
Submitted:
21 November 2023
Posted:
22 November 2023
You are already at the latest version
Abstract
This review covers the last 25 years literature on analogs of suberoylanilide hydroxamic acid (SAHA, known also as Vorinostat) acting as HDAC inhibitor. In particular, the topic has been focused on the synthesis and biological activity of compounds where the phenyl group (the surface recognition moiety, CAP) of SAHA has been replaced by an azaheterocycle through a direct bond with amide nitrogen atom, and the methylene chain in the linker region is of variable length. Most of the compounds reported displayed good to excellent inhibitory activity against HDACs and in many cases showed anti-proliferative activity against human cancer cell lines.
Keywords:
HDAC
; SAHA
; azaheterocycles
; synthesis
; anticancer
1. Introduction
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) catalyze, respectively, deacetylation and acetylation of specific lysine residues situated on the amino-terminal tails of histone proteins. These enzymes play a key role in gene transcription [1] since acetylation is associated with an open chromatin configuration resulting in enhancing transcription [2] whereas the deacetylation process induces condensed and transcriptionally inactive heterochromatin [3].
Normally, it exists a balance between histone acetylation and deacetylation in normal cells, however, it has also demonstrated that these two enzymes are not only involved in the regulation of chromatin structure and gene expression, but can also regulate cell-cycle progression and carcinogenic processes [4]. Inhibition of HDACs can lead to cell differentiation, apoptosis and cell-cycle arrest both in several cancer cell lines and in vivo, thus making HDAC inhibitors (HDACi) a very important class of anticancer agents [5,6]. Besides to their anticancer effects, some HDACi also exhibit valuable neuroprotective properties in brain injuries such as stroke [7] and ischemia [8]. Further, some studies have reported the potential of HDACi to treat chronic neurological disorders such as amyotrophic lateral sclerosis [9], and Alzheimer’s disease [10].
The common classification of HDACs is based on molecular phylogenetic analysis of primary structure. They are grouped (based on homology to yeast enzymes [11] in distinct classes: class I (HDAC1, HDAC2, HDAC3 and HDAC8), class IIa (HDAC4, HDAC5, HDAC7 and HDAC9), class IIb (HDAC6 and HDAC10) and class IV (HDAC11), these classes contain zinc-dependent domains. The class III belongs to a structurally and mechanistically distinct class of NAD+-dependent hydrolases (sirtuins, Sirt1–Sirt7) [12].
To date, several HDAC inhibitors (HDACis) were addressed for cancer treatment, many of them contain an amide-alkyl-hydroxamic acid framework, such as that present in suberoylanilide hydroxamic acid (SAHA), well-known as Vorinostat (Figure 1). SAHA was the first HDAC inhibitor approved by US Food and Drug Administration in 2006 for the treatment of cutaneous T cell lymphoma [13].
SAHA structure, as well as that of many HDACis, conforms to the well-known HDAC inhibition pharmacophore A-B-C, where A is a cap group (CAP) for protein surface interactions, C is a zinc coordinating group (ZBG) that repress the hydrolysis of acetyl group in the lysine residue, and B is a linker group that connects CAP with ZBG [14].
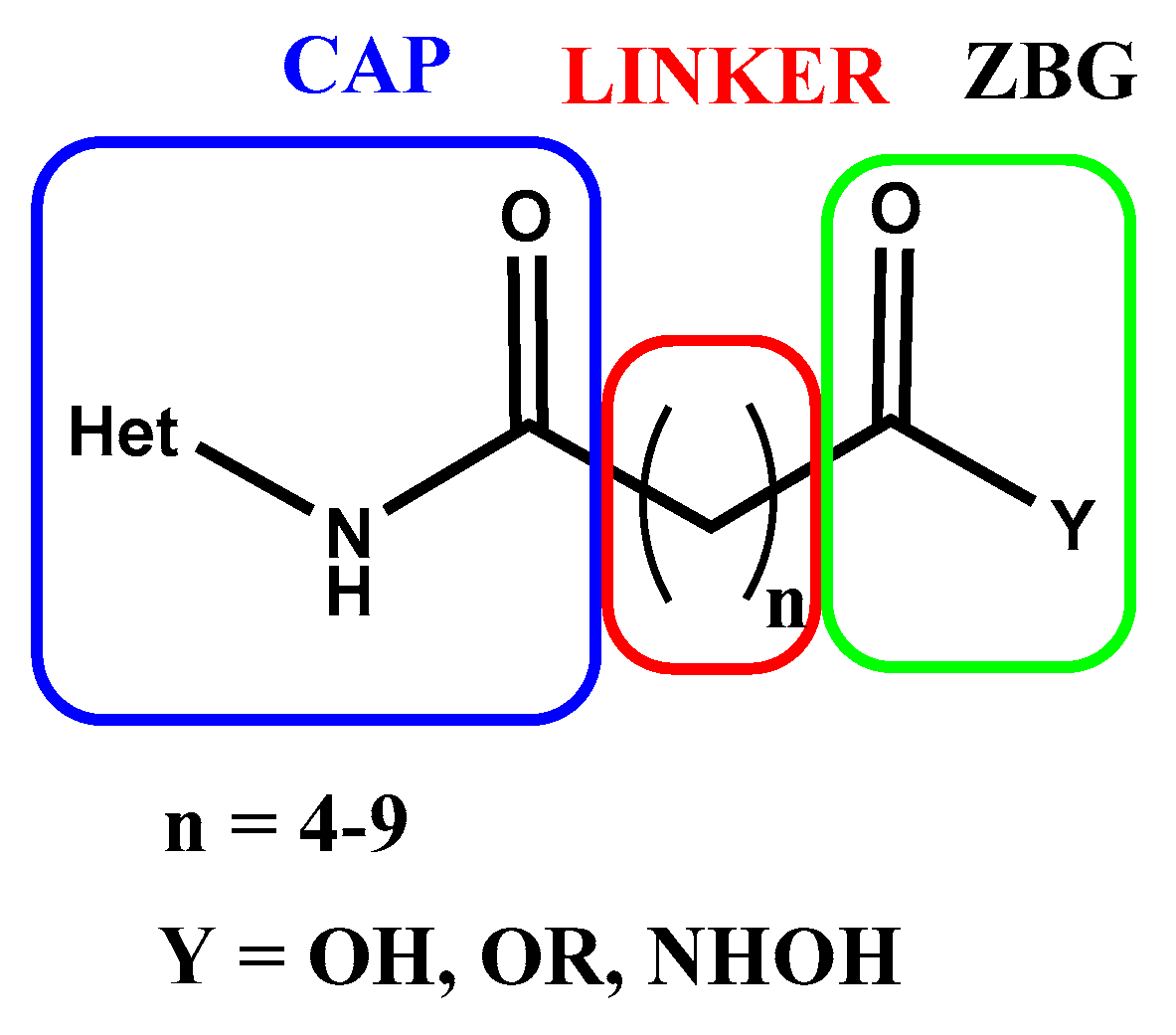
Along the years, many SAHA analogues have been synthesized and tested as HDACi. The present review reports the synthesis and biological activity of HDACi analogues of Vorinostat focusing attention on those bearing an aza-heteroaromatic instead of phenyl group in CAP fragment, a linear aliphatic chain of different length as linker, and carboxy-, ester-, or hydroxamic group as ZBG (Figure 2). This review, excluding patent literature, covers literature articles of the last 25 years.
For the sake to make easier the reading of this contribute we divided the review in sub-headings, depending on the length chain of the aliphatic linker. In turn, each sub-heading has been structured based on the class of heterocycle bound to amide nitrogen atom of the CAP group.
2. Two-carbon linker chain
2.1. 2-amino-1,3,4-thiadiazoles in CAP group
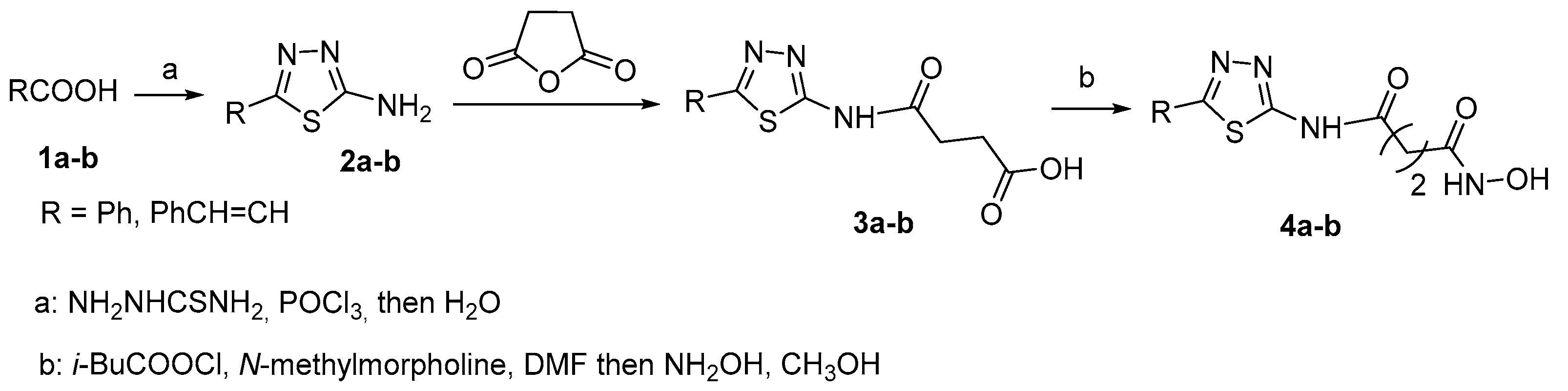
The only reported HDACi bearing a C-2 alkyl chain are 2-amino-1,3,4-thiadiazole-based hydroxamates 4a and 4b [15]. They were synthesized as depicted in Scheme 1.
HDAC inhibitory activity of compounds 4a and 4b was assessed by the Color de Lys assay and the results showed in both cases IC50 values > 5 μM, lower than that of SAHA (IC50 = 0.15 ± 0.02).
3. Three-carbon linker chain
3.1. 2-amino-1,3,4-thiadiazoles in CAP group
Hydroxamates bearing 2-amino-1,3,4-thiadiazole-derivatives in the CAP group and a C-3 linker chain were obtained as depicted in Scheme 2. Intermediates 5a-d were obtained by reaction between 2-amino-1,3,4-thiadiazoles 2a-d and methyl 5-chloro-5-oxopentanoate (in turn obtained from dimethyl glutarate after partial hydrolysis and treatment with SOCl2). Treatment of 5a-d with NH2OK in methanol gave compounds 6a-d [15].
Among 6a-d, only 6b showed HDAC inhibition activity (IC50 =0.16 ± 0.03) close to that of SAHA (IC50 = 0.15 ± 0.02). Moreover, the effect of 6b on the cell viability in MDA-MB-231 breast cancer cells and K562 chronic myelogenous leukemia cells was evaluated, resulting in a IC50 value = 5.90 ± 2.75 and 6.75 ± 2.37, respectively.
2.2. Indazoles in CAP group
Among the series of indazole derivatives 16a-s characterized by different spacer length and substituents on the heterocyclic ring, prepared as shown in Scheme 3, the only reported compound with a 3-carbon linker chain is 16a (n =3, R = 3-methoxyphenyl) and related precursors [16].
The biological activity as HDACi of compound 16a was tested against HDAC1, HDAC2, and HDAC8 (IC50 = 76nM, 168nM, and 54 nM, respectively) and compared with that of SAHA (IC50 = 13nM, 70nM, and 44 nM, respectively). Moreover, 16a was administered to solid cancer cell lines HCT116 (human colorectal cancer cells, IC50 > 50μM), MCF-7 (human breast cancer cells, IC50 > 41.5mM) and HeLa (human cervical cancer cells, IC50 > 50mM) but its activity was lower than that found for SAHA (IC50 = 4.9 mM, 0.8 mM, 5.0 μM for the three cell lines, respectively).
3. Four-carbon linker chain (4-C spacer)
3.1. 2-amino-1,3,4-thiadiazoles in CAP group
Compounds 17 a-d (Figure 3) were obtained with the same synthetic sequence shown in Scheme 2, using in this case 6-chloro-6-oxohexanoic acid as acyl chloride [15].
The HDAC inhibitory activity of the compounds was assayed: in all four cases it was lower compared with that SAHA chosen as positive control.
3.2. Indazoles in CAP group
With the synthetic sequence depicted in Scheme 3, when n=4 and R = 3-methoxyphenyl, indazole derivative 16b (Figure 4) was obtained [16]. This compound showed activity towards HDAC1 (IC50 = 13 nM), HDAC2 (IC50 = 62 nM), and HDAC8 (IC50 = 41 nM) equal or a little better than that of SAHA (HDAC1 IC50 = 13 nM; HDAC2 IC50 = 70 nM; HDAC8 IC50 = 44 nM).
3.3. Benzothiazoles in CAP group
Benzothiazole derivatives 19a-f [17] were obtained in good yields (from 70 to 90%) by reaction between 5-substituted 2-aminobenzothiazoles and adipic acid monomethyl ester in the presence of 1,1'-carbonyldiimidazole and triethylamine in THF followed by conversion of the ester intermediates to the corresponding hydroxamic acids (Scheme 4).
Cytotoxicity assays of compounds 19a-f against five cancer cell lines, namely SW620, MCF-7, PC3, AsPC-1, and NCI-H460 revealed that compounds 19a–d exhibited cytotoxicity against all tested cancer cell lines with IC50 values from 7.90 to 15.12 μg/ml whereas compounds 19e and 19f were not cytotoxic (IC50 of >30 μg/ml).
However, when the effect of 19a-f on histone acetylation in SW620 cells was examined, the HDAC inhibition at concentration of 1 μg/mL was not significant.
3.4. 4-Anilinothieno[2,3-d]pyrimidine derivatives in CAP group
Thieno[2,3- d]pyrimidine-based HDACs inhibitors with different length of the spacer (n = 2, 3, 4) [18] were synthesized from the thieno[3,2-d]pyrimidin-4(3H)-one 20 after nitration in alpha position to thiophene ring, chlorination on the pyrimidine ring, treatment with different anilines and reduction of the nitro group to amino group. The latter was reacted with the acyl chloride MeOCO(CH2)nCOCl (n = 4-6) to form the methyl ester intermediates 25 that were subjected to hydrolysis. Subsequent treatment of the obtained acid with hydroxylamine hydrochloride in the presence of BOP and DMAP afforded the targeted compounds 26 (Scheme 5).
Focusing attention on the C-4 spacer, compounds 26a (R = 3-Cl, 4-F) and 26b (R = 3-CF3, 4-Cl) their in vitro inhibitory activity against HDAC1, HDAC3, and HDAC6 was lower with respect to that of SAHA, except for 26a towards HDAC3 (26a IC50 = 126.56 ± 9.04; SAHA IC50 = 158.17 ± 6.66).
3.5. Thiazolyl-coumarin derivatives in CAP group
The in vitro inhibitory activity against HDACs of thiazolyl-coumarins linked, through a C-6 alkyl spacer, to classic zinc binding groups, such as hydroxamic and carboxylic acid moieties, was evaluated [19].
In particular, compounds 32a-c and 33a-c were synthesized as shown in Scheme 6.
The first step was a Knoevenagel-type condensation between from salicyl aldehydes 27a-c and ethyl acetoacetate. After bromination of the cumarin acetyl group, the Hantzsch synthesis gave the thiazole intermediates 30a-c that was reacted with the acyl chloride derived from adipic acid methyl ester to give 31a-c. From the latter, acids 32a-c and hydroxamic acids 33a-c were obtained. Compounds 33a-c were the most active inhibitory compounds of HDACs towards HeLa cells
In addition, it has been shown that the expression and activity of distinct histone deacetylases (HDACs) are strongly correlated to the cardiac fibrosis (CF) development. In particular, HDAC1 and HDAC2 are mainly associated with the regulation of the biology of CFs in the heart; in this context, compound 33a showed significant inhibition on CFs proliferation at 1 µM and also a decrease of procollagen type I and α–smooth muscle actin (α–SMA) expression levels.
4. Five-carbon linker chain (5-C spacer)
4.1. 2-amino-1,3,4-thiadiazoles in CAP group
Compounds 34 a-d (Figure 5) were obtained as shown in Scheme 2, using in this case 7-chloro-7-oxohexanoic acid as acyl chloride [15].
The HDAC inhibitory activity evaluation of compound 34a (R = Ph, IC50 = 0.089 ± 0.005 μM), resulted better than that of SAHA (IC50 = 0.15 ± 0.02 μM), similar in cases 34c (R = PhCH2, IC50 = 0.22 ± 0.04 μM) and 34d (R = PhCH2CH2, IC50 =0.33 ± 0.05 μM), while for 34b (R = PhCH=CH) was >5μM.
Other compounds bearing 1,3,4-thiadiazole ring as the surface recognition motif, obtained with a sequence very similar to that depicted in Scheme 2, were compounds 34e-y (Figure 5) [20].
Among them, only 34g (R = 4-OCH3), 34i (R = 4-CH3), 34v (R = pyridin-3-yl), 34w (R = pyridin-4-yl), 34x (furan-2-yl), 34y (thiophen-2.yl) showed IC50 values IC50 of HDACs close to that of SAHA.
4.2. 4-Anilinothieno[2,3-d]pyrimidine derivatives in CAP group
The inhibitory activity of compounds 26c-g, synthesized as depicted in Scheme 5, was tested for HDAC1, HDAC3, and HDAC6. In all cases it was higher than that of SAHA, as reported in Table 1 [18]
4.3. Indazole nucleus in CAP group
Indazolyl compound 16c (Figure 6) was obtained through the synthetic sequence depicted in Scheme 3, when n=5 and R = 3-methoxyphenyl [16]
This compound showed activity towards HDAC1 (IC50 = 2.6 nM), HDAC2 (IC50 = 6.3 nM), and HDAC8 (IC50 = 4.5 nM) very higher than that observed for the corresponding homologues 16a and 16b, thus indicating a strong effect of the chain length in inducing biological activity.
