infection.
In fact, diabetes has been found to cause immune deficiencies in several essential compartments to establish an adequate immune response. Chronic hyperglycemia is accompanied by the production of advanced glycation end products and ROS. This pro-oxidant status promotes chronic low-grade inflammation, which is detrimental to the immune response. More specifically, T2D is accompanied by the recruitment of M1 polarized macrophages within the adipose tissue where they will secrete significant amounts of pro-inflammatory mediators (Tumor necrosis factor α - TNF-α, C-reactive protein - CRP, interleukin-1β - IL-1β, CCL2, CCL3, CXCL8, IL-6, and IL-12) [44,45]. However, under bacterial stimulation, peripheral blood mononuclear cells (PBMCs) from diabetic patients have been found to have a decrease in functional capabilities compared to healthy individuals. Thus, a decrease in the secretion of cytokines such as IL-1β, IL-6 and IL-2 is observed [46,47,48]. Of note, IL-6 plays an important role in protection against pathogens and in the adaptive immune response by inducing antibody production and the development of effector T cells [49]. Therefore, these studies reveal that inhibition of these cytokines in hyperglycemia can affect the immune response development against pathogens. In addition, a suppression of type I interferon (IFN) production is reported in PBMC cultured under high glucose conditions and stimulated with viral RNA mimetic, namely polyinosinic:polycytidylic acid - poly:IC [50]. It was also demonstrated that leukocyte recruitment is inhibited in a diabetic context, notably due to an alteration in the expression of adhesion molecules [51]. More specifically, for macrophages, there is a decrease in Fcγ receptor expression, as well as in the antimicrobial and phagocytosis capacities [52,53]. Similarly, for neutrophils, a reduction in antimicrobial capacities was noted. This is due to an alteration of the pro-oxidant and degranulation capacities [54,55,56]. Furthermore, the production of neutrophil extracellular traps (NETs) or NETose is decreased under hyperglycemic conditions, leading to increased susceptibility to infections [57]. Deficiencies in degranulation have also been demonstrated in NK cells from T2D subjects, resulting from a defect in NKG2D and NKp46 activating receptors [58]. Of note, complement activation is impacted in hyperglycemia, due to dysfunction of opsonization by C4 fragments of complement [59].
In addition to effector cells, T2D is accompanied by altered presentation and thus pathogen detection. Therefore, diabetic mice showed reduced expression of the adaptor protein containing the Toll-like receptor 2 (TLR) and the Toll / IL-1R domain (TIRAP) [60], involved in the detection of pathogens. However, contradictory data exist since it has been shown that the expression of TLRs by monocytes and neutrophils from diabetic subjects was increased [61]. In fact, it is now known that the quality of blood glucose control has an impact on TLRs expression. A subject with hyperglycemia and poor control will have decreased TLRs expression [61]. While a subject with controlled hyperglycemia will have higher TLRs expression [61]. Dendritic cells, the main antigen-presenting cells (APCs), showed delayed recruitment to the site of infection in diabetic mice in the course of M. tuberculosis infection [62]. This delay could be attributed, as previously for monocytes/macrophages, to reduced levels of CCL2 and CCL5 [62]. In addition, in the context of intracellular bacterial infection, this delay in recruitment has been shown to be accompanied by impaired phagocytosis [63].
Diabetes has been repeatedly shown to be a risk factor for developing bacterial and fungal infections, and this has already been reviewed [64,65]. However, viral infections have less been reported. Nevertheless, a study including nearly 190,000 people in China showed that diabetic individuals have a 1.5 times higher relative risk of developing hepatitis B virus infection than non-diabetic individuals [66]. Associated with the immunological disorders discussed above, Middle East Respiratory Syndrome coronavirus (MERS-CoV) infection in diabetic mice is accompanied by unresolved lung inflammation leading to exacerbated pathology [67]. These mice were also shown to have fewer monocytes/macrophages and CD4+ T cells, without affecting viral clearance [67]. For SARS-CoV-2, chronic inflammation found in T2D appears to be a risk factor for developing severe forms of COVID-19 [68]. Furthermore, strengthening the link between TD2 and viral infection, it is now known that SARS-CoV-2 can infect adipocytes, leading to a decrease in adiponectin secretion. This contributes to the establishment of an inflammatory state conducive to the development of insulin resistance [69], explaining the increased risk of transitioning from a prediabetic state to diabetes in COVID-19 patients. Finally, complications of diabetes include cardiovascular damage (atherothrombosis, stroke, myocardial infarction). Many of these cardiovascular complications are related to endothelial damage. Moreover, severe forms of DENV are known to be associated with damage to the endothelial barrier. Therefore, it is hypothesized in the literature that severe forms of DENV infection are more likely to occur in diabetic individuals due to pre-existing endothelial damage [70,71].
ii. Obesity
Overweight and obesity are defined as an excessive accumulation of fat in an individual. Abnormal accumulation of visceral adipose tissue (VAT) is a risk factor for developing cardiovascular events and is often associated with the development of insulin resistance [72]. Previously limited to high-income countries, overweight and obesity are now also increasing rapidly in low- and middle-income countries. According to the WHO, today more people are suffering from obesity than from malnutrition [73]. In 2016, there were 1.9 billion overweight people, of whom over 650 million were obese [73].
Obesity is a multifactorial metabolic disorder combining genetic, environmental and developmental factors [74], whose simplest cause corresponds to a caloric intake that is not consistent with the individual's energy expenditure. However, more complex mechanisms can be underlined, such as an impairment on the neurons of the arcuate nucleus of the hypothalamus involved in the control of food intake. This is mediated by the expression of orexigenic factors (neuropeptide Y, agouti-related protein) or anorexigenic factors (proopiomelanocortin, Cocaine and amphetamine regulated transcript - CART) under the hormonal control of leptin and insulin secreted by adipocytes and β-cells of the islets of Langerhans, respectively [74]. More recently, the impact of dysbiosis of the intestinal microbiota on the risk of obesity has been discussed. Indeed, the abundance of firmicutes bacteria seems to be linked to the development of obesity, due to the ability of these bacteria to extract energy more efficiently, thus promoting calorie absorption and weight gain [75].
Beyond the cardiovascular and endocrine complications associated with obesity, obese individuals exhibit immune disorders. In addition to its important storage role, VAT is a tissue that is strongly linked to immunity, in particular through the secretion of adipokines (leptin, adiponectin), cytokines (TNF-α, lL-6) and chemokines (CCL2). They exhibit activity not only in the inflammatory state, but also in the immune system [76]. Thus, hyperplasia and hypertrophy in obesity are accompanied by a deregulation of the immune system, which we will describe here. Adipose tissue contains different cell types including, in addition to adipocytes, endothelial cells, fibroblasts, pre-adipocytes, but especially leukocytes [77]. First, in obese subjects, there is an alteration in the recruitment, proliferation and polarization of macrophages within the adipose tissue [78]. Increased adipocyte size has been shown to trigger a stress response and the release of chemokines (CCL2 and CCL5) [79], leading to the recruitment and accumulation of monocytes and macrophages in adipose tissue [78]. On the other hand, the increased presence of free and non-esterified fatty acids in obesity results in the activation of TLR4 and TLR2, leading to the production of pro-inflammatory cytokines (e.g., IL-6) and reduced expression of anti-inflammatory cytokines (e.g., IL-10) [80,81]. This pro-inflammatory state may ultimately be responsible for the development of insulin resistance and T2D, as previously explained [81]. Finally, while most M2 polarized macrophages are found in healthy subjects' adipose tissue, macrophage polarization in obese individuals is oriented toward the M1 phenotype [77]. This accumulation is, on the one hand, due to the recruitment of blood monocytes, but also to the proliferation and polarization shift of resident macrophages in adipose tissue [82]. Leukocytosis in adipose tissue in obesity also involves neutrophils. Similarly, to monocytes, neutrophils from obese subjects show increased migration within adipose tissue, as well as increased basal superoxide production, thus participating in low-grade inflammation [83]. Finally, NK cells follow this trend, with increased activation, proliferation, and IFN-γ secretion [84]. Cells involved in adaptive immunity are also not spared in obesity. Indeed, adipose tissue from obese subjects showed a higher presence of CD4+ helper 1 and CD8+ cytotoxic T cells [85]. Although this pro-inflammatory status is the one found in case of infection, its chronicity is accompanied by an enhanced susceptibility to infections and a lower reactivity to pathogens in obese subjects [83,86].
The immune alterations in obesity are responsible for a heightened risk of developing infections. These infections often occur opportunistically or nosocomially in tissues harboring a commensal microbial flora, at the interface between the internal and external medium. Consequently, obese mice have shown insufficient antimicrobial activity to combat Staphylococcus aureus during a cutaneous infection [87]. Fungal infections with Candida albicans are also more prevalent in obese individuals [88]. Furthermore, there is an increased risk of developing Escherichia coli infections in the urinary tract of these subjects [89]. However, it is in the respiratory system that the most data have been reported. Indeed, obese subjects are at a higher risk of contracting upper or lower respiratory system infections [90]. Miceob/ob have shown alterations in their immune response when exposed to pathogens of bacterial origin, including Mycobacterium tuberculosis, Listeria monocytogenes, and Klebsiella pneumoniae [91,92,93]. Due to poor control of these infections by the immune system, they are more likely to breach the endothelial barrier, leading to septicemia [91]. The inability to control infection under these conditions has been partly attributed to defects in antigen-specific T cell immune responses [93], as well as deficiencies in the phagocytic activity of neutrophils and macrophages [92]. The severity of infections in obese subjects is also heightened in the case of viral infection. Consequently, successive epidemics of H1N1 influenza and SARS-CoV-2 have been correlated with an increased risk of mortality and morbidity in obese patients [94,95]. In the case of Influenza virus infection, it has been demonstrated in a murine model that the increased susceptibility of obese individuals is linked to a defect in antigen presentation to T lymphocytes by dendritic cells, leading to deficient cytotoxic and memory responses [96,97]. It is noteworthy that obesity has recently been associated with greater severity of West Nile virus (WNV) infection in a murine model [98]. This increase in severity was found in females and is linked to dysfunctions of the adaptive immunity in the acute phase, as well as in the late phase with a decrease in the production of neutralizing antibodies in these individuals [98]. Correspondingly, with this inability to produce a humoral response, it is known that obesity has a negative impact on immune response in the case of vaccination. Thus, the immunogenicity induced by vaccines targeting the Hepatitis B and H1N1 influenza viruses is reduced in obese individuals [99,100].
b. Exercise
Much like nutrition, physical activity has an impact on an individual's metabolic status; however, its influence on immune function remains a subject of debate (Figure 5). Epidemiological evidence suggests that regular physical activity reduces the incidence of chronic diseases and the risk of viral and bacterial infections [101]. In fact, frequent physical exercise may enhance immune competence. A prospective cohort study of 1,509 Swedish men and women aged between 20 to 60 years demonstrated that higher levels of physical activity were associated with a lower incidence of self-reported upper respiratory tract infections [102]. Acute exercise sessions of less than 60 minutes have been shown to result in a transient alteration in leukocyte counts and lymphocyte proliferation capabilities [103]. Although these changes remained modest, they could be considered as an enhancement of immunosurveillance during moderate physical activity. This type of exercise serves as a crucial adjunct to the immune system, facilitating the continuous turnover of leukocytes between circulation and tissues [104]. Moreover, physical exercise has been suggested as a valuable tool for COVID-19 prevention by improving immune function, particularly innate and adaptive immunity, and modulating the anti-inflammatory state [105].
