
Submitted:
20 November 2023
Posted:
22 November 2023
You are already at the latest version
Abstract
Phα1β a spider peptide, purified from the venom of Phoneutria nigriventer inhibits high voltage calcium channels and is antagonist of TRPA1 receptor two important pain transduction path-ways. In the last 15 years, we have showed, in preclinical tests with Phα1β native and recombi-nant in rodent models of pain the potential of Phα1β to become a new analgesic drug. Evaluation of new drug on human ERG channel is requirement of the regulatory agencies. For the hERG channel inhibition assay it was used the commercial kit FLIPR® Potassium Assay. It was tested the effect of Phα1β and as positive control dofetilide, a well characterized hERG block-er. Phα1β 56, 225, 450 and 900 pMol caused at a higher concentration a very weak inhibition of the hERG channel activity of about 13.47 % with channel inhibition at 50%, IC50 > 900 pMol. Dofetilide (0.0001 - 10 µM), a known hERG inhibitor, caused a well-known, concentra-tion-dependent inhibition of the hERG channel with a mean IC50 of 0.1642 µM (0.1189 – 0.2282 µM. For the cytotoxicity assay, Phα1β concentrations of 56, 225, 450 and 900 pMol incubated for 24 h with HEK293-hERG cells did not alter the viability of the HEK293-hERG cells. It is con-cluded that Phα1β even at high concentrations does not interfere with hERG channels.
Keywords:
Phα1β
; analgesic
; dofetilide
; Kv11.1 potassium channel
; hERG channel interaction
; HEK293-hERG
; cells viability
1. Introduction
In the last two decades, considerable research has focused on N-type calcium channel inhibitors to develop novel analgesic drugs [1]. The ω-conotoxin MVIIA, derived from the snail Conus magnus, underwent synthesis resulting in the compound known as ziconotide, which is commercially available under the name Prialt®. Ziconotide is a selective, reversible, and potent blocker of N-type high-voltage-sensitive calcium channels, HVCCs [2] and is an efficient agent for pain control, however the drug produces maximal analgesia at doses near to its toxic dose causing severe side effects. Ziconotide (Prialt®) was developed as a first-class analgesic drug for neuropathic pain. Yet, the narrow therapeutic window and the adverse effects of ziconotide limit its clinical use in patients [3,4,5]. Pharmacological management of severe chronic pain is difficult to achieve with currently available analgesic drugs, and remains a large unmet therapeutic need [5]. Currently, neuropathic pain management is unsatisfactory and remains a challenge in clinical practice [6] and the search for new effective and safe analgesic drugs is imperial.
Animal venoms represent a rich source of novel drugs. Phα1β, a spider peptide purified from the venom of Phoneutria nigriventer [7], demonstrates inhibitory effects on high voltage calcium channels (HVCCs) [8], with a specific preference for N-type channels. Additionally, it acts as an antagonist of the TRPA1 receptor, a significant pathway involved in pain transduction [9]. The dual activity of Phα1β, both in its native and recombinant forms, on both TRPA1 channels and HVCCs suggests a potential advantage. This dual action could potentially broaden its efficacy in various pain-related conditions [10,11].
Pharmaceutical companies embark on the development of new drugs by first conducting pre-clinical tests on animals. These tests serve the purpose of demonstrating the drug's efficacy in combating the targeted disease and subsequently assessing its safety profile. Thus, our research group has compared the analgesic and side effects of native and recombinant Phα1β with ω-Conotoxin MVIIA (ziconotide, Prialt®), administered intrathecally, in several rodents models of pain, including neuropathic pain [12,13]. The obtained results indicated that native and the recombinant Phα1β had a higher analgesic profile than ziconotide [14,15] and more importantly, its analgesic property was associated with fewer side effects [13]. Notably, it was also observed that Phα1β potentiated the analgesic effect of morphine, reducing the adverse effect of the opioid in neuropathic pain treatment in rodents [12], enabling its use as an adjuvant drug to morphine in the treatment of pain. We also investigated the effects of Phα1β recombinant (CTK 01512-2) in the mouse model of experimental autoimmune encephalomyelitis (EAE) [16]. The effects of CTK 01512-2 were compared to those displayed by ziconotide and fingolimod, a drug employed for multiple sclerosis (MS) treatment. The intrathecal (it) or intravenous (iv) administration of the recombinant Phα1β reduced the EAE-elicited by Multiple Sclerosis-like symptoms similarly to that seen in animals that received fingolimod orally in MS treatment [17]. Ziconotide lacked any significant effect when dosed by intravenous route. Our results indicate that the recombinant Phα1β greatly improved the neuroinflammatory responses in a mouse model of MS with higher efficacy when compared to ziconotide, pointing out this molecule as a promising adjuvant for MS management [17].
