
Submitted:
20 November 2023
Posted:
21 November 2023
You are already at the latest version
Abstract
Background: With continuous advances in next-generation sequencing (NGS), novel hepadnaviruses have been discovered in many species over the last 10 years. Methods: In this study, the cloud search and analysis of NGS data published in Nature by Edgar et al. was used as a basis to re-mine and reanalyze public NGS data for new hepadnaviruses. Results: Ultimately, at least 41 new species of hepadnaviruses were identified, including hepadnaviruses from the model animals hamster and mouse, frog hepadnavirus with pan-species infectivity, and diverse African cichlid hepadnaviruses that circulate within populations. Conclusions: The discovery of the new species of hepadnaviruses not only provides new clues for the study of the origin and evolution of hepadnaviruses, but also can be used to construct new hepadnavirus animal infection models, which will be helpful for the research of eradicative drugs for hepatitis B virus (HBV).
Keywords:
new hepadnaviruses
; hepatitis B virus
; animal model
; HBV cure
; viral infection
; metagenomics
; next-generation sequencing (NGS)
1. Introduction
Currently, there are more than 257 million chronic HBV patients globally, including 80–100 million patients in China of whom approximately 40% will progress to develop cirrhosis and liver cancer [1,2,3]. HBV remains a major global public health challenge. Hepadnaviruses are members of the Hepadnaviridae family, which can be divided into 6 genera [4], including orthohepadnaviruses that infect mammals, avihepadnaviruses that infect duck and birds, metahepadnaviruses and parahepadnaviruses that infects fish, herpetohepadnaviruses that infect reptiles and frogs, and non-enveloped nackednaviruses that infects fish. The origin of hepadnaviruses can be traced to as early as 400 million years ago [4].
Next-generation sequencing (NGS) analyses can be utilized to discover viral sequences in hosts, especially in cases of novel viruses with no reference sequences. With advances in NGS technology in recent years, new hepadnaviruses have been successively discovered in numerous animal species (metagenomic HBV), such as fish [4,7,8], frogs [6,9,10], bats [5,11], cats [12], dogs [13], antelope [14], raccoons [15], horses [16], and shrews [17,18,19]. These findings have yielded a deeper understanding of the origin and evolution of hepadnaviruses [20] together with studies on ancient HBV [21,22] and endogenous HBV in birds [23,24].
The lack of any suitable animal model of hepadnaviral infection severely limits HBV studies [25,26]. Hepadnavirus infections such as those caused by human HBV exhibit very strong host specificity, only infecting humans and chimpanzees but not common model animal species such as mice, rats, rabbits, or monkeys. After the discovery of the human HBV receptor sodium taurocholate co-transporting polypeptide (NTCP) [27,28], it was found that the mice hepatocytes expressing human NTCP were not susceptible to hepadnaviral infection, suggesting that additional host factors can restrict such infections in mice. At present, research on hepadnaviruses in mice has been restricted to HBV transgenic mice, AAV/AdV vector-medicated HBV infection, and the human liver chimeric mouse model [29,30,31,32], which severely limit anti-HBV drug research. It is necessary to discover more novel hepadnaviruses in new species in order to establish ideal animal models of hepadnaviral infection.
With advances in NGS technologies, the accumulated volume of nucleic acid sequences in public databases has surpassed 20 PB and is growing exponentially. In 2022, Edgar et al. published the Serratus cloud computing platform in Nature [33] and fully utilized existing commercialized cloud computing power and storage space for the mining and reutilization of NGS data. A total of 2849 vertebrate viruses were discovered, including hepadnaviruses.
In this study, Serratus platform were used as a basis to further re-mine and reanalyze raw NGS data available in public databases for hepadanviruses. Dozens of hepadnavirus genomes were obtained from new species, including hepadnaviruses from hamster, frog hepadnavirus with pan-species infectivity, and diverse African cichlid hepadnaviruses circulating within populations.
2. Materials and Methods
2.1. Search of viral genomes
BLASTN [34] was used to screen the Sequence Read Archive (SRA) database, which is accessible at the National Center to Biotechnology Information (NCBI). The multiple nucleotide sequences of the Hepadnaviridae genome were used as bait to query. A Blast hit with an E-value of 10−20 was considered generally, and all hits were verified manually by inspection of the Blast outputs. The main data, that is, samples with hepadnavirus sequences in the metagenome, come from the study of Edgar et al. [33], and they can be obtained from the Serratus Explorer website (serratus.io).
2.2. Assembly of viral genomes
Raw sequencing reads were downloaded from NCBI or ENA. Fastp [35] was used for quality control and Cutadapt for trimming adapter sequences and low-quality bases. MEGAHIT [36] was utilized for de novo assembly of a viral genome. MMseqs2 [37] was used to search nucleotide sequence sets. There are two methods for assembling virus genomes. (1) No reference template assembly method by MEGAHIT. A series of assembled fragments were obtained after using MEGAHIT to preprocess NGS data. Using MMseqs2, all of these assembled fragments were analyzed with known Hepadnaviridae genomes as bait to obtain newly assembled hepadnavirus-related fragments. (2) Search-first, assembly-second method. MMseqs2 involves analyzing NGS data with all of the known Hepadnaviridae genomes as bait, thus directly conducting blast analysis to obtain fragment sequences related to the hepadnaviruses. The obtained relevant fragments were assembled to the closest Hepadnaviridae genome template with Geneious software [38]. When necessary, multiple NGS samples with template references were combined for assembly. Both ends of the linear assembly were completed manually to account for the circularization of the viral genomes. For cases of very low read coverage resulting in fragmental genomes, the fragments were manually joined using closely related genomes as the reference.
2.3. Annotation of viral genomes
Unlike the traditional EcoRI restriction enzyme cleavage sites of the Hepadnaviridae genome, all Hepadnaviridae genome initiation sites were selected from the start position of the HBc gene. For each newly discovered virus genome, open reading frames were predicted with Geneious software [38], and they were annotated based on those from the related reference virus genomes.
2.4. Sequence alignments
Sequence alignment was conducted with Geneious software using the MAFFT [39] plugin with default settings: automatic algorithm selection, a 200PAM/k=2 scoring matrix with a gap-open penalty, and offset values set to 1.53 and 0.123.
2.5. Phylogenetic analysis
A phylogenetic tree was created using the Geneious tree builder [38]. The genetic distance model based on the Tamura–Nei model and the neighbor-joining method for constructing a tree were used to analyze the relationships among individuals with no outgroup.
2.6. Metagenomic analysis
2.7. In vitro virological assays
Harvesting and ultracentrifugation of the hepadnaviruses were performed as described previously [42]. Based on the full genome analysis of mouse (Mouse-mus490-Frog) and hamster (Hamster-olig706) hepadnaviruses, a 1.1-fold replicating plasmid was constructed to study the replication of hepadnaviruses. PEI transfection reagent was used to transfect Huh7 cells, and cell supernatants were collected after 3 and 6 days. After ultracentrifugal sedimentation, the supernatants were further separated by CsCl density gradient ultracentrifugation. Real-time PCR was performed to quantify the hepadnavirus in the fractions. Transmission electron microscopy negative staining was performed by Wuhan Servicebio Technology, China. Virus suspensions were dropped on a copper grid, and the grids were observed using a transmission electron microscope (HT7800/HT7700, HITACHI).