However, prolonged and intensive exercise in athletes is associated with immune dysfunction, inflammation, and oxidative stress [106,107]. Exercise may transiently impair immune protection, thereby increasing the risk of infection [108]. Consequently, athletes may experience a temporary or sustained reactivation of viruses, such as the Epstein-Barr virus, due to the suppression of immune parameters during years of intense training [109,110,111].
Figure 5.
Benefice and risk of physical activity in the context of infection. The role of physical activity on immunity in case of viral infection remains a subject of debate in the literature. The data available to date seem to point towards the description of a hormesis phenomenon. Physical activity has a beneficial effect, illustrated by a reduction in the incidence of infection and an increase in immune-surveillance, in the case of regular and moderated activity. On the other hand, when the “dose” of exercise becomes intense and prolonged, deleterious effects and a context conducive to infection are found, namely an increase of inflammation, oxidative stress and alteration of the immune system
Figure 5.
Benefice and risk of physical activity in the context of infection. The role of physical activity on immunity in case of viral infection remains a subject of debate in the literature. The data available to date seem to point towards the description of a hormesis phenomenon. Physical activity has a beneficial effect, illustrated by a reduction in the incidence of infection and an increase in immune-surveillance, in the case of regular and moderated activity. On the other hand, when the “dose” of exercise becomes intense and prolonged, deleterious effects and a context conducive to infection are found, namely an increase of inflammation, oxidative stress and alteration of the immune system
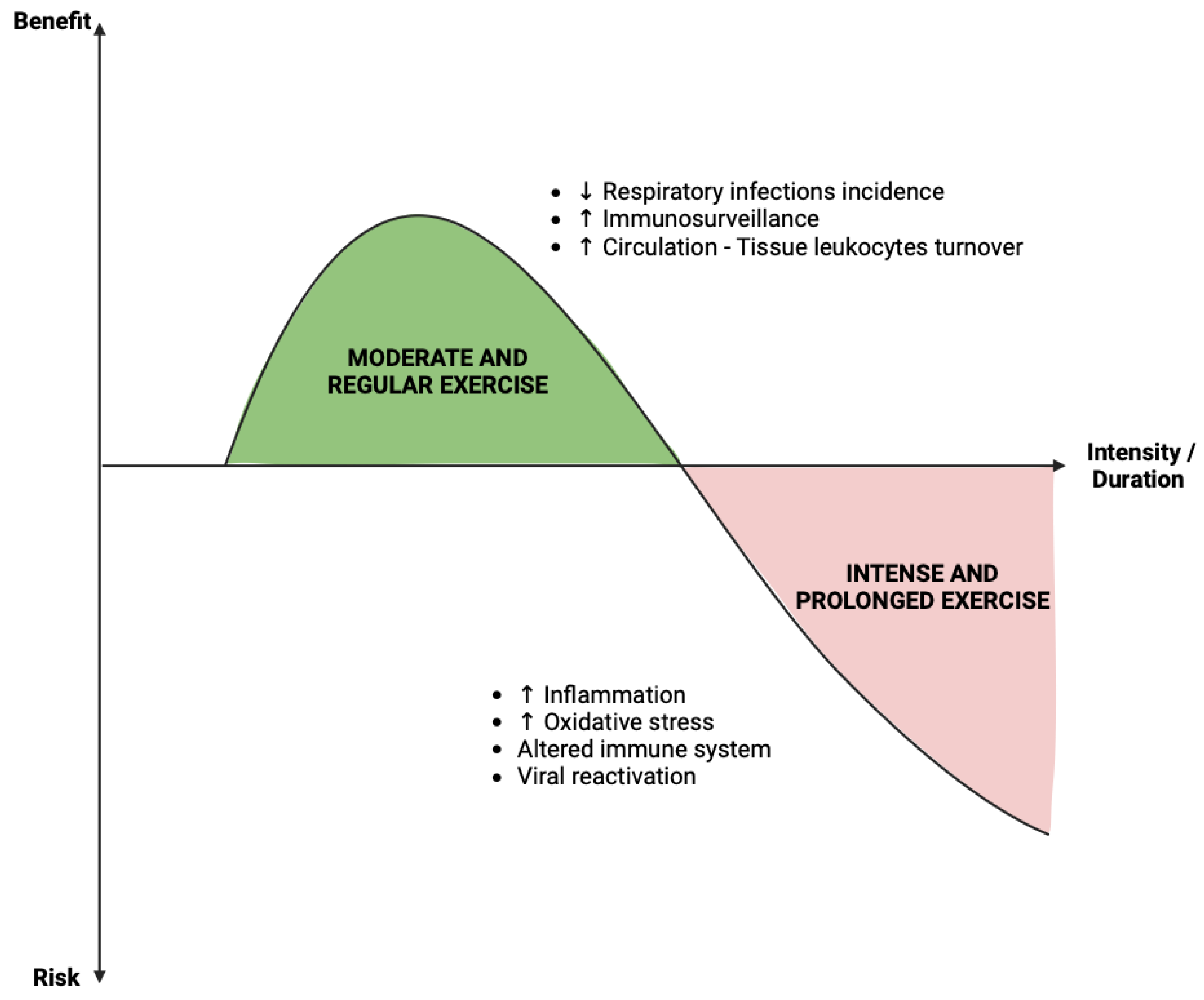
Collectively, these studies demonstrate that physical exercise is beneficial for maintaining functional immunity. However, when practiced at a very high intensity, it is suggested to alter immune function and increase susceptibility to infections (Figure 5). It is also crucial to bear in mind that the connection between exercise and immune function is undoubtedly a multifactorial concept involving other aspects such as nutrition and changes in the microbiome, for example [112]. To this end, further studies using a multifactorial approach will be necessary to enhance our understanding of the immune changes induced by exercise and their underlying mechanisms.
3. At the cellular level: Metabolism involvement in antiviral innate response and sensing
Until now, immunometabolism has been defined as the impact that cellular metabolism may have on the development of the immune response, focusing mainly on cells that are part of the immune system, whether adaptive or innate. Indeed, many reviews focus on immune cells and the metabolic pathways involved in their activation and functionality. However, immunometabolism should also include the early responses established by parenchymal and connective cells after damage or infection. These responses are essential for the immune control of pathogens and the subsequent development of the immune responses themselves. That is why we are now going to shed light on the cells at the forefront during infection and how their metabolism, and its inherent control during the infection, can contribute to a more or less effective antiviral response.
a. Metabolic-dependent sensing of the viral genome and interferon secretion
During a viral infection, cells at the infection site develop an antiviral response based on the secretion of type I interferons (including IFN-α and β) and type III interferons (structurally related to the IL-10 family) [113]. About type I interferons, infected cells will predominantly secrete IFN-β, while IFN-α subtypes concern immune cells and more particularly monocytes and dendritic cells.
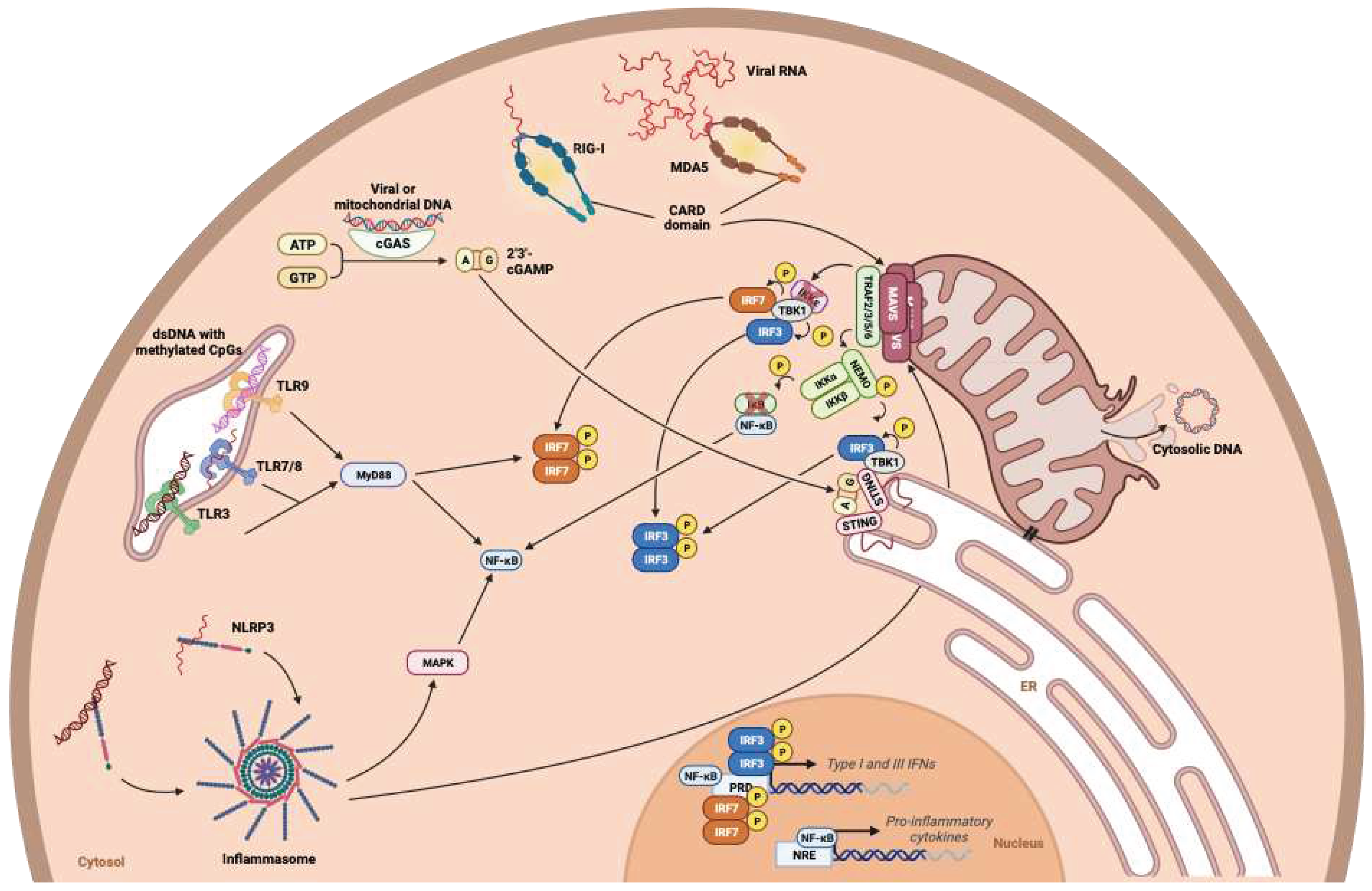
The secretion of IFNs and the resulting antiviral response depend on the recognition of viral PAMPs by cellular PRRs. Viral nucleic acids are the most powerful mediators among the PAMPs capable of alerting the infected cell. The PRRs involved in the detection of these viral nucleic acids vary according to their type. The recognition of viral RNA in the cytosol of infected cells is based on the RIG-I-like receptor (RLR) family, which includes RIG-I and MDA5. RIG-I has shown the ability to bind RNA with motifs that are not found in mammals, such as 5'-PPP or 5'-PP caps [114,115]. On the other hand, detection through MDA5 remains to be explored, although MDA5 has been shown to be essential as a sensor for synthetic double-stranded RNA, such as the poly:IC and the picornaviruses [116,117]. In both cases, recognition is followed by a conformational change and oligomerization of RLRs via their Caspase Activation and Recruitment Domain (CARD). Subsequently, this domain becomes available to bind to the Mitochondrial Antiviral Signaling protein (MAVS) CARD domain, localized at the external mitochondrial membrane (Figure 6) [118,119]. The RLR – MAVS interaction thus leads to the activation of IFN regulatory factors 3, 7 (IRF3, IRF7) and NF-κB. This activation occurs via a signaling cascade involving TNF3 receptor-associated factor (TRAF3), TANK-binding kinase 1 (TBK1) and IκB kinase-ε (IKKε), resulting in the secretion of type I and III IFNs (Figure 6) [120]. The detection of cytosolic DNA is based on the cGAS/STING axis, which, despite the introduction of other sensors, remains the most widely reported pathway in the literature. By detecting cytosolic dsDNA, cyclic GMP-AMP Synthase (cGAS) produces cyclic GMP-AMP (cGAMP) leading to the activation of Stimulator of Interferon Genes (STING) [121]. In turn, STING forms a signaling complex with TBK1 inducing IRF3 and the expression of type I and III IFNs (Figure 6) [121]. At the endosomal level, TLRs 3, 7/8 and 9 respectively detect double-stranded DNA, single-stranded RNA and DNA containing unmethylated CpGs [122]. Signaling by TLRs leads, via their own adaptor proteins (TIR domain Containing Adaptator Molecule 1 - TICAM1 and Myeloid Differentiation primary response 88 - MYD88), to the same transcriptional factors as for the RLR and cGAS/STING pathways, namely activation and dimerization of IRFs 3 and 7 [122]. Finally, the viral genome can also be detected via sensors associated with the inflammasome, the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and particularly NLRP3 NOD-Like Receptor family, Pyrin domain containing 3 (NLRP3). This detection induces MAPK-dependent signaling, associated with the implementation of unspecific inflammatory signals [123]. Additionally, signaling via MAVS leads as previously mentioned, to a more specific antiviral response [122].