Some of the advantages of the antinociceptive action of Phα1β over ω-conotoxin MVIIA (ziconotide, Prialt®) a first-class analgesic drug: 1- Phα1β presents a greater therapeutic window than that of ω-conotoxin MVIIA (ziconotide, Prialt®), [13]. The therapeutic window of Phα1β is 16 and for ω-conotoxin MVIIA (ziconotide, Prialt®) is 4 (16:4), [13]. 2-In pain relief, Phα1β induces less adverse effects than Conotoxin MVIIA, ziconotide Prialt®) [13]. Conotoxin MVIIA is 2.7 times more toxic than Phα1β. The DT50 (50% of the toxic dose) for Phα1β is 788 pMmoles while Conotoxin MVIIA, Ziconotide Prialt® is 287 pMmol, very close to its antinociceptive action [13]. 3- Phα1β induces analgesia at doses far below its toxic dose while ω-conotoxin MVIIA (Prialt®) only induces analgesia at doses very close to the toxic dose, inducing adverse effects that limit its clinical use [5]. 4- Phα1β has a greater capacity to induce analgesia in an already installed nociceptive process in comparison to ω-conotoxin MVIIA (Prialt®) [13]. 5- Phα1β is capable of reverse morphine tolerance [18], while ω-conotoxin MVIIA (ziconotide, Prialt®) does not [19]. 6-The IC50 (50% of the inhibitory dose) for Phα1β on the release of glutamate induced by capsaicin in nerve ending is 3 times lower than the dose used for ω-Conotoxin MVIIA (Phα1β, 2.1µM and ω-Conotoxin MVIIA 6.2 µM) [13]. 7-Phα1β improved neuroinflammatory responses in the multiple Sclerosis mouse model with higher efficacy than ziconotide [17]. 8- Astrocyte proliferation is a pathological hallmark of peripheral inflammation, which can be reversed by the Phα1β toxin treatment while ω-conotoxin MVIIA toxin has no effect [20]. 9- In rats, intravenous administration of ω-conotoxin MVIIA decreased blood pressure while Phα1β recombinant intravenous induced analgesia of the neuropathic pain, causing negligible cardiac problems [21]. Cardiotoxicity observed during the early stages of discovery and development of new drugs is a very a frequent problem, and in many cases, for the early interruption of new candidates for drug development. The interaction of drug candidate molecule with the potassium channel of the human ether -a-go-go- related gene (hERG), a voltage-gated potassium channel involved in the repolarization of the cardiac action potential, can generate serious cardiac arrhythmias that may cause sudden death [22,23]. This is because blockade of the hERG -like potassium channel by a drug causes an increase in cardiac action potential duration, associated with prolongation of the QT interval of the cardiac action potential, which can result in fatal arrhythmias [24]. These ventricular arrhythmias are called torsades de pointes (TdP). Since the 1990's around 10 drugs, after being approved by the regulatory agencies, had to be withdrawn from the market due to their cardiac toxicities resulting in sudden deaths. The best-known examples include Seldane® (terfenadine - antihistamine), Hismanal® (astemizole - antihistamine), Propulsid® (cisapride - prokinetic), Serlect® (sertindole - antipsychotic), Raxar® (grepafloxin - antibiotic), Zagam® (sparfloxicin – antibiotic) [25]. However, the prevalence of TdP caused by drugs is a rather rare phenomenon, and therefore difficult to be detected during the initial stages of development of a new drug. These serious side effects result from the coincidence of risk factors including exposure to a sufficient concentration of the drug in cardiac tissue, hERG channel blockade in addition to the patient's susceptibility.
The hERG gene channel was a critical step in understanding how to develop appropriate methodologies to conduct pre-clinical safety pharmacological investigations. Subsequent studies determined that the drugs withdrawn from the market had a common feature of inhibiting hERG channel function. The hERG channel mediates the major external potassium current known as IKr, a predominant repolarization current that drives ventricular repolarization. Thus, a significant effort has been made to develop non-clinical strategies to assess and minimize the risk of QT prolongation before administering a compound to man. These rare, but very serious events, led regulatory agencies, academia and the pharmaceutical industry to unite and propose safety pharmacology studies, both in vitro and in vivo, which include the cardiovascular system as one of the systems that should be carefully investigated, to assess the risk/benefit ratio and eliminate, already in the early stages of discovery and pre-clinical development of new drug candidates with the potential to cause TdP. In vitro electrophysiology studies performed in CHO cells, (Chinese hamster ovary) or in HEK 293 (human embryonic kidney) transfected with the hERG potassium channel were considered the gold standard for evaluating the possibilities of drug candidate molecules interacting with the hERG channel and causing ventricular arrhythmia. These studies must be complemented with safety studies performed in vivo in rats and dogs using telemetry. Using this technique, it is possible to evaluate different parameters in the cardiovascular system in real time and confirm the potential adverse effects of a new molecule before its administration to humans [26,27].
The results presented for the Phα1β suggest it may have potential to become a new analgesic drug. The regulatory agencies, such as the Food and Drug administration (FDA) and European Medicine Agency (EMA), that control the registry of new drugs, have several requirements for the approval of a new drug. The FDA has issued guidelines (ICH S6 and S7A) that require evaluation of novel chemical drugs for their potential to induce QT prolongation in drug development which is an indicator of a serious adverse effect causing ventricular arrhythmia and which may lead to sudden death [28,29]. It has been estimated that almost 70% of new compounds are eliminated at early stages mostly due to ERG-related safety issues [30] thereby limiting the number of drugs that enter the development pipeline. The present study describes the evaluation of Phα1β recombinant interaction on the KV11.1 potassium channel in HEK293 cells transfected with the human ERG channel. For the hERG channel inhibition assay it we used the commercial kit FLIPR® Potassium Assay.
