
Submitted:
21 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
Epigenetic changes are changes in gene expression that do not involve alterations to the basic DNA sequence. These changes lead to establishing a so-called epigenetic code that dictates which and when genes are activated, thus orchestrating gene regulation and playing a central role in development, health, and disease. The brain, being for the most formed by cells that do not undergo a renewal process through life, is highly prone to the risk of alterations leading to neuronal death and neurodegenerative disorders, mainly at late age. Here we review the main epigenetic modifications that have been described in the brain, with particular attention to those related to the onset of developmental anomalies or neurodegenerative conditions and/or occurring in old age. DNA methylation and several types of histone modifications (acetylation, methylation, phosphorylation, ubiquitination, sumoylation, lactylation, and crotonylation) are major players in these processes. They are directly or indirectly involved in the onset of neurodegeneration in Alzheimer’s or Parkinson’s disease. Therefore, this review briefly describes the role of these epigenetic changes in the mechanisms of brain development, maturation, and aging and some of the most important factors dynamically regulating or contributing to these changes such as oxidative stress, inflammation, and mitochondrial dysfunction.
Keywords:
Epigenetics
; DNA
; Histones
; Brain
; Neurons
; Development
; Neurodegeneration
1. Introduction
Understanding the cellular and molecular mechanisms underlying aging is of paramount importance due to the profound impact that aging has on human health and society. Under this perspective, epigenetics provides a crucial framework for comprehending its molecular underpinnings. The genetic code, inscribed within the DNA sequence, serves as the blueprint for life. Yet, within each cell, an additional layer of information, the epigenetic code, dictates which and when genes are activated, thus orchestrating gene regulation and playing a central role in development, health, and disease.
Epigenetics, a term coined by the British developmental biologist Conrad Waddington in the mid-20th century [1], is the study of the changes in gene expression that do not involve alterations to the underlying DNA sequence. Epigenetic changes can be influenced by a variety of factors, including developmental processes, environmental exposures, and lifestyle choices. Over time, epigenetic modifications can accumulate, leading to the establishment of an “epigenetic landscape” unique to an individual’s aging process. During aging, changes in these epigenetic marks can lead to alterations in gene expression patterns, contributing to age-related phenotypes and diseases. Among the several types of epigenetic modifications, specific DNA methylation patterns correlate with chronological age. On these premises, researchers have developed a series of so-called epigenetic clocks that provide a molecular measure of aging and can be used to assess the biological age of an individual, which may differ from his chronological age [2]. Understanding the epigenetic changes associated with aging opens avenues for potential interventions to slow down or reverse age-related conditions. Starting from this understanding, epigenetic therapies are being explored to rejuvenate tissues and combat age-related diseases.
1.1. Types of epigenetic modifications
There are three main groups of epigenetic modifications: DNA methylation, histone modifications, and non-coding RNAs (Figure 1).
DNA Methylation: The addition of a methyl group to a cytosine base in the DNA, typically occurring in the context of CpG dinucleotides, is known as DNA methylation, and often leads to gene silencing. The CpG sites, also known as CG sites, refer to certain regions inside the DNA molecule where a cytosine nucleotide is immediately followed by a guanine nucleotide in the linear arrangement of bases along its 5‘ → 3‘ orientation. CpG sites are found at a high frequency inside genomic areas known as CpG islands, also referred to as CG islands. DNA methylation acts as a molecular “off switch” by preventing transcription factors and RNA polymerase from accessing the promoter region of the gene, thereby inhibiting gene expression [3]. DNA methyltransferases (DNMTs), methyl-CpG binding proteins (MBPs), and ten-eleven translocation proteins enable the maintenance, interpretation, and removal of DNA methylation [4] (Table 1). Different forms of methylation, including 5-methylcytosine (5mC), 5-hydroxymethylcytosine, and other oxidized forms, have been detected by recently developed sequencing technologies.
Histone Modifications: Histone modifications are central regulators of gene expression, as they determine which genes are turned on or off in each cell or tissue, affecting cellular function and, in the general frame of this paper, contributing to the aging phenotype. Histones are protein spools around which DNA is wound. Chemical modifications, such as acetylation, methylation, phosphorylation, ubiquitination, sumoylation, lactylation, and crotonylation can alter the structure of histones and, consequently, regulate DNA transcription. The interplay of these modifications at specific histone residues creates a dynamic “chromatin landscape” that can either promote or inhibit gene transcription. Thus, by modifying chromatin structure and accessibility the different types of histone modifications provide a complex regulatory framework that governs gene expression. The precise combinations of these modifications at specific histone residues create a “histone code” that can be read and interpreted by various cellular machinery to dictate gene transcription outcomes [5].