Figure 6.
Molecular mechanisms leading to the detection of viral genome. Detection of RNA or DNA from viral origin is dependent on several pathways acting in concert to establish a rapid and efficient antiviral response. These include the activation of RIG-I like (RLR) receptors, Toll-like (TLR) receptors but also of the inflammasome via NLRP3. In addition, cytosolic viral DNA or released DNA resulting from mitochondrial damages lead up to a detection through the cGAS/STING pathway. All these warning signals are integrated and lead to the activation of transcription factors common to these detection pathways, namely IRF3, IRF7 and NF-κB. These transcription factors interact with their promoters to enable the expression of type I and III interferons, on the one hand, and pro-inflammatory cytokines on the other.
Figure 6.
Molecular mechanisms leading to the detection of viral genome. Detection of RNA or DNA from viral origin is dependent on several pathways acting in concert to establish a rapid and efficient antiviral response. These include the activation of RIG-I like (RLR) receptors, Toll-like (TLR) receptors but also of the inflammasome via NLRP3. In addition, cytosolic viral DNA or released DNA resulting from mitochondrial damages lead up to a detection through the cGAS/STING pathway. All these warning signals are integrated and lead to the activation of transcription factors common to these detection pathways, namely IRF3, IRF7 and NF-κB. These transcription factors interact with their promoters to enable the expression of type I and III interferons, on the one hand, and pro-inflammatory cytokines on the other.
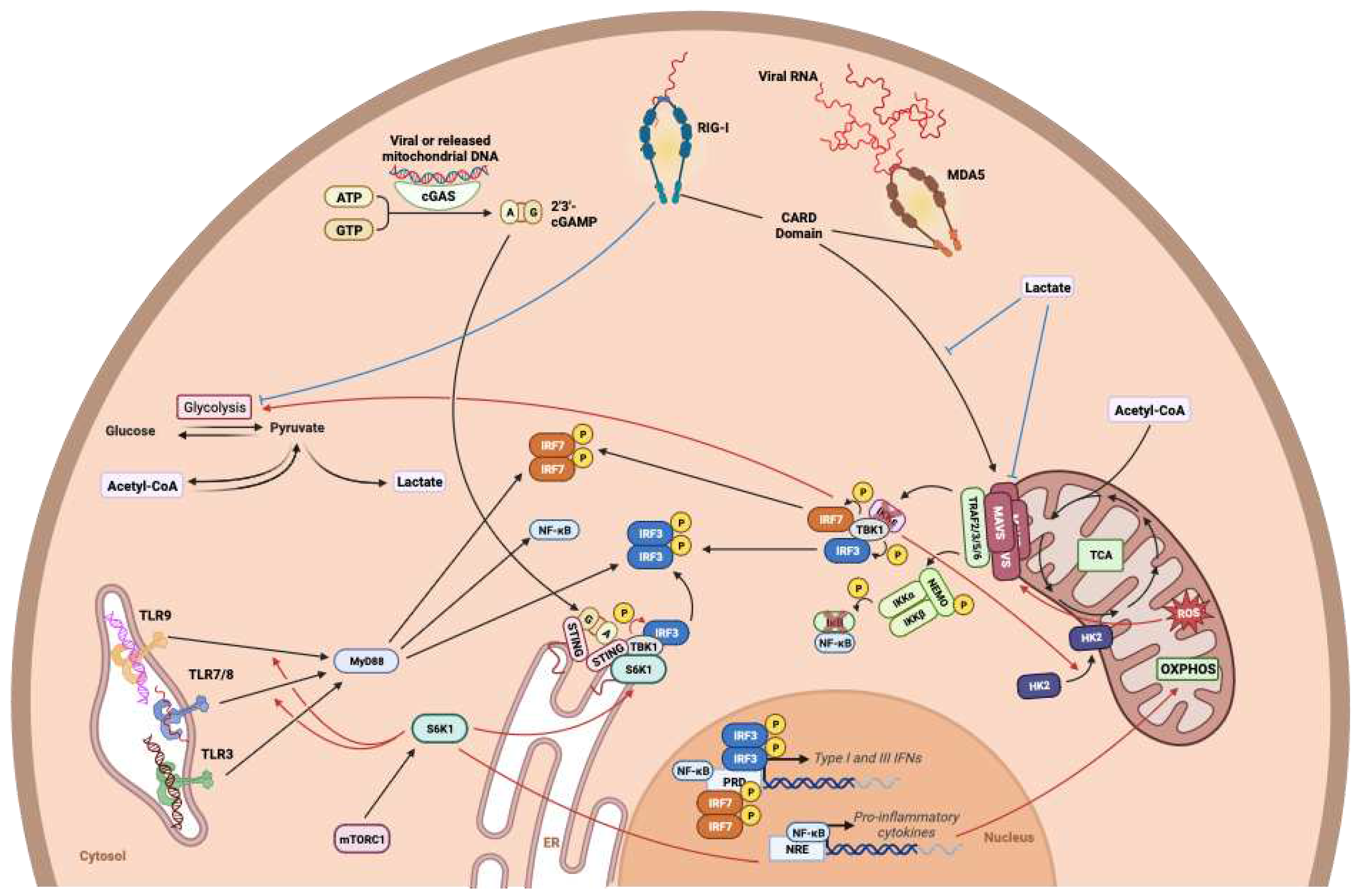
Interestingly, the recent discovery of the interplay between metabolism in signaling and the detection of the viral genome has been observed. For example, the role of lactate production in IFN regulation has recently been highlighted [124]. Using a metabolomic approach in a model of human embryonic kidney cells (HEK293) stimulated with poly:IC, the down-regulation of glycolysis, illustrated by the decrease in all its intermediates, was demonstrated during the establishment of type I IFN production, dependent on the RLR pathway [124]. In particular, the reduction in glycolysis observed during RLR signaling was associated with a reduction in mitochondrial localization of hexokinase 2 (HK2), which is essential for hexokinase to exert its full functionality. These data seem to suggest an inverse relationship between glycolysis and antiviral signaling, since inhibition of glycolysis, either by HK2 knockout or pharmacologically with 2-deoxyglucose inevitably, leads to an increase in TBK1-IRF3 signaling and IFN-β production [124]. To support this idea, the same work highlighted the intrinsic inhibitory function of lactate on RLR activation. Knockdown of the PDHc subunit of pyruvate dehydrogenase led to the impairment of oxidative phosphorylation and accumulation of lactate. This lactate has been shown to interact with MAVS, limiting its mitochondrial localization, its association with RIG-I and its aggregation, all of which are essential for signaling in the IFN pathway (Figure 7) [124]. Furthermore, while lactate has a direct effect on MAVS, no deficits in the cGAS/STING pathway have been reported in these models [124]. In parallel, and supporting the idea that oxidative phosphorylation, unlike glycolysis, favors MAVS signaling, the production of mitochondrial ROS has been shown to be essential for MAVS oligomerization (Figure 7). Inhibition of mitochondrial ROS formation by MitoQ, an antioxidant that specifically targets mROS, results in a reduction in MAVS oligomerization and a subsequent lower secretion of IFN-β [125]. Mitochondrial ROS are thought to be involved in the peroxidation of mitochondrial membrane lipids, in turn, the state of the mitochondrial membrane influences MAVS oligomerization [126].