3. Results
3.1. Discovery of a large number of novel hepadnaviruses
This study found dozens of novel hepadnaviruses (Supplementary Table S4) based on Edgar's study [33]. The Hepadnaviridae family can be divided into 6 genera [4], namely, the genera nackednavirus represented by rockfish hepadnavirus, herpetohepadnavirus represented by frog hepadnavirus, Avihepadnavirus represented by duck hepatitis B virus (DHBV), metahepadnavirus represented by white sucker hepadnavirus, parahepadnavirus represented by bluegill hepadnavirus, and orthohepadnavirus represented by human HBV (Figure 1A,B). In order to better analyze and visualize hepadnavirus genomic characteristics, these genomes were displayed from the origin of the HBc gene (HBc-origin), which is distinct from the traditional EcoRI cleavage site (EcoRI-origin) [43] (Figure 1C). The use of HBc-origin as the display method yields the following advantages: (1) Some hepadnavirus genomes do not contain a traditional corresponding EcoRI cleavage site. (2) The new presentation ensures that neither the preS gene nor the HBp gene is truncated at the start position or spans both sides of it. (3) The hepadnavirus HBc gene is conserved and the HBc-origin site can be successfully found under most circumstances. The HBp-origin display is another potential option; however, it may cause HBc truncation. Overall, the selected HBc-origin method for new hepadnaviruses displayed the HBc-HBp gene structure clearly and was consistent with the pgRNA gene structure of hepadnaviruses.
3.2. Discovery of frog hepadnaviruses in mammals
With respect to frog hepadnaviruses, this study discovered several lizard and toad hepadnaviruses (Figure 2). Frog hepadnaviruses have previously been discovered in amphibians and reptiles such as frogs [6,10] and lizards [4]. Surprisingly, we found frog hepadnaviruses in mice and dogs, and these sequences were extremely close to the previously discovered frog hepadnavirus Frog-KX058435 (Figure 2A,B). This is the first report of the discovery of frog hepadnaviruses in mammals. Further studies revealed that frog hepadnavirus fragments were present in other animals, such as snub-nosed monkeys, giant pandas, pigs, and even Japanese sea cucumbers (Figure 2C). In the PRJNA248058 project, NGS analyses of 12 tissue samples from a 32-year-old cancer-free black snub-nosed monkey (Rhinopithecus bieti) suggests that hepadnavirus fragments were mainly found in the kidneys and small intestine, while few or none were present in the liver or blood (Figure 2D). This shows that snub-nosed monkey frog hepadnavirus may not mainly infect the liver, suggesting that the mode of transmission for these viruses may be different from the traditional bloodborne transmission route. The discovery of monkey frog hepadnavirus suggests that frog hepadnaviruses can be used for primate models in addition to mouse models. The variation in frog hepadnavirus sequences seems to be limited and its mode of infection seems to be distinct from that of traditional hepadnaviruses with strict host tropism such that it may be a non-species-specific hepadnavirus. Perplexingly, frog hepadnaviruses were also found in plants such as cherry, soybean, wild cabbage, and yeast, which may be the result of animal fecal contamination or some other unknown reasons. However, this also demonstrates that frog hepadnaviruses are widely distributed in nature (Supplementary Figure S1).
3.3. Mammals harbor many orthohepadnaviruses
This study discovered two new orthohepadnaviruses in buffalos and hamsters (Figure 3A). The buffalo hepadnavirus was the closest to the previously reported sheep hepadnavirus [14]. Hamster hepadnavirus was similar to shrew hepadnaviruses [17,18,19], as they both have similar genomic structures, whereas it was distinct from the newly discovered frog hepadnavirus in mice (Figure 3B). The hamster in which the virus was identified was a long-tailed pygmy rice rat (Oligoryzomys longicaudatus) from Cricetidae family (Figure 3C). Whether rodent model animals such as Syrian hamsters or Chinese striped hamsters from the same Cricetidae family can be infected by this hamster hepadnavirus thus represents an extremely meaningful question. To assess this possibility, the whole genomic sequences of mouse frog hepadnavirus and hamster hepadnavirus were synthesized and hepadnavirus 1.1-fold replicons were constructed [42]. In the cell supernatant of Huh7 cells, a peak similar to the hepadnavirus Dane particle peak could be detected at a density of approximately 1.18–1.22 g/mL (Figure 3D,E). The mouse frog hepadnavirus did not exhibit a corresponding naked particle peak to that observed for human HBV and hamster hepadnavirus at a density of 1.28–1.32 g/mL, indicating that this frog hepadnavirus cannot be released in the form of naked virions. Negative stain electron microscopy showed that hamster hepadnavirus and mouse frog hepadnavirus particles are slightly larger than human HBV, spherical, and 40–60 nm in diameter. However, preliminary mouse and hamster viral infection experiments were unsuccessful for unknown reasons (data not shown). In addition to the hamster hepadnavirus, it is worth noting that a common novel unknown insert sequence in HBV was present in rat, pig, gammarid, yellow croaker, and Varunidae samples (Figure 3A). As this 39-bp insert sequence (CCCCAACTGGGGTAACCTTTGGGCTCCCCGGGCGCGACC) was first found in gammarids (Gammarus pisinnus), it was named as gamm-insert (Figure 3F). The gamm-insert was located before the ATG start codon in the preS gene. To the best of our knowledge, there have been no studies on engineering modifications of the gamm-insert in the preS gene despite its presence in samples from many animals. In addition, the D-genotype HBV preS1 gene exhibited a 33-bp sequence that was absent when compared with other HBV genotypes, corresponding to an 11 aa polypeptide, but the reason for this is currently unknown (Figure 3H). In this study, two D-genotype HBV genomes were simultaneously identified from a single soil sample. Specifically, soil-soil880-D-aspilia contained the 33-bp insert, while soil-soil880 lacked the insert. The 33-bp insert (TCATGGGAGGTTGGTCATCAAAACCTCGCAAAG) was designated as the aspilia-insert due to its discovery in soil near the flowering plant Aspilia grazielae. It is possible that the D-genotype HBV found in humans originated from a virus with the aspilia-insert.
3.4. African cichlids harbor many novel hepadnaviruses
There are diverse novel hepadnaviruses in fish that mainly include non-enveloped stickleback and elephantfish nackednaviruses, porgy and Aulopiformes parahepadnaviruses, and common carp and icefish metahepadnaviruses (Figure 4A). Surprisingly, a large number of novel hepadnaviruses were found in African cichlids and the sequences of these viruses were extremely diverse and included both nackednaviruses and metahepadnaviruses (Figure 4B). The variety of hepadnaviruses discovered in African cichlids was surprising (Figure 4A). In the PRJNA552202 project, researchers sequenced 2242 African cichlids samples from 5 tissues in 76 cichlid species from Lake Tanganyika [44] and a large number of hepadnaviruses were detected in these samples. In the PRJNA845781 project, RNA sequencing was carried out on 140 elephantfish samples, and hepadnaviruses were widely found to be present in 12/13 elephantfish. Specifically, 13 tissues or organs from elephantfish BB366 were sequenced (Figure 4C), revealing that the tissue with the highest abundance of hepadnavirus was not the liver but the skin and electric organs, indicating that elephantfish hepadnaviruses are also not liver tropism. Elephantfish hepadnaviruses do not encode an envelope protein such that they are nackednaviruses, and the route of transmission may be distinct from traditional bloodborne transmission. In addition, potential linear hepadnaviruses were also found in dragonfish and elephantfish, and these were designated dragonfish-akar681-l and elephantfish-brie291-l (Figure 4D). These viruses were detected as completely different flanking nucleic acid sequences that are distinct from traditional circular hepadnavirus genomes. It cannot be completely ruled out that the differences in the flanking nucleic acid sequences are a result of integration into the host genome, and further study is required to assess this possibility. Furthermore, it was found that the sturgeon-acip200 and stickleback-gast636 were highly conserved, with only one base pair difference between their genomes (Figure 4A).
