
Submitted:
21 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
There is no routine approach to identify patients with gynecologic malignancies at high risk for chemotherapy-resistance. Circulating tumor DNA (ctDNA) tumor fraction (TFx) represents a minimally-invasive approach to tumor profiling, with as yet limited data on utility in gynecologic malignancies. The objective was to investigate the use of ctDNA TFx in a cohort of patients with ovarian and endometrial cancer. Plasma samples from patients with biopsy-proven ovarian or endometrial cancer collected between 4/2018 and 4/2020 were subjected to shallow whole genome sequencing with determination of TFx via ichorCNA package. The association of TFx to continuous and categorical baseline clinicopathologic factors and progression-free survival was assessed. 210 plasma samples from 78 patients with gynecologic cancers were analyzed. Mean TFx for ovarian cancer was 5.5% and endometrial cancer 2.4% and there was no significant difference in TFx among histology either for endometrial or ovarian cancers. Grade was associated with significant difference in ‘sentinel’ TFx among ovarian cancers but not endometrial cancers. ctDNA TFx dynamics over time demonstrated rapid clearance of ctDNA in most patients with ovarian cancer, while endometrial cancer had consistently low TFx. ctDNA TFx is feasible in ovarian and endometrial cancers and may be a valuable important tool for prognostication.
Keywords:
ovarian cancer
; circulating tumor dna
; biomarker
Introduction
About 107,500 women were diagnosed with gynecologic cancer and 31,600 women died of the disease in the U.S. in 2017 [1].. Ovarian cancer is the most lethal gynecologic cancer [1]. The majority of ovarian cancers are diagnosed at an advanced stage with no effective screening tests and in patients with advanced stage disease, the survival is significantly lower than patients with early stage disease [1]. Surgical debulking and platinum based chemotherapy have become the standard primary treatment of advanced stage ovarian cancer [2,3,4]. Over the last decade, outcomes have significantly improved with aggressive surgical debulking and the introduction of new chemotherapy regimens and targeted therapies like bevacizumab and PARP inhibitors (Niraparib, Olaparib, and Rucaparib) [4]. After completing therapy, >80-90% of the patients go into complete remission using the current clinical diagnostic strategy. Unfortunately, however, about two-thirds of patients will relapse within a median time of 18-24 months [5,6]. The standard practice after completing primary therapy is surveillance with serial follow up of CA-125 and/or imaging [7,8]. Ovarian cancer is characterized by high frequency of P53 mutation, high somatic copy number alteration (SCNAs) and genomic instability. These data indicate a critical role of genomic instability and SCNAs in ovarian cancer prognosis and tumorigenesis [9,10,11].
These data support the need for a better diagnostic marker that can identify high-risk patients who are likely to have platinum resistant or refractory disease who still have molecular residual cancer at the time of completing primary treatment or at the time of recurrence. The current clinical strategy using clinical evaluation with imaging and/or serum CA-125 can detect only patients with macroscopic residual or recurrent disease. With the current surveillance strategy, the critical time period to identify patients with aggressive disease who might benefit from more or different treatment is lost. Further, there is a need for a new marker that allows for more individualized care of patients with such aggressive disease by utilizing novel approaches to identify genomic alteration that can provide rationale for personalized targeted therapy.
Circulating tumor DNA (ctDNA) refers to DNA that tumor sheds into systemic circulation. Next generation sequencing analysis of ctDNA provides a non-invasive approach or tumor profiling without requiring invasive tissue biopsy [12]. The use of ctDNA for the diagnosis, surveillance and treatment of certain malignancies has expanded in recent years and offers a promising tool to provide personalized therapies to patients with malignancy [13,14,15,16]. Initial studies on the use of ctDNA have focused on investigating specific mutations or sequencing specific panels of cancer-related genes [17]. However, recent work by Stover et al demonstrated the utility of quantifying the tumor fraction (TFx) via low coverage (0.1x) whole genome sequencing without prior knowledge of tumor mutations in triple negative breast cancer [18]. The majority of studies to date that evaluate the use of ctDNA and TFx have been conducted primarily in breast, lung and colon cancers [19,20,21]. There is a paucity of data regarding their role in the treatment and surveillance of gynecologic malignancies; however, these data are promising and have the potential to facilitate the provision of personalized care for patients diagnosed with gynecologic malignancies [22]. The objective of this study was to investigate the use of ctDNA and TFx in a cohort of patients with ovarian and endometrial cancer.
Methods
Patient Identification and Clinicopathologic Data
Patients with pathologic diagnosis of ovarian or endometrial cancers were consented for collection of plasma for ctDNA analyses. The study was approved by the Cleveland Clinic Institutional Review Board and patients signed informed consent for the study. Patient characteristics and clinical and treatment data of our patients are summarized in Table 1. Patients with a partial or complete response by RECIST 1.1 when evaluated by CT scan per judgment of their treating physician were classified as ‘responders’, while patients with best response as stable disease or progression. Progression-free survival was defined as the time from diagnosis (for primary setting) or start of treatment (for recurrent tumors) to disease progression, last follow up, or death. Overall survival was calculated from the time of diagnosis for primary setting or start of treatment in recurrent setting to death or last follow up.

Table 1.