The link between metabolic pathways and viral genome detection has also been demonstrated in the cGAS/STING and TLRs pathways (Figure 7). In T cells, inhibition of mTORC1, a complex involved in the control of metabolism and cell growth, led to a drastic reduction in the production of type I IFN compared to control cells, despite stimulation with cGAMP and a co-stimulation signal [127]. The importance of mTORC1 on the activity of the cGAS/STING pathway was demonstrated pharmacologically using rapamycin but also in T cell from KO mice for Raptor, a subunit of the mTORC1, leading to a loss of activity of the complex [127]. Investigation of the mechanism by which mTORC1 influences cGAS/STING led to evidence that two downstream factors of mTORC1 (i.e., 4E-BP1 and S6K1) would play a role in the antiviral axis, with S6K1 being preponderant (Figure 7) [127]. However, it has not been explored whether this effect was due to a variation in metabolism in particular, even though mTORC1 finely regulates T cell metabolism, notably during their activation. On the other hand, it has been previously reported that specific inhibition of S6K1 by PF-4708671 resulted in exacerbated glycolysis and inhibition of mitochondrial complex I, illustrating the intimate relationship between S6K1, oxidative phosphorylation and mitochondrial metabolism [128,129]. These data again support the importance of oxidative phosphorylation in the secretion of type I IFNs. In addition to its role in cellular metabolism, S6K1 interacts directly with STING via its kinase domain to form a tripartite S6K1-STING-TBK1 complex required for IRF3 activation [130]. Similarly, the detection of the endosomal genome by TLRs appears to depend on mTORC1. In mouse pDCs, stimulation of TLR9 by its ligand in the presence of rapamycin resulted in lower type I IFN production, and this decrease was correlated with a reduction in IRF7 phosphorylation in rapamycin-treated cells [131]. As previously mentioned, the pathway involving S6K downstream of mTORC1 appears to be involved [130]. Endosomal RNA detection also seems to be affected when mTORC1 is inhibited, since immunization of mice with the Yellow Fever Virus vaccine strain (17D) in the presence of rapamycin results in a decrease in IFN-α/β secretion, this time linked to downregulation of signaling via TLR7 [131]. Interestingly, the effect of mTORC1 appears to be limited to the cGAS/STING and TLR/MYD88 axes, since the use of poly:IC as an antagonist of RLRs in the presence of rapamycin does not result in a difference in IFN production compared to the control situation [131]. Although these data were obtained in immune cells, it is reasonable to speculate that a similar function could be found in non-immune cells. Thus, the metabolic context at the time of infection influences the efficiency of interferon secretion.
Figure 7.
Involvement of metabolism in viral genome detection. The study of the influence of metabolic pathways on the detection of viral pathogens reveals the intricate relationship between metabolic players and viral genome detection. The mTORC1 pathway and S6K, which is a part of this major signaling pathway, have shown their role in promoting cGAS/STING and TLR-dependent detection pathways. On the other hand, aerobic glycolysis and its final metabolite, lactate, interact negatively with the RLR-dependent detection pathway, notably by inhibiting MAVS.
Figure 7.
Involvement of metabolism in viral genome detection. The study of the influence of metabolic pathways on the detection of viral pathogens reveals the intricate relationship between metabolic players and viral genome detection. The mTORC1 pathway and S6K, which is a part of this major signaling pathway, have shown their role in promoting cGAS/STING and TLR-dependent detection pathways. On the other hand, aerobic glycolysis and its final metabolite, lactate, interact negatively with the RLR-dependent detection pathway, notably by inhibiting MAVS.
b. Metabolic-dependent antiviral response
In fact, a metabolism suited to the secretion of type I and III IFNs is important, since the IFNs thus secreted, via an autocrine activation loop and a paracrine action, lead to the production of antiviral effectors [132]. Because these two types of IFNs are secreted concomitantly, it is difficult to determine their intrinsic effect on the production of interferon stimulated genes (ISGs). Type I and III IFNs bind to their respective receptors, IFNAR1/2 and IFNLR1/IL-10Rβ, resulting in the phosphorylation and homodimerization of STAT1 or Gamma interferon Activation Factor (GAF). Additionally, it results in the formation of the Interferon Stimulated Genes Factor 3 (ISGF3) complex composed of IRF9 and phosphorylated STAT1 and 2 [132]. ISGF3 then interacts with interferon-sensitive responsive element (ISRE), while GAF interacts with gamma interferon activation site (GAS), both leading to the expression of ISGs (Figure 8) [132]. ISGs include several antiviral effectors with different functions. While attachment has so far found few limiting factors, apart from heparanase [133], entry is the target of several antiviral effectors, including Myxovirus resistance genes (MX1 and MX2) or proteins of the IFN-inducible transmembrane (IFITM) and tripartite motif (TRIM) families. MX1 and MX2 appear to limit the arrival of viral components in their destination compartment [134,135], while members of the IFITM family, found mainly in late endosomes and lysosomes, inhibit the entry of viruses using these pathways [136,137,138]. However, their mechanism of action remains unclear, with some suggesting that they alter the endosomal acidification essential to the fusion process, while others argue that IFITMs may directly influence the physical properties of endosomal and lysosomal membranes [139].
Figure 8.
Antiviral effectors and cell metabolism interactions. The binding of type I and III interferons (IFNs) to their respective receptors results, through the JAK/STAT pathway, in the nuclear translocation of ISGF3 and GAF factors. These factors interact with their respective promoters, ISRE and GAS, leading to the expression of interferon-stimulated genes (ISGs), which act as effectors in the antiviral response. During viral infection, several of these ISGs have exhibited antiviral activity, whether during virus entry, replication, or assembly. Other ISGs, such as ISG15, play a role in promoting the antiviral response by enhancing the activity of certain factors involved in this response. Metabolism and the antiviral response are intricately linked, particularly in the expression of antiviral effectors. Oxidative phosphorylation, for instance, tends to enhance the expression of certain interferon-stimulated genes (ISGs) like ISG15, ISG54, or ISG56. However, a duality in this response appears to exist, as some genes encoding antiviral effectors are more expressed during aerobic glycolysis, suggesting they might serve as a backup mechanism in the event of metabolic reprogramming during infection.
Figure 8.
Antiviral effectors and cell metabolism interactions. The binding of type I and III interferons (IFNs) to their respective receptors results, through the JAK/STAT pathway, in the nuclear translocation of ISGF3 and GAF factors. These factors interact with their respective promoters, ISRE and GAS, leading to the expression of interferon-stimulated genes (ISGs), which act as effectors in the antiviral response. During viral infection, several of these ISGs have exhibited antiviral activity, whether during virus entry, replication, or assembly. Other ISGs, such as ISG15, play a role in promoting the antiviral response by enhancing the activity of certain factors involved in this response. Metabolism and the antiviral response are intricately linked, particularly in the expression of antiviral effectors. Oxidative phosphorylation, for instance, tends to enhance the expression of certain interferon-stimulated genes (ISGs) like ISG15, ISG54, or ISG56. However, a duality in this response appears to exist, as some genes encoding antiviral effectors are more expressed during aerobic glycolysis, suggesting they might serve as a backup mechanism in the event of metabolic reprogramming during infection.
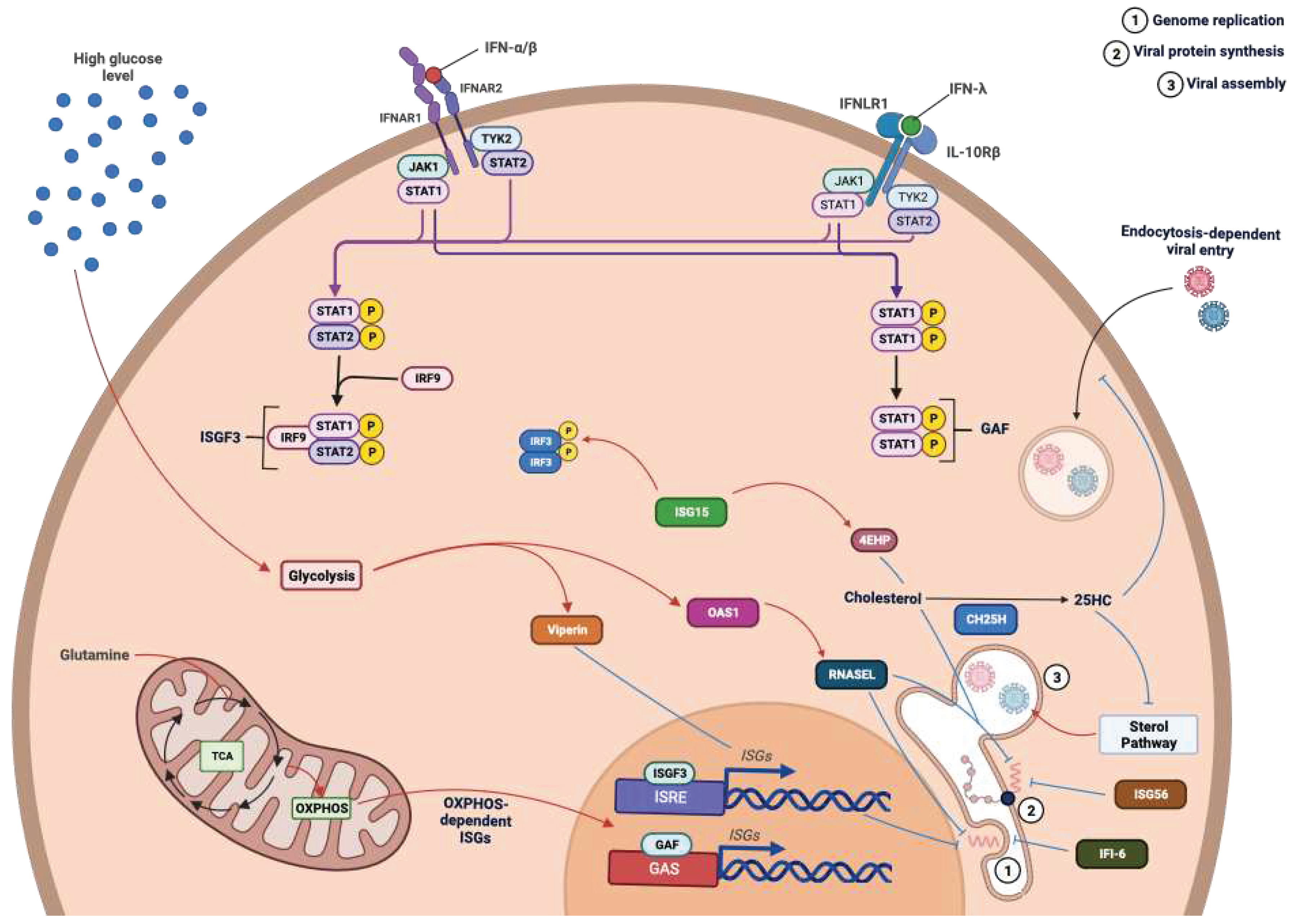
Among the ISGs inhibiting viral entry, the cholestrol-25-hydroxylase (CH25H) has been reported for several viruses, including DENV and ZIKV [140]. The CD25H catalyzes the conversion of cholesterol into 25-hydroxy-cholesterol (25HC), an intermediate thought to be involved in the alteration of physicochemical properties of the membrane and thus, inhibiting the fusion of enveloped viruses [9]. But 25HC possibly plays a post-entry role as an inhibitor of the sterol synthesis pathway, which is essential for viral progeny [141,142,143,144]. The translation of viral proteins is also a target for antiviral effectors, including proteins of the Interferon Induced Protein with Tetratricopeptide repeats family - IFIT (ISG56/IFIT1, ISG54/IFIT2), the protein kinase R (PKR) or ISG15. PKR and IFIT proteins regulate translation initiation by interacting with the initiation factors eiF2α, eiF3C or eiF3E [145,146,147,148]. In addition, certain proteins of the IFIT family have the ability to recognize type 0 caps (without 2-O-methylation) or type 1 caps (m7GpppAmN) leading to inhibition of translation [149,150]. ISG15 protein, a key player in ISGylation at the post-translational stages of viral and host proteins production, has shown multiple functions depending on the ISGylated target [151,152,153]. Going from IRF3 stabilization, maintaining downstream signaling, to increasing the affinity of 4EHP, a negative regulator of translation, for the 5’ cap of messenger RNAs, promoting its ability to inhibit translation [152,153]. The establishment of the replicative complex by deformation of the endoplasmic reticulum (ER) is an essential element in flavivirus replication. This phenomenon is complicated by the expression of Interferon α inducible protein 6 (IFI-6), which prevents the ER membrane invaginations required for the replicative complex [154].