2. Results
2.1. Evaluation of dofetilide or Phα1β interaction on the Kv11.1 potassium channel in HEK293 cells transfected with the human ERG channel
Phα1β at high concentrations caused a discreet inhibition of the hERG channel activity (13.47 %) Figure 1A. The concentration of Phα1β that presents channel inhibition at 50% (IC50 > 900 pMol) is estimated to be above the concentration of 900 pMol used in the test. On the other hand, the dofetilide (0.0001 - 10 µM), a known hERG inhibitor, caused concentration-dependent inhibition of the hERG channel with a mean IC50 of 0.1642 µM (Figure 1B).
2.2. Evaluation of cytotoxicity of Phα1β on HEK293-hERG cells.
For the cytotoxicity assay, Phα1β concentrations of 56, 225, 450 and 900 pMol used for the test of hERG channel inhibition assay were incubated with HEK293-hERG cells. After a 24 hours incubation period Phα1β did not alter the viability of the HEK293-hERG cells (Figure 2).
3. Discussion
Compounds that cause long QT syndrome have been discontinued in the early stages of preclinical development in the evaluation of safety tests [31]. In this study we evaluated the Phα1β effect with the hERG channel in vitro, using the HEK-293 cell line transfected with the human hERG channel. The interaction of Phα1β with the hERG channel was evaluated at concentrations between 56 and 900 pMol. Dofetilide was used as a positive control in the study, which was incubated at concentrations between 0.0001 and 10 μM. Both compounds were maintained for 30 minutes in contact with the cells. To evaluate cell viability after treatment with Phα1β for the same period, the MTT method viability test was used.
The results obtained in this study demonstrate that Phα1β when used in high concentrations between 56 and 900 pMol caused only a slight inhibition (about 13.47%) of the activity of the hERG channel, with its inhibitory concentration 50% (IC50) estimated to be higher than 900 pMol. On the other hand, at concentrations from 0.0001 to 10 μM, dofetilide inhibited the hERG channel in a concentration-dependent manner (maximum inhibition of 80.59 %) the activity of the hERG channel with an IC50 of 0.1642μM. In addition. Phα1β at a concentration of 900 pMol did not alter cell viability. In conclusion, based on the results obtained in this study that Phα1β in the concentrations and conditions tested does not interfere with the hERG channel. The inhibition observed for dofetilide in the hERG channel in vitro, is in accordance with tests previously performed at CIEnP and with data from the literature [32,33], validating the test performed.
Traditionally, patch clamp electrophysiology has been used as the gold standard for ion channel studies. However, the method is a labor-intensive with low yield. Assay validation demonstrated that electrophysiology "gold standard" assay for studying biophysical properties and the thallium methodology produced equivalent results, proving to be an appropriate methodology to evaluate the safety of new compounds on the hERG channel [32,34,35]. The qualities of the thallium flux allow it to play an important role in identifying and assessing the activity of potassium channel modulators [32,35]. The Potassium Channel Assay used FLIPR® Potassium Assay – Molecular Devices consists of a screening of potassium channel activity that allows rapid and robust assay. The test is based on the permeability of potassium channels to thallium [32]. The fluorogenic signal quantitatively reflects the activity of ion channels that are permeable to thallium and is capable of detecting modulators of voltage gated potassium channel expressed in mammalian cells including hERG [33]. The human ether-á-go-go related gene hERG encodes a voltage-gated potassium channel, which plays a critical role in the repolarization of the cardiac action potential [36,37]. Disruption of the hERG channel activity may cause QT prolongation, which might result in fatal ventricular arrhythmia such as Torsade de Pointes [28,32,34,35]. Technically, hERG is the name of a human gene for human ether á-go-go-related gene that encodes the pore-forming subunit of the delayed rectifier IKr channel in humans. The hERG binding assay provides valuable information during drug discovery and their malfunction causes a variety of human diseases and represents a class of drugs that increase the risk arrhythmogenic QT prolongation [36,38]. In 2001, 30% of pharmaceutical drugs tested clinically were abandoned because of the lack of efficacy and 30% of others were also abandoned because of safety concerns such as cardiotoxicities including ion channel inhibition [39,40]. Drug wear in tests due to cardiotoxic effect and pro-arrhythmias represents a major problem in drug discovery in the pharmaceutical industry [41]. In the last 10 years of safety studies in the use of medicinal drugs, the lack of clinical and non-clinical combinations of drugs was responsible for 30% of drug suspensions. The classic example is terfenadine, an antihistamine, that blocked the cardiac hERG channel, causing fatal arrhythmia and therefore withdrawn from markets worldwide [42].