Non-coding RNAs (ncRNAs): Small RNA molecules, like microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), can bind to messenger RNAs (mRNAs) and inhibit their translation or promote their degradation. This post-transcriptional gene regulation is a fundamental part of epigenetic control [6].
1.1.1. DNA Methylation
Remarkably, the brain exhibits a notable concentration of DNA methylation, but it is important to note that 5mC only constitutes around 1% of the total nucleic acids present in the mammalian genome, which, in general, shows a scarcity of CpG sites. CpG sites that are observed across the genome exhibit a significant level of methylation, except in CpG islands. It is noteworthy that non-CpG methylation has been observed in both mouse and human embryonic stem cells. However, it is important to note that this methylation is not present in mature tissues [7]. Recent research has provided a more comprehensive examination of the murine frontal cortex. This investigation has demonstrated that while the bulk of methylation events take place at CpG sites, a notable proportion of methylation also happens at non-CpG sites.
Significantly, the impact of DNA methylation on gene activity can vary depending on the specific genomic areas and the underlying genetic sequence. DNA methylation can occur in intergenic regions, CpG islands, and the gene body [7]. An estimated 45% of the mammalian genome is comprised of transposable and viral elements, which are rendered inactive by the process of bulk methylation in the DNA intergenic regions. The potential danger of these elements lies in their ability to cause gene disruption and DNA mutation when replicated and inserted. A significant proportion of gene promoters (frequently of housekeeping genes), around 70%, are located within CpG islands. CpG islands, particularly those linked to promoters, exhibit a significant degree of conservation across both mice and humans. The presence and conservation of CpG islands over evolutionary time suggest that these genomic areas hold significant functional significance. The gene body is commonly defined as the region of the gene that extends beyond the initial exon, as it has been observed that methylation of the first exon, like promoter methylation, can result in the suppression of gene expression. Multiple studies have provided evidence indicating that the process of DNA methylation occurring within the gene body is positively correlated with an increased level of gene expression in cells undergoing division. Nevertheless, in cells that divide at a slow rate or do not divide at all, such as those found in the brain, it has been observed that gene body methylation does not correlate with upregulation of gene expression [7].
1.1.2. Histone epigenetic modifications
Histones are proteins found in the cell nucleus that play a critical role in packaging and organizing DNA into a compact structure called chromatin. Epigenetic modifications of histones involve chemical alterations to these proteins, which can have profound effects on gene expression and, consequently, various cellular processes. These modifications are essential for the regulation of gene activity and have far-reaching implications in development, health, and disease.

Table 2.
Histone modifications and their biological effects. Amino acid residues are indicated by one-letter notation. Abbreviations: AC =acetylation; Cr = crotonylation; Las =lactylation; Me = methylation; P = phosphorylation; Su = sumoylation; Ub = ubiquitination.
Table 2.
Histone modifications and their biological effects. Amino acid residues are indicated by one-letter notation. Abbreviations: AC =acetylation; Cr = crotonylation; Las =lactylation; Me = methylation; P = phosphorylation; Su = sumoylation; Ub = ubiquitination.
| Histone | Type of modification | Residue(s) | Biological effect |
|---|---|---|---|
| H1 | Su | - | Gene repression, chromatin compaction |
| H2A | Ac | K5 | Gene activation |
| P | S1 | Mitosis | |
| P | T120 | Mitosis, gene activation | |
| Su | - | Gene repression, chromatin compaction | |
| Ub | K119 | Gene repression | |
| H2AX | P | S139 | DNA repair |
| Su | K5, K9, K13, K15, K118, K119, K127, K133, K134 | Gene repression, chromatin compaction | |
| H2B | Ac | K5, K12, K15, K20 | Gene activation |
| P | S14 | Apoptosis | |
| Su | - | Gene repression, chromatin compaction | |
| Ub | K12 | Gene activation | |
| H3 | Ac | K4, K9, K14, K18, K23, K27, K36 | Gene activation |
| Ac | K56 | Histone deposition | |
| Cr | K9 | DNA repair | |
| Cr | K4, K14, K18, K27 | Gene activation | |
| La | K4, K18, K79 | Gene activation | |
| Me | K9, K27 | Gene repression | |
| Me | R2, R8, R17, R26 | Gene activation | |
| P | T6 | Gene activation | |
| P | S10, S28, T3, T11 | Mitosis, DNA repair | |
| P | T45 | DNA replication | |
| Ser | Q5 | Gene activation | |
| Su | K18 | Gene repression, chromatin compaction | |
| Ub | K23 | Maintenance of DNA methylation | |
| H4 | Me | R3 | Gene activation |
| P | S1 | Mitosis, gene activation | |
| Ac | K12, K91 | Histone deposition | |
| Ac | K5, K8, K16 | Gene activation | |
| Me | K20 | Gene repression | |
| Su | K12 | Gene repression, chromatin compaction |
Several key histone modifications have been extensively studied. These include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, lactylation, and crotonylation (Table 2 and Figure 1 and Figure 2).