In a similar way to type I IFN secretion, we showed that the promotion of oxidative phosphorylation was favorable to the production of antiviral effectors in response to poly:IC stimulation. Indeed, the stimulation of galactose-cultured cells to increase their oxidative phosphorylation resulted in greater expression of ISG54 and ISG56, two antiviral effectors belonging to the IFIT family (Figure 8) [155]. This positive effect on ISGs expression was abolished in response to rotenone treatment, undeniably linking the observed effect to mitochondrial chain activity [155]. Recently, we showed that up-regulation of antiviral effector production in galactose-grown cells was actually associated with anaplerotic feeding of TCA and OXPHOS by L-glutamine [156]. Surprisingly, other colleagues have shown that high glucose, generally propitious to glycolysis, promotes the expression of an essential ISG for the control of Zika virus infection, namely viperin [157]. These results contrast with our own. However, in this work, the expression of other ISG was not explored [157]. We reconciled these data with our work, showing that cells cultured without L-glutamine, and therefore whose metabolism relies on glycolysis, expressed viperin and OAS1 to a greater extent in response to poly:IC. This observation was indeed related to the glycolytic metabolism of these cells, since the expression of viperin and OAS1 was reduced in the presence of 2-deoxyglucose, an inhibitor of glycolysis (Figure 8) [156]. Therefore, these two ISGs appear to be apart when compared with the other effectors we have studied. This reveals the possibility of a dichotomy in ISGs expression based on cellular metabolism, with some ISGs having greater expression in glycolytic context, while others are favored by oxidative phosphorylation and TCA. In addition, the extent to which a virus or a host relies on L-glutamine during infection remains an open and unresolved question in the literature [158]. Is L-glutamine indispensable to the virus during replication, or does the host need it to produce an effective antiviral response [158]? Our work has shown that even without a productive viral infection, L-glutamine is essential for mounting an antiviral response. However, some antiviral effectors do not require L-glutamine to be produced (such as viperin and OAS1). From a dependency point of view, it seems that viruses may have a greater requirement of L-glutamine for their progeny. It is also possible that this bivalence of the antiviral response is an evolutionary adaptation that enables a response to infection regardless of the metabolic context of the infected cell, particularly in the case of virus-induced metabolic reprogramming.
c. Viral-induced metabolic reprogramming and associated immune evasion
During infection, a virus uses all the cellular machinery and components at its disposal to replicate actively. Among the mechanisms employed by most viruses is the hijacking of the host's cellular metabolism. This enables the rapid production of large quantities of biomass (nucleotides, lipids, amino acids) and energy in the form of ATP. This ability to take control of host metabolism has recently been the focus of several in-depth reviews in the literature. Briefly, to meet their need for resources and energy, viruses induce in the infected cell an effect similar to that already identified by Warburg for cancer cells [5]. Namely, the promotion of glycolysis and the diverting of pyruvate towards lactate production, rather than into the TCA, despite the aerobic context – the so-called aerobic glycolysis or Warburg effect. This results in the rapid production of energy, but also of intermediates that may be involved in other metabolic pathways essential for biomass formation, such as the PPP for nucleic acids [4]. Alongside this Warburg-like effect, there is often an increase in glutaminolysis, with glutamine then anaplerotically feeding the TCA, and its intermediates also enabling biomass production [6]. A noteworthy example is the Dengue virus, which, through its NS1 and NS3 nonstructural proteins, can increase glycolytic activity and cellular lipogenesis [159,160,161]. ZIKV could also disrupt mitochondrial activity by redirecting the use of glycolysis intermediates towards the PPP. This results in mitochondrial dysfunction that may contributes to pathophysiological processes leading to the congenital defects observed in newborns born to infected mothers [162,163]. The significance of this impact on mitochondrial activity needs to be considered in relation to the cellular model [164]. Additionally, there is a viral-induced action on nutrient receptors, influencing the availability of these nutrients and, necessarily, the metabolic pathways associated with them. The expression of transporters such as GLUT1 can thus be upregulated in various cell types (e.g., epithelial and immune cells), as seen in infections by HIV and the Epstein-Barr virus [165,166,167].
Moreover, since the antiviral response seems to rely on appropriate metabolism to be more effective, targeting metabolism becomes an attractive strategy for immune evasion. Therefore, Poxviruses have been shown to produce the F17 protein that has the ability to interact with the Rictor and Raptor subunits of the mTORC1 and mTORC2 complexes, leading to their sequestration and thus dysregulation of mTOR [168]. As mentioned previously, mTOR is also involved in the regulation of the cGAS / STING-dependent antiviral response via S6K [127]. Sequestration of Rictor and Raptor by F17 thus ultimately leads to a decrease in ISG secretion [168]. Similarly, ZIKV NS4A and NS4B proteins inhibit the AKT-mTOR signaling pathway, even if no exploration of this effect on antiviral gene expression was achived [169]. Another study showed that the human immunodeficiency virus (HIV) replication relies on metabolic reprogramming of the host cells. In contrast, cells of individuals known as "controllers" were refractory to this reprogramming, which could explain the low virus replication in these individuals [170]. Despite antiretroviral treatment, the metabolism of non-controller LTs is glucose-dependent, whereas controllers LTs have a variety of metabolic sources to ensure their survival and function [170]. The controllers’ cells also showed a better effector profile, with higher expression of IFNB1 and TNF, strengthening the hypothesis of HIV's ability to hijack the antiviral response by monitoring cell metabolism [170]. Interestingly, the deleterious effect of metabolic reprogramming on the control of viral replication was overcome by treating cells from non-controllers with interleukin 15. This treatment diversified their cellular metabolism, leading to a significant increase in cellular respiration through enhanced lipolysis [170]. Furthermore, supporting the role of mitochondria in the establishment of the innate immune response, it has been established that mitochondrial elongation associated with inhibition of dynamin-related protein 1 (DRP1)-mediated fission, via the NS4B protein of the Dengue virus, limits the development of the RIG-I-dependent antiviral response in favor of viral replication. While mitochondrial elongation is commonly linked to increased OXPHOS and ROS production, in this context, the alteration of the mitochondria–ER platform appears to be the cause of the inhibition of the antiviral response [171].
4. Concluding remarks
In conclusion, although viral control of cellular metabolism for its own benefit appears to be a common consequence of infection that affects the antiviral response, our work tends to show that immune evasion through metabolic control appears to have been thwarted by the host. Regardless of the metabolic context, whether glycolytic or OXPHOS-dependent, the host cell is able to shape its response and produce antiviral effectors; on the one hand, viperin, and on the other, ISGs such as ISG15, 54 or 56. However, as witnessed in the course of several successive epidemics, viruses consistently employ strategies to disrupt the antiviral response during replication, in a frenetic evolutionary race. This crosstalk between virus and immunometabolism, once again, illustrates the red queen effect in infectious processes [172].
Author Contributions
Writing—original draft preparation, G.L., M.H., A.P.-R., D.E.S.; writing—review and editing, G.L., M.H., A.P.-R., D.E.S., P.K.-T., W.V.; conceptualization, G.L., W.V. All authors have read and agreed to the published version of the manuscript.