To improve the accuracy of preclinical cardiotoxicity, screening assays utilizing human cardiac myocytes isolated from human hearts were validated to predict the adverse effect of drugs [43]. This technology is believed to create new opportunities for the studies to reveal drug sensitivities and has been recommended in the revised ICH guidelines in the near future [44]. Blockade of hERG channels in the heart is an unintenonal side effect of many drugs and can induce cardiac arrhythmia and sudden death. It has become a common practice in the past few years to screen compounds for hERG channel activity early during the drug discovery process. The regulatory guidelines (ICHS7B) recommending inhibition of the delayed rectifier current (IKr), carried by human ether-a-go-go-related gene (hERG) channels in cardiac cells (the hERG test), showed in this publication, as a ‘first line test for identifying compounds inducing QT prolongation. However later studies demonstrated that hERG channel testing alone might not be sufficient to eliminate cardiac arrhythmia induced by drugs that affect other cardiac ion channels [45]. Thus, following the hERG channel test of the present study, we will perform experiments for the in vivo effect of Phα1β in the cardiovascular system of rats by telemetric analysis. After, in which the development and clinical trials with Phα1β in the management of several conditions of pain in humans will pave the way to use Phα1β in large groups of patients. In conclusion the results of the present study support further advance of the preclinical development of Phα1β to permit its clinical studies as a new option to treat chronic pain alone or as an adjuvant drug in opioid treatments [46]. Further investigations are still required to assess the therapeutic efficacy and safety profile of Phα1β in humans.
4. Material and Methods
4.1. Phα1β recombinant
Giotto Biotech® (www.giottobiotech.com) synthesized the recombinant version of Phα1β via Escherichia coli expression CTK-01512-2 used in the evaluation. It was purified through a proprietary production process, with a combination of ion exchange and size exclusion chromatography. The yield of the process was 0.5 mg/ml. The peptide molecular weight (Mw) was 6,045 kDa. Both the native and the recombinant Phα1β have the same 55 amino acids (sequence: ACIPRGEICTDDCECCGCDNQCYCPPGSSLGIFKCSCAHANKYFCNRKKEKCKKA). The sequence of the recombinant and the natural Phα1β peptides are identical, with the exception of a methionine added at the N terminal portion of the recombinant peptide (the addition of the starting methionine is a common practice in the heterologous protein expression). The purity of the recombinant toxin is higher than 90% presented in an SDS PAGE assay. The inhibitory effects on the rodents nociception induced by native and the recombinant Phα1β were not statistically different, developing with no side effects [15,21].
4.2. Evaluation of Phα1β and dofetilide interaction on the Kv11.1 potassium channel in HEK293 cells transfected with the human ERG channel.
The recombinant HEK293 cell line expressing human ERG potassium channel (ether-a- go go-related gene, also known as KCNH2 or Kv11.1) from BPS Bioscience, accession number NM_000238, growth medium 1B BPS # 79531 and thaw medium 1 # 60187 was used in this study. Cells were thawed and cultured according to the supplier's specifications: hERG (Kv11.1)-HEK-293 Recombinant Cell line # 60619 and used as described. Cells transfected with the human hERG channel were subcultured in 96-well plates and maintained under controlled conditions for 24 h. They were then incubated with the probe for 1 h before adding the 30 min treatments, and in sequence read on the FlexStation.
The evaluation of the interaction of Phα1β recombinant and dofetilide with the hERG channel used the commercial kit FLIPR® Potassium Assay (Molecular Devices) and the assay was performed according to the manufacturer's specifications. The kit FLIPR® Potassium Assay contains a thallium sensitive indicator dye. During the initial dye-loading step the thallium as acetoximethyl ester (AM), enters the cells by passively diffusion across the cell membrane. Cytoplasm esterase cleave the AM ester relieving its active fluorogenic form. A masking dye is applied extracellularly to reduce the background fluorescence. To activate potassium channel, the cells are stimulated with either a mixture of K+ and TI+ or a ligant in the presence of TI+. The increase in fluorescent signal represent the influx of TI+ into the cell specifically through potassium channels, representing a functional measurement of the potassium channel activity. For the assay, cells were plated in 96-wells, 5 x 10 4 cells/well of the HEK -293-cells line (BPS Bioscience), expressing human ERG Human Ether-`a-go-go Related Gene Potassium Channel Kv11.1. After 24 hours, the plate culture medium was aspirated and replaced by 50 μL of calcium and magnesium free HBSS. Then, the cells were incubated with 50 μL of the fluorescent probe present in the commercial kit, containing probenecid (Sigma-Aldrich) at a final concentration of 2.5 mM per well. After 1 hour of incubation at room temperature and in the absence of light, 25 μL of Phα1β at concentrations of 56, 225, 450 and 900 pMol or dofetilide (Sigma-Aldrich), hERG channel inhibitor, at concentrations of 0.0001 to 10 μM were incubated with the cells for 30 minutes. The stimulus buffer was added to each column through automated pipetting present in the FlexStation 3 equipment. Data were obtained using the SoftMax® Pro Software, at a wavelength of 485/525 nm. Data analysis was performed using SoftMax Pro Software and GraphPad Prism®. Results were expressed as percentage inhibition of the hERG channel.
4.3. Evaluation of Phαβ on the viability of HEK-293 cells.
The viability assay of HEK-293 cells was performed using the colorimetric MTT reduction test (3-(4, 5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide), through which viable cells reduce the MTT salt forming a formazan complex within their mitochondria, as previously described by Mosmann [47]. For this assay, after treating HEK-293 cells (5 x 10 4 cells/well) with Phα1β (56-900 pMol), the culture medium was replaced with medium containing MTT (0.5 mg/mL), and the cells were incubated for approximately 1 hour and 30 minutes at 37 ºC ± 0.1 °C in a humidified atmosphere containing 5% ± 0.1% CO2. Subsequently, the MTT solution was removed and 200 μL of dimethyl sulfoxide was added. Absorbance was measured at 570 nm using the spectrophotometer (SpectraMax MiniMax 300, Imaging Cytometer, Molecular Device). Data analysis was performed using SoftMax Pro Software and GraphPad Prism®. The results were expressed as a percentage of viable cells in relation to the control (vehicle group).