5. Six-carbon linker chain (6-C spacer)
5.1. 2-amino-1,3,4-thiadiazoles in CAP group
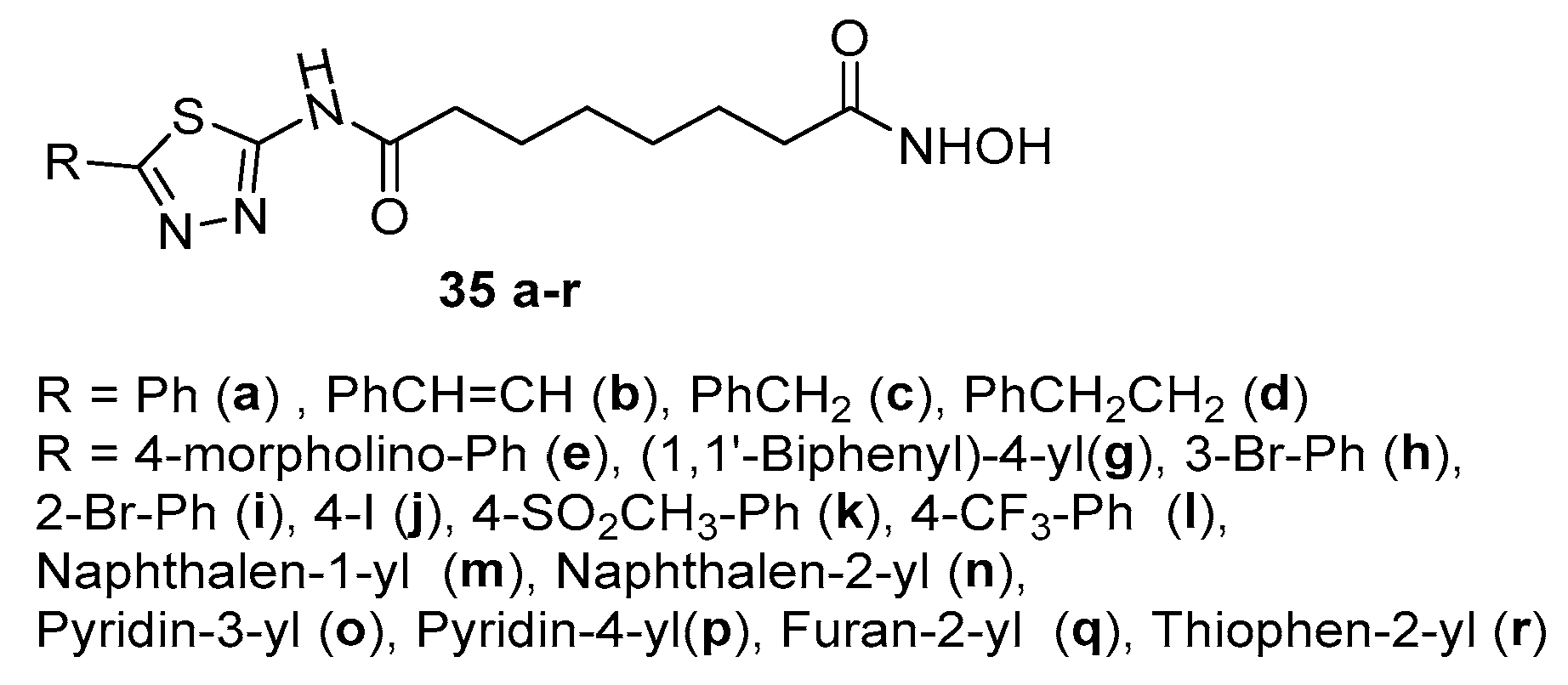
Compounds 35 a-d (Figure 7) were obtained as shown in Scheme 2, using in this case 8-chloro-8-oxohexanoic acid as acyl chloride. [15] and compounds 35 e-r were obtained in a similar manner [20].
Among them, only compounds 35o (R = pyridin-3-yl), 35p (R = pyridin-4-yl), and 35r (R = thiophen-2-yl) showed an IC50 (referred to HDACs) value lower that that found for SAHA. This recalls the behavior found for 34v, 34w and 34y, bearing the same CAP group but with a 5-C spacer, indicating the efficacy of pyridine and thiophene derivatives. Moreover, the viability of cancer cells MDA-MB-231, K562, and PC3 was measured by MTT assay for compounds 34v, 34w, 34y, 35o, 35p, and 35r. The results showed that 35r is more efficacy than SAHA on all cell lines tested, whereas 35o only on MDA-MB-231 cells.
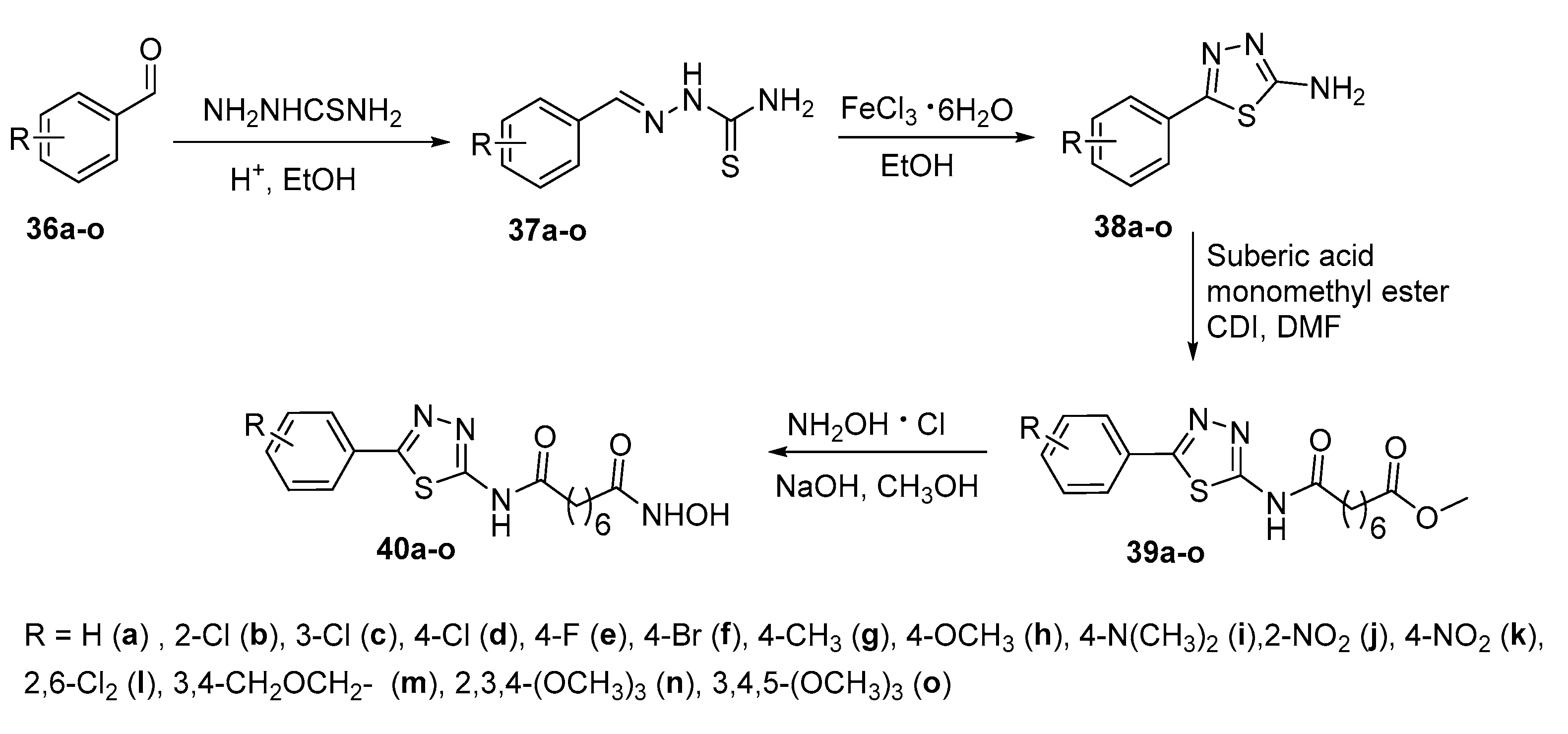
Thiadiazole derivatives 40a-o were synthesized as depicted in Scheme 7 starting from benzaldehyde 36a (or differently substituted benzaldehydes 36b-o) and thiosemicarbazide followed by cyclization to thiadiazole derivatives 38a-o. The latter were reacted with 1,10-carbodiimidazole (CDI) and suberic monomethyl ester acid to obtain derivatives 39a-o, from whose final hydroxamates 40a-o were obtained [21].
In this series, compounds 40b, 40c and 40d were found to possess potent anticancer cytotoxicity and HDAC inhibition effects. They were generally two- to five-fold more potent in terms of cytotoxicity compared to SAHA against five cancer cell lines tested (SW620, colon cancer; MCF-7, breast cancer; PC3, prostate cancer; AsPC-1, pancreatic cancer; and NCI-H460, lung cancer). Docking studies revealed that these hydroxamic acids displayed higher affinities than SAHA toward HDAC8.
5.2. Thiazoles in CAP group
Phenylthiazole-bearing hydroxamates [22] Ortho- and meta-amino-substituted phenylthiazole derivatives 41-49 were synthesized starting from commercial 4-(2-nitrophenyl)thiazol-2-ylamine and 4-(3-nitrophenyl)thiazol-2-ylamine (Scheme 8).
In the same paper, phenylthiazoles 50-55 bearing an amide or urethane residue on the benzene ring in linkage with a bulkier alkyl group have been reported. They were synthesized starting from compounds 43a and 43b through the sequence shown in Scheme 9.
The inhibitory activity of the above compounds has been tested towards HDAC1, HDAC2, HDAC3, HDAC8, HDAC10, and HDAC6. In comparison with the unsubstituted phenylthiazole analogue, the introduction of an amino group as in 45a and 45b or a glycineamide residue as in 47 do not produce significant changes in both, activity and isoform selectivity. The ortho-nitro compound 44a is almost 10-fold less potent than the corresponding amine analogue 45a. The meta-substituted ethyl carbamate 49 showed an activity against HDAC1 and HDAC2 very close to that of its amine analogue 45b, but it showed a 3-fold improvement in its HDAC6 inhibitory activity. Ongoing from the ethyl- (49) to the tert-butyl- (50b) carbamate an increase in HDAC6 inhibitory activity was observed, but no changes in inhibitory activity toward HDAC1 and HDAC2. Moreover, the introduction of a Boc protecting group leads to an enhancement in the inhibitory activity toward HDAC6 (>15-fold in 51b in comparison with 45b). Interestingly, replacement of the tert-butyloxy group of 51b by a cyclohexyl group as in 55 leads to subnanomolar potency against both HDAC2 and HDAC3 (IC50 values 200-fold increase against HDAC2 and >20-fold increase against HDAC3), while the IC50 value for HDAC6 was still below 0.2 nM. Compound 51a showed a 2-fold decrease in activity toward HDAC1 and HDAC2, with similar inhibitory potency against HDAC6 relative to the unprotected ortho-NH2 ligand 45a. Also, conversion of 51b to the pivaloyl derivative 53 produced a >10-fold decrease in HDAC6 inhibition. Inhibitory data of compound 44b have been also reported in a previous work [23].
Compounds 44a, 45a, 45b, and 49 have also been tested towards five pancreatic cancer cell lines and their antiproliferative activity was compared with that of SAHA and showed similar or improved potencies relative to SAHA. Among them, the meta-amino-substituted phenylthiazole 45b gave the best IC50 value against the Mia Paca-2 cell line (IC50=10 nM), while its carbamate analogue 49 showed the best overall inhibitory activity against all five pancreatic cancer cell lines.
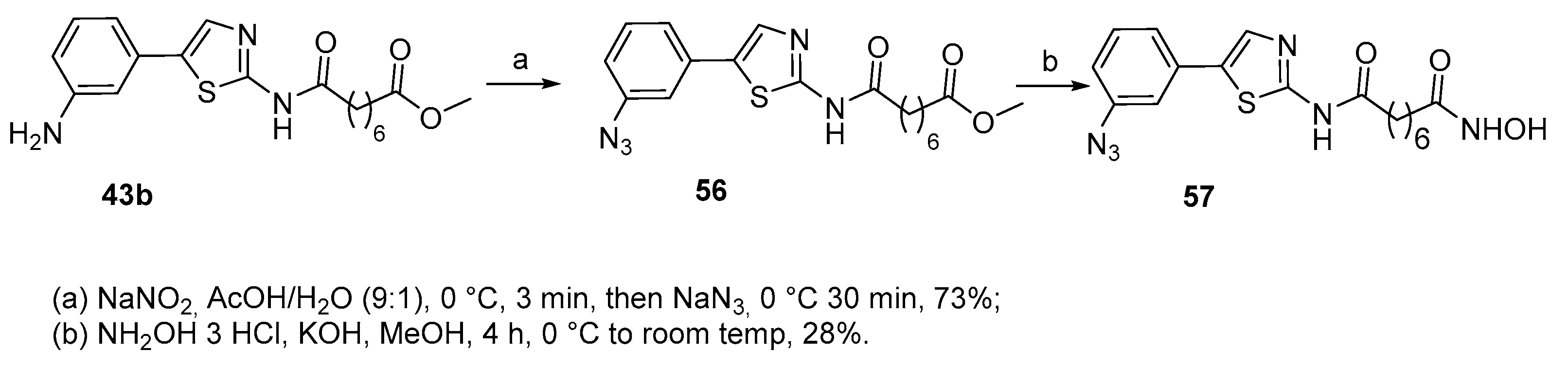
In another study [24] the phenylthiazole-based probe 57, with an azide group on the phenyl ring, was designed to mimic the scaffold of SAHA. The synthesis is shown in Scheme 10.
Compound 57 resulted 6.1-fold less active in inhibiting HDAC8 compared to SAHA and this behavior has been attributed to an increase of the lipophilic nature of the solvent exposed surface binding group that influences to the overall binding affinity.
Finally, hydroxamic derivatives bearing both unsubstituted- and p-N-pyrrolidinyl-substituted phenylthiazole amide functionality showed better HDAC potency and cellular activity (towards HT1080 and MDA435 cells) with respect to SAHA [25].
5.3. Pyrazole nucleus in CAP group
HDACi bearing pyrazole and isoxazole derivatives in CAP group have been synthesized and studied by Petukhov et al. [26,27].
In particular, compounds with pyrazole nucleus 61, 67 and 68a-d have been synthesized according to Scheme 11, starting from commercially available 4-nitropyrazole (58). The synthetic strategy involves as first step an amide coupling between 4-aminopyrazole, obtained from 58 through hydrogenolysis, and monomethyl suberate to give (59). Alkylation of 59 with toluene-4-sulfonic acid 3-azido-5-azidomethylbenzyl ester in the presence of K2CO3 afforded compound 60. Compounds 62 and 63 were obtained by alkylation of 59 with benzyl bromide or 4-nitrobenzyl bromide in the presence of NaH in DMF, respectively. Reduction of the nitro group of 63 gave aniline 64, a key intermediate for compounds 65 and 66. Diazotization of amino group of the aniline derivative 64 followed by azide displacement reaction with NaN3 gave the corresponding azido compound 65. Treatment of 64 with Boc anhydride furnished the carbamate 66. Compound 67 was obtained by an amide coupling between 64 and 3-azido-5-azidomethylbenzoic acid followed by treatment with KOH/NH2OH in MeOH. The same treatment on the methyl esters 60, 62, 63, 65, and 66 gave the corresponding hydroxamates 61 and 68a-d, respectively.
Pyrazoles 61, 67, and 68a-d were tested for inhibition of HDAC3 and HDAC8 isoforms. The inhibition of HDAC8 was measured using the fluorescent acetylated HDAC substrate Fluor de Lys and the commercially available recombinant human HDAC8, whereas the inhibition of HDAC3 was measured using the fluorescent HDAC substrate Boc-L-Lys(Ac)-AMC and the commercial recombinant human HDAC3/NCoR2.
The results are summarized in Table 2.
The simplest benzyl substituted pyrazole 68a inhibited HDAC3 and HDAC8 with IC50s of 44 and 76 nM, respectively. Introduction of a nitro group at the 4-position of the benzyl group of 68a gave compound 68b that showed slightly lower activity for both isoforms, whereas the corresponding azido compound 68c exhibited a 2.0- and 2.7-fold better potency, being its IC50 values 22 and 28 nM for HDAC3 and HDAC8, respectively. Overall, compounds 68a-68c exhibited an inhibitory activity against HDAC3 comparable to that of SAHA but a better double digit nanomolar activity against HDAC8. Introduction of a bulky Boc-protected amino group in 68d decreased the HDAC activity by about 10-fold. Replacement of the Boc group with a lipophilic aromatic diazide as in 67 further decreased the activity for both HDAC3 and HDAC8 to 432 and 487 nM, respectively. Comparison of the activity data of 68b-68c with 68d and 67 shows that the presence of the bulky substituent in the para position of the terminal phenyl ring leads to the lower activities for both HDAC3 and HDAC8 isoforms. The replacement of the phenyl group with a 3-azido-5-azidomethyl phenyl group, resulting in 61 revealed that this compound was 8-fold more active toward HDAC8 than for HDAC3, with IC50s equal to 17 and 128 nM, respectively. The activity of the methyl ester 60 towards HDAC8 was 36.0 ± 2.20 μM [27].
Compound 61, also called SAHA diazide, was also tested against HDAC1 and HDAC4; compared with the activity of SAHA (Ki = 0.051 and >30 μM for HDAC1 and HDAC4, respectively), Ki values for 61 were Ki = 0.14 and 13.05 μM for HDAC1 and HDAC4, respectively.
5.4. Pyridine and pyrimidine nucleus in CAP group
The synthesis and biological activity of compounds 69 a-c, bearing a pyridinyl substituent in the CAP group (Figure 8) have been reported, but their activity towards HDAC1 was much lower than that of SAHA [29].
Similar behavior was also found for the pyrimidine derivative 70.