Funding
G.L., D.E.S. and A.P.-R. have PhD degree scholarship from Réunion University (Ecole doctorale STS) funded by DIRED/2021-0161, DIRED/2021-1115 and DIRED/2021-1124 from Région Réunion, respectively.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
All figures were created with Biorender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and Its Impact on the Immune System. CDR 2020, 16, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Palmer, Clovis. S. Innate Metabolic Responses against Viral Infections. Nat Metab 2022, 4, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat Rev Mol Cell Biol 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral Hijacking of Cellular Metabolism. BMC Biol 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.N.S.; Arjona, S.P.; Santerre, M.; Sawaya, B.E. Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses 2022, 14, 602. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Bui, L.; Fedele, A.L.; Di Mario, C.; Benvenuto, R.; Federico, F.; Ferraccioli, G.; et al. Overweight/Obesity Affects Histological Features and Inflammatory Gene Signature of Synovial Membrane of Rheumatoid Arthritis. Sci Rep 2019, 9, 10420. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Zhu, Z.; Oliveira, M.F.; Smith, D.M.; Rich, J.N.; Bernatchez, J.A.; Siqueira-Neto, J.L. Mechanism of Action of Methotrexate Against Zika Virus. Viruses 2019, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of Immune Cells in Cancer. Nat Rev Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.-C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.-H. Immune Infiltration in Human Tumors: A Prognostic Factor That Should Not Be Ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; De Bruyn, M.; Nijman, H.W. Tumor-Infiltrating Lymphocytes in the Immunotherapy Era. Cell Mol Immunol 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Voelxen, N.F.; Blatt, S.; Knopf, P.; Henkel, M.; Appelhans, C.; Righesso, L.A.R.; Pabst, A.; Goldschmitt, J.; Walenta, S.; Neff, A.; et al. Comparative Metabolic Analysis in Head and Neck Cancer and the Normal Gingiva. Clin Oral Invest 2018, 22, 1033–1043. [Google Scholar] [CrossRef]
- Ottensmeier, C.H.; Perry, K.L.; Harden, E.L.; Stasakova, J.; Jenei, V.; Fleming, J.; Wood, O.; Woo, J.; Woelk, C.H.; Thomas, G.J.; et al. Upregulated Glucose Metabolism Correlates Inversely with CD8+ T-Cell Infiltration and Survival in Squamous Cell Carcinoma. Cancer Research 2016, 76, 4136–4148. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of Tissue Macrophages in Homeostasis and Pathology. Cell Mol Immunol 2022, 19, 384–408. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages. Journal of Biological Chemistry 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. The Journal of Immunology 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metabolism 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-Inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. Journal of Biological Chemistry 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. Journal of Biological Chemistry 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; Prange, K.H.M.; Van Weeghel, M.; Van Der Velden, S.; Ryan, D.G.; Wüst, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A Defective Pentose Phosphate Pathway Reduces Inflammatory Macrophage Responses during Hypercholesterolemia. Cell Reports 2018, 25, 2044–2052.e5. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative Metabolism and PGC-1β Attenuate Macrophage-Mediated Inflammation. Cell Metabolism 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat Immunol 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Namgaladze, D.; Brüne, B. Fatty Acid Oxidation Is Dispensable for Human Macrophage IL-4-Induced Polarization. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2014, 1841, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Liu, J.; Rovira, I.I.; Gonzalez-Hurtado, E.; Lee, J.; Wolfgang, M.J.; Finkel, T. Fatty Acid Oxidation in Macrophage Polarization. Nat Immunol 2016, 17, 216–217. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules That Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-Ketoglutarate Orchestrates Macrophage Activation through Metabolic and Epigenetic Reprogramming. Nat Immunol 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira Da Costa, M.; Reis E Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Sathaliyawala, T.; O’Gorman, W.E.; Greter, M.; Bogunovic, M.; Konjufca, V.; Hou, Z.E.; Nolan, G.P.; Miller, M.J.; Merad, M.; Reizis, B. Mammalian Target of Rapamycin Controls Dendritic Cell Development Downstream of Flt3 Ligand Signaling. Immunity 2010, 33, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Woltman, A.M.; Van Der Kooij, S.W.; Coffer, P.J.; Offringa, R.; Daha, M.R.; Van Kooten, C. Rapamycin Specifically Interferes with GM-CSF Signaling in Human Dendritic Cells, Leading to Apoptosis via Increased p27KIP1 Expression. Blood 2003, 101, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; O’Brien, T.F.; Wright, G.; Yang, J.; Shin, J.; Wright, K.L.; Zhong, X.-P. Critical Role of the Tumor Suppressor Tuberous Sclerosis Complex 1 in Dendritic Cell Activation of CD4 T Cells by Promoting MHC Class II Expression via IRF4 and CIITA. The Journal of Immunology 2013, 191, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.J.; Everts, B. Dendritic Cell Metabolism. Nat Rev Immunol 2015, 15, 18–29. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J.W.; et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-IKKɛ Supports the Anabolic Demands of Dendritic Cell Activation. Nat Immunol 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and Hypoxia-Inducible Factor-1α Modulate Lipopolysaccharide-Induced Dendritic Cell Activation and Function. The Journal of Immunology 2008, 180, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKα1 Deficiency Amplifies Proinflammatory Myeloid APC Activity and CD40 Signaling. Journal of Leukocyte Biology 2013, 94, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Research and Clinical Practice 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, K.; Morris, J.; Bridson, T.; Govan, B.; Rush, C.; Ketheesan, N. Immunological Mechanisms Contributing to the Double Burden of Diabetes and Intracellular Bacterial Infections. Immunology 2015, 144, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Reed, R.L.; Meredith, K.E.; Scuderi, P. Serum Levels of Tumor Necrosis Factor and IL-1α and IL-1β in Diabetic Patients. Diabetes Care 1991, 14, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Aoki, N.; Nishimura, A. In Vitro Production of Interleukin-1, Interleukin-6, and Tumor Necrosis Factor-Alpha in Insulin-Dependent Diabetes Mellitus. The Journal of Clinical Endocrinology & Metabolism 1993, 77, 1072–1077. [Google Scholar] [CrossRef]
- Reinhold, D.; Ansorge, S. Elevated Glucose Levels Stimulate Transforming Growth Factor-Β1 (TGF-Β1), Suppress Interleukin IL-2, IL-6 and IL-10 Production and DNA Synthesis in Peripheral Blood Mononuclear Cells. Horm Metab Res 1996, 28, 267–270. [Google Scholar] [CrossRef]
- Okada, M.; Kitahara, M.; Kishimoto, S.; Matsuda, T.; Hirano, T.; Kishimoto, T. IL-6/BSF-2 Functions as a Killer Helper Factor in the in Vitro Induction of Cytotoxic T Cells. J Immunol 1988, 141, 1543–1549. [Google Scholar] [CrossRef]
- Hu, R.; Xia, C.-Q.; Butfiloski, E.; Clare-Salzler, M. Effect of High Glucose on Cytokine Production by Human Peripheral Blood Immune Cells and Type I Interferon Signaling in Monocytes: Implications for the Role of Hyperglycemia in the Diabetes Inflammatory Process and Host Defense against Infection. Clinical Immunology 2018, 195, 139–148. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; Nerurkar, P.V.; Orillo, B.; Thompson, K.S.; Verma, S.; Nerurkar, V.R. Reduced Immune Cell Infiltration and Increased Pro-Inflammatory Mediators in the Brain of Type 2 Diabetic Mouse Model Infected with West Nile Virus. J Neuroinflammation 2014, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Lecube, A.; Pachón, G.; Petriz, J.; Hernández, C.; Simó, R. Phagocytic Activity Is Impaired in Type 2 Diabetes Mellitus and Increases after Metabolic Improvement. PLoS ONE 2011, 6, e23366. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Twahirwa, M.; Rahbar, M.H.; Schlesinger, L.S. Phagocytosis via Complement or Fc-Gamma Receptors Is Compromised in Monocytes from Type 2 Diabetes Patients with Chronic Hyperglycemia. PLoS ONE 2014, 9, e92977. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.-C.; Yen, C.-L.; Wu, Y.-H.; Chen, S.-Y.; Hsieh, C.-Y.; Chang, T.-C.; Ou, H.-Y.; Shieh, C.-C. Increased Resistin May Suppress Reactive Oxygen Species Production and Inflammasome Activation in Type 2 Diabetic Patients with Pulmonary Tuberculosis Infection. Microbes and Infection 2015, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Perner, A.; Nielsen, S.E.; Rask-Madsen, J. High Glucose Impairs Superoxide Production from Isolated Blood Neutrophils. Intensive Care Med 2003, 29, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Stegenga, M.E.; van der Crabben, S.N.; Blümer, R.M.E.; Levi, M.; Meijers, J.C.M.; Serlie, M.J.; Tanck, M.W.T.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia Enhances Coagulation and Reduces Neutrophil Degranulation, Whereas Hyperinsulinemia Inhibits Fibrinolysis during Human Endotoxemia. Blood 2008, 112, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High Glucose Modulates IL-6 Mediated Immune Homeostasis through Impeding Neutrophil Extracellular Trap Formation. FEBS Letters 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Berrou, J.; Fougeray, S.; Venot, M.; Chardiny, V.; Gautier, J.-F.; Dulphy, N.; Toubert, A.; Peraldi, M.-N. Natural Killer Cell Function, an Important Target for Infection and Tumor Protection, Is Impaired in Type 2 Diabetes. PLoS ONE 2013, 8, e62418. [Google Scholar] [CrossRef]
- Mauriello, C.T.; Hair, P.S.; Rohn, R.D.; Rister, N.S.; Krishna, N.K.; Cunnion, K.M. Hyperglycemia Inhibits Complement-Mediated Immunological Control of S. Aureus in a Rat Model of Peritonitis. Journal of Diabetes Research 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Martinez, N.; Ketheesan, N.; Martens, G.W.; West, K.; Lien, E.; Kornfeld, H. Defects in Early Cell Recruitment Contribute to the Increased Susceptibility to Respiratory Klebsiella Pneumoniae Infection in Diabetic Mice. Microbes and Infection 2016, 18, 649–655. [Google Scholar] [CrossRef]
- Gupta, S.; Maratha, A.; Siednienko, J.; Natarajan, A.; Gajanayake, T.; Hoashi, S.; Miggin, S. Analysis of Inflammatory Cytokine and TLR Expression Levels in Type 2 Diabetes with Complications. Sci Rep 2017, 7, 7633. [Google Scholar] [CrossRef] [PubMed]
- Vallerskog, T.; Martens, G.W.; Kornfeld, H. Diabetic Mice Display a Delayed Adaptive Immune Response to Mycobacterium Tuberculosis. J.I. 2010, 184, 6275–6282. [Google Scholar] [CrossRef]
- Williams, N.L.; Morris, J.L.; Rush, C.; Govan, B.L.; Ketheesan, N. Impact of Streptozotocin-Induced Diabetes on Functional Responses of Dendritic Cells and Macrophages towards Burkholderia Pseudomallei. FEMS Immunol Med Microbiol 2011, 61, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Carey, I.M.; Critchley, J.A.; DeWilde, S.; Harris, T.; Hosking, F.J.; Cook, D.G. Risk of Infection in Type 1 and Type 2 Diabetes Compared With the General Population: A Matched Cohort Study. Diabetes Care 2018, 41, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Critchley, J.A.; Carey, I.M.; Harris, T.; DeWilde, S.; Hosking, F.J.; Cook, D.G. Glycemic Control and Risk of Infections Among People With Type 1 or Type 2 Diabetes in a Large Primary Care Cohort Study. Diabetes Care 2018, 41, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, X.; Ji, Y.; Li, H.; Hou, F.; Xiao, C.; Yuan, P. Increased Risk of Hepatitis B Virus Infection amongst Individuals with Diabetes Mellitus. Bioscience Reports 2019, 39, BSR20181715. [Google Scholar] [CrossRef] [PubMed]
- Kulcsar, K.A.; Coleman, C.M.; Beck, S.E.; Frieman, M.B. Comorbid Diabetes Results in Immune Dysregulation and Enhanced Disease Severity Following MERS-CoV Infection. JCI Insight 2019, 4, e131774. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Norouzi, S.; Ruggiero, A.; Khan, M.S.; Myers, S.; Kavanagh, K.; Vemuri, R. Type-2 Diabetes as a Risk Factor for Severe COVID-19 Infection. Microorganisms 2021, 9, 1211. [Google Scholar] [CrossRef]
- Reiterer, M.; Rajan, M.; Gómez-Banoy, N.; Lau, J.D.; Gomez-Escobar, L.G.; Ma, L.; Gilani, A.; Alvarez-Mulett, S.; Sholle, E.T.; Chandar, V.; et al. Hyperglycemia in Acute COVID-19 Is Characterized by Insulin Resistance and Adipose Tissue Infectivity by SARS-CoV-2. Cell Metabolism 2021, 33, 2174–2188.e5. [Google Scholar] [CrossRef]
- Htun, N.S.N.; Odermatt, P.; Eze, I.C.; Boillat-Blanco, N.; D’Acremont, V.; Probst-Hensch, N. Is Diabetes a Risk Factor for a Severe Clinical Presentation of Dengue? - Review and Meta-Analysis. PLoS Negl Trop Dis 2015, 9, e0003741. [Google Scholar] [CrossRef]
- Sekaran, S.D.; Liew, Z.M.; Yam, H.C.; Raju, C.S. The Association between Diabetes and Obesity with Dengue Infections. Diabetol Metab Syndr 2022, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Després, J.-P. Body Fat Distribution and Risk of Cardiovascular Disease: An Update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef]
- World Health Organisation Obesity and Overweight Available online:. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 30 September 2023).