Cohort clinical and pathologic characteristics.
| Endometrial (N=20) |
Ovarian (N=51) |
Total (N=73) |
|
| Histology | |||
| Carcinosarcoma | 2 (10.0%) | 0 (0%) | 2 (2.7%) |
| Endometrioid | 11 (55.0%) | 2 (3.9%) | 13 (17.8%) |
| Mixed Clear Cell/Endometrioid | 1 (5.0%) | 0 (0%) | 1 (1.4%) |
| Serous | 4 (20.0%) | 46 (90.2%) | 50 (68.5%) |
| Clear cell | 0 (0%) | 3 (5.9%) | 3 (4.1%) |
| Missing | 2 (10.0%) | 0 (0%) | 4 (5.5%) |
| Stage (Primary/Recurrent) | |||
| Primary | 11 (55.0%) | 32 (62.7%) | 43 (58.9%) |
| Recurrent | 9 (45.0%) | 19 (37.3%) | 28 (38.4%) |
| Missing | 0 (0%) | 0 (0%) | 2 (2.7%) |
| Grade | |||
| Dedifferentiated | 3 (15.0%) | 0 (0%) | 3 (4.1%) |
| High Grade | 10 (50.0%) | 48 (94.1%) | 58 (79.5%) |
| Intermediate Grade | 3 (15.0%) | 2 (3.9%) | 5 (6.8%) |
| Low Grade | 4 (20.0%) | 0 (0%) | 4 (5.5%) |
| Missing | 0 (0%) | 1 (2.0%) | 3 (4.1%) |
| Number of Cycles | |||
| <= 6 | 20 (100%) | 21 (41.2%) | 41 (56.2%) |
| >6 | 0 (0%) | 19 (37.3%) | 19 (26.0%) |
| Missing | 0 (0%) | 11 (21.6%) | 13 (17.8%) |
| Status | |||
| Alive with Disease | 7 (35.0%) | 17 (33.3%) | 24 (32.9%) |
| Deceased with Disease | 7 (35.0%) | 9 (17.6%) | 16 (21.9%) |
| No Evidence of Disease | 3 (15.0%) | 22 (43.1%) | 25 (34.2%) |
| Missing | 3 (15.0%) | 3 (5.9%) | 8 (11.0%) |
Sample Processing and Ultra Low-Pass Whole-Genome Sequencing
Venous blood samples were collected in EDTA (BD) or Cell-Free DNA BCT (Streck) tubes. Blood processing to component parts, cell-free DNA extraction from plasma, and DNA quantification was performed as described previously [23]. Library construction of cell-free DNA was performed using the Kapa HyperPrep kit with custom adapters (IDT). 5-50 ng of cfDNA input (1000-20,000 haploid genome equivalents) was used for ultra low-pass whole-genome sequencing (ULP-WGS). Constructed sequencing libraries were pooled (2 uL of each x 96 per pool) and sequenced using 100bp paired-end runs over 1 x lane on a HiSeq2500 (Illumina) for ULP-WGS. ULP-WGS of cfDNA was performed to average genome-wide fold coverage of 0.1X. Segment copy number and TFx were derived via ichorCNA [23]. Samples were excluded if the median absolute deviation (MAD) of the copy ratios (2log2 ratio) between adjacent bins, genome-wide, was greater than 0.20 suggesting poor quality sequence data. Genome-wide copy number plots were generated by ichorCNA.
Statistical Analyses and Data Visualization
All statistical analyses and data visualizations were performed in R version 3.3.1. The association of TFx to continuous and categorical clinicopathologic factors was evaluated using Wilcoxon rank-sum and chi-square test or analysis of variance, respectively. Multiple linear regression models were constructed using the ‘lm’ function in R. Association with progression-free survival was assessed via log-rank test and Kaplan-Meier visualization constructed using the ‘packHV’ package in R.
Results
Ovarian and endometrial cancer cohorts
We identified 210 plasma samples from 78 patients with biopsy-proven ovarian cancer or endometrial cancer collected between 04/2018 and 04/2020 under IRB-approved protocols at a single institution and abstracted detailed clinicopathologic information (Table 1). Among ovarian cancers, 90.2% (46/51) were serous histology and 94.1% (48/51) high grade. Among endometrial cancers, 55% (11/20) were endometrioid and 50% (10/20) high grade. Among both cancer types, there were samples collected from patients in both the primary (ovarian 62.7%, 32/51; endometrial 55%, 11/20) and recurrent (ovarian 37.3%, 19/51; endometrial 45%, 9/20) settings. At the time of data freeze, 17.6% (9/51) ovarian patients and 35% (7/20) endometrial patients had died of disease. Median first progression-free survival (PFS1) was 15 months for ovarian and 9 months for endometrial cancer.
Tumor fraction association with clinical and pathologic variables in endometrial and ovarian cancer
Our approach offers a ‘tumor fraction’ calculation based on SCNAs detected in cfDNA without a priori knowledge of tumor mutation status [23]. Tumor fraction measurement using ichorCNA has a broad dynamic range, both within individuals and among distinct patients and among the 210 samples analyzed, TFx range was 0 to 76%.
We evaluated the association of the first ‘sentinel’ blood draw with cancer and clinicopathologic characteristics (Figure 1; Table 2). The mean tumor fraction for ovarian cancer was 5.5% and for endometrial cancer was 2.4%, not significantly different between cancer types (t-test p=0.065; Figure 1A, Supp Figure 1A). Evaluating primary versus recurrent settings within each individual cancer type demonstrated no significant difference in ‘sentinel’ TFx in either endometrial (t-test p=0.64) or ovarian (t-test p=0.84) cancers (Figure 1B, Supp Figure 1B). We also evaluated ‘sentinel’ TFx by tumor histology within each cancer type and, similarly, there was not a significant difference in TFx among histology either for endometrial (Wilcoxon rank-sum p=0.62) or ovarian (Wilcoxon rank-sum p=0.24) cancers (Figure 1C-D, Supp Figure 1C-D). Interestingly, grade was associated with significant difference in ‘sentinel’ TFx among ovarian cancers (t-test p=0.01) but not endometrial cancers (ANOVA p=0.48; Figure 1E, Supp Figure 1E). We investigated if a single ‘sentinel’ TFx value was associated with best overall therapy response (Figure 1F, Supp Figure 1F). There was no significant association for either endometrial (ANOVA p=0.99) or ovarian cancer (ANOVA p=0.40). We evaluated the characteristics of ‘sentinel’ TFx by sites of metastatic disease (Table 2). For metastatic sites with at least two representative patients, mean TFx ranged from 1.6% (lung/pleural effusion metastases in endometrial cancer) to 5.2% (peritoneal metastases in ovarian cancer). A summary of tumor fraction association with clinical and pathologic variables in endometrial and ovarian cancer demonstrated that no association was significant after multiple test correction for either endometrial or ovarian cancer (Supp Table 1).