3.5. Identification of multiple integrated avian hepadnaviruses
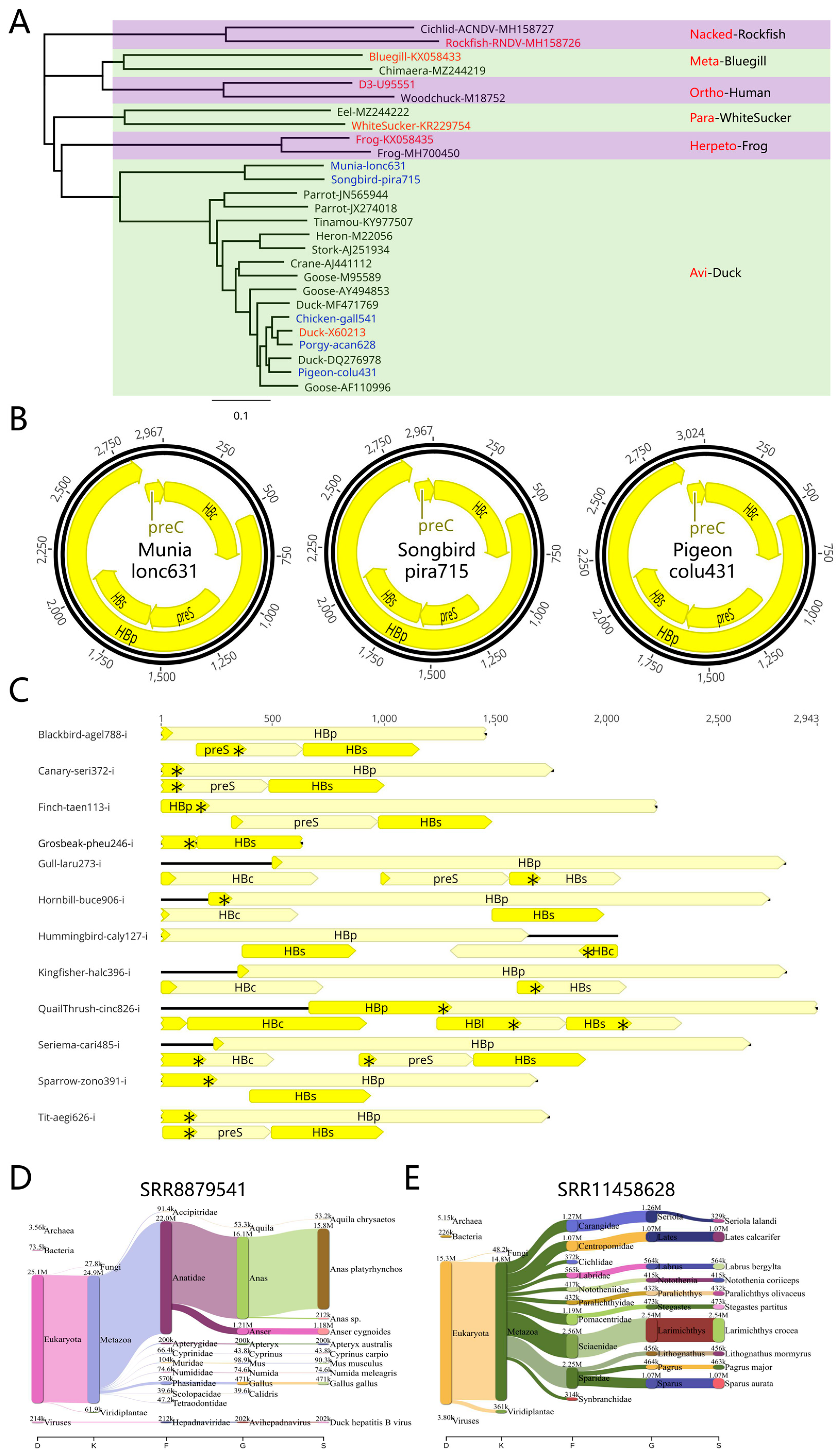
With respect to avian hepadnaviruses, white-rumped munia, American songbird, and pigeon hepadnaviruses were newly discovered in this study, while chicken and porgy hepadnaviruses were also detected (Figure 5A,B). Here, the DHBV genome was found to be similar to the porgy fish hepadnavirus porgy-acan628 (Figure 5A). Porgy fish hepadnavirus also exhibited rich diversity, including the porgy-acan628 hepadnavirus that is similar to avian hepadnavirus and the porgy-pagr765 hepadnavirus that is more similar to sucker and eel fish hepadnaviruses. This is particularly true for porgy-acan628 which belongs to the group of avian hepadnaviruses, making it the first avian hepadnavirus to be found in fish. It is possible that porgy-acan628 hepadnavirus entered birds from fish via the food chain. However, the vast majority of avian hepadnaviruses discovered in this study were present in an integrated form (Figure 5C). Integration patterns were as follows: 1) Most HBs genes were integrated and can be transcribed; 2) The 5' terminus within the HBp and preS genes often contain stop codons; 3) The 3' terminus of most HBp genes is intact, while the 5' terminus is missing; and 4) A few HBc genes contain complete open reading frames, as in the case of QuailThrush-cinc826-i. A hepadnavirus Chicken-gall541 from chicken samples is extremely close to that of DHBV (Figure 5A). However, this may be due to contamination by the duck samples, as DHBV exhibits strict host specificity and is unable to infect chickens. Indeed, species analyses of the chicken sample NGS sequences suggest that the sample may have been derived from ducks (Figure 5D). In contrast, species analyses of the NGS sequences of avian hepadnavirus Porgy-acan628's sample indicated that most sequencing results were from croaker fish (Larimichthys crocea) or porgy fish (Sparidae family) instead of avian duck (Figure 5E). In addition, a large number of DHBV viruses were detected in duck and goose samples (Supplementary Figure S2). To the best of our knowledge, DHBV-chen299 is the first DHBV virus found in the Australian wood duck (Chenonetta jubata).
4. Discussion
4.1. Discovery of a large number of novel hepadnaviruses
In this study, bioinformatics analyses of NGS data were researched and reanalyzed primarily based on Edgar's study [33], and dozens of hepadnavirus genomes from new hosts were assembled. At least 41 novel hepadnaviruses with complete genomes were identified, marking the highest number of novel hepadnavirus discoveries in a single research. For ease of description and discussion, species names from NGS sample annotation were utilized to refer to the new species host of hepadnaviruses. Some DHBV found in rats or human HBV found in fishes may be attributable to species or laboratory contamination, which need to be confirmed to determine if hepadnavirus infections are present in a given species in the future (Supplementary Figure S3). Certain new hepadnavirus genomes were consistent with those published by Lauber et al. [4]. These genomes have the same abbreviations as in their study (Salmon-onco573-SSNDV, Cichlid-asta966-AMDV, Killifish-luca002-KNDV-Lg, Killifish-luca778-KNDV-1, Elephantfish-brie518-BWNDV1, and Elephantfish-brie445-BWNDV2). The porgy hepadnavirus partially derived from the Australasian snapper polymerase gene (MH716821) and a complete hepadnavirus genome Porgy-pagr765 were reanalyzed and assembled from data SRR7527765 [45].
With respect to orthohepadnaviruses, hamster and bovine hepadnaviruses are newly discovered. Novel metahepadnaviruses including cichlid, herring, common carp, eel, whale, icefish, Caranginaem, and Japanese halfbeak hepadnaviruses were also discovered, while new parahepadnaviruses including porgy and aulopiformes hepadnaviruses were identified for the first time. With respect to nackednaviruses, dragonfish, sturgeon, and stickleback hepadnaviruses were newly discovered. Among frog hepadnaviruses, lizard, toad, dog, and mouse hepadnaviruses were newly discovered. Newly identified avian hepadnaviruses included white-rumped munia, American songbird, pigeon, and porgy hepadnaviruses. Of these, the most important viruses are hamster hepadnavirus, mouse frog hepadnavirus, human HBV with the gamm-insert from pigs and rats, and cichlid hepadnavirus. Some approximate interpretations were performed according to the research and evolution of hepadnavirus. For example, the following three phenomena are all roughly consistent with the evolution of viruses: hamster hepadnavirus is similar to bovine hepadnavirus; many hepadnaviruses with substantial sequence variability were detected in the African cichlid family; and hepadnaviruses with similar sequences were identified in different experiments using a range of methods.