Acetylation, the addition of an acetyl group typically on lysine residues on histone tails, is generally associated with gene activation. Acetyl groups are added to histones by histone acetyltransferases (HATs). This process neutralizes the positive charge on histones, leading to a relaxed chromatin structure that is more open and accessible to transcription factors and RNA polymerase [8]. The ensuing changes affect the accessibility of DNA to cellular machinery [9].
Methylation, the addition of 1-3 methyl groups to lysine or arginine residues on histone proteins, is carried out by histone methyltransferases (HMTs) [10] and can be associated with both gene activation (by recruiting chromatin-modifying complexes) and repression (forming barriers to inhibit transcription), depending on the specific histone residue and the degree of methylation [11].
Phosphorylation is associated with changes in chromatin structure during various cellular processes, including DNA replication, repair, and mitosis. Histone phosphorylation is the addition of phosphate groups to serine, tyrosine, or threonine residues on histone tails. The modification can alter chromatin structure and facilitate gene activation or repression [12].
Ubiquitination, the addition of ubiquitin molecules to specific lysine residues on histone tails, is carried out by ubiquitin ligases. Ubiquitination can affect gene expression as it alters chromatin structure by recruiting proteins that either activate or repress transcription. It plays a role in transcriptional regulation and DNA repair [13].
Sumoylation, the addition of small ubiquitin-like modifier (SUMO) proteins to specific lysine residues on histone tails, is catalyzed by SUMO ligases. Sumoylation can affect chromatin structure and gene expression by recruiting proteins that modulate transcriptional activity and contribute to genome stability [14].
Lactylation is the addition of lactate to histone molecules. Lactylation can accelerate transcription and promote gene expression. It has been implicated in several disease model molecules [15].
Crotonylation is the addition of crotonyl groups to histone lysine residues. Crotonylation has a role in DNA damage and repair, and gene activation [16].
The above histone modifications are pivotal for gene regulation and genome stability. They contribute to various cellular processes, including gene expression, cell differentiation, and DNA repair, and are thus crucial in cell fate determination during development contributing to the establishment of lineage-specific gene expression patterns. Remarkably, some histone modifications can be passed on to daughter cells during cell division, contributing to the so-called “epigenetic inheritance” [17].
1.1.3. ncRNAs
ncRNAs that do not undergo translation to produce proteins can be categorized into two main groups: housekeeping ncRNAs and regulatory ncRNAs. RNA molecules with regulatory functions can be broadly classified into two main categories according to their size: short-chain non-coding RNAs, which encompass small interfering RNAs (siRNAs), miRNAs, and PIWI-interacting RNAs (piRNAs), and lncRNAs [18]. siRNAs have a size of 19-24 bp, derive from double-strand DNA, and silence gene transcription. miRNAs are 19-24 bp long, originate from hairpin-containing primary transcripts (pri-miRNA), and silence gene transcription. piRNAs are of larger size (26-31 bp), derive from a long chain size precursor, and repress transposons via transcriptional or posttranscriptional mechanisms. lncRNAs have a size of more than 200 bp, derive from multiple sources, and regulate gene expression in various ways, including epigenetic, transcriptional, post-transcriptional, translational, and protein location mechanisms.
2. Epigenetic regulation in the developing and mature brain
Epigenetic modifications are central players in the intricate processes of brain development and function. The dynamic interplay between genetics and epigenetics shapes the complexity of the brain and its ability to adapt to a constantly changing environment. Broadly speaking, epigenetic and epitranscriptomic changes, i.e., the RNA editing that affects mRNA functions, regulates neuronal lineage, differentiation, and connectivity with obvious consequences on the structure and function of synapses. In addition, both types of changes have been associated with several neurodevelopmental disorders [19]. Interestingly, dysregulation of epigenetic processes has been implicated in autism spectrum disorder (ASD) and intellectual disability, further highlighting the significance of epigenetics in brain development and maturation [20].