Figure 1.
Association of Tumor Fraction with Clinicopathologic Characteristics in Patients with Ovarian and Endometrial Cancers. To avoid duplicate sample bias, the association of the tumor fraction (TFx) of the first ‘sentinel’ blood draw was evaluated relative to clinicopathologic characteristics, including: (A) ovarian versus endometrial cancer; (B) primary versus recurrent settings within each individual cancer type; (C-D) tumor histology within each cancer type; (E) tumor grade; (F) best overall therapy response. TFx is presented as log10 transformed in each y-axis.
Figure 1.
Association of Tumor Fraction with Clinicopathologic Characteristics in Patients with Ovarian and Endometrial Cancers. To avoid duplicate sample bias, the association of the tumor fraction (TFx) of the first ‘sentinel’ blood draw was evaluated relative to clinicopathologic characteristics, including: (A) ovarian versus endometrial cancer; (B) primary versus recurrent settings within each individual cancer type; (C-D) tumor histology within each cancer type; (E) tumor grade; (F) best overall therapy response. TFx is presented as log10 transformed in each y-axis.
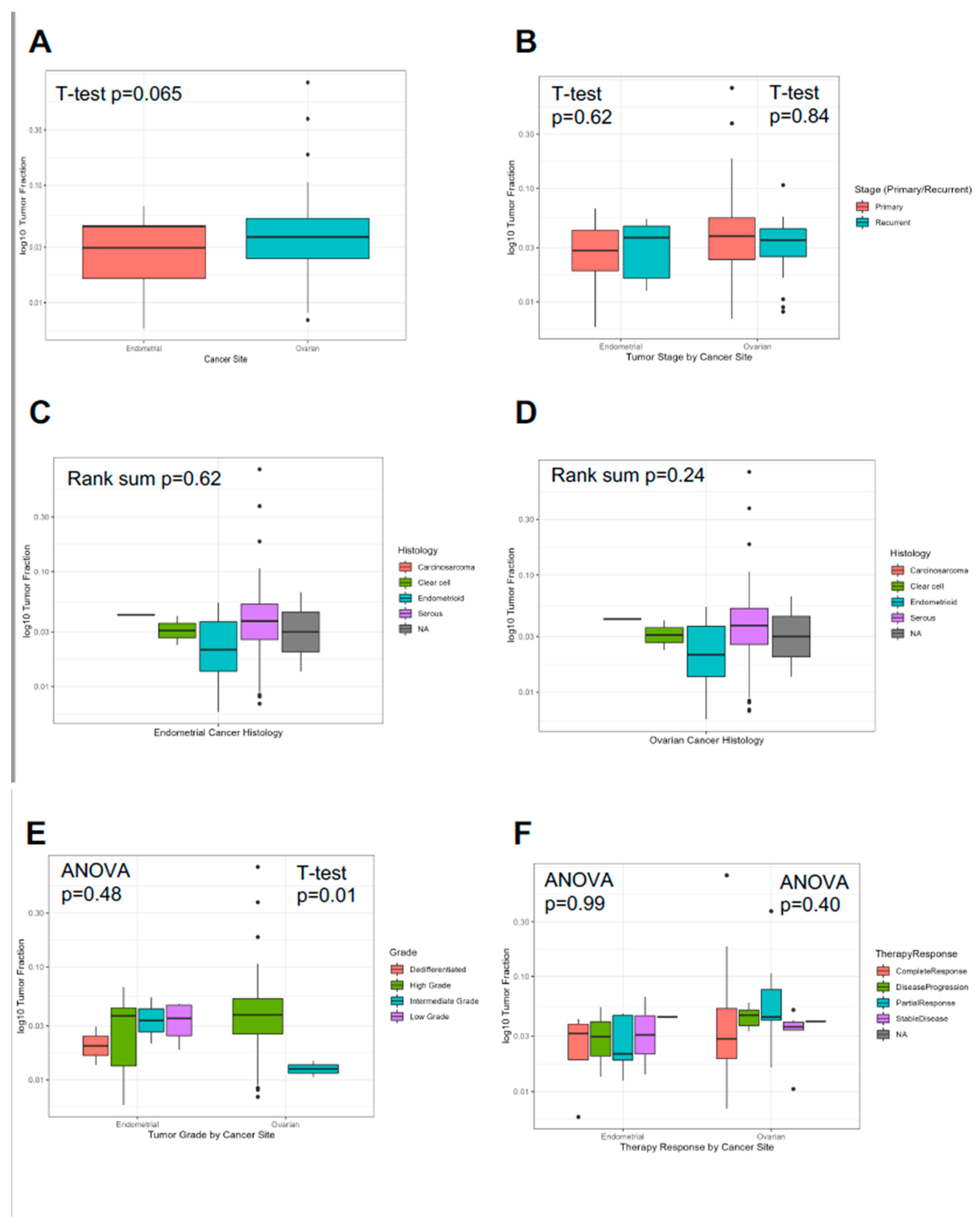
Table 2.