The more simple pyridinyl derivatives 71a-c (Figure 8) were profiled partially purified HDAC enzyme obtained from H1299 cell lysate, in antiproliferative assays (towards H1299 and HCT116) and in a p21 promoter induction assay. [30]
In these cases, the activity towards enzyme was comparable to that of SAHA. The 2-pyridyl isomer 71a was essentially equipotent to SAHA in the promoter assay, but 3-fold less potent in HCT116 growth inhibition, and >10 μM in H1299 growth inhibition. The 3- and 4-pyridyl isomers 71b and 71c were less potent than SAHA. The difference in cellular activity of these positional isomers has been hypothesized due to differences in cellular permeability or intracellular metabolism of the compounds.
5.5. Thienopyrimidine nucleus in CAP group
The biological activity of thienopyrimidine derivatives 26h-y, synthesized as reported in Ref. 18 and depicted in above Scheme 5, have been tested as inhibitors of HDAC1, HDAC3, and HDAC6, and of proliferation of RMPI8226 and HCT-116 cancer cells. In all cases the activity found was comparable with that of SAHA.
In the same paper also the biological activity of compound 72 (Figure 9) was tested and the results showed poor inhibitory activity in many cases, suggesting that the presence of the 4-aniline fragment could increase the lipophilic interaction with HDACs to induce good inhibitory activities against them.
The above cited paper was followed by a second [31], only focused on the C-6 spacer, in which the fifteen novel compounds 76 a-o, bearing the thienopyrimidine fragment on the CAP group were synthesized from methyl 3-aminothiophene-2-carboxylate that, after cyclization with formamidine acetate under microwave conditions, gave the thieno[3,2-d]pyrimidin-4(3H)-one (20) in similar conditions to those already reported in Scheme 5. The latter was subjected to nitration and subsequent chlorination then coupled with a series of anilines to give compounds 73a-o. Reduction of nitro group to amino group afforded the key precursors 74 a-o. After treatment with acyl chlorides amides 75a-o were obtained. Lastly, the target products 76 a-o were obtained after reaction with hydroxylamine hydrochloride (Scheme 12).
The ability of compounds 76 a-o to inhibit recombinant human HDAC1, HDAC3, and HDAC6 isoforms and ‘in vitro’ activity against cancer cell lines RMPI 8226 and HCT 116 was tested. Most of them displayed good inhibitory and anticancer activities, particularly compound 76j that showed IC50 values (29.81 ± 0.52 nM, 24.71 ± 1.16 nM, and 21.29 ± 0.32 nM for HDAC1, HDAC3, and HDAC6, respectively), much lower than those found for SAHA (195.00 ± 16.12 181.05 ± 28.92 105.10 ± 25.46). Moreover, the IC50 values of compound 76j against RPMI 8226 and HCT 116 proliferation were 0.97 ± 0.072 mM and 1.01 ± 0.033 mM, respectively, and it up-regulated the level of histone H3 acetylation at the concentration of 0.3 mM.
5.6. Indazole nucleus in CAP group
In Figure 10 are shown indazolyl derivatives 16d-s, synthesized through the approach above depicted in Scheme 3 [16].
Among compounds 16d-s, compounds 16n and 16p emerged as excellent inhibitors of HDAC1 (IC50 = 2.7 nM and IC50 = 3.1 nM), HDAC2 (IC50 = 4.2 nM and IC50 = 3.6 nM) and HDAC8 (IC50 = 3.6 nM and IC50 = 3.3 nM). Antiproliferation assays revealed that these compounds also showed anti-proliferative activities against HCT-116 and HeLa cells better than SAHA. Moreover, compounds 16n and 16p up-regulated the level of acetylated α-tubulin and histone H3 and promote cell apoptosis.
According to a similar synthetic route similar to that of Scheme 3, 1H-pyrazolo [3,4-b] pyridine derivatives 77a-b (Figure 11), bioisosters of compounds 16e and 16n, respectively, were obtained from 2,6-dichloronicotinonitrile through a multistep sequence [16].
The inhibitory activities of 77a and 77b towards HDACs slightly decreased indicating that the presence of the 6-phenyl-1H-indazole scaffold is important to affect the biological activity.
5.7. Benzothiazole moiety in CAP group
Compounds 78a-f (Figure 12) were obtained from 2-aminobenzothiazole derivatives with the sequence depicted in Scheme 4, with the difference to use suberic acid monomethyl ester instead of adipic acid monomethyl ester [17].
It was observed that several compounds showed good inhibition against HDAC3 and HDAC4. The amount of enhanced acetylation of histone-H3 and -H4 in SW620 cells by 78a–c, and 78f was similar to that found for SAHA.
Moreover, all six compounds displayed cytotoxicity against five cancer cell lines (SW620, colon cancer; MCF-7, breast cancer; PC3, prostate cancer; AsPC-1, pancreatic cancer; NCI-H460, lung cancer), with average IC50 values range from 0.59 to 11.08μg/mL
Homologues 4C-bridged compounds showed slight or no increase in histone acetylation, suggesting that the linker length between the benzothiazol and hydroxamic moieties required for good HDAC inhibition of this compound series was similar to that of SAHA. In addition, the size of the 6-substituents on the benzene ring rather than their electronic effects was important for HDAC binding, for example, 78d and 78e bearing relatively larger substituents (–OC2H5 and –SO2CH3) compared to the other compounds in the series did not inhibit HDAC activity. Actually, compounds 78c (bearing –OCH3, an electron-donating group) and 78f (bearing –NO2, an electron-withdrawing group) showed similar HDAC inhibitor power and were almost equally cytotoxic.
5.8. Benzoxazole moiety in CAP group
From the reaction between 2-aminobenzoxazole and suberic acid monomethyl ester and subsequent transformation of the methyl ester to hydroxamic group, compound 80, that can be considered a bioisoster of 78a, was obtained [32].
Scheme 13.
Benzoxazole analogue of SAHA from 2-aminobenzoxazole.
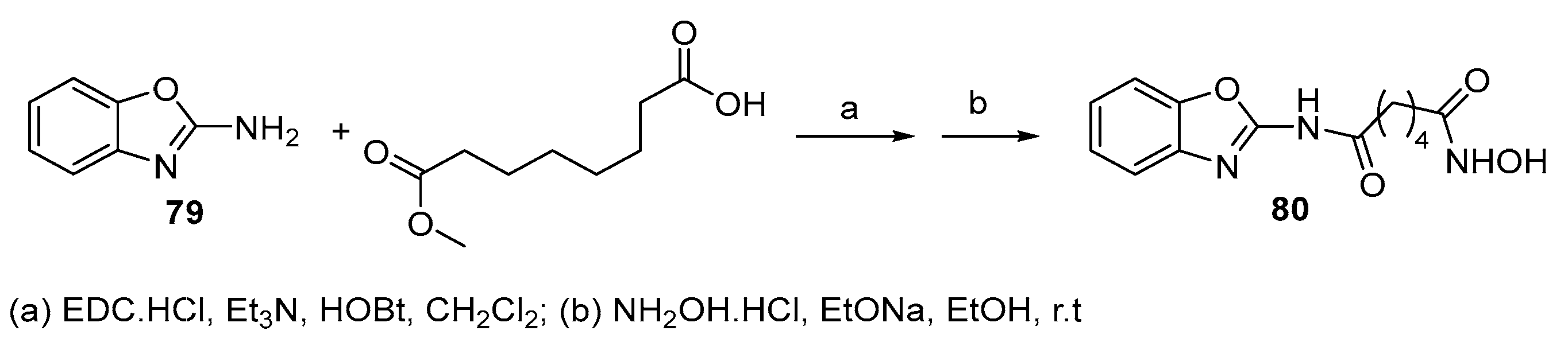
Compound 80 was an inhibitor of human HDAC1, HDAC2 more potent than Vorinostat and comparable as an inhibitor of HDAC6. It was a slightly more potent inhibitor than Vorinostat of the growth of A549, Caco-2 and SF268 cells and was chosen for further studies against two colon cancer cell lines HCT116 GNAS R201C/+ and LS174T cells, that genetically resemble PMP tumor cells and it proved to be a more potent antiproliferative compound than Vorinostat in both cases.
5.9. Isoquinoline moiety in CAP group
Novel HDACi bearing isoquinoline fragment in CAP group have been synthesized starting from 2-methyl-5-nitrobenzoic acid (81) [33]. After esterification to 82 followed by treatment with DMA-DMF and cyclisation with 3,4-dimethoxylbenzylamine intermediate 84 was obtained. The latter was deprotected to 85 then chlorinated to 86 which was coupled with a series of anilines to generate compounds 87. Reduction of the nitro group followed by reaction with 8-methoxy-8-oxooctanoic acid afforded the amides 89 which, after treatment with freshly prepared hydroxylamine gave compounds 90a–h. (Scheme 14) [33].
Compounds 90a-h were tested against HDAC1, HDAC3, and HDAC6 and all showed better activity than SAHA, used as positive control. The best active compound resulted to be 90c, showing IC50 values 4.17 nM, 4.00 nM and 3.77 nM against HDAC1, HDAC3, and HDAC6, respectively. Furthermore, the antiproliferative activity of compounds 90a-h against multiple myeloma cell line RPMI 8226 was tested and the more active were 90a, 90f, 90g with IC50 values 0.46 μM, 0.52 μM and 0.47 μM, respectively.
When intermediate 86 was reacted with aliphatic amines under microwaves conditions, after reduction of the nitro group to amino group and subsequent treatment as reported in steps h and i of Scheme 14, isoquinolines 91a-d (Figure 13) were obtained.
Compound 91a with a large substituent at C-1 position of the isoquinoline ring significantly decreased with respect to 91b-d the inhibitory activities against HDACs as well as the proliferation of RPMI 8226 cells. Compounds 91b–d displayed similar enzymatic activities suggesting that small aliphatic amines at the C-1 position do not significantly affect the inhibitory activities against HDACs enzyme in vitro and the proliferation of the cancer cells with respect to compounds 90a-h, bearing an aromatic substituent at C-1 position.
Finally, to test the effects of spatial orientation of the N-substituents, compound 83 depicted in Scheme 14 was reacted with different aliphatic amines in toluene at 110 °C and the obtained intermediate subjected to steps g, h, and i (reported Scheme 14), thus obtaining isoquinoline-1(2H)-one derivatives 92a-d (Figure 14)
The inhibitory activity of the series 92a-d towards HDAC1, 3, 6 isoforms and cancer cell proliferation were evaluated. These compounds exhibited weaker inhibitory activities against HDACs indicating that the binding affinity between N-substituent isoquinoline-1-one scaffold and HDACs surface was decreased with respect to 91a-d series.
In a paper focused on the study of the influence of substitution of the phenyl SAHA capping group with various substituents [34], two compounds bearing heterocyclic rings have been reported, one (93) with a isoquinolinyl group and the other (94)with a pyrimidin-2(1H)-one moiety (Figure 15) but both displayed a very weak antiproliferative and histone deacetylation activities.
5.10. Quinazoline moiety in CAP group
Taking into account the known role of hydroxamic acids as HDAC inhibitors and that of quinazolines as EGFR/HER2 inhibitors, some authors designed to synthesize compounds bearing both functionalities, in order to find efficient multitarget inhibitors [35]. Thus, among various compounds, they prepared quinazoline derivative 101, starting from 95 with the multistep procedure depicted in Scheme 15.
The HDAC inhibitory activity of the quinazoline SAHA analogue 101 was determined using the Biomol Color de Lys system and the IC50 value was 15.3 nM. This compound showed to possess also EGFR and HER2 kinase activity.
6. Seven-carbon linker chain (7-C spacer)
6.1. Pyridine and pyrimidine moiety in CAP group
A series of compounds bearing pyridine or pyrimidine moiety bound to an azelayl scaffold through Schotten-Bauman-like reaction was synthesized as reported in Scheme 16 [36]. The series was subjected to biological screening on a panel of tumor cell lines: noticeably, none of the compounds induced cytotoxicity in the normal fibroblast cell line, while only osteosarcoma cells (U2OS) appeared to be sensitive to compound 106a.
Compound 106a was studied ‘in silico’, by using histone deacetylases as molecular target, which revealed that it is able to interact with HDAC 7, in agreement with studies which have disclosed an unexpected function for HDAC7 in osteoclasts.
Author Contributions
Conceptualization, C.B. and G.M.; methodology, N.C.; formal analysis, S.B.; data curation, G.D.; writing—original draft preparation, C.B. and G.M.; writing—review and editing, S.B., G.D. and N.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8-21. [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and Co-regulates major cellular functions. Science 2009, 325, 834e840. [CrossRef]
- Vaijayanthi, T.; Pandian, G.N.; Sugiyama, H. Chemical control system of epigenetics. Chem. Rec. 2018, 18, 1833e1853. [CrossRef]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737-749. [CrossRef]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone Deacetylase Inhibitors J. Cell. BioChem. 2005, 96, 293-304. [CrossRef]
- Glaser, K. B. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659-671. [CrossRef]
- Langley, B.; D’Annibale, M.A.; Suh, K.; Ayoub, I.; Tolhurst, A.; Bastan, B.; Yang, L.; Ko, B.; Fisher, M.; Cho, S.; Beal, M.F.; Ratan, R.R. Pulse Inhibition of Histone Deacetylases Induces Complete Resistance to Oxidative Death in Cortical Neurons without Toxicity and Reveals a Role for Cytoplasmic p21waf1/cip1 in Cell Cycle-Independent Neuroprotection. J. Neurosci. 2008, 28, 163-176. [CrossRef]
- Sinn, D.I.; Kim, S.J.; Chu, K.; Jung, K.H.; Lee, S.T.; Song, E.C.; Kim, J.M.; Park, D.K.; Kun Lee, S.; Kim, M.; Roh, J.K. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol. Dis. 2007, 26, 464-472. [CrossRef]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40-49. [CrossRef]
- Hahnen, E.; Hauke, J.; Trankle, C.; Eyupoglu, I.Y.; Wirth, B.; Blumcke, I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders Expert Opin. Invest. Drugs 2008, 17, 169-184. [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [CrossRef]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. Mechanisms and molecular probes of sirtuins. Chem. Biol. 2008, 15, 1002–1013. [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Nature 1999, 401, 188-193. [CrossRef]
- Guan, P.; Sun, F.; Hou, X.; Wang, F.; Yi, F.; Xu, W.; Fang, H. Design, synthesis and preliminary bioactivity studies of 1,3,4-thiadiazole hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem., 2012, 20, 3865-3872. [CrossRef]
- Liu, J.; Zhou, J.; He, F.; Gao, L.; Wen, Y.; Gao, L.; Wang, P.; Kang, D.; Hu, L. Design, synthesis and biological evaluation of novel indazole-based derivatives as potent HDAC inhibitors via fragment-based virtual screening. Eur. J. Med. Chem. 2020, 192, 112189. [CrossRef]
- Oanh, D.T.K.; Hai, H.V.; Park, S.H.; Kim, H.-J.; Han, B.-W.; Kim, H.-S.; Hong, J.-T.; Han, S.-B.; Hue, V.T.M.; Nam, N.-H.. Benzothiazole-containing hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorg.Med. Chem. Lett. 2011, 21, 7509–7512. [CrossRef]
- Yang, W.; Li, L.; Ji, X.; Wu, X.; Su, M.; Sheng, L.; Zang, Y.; Li, J.; Liu, H. Design, synthesis and biological evaluation of 4-anilinothieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem. 2014, 22, 6146–6155. [CrossRef]
- Pardo-Jiménez, V.; Navarrete-Encina, P.; Díaz-Araya, G. Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity. Molecules 2019, 24, 739; [CrossRef]
- Guan, P.; Wang, L.; Hou, X.; Wan, Y.; Xu, W.; Tang, W.; Fang, H. Improved antiproliferative activity of 1,3,4-thiadiazole-containing histone deacetylase (HDAC) inhibitors by introduction of the heteroaromatic surface recognition motif. Bioorg. Med. Chem. 2014, 22, 5766–5775. [CrossRef]
- Nam, N.-H.; Huong, T.L.; Dung, D.T.M.; Dung, P.T.P.; Oanh, D.T.K.; Park, S.H.; Kim, K.; Han, B.W.; Yun, J.; Kang, J.S.; Kim, Y.; Han, S.-B. Synthesis, bioevaluation and docking study of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. J. Enzyme Inhib. Med. Chem. 2014; 29, 611-618. [CrossRef]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.M.; Savoy, D.N.; Billadeau, D.D.; Kim, K.H. Chemistry, Biology, and QSAR Studies of Substituted Biaryl Hydroxamates and Mercaptoacetamides as HDAC Inhibitors—Nanomolar-Potency Inhibitors of Pancreatic Cancer Cell Growth. ChemMedChem 2008, 3, 487–501. [CrossRef]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D'Annibale, M.A.; Suto, C.M.; Brett C. Langley, B.C. Functional Differences in Epigenetic Modulators Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies J. Med. Chem. 2007, 50, 3054-3061. [CrossRef]
- He, B.; Velaparthi, S.; Pieffet, G.; Pennington, C.; Mahesh, A.; Holzle, D.L.; Brunsteiner, M.; van Breemen, R.; Blond, S.Y.; Petukhov, P.A. Binding Ensemble Profiling with Photoaffinity Labeling (BEProFL) Approach: Mapping the Binding Poses of HDAC8 Inhibitors. J. Med. Chem. 2009, 52, 7003–7013. [CrossRef]
- Glaser, K.B.; Li, J.; Pease, L.J.; Staver, M.J.; Marcotte, P.A.; Guo, J.; Frey, R.R.; Garland, R.B.; Heyman, H.R.; Wada, C.K.; Vasudevan, A.; Michaelides, M.R.; Davidsen, S.K.; Curtin, M.L. Differential protein acetylation induced by novel histone deacetylase inhibitors. Biochem Biophys Res Commun. 2004, 325, 683–690. [CrossRef]
- Neelarapu, R.; Holzle, D.L.; Velaparthi, S.; Bai, H.; Brunsteiner, M.; Blond, S.Y.; Petukhov, P.A. Design, Synthesis, Docking, and Biological Evaluation of Novel Diazide-Containing Isoxazole-and Pyrazole-Based Histone Deacetylase Probes. J. Med. Chem. 2011, 54, 4350–4364. [CrossRef]
- Vaidya, A.S.; Neelarapu, R.; Madriaga, A.; Bai, H.; Mendonca, E.; Abdelkarim, H.; van Breemen, R.B.; Blond, S.Y.; Petukhov, P.A. Novel histone deacetylase 8 ligands without a zinc chelating group: Exploring an 'upside-down' binding pose. Bioorg. Med. Chem. Lett. 2012, 22, 6621–6627. [CrossRef]
- Albrow, V.E.; Grimley, R.L.; Clulow, J.; Rose, C.R.; Sun, J.; Warmus, J.S.; Tate, E.W.; Jonesd, L.H.; Storer, R.I.. Design and development of histone deacetylase (HDAC) chemical probes for cell-based profiling. Mol. BioSyst., 2016, 12, 1781—1789. [CrossRef]
- Li, Y.; Luo, X.; Guo, Q.; Nie, Y.; Wang, T.; Zhang, C.; Huang, Z.; Wang, X.; Liu, Y.; Chen, Y.; Zheng, J.; Yang, S.; Yan Fan, Y.; Xiang, R. Discovery of N1-(4-((7-Cyclopentyl-6-(dimethylcarbamoyl)-7 H-pyrrolo[2,3- d]pyrimidin-2-yl)amino)phenyl)- N8-hydroxyoctanediamide as a Novel Inhibitor Targeting Cyclin-dependent Kinase 4/9 (CDK4/9) and Histone Deacetlyase1 (HDAC1) against Malignant Cancer. J. Med. Chem. 2018, 61, 3166-3192. [CrossRef]
- Remiszewski, S.W:, Sambucetti, L.C.; Atadja, P.; Bair, K.W.; Cornell, W.D.; Green, M.A.; Howell, K.L.; Jung, M.; Kwon, P.; Trogani, N.; Walker, H. Inhibitors of Human Histone Deacetylase: Synthesis and Enzyme and Cellular Activity of Straight Chain Hydroxamates. J. Med. Chem. 2002, 45, 753–757. [CrossRef]
- Wang, J.; Su, M.; Li, T.; Gao, A.; Yang, W.; Sheng, L.; Zang, Y.; Li, J.; Liu, H. Design, synthesis and biological evaluation of thienopyrimidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Eur. J. Med. Chem. 2017, 128, 293-299. [CrossRef]
- Mantzourani, C.; Gkikas, D.; Kokotos, A.; Nummela, P.; Theodoropoulou, M.A.; Wu, K.-C.; Fairlie, D.P.; Politis, P.K.; Ristimäki, A.; Kokotos, G. Synthesis of benzoxazole-based vorinostat analogs and their antiproliferative activity. Bioorg. Chem. 2021, 114, 105132. [CrossRef]
- Yang, W.; Li, L.; Wang, Y.; Wu, X.; Li, T.; Yang, N.; Su, M.; Sheng, L.; Zheng, M.; Zang, Y.; Li, J.; Liu, H. Design, synthesis and biological evaluation of isoquinoline-based derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem. 2015, 23, 5881-5890. [CrossRef]
- Salmi-Smail, C.; Fabre, A.; Dequiedt, F.; Restouin, A.; Castellano, R.; Garbit, S.; Roche, P.; Morelli, X.; Brunel, J.M.; Collette, Y. Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J. Med. Chem. 2010, 53, 3038–3047. [CrossRef]
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [CrossRef]
- Boga, C.; Micheletti, G. Design and Synthesis of Organic Molecules as Antineoplastic Agents. Molecules 2020, 25, 2808. [CrossRef]
Figure 1.