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocrine Reviews 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.; Kang, D.-W.; DiBaise, J.K. Effects of Gut Microbes on Nutrient Absorption and Energy Regulation. Nutr Clin Pract 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. The Journal of Clinical Endocrinology & Metabolism 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D.; Dixit, V.D. Immunological Complications of Obesity. Nat Immunol 2012, 13, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. The Journal of Clinical Endocrinology & Metabolism 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.-K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A Subpopulation of Macrophages Infiltrates Hypertrophic Adipose Tissue and Is Activated by Free Fatty Acids via Toll-like Receptors 2 and 4 and JNK-Dependent Pathways. Journal of Biological Chemistry 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid–Induced Insulin Resistance. J. Clin. Invest. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local Proliferation Initiates Macrophage Accumulation in Adipose Tissue during Obesity. Cell Death Dis 2016, 7, e2167–e2167. [Google Scholar] [CrossRef] [PubMed]
- Brotfain, E.; Hadad, N.; Shapira, Y.; Avinoah, E.; Zlotnik, A.; Raichel, L.; Levy, R. Neutrophil Functions in Morbidly Obese Subjects. Clinical and Experimental Immunology 2015, 181, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Kim, M.-S.; Pae, M.; Yamamoto, Y.; Eberlé, D.; Shimada, T.; Kamei, N.; Park, H.-S.; Sasorith, S.; Woo, J.R.; et al. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metabolism 2016, 23, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat Med 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M. Reassessing Human Adipose Tissue. N Engl J Med 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guerrero-Juarez, C.F.; Chen, S.X.; Zhang, X.; Yin, M.; Li, F.; Wu, S.; Chen, J.; Li, M.; Liu, Y.; et al. Diet-Induced Obesity Promotes Infection by Impairment of the Innate Antimicrobial Defense Function of Dermal Adipocyte Progenitors. Sci. Transl. Med. 2021, 13, eabb5280. [Google Scholar] [CrossRef] [PubMed]
- Shipman, A.R.; Millington, G.W.M. Obesity and the Skin: Obesity and the Skin. British Journal of Dermatology 2011, 165, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Liccardi, A.; Graziadio, C.; Barrea, L.; Muscogiuri, G.; Colao, A. Obesity and Infectious Diseases: Pathophysiology and Epidemiology of a Double Pandemic Condition. Int J Obes 2022, 46, 449–465. [Google Scholar] [CrossRef]
- Kaspersen, K.A.; Pedersen, O.B.; Petersen, M.S.; Hjalgrim, H.; Rostgaard, K.; Møller, B.K.; Juul-Sørensen, C.; Kotzé, S.; Dinh, K.M.; Erikstrup, L.T.; et al. Obesity and Risk of Infection: Results from the Danish Blood Donor Study. Epidemiology 2015, 26, 580–589. [Google Scholar] [CrossRef]
- Las Heras, V.; Clooney, A.G.; Ryan, F.J.; Cabrera-Rubio, R.; Casey, P.G.; Hueston, C.M.; Pinheiro, J.; Rudkin, J.K.; Melgar, S.; Cotter, P.D.; et al. Short-Term Consumption of a High-Fat Diet Increases Host Susceptibility to Listeria Monocytogenes Infection. Microbiome 2019, 7, 7. [Google Scholar] [CrossRef]
- Mancuso, P.; Gottschalk, A.; Phare, S.M.; Peters-Golden, M.; Lukacs, N.W.; Huffnagle, G.B. Leptin-Deficient Mice Exhibit Impaired Host Defense in Gram-Negative Pneumonia. J Immunol 2002, 168, 4018–4024. [Google Scholar] [CrossRef] [PubMed]
- Wieland, C.W.; Florquin, S.; Chan, E.D.; Leemans, J.C.; Weijer, S.; Verbon, A.; Fantuzzi, G.; van der Poll, T. Pulmonary Mycobacterium Tuberculosis Infection in Leptin-Deficient Ob/Ob Mice. International Immunology 2005, 17, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.K.; Acosta, M.; Samuel, M.C.; Schechter, R.; Vugia, D.J.; Harriman, K.; Matyas, B.T. the California Pandemic (H1N1) Working Group A Novel Risk Factor for a Novel Virus: Obesity and 2009 Pandemic Influenza A (H1N1). Clinical Infectious Diseases 2011, 52, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Popkin, B.M.; Du, S.; Green, W.D.; Beck, M.A.; Algaith, T.; Herbst, C.H.; Alsukait, R.F.; Alluhidan, M.; Alazemi, N.; Shekar, M. Individuals with Obesity and COVID-19: A Global Perspective on the Epidemiology and Biological Relationships. Obesity Reviews 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.A.; Sheridan, P.A.; Beck, M.A. Diet-Induced Obesity Impairs the T Cell Memory Response to Influenza Virus Infection. J.I. 2010, 184, 3127–3133. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Sheridan, P.A.; Tseng, R.J.; Sheridan, J.F.; Beck, M.A. Selective Impairment in Dendritic Cell Function and Altered Antigen-Specific CD8 + T-Cell Responses in Diet-Induced Obese Mice Infected with Influenza Virus. Immunology 2009, 126, 268–279. [Google Scholar] [CrossRef]
- Geerling, E.; Stone, E.T.; Steffen, T.L.; Hassert, M.; Brien, J.D.; Pinto, A.K. Obesity Enhances Disease Severity in Female Mice Following West Nile Virus Infection. Front. Immunol. 2021, 12, 739025. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Kim, J.-K.; Kim, D.-J.; Nam, J.-H.; Shim, S.-M.; Choi, Y.-K.; Lee, C.-H.; Poo, H. Diet-Induced Obesity Dramatically Reduces the Efficacy of a 2009 Pandemic H1N1 Vaccine in a Mouse Model. The Journal of Infectious Diseases 2012, 205, 244–251. [Google Scholar] [CrossRef]
- Weber, D.; Rutala, W.; Samsa, G.; Bradshaw, S.; Lemon, S. Impaired Immunogenicity of Hepatitis B Vaccine in Obese Persons. New England Journal of Medicine 1986, 314, 1393–1393. [Google Scholar] [CrossRef]
- Pape, K.; Ryttergaard, L.; Rotevatn, T.A.; Nielsen, B.J.; Torp-Pedersen, C.; Overgaard, C.; Bøggild, H. Leisure-Time Physical Activity and the Risk of Suspected Bacterial Infections. Medicine & Science in Sports & Exercise 2016, 48, 1737–1744. [Google Scholar] [CrossRef]
- Fondell, E.; Lagerros, Y.T.; Sundberg, C.J.; Lekander, M.; Bälter, O.; Rothman, K.J.; Bälter, K. Physical Activity, Stress, and Self-Reported Upper Respiratory Tract Infection. Medicine & Science in Sports & Exercise 2011, 43, 272–279. [Google Scholar] [CrossRef]
- Nieman, D.C.; Henson, D.A.; Austin, M.D.; Brown, V.A. Immune Response to a 30-Minute Walk: Medicine & Science in Sports & Exercise 2005, 37, 57–62. [CrossRef]
- Adams, G.R.; Zaldivar, F.P.; Nance, D.M.; Kodesh, E.; Radom-Aizik, S.; Cooper, D.M. Exercise and Leukocyte Interchange among Central Circulation, Lung, Spleen, and Muscle. Brain, Behavior, and Immunity 2011, 25, 658–666. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; González-Bernal, J.J.; Sánchez-Serrano, N.; Navascués, L.J.; Ascaso-del-Río, A.; Mielgo-Ayuso, J. Physical Exercise as a Multimodal Tool for COVID-19: Could It Be Used as a Preventive Strategy? International Journal of Environmental Research and Public Health 2020, 17, 8496. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C. Immune Response to Heavy Exertion. Journal of Applied Physiology 1997, 82, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Northoff, H.; Berg, A. Immunologic Mediators as Parameters of the Reaction to Strenuous Exercise. Int J Sports Med 1991, 12, S9–S15. [Google Scholar] [CrossRef]
- Nieman, D.C.; Wentz, L.M. The Compelling Link between Physical Activity and the Body’s Defense System. Journal of Sport and Health Science 2019, 8, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Clancy, R.L. Reversal in Fatigued Athletes of a Defect in Interferon Secretion after Administration of Lactobacillus Acidophilus. British Journal of Sports Medicine 2006, 40, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.; Pyne, D.B.; P. Austin, J.; Lynn Francis, J.; Clancy, R.L.; Mcdonald, W.A.; Fricker, P.A. Epstein-Barr Virus Reactivation and Upper-Respiratory Illness in Elite Swimmers: . Medicine & Science in Sports & Exercise 2002, 34, 411–417. [CrossRef]
- Reid, V.L. Clinical Investigation of Athletes with Persistent Fatigue and/or Recurrent Infections. British Journal of Sports Medicine 2004, 38, 42–45. [Google Scholar] [CrossRef]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The Microbiome of Professional Athletes Differs from That of More Sedentary Subjects in Composition and Particularly at the Functional Metabolic Level. Gut, 2017; gutjnl-2016-313627. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5’-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis E Sousa, C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5’-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential Role of Mda-5 in Type I IFN Responses to Polyriboinosinic:Polyribocytidylic Acid and Encephalomyocarditis Picornavirus. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural Basis for the Activation of Innate Immune Pattern-Recognition Receptor RIG-I by Viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-G.; Wang, Y.-Y.; Han, K.-J.; Li, L.-Y.; Zhai, Z.; Shu, H.-B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling. Molecular Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Paz, S.; Vilasco, M.; Werden, S.J.; Arguello, M.; Joseph-Pillai, D.; Zhao, T.; Nguyen, T.L.-A.; Sun, Q.; Meurs, E.F.; Lin, R.; et al. A Functional C-Terminal TRAF3-Binding Site in MAVS Participates in Positive and Negative Regulation of the IFN Antiviral Response. Cell Res 2011, 21, 895–910. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA Sensing by the cGAS–STING Pathway in Health and Disease. Nat Rev Genet 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.T.; Gale, M.; Loo, Y.-M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef] [PubMed]
- Buskiewicz, I.A.; Montgomery, T.; Yasewicz, E.C.; Huber, S.A.; Murphy, M.P.; Hartley, R.C.; Kelly, R.; Crow, M.K.; Perl, A.; Budd, R.C.; et al. Reactive Oxygen Species Induce Virus-Independent MAVS Oligomerization in Systemic Lupus Erythematosus. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Nobre, L.; Wise, D.; Ron, D.; Volmer, R. Modulation of Innate Immune Signalling by Lipid-Mediated MAVS Transmembrane Domain Oligomerization. PLoS ONE 2015, 10, e0136883. [Google Scholar] [CrossRef]