In the developing mammalian cortex, radial glial cells (RGCs) act as primary neural stem cells (NSCs) and give rise to a variety of neurons and glial cells following intricate developmental programs with astounding spatiotemporal accuracy. Controlling RGCs’ temporal competency is a crucial mechanism for the cerebral cortex’s highly preserved and predictable structure. Remarkably, the pattern of gene expression of RGCs is largely shaped by several epigenetic controls, including DNA methylation, histone modifications such as H3K4me3 and H3K27me3, and 3D chromatin architecture [21]. Epitranscriptomic changes, such as m6A-eRNA methylation and m5C RNA methylation also control the function and turnover of cell-type-specific transcripts, which in turn regulates the temporal pre-patterning of RGCs [21]. DNA methylation patterns have a well-recognized role in NSC proliferation and differentiation and help establish and maintain the specific identity of neurons, contributing to the diversity of neuronal subtypes in the brain. It was demonstrated that during the transition from fetal to young adult development, there is a significant rearrangement of the methylome, which is closely associated with the process of synaptogenesis. During this temporal phase, there is a notable accumulation of highly conserved non-CG methylation (mCH) specifically in neurons, but glial cells do not exhibit a similar pattern [22]. Consequently, mCH emerged as the prevailing form of methylation within the human neuronal genome. Other studies provided comprehensive and high-resolution maps of 5-hydroxymethylcytosine (hmC) at the single-base level. These maps have revealed that hmC is present in the genomes of fetal brain cells, specifically marking sites that are believed to be involved in regulatory processes [22]. Growing evidence also suggests that DNA cytosine and hydroxyl cytosine methylation carried by DNMTs and/or MBPs plays a pivotal role in neurogenesis, neuronal differentiation, synaptogenesis, learning, and memory [23] (Table 2). It has been also recently demonstrated that DNA methylation regulates the differentiation of oligodendrocytes and Schwann cells during development and repair [24]. Remarkably, experience-dependent DNA methylation, can modify gene expression and contribute to the brain’s ability to adapt to environmental challenges [25]. On the other hand, the mechanisms of deviant DNA methylation in neurodegenerative diseases continue to be unclear. Remarkably, however, DNA methylation modifies and potentially restores youthful gene expression patterns as drugs targeting this epigenetic modification, such as 5-azacytidine and decitabine, can reverse age-related neurodegeneration [26].
Histone methylation and acetylation guide the differentiation of NSCs into various neural cell types, including neurons and glial cells [27]. In contrast to the reversible and dynamic nature of acetylation, which is primarily linked to the expression of specific genes, histone methylation is characterized by its stability and potential involvement in the long-term maintenance of specific genomic areas [27]. Histone methylation is critical for the regulation of neurodevelopmental processes, synaptic plasticity, and the formation of long-term memories [28]. In particular, lysine methylation has been shown as a direct contributor to epigenetic inheritance, and H3K4me has been found to promote transcriptional activation, while H3K9me is related to transcriptional suppression [27] (Table 1). Histone acetylation, particularly at genes associated with synaptic plasticity, plays a critical role in memory formation and the ability of neurons to strengthen or weaken their connections [29]. Previous research demonstrated that there was an elevation in histone acetylation inside the hippocampus following training, in contrast to untrained control subjects, a reduction in histone acetylation was observed in other brain areas, such as the cortex (reviewed in [29]). Lysine deacetylase (KDAC) inhibitors, such as trichostatin A (TSA) and sodium butyrate (NaB), have been shown to augment long-term potentiation (LTP). Additionally, the administration of NaB through systemic injection has been demonstrated to enhance memory in vivo. The administration of TSA through intrahippocampal injection immediately following the learning process leads to improvements in long-term memory, while having no impact on short-term memory. This finding suggests that histone acetylation plays a crucial role in the consolidation of memory. Research has also indicated that the administration of NaB can promote the consolidation of long-term memory in response to mild stimuli and prolong the persistence of long-term memory. Therefore, it can be inferred that histone acetylation plays a significant part in the process of long-term memory formation [29]. Remarkably, histone acetylation which has been associated with the establishment of long-term memory and synaptic plasticity, may take place at many lysine residues located within the four core histone proteins. It is worth noting that alterations in histone acetylation are associated with AD and may serve as potential diagnostic and therapeutic targets [30].