Structure of SAHA with indication of his specific CAP-LINKER-ZBG framework.
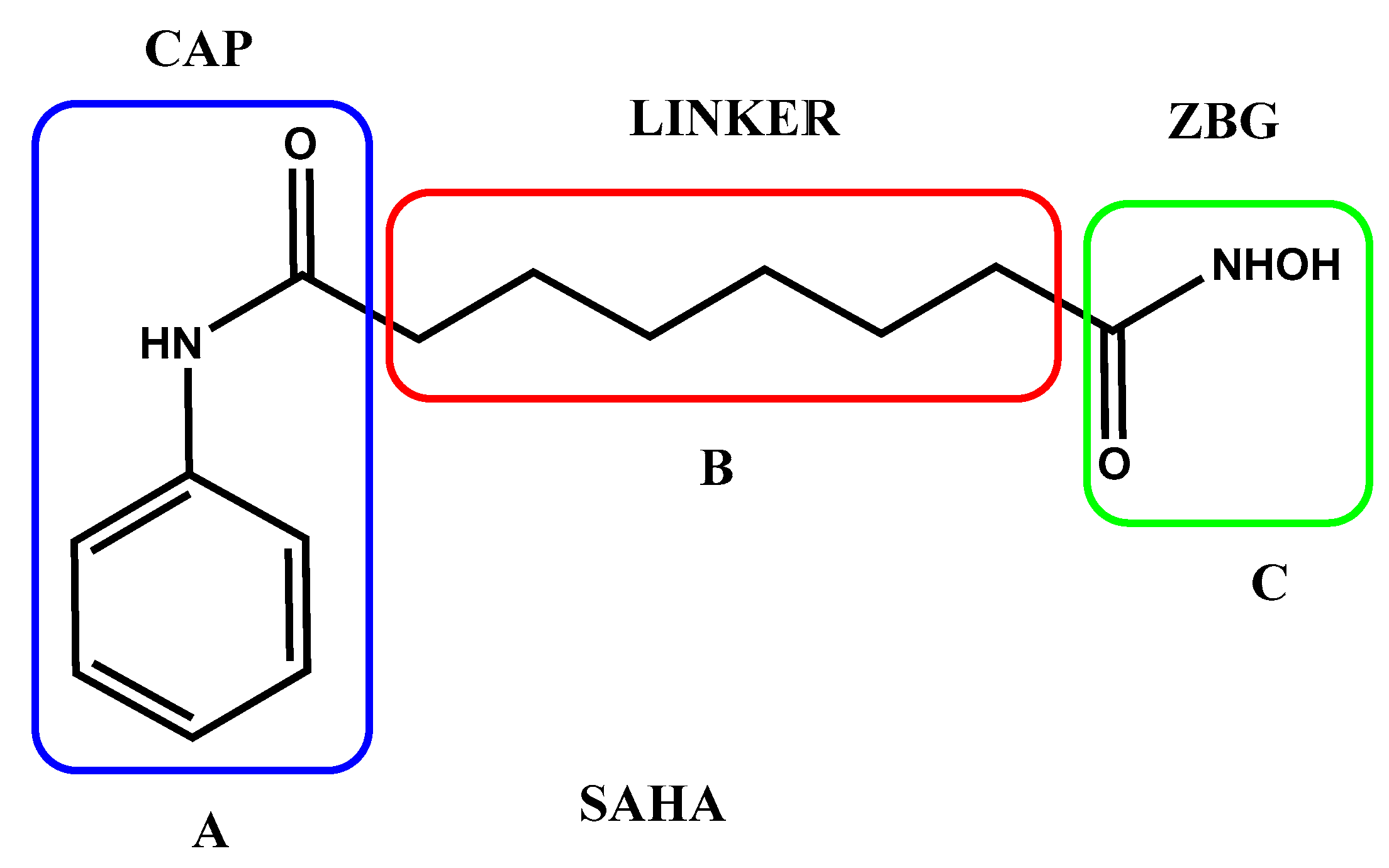
Figure 2.
Structure of SAHA analogues HDACi reported in this review.
Scheme 1.
Synthetic pathway to 1,3,4-thiadiazole derivatives 4a-b.
Scheme 2.
Synthetic route for preparation of compounds 6a-d.
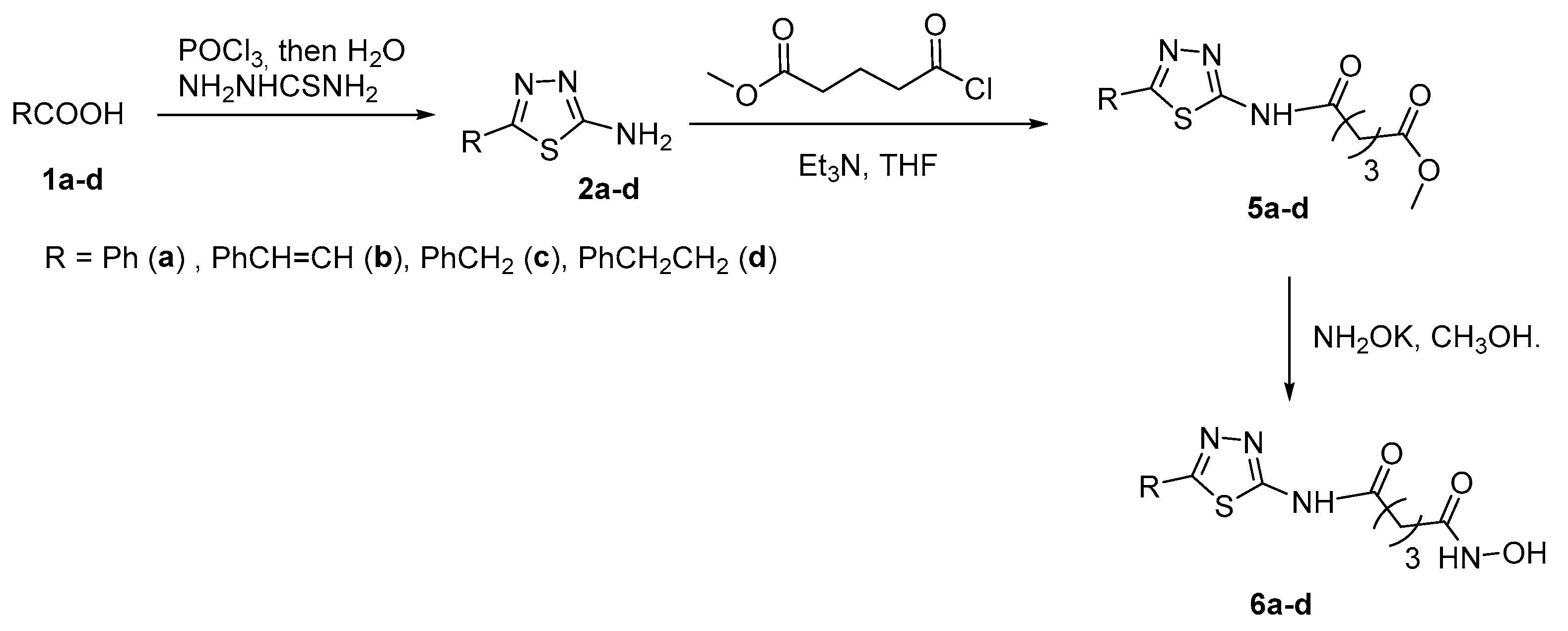
Scheme 3.
Preparation of indazole-based series 16a-s.
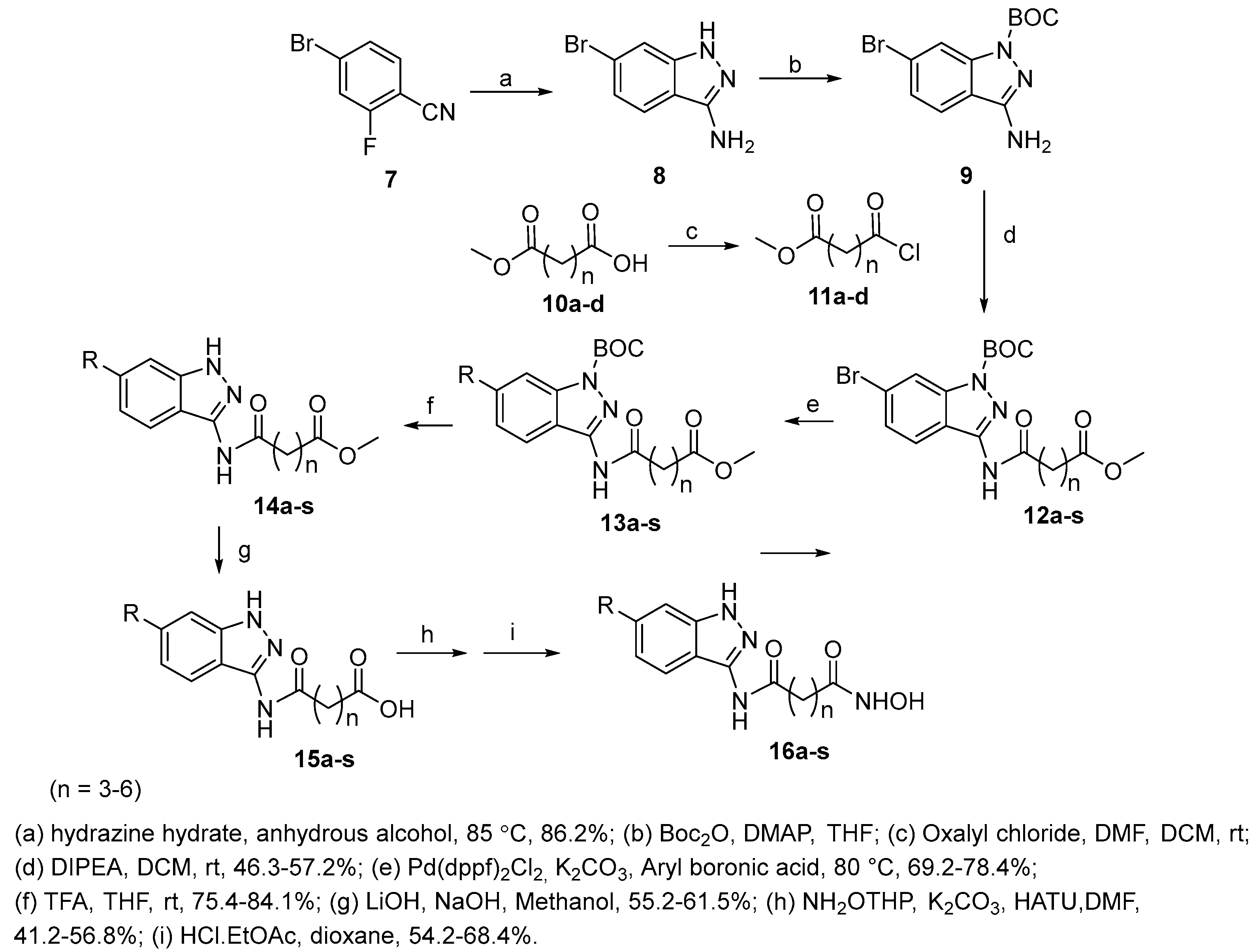
Figure 3.
Thiadiazole hydroxamic acids with a four-methylene chain.
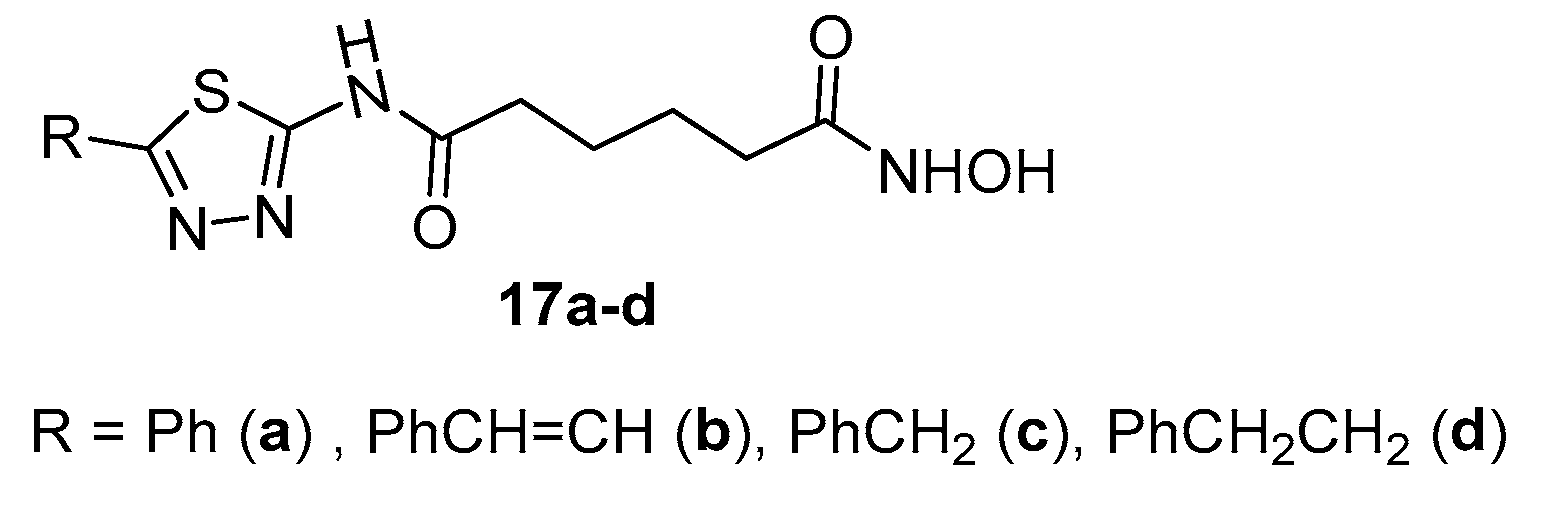
Figure 4.
Indazole derivative 16b.