4.2. Frog hepadnaviruses exhibit pan-species infectivity
As the discovered hepadnaviruses in this study exhibit strict host specificity, there remains a lack of suitable animal models of hepadnavirus infection [25,26]. The various frog hepadnaviruses discovered in this study indicated that frog hepadnaviruses appear to possess pan-species infectivity and can infect many mammals in addition to amphibians and reptiles. In particular, frog hepadnaviruses were found in common animals such as snub-nosed monkeys, pigs, dogs, and mice. Whether these viruses possess high levels of infectivity and a high replication capacity in mammals such as monkeys, mice, and other common model animals; whether these viruses can form cccDNA similar to human HBV [46]; and whether the frog hepadnavirus infection animal model can be used to evaluate anti-HBV drugs all represent important topics for future research.
4.3. The pressing need for animal models of orthohepadnavirus infection
Animal models of hepadnavirus infection remain a persistent challenge in the field, particularly with respect to human HBV [25,26]. Many hepadnaviruses detected in this study may cause sporadic infections, indicating that animals can be infected by these hepadnaviruses but that there are some barriers to infection and replication efficiency. There are three potential approaches to the construction of an ideal animal model of hepadnavirus infection.
The first relies on starting with highly abundant hepadnaviruses, such as cichlid hepadnavirus or elephantfish hepadnavirus for which a high abundance of hepadnavirus fragments were detected. However, the hosts for these hepadnaviruses are fish, which are uncommon experimental animals.
The second approach requires the establishment of high replication infections under controlled conditions. NGS data are extremely sensitive and can detect very low-level infections, such as infections with snub-nosed monkey, giant panda, and pig-frog hepadnaviruses, which represent conditional infections that can be achieved in some instances, particularly in specific knockout or overexpression animal models that result in host immunosuppression or enhanced hepadnavirus replication, allowing for the establishment of a model of conditional infection. This approach is the most promising as it entails the use of hamster hepadnavirus or mouse frog hepadnavirus to achieve the in vitro assembly and secretions of virus-like particles. However, for unknown reasons, mouse and hamster in vivo infections could not be achieved in the present study (data not shown). The host animals may thus require certain immune modifications. For example, feline hepadnavirus infections are mostly present in the context of a comorbid retroviral infection [12].
The third approach involves the engineering modification of human HBV. Human HBV containing the gamm-insert is found in many species and may hold potential for this approach, although this was not studied owing to time limitations. The reasons for the presence of the gamm-insert in both human HBV and other species are not clear. One possibility is that human HBV with the gamm-insert may have entered the wastewater, thereby contaminating the fish, shellfish, and prawns therein. An alternative scenario is that viruses with the gamm-insert may already be present in fish, shellfish, and prawns, and thereby infecting humans via the food chain. While the possibility of this latter option is low, it is still a very significant concern as these species are important food sources for humans, especially eels and yellow croakers in East Asia.
4.4. Diverse fish hepadnaviruses
Among all hepadnaviruses, fish hepadnaviruses are the most primitive, diverse, and widely distributed in different genera (meta, para, and nacked) in the Hepadnaviridae family [4]. Some fishes contain different hepadnaviruses with substantial nucleic acid sequence variation, such as African cichlid, elephantfish, eel, and killfish hepadnaviruses, which may be attributable to their long evolutionary history. African cichlids yielded the most surprising findings, revealing that different genera of cichlids are widely infected with hepadnaviruses. Cichild-cypr446, cichild-lamp675, cichild-ACNDV-MH158727, and cichild-nacked-ANDV are the most primitive non-enveloped nackednaviruses, while traditional cichlid hepadnaviruses more closely resemble mammalian hepadnaviruses in the metahepadnaviruses genus. African cichlids reside in three great lakes in East Africa, including Lake Malawi, Lake Tanganyika, and Lake Victoria, all of which are located near the East African Great Rift Valley. The possibility of these viruses entering humans through the food chain cannot be excluded. The significance of these viruses in the evolutionary history of hepadnaviruses will require further elucidation.
4.5. Multiple integrated avian hepadnaviruses
Munia (Lonchura striata) and American songbird (Piranga ludoviciana) hepadnaviruses were located in the same evolutionary branch. White-rumped munia are native to South Asia and southern China while the American songbird lives in Central America and North America. This suggests that the ancestor of these munia and American songbird hepadnaviruses may have once been widely distributed throughout the world. The pigeon (Columba livia) hepadnavirus was found to be phylogenetically close to duck hepadnavirus with both being located in the duck hepadnavirus evolutionary branch, while the DHBV-porgy-acan628 hepadnavirus discovered in the porgy fish (Acanthopagrus latus) was situated in the avian hepadnavirus branch in close proximity to duck hepadnavirus. This is the first avian hepadnavirus found in fish. It is worth examining whether fish hepadnaviruses enter birds through the food chain and whether this may be an origin of these avian hepadnaviruses. This study also revealed that in addition to duck hepadnavirus, many other avian hepadnaviruses have integrated into host genomes, consistent with previous literature reports [23,47,48,49].
4.6. Study limitations and shortcomings
It is perplexing that hepadnaviruses were also detected in the NGS analyses of plants in this study (Supplementary Figure S1). For example, frog hepadnaviruses were found in soybeans, cherries, yeast, and Japanese sea cucumbers, while human HBV was detected in sesame, calabash, and woad. This may be a result of laboratory contamination, fecal contamination, or other unknown reasons, highlighting a need for further validation. While this study discovered many novel hepadnaviruses, we only evaluated the replication and infectious capacity of mouse frog hepadnavirus and hamster hepadnavirus.
5. Conclusions
In summary, this study discovered many novel hepadnaviruses by evaluating a large volume of NGS data in public databases. This not only provides new clues regarding the origins and evolution of hepadnaviruses, but can be used to construct new hepadnavirus animal infection models to aid in the further elucidation of the pathogenesis of HBV infection and the development of HBV cure drugs [50,51].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: novel hepadnaviruses discovered in plant NGS data; Figure S2: newly discovered DHBV and primate HBV discovered in the NGS data; Figure S3: possible contaminating HBV and DHBV discovered in the NGS data; Table S4: novel hepadnavirus statistics; File S5: the annotated genome sequences of all novel hepadnaviruses.
Author Contributions
Conceptualization, H.B. and D.C.; methodology, H.B. and L.L.; software, H.B.; validation, H.B., X.W., P.Y., P.L. Y.G., Y.W., Y.L. and C.H.; formal analysis, H.B.; investigation, H.B.; resources, H.B.; data curation, H.B.; writing—original draft preparation, H.B.; writing—review and editing, H.B., X.W. and D.C.; visualization, H.B.; supervision, D.C.; project administration, D.C.; funding acquisition, H.B., X.W., Y.W. and D.C. This study is part of the postdoctoral research of H.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Beijing YouAn Hospital Youth Innovation Foundation [grant number YNKTQN2021020], the Beijing Postdoctoral Research Foundation [grant number 2022-ZZ-039], Beijing Nova Program [grant Z171100001117119], the Scientific Research Project of Beijing YouAn Hospital [grant number BJYAYY-YN2022-19], the National Natural Science Foundation of China [grant number 82073676], the Key Programs of Beijing Municipal Education Commission of China [grant number KZ202010025037], the Chinesisch-Deutsche Zentrum für Wissenschaftsförderung [grant number C-0012], and the pilot project of public welfare development and reform of Beijing-affiliated medical research institutes [grant numbers JingYiYan2021-10 and 2019-6].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The annotated genome sequences of all of the novel hepadnaviruses presented in this study are openly available in the Supplementary file S5 or GenBase database [52] in the National Genomics Data Center of China (C_AA050214-C_AA050361).
Acknowledgments
We thank Wei Liu from University of Pennsylvania for his valuable discussions and insightful comments. We thank LetPub for its linguistic assistance during the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Glebe D, Goldmann N, Lauber C, et al. HBV evolution and genetic variability: impact on prevention, treatment and development of antivirals. Antiviral Res. 2021, 186, 104973.
- Tong S, Li J, Wands JR, et al. Hepatitis B virus genetic variants: biological properties and clinical implications. Emerg Microbes Infect. 2013, 2, e10.
- Guo X, Wu J, Wei F, et al. Trends in hepatitis B virus resistance to nucleoside/nucleotide analogues in North China from 2009–2016: a retrospective study. Int J Antimicrob Agents. 2018, 52, 201–209.