Histone phosphorylation is associated with synaptic plasticity and learning contributing to the regulation of immediate early genes in response to neuronal activity, which is crucial for memory consolidation [31]. Histone H2B ubiquitination recruits H3K4me3 and plays a role in the regulation of several genes involved in neurodevelopment and synaptic plasticity [32]. Histone sumoylation contributes to the epigenetic regulation of genes involved in neuronal differentiation and synaptic plasticity by modulating N-methyl-D-aspartate (NMDA) receptors, and L- and N-type voltage-gated calcium channels [33]. Crotonylation is another epigenetic modification that has been demonstrated in NSCs. This type of epigenetic mark is involved in NSC self-renewal and differentiation (by protecting pluripotency factors), as well as telomere protection [16].
ncRNAs, including miRNAs, regulate gene expression at synapses, influencing synaptic plasticity and learning processes [34]. Among the regulated genes is cAMP response element binding protein 2 (CREB2) which is crucial for long-term synaptic plasticity. The identification of distinctive DNA non-coding regulatory sequences that are important in brain cell differentiation, maturity, and plasticity has also been made possible by genome-wide analysis of epigenetic changes. Genomic enhancer elements are brief DNA regulatory sequences that bind transcription factors and work with gene promoters to increase transcriptional activity. This mechanism regulates gene expression programs crucial for determining the fate and function of neurons and is linked to many brain disease states [35]. Neurons are mostly rich in enhancers, which undergo bidirectional transcription to generate non-coding enhancer RNAs (eRNAs) and underlie dynamic gene expression patterns and cell-type-specificity [35]. A list of references on the enhancers linked to neuronal development can be found in Supplementary Table S1 from [35].
Dynamic regulation of epigenetic modifications in response to environmental factors
DNA methylation is a stable epigenetic modification; however, it can be dynamically altered in response to environmental factors such as diet, stress, toxins, and early-life experiences [36]. Studies have e.g., shown that maternal diet during pregnancy can lead to changes in DNA methylation patterns in offspring, affecting long-term health outcomes [37].
Histone modifications can also be dynamically regulated in response to environmental factors, a phenomenon known as epigenetic plasticity. This process allows the genome to adapt to changing conditions and underscores the interaction between genes and the environment. Positive environmental factors, such as cognitive stimulation and physical activity, can promote histone modifications associated with synaptic plasticity and learning. Studies in rodents have shown that environmental enrichment can lead to increased histone acetylation and improved cognitive function [38]. On the other hand, exposure to drugs and environmental toxins also affects histone acetylation [39]. Chronic exposure to addictive substances, such as cocaine, can lead to changes in histone acetylation patterns in reward-related brain regions, contributing to addiction-related behaviors [40]. Histone methylation can also be dynamically regulated by stressors, including physical and psychological stress. Stress-induced changes in histone methylation can affect gene expression patterns in several areas of the brain [41], and chronic stress can lead to alterations in histone methylation in genes associated with mood regulation, contributing to the development of mood disorders [42].
3. Epigenetic modifications in the aging brain
The decline in physiological functions that characterize aging is particularly apparent in the brain, which is mainly populated by postmitotic neurons that cannot be renewed and are therefore at risk of alterations leading to neurodegenerative disorders and/or neuronal death. One of the hallmark structural changes in the aging brain is a decrease in brain volume (atrophy), particularly in regions associated with memory and higher-order cognition, such as the hippocampus and the prefrontal cortex, which are critical for recall and executive function. While not as dramatic as in neurodegenerative diseases, the physiologically aging brain, particularly in selected regions, experiences some degree of neuronal loss, which can contribute to the cognitive deficits observed in the elderly [43]. Another prominent feature of brain aging is the ability of dendritic spines to change their structure. Age-related reductions in spine number and maturity, as well as changes in synaptic transmission, maybe a direct result of abnormal neural plasticity that affects the aged brain [44].
In addition, age-related alterations in the white matter, including demyelination and reduced integrity of white matter tracts, can lead to slowed information processing and cognitive decline [45].
Functional changes comprise cognitive decline with a reduction in processing speed, working memory, which is responsible for temporarily holding and manipulating information [46], episodic memory, which involves the ability to recall specific events and details [47], and changes in attention, including reduced ability to filter out irrelevant information, which can affect task performance [48]. Other functional alterations disturb neurotransmitter systems, particularly the decline in dopamine and acetylcholine levels [49], and functional connectivity patterns within the brain’s networks, altering information processing and integration [50].