Tumor Fraction by Metastatic Site.
| Primary Site | Metastatic Site | Total | Mean | Median | Standard Deviation | Standard Error |
|---|---|---|---|---|---|---|
| Endometrial | Peritoneal | 13 | 0.023 | 0.021 | 0.02 | 0.006 |
| Endometrial | Liver | 3 | 0.029 | 0.018 | 0.022 | 0.013 |
| Endometrial | Lung/Pleural Effusion | 3 | 0.016 | 0 | 0.027 | 0.016 |
| Endometrial | Node | 3 | 0.022 | 0.021 | 0.008 | 0.004 |
| Endometrial | Other | 2 | 0 | 0 | 0 | 0 |
| Ovarian | Peritoneal | 41 | 0.052 | 0.034 | 0.118 | 0.018 |
| Ovarian | Liver | 3 | 0.044 | 0.046 | 0.003 | 0.001 |
| Ovarian | Lung/Pleural Effusion | 7 | 0.033 | 0.035 | 0.008 | 0.003 |
| Ovarian | Node | 1 | 0.107 | 0.107 | - | - |
| Ovarian | Other | 1 | 0.041 | 0.041 | - | - |
Circulating tumor dynamics in ovarian and endometrial cancers
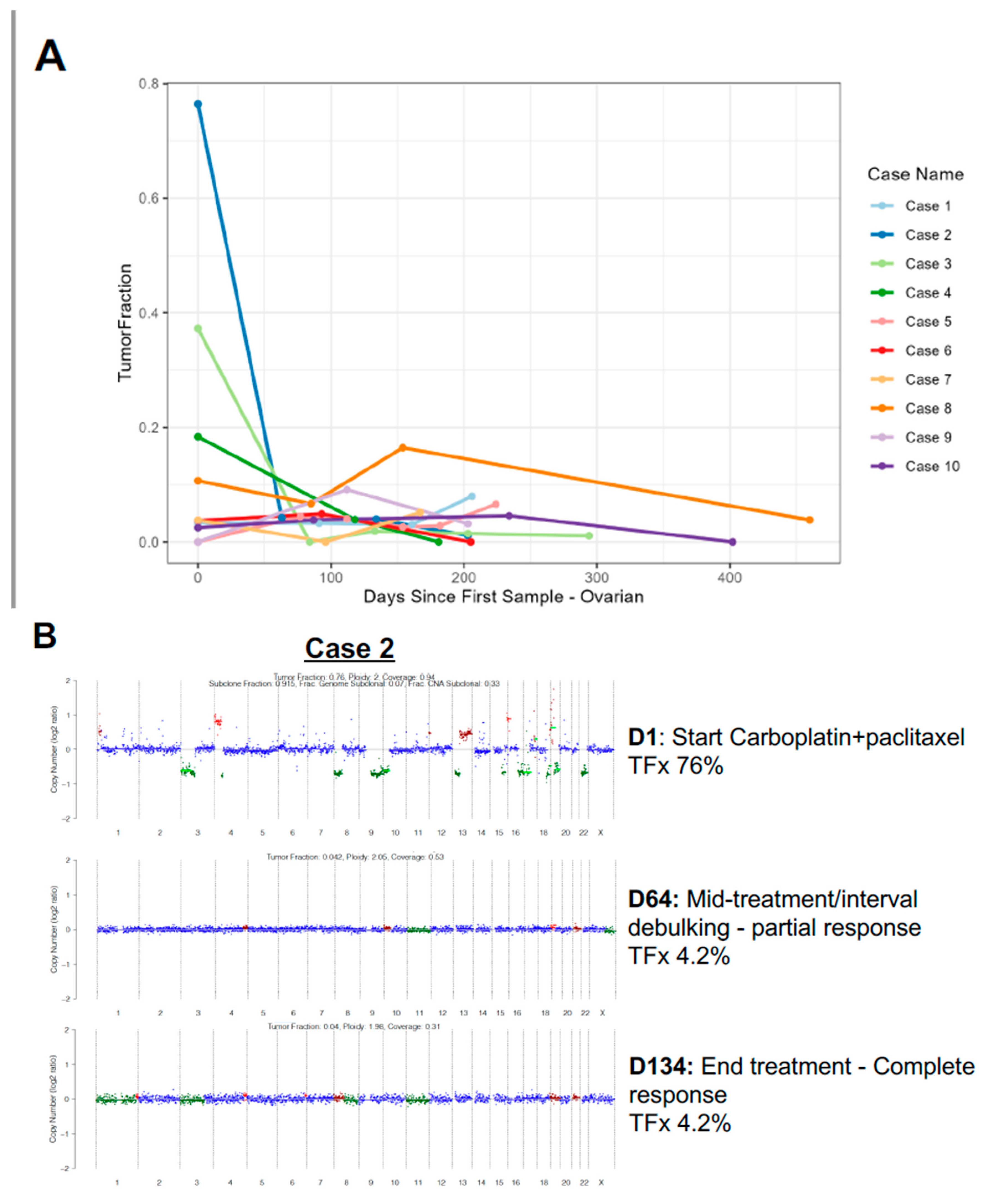
A unique aspect of this cohort was the rich collection of serial samples, with average of nearly three samples per individual (range 1 – 13 samples per patient). In the majority of cases, most samples had persistently low TFx, often near or below the threshold of ‘detectable’ ctDNA of 3% [23]. For example, the patient with 13 samples collected over 234 days demonstrated TFx range from 0 – 8.9%, with 5/13 samples ‘undetectable’ (<3%), primarily mid-treatment samples. We visualized the dynamics of ctDNA TFx for the ten patients with the greatest dynamic range and at least three samples (Figure 2A, Supp Figure 1G). For ovarian cancer, multiple patients demonstrate very high initial TFx, associated with rapid decline then persistently low subsequent TFx values (e.g. Cases 2/3/4 Figure 2A). Alternatively, for endometrial cancer there did not appear to be a consistent trend, with modest increases and decreases all at relatively low TFx<6% (Supp Figure 1G).