- Lauber C, Seitz S, Mattei S, et al. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe. 2017, 22, 387–399.e6.
- He B, Fan Q, Yang F, et al. Hepatitis virus in long-fingered bats, Myanmar. Emerg Infect Dis. 2013, 19, 638–640.
- Dill JA, Camus AC, Leary JH, et al. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J Virol. 2016, 90, 7920–7933.
- Hahn CM, Iwanowicz LR, Cornman RS, et al. Characterization of a novel hepadnavirus in the white sucker (Catostomus commersonii) from the Great Lakes Region of the United States. J Virol. 2015, 89, 11801–11811.
- Adams CR, Blazer VS, Sherry J, et al. Phylogeographic genetic diversity in the white sucker hepatitis B virus across the Great Lakes Region and Alberta, Canada. Viruses. 2021, 13, 285.
- Chen X-X, Wu W-C, Shi M. Discovery and characterization of actively replicating DNA and retro-transcribing viruses in lower vertebrate hosts based on RNA sequencing. Viruses. 2021, 13, 1042.
- Debat HJ, Ng TFF. Complete genome sequence of a divergent strain of Tibetan frog hepatitis B virus associated with a concave-eared torrent frog (Odorrana tormota). Arch Virol. 2019, 164, 1727–1732.
- de Souza AJS, Malheiros AP, Chagas AACd, et al. Orthohepadnavirus infection in a neotropical bat (Platyrrhinus lineatus). Comp Immunol Microbiol Infect Dis. 2021, 79, 101713.
- Pesavento PA, Jackson K, Hampson TSTTB, et al. A novel hepadnavirus is associated with chronic hepatitis and hepatocellular carcinoma in cats. Viruses. 2019, 11, 969.
- Diakoudi G, Capozza P, Lanave G, et al. A novel hepadnavirus in domestic dogs. Sci Rep. 2022, 12, 2864.
- Gogarten JF, Ulrich M, Bhuva N, et al. A novel orthohepadnavirus identified in a dead Maxwell's duiker (Philantomba maxwellii) in Taï National Park, Côte d'Ivoire. Viruses. 2019, 11, 279.
- Jo WK, Alfonso-Toledo JA, Salas-Rojas M, et al. Natural co-infection of divergent hepatitis B and C virus homologues in carnivores. Transbound Emerg Dis. 2022, 69, 195–203.
- Rasche A, Lehmann F, Goldmann N, et al. A hepatitis B virus causes chronic infections in equids worldwide. Proc Natl Acad Sci U S A. 2021, 118, e2013982118.
- He W-Q, Chen X-J, Wen Y-Q, et al. Detection of hepatitis B virus-like nucleotide sequences in liver samples from murine rodents and Asian house shrews. Vector-Borne Zoonotic Dis. 2019, 19, 781–783.
- Nie F-Y, Tian J-H, Lin X-D, et al. Discovery of a highly divergent hepadnavirus in shrews from China. Virology. 2019, 531, 162–170.
- Rasche A, Lehmann F, König A, et al. Highly diversified shrew hepatitis B viruses corroborate ancient origins and divergent infection patterns of mammalian hepadnaviruses. Proc Natl Acad Sci U S A. 2019, 116, 17007–17012.
- Locarnini S, Littlejohn M, Aziz MN, et al. Possible origins and evolution of the hepatitis B virus (HBV). Semin Cancer Biol. 2013, 23 Pt B, 561–575. [Google Scholar] [CrossRef]
- Locarnini SA, Littlejohn M, Yuen LKW. Origins and evolution of the primate hepatitis B virus. Front Microbiol. 2021, 12, 653684.
- Datta, S. Excavating new facts from ancient Hepatitis B virus sequences. Virology. 2020, 549, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Liu W, Pan S, Yang H, et al. The first full-length endogenous hepadnaviruses: identification and analysis. J Virol. 2012, 86, 9510–9513.
- Suh A, Weber CC, Kehlmaier C, et al. Early mesozoic coexistence of amniotes and hepadnaviridae. PLoS Genet. 2014, 10, e1004559.
- Ploss A, Strick-Marchand H, Li W. Animal models for hepatitis B: does the supply meet the demand? Gastroenterology. 2021, 160, 1437–1442.
- Zhang X, Wang X, Wu M, et al. Animal models for the study of hepatitis B virus pathobiology and immunity: past, present, and future. Front Microbiol. 2021, 12, 715450.
- Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012, 1, e00049.
- Li W, Urban S. Entry of hepatitis B and hepatitis D virus into hepatocytes: basic insights and clinical implications. J Hepatol. 2016, 64 (Suppl. S1), S32–S40.
- Burwitz BJ, Zhou Z, Li W. Animal models for the study of human hepatitis B and D virus infection: new insights and progress. Antiviral Res. 2020, 182, 104898.
- Li D, He W, Liu X, et al. A potent human neutralizing antibody Fc-dependently reduces established HBV infections. Elife. 2017, 6, e26738.
- Rizzetto M, Canese MG, Gerin JL, et al. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J Infect Dis. 1980, 141, 590–602.
- Zhang T-Y, Yuan Q, Zhao J-H, et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut. 2016, 65, 658–671.
- Edgar RC, Taylor B, Lin V, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature. 2022, 602, 142–147.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990, 215, 403–410.
- Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, 34, i884–i890.
- Li D, Liu C-M, Luo R, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015, 31, 1674–1676.
- Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017, 35, 1026–1028.
- Kearse M, Moir R, Wilson A, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 28, 1647–1649.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013, 30, 772–780.
- Kim D, Song L, Breitwieser FP, et al. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729.
- Breitwieser FP, Salzberg SL.. Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics. 2020, 36, 1303–1304.
- Wang X-J, Zhang X-J, Hu W, et al. A simple and efficient strategy for the de novo construction of greater-than-genome-length hepatitis B virus replicons. J Virol Methods. 2014, 207, 158–162.
- Galibert F, Mandart E, Fitoussi F, et al. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature. 1979, 281, 646–650.
- El Taher A, Böhne A, Boileau N, et al. Gene expression dynamics during rapid organismal diversification in African cichlid fishes. Nat Ecol Evol. 2021, 5, 243–250.
- Geoghegan JL, Di Giallonardo F, Cousins K, et al. Hidden diversity and evolution of viruses in market fish. Virus Evol. 2018, 4, vey031.
- Nassal, M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Suh A, Brosius J, Schmitz J, et al. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat Commun. 2013, 4, 1791.
- Gilbert C, Feschotte C. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol. 2010, 8, e1000495.
- Tu T, Budzinska MA, Shackel NA, et al. HBV DNA integration: molecular mechanisms and clinical implications. Viruses. 2017, 9, 75.
- Revill PA, Chisari FV, Block JM, et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol. 2019, 4, 545–558.
- Wang X-Y, Wen Y-M. A "sandwich" strategy for functional cure of chronic hepatitis B. Emerg Microbes Infect. 2018, 7, 91.
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 2023, 51, D18–D28.
Figure 1.
Hepadnavirus classification and new display method. (A) Phylogenetic trees of 6 hepadnaviruses. Representative hepadnaviruses and their classification are shown in red. (B) Unrooted tree layout for 6 hepadnaviruses. (C) HBc origin-based display of the hepadnavirus genome for Human HBV-U95551, an orthohepadnavirus; frog hepadnavirus-KX058435, a frog hepadnavirus; and rockfish hepadnavirus-MH158726, a nackednavirus.
Figure 1.
Hepadnavirus classification and new display method. (A) Phylogenetic trees of 6 hepadnaviruses. Representative hepadnaviruses and their classification are shown in red. (B) Unrooted tree layout for 6 hepadnaviruses. (C) HBc origin-based display of the hepadnavirus genome for Human HBV-U95551, an orthohepadnavirus; frog hepadnavirus-KX058435, a frog hepadnavirus; and rockfish hepadnavirus-MH158726, a nackednavirus.