Aging has an obvious impact on neurological disorders, being the primary risk factor for neurodegenerative disorders such as
Recent research has shown that epigenetic changes, specifically modifications to histones and DNA, play a pivotal role in the aging process and the development of age-related neurological conditions such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [51]. Aging also increases susceptibility to stroke and cerebrovascular diseases due to vascular changes including reduced cerebral blood flow and the development of small vessel disease [52]. There are several ways by which epigenetic modifications can contribute to age-related cognitive decline. As mentioned in the previous section, the influence of epigenetic marks on the expression of genes associated with neuroplasticity and synaptic function may be effective in old age leading, broadly speaking, to impairment of brain structure and function [53]. For example, alterations in histone acetylation and DNA methylation are associated with AD and may serve as potential diagnostic and therapeutic targets [30]. Likewise, H3K27cr has been observed in AD to regulate exocytotic mechanisms of amyloid β clearance [54] and H4K12la is specifically activated in plaques of the 5XFAD mouse [55] Epigenetic modifications in the aging brain can also serve as biomarkers for predicting age-related cognitive decline and the risk of developing neurodegenerative disorders [56]. Epigenetic clocks, which estimate biological age based on DNA methylation patterns, have shown promise in this regard [57]. Epigenetic clocks have indeed revealed that the epigenetic age of brain tissues can differ from chronological age and that at least certain areas of the brain may undergo accelerated aging [58]. Therefore, epigenetic clocks have been linked to longevity and age-related health outcomes, and further research in the field may provide insights into the mechanisms underlying healthy aging [59].
An important feature of epigenetic modifications is that they are reversible, making them attractive targets for therapy [60]. Therefore, developing drugs or interventions that can modify epigenetic marks may offer avenues for slowing down the aging process or mitigating age-related neurodegenerative diseases [61]. Also of importance is that epigenetic changes in the aging brain are influenced by environmental factors, including diet, physical activity, and stress. Thus, understanding how these factors impact epigenetic modifications can inform lifestyle interventions that promote healthy brain aging [62].
Oxidative stress, chronic inflammation, changes in chromatin remodeling, dysregulation of the enzymes involved in histone regulation, senescent cells, and telomere shortening are among the several factors that may contribute to histone epigenetic changes in the aging brain.
3.1. Contribution of oxidative stress to epigenetic changes in the aging brain
Oxidative stress is a prominent factor in the aging process and a hallmark of aging. Among its several consequences, oxidative stress may lead to different types of epigenetic changes from DNA methylation to histone modifications, and non-coding RNA profiles that can influence gene expression and contribute to age-related neurodegenerative conditions. Overall, oxidative stress-induced DNA damage can impair the enzymes responsible for maintaining the epigenetic marks, leading to their dysregulation [63]. DNA damage can trigger changes in histone modifications, including increased histone H3K9 acetylation, which is associated with DNA repair processes [64]. Several effects have been described because of oxidative stress, including aberrant methylation of CpG sites, resulting in DNA hypomethylation or hypermethylation. This can in turn affect the expression of genes involved in neuroprotection, synaptic plasticity, and inflammation [65]. Oxidative stress can also disrupt the balance of histone modifications. For instance, increased levels of oxidative stress may reduce acetylation and promote deacetylation, leading to transcriptional repression of neuroprotective genes [66]. Oxidative stress can likewise influence the expression of non-coding RNAs, including miRNAs and lncRNAs. These non-coding RNAs can regulate the expression of genes associated with neurodegenerative processes [67].
3.2. Contribution of inflammation to epigenetic changes in the aging brain
Inflammation is a central feature of aging-related neurodegenerative diseases, and it is increasingly recognized as a contributor to epigenetic changes in the aging brain. Epigenetic modifications, including DNA methylation and histone acetylation, can regulate the expression of pro-inflammatory genes and trigger a vicious circle that contributes to the sustained activation of inflammatory pathways [68].
Alterations in DNA methylation patterns may affect the regulation of genes involved in immune responses, oxidative stress, and neuroinflammation [69]. Chronic inflammation sustained by pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα), can lead to histone modifications that promote gene expression changes associated with inflammatory responses [70]. Thus, inflammation can increase histone acetylation at pro-inflammatory gene promoters, with sustained activation of inflammatory pathways [42]. Inflammatory processes can also alter the expression of miRNAs that target genes involved in neuroinflammation and neurodegeneration [71].
Another source of inflammation in the aging brain derives from senescent cells. Cells undergo a process known as cellular senescence in which they alter their normal phenotype in response to stress and enter a prolonged cell cycle arrest state accompanied by a distinctive secretory phenotype [72], referred to as senescence-associated secretory phenotype (SASP) with secretion, among others, of pro-inflammatory cytokines, growth factors, matrix-remodeling enzymes, and miRNAs. Additionally, senescent cells exhibit altered morphology and proteostasis, decreased propensity to go through apoptosis, impaired autophagy, accumulation of lipid droplets, and increased activity of senescence-associated-galactosidase (SA-gal). It is worth noting that SASP components can influence epigenetic changes such as DNA methylation, chromatin remodeling, and histone post-translational modifications in nearby cells [73] and that senolytic drugs selectively targeting and eliminating senescent cells have the potential to reduce inflammation and oxidative stress [44].