We investigated a dramatic example in greater detail. Case 2 represented a patient with newly diagnosed stage IIIC high grade serous ovarian cancer, germline BRCA mutated. The baseline ctDNA sample was collected at time of initiation of carboplatin+paclitaxel and demonstrated extremely high TFx of 76%, or more than three quarters of free DNA in circulation tumor-derived (Figure 2B, top panel). Upon repeat ctDNA assessment after four cycles of carboplatin+paclitaxel (D64) at time of planned interval debulking, there was a dramatic decline in TFx to 4.2% (Figure 2B, middle panel), just above the detectable threshold. A follow-up ctDNA assessment after interval debulking and a subsequent four cycles of carboplatin+paclitaxel (D134) again demonstrated a low TFx of 4.0% (Figure 2B, bottom panel). The copy number alterations evident at high TFx were no longer detectable for either low TFx samples. The patient remains with no evidence of disease.
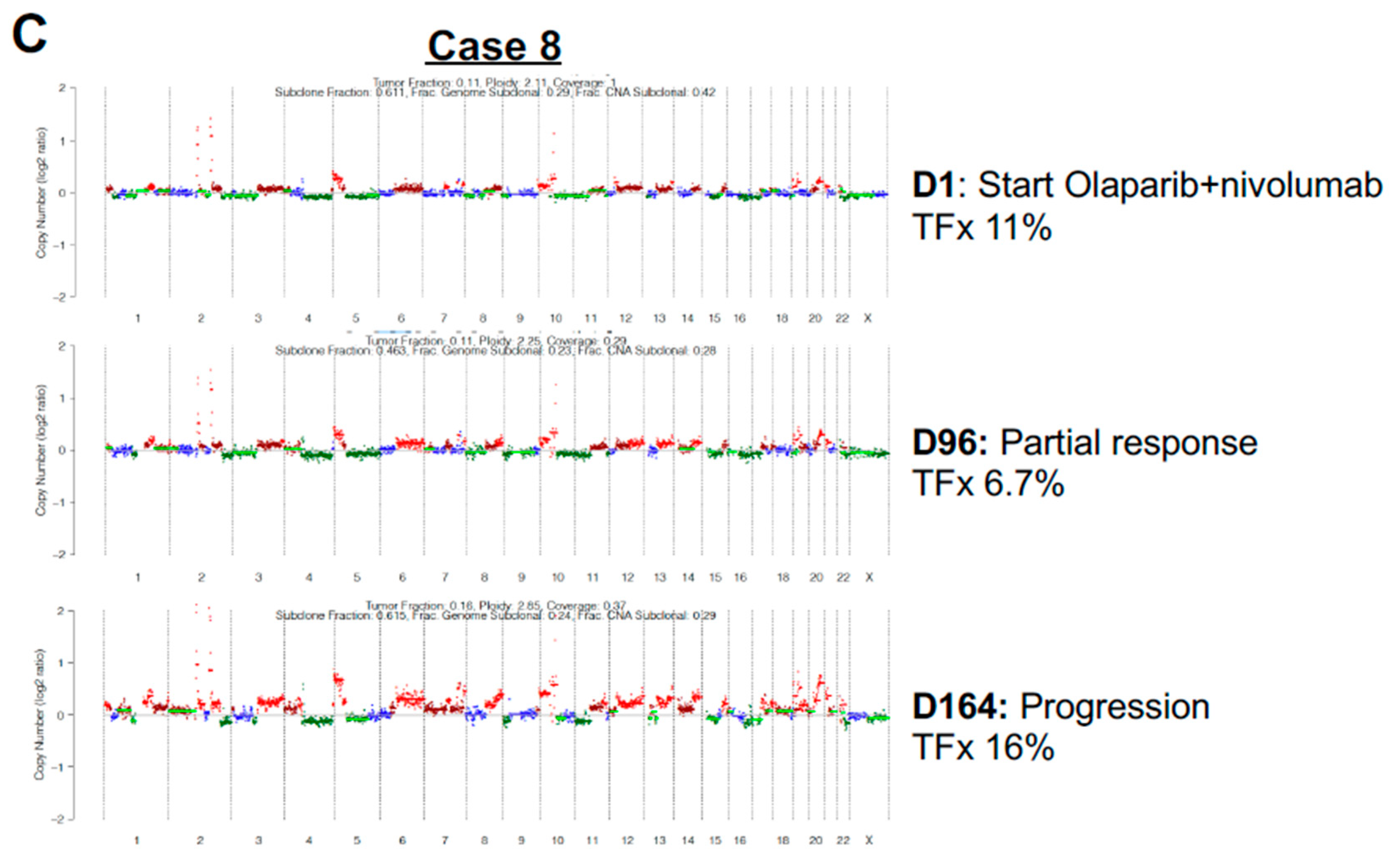
A second case of interest, Case 8, demonstrated a modest decline in TFx then rebound (Figure 2A, 2C). Case 8 represented a patient with recurrent high grade serous ovarian cancer, germline BRCA wild-type who was initiating salvage therapy with Olaparib and nivolumab. The baseline ctDNA sample was collected at time of Olaparib+nivolumab and TFx of 11% with complex copy number profile, including both focal (chromosome 2) and large, arm-level gain/amplification events (Figure 2C, top panel). Repeat ctDNA assessment at D96 of Olaparib+nivolumab at time of imaging partial response demonstrated a modest decline in TFx to 6.7% (Figure 2C, middle panel), with similar appearance of copy number profile. A follow-up ctDNA assessment at disease progression (D164) demonstrated rebound of TFx to 16% (Figure 2C, bottom panel). The copy number alterations detectable at the first two time points were more dramatically evident at a higher TFx, without definitive development of new copy number events.