In order to reduce as much as possible deviations that may interfere with the reliability, traceability and reproducibility of the results, the following procedures were rigorously applied from experimental planning to carrying out the experiments: 1) The study was conducted as described in SOP B.06 and has been described in Portuguese according to POP B.02; 2) can be translated exactly and completely into English; 3) The Final Report was described in Portuguese according to POP B.05 and, if necessary, could be translated into English; 4) All reagents that were used were received and stored at Centro de Investigação e Estudos Préclinicos( CIEnP), as described in POP G.01 and were used within the expiration date indicated by the manufacturer; 5) No data was omitted for the calculation of means, statistical analysis and/or analysis of results; 6) The experiments were conducted jointly with their respective control (Reference Item) for method validation; 7) All information regarding the quality of cells, cell lines and/or in vitro systems used, ideal conditions for cultivation and maintenance, as well as factors that may change and/or influence the quality of the results obtained; 8) The statistical tests for analyzing the results were previously selected and are described in item 9. Statistical analysis: Data were analyzed using GraphPad Prism® software (GraphPad Software Inc., San Diego, CA, USA). The results were expressed as Mean ± Error Average Standard (EPM)
References
- S. S. Bowersox, T. Gadbois, T. Singh, M. Pettus, Y. X. Wang, and R. R. Luther, "Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain," J Pharmacol Exp Ther, vol. 279, pp. 1243-9, Dec 1996.
- G. P. Miljanich and J. Ramachandran, "Antagonists of neuronal calcium channels: structure, function, and therapeutic implications," Annu Rev Pharmacol Toxicol, vol. 35, pp. 707-34, 1995.
- R. D. Penn and J. A. Paice, "Adverse effects associated with the intrathecal administration of ziconotide," Pain, vol. 85, pp. 291-6, Mar 2000.
- A. Schmidtko, J. Lotsch, R. Freynhagen, and G. Geisslinger, "Ziconotide for treatment of severe chronic pain," Lancet, vol. 375, pp. 1569-77, May 1 2010.
- P. S. Staats, T. Yearwood, S. G. Charapata, R. W. Presley, M. S. Wallace, M. Byas-Smith, et al., "Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial," JAMA, vol. 291, pp. 63-70, Jan 7 2004.
- N. Attal, G. Cruccu, R. Baron, M. Haanpaa, P. Hansson, T. S. Jensen, et al., "EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision," Eur J Neurol, vol. 17, pp. 1113-e88, Sep 2010.
- N. Cordeiro Mdo, S. G. de Figueiredo, C. Valentim Ado, C. R. Diniz, V. R. von Eickstedt, J. Gilroy, et al., "Purification and amino acid sequences of six Tx3 type neurotoxins from the venom of the Brazilian 'armed' spider Phoneutria nigriventer (Keys)," Toxicon, vol. 31, pp. 35-42, Jan 1993.
- L. B. Vieira, C. Kushmerick, M. E. Hildebrand, E. Garcia, A. Stea, M. N. Cordeiro, et al., "Inhibition of high voltage-activated calcium channels by spider toxin PnTx3-6," J Pharmacol Exp Ther, vol. 314, pp. 1370-7, Sep 2005.
- R. Tonello, C. Fusi, S. Materazzi, I. M. Marone, F. De Logu, S. Benemei, et al., "The peptide Phalpha1beta, from spider venom, acts as a TRPA1 channel antagonist with antinociceptive effects in mice," Br J Pharmacol, vol. 174, pp. 57-69, Jan 2017.
- E. L. Andrade, F. C. Meotti, and J. B. Calixto, "TRPA1 antagonists as potential analgesic drugs," Pharmacol Ther, vol. 133, pp. 189-204, Feb 2012.
- R. Nassini, S. Materazzi, S. Benemei, and P. Geppetti, "The TRPA1 channel in inflammatory and neuropathic pain and migraine," Rev Physiol Biochem Pharmacol, vol. 167, pp. 1-43, 2014.
- F. Rosa, G. Trevisan, F. K. Rigo, R. Tonello, E. L. Andrade, M. do Nascimento Cordeiro, et al., "Phalpha1beta, a peptide from the venom of the spider Phoneutria nigriventer shows antinociceptive effects after continuous infusion in a neuropathic pain model in rats," Anesth Analg, vol. 119, pp. 196-202, Jul 2014.
- A. H. Souza, J. Ferreira, M. D. N. Cordeiro, L. B. Vieira, C. J. De Castro, G. Trevisan, et al., "Analgesic effect in rodents of native and recombinant Ph alpha 1beta toxin, a high-voltage-activated calcium channel blocker isolated from armed spider venom," Pain, vol. 140, pp. 115-126, Nov 15 2008.
- J. F. da Silva, N. S. Binda, E. M. R. Pereira, M. S. L. de Lavor, L. B. Vieira, A. H. de Souza, et al., "Analgesic effects of Phalpha1beta toxin: a review of mechanisms of action involving pain pathways," J Venom Anim Toxins Incl Trop Dis, vol. 27, p. e20210001, 2021.