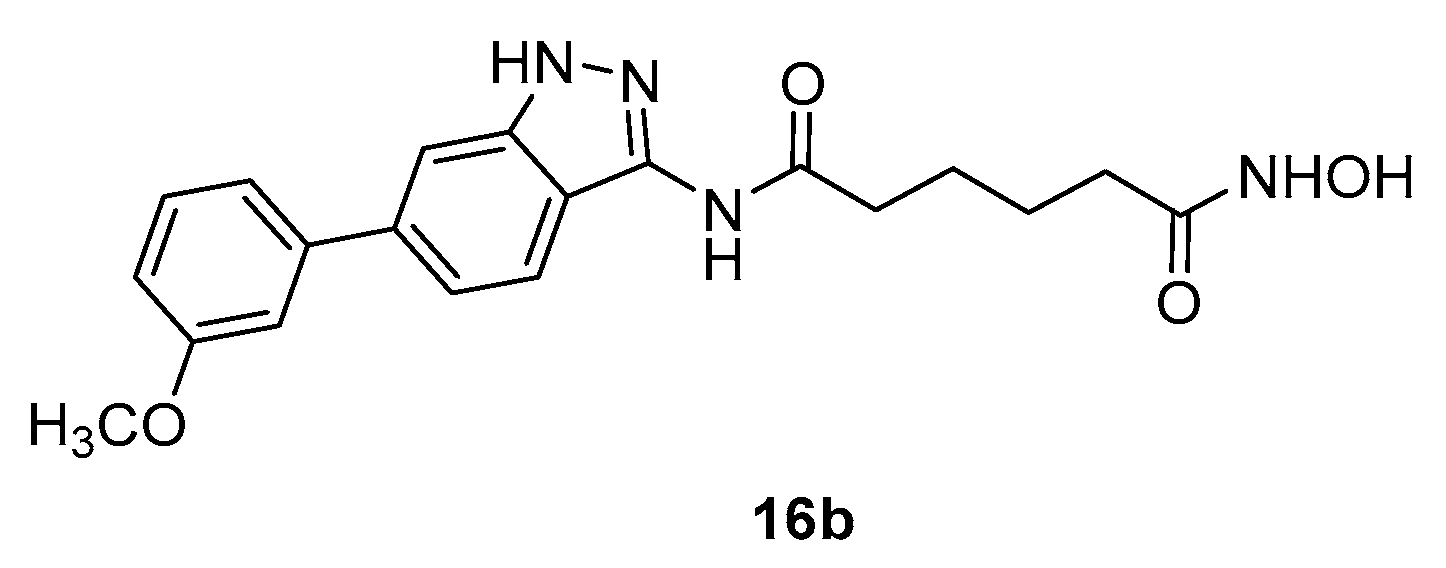
Scheme 4.
Synthesis of benzothiazolyl analogues of SAHA with C-4 methylene linker.
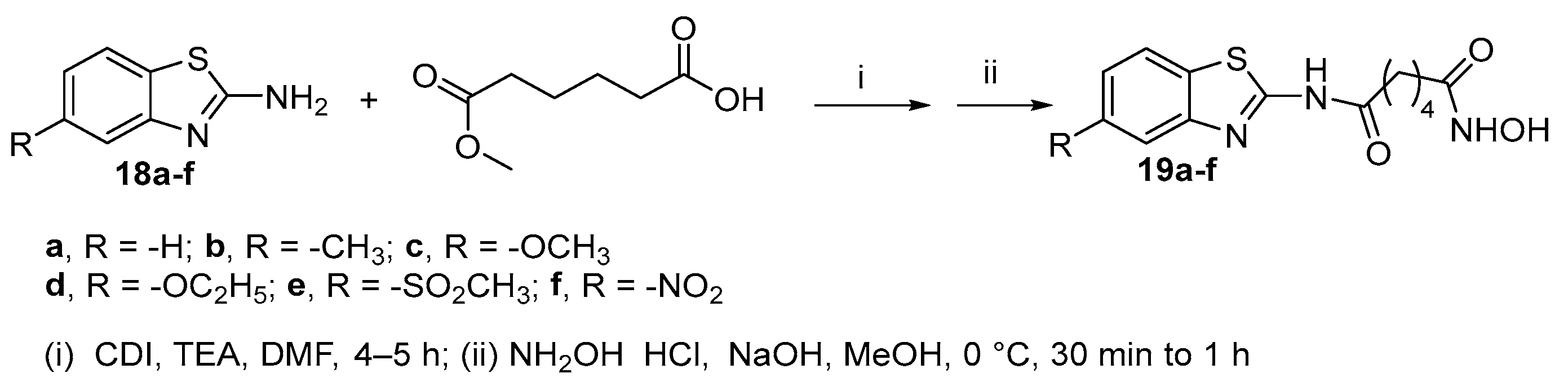
Scheme 5.
Synthetic approach to thieno[2,3- d]pyrimidine-based HDACs inhibitors with different length of the spacer.
Scheme 5.
Synthetic approach to thieno[2,3- d]pyrimidine-based HDACs inhibitors with different length of the spacer.
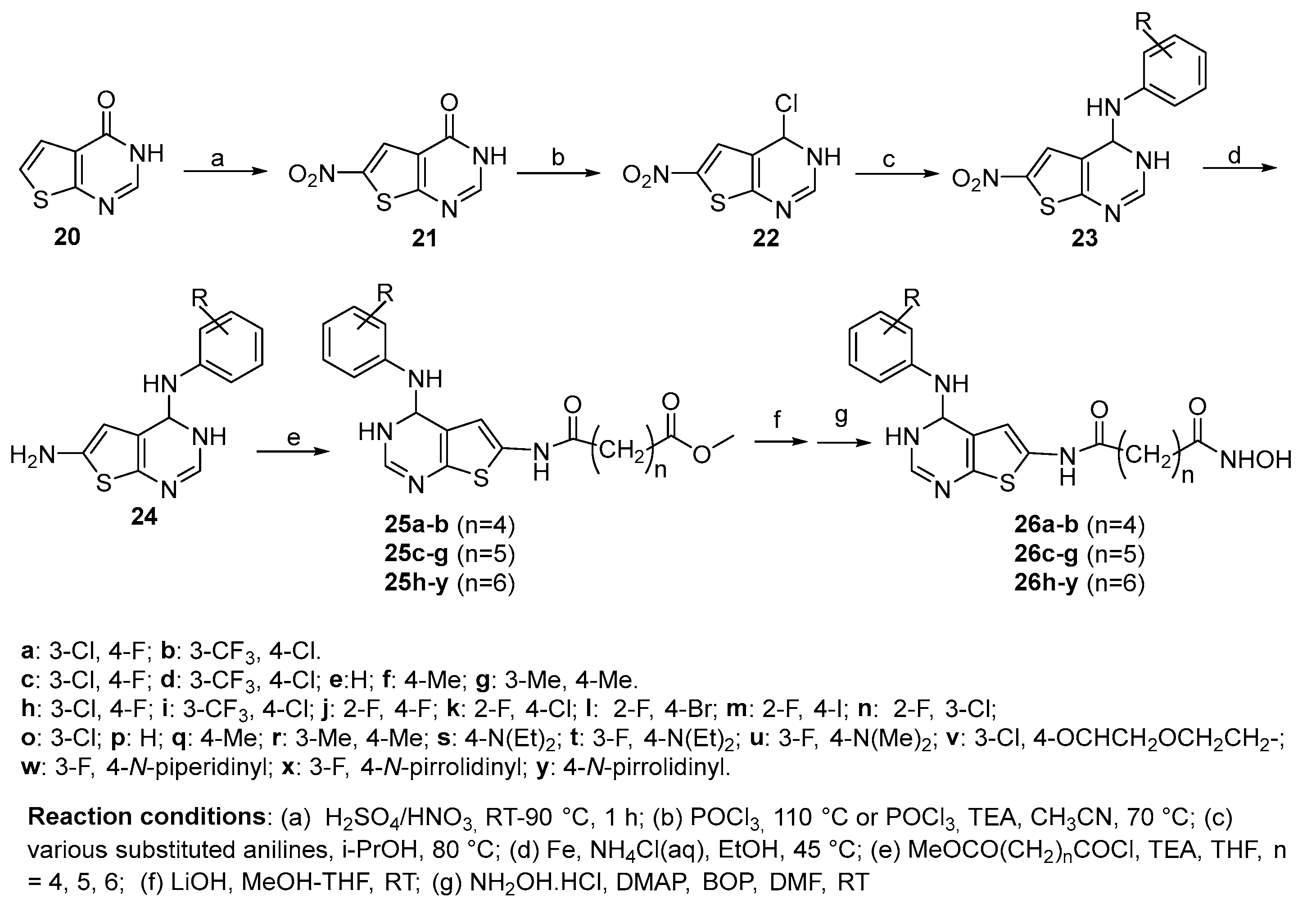
Scheme 6.
Synthetic route to thiazolyl derivatives 32 and 33.
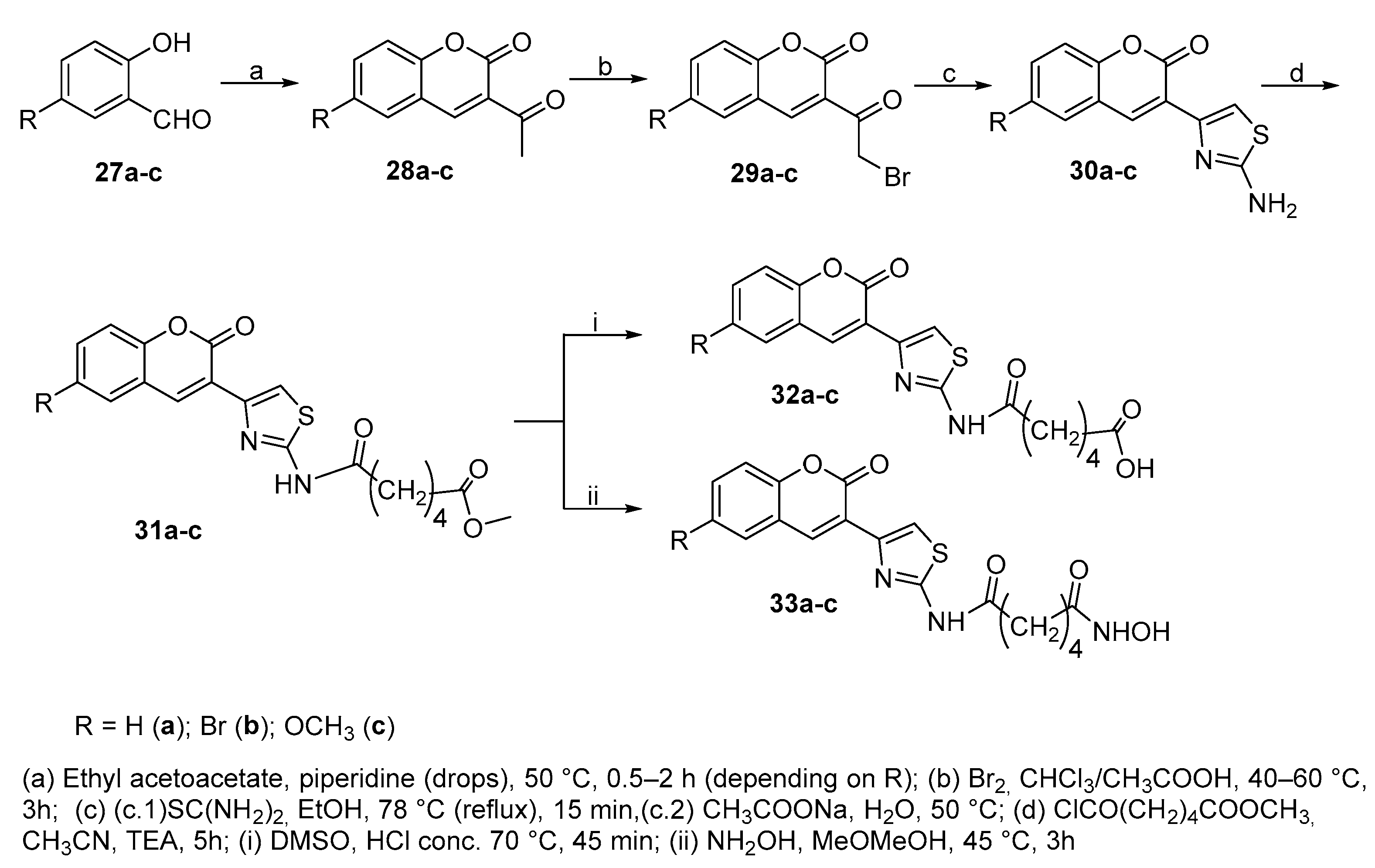
Figure 5.
2-Amino-1,3,4-thiadiazoles connected to a C-5 spacer through amide bond.
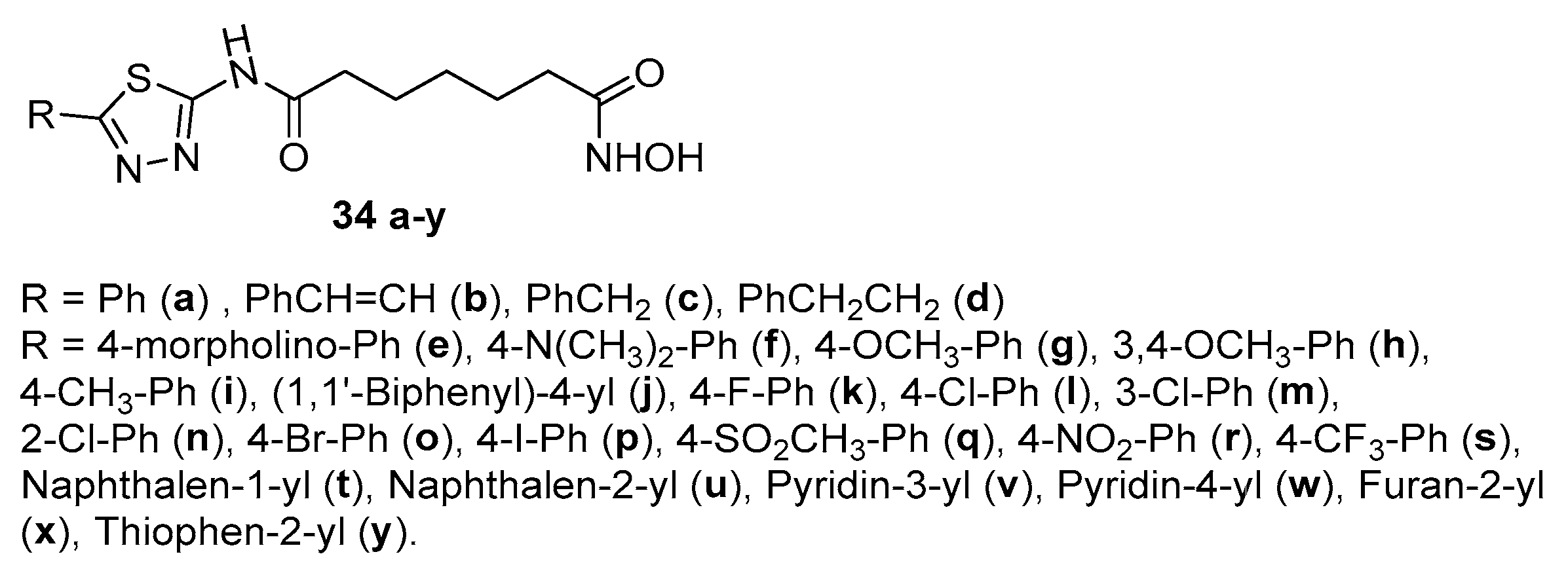
Figure 6.
Indazolyl SAHA analogue 16c.
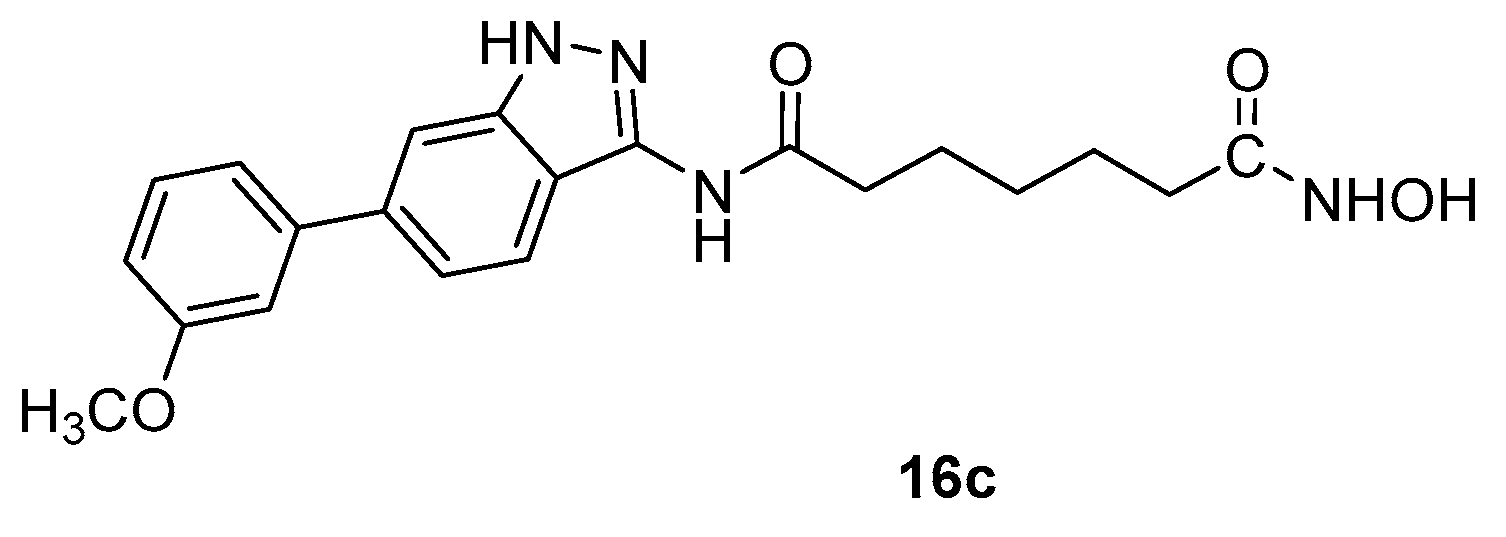
Figure 7.
2-amino-1,3,4-thiadiazoles connected to a C-6 spacer through amide bond, with hydroxamic acid as ZBG group.
Figure 7.