3.3. Contribution of mitochondrial dysfunction to epigenetic changes in the aging brain
Mitochondrial dysfunction, including mitochondrial stress, increased oxidative damage, and reduced ATP production, can lead to alterations in DNA methylation patterns. These changes may affect the regulation of genes involved in energy metabolism, oxidative stress responses, and neuronal survival [74]. Mitochondrial dysfunction can also lead to altered expression of non-coding RNAs, including miRNAs and lncRNAs. Dysregulated non-coding RNAs can target genes involved in mitochondrial biogenesis, oxidative stress responses, and neuronal maintenance [75]. Epigenetic modifications regulate the expression of mitochondrial genes. These modifications can affect the efficiency of mitochondrial energy production and oxidative stress responses [76]. Mitochondrial dysfunction can lead to the release of mitochondrial-derived signals, such as reactive oxygen species (ROS) and mitochondrial DNA fragments. These signals can influence epigenetic changes in nearby cells, including neurons, leading to altered gene expression patterns [77].
3.4. Other factors contributing to epigenetic changes in the aging brain
Age-related changes in chromatin remodeling complexes can influence histone modifications. For instance, reduced activity of ATP-dependent chromatin remodeling complexes can lead to changes in histone acetylation and methylation patterns [78]. Likewise, age-related dysregulation of enzymes responsible for adding (writers) or removing (erasers) histone modifications can result in imbalanced histone marks. For example, altered activity of HATs or histone deacetylases (HDACs) can affect histone acetylation levels [79]. In keeping with these observations, small molecules targeting epigenetic enzymes, such as DNMTs and HDACs, are being investigated as potential interventions to reverse age-related epigenetic changes and restore youthful gene expression patterns [80]. Finally, telomere shortening, a characteristic of aging, can trigger chromatin alterations and changes in histone modifications at telomeric regions. These alterations can affect gene expression near telomeres [81].
4. Histone modifications and brain aging
Recent research has shed light on the specific histone modifications linked to brain aging (Table 3). Studies have shown that a decrease in histone acetylation, particularly at genes associated with memory and synaptic plasticity, is associated with cognitive decline in aging individuals [82]. Age-related changes in histone methylation patterns have been observed in the brains of older individuals, and alterations in methylation at specific genes are linked to neurodegenerative diseases and cognitive decline [83]. We have recently shown that in the old mouse brain histone H2AXγ phosphorylation is associated with caspase-dependent cell death and abortive cell cycle re-entry [84, 85]. Developing small molecules that selectively target specific histone modifications, such as H3K4me3 or H3K27me3, may allow for precise modulation of gene expression relevant to cognitive function and neuroprotection [86]. It is worth mentioning that most studies examine bulk brain tissue, which may mask cell-type-specific epigenetic changes. Investigating histone modifications at the cellular level, especially in specific neuronal subtypes, can provide further insights into their roles in brain aging. In addition, much of the focus of current research has been on promoter regions, but understanding the role of histone modifications in enhancers, non-coding RNAs, and other non-coding regions is crucial for a comprehensive view of epigenetic regulation in brain aging [35].
As mentioned, dysregulation of histone modifications has been implicated in neurodegenerative diseases such as AD and PD which display a typical old-age onset. However, the exact role of histone modifications in disease pathogenesis and progression is not fully understood [87]. Studies have revealed alterations in histone modifications in the brains of individuals with AD. These changes include global reductions in histone acetylation levels and alterations in histone methylation patterns [82]. In AD, reduced histone acetylation, particularly at genes associated with memory and synaptic function, is linked to cognitive decline, and aberrant histone acetylation and methylation patterns are associated with disease progression [88]. Epigenetic drugs, such as histone deacetylase inhibitors (HDACIs), have shown promise in preclinical studies for their ability to reverse cognitive deficits and reduce amyloid-beta levels in animal models of Alzheimer’s disease [88]. The use of HDACIs to restore histone acetylation levels is thus a potential therapeutic approach to mitigate cognitive deficits in AD [89]. Aberrant histone methylation patterns are associated with tau pathology, one of the hallmarks of AD. Histone methylation marks have been found at specific tau gene promoters, affecting tau protein expression [90]. Targeting HMTs involved in tau regulation could thus represent another potential therapeutic strategy to reduce tau pathology.