Figure 2.
Circulating tumor fraction dynamics in ovarian cancer. (A) Circulating tumor DNA tumor fraction (TFx) dynamics for the ten patients with ovarian cancer representing the greatest dynamic range and at least three samples. Each individual patient represented by a different color, as indicated. (B) Example case of newly diagnosed stage IIIC high grade serous ovarian cancer, germline BRCA mutated with baseline TFx at time of initiation of carboplatin+paclitaxel demonstrating extremely high TFx of 76% with multiple copy number alterations (top panel). Tepeat TFx after four cycles of carboplatin+paclitaxel (D64; middle panel) at time of planned interval debulking demonstrated a dramatic decline in TFx to 4.2% with follow-up TFx after interval debulking and subsequent four cycles of carboplatin+paclitaxel (D134; bottom panel) demonstrated a low TFx of 4.0%. The copy number alterations evident at high TFx were no longer detectable for either low TFx samples. The patient remains with no evidence of disease. (C) Example case of recurrent high grade serous ovarian cancer, germline BRCA wild-type. Baseline TFx sample at time of Olaparib+nivolumab initiation demonstrated TFx of 11% with complex copy number profile (top panel). Repeat ctDNA assessment at D96 (middle panel) at time of imaging partial response demonstrated a modest decline in TFx to 6.7%, yet follow-up ctDNA assessment at disease progression (D164; bottom panel) demonstrated rebound of TFx to 16%. The copy number alterations detectable at the first two time points were more dramatically evident at a higher TFx, without definitive development of new copy number events.
Figure 2.
Circulating tumor fraction dynamics in ovarian cancer. (A) Circulating tumor DNA tumor fraction (TFx) dynamics for the ten patients with ovarian cancer representing the greatest dynamic range and at least three samples. Each individual patient represented by a different color, as indicated. (B) Example case of newly diagnosed stage IIIC high grade serous ovarian cancer, germline BRCA mutated with baseline TFx at time of initiation of carboplatin+paclitaxel demonstrating extremely high TFx of 76% with multiple copy number alterations (top panel). Tepeat TFx after four cycles of carboplatin+paclitaxel (D64; middle panel) at time of planned interval debulking demonstrated a dramatic decline in TFx to 4.2% with follow-up TFx after interval debulking and subsequent four cycles of carboplatin+paclitaxel (D134; bottom panel) demonstrated a low TFx of 4.0%. The copy number alterations evident at high TFx were no longer detectable for either low TFx samples. The patient remains with no evidence of disease. (C) Example case of recurrent high grade serous ovarian cancer, germline BRCA wild-type. Baseline TFx sample at time of Olaparib+nivolumab initiation demonstrated TFx of 11% with complex copy number profile (top panel). Repeat ctDNA assessment at D96 (middle panel) at time of imaging partial response demonstrated a modest decline in TFx to 6.7%, yet follow-up ctDNA assessment at disease progression (D164; bottom panel) demonstrated rebound of TFx to 16%. The copy number alterations detectable at the first two time points were more dramatically evident at a higher TFx, without definitive development of new copy number events.
Association of circulating tumor DNA TFx with outcome in ovarian and endometrial cancers
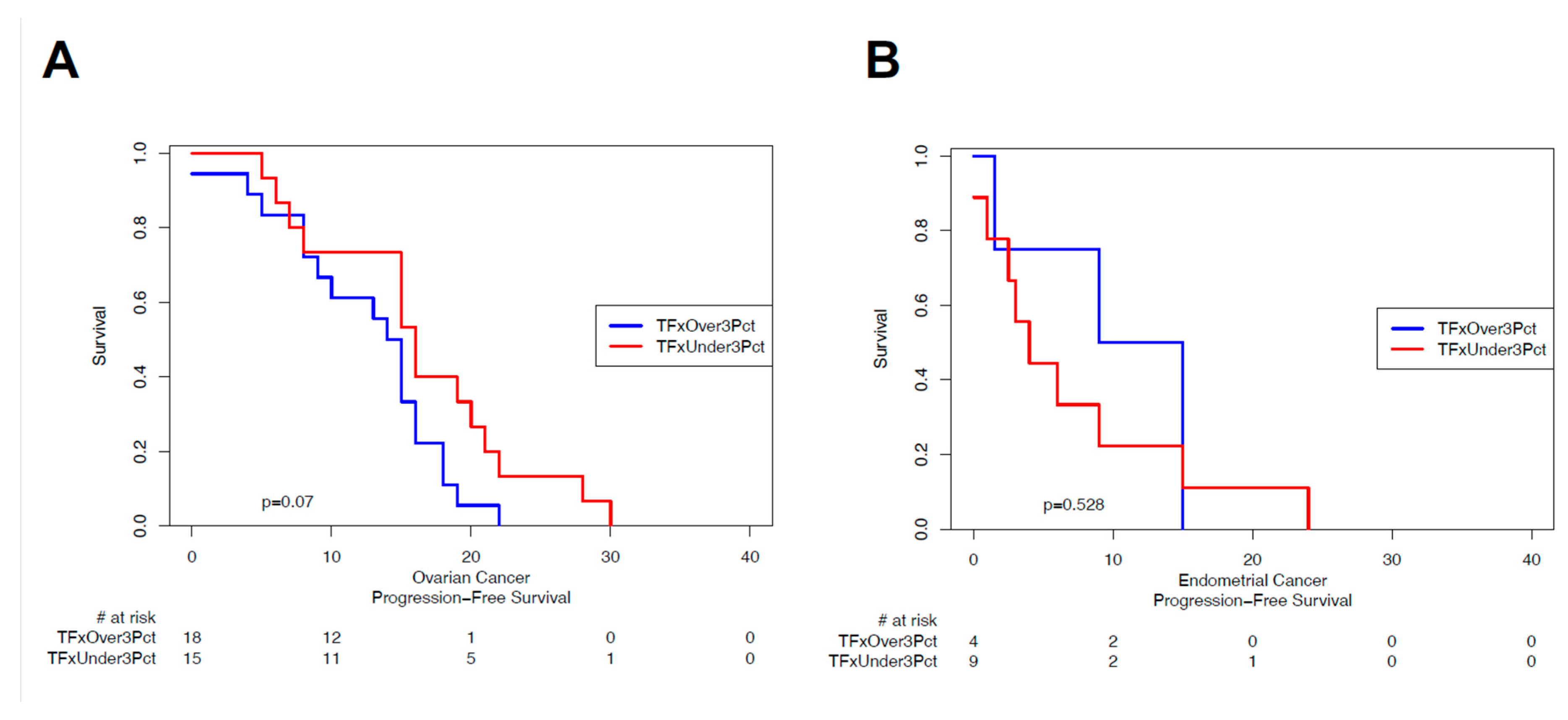
Circulating tumor DNA TFx or similar metrics (e.g. highest variant allele fraction/VAF of detectable mutations) has been associated with prognosis across a variety of cancers [13,17,18,20,24]. Given the heterogeneity of the sample cohort, we investigated the association of ‘sentinel’ TFx with progression-free survival for patients with a sample collected at initiation of therapy for either primary or recurrent ovarian (Figure 3A) or endometrial (Figure 3B) cancers. While various TFx thresholds have been investigated, given the low overall TFx among the cohort we a priori established the 3% TFx ‘detectable’ level as our threshold. For ovarian cancer, there was a non-significant trend (log-rank p=0.07) toward shorter PFS for patients with detectable ctDNA (TFx>3%; Figure 3A). For endometrial cancer, with a limited number of evaluable patients there was no appreciable trend (log-rank p=0.53; Figure 3B).
Figure 3.