- F. K. Rigo, G. Trevisan, S. D. De Pra, M. N. Cordeiro, M. H. Borges, J. F. Silva, et al., "The spider toxin Phalpha1beta recombinant possesses strong analgesic activity," Toxicon, vol. 133, pp. 145-152, Jul 2017.
- R. C. Dutra, A. F. Bento, D. F. Leite, M. N. Manjavachi, R. Marcon, M. A. Bicca, et al., "The role of kinin B1 and B2 receptors in the persistent pain induced by experimental autoimmune encephalomyelitis (EAE) in mice: evidence for the involvement of astrocytes," Neurobiol Dis, vol. 54, pp. 82-93, Jun 2013.
- R. B. M. Silva, S. Greggio, G. T. Venturin, J. C. da Costa, M. V. Gomez, and M. M. Campos, "Beneficial Effects of the Calcium Channel Blocker CTK 01512-2 in a Mouse Model of Multiple Sclerosis," Mol Neurobiol, vol. 55, pp. 9307-9327, Dec 2018.
- F. K. Rigo, G. Trevisan, F. Rosa, G. D. Dalmolin, M. F. Otuki, A. P. Cueto, et al., "Spider peptide Phalpha1beta induces analgesic effect in a model of cancer pain," Cancer Sci, vol. 104, pp. 1226-30, Sep 2013.
- Y. X. Wang, M. Pettus, D. Gao, C. Phillips, and S. Scott Bowersox, "Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain," Pain, vol. 84, pp. 151-8, Feb 2000.
- H. Tenza-Ferrer, L. A. V. Magno, M. A. Romano-Silva, J. F. da Silva, and M. V. Gomez, "Phalpha1beta Spider Toxin Reverses Glial Structural Plasticity Upon Peripheral Inflammation," Front Cell Neurosci, vol. 13, p. 306, 2019.
- F. K. Rigo, M. F. Rossato, V. Borges, J. F. da Silva, E. M. R. Pereira, R. A. M. de Avila, et al., "Analgesic and side effects of intravenous recombinant Phalpha1beta," J Venom Anim Toxins Incl Trop Dis, vol. 26, p. e20190070, Apr 17 2020.
- L. Belardinelli, C. Antzelevitch, and M. A. Vos, "Assessing predictors of drug-induced torsade de pointes," Trends Pharmacol Sci, vol. 24, pp. 619-25, Dec 2003.
- C. E. Pollard, J. P. Valentin, and T. G. Hammond, "Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective," Br J Pharmacol, vol. 154, pp. 1538-43, Aug 2008.
- C. E. Pollard, N. Abi Gerges, M. H. Bridgland-Taylor, A. Easter, T. G. Hammond, and J. P. Valentin, "An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk," Br J Pharmacol, vol. 159, pp. 12-21, Jan 2010.
- P. Mamoshina, B. Rodriguez, and A. Bueno-Orovio, "Toward a broader view of mechanisms of drug cardiotoxicity," Cell Rep Med, vol. 2, p. 100216, Mar 16 2021.
- M. K. Pugsley, S. Authier, and M. J. Curtis, "Principles of safety pharmacology," Br J Pharmacol, vol. 154, pp. 1382-99, Aug 2008.
- R. M. Wallis, "Integrated risk assessment and predictive value to humans of non-clinical repolarization assays," Br J Pharmacol, vol. 159, pp. 115-21, Jan 2010.
- M. L. De Bruin, M. Pettersson, R. H. Meyboom, A. W. Hoes, and H. G. Leufkens, "Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death," Eur Heart J, vol. 26, pp. 590-7, Mar 2005.
- J. C. Hancox, M. J. McPate, A. El Harchi, and Y. H. Zhang, "The hERG potassium channel and hERG screening for drug-induced torsades de pointes," Pharmacol Ther, vol. 119, pp. 118-32, Aug 2008.
- S. L. Wynia-Smith, A. L. Gillian-Daniel, K. A. Satyshur, and G. A. Robertson, "hERG gating microdomains defined by S6 mutagenesis and molecular modeling," J Gen Physiol, vol. 132, pp. 507-20, Nov 2008.
- M. C. Sanguinetti and M. Tristani-Firouzi, "hERG potassium channels and cardiac arrhythmia," Nature, vol. 440, pp. 463-9, Mar 23 2006.
- F. Potet, A. N. Lorinc, S. Chaigne, C. R. Hopkins, R. Venkataraman, S. Z. Stepanovic, et al., "Identification and characterization of a compound that protects cardiac tissue from human Ether-a-go-go-related gene (hERG)-related drug-induced arrhythmias," J Biol Chem, vol. 287, pp. 39613-25, Nov 16 2012.
- W. A. Schmalhofer, A. M. Swensen, B. S. Thomas, J. P. Felix, R. J. Haedo, K. Solly, et al., "A pharmacologically validated, high-capacity, functional thallium flux assay for the human Ether-a-go-go related gene potassium channel," Assay Drug Dev Technol, vol. 8, pp. 714-26, Dec 2010.
- H. B. Yu, M. Li, W. P. Wang, and X. L. Wang, "High throughput screening technologies for ion channels," Acta Pharmacol Sin, vol. 37, pp. 34-43, Jan 2016.