2-amino-1,3,4-thiadiazoles connected to a C-6 spacer through amide bond, with hydroxamic acid as ZBG group.
Scheme 7.
Synthetic pathway for N1-hydroxy-N8-(5-substituted phenyl-1,3,4-thiadiazol-2-yl)octandiamides 40a–o.
Scheme 7.
Synthetic pathway for N1-hydroxy-N8-(5-substituted phenyl-1,3,4-thiadiazol-2-yl)octandiamides 40a–o.
Scheme 8.
Synthetic sequence for preparation of phenylthiazole derivatives.
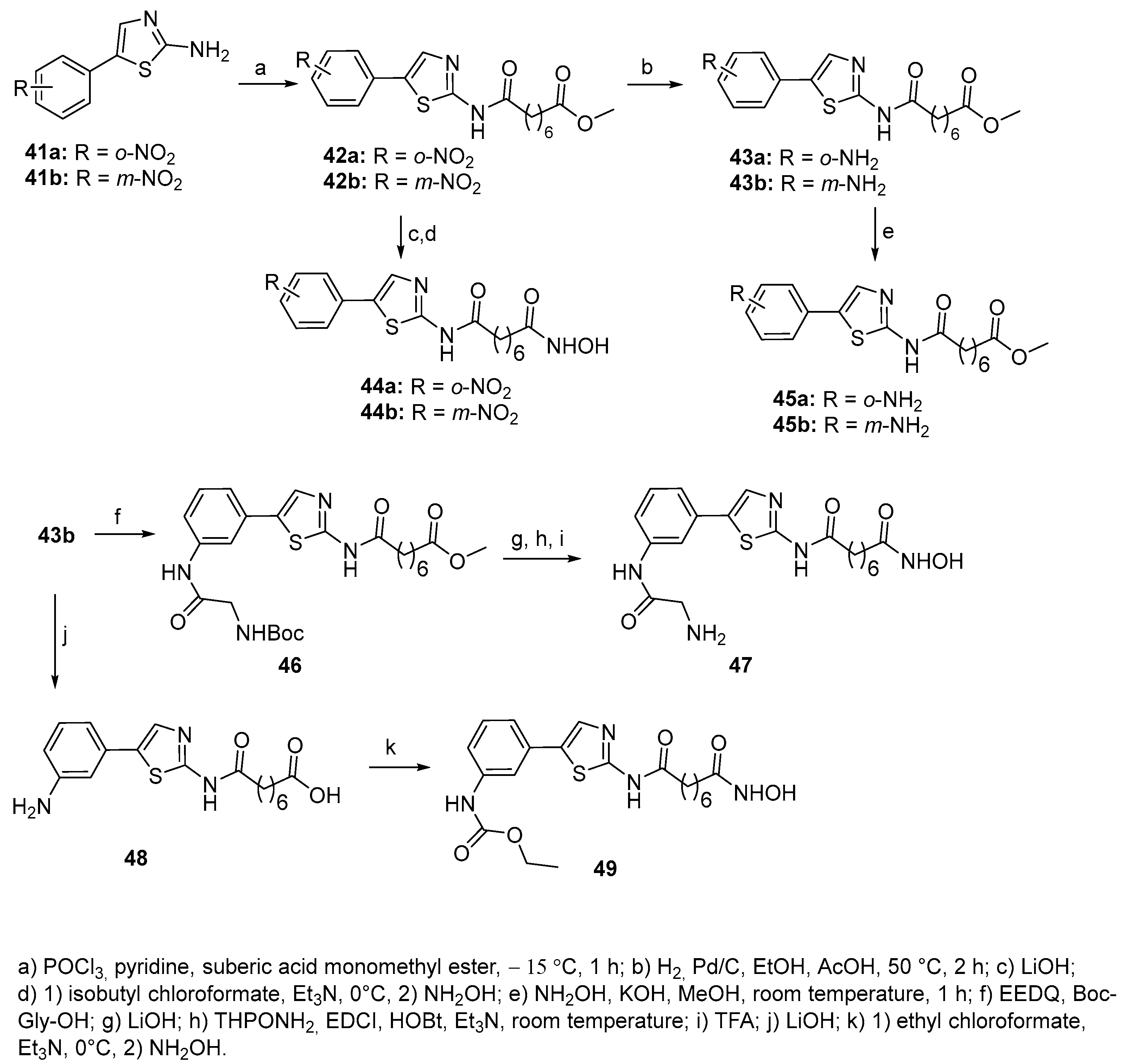
Scheme 9.
Synthesis of compounds 50-55.
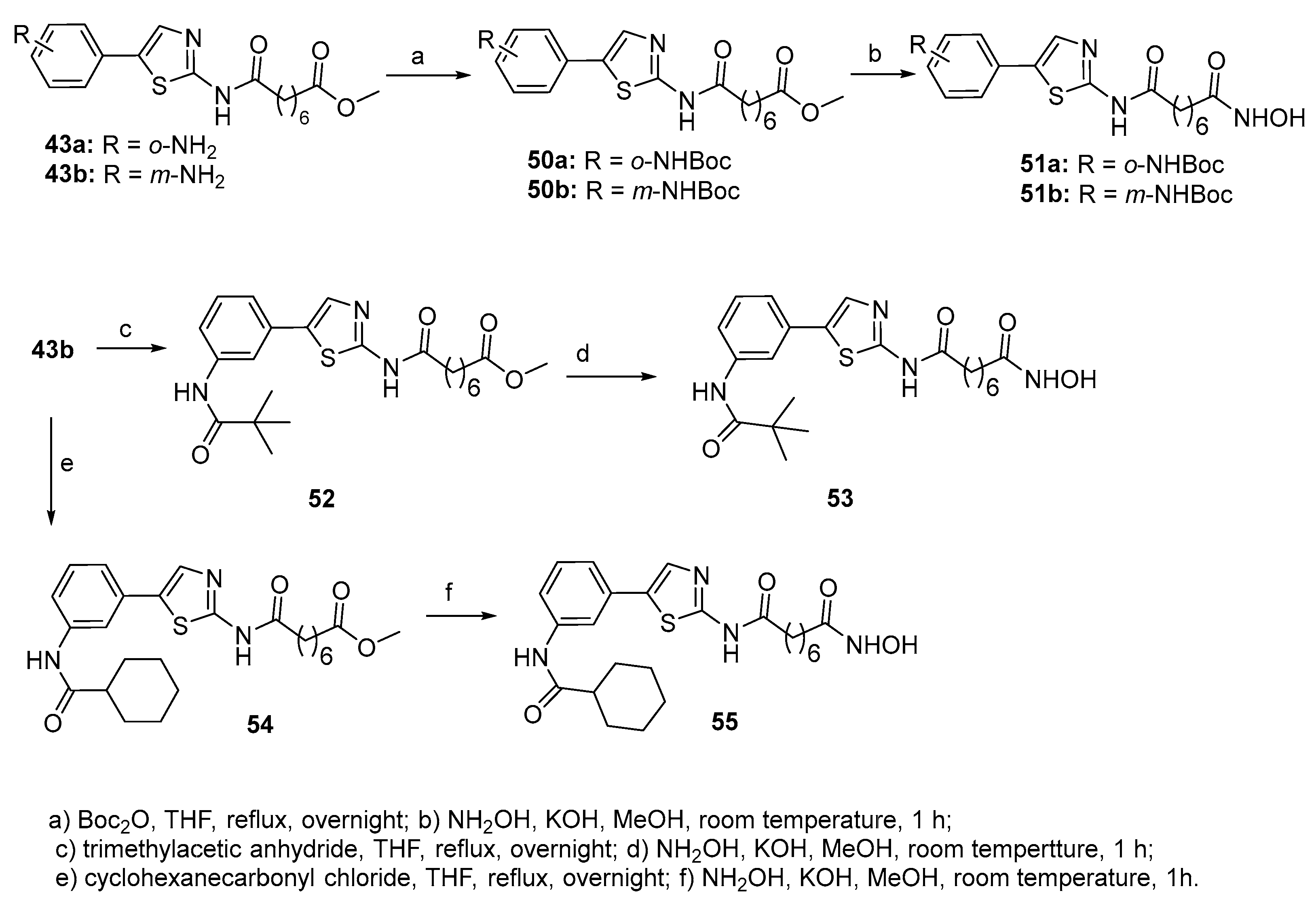
Scheme 10.
Synthesis of azide-containing compound 57.
Scheme 11.
Multi-step route to SAHA analogues 61, 67 and 68a-d with pyrazole nucleus in CAP group.
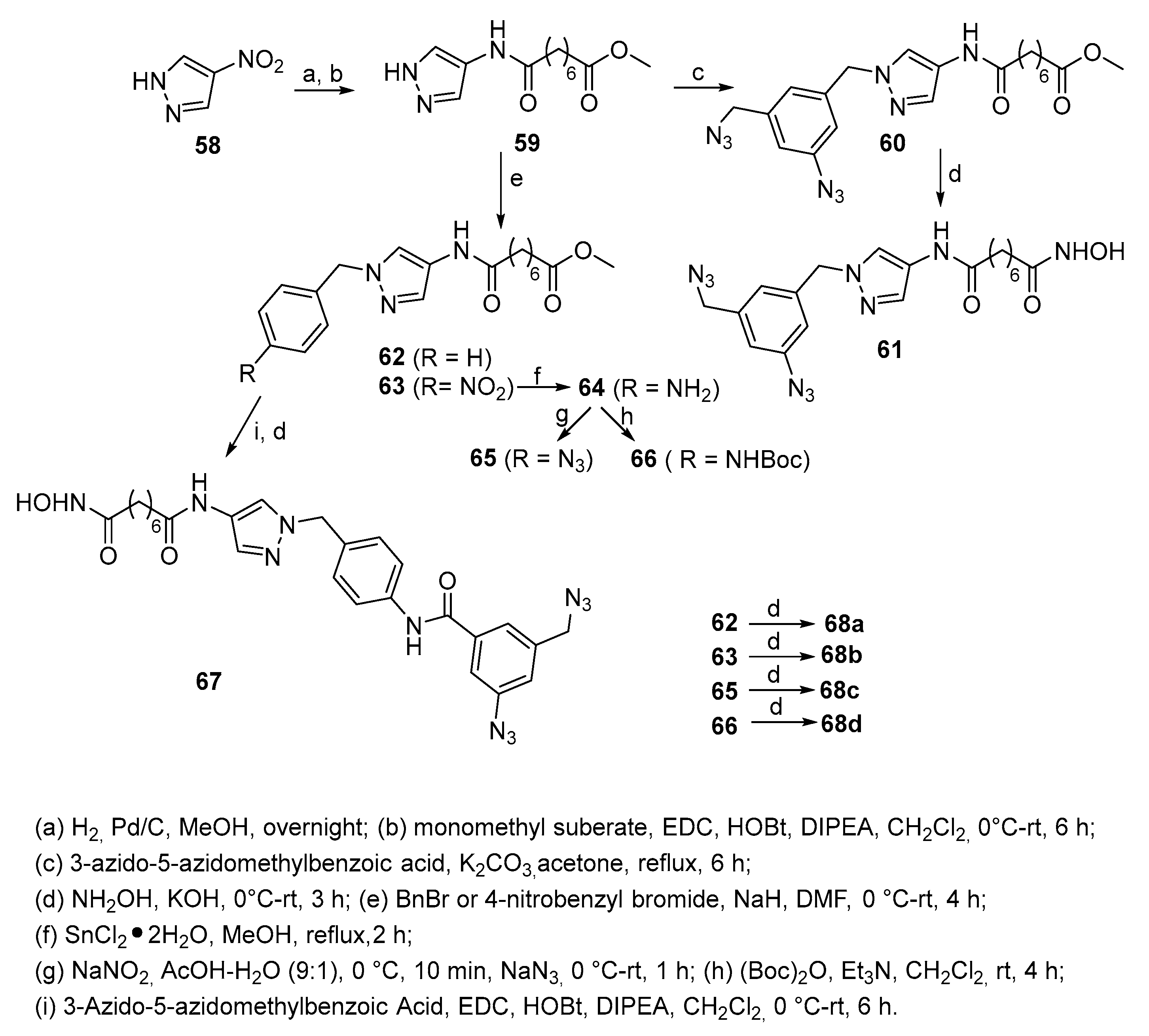
Figure 8.
Pyridine derivatives tested against HDAC1.
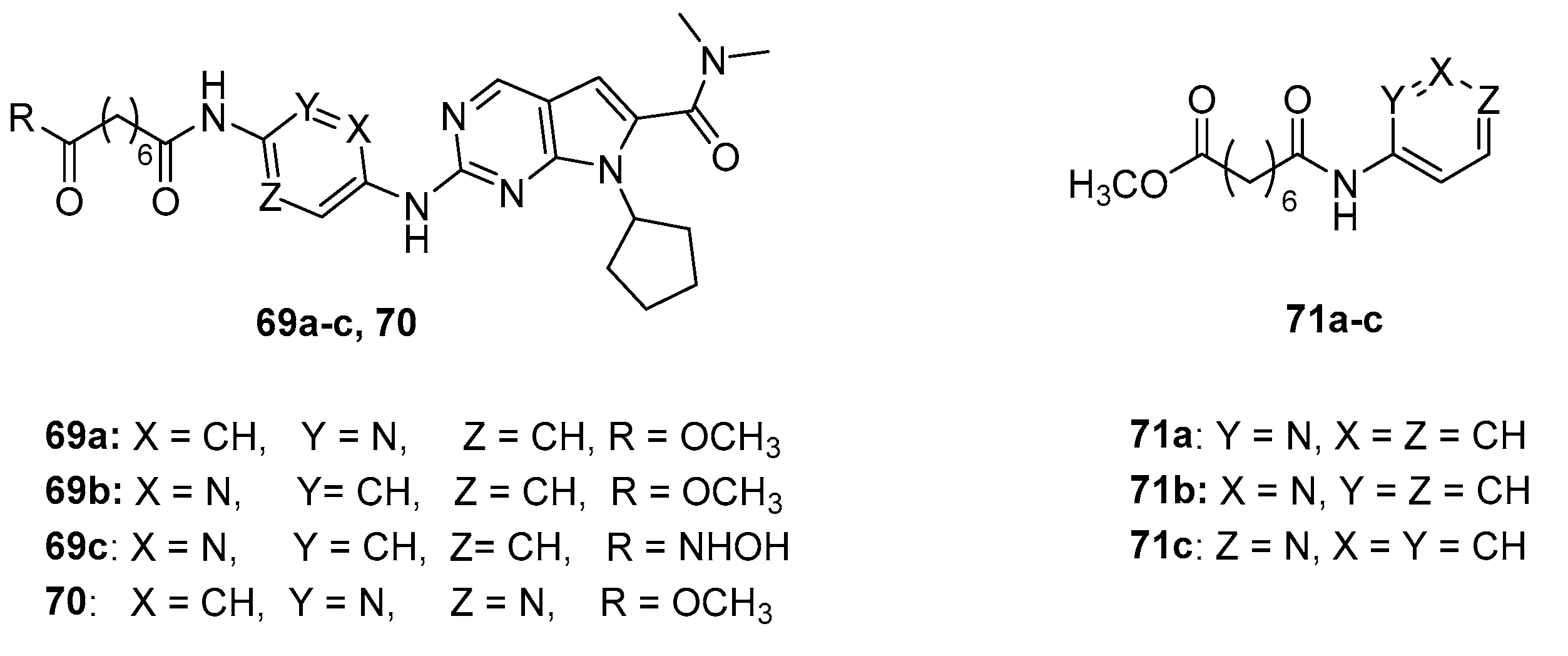
Figure 9.
Structure of compound 72.
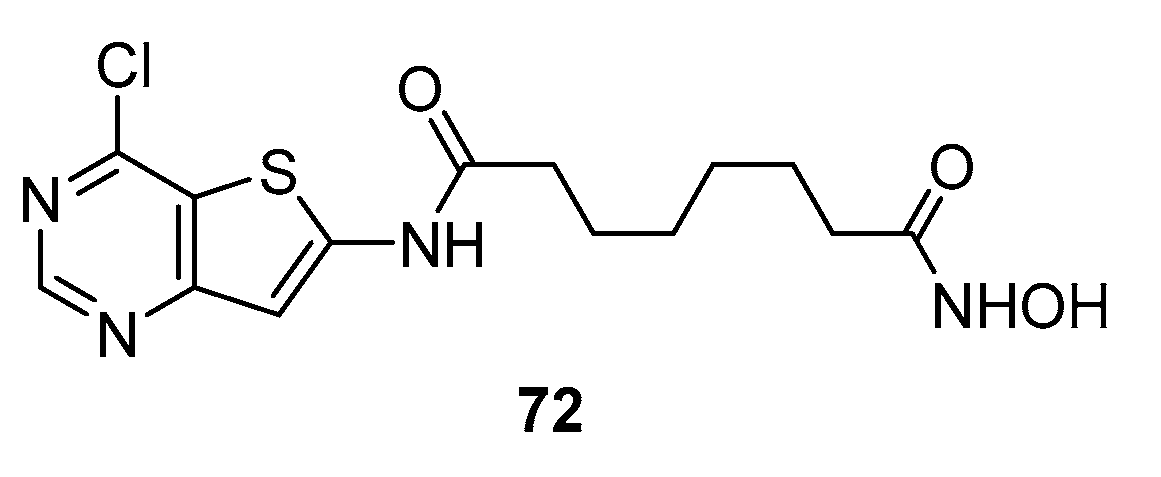
Scheme 12.
SAHA analogues with thienopyrimidine nucleus in CAP group.