Association of circulating tumor DNA tumor fraction with progression-free survival in ovarian and endometrial cancers. Kaplan-Meier visualization of progression-free survival (PFS; x-axis) and proportion free of progression (y-axis) for patients with ovarian cancer (A) and endometrial cancer (B), stratified by tumor fraction (TFx) above versus below 3% at time of ‘sentinel’ blood draw. PFS is determined as time from blood draw to progression or death of any cause. Log-rank p-value indicated.
Figure 3.
Association of circulating tumor DNA tumor fraction with progression-free survival in ovarian and endometrial cancers. Kaplan-Meier visualization of progression-free survival (PFS; x-axis) and proportion free of progression (y-axis) for patients with ovarian cancer (A) and endometrial cancer (B), stratified by tumor fraction (TFx) above versus below 3% at time of ‘sentinel’ blood draw. PFS is determined as time from blood draw to progression or death of any cause. Log-rank p-value indicated.
Discussion
The treatment of advanced gynecologic malignancies has significantly improved patient outcomes in the last decade due to advancements in surgical debulking as well as the development of novel therapeutic targets. However, the treatment of platinum refractory and resistant disease remains challenging. Better diagnostic markers that can identify high-risk patients with molecular residual cancer at the time of primary treatment completion or at the time of recurrence are needed. Our data demonstrates that ctDNA and TFx is a feasible platform to identify patients with disease recurrence and also provides prognostic information in patients with gynecologic malignancy.
Next generation sequencing analysis of ctDNA allows for a non-invasive approach for genomic profiling of tumor without tumor biopsy. Prior studies have demonstrated the role of ctDNA in identifying molecular residual disease and early recurrence in breast, lung and colon cancers [9,10,11].. Chaudhuri et al. recently assessed the role of ctDNA using second generation sequencing (CAPP-Seq) in 40 patients with stage I-III lung cancer and 54 healthy individuals. The authors reported a significant correlation between post-treatment ctDNA and molecular residual disease. Detection of ctDNA preceded radiographic detection in 72% of the patients by 5.2 months [12].. Prior studies have focused on investigating specific mutation or sequencing specific panel of cancer-related genes. There are existing studies that evaluate the utility of ctDNA in the diagnosis, prognostication, detection of residual disease and monitoring of disease in ovarian cancer. However, these studies similarly focus on specific mutations or panels of cancer-related genes. Limitations of this technique include the potential for small ctDNA yield particularly in the setting of low tumor burden, as well as the possibility of false positives due to non-cancer derived mutations [22]. Increasingly, more broadly applicable methods off the capability to quantify TFx via low coverage (0.1x) whole genome sequencing without prior knowledge of tumor mutations, including via the ichorCNA package. In one study of triple-negative breast cancer, the authors reported that ctDNA is feasible in nearly all patients and that tumor fraction >10% was associated with significantly worse survival [13].. This represents a promising tool, but its role has yet to be elucidated in gynecologic malignancies.
The present study evaluated the feasibility and utility of ctDNA TFx in ovarian and endometrial cancer. A major strength of this study was the high number of serial samples from individual patients which allowed for a description of the ctDNA TFx trend over time and correlation with clinical disease status in individual patients. Multiple patients with ovarian cancer demonstrated a high TFx at the time of initial collection followed by rapid decline and persistently low TFx values, corresponding to treatment response. In one patient with recurrent high grade serous ovarian cancer, detection of increasing TFx correlated with disease progression, suggesting that TFx may be a useful tool in the early detection of progression of disease in ovarian cancer. These findings need to be validated in a larger sample size but may be an important tool to aid in patient counseling and clinical decision-making.