- B. Zou, H. Yu, J. J. Babcock, P. Chanda, J. S. Bader, O. B. McManus, et al., "Profiling diverse compounds by flux- and electrophysiology-based primary screens for inhibition of human Ether-a-go-go related gene potassium channels," Assay Drug Dev Technol, vol. 8, pp. 743-54, Dec 2010.
- M. E. Curran, I. Splawski, K. W. Timothy, G. M. Vincent, E. D. Green, and M. T. Keating, "A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome," Cell, vol. 80, pp. 795-803, Mar 10 1995.
- C. D. Weaver, D. Harden, S. I. Dworetzky, B. Robertson, and R. J. Knox, "A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells," J Biomol Screen, vol. 9, pp. 671-7, Dec 2004.
- M. C. Trudeau, J. W. Warmke, B. Ganetzky, and G. A. Robertson, "HERG, a human inward rectifier in the voltage-gated potassium channel family," Science, vol. 269, pp. 92-5, Jul 7 1995.
- I. Kola and J. Landis, "Can the pharmaceutical industry reduce attrition rates?," Nat Rev Drug Discov, vol. 3, pp. 711-5, Aug 2004.
- D. Laustriat, J. Gide, and M. Peschanski, "Human pluripotent stem cells in drug discovery and predictive toxicology," Biochem Soc Trans, vol. 38, pp. 1051-7, Aug 2010.
- N. Stockbridge, J. Morganroth, R. R. Shah, and C. Garnett, "Dealing with global safety issues : was the response to QT-liability of non-cardiac drugs well coordinated?," Drug Saf, vol. 36, pp. 167-82, Mar 2013.
- B. O. Villoutreix and O. Taboureau, "Computational investigations of hERG channel blockers: New insights and current predictive models," Adv Drug Deliv Rev, vol. 86, pp. 72-82, Jun 23 2015.
- K. Blinova, Q. Dang, D. Millard, G. Smith, J. Pierson, L. Guo, et al., "International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment," Cell Rep, vol. 24, pp. 3582-3592, Sep 25 2018.
- E. G. Navarrete, P. Liang, F. Lan, V. Sanchez-Freire, C. Simmons, T. Gong, et al., "Screening drug-induced rrhythmia using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays," Circulation, vol. 128, pp. S3-13, Sep 10 2013.
- H. R. Lu, E. Vlaminckx, A. N. Hermans, J. Rohrbacher, K. Van Ammel, R. Towart, et al., "Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B Guidelines," Br J Pharmacol, vol. 154, pp. 1427-38, Aug 2008.
- R. Tonello, F. Rigo, C. Gewehr, G. Trevisan, E. M. Pereira, M. V. Gomez, et al., "Action of Phalpha1beta, a peptide from the venom of the spider Phoneutria nigriventer, on the analgesic and adverse effects caused by morphine in mice," J Pain, vol. 15, pp. 619-31, Jun 2014.
- T. Mosmann, "Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays," J Immunol Methods, vol. 65, pp. 55-63, Dec 16 1983.
Figure 1A.
Concentration-response curve of Phα1β on the hERG channel by Potassium Assay Kit. The HEK293 cells transfected with hERG were incubated with Phα1β 56, 225, 450 and 900 pMol for 30 minutes. Then, Flex Station 3 carried out the addition of 1 mM Thallium + 10 mM Potassium. The y-axis is the percentage of the relative inhibition of Phα1β on the hERG channel using the SoftMax Pro 7.1 software, (excitation 485 nm, emission 538 nm). Data analysis performed using GraphPad Prism 9 show the IC50 > 900 pMol, for the inhibitory effect of Phα1β on the hERG channel. The data in the graph were expressed as mean ± standard error of the mean of three independent experiments.
Figure 1A.
Concentration-response curve of Phα1β on the hERG channel by Potassium Assay Kit. The HEK293 cells transfected with hERG were incubated with Phα1β 56, 225, 450 and 900 pMol for 30 minutes. Then, Flex Station 3 carried out the addition of 1 mM Thallium + 10 mM Potassium. The y-axis is the percentage of the relative inhibition of Phα1β on the hERG channel using the SoftMax Pro 7.1 software, (excitation 485 nm, emission 538 nm). Data analysis performed using GraphPad Prism 9 show the IC50 > 900 pMol, for the inhibitory effect of Phα1β on the hERG channel. The data in the graph were expressed as mean ± standard error of the mean of three independent experiments.