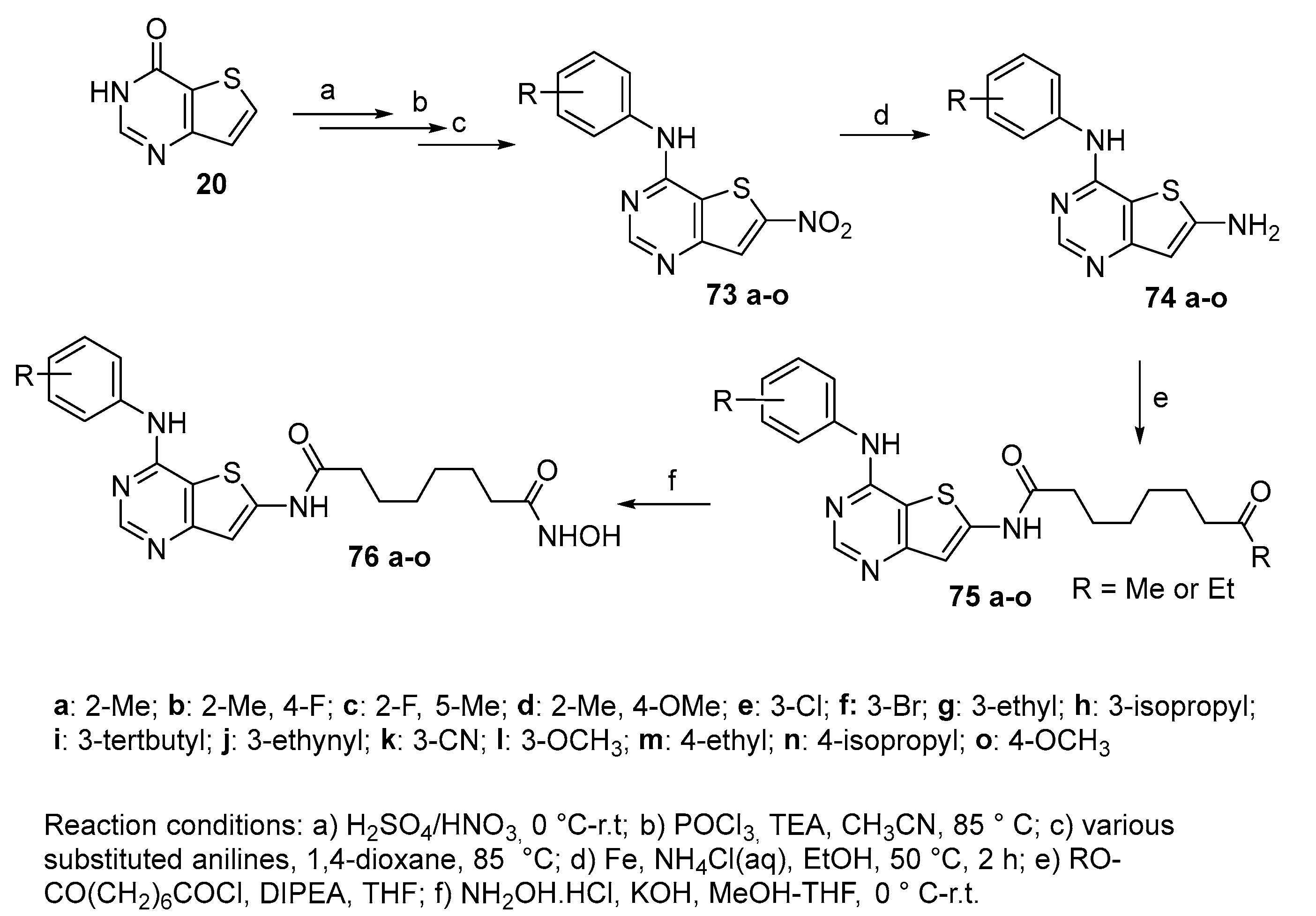
Figure 10.
Indazolyl SAHA analogues 16d-s.
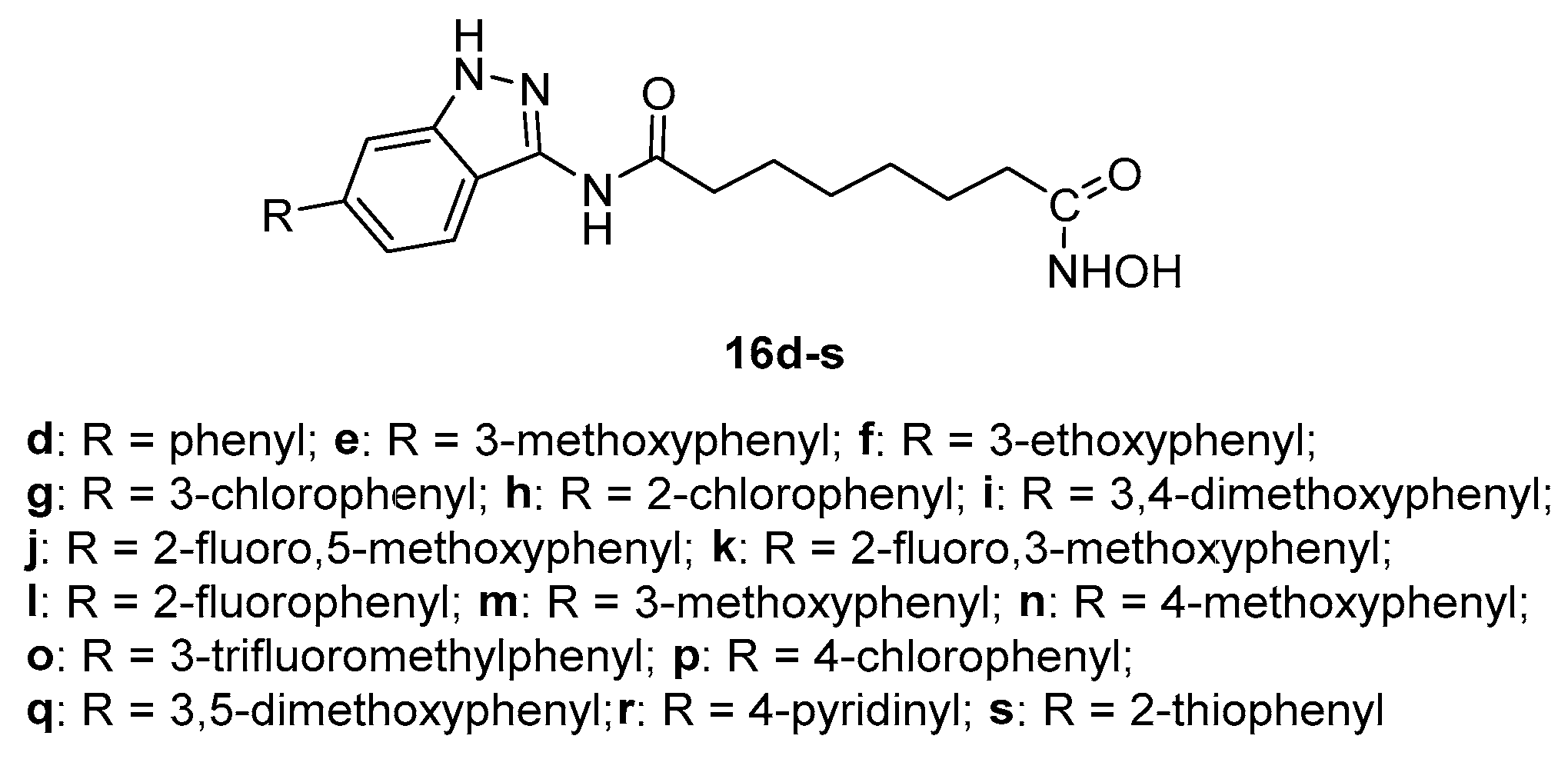
Figure 11.
structure of compounds 77a,b.
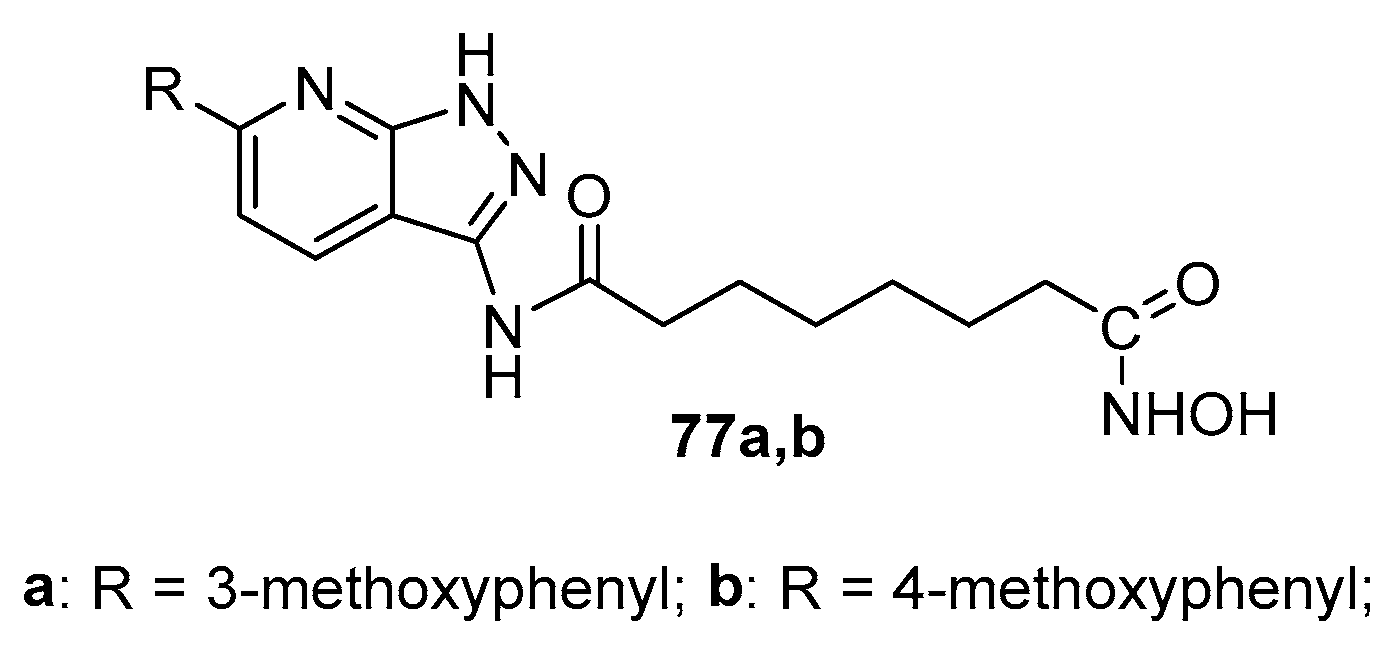
Figure 12.
SAHA analogues with benzothiazolyl scaffold in CAP group.
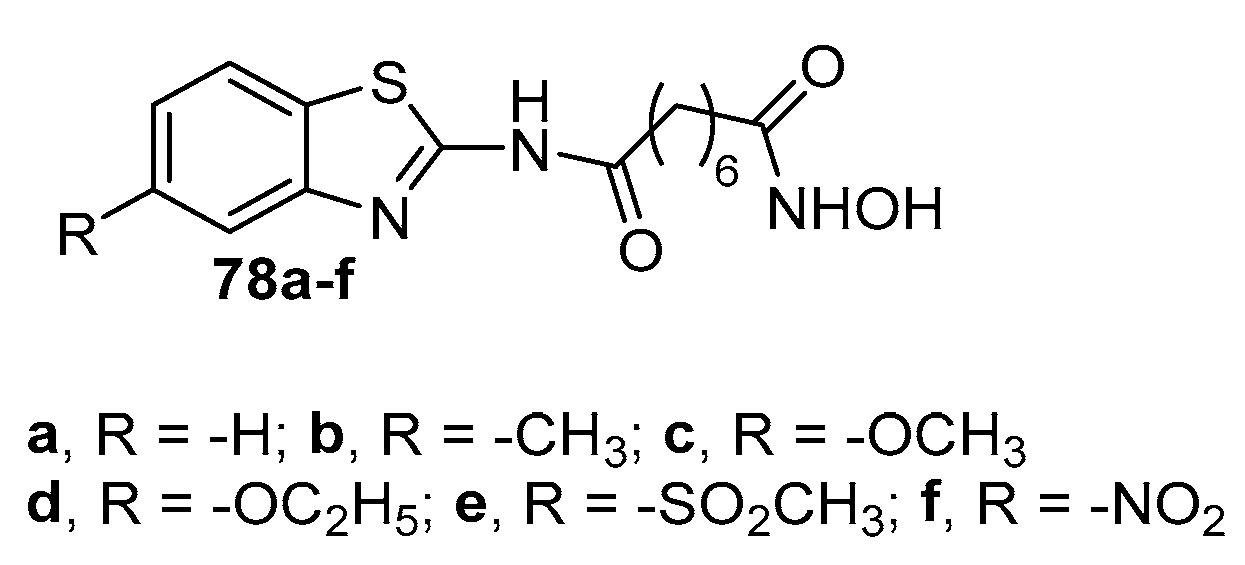
Scheme 14.
Synthetic way to SAHA analogues 89 and 90 with isoquinoline scaffold in CAP group.
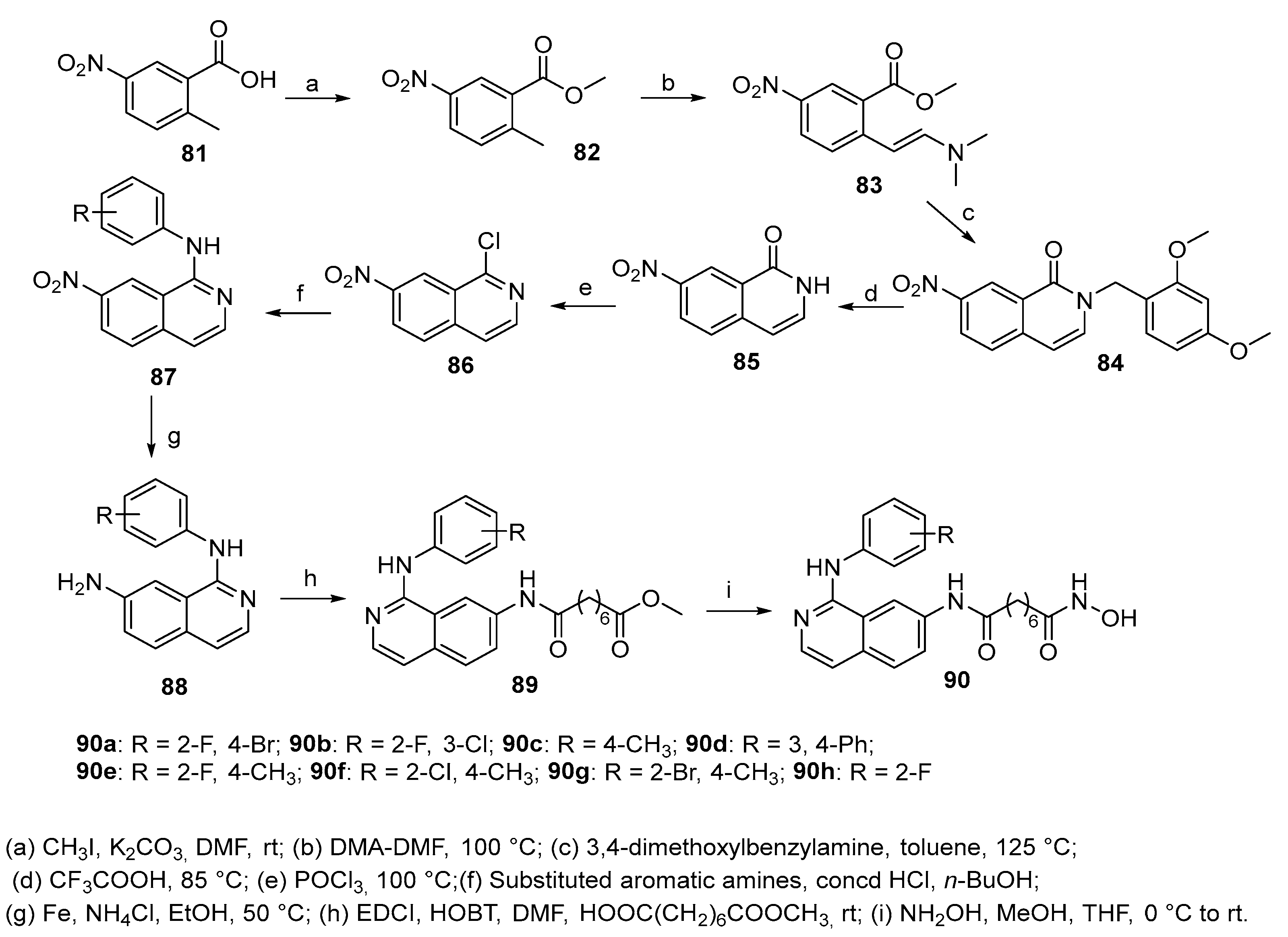
Figure 13.
Isoquinoline derivatives 91a-d.