- Imanishi, T.; Unno, M.; Kobayashi, W.; Yoneda, N.; Matsuda, S.; Ikeda, K.; Hoshii, T.; Hirao, A.; Miyake, K.; Barber, G.N.; et al. Reciprocal Regulation of STING and TCR Signaling by mTORC1 for T-Cell Activation and Function. Life Sci. Alliance 2019, 2, e201800282. [Google Scholar] [CrossRef] [PubMed]
- Shum, M.; Bellmann, K.; St-Pierre, P.; Marette, A. Pharmacological Inhibition of S6K1 Increases Glucose Metabolism and Akt Signalling in Vitro and in Diet-Induced Obese Mice. Diabetologia 2016, 59, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Shum, M.; Houde, V.P.; Bellemare, V.; Junges Moreira, R.; Bellmann, K.; St-Pierre, P.; Viollet, B.; Foretz, M.; Marette, A. Inhibition of Mitochondrial Complex 1 by the S6K1 Inhibitor PF-4708671 Partly Contributes to Its Glucose Metabolic Effects in Muscle and Liver Cells. Journal of Biological Chemistry 2019, 294, 12250–12260. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Alain, T.; Szretter, K.J.; Stephenson, K.; Pol, J.G.; Atherton, M.J.; Hoang, H.-D.; Fonseca, B.D.; Zakaria, C.; Chen, L.; et al. S6K-STING Interaction Regulates Cytosolic DNA–Mediated Activation of the Transcription Factor IRF3. Nat Immunol 2016, 17, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Manicassamy, S.; Tang, H.; Kasturi, S.P.; Pirani, A.; Murthy, N.; Pulendran, B. Toll-like Receptor–Mediated Induction of Type I Interferon in Plasmacytoid Dendritic Cells Requires the Rapamycin-Sensitive PI(3)K-mTOR-p70S6K Pathway. Nat Immunol 2008, 9, 1157–1164. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-Viral Specificity of IFN-Induced Genes Reveals New Roles for cGAS in Innate Immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 Is an Interferon-Induced Inhibitor of HIV-1 Infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Janzen, C.; Hohenberg, H.; Haller, O. Antivirally Active MxA Protein Sequesters La Crosse Virus Nucleocapsid Protein into Perinuclear Complexes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.-M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 Inhibits Influenza A Virus Infection by Preventing Cytosolic Entry. PLoS Pathog 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Woodward, J.; Lau, D.T.-Y.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M. IFITM1 Is a Tight Junction Protein That Inhibits Hepatitis C Virus Entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Wee, Y.S.; Roundy, K.M.; Weis, J.J.; Weis, J.H. Interferon-Inducible Transmembrane Proteins of the Innate Immune Response Act as Membrane Organizers by Influencing Clathrin and v-ATPase Localization and Function. Innate Immun 2012, 18, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, Y.-Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X.-Y.; Zhang, N.-N.; Watanabe, M.; Dong, H.-L.; Liu, P.; et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef]
- Bordier, B.B.; Marion, P.L.; Ohashi, K.; Kay, M.A.; Greenberg, H.B.; Casey, J.L.; Glenn, J.S. A Prenylation Inhibitor Prevents Production of Infectious Hepatitis Delta Virus Particles. J Virol 2002, 76, 10465–10472. [Google Scholar] [CrossRef] [PubMed]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.-Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Casey, J.L.; et al. In Vivo Antiviral Efficacy of Prenylation Inhibitors against Hepatitis Delta Virus. J. Clin. Invest. 2003, 112, 407–414. [Google Scholar] [CrossRef]
- Chang, T.Y.; Limanek, J.S. Regulation of Cytosolic Acetoacetyl Coenzyme A Thiolase, 3-Hydroxy-3-Methylglutaryl Coenzyme A Synthase, 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase, and Mevalonate Kinase by Low Density Lipoprotein and by 25-Hydroxycholesterol in Chinese Hamster Ovary Cells. Journal of Biological Chemistry 1980, 255, 7787–7795. [Google Scholar] [CrossRef]
- Kandutsch, A.A.; Chen, H.W.; Heiniger, H.-J. Biological Activity of Some Oxygenated Sterols. Science 1978, 201, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Guo, J. A New Pathway of Translational Regulation Mediated by Eukaryotic Initiation Factor 3. The EMBO Journal 2000, 19, 6891–6899. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.J.; Bhasker, C.R.; Merrick, W.C.; Sen, G.C. Viral Stress-Inducible Protein P56 Inhibits Translation by Blocking the Interaction of eIF3 with the Ternary Complex eIF2·GTP·Met-tRNAi. Journal of Biological Chemistry 2003, 278, 39477–39482. [Google Scholar] [CrossRef] [PubMed]
- Pindel, A.; Sadler, A. The Role of Protein Kinase R in the Interferon Response. Journal of Interferon & Cytokine Research 2011, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Hui, D.J.; Merrick, W.C.; Sen, G.C. Distinct Induction Patterns and Functions of Two Closely Related Interferon-Inducible Human Genes, ISG54 and ISG56. Journal of Biological Chemistry 2006, 281, 34064–34071. [Google Scholar] [CrossRef] [PubMed]
- Fleith, R.C.; Mears, H.V.; Leong, X.Y.; Sanford, T.J.; Emmott, E.; Graham, S.C.; Mansur, D.S.; Sweeney, T.R. IFIT3 and IFIT2/3 Promote IFIT1-Mediated Translation Inhibition by Enhancing Binding to Non-Self RNA. Nucleic Acids Research 2018, 46, 5269–5285. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.T.; Pestova, T.V. Inhibition of Translation by IFIT Family Members Is Determined by Their Ability to Interact Selectively with the 5′-Terminal Regions of Cap0-, Cap1- and 5′ppp- mRNAs. Nucleic Acids Research 2014, 42, 3228–3245. [Google Scholar] [CrossRef]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Molecular Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Okumura, F.; Zou, W.; Zhang, D.-E. ISG15 Modification of the eIF4E Cognate 4EHP Enhances Cap Structure-Binding Activity of 4EHP. Genes Dev. 2007, 21, 255–260. [Google Scholar] [CrossRef]
- Shi, H.-X.; Yang, K.; Liu, X.; Liu, X.-Y.; Wei, B.; Shan, Y.-F.; Zhu, L.-H.; Wang, C. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Molecular and Cellular Biology 2010, 30, 2424–2436. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR Screen Identifies IFI6 as an ER-Resident Interferon Effector That Blocks Flavivirus Replication. Nat Microbiol 2018, 3, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; El Safadi, D.; Paulo-Ramos, A.; Hoareau, M.; Desprès, P.; Krejbich-Trotot, P.; Chouchou, F.; Roche, M.; Viranaicken, W. The Efficient Antiviral Response of A549 Cells Is Enhanced When Mitochondrial Respiration Is Promoted. Pathogens 2022, 11, 1168. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Paulo-Ramos, A.; Hoarau, M.; El Safadi, D.; Meilhac, O.; Krejbich-Trotot, P.; Roche, M.; Viranaicken, W. Metabolic Dependency Shapes Bivalent Antiviral Response in Host Cells: The Role of Glutamine, Immunology. 2023.
- Reslan, A.; Haddad, J.G.; Desprès, P.; Bascands, J.-L.; Gadea, G. High Glucose Induces in HK2 Kidney Cells an IFN–Dependent ZIKV Antiviral Status Fueled by Viperin. Biomedicines 2022, 10, 1577. [Google Scholar] [CrossRef] [PubMed]
- Koufaris, C.; Nicolaidou, V. Glutamine Addiction in Virus-Infected Mammalian Cells: A Target of the Innate Immune System? Medical Hypotheses 2021, 153, 110620. [Google Scholar] [CrossRef]
- Allonso, D.; Andrade, I.S.; Conde, J.N.; Coelho, D.R.; Rocha, D.C.P.; Da Silva, M.L.; Ventura, G.T.; Silva, E.M.; Mohana-Borges, R. Dengue Virus NS1 Protein Modulates Cellular Energy Metabolism by Increasing Glyceraldehyde-3-Phosphate Dehydrogenase Activity. J Virol 2015, 89, 11871–11883. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J Virol 2015, 89, 2358–2366. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; LaCount, D.J.; Kuhn, R.J.; Randall, G. Dengue Virus Nonstructural Protein 3 Redistributes Fatty Acid Synthase to Sites of Viral Replication and Increases Cellular Fatty Acid Synthesis. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Karmirian, K.; Pedrosa, C.D.S.G.; Souza, L.R.Q.; Assis-de-Lemos, G.; Martins, T.M.; Ferreira, J.D.C.C.G.; De Azevedo Reis, G.F.; Silva, E.S.; Silva, D.; et al. Zika Virus Infection Leads to Mitochondrial Failure, Oxidative Stress and DNA Damage in Human iPSC-Derived Astrocytes. Sci Rep 2020, 10, 1218. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Low, J.Z.H.; Gan, E.S.; Kwek, S.S.; Cui, L.; Tan, H.C.; Mok, D.Z.L.; Chan, C.Y.Y.; Sessions, O.M.; Watanabe, S.; et al. Dysregulated Metabolism Underpins Zika-Virus-Infection-Associated Impairment in Fetal Development. Cell Reports 2021, 37, 110118. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Crook, A.A.; Pattnaik, A.; Torres-Gerena, A.D.; Khalimonchuk, O.; Powers, R.; Franco, R.; Pattnaik, A.K. Redox Regulation and Metabolic Dependency of Zika Virus Replication: Inhibition by Nrf2-Antioxidant Response and NAD(H) Antimetabolites. J Virol 2023, 97, e01363–22. [Google Scholar] [CrossRef]
- Anzinger, J.J.; Butterfield, T.R.; Gouillou, M.; McCune, J.M.; Crowe, S.M.; Palmer, C.S. Glut1 Expression Level on Inflammatory Monocytes Is Associated With Markers of Cardiovascular Disease Risk in HIV-Infected Individuals. JAIDS Journal of Acquired Immune Deficiency Syndromes 2018, 77, e28–e30. [Google Scholar] [CrossRef] [PubMed]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A.; et al. Glut1-Mediated Glucose Transport Regulates HIV Infection. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 2549–2554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jia, L.; Lin, W.; Yip, Y.L.; Lo, K.W.; Lau, V.M.Y.; Zhu, D.; Tsang, C.M.; Zhou, Y.; Deng, W.; et al. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 Upregulates Glucose Transporter 1 Transcription via the mTORC1/NF-κB Signaling Pathways. J Virol 2017, 91, e02168–16. [Google Scholar] [CrossRef] [PubMed]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157.e17. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Angin, M.; Volant, S.; Passaes, C.; Lecuroux, C.; Monceaux, V.; Dillies, M.-A.; Valle-Casuso, J.C.; Pancino, G.; Vaslin, B.; Le Grand, R.; et al. Metabolic Plasticity of HIV-Specific CD8+ T Cells Is Associated with Enhanced Antiviral Potential and Natural Control of HIV-1 Infection. Nat Metab 2019, 1, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host & Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef]
- Van Valen, L. A New Evolutionary Law. EVOL. THEORY 1973, 1, 1–30. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