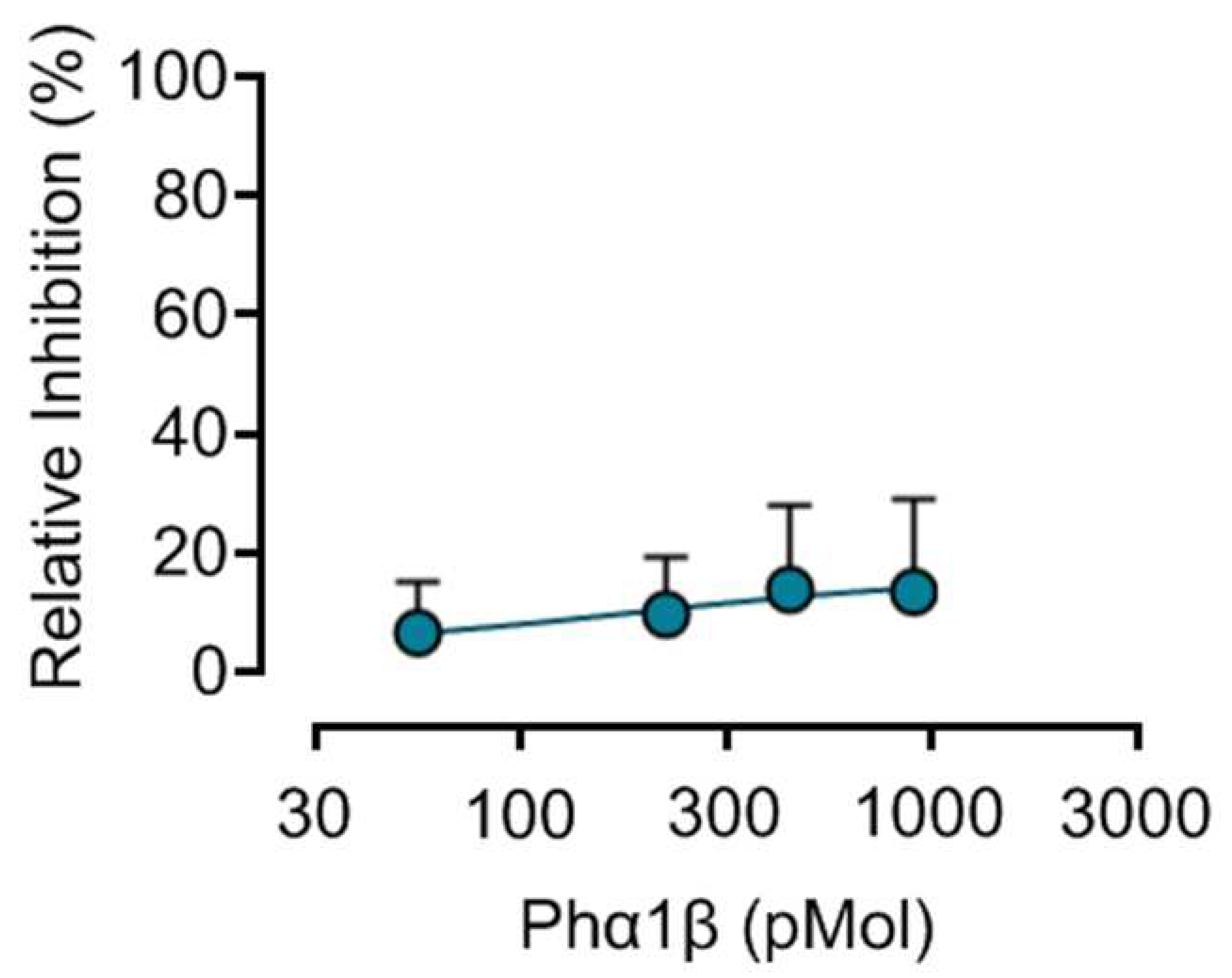
Figure 1B.
Concentration-response curve of the inhibitor dofetilide on the hERG channel by Potassium Assay Kit. HEK293 cells transfected with hERG were incubated with the dofetilide 0.0001 - 10 µM for 30 minutes. Then, FlexStation 3 carried out the addition of 1 mM Thallium + 10 mM Potassium. The y-axis is the percentage of the relative inhibition of dofetilide on the hERG channel using the SoftMax Pro 7.1 software, (excitation 485 nm, emission 538 nm). Data analyzes performed using GraphPad Prism 9 show the IC50 = 0.1642 µMol for the inhibitory effect of dofetilide on the hERG channel. The data in the graph were expressed as mean ± standard error of the mean of three independent experiments.
Figure 1B.
Concentration-response curve of the inhibitor dofetilide on the hERG channel by Potassium Assay Kit. HEK293 cells transfected with hERG were incubated with the dofetilide 0.0001 - 10 µM for 30 minutes. Then, FlexStation 3 carried out the addition of 1 mM Thallium + 10 mM Potassium. The y-axis is the percentage of the relative inhibition of dofetilide on the hERG channel using the SoftMax Pro 7.1 software, (excitation 485 nm, emission 538 nm). Data analyzes performed using GraphPad Prism 9 show the IC50 = 0.1642 µMol for the inhibitory effect of dofetilide on the hERG channel. The data in the graph were expressed as mean ± standard error of the mean of three independent experiments.

Figure 2.
Assessment of viability of HEK293-hERG cells after incubation with Phα1β 56, 225, 450 and 900 pMol for 24 hours and then cell viability assay was performed by MTT method. The y-axis represents the % of the cell viability for the mean ± standard error of 3 separate assays in duplicate. One-way analysis of variance (ANOVA) was used for statistical analysis, followed by Dunnett's test. The viability percentage was calculated in relation to the Vehicle control.
Figure 2.
Assessment of viability of HEK293-hERG cells after incubation with Phα1β 56, 225, 450 and 900 pMol for 24 hours and then cell viability assay was performed by MTT method. The y-axis represents the % of the cell viability for the mean ± standard error of 3 separate assays in duplicate. One-way analysis of variance (ANOVA) was used for statistical analysis, followed by Dunnett's test. The viability percentage was calculated in relation to the Vehicle control.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