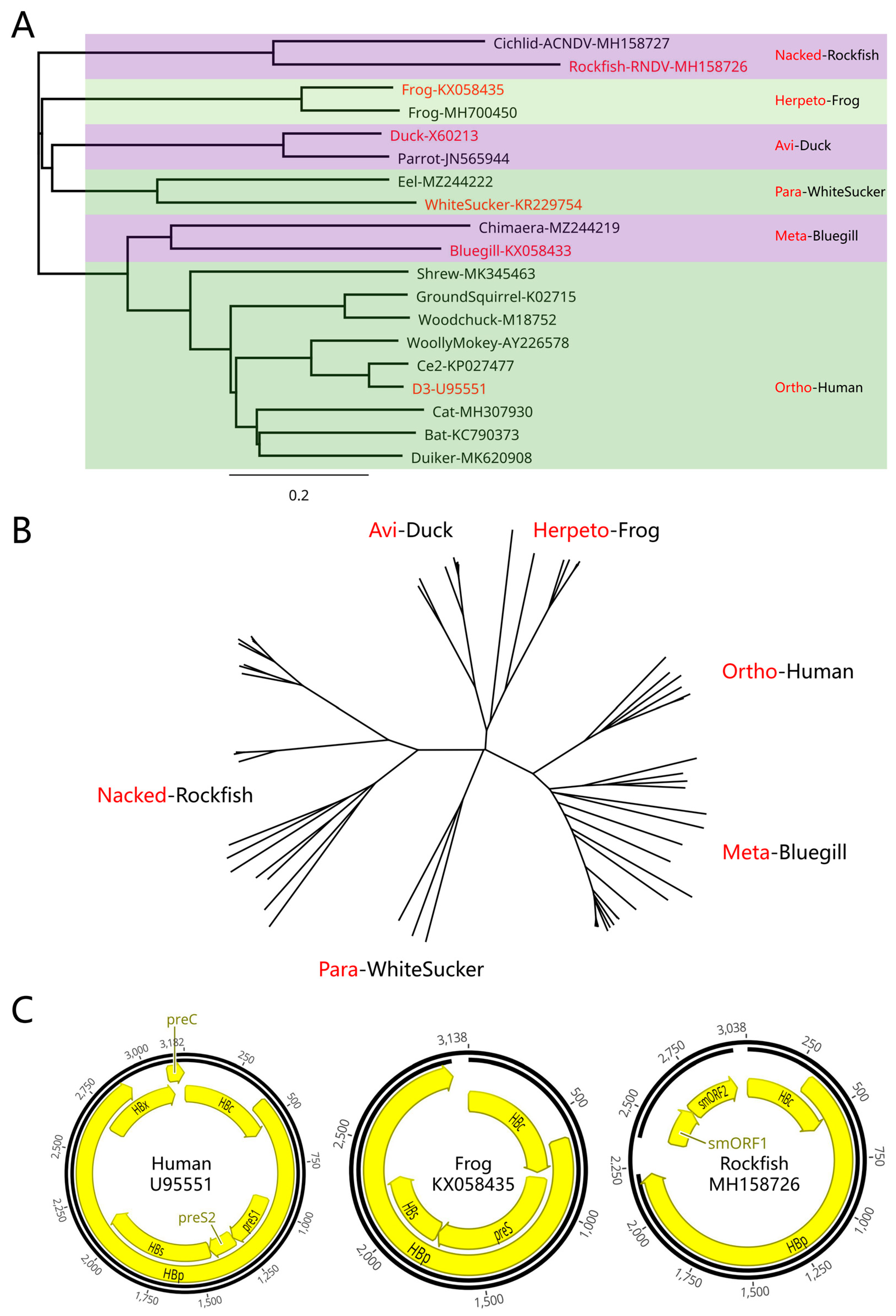
Figure 2.
Novel frog hepadnaviruses. (A) Newly discovered frog hepadnaviruses. Representative hepadnaviruses and corresponding classification details are shown in red, while newly discovered hepadnaviruses are shown in blue. Newly discovered hepadnavirus were named according to the first 4 letters of the species name + the last 3 numbers of the SRA sequence number. (B) Representative frog hepadnavirus genomes and the newly discovered representative Mouse-mus490, Dog-cani315, and Lizard-sapr381 viruses. (C) Comparison of the whole genomes of mammal-frog hepadnaviruses. Frog hepadnaviruseses were discovered in Mouse-mus490, Dog-cani315, snub-nosed monkey, pig, giant panda, and sea cucumber, and their sequences were extremely similar. g: gap. (D) Tissue distribution of snub-nosed monkey hepadnaviruses. A black snub-nosed monkey (Rhinopithecus bieti) from Yunnan province in which some fragments were found in the kidneys and small intestine, a few fragments were found in the liver and oral cavity, and no fragments were detected in other tissues such as the blood.
Figure 2.
Novel frog hepadnaviruses. (A) Newly discovered frog hepadnaviruses. Representative hepadnaviruses and corresponding classification details are shown in red, while newly discovered hepadnaviruses are shown in blue. Newly discovered hepadnavirus were named according to the first 4 letters of the species name + the last 3 numbers of the SRA sequence number. (B) Representative frog hepadnavirus genomes and the newly discovered representative Mouse-mus490, Dog-cani315, and Lizard-sapr381 viruses. (C) Comparison of the whole genomes of mammal-frog hepadnaviruses. Frog hepadnaviruseses were discovered in Mouse-mus490, Dog-cani315, snub-nosed monkey, pig, giant panda, and sea cucumber, and their sequences were extremely similar. g: gap. (D) Tissue distribution of snub-nosed monkey hepadnaviruses. A black snub-nosed monkey (Rhinopithecus bieti) from Yunnan province in which some fragments were found in the kidneys and small intestine, a few fragments were found in the liver and oral cavity, and no fragments were detected in other tissues such as the blood.
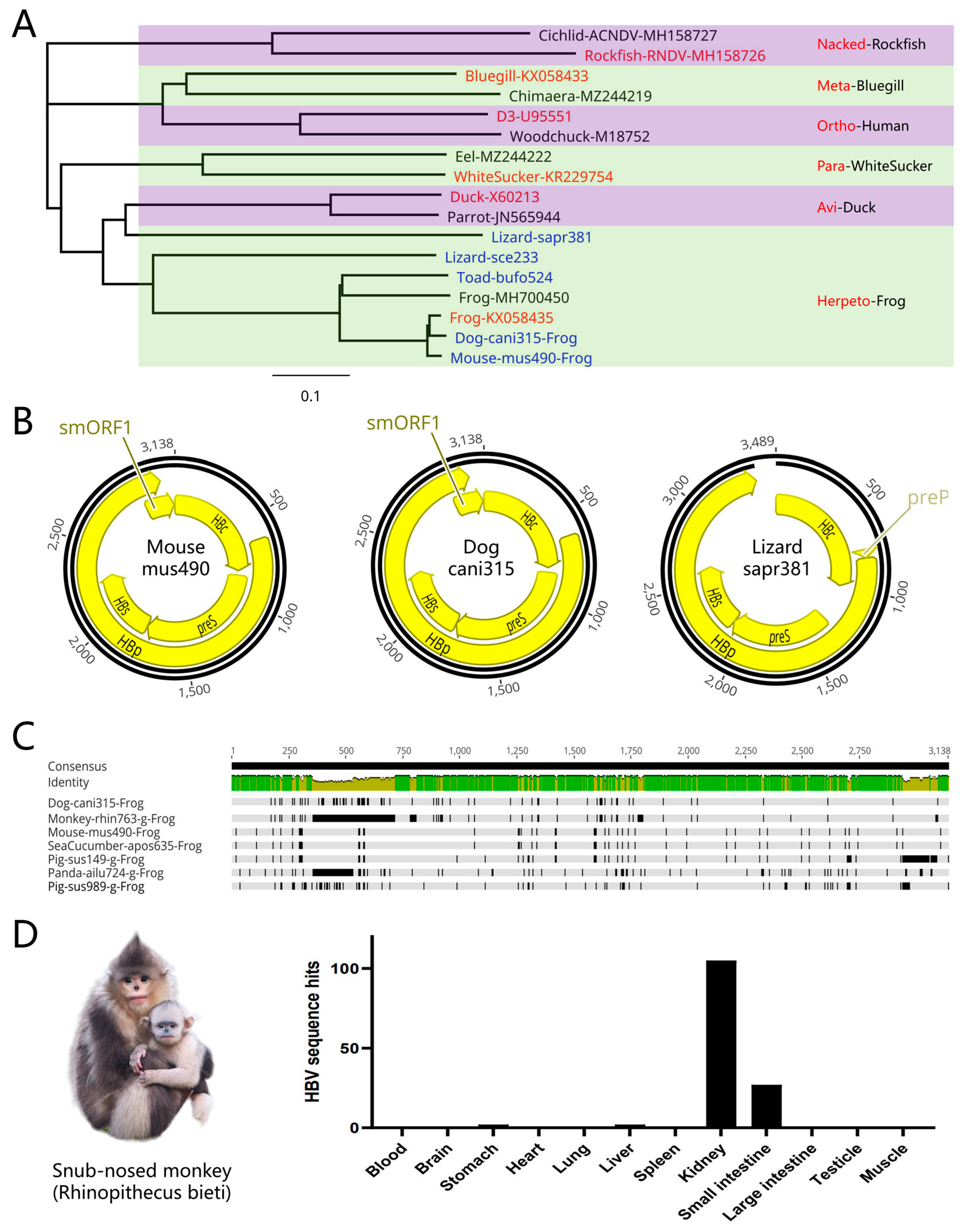
Figure 3.