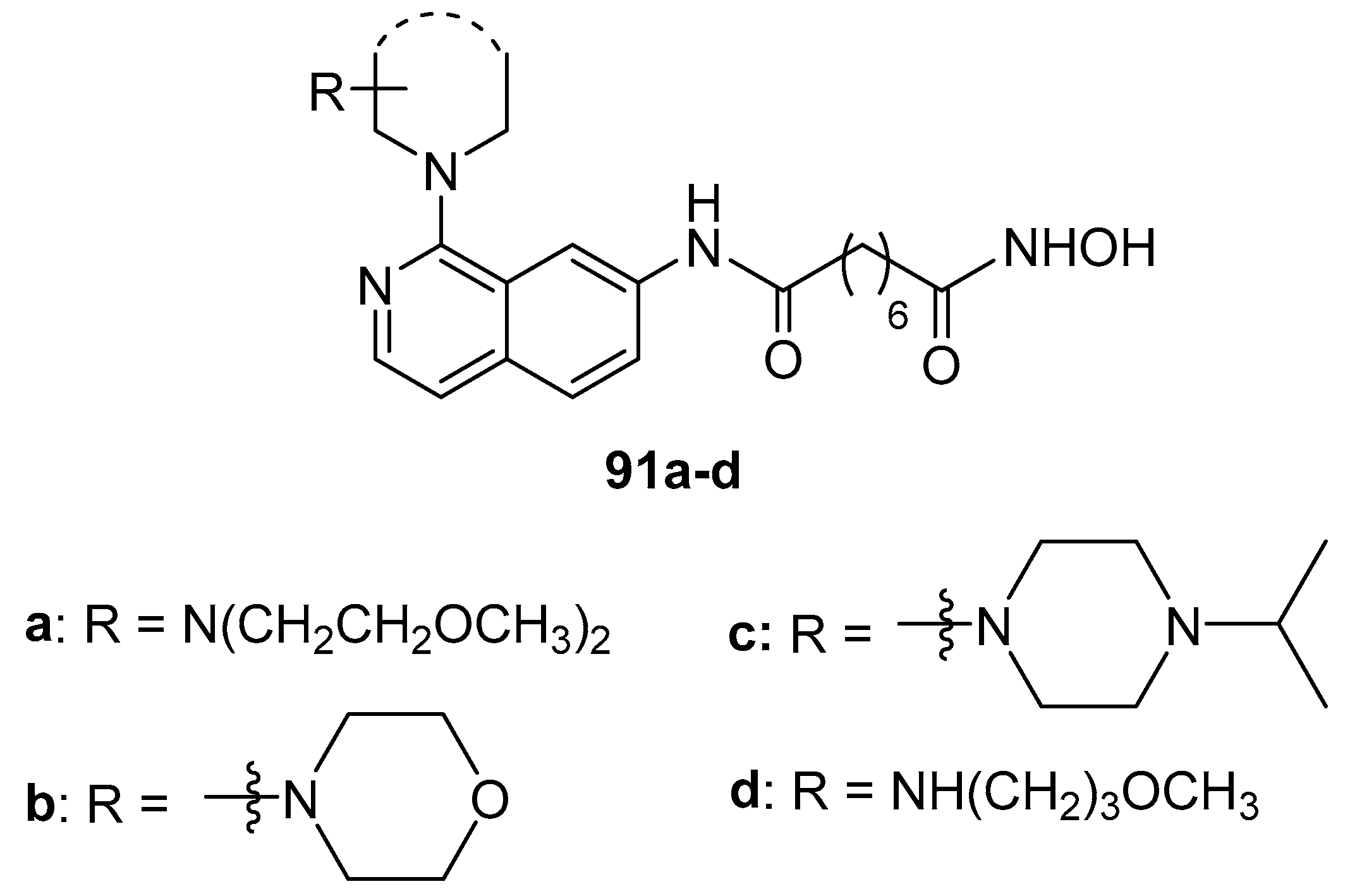
Figure 14.
Isoquinoline-1(2H)-one derivatives 92a-d.
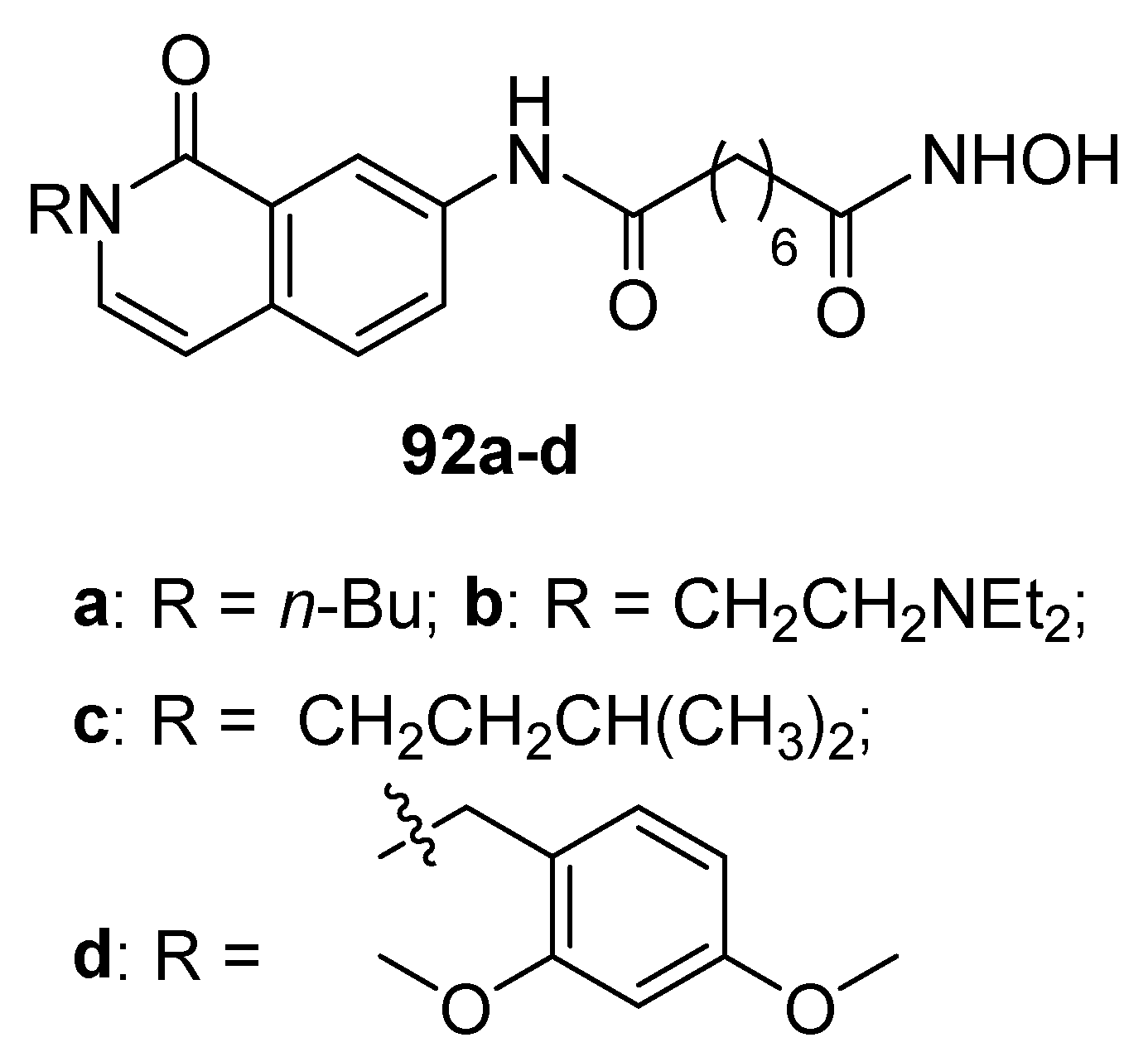
Figure 15.
Isoquinoline and pyrimidinone derivatives 93 and 94.
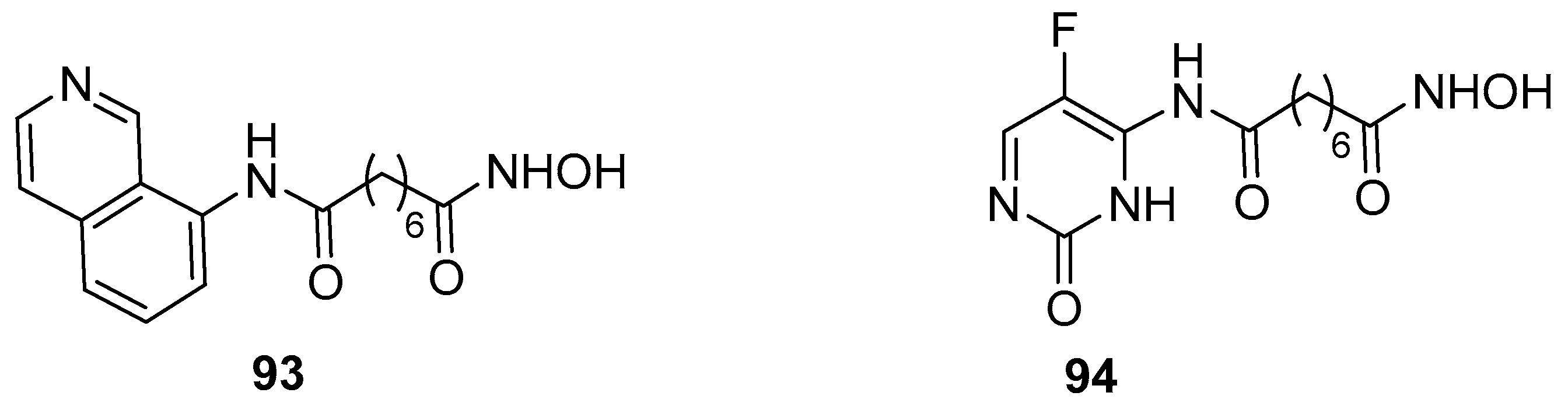
Scheme 15.
Synthetic scheme to quinazoline derivative 101.
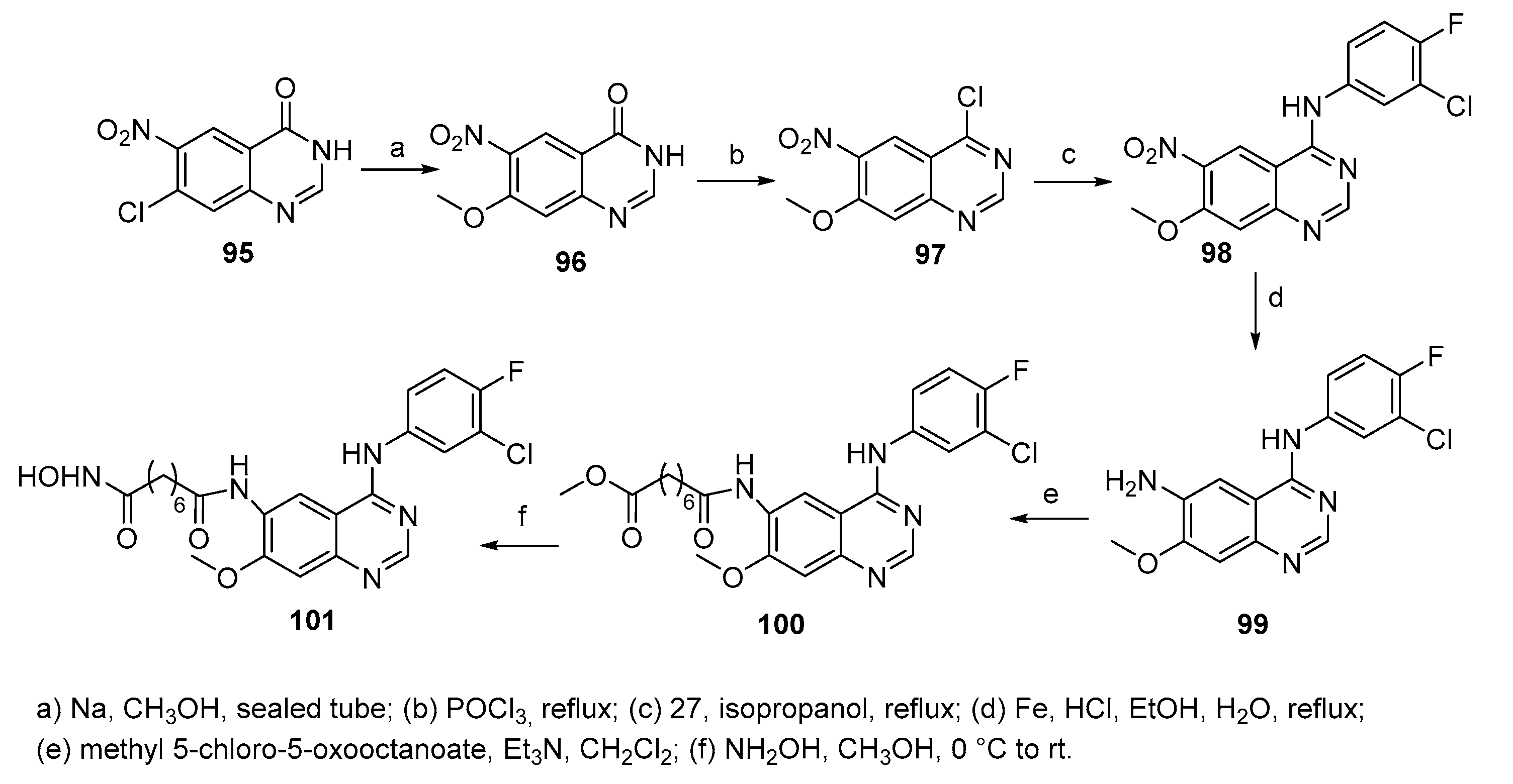
Scheme 16.
Synthesis of pyridine and pyrimidine derivatives 105 and 106.
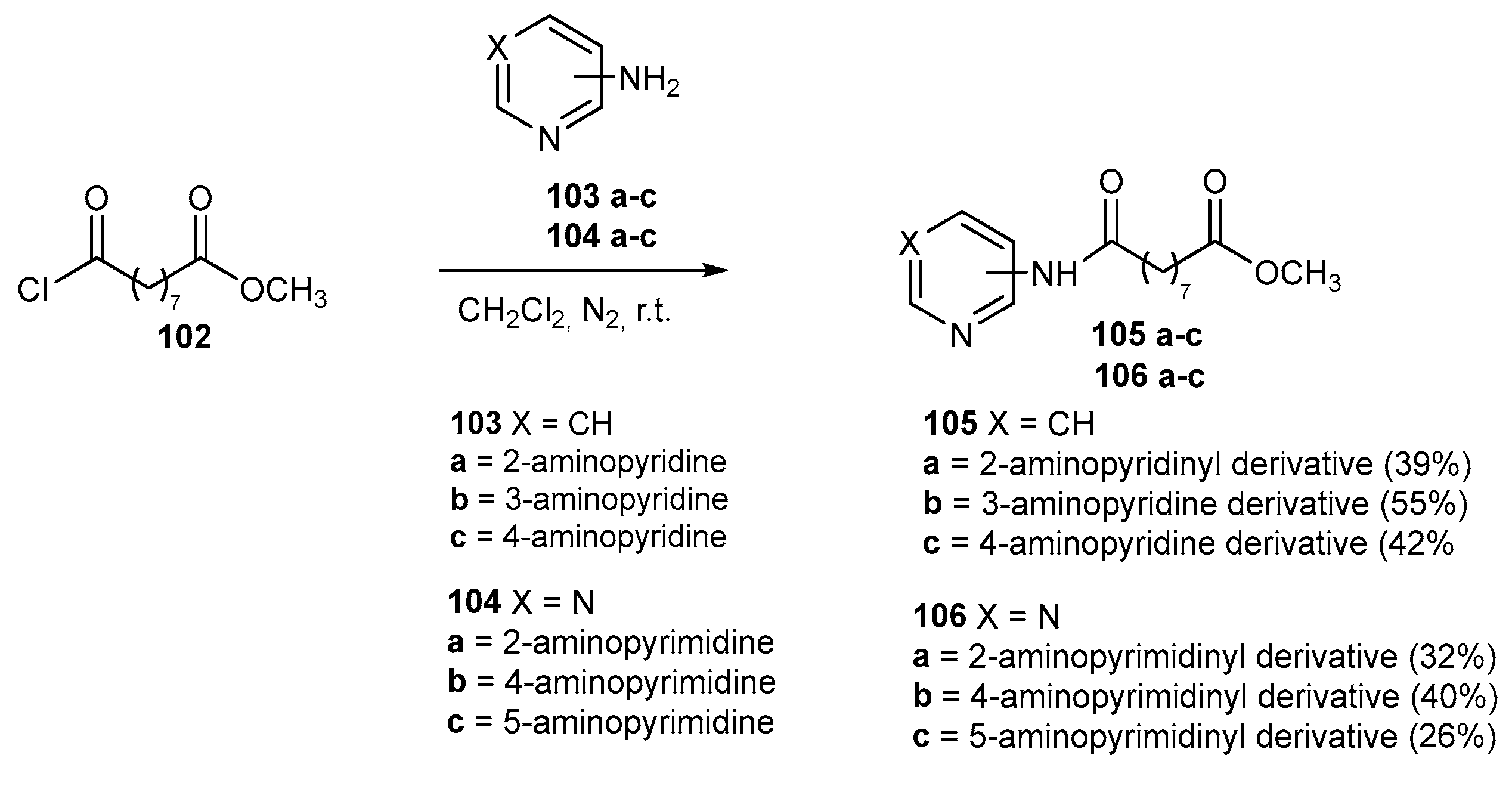

Table 1.
‘In vitro’ inhibitory activity against HDACs of compounds 26c-26g.
| Compound | R | HDAC1 IC50 (nM) |
HDAC3 IC50(nM) | HDAC6 IC50(nM) |
|---|---|---|---|---|
| 26c | 3-Cl, 4-F | 35.89 ± 16.34 | 37.67 ± 1.61 | 23.99 ± 0.72 |
| 26d | 3-CF3, 4-Cl | 40.84 ± 8.23 | 48.26 ± 1.78 | 30.00 ± 1.14 |
| 26e | H | 11.77 ± 0.50 | 20.77 ± 0.64 | 26.99 ± 4.95 |
| 26f | 4-CH3 | 14.01 ± 1.32 | 9.33 ± 0.10 | 19.68 ± 1.96 |
| 26g | 3-CH3, 4-CH3 | 29.82 ± 11.51 | 14.74 ± 0.03 | 16.87 ± 3.02 |
| SAHA | 93.34 ± 2.78 | 158.17 ± 6.66 | 78.98 ± 13.19 |
Table 2.
Inhibition of HDAC3 and HDAC8 isoforms by pyrazoles 61, 67, and 68a-d compared with that of SAHA.
Table 2.
Inhibition of HDAC3 and HDAC8 isoforms by pyrazoles 61, 67, and 68a-d compared with that of SAHA.
| Compound | HDAC3 IC50 ± SD (nM) |
HDAC8 IC50 ± SD (nM) |
|---|---|---|
| SAHA | 27± 1.0 | 440± 21 |
| 61 | 128 ± 9.8 | 17± 3 |
| 67 | 432± 52 | 487± 80 |
| 68a | 44± 5.8 | 76± 5.0 |
| 68b | 59± 1.0 | 82± 9.0 |
| 68c | 22± 1.3 | 28± 3.0 |
| 68d | 191± 18 | 147± 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