Moreover, this study demonstrated that for ovarian cancer patients with a detectable ctDNA TFx>3%, there was a trend toward shorter PFS when compared to those with TFX<3%. This trend was not appreciable in the endometrial cancer cohort, however, most endometrial cases exhibited many or all samples below the 3% TFx detection threshold, possibly indicating tumors less prone to shedding DNA. This potential correlation of TFx and survival and prognosis carries significant importance and needs to be investigated on a larger scale.
There are several limitations to this study. First, this study is limited by its small sample size. Future studies evaluating the use of TFx in gynecologic malignancies using larger cohorts are necessary to validate these findings. Further, direct comparison of cases with uneven number and/or times of sampling events is difficult, and subsequent investigations will benefit from standardization of TFx sampling across cases. Additionally, the use of TFx in ovarian and endometrial cancer will need to be correlated with targeted panel or whole exome sequencing, which will be the focus of a forthcoming study.
Conclusions
This study demonstrates that collection of ctDNA TFx is not only feasible in ovarian and endometrial cancers but also may be an important tool for early disease recurrence detection and prognostication. Future studies with larger patient cohorts are needed to validate these findings.
Author Contributions
Conceptualization, DGS, HM.; Methodology, MV, DGS, HM.; Formal Analysis, MV, DGS, HM; Data Curation, AG, PR, HM; Writing – Original Draft Preparation, MV, AG, KC, DGS, HM; Writing – Review & Editing, all authors.; Visualization, MV, DGS.; Supervision, HM; Funding Acquisition, DGS, HM.
Funding
This research was funded by Cleveland Clinic Philanthropy.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of Cleveland Clinic.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to restrictions secondary to information that could compromise the privacy of research participants.
Acknowledgments
The authors would like to acknowledge the Broad Institute Genomics Platform for sequencing support.
Conflicts of Interest
DGS has served on an advisory board for Novartis.
References
- American Cancer Society I. Cancer Facts & Figures 2023. Vol 2023. Atlanta.
- Pierce SR, Clark LH. Current First-line Therapy for Ovarian Cancer: A Comprehensive Review. Obstet Gynecol Surv. 2018;73:650-657. [CrossRef]
- Luvero D, Plotti F, Aloisia A, et al. Ovarian cancer relapse: From the latest scientific evidence to the best practice. Crit Rev Oncol Hematol. 2019;140:28-38. [CrossRef]
- Fotopoulou C, Hall M, Lord R, et al. Perspectives of Healthcare Professionals on the Management and Treatment of Advanced Ovarian Cancer in the UK: Results From the KNOW-OC Survey. Clin Oncol (R Coll Radiol). 2023.
- Wang Q, Zheng Y, Wang P, et al. The prognostic factor for recurrence in advanced-stage high-grade serous ovarian cancer after complete clinical remission: a nested case-control study. J Ovarian Res. 2021;14:179. [CrossRef]
- Piedimonte S, Kim R, Bernardini MQ, et al. Validation of the KELIM score as a predictor of response to neoadjuvant treatment in patients with advanced high grade serous ovarian cancer. Gynecol Oncol. 2022;167:417-422. [CrossRef]
- Pignata S, Cannella L, Leopardo D, Bruni GS, Facchini G, Pisano C. Follow-up with CA125 after primary therapy of advanced ovarian cancer: in favor of continuing to prescribe CA125 during follow-up. Ann Oncol. 2011;22 Suppl 8:viii40-viii44. [CrossRef]
- Zachou G, El-Khouly F, Dilley J. Evaluation of follow-up strategies for women with epithelial ovarian cancer following completion of primary treatment. Cochrane Database Syst Rev. 2023;8:CD006119. [CrossRef]
- Bell D, Berchuck A, Birrer M, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-615. [CrossRef]
- Zhang Q, Burdette JE, Wang J-P. Integrative network analysis of TCGA data for ovarian cancer. BMC Systems Biology. 2014;8:1338. [CrossRef]
- Iijima M, Banno K, Okawa R, et al. Genome-wide analysis of gynecologic cancer: The Cancer Genome Atlas in ovarian and endometrial cancer. Oncology letters. 2017;13:1063-1070. [CrossRef]
- Alix-Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annual review of medicine. 2012;63:199-215. [CrossRef]
- Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nature communications. 2017;8:1324. [CrossRef]
- Choudhury AD, Werner L, Francini E, et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI insight. 2018;3. [CrossRef]
- Merker JD, Oxnard GR, Compton C, et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36:1631-1641. [CrossRef]
- Zill OA, Greene C, Sebisanovic D, et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer discovery. 2015;5:1040-1048. [CrossRef]
- O'Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nature communications. 2018;9:896. [CrossRef]
- Stover DG, Parsons HA, Ha G, et al. Association of Cell-Free DNA Tumor Fraction and Somatic Copy Number Alterations With Survival in Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2018;36:543-553. [CrossRef]
- Dandachi N, Posch F, Graf R, et al. Longitudinal tumor fraction trajectories predict risk of progression in metastatic HR+ breast cancer patients undergoing CDK4/6 treatment. Molecular Oncology. 2021;15:2390-2400. [CrossRef]
- Reichert ZR, Morgan TM, Li G, et al. Prognostic value of plasma circulating tumor DNA fraction across four common cancer types: a real-world outcomes study. Ann Oncol. 2023;34:111-120. [CrossRef]
- Van Roy N, Van Der Linden M, Menten B, et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin Cancer Res. 2017;23:6305-6314. [CrossRef]
- Asante DB, Calapre L, Ziman M, Meniawy TM, Gray ES. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020;468:59-71. [CrossRef]
- Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals 1 high concordance with metastatic tumors. Nature Communications. 2017:In Press.
- Choudhury AD, Werner L, Francini E, et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI Insight. 2018;3. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