Novel orthohepadnaviruses. (A) Newly discovered orthohepadnaviruses. Representative hepadnaviruses and corresponding classification details are shown in red, while newly discovered hepadnaviruses are shown in blue. In addition to the conventional Buffalo-buff360 and Hamster-olig706 viruses, a large number of genotype B and D human HBV sequences with the gamm-insert were present in mice, gammarids, and yellow croakers. (B) Representative orthohepadnavirus genomes and the newly discovered Buffalo-buff360 and Hamster-olig706 viruses. (C) Longtail pygmy rice rat (Oligoryzomys longicaudatus) in which hepadnaviruses were discovered. (D) Ultracentrifugation-based identification of hepadnaviruses. Two DNA peaks were evident at 1.18–1.22 and 1.28–1.32 g/mL for HBV and hamster hepadnavirus, while only the first peak was detected for the mouse frog hepadnavirus. (E) Negative stain electron microscopy identification of hepadnaviruses. When imaged via negative stain electron microscopy, the diameters of HBV, hamster, and mouse frog hepadnavirus particles progressively increased, with diameters ranging from 40–60 nm. (F) The gamm-insert in genotype B and D human HBV. A 39 bp gamm-insert was present in genotypes B and D and located before the ATG codon in the preS gene. (G) The aspilia-insert in genotype D. This D-aspilia-insert sequence was 33 bp in length and located 2 bp before the ATG codon in the preS gene.
Figure 3.
Novel orthohepadnaviruses. (A) Newly discovered orthohepadnaviruses. Representative hepadnaviruses and corresponding classification details are shown in red, while newly discovered hepadnaviruses are shown in blue. In addition to the conventional Buffalo-buff360 and Hamster-olig706 viruses, a large number of genotype B and D human HBV sequences with the gamm-insert were present in mice, gammarids, and yellow croakers. (B) Representative orthohepadnavirus genomes and the newly discovered Buffalo-buff360 and Hamster-olig706 viruses. (C) Longtail pygmy rice rat (Oligoryzomys longicaudatus) in which hepadnaviruses were discovered. (D) Ultracentrifugation-based identification of hepadnaviruses. Two DNA peaks were evident at 1.18–1.22 and 1.28–1.32 g/mL for HBV and hamster hepadnavirus, while only the first peak was detected for the mouse frog hepadnavirus. (E) Negative stain electron microscopy identification of hepadnaviruses. When imaged via negative stain electron microscopy, the diameters of HBV, hamster, and mouse frog hepadnavirus particles progressively increased, with diameters ranging from 40–60 nm. (F) The gamm-insert in genotype B and D human HBV. A 39 bp gamm-insert was present in genotypes B and D and located before the ATG codon in the preS gene. (G) The aspilia-insert in genotype D. This D-aspilia-insert sequence was 33 bp in length and located 2 bp before the ATG codon in the preS gene.
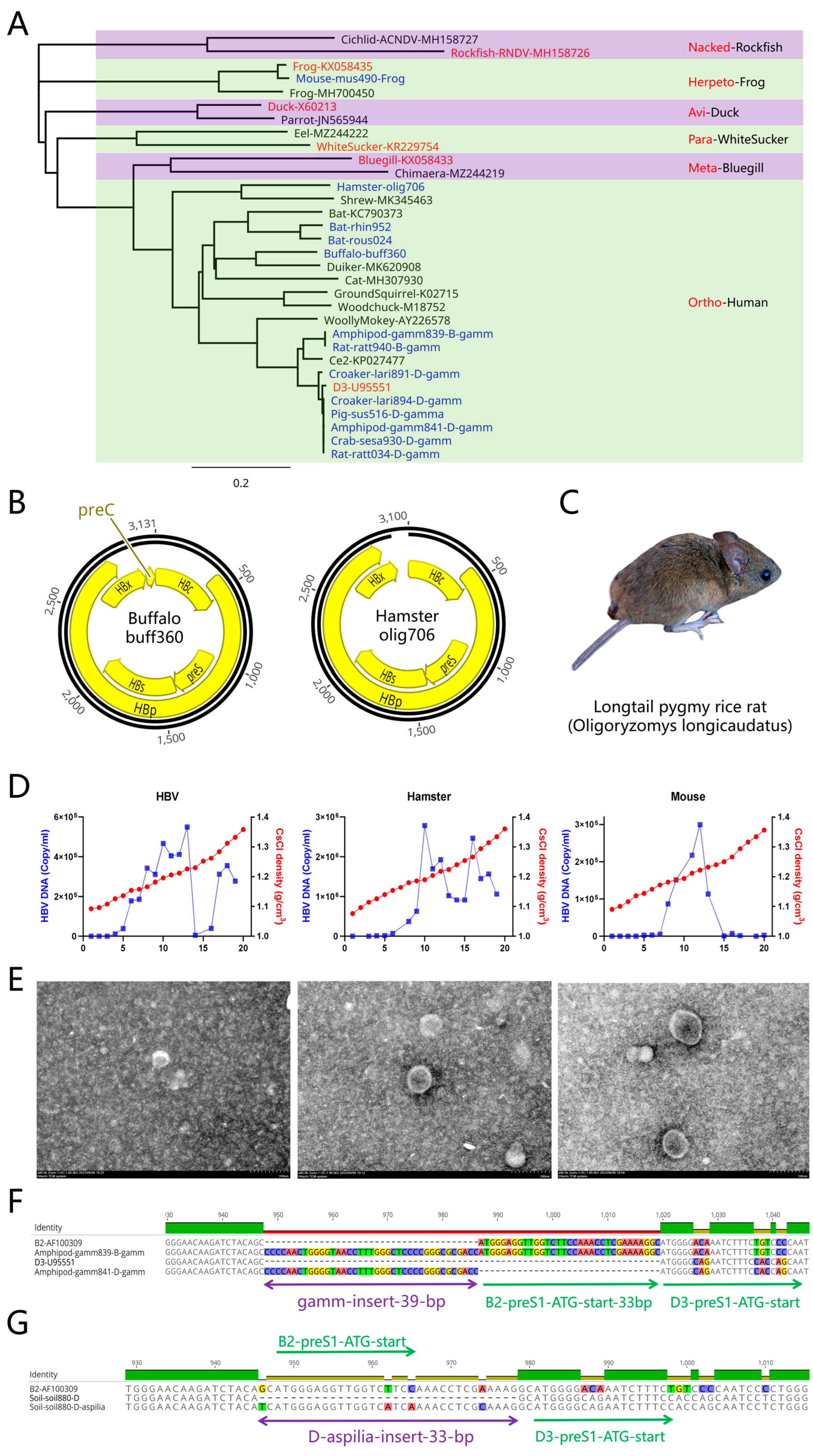
Figure 4.
Novel fish hepadnaviruses. (A) Newly discovered fish hepadnaviruses including caranginae Jack-trac480-g, killifish Killifish-luca002-KNDV-Lg, cichlid Cichlid-trop300, cichlid Cichlid-trop302, and cichlid Cichlid-oreo330 (Tilapia). To facilitate these analyses, a deleted sequence represented by 3N was added before and after the viral genome, and a total of 6Ns represents the linker. (B) Representative fish hepadnavirus genomes including nackednaviruses Stickleback-gast636 and Cichlid-cypr446, as well as metahepadnavirus Cichlid-simo376. (C) Tissue distribution of elephantfish (Brienomyrus brachyistius) hepadnavirus fragments. Brain tissues were found to contain the highest number of fragments at up to 104, followed by flank skin, head skin, electric organ, skeletal muscle, and kidneys. (D) Possible linear hepadnavirus genomes. Dragonfish Dragonfish-akar681-l and elephantfish Elephantfish-brie291-l may be linear genomes with different cutoff points. The dragonfish hepadnavirus is in the form of accessory gene-HBc-HBp while the elephantfish hepadnavirus is in the form of HBc-HBp-accessory gene.
Figure 4.
Novel fish hepadnaviruses. (A) Newly discovered fish hepadnaviruses including caranginae Jack-trac480-g, killifish Killifish-luca002-KNDV-Lg, cichlid Cichlid-trop300, cichlid Cichlid-trop302, and cichlid Cichlid-oreo330 (Tilapia). To facilitate these analyses, a deleted sequence represented by 3N was added before and after the viral genome, and a total of 6Ns represents the linker. (B) Representative fish hepadnavirus genomes including nackednaviruses Stickleback-gast636 and Cichlid-cypr446, as well as metahepadnavirus Cichlid-simo376. (C) Tissue distribution of elephantfish (Brienomyrus brachyistius) hepadnavirus fragments. Brain tissues were found to contain the highest number of fragments at up to 104, followed by flank skin, head skin, electric organ, skeletal muscle, and kidneys. (D) Possible linear hepadnavirus genomes. Dragonfish Dragonfish-akar681-l and elephantfish Elephantfish-brie291-l may be linear genomes with different cutoff points. The dragonfish hepadnavirus is in the form of accessory gene-HBc-HBp while the elephantfish hepadnavirus is in the form of HBc-HBp-accessory gene.
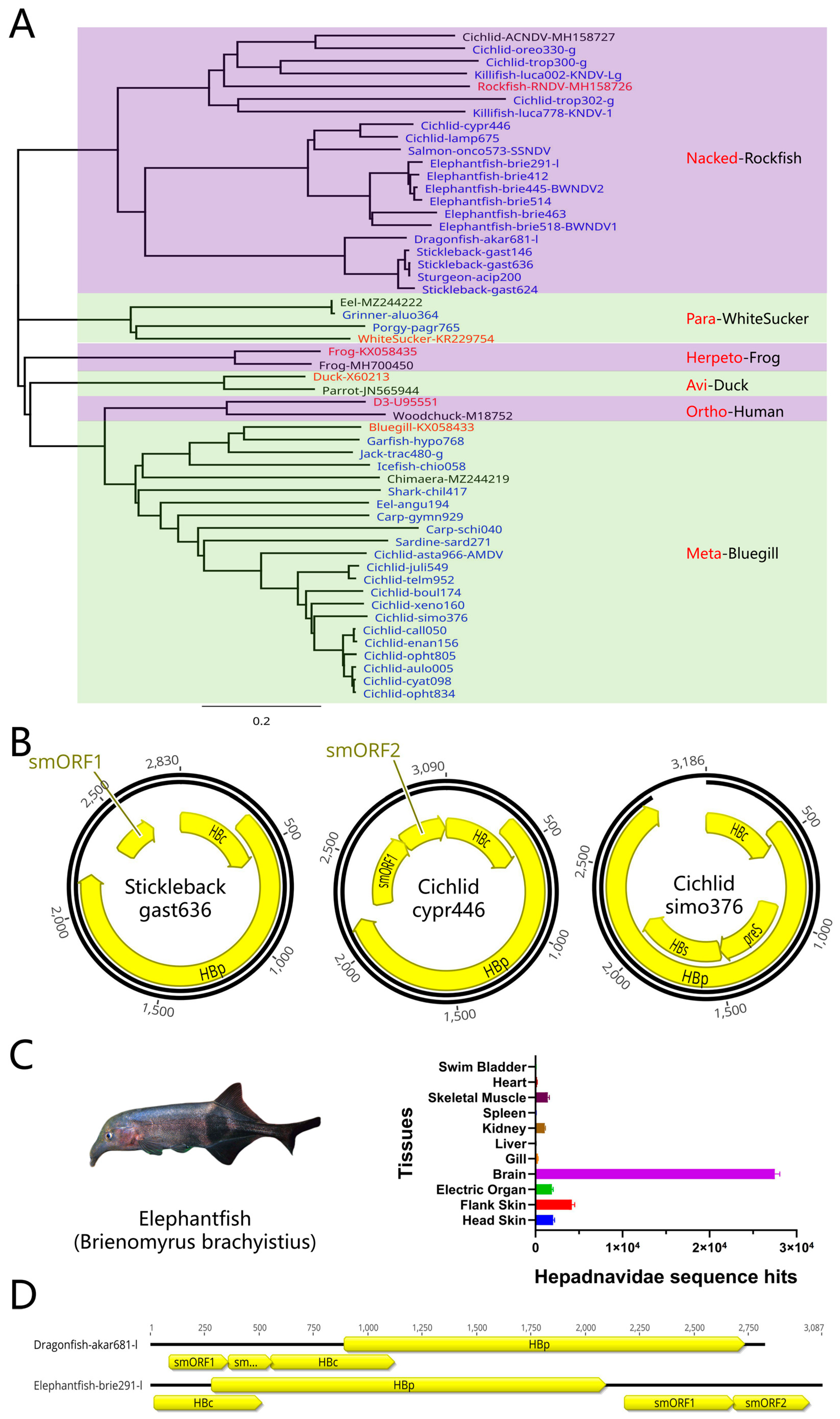
Figure 5.
Novel avian hepadnaviruses. (A) Newly discovered avian hepadnaviruses. (B) Representative avian hepadnavirus genomes including Munia-lonc631, Songbird-pira715, and Pigeon-colu431. (C) Avian hepadnaviruses that had integrated into the genome. Schematic diagram of Blackbird-agel788-i. The flanking host gene sequence that was integrated has been removed, i represents integration, and * represents a stop codon. Most viral sequences harbored complete HBs gene structures and an incomplete HBp gene structure. (D, E) Species composition analysis using NGS data. Sequence species analyses of SRR8879541 revealed that most sequences belonged to Anas platyrhynchos and not Gallus gallus. Sequence species analyses of SRR11458628 revealed that most sequences belonged to Larimichthys crocea.
Figure 5.
Novel avian hepadnaviruses. (A) Newly discovered avian hepadnaviruses. (B) Representative avian hepadnavirus genomes including Munia-lonc631, Songbird-pira715, and Pigeon-colu431. (C) Avian hepadnaviruses that had integrated into the genome. Schematic diagram of Blackbird-agel788-i. The flanking host gene sequence that was integrated has been removed, i represents integration, and * represents a stop codon. Most viral sequences harbored complete HBs gene structures and an incomplete HBp gene structure. (D, E) Species composition analysis using NGS data. Sequence species analyses of SRR8879541 revealed that most sequences belonged to Anas platyrhynchos and not Gallus gallus. Sequence species analyses of SRR11458628 revealed that most sequences belonged to Larimichthys crocea.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