Emerging evidence suggests that epigenetic dysregulation, including histone modifications, also contributes to the pathogenesis of PD. These changes can affect gene expression patterns in the brain, influencing dopaminergic neuronal function and survival [91]. Altered histone acetylation patterns have been observed in animal models and post-mortem brains of PD patients. These changes can affect the expression of genes involved in neuroinflammation and mitochondrial dysfunction, contributing to PD pathogenesis [92]. Sirtuin 1 (SIRT1), a histone deacetylase, plays a crucial role in regulating aging-related processes. SIRT1 can be activated through compounds like resveratrol that may promote neuroprotection and cognitive function [93]. As in the case of AD, targeting HDACs to modulate histone acetylation levels is being explored as a potential therapeutic approach for PD [94]. Interestingly, aberrant histone methylation patterns have been linked to alpha-synuclein aggregation, a hallmark of PD, as they influence the expression of genes associated with alpha-synuclein metabolism and protein clearance [92]. Epigenetic modulators, such as HDACIs and HMT inhibitors, have shown promise in preclinical models of PD. These compounds can mitigate neuroinflammation, enhance protein clearance mechanisms, and protect dopaminergic neurons [95]. HDACIs such as vorinostat and valproic acid, were reported to increase histone acetylation, promoting gene expression associated with synaptic plasticity and memory formation [96].
5. Epigenetic clocks and their relevance for aging
Epigenetic clocks have gained significant attention in the field of aging research due to their precision in predicting an individual’s biological age, which may differ from their chronological age [2]. DNA methylation-based clocks, such as the Horvath and Hannum clocks, use specific patterns of DNA methylation at CpG sites to estimate biological age. These clocks have been validated in various tissues and populations [97]. Epigenetic clocks have been associated with health outcomes, including the risk of age-related diseases, such as cardiovascular disease and cancer, as well as overall mortality [98]. Epigenetic clocks can also measure “age acceleration,” which indicates whether individuals are aging faster or slower than expected based on their chronological age. Epigenetic clocks may serve as valuable tools for assessing the effectiveness of anti-aging interventions. They can be used to monitor changes in biological age in response to lifestyle modifications or medical treatments [99], as factors such as diet and exercise, can influence age acceleration [59]. Despite their promise, there are challenges and debates surrounding the use of epigenetic clocks, including their biological interpretation and accuracy. Ongoing research aims to refine these clocks and enhance their predictive power [100].
6. Conclusions
The brain epigenetic landscape is emerging as a very important factor in the regulation of brain structure and function from development to old age. Although much progress has been made in understanding the role of the main epigenetic modifications in the normal and pathological brain, most molecular changes have been discovered using biochemical and immunochemical approaches that not always have permitted linking these changes to specific brain areas and/or cell types. This will be the primary challenge in future research aiming to further proceed toward the translational use of these results in clinical practice.
Author Contributions
Conceptualization, L.L., and A.M.; data curation, L.L., and A.M.; writing—original draft preparation, A.M.; writing—review and editing, L.L., and A.M..; visualization, X.X.; funding acquisition, L.L., and A.M. All authors have read and agreed to the published version of the manuscript.
Funding
The experimental work described in this paper was funded by local grants from the University of Turin. The APC was waived by MDPI.
Acknowledgments
We wish to thank Prof. Paolo Cascio for his critical reading and suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
List of abbreviations
2MeCP2 = methyl-CpG-binding protein 2
5mC = 5-methylcytosine
AD = Alzheimer’s disease
ASD = autism spectrum disorder
BDNF = brain-derived neurotrophic factor
CREB2 = cAMP response element binding protein 2
DNMT1 = DNA methyltransferase 1
DNMT3A = DNA methyltransferase 3A
DNMT3B = DNA methyltransferase 3B
DNMTs = DNA methyltransferases
eRNAs = enhancer RNAs
GATA3 = GATA binding protein 3
HAT = histone acetyltransferase
HDACs = histone deacetylases
HDACIs = histone deacetylase inhibitors
hmC = 5-hydroxymethylcytosine
HMT = histone methyltransferase
IEGs = immediate early genes
KDAC = lysine deacetylase
KMT = histone lysine methyltransferase
lncRNAs = long non-coding RNAs
LTP = long-term potentiation
MBPs = methyl-CpG binding proteins
mCH = non-CG methylation
MBD = methyl-binding domain
MBD1-5 = Methyl-CpG-binding domain protein 1 to 5
mRNA = messenger RNA
miRNAs = microRNAs
NaB = sodium butyrate
ncRNAs = non-coding RNAs
NMDA = N-methyl-D-aspartate
NSCs = neural stem cells
PD = Parkinson’s disease
piRNAs = PIWI-interacting RNAs
pri-miRNA= hairpin-containing primary transcripts
RGCs = radial glial cells
ROS = reactive oxygen species
SA-gal = senescence-associated-galactosidase
SASP = senescence-associated secretory phenotype
siRNAs = small interfering RNAs
SIRT1 = sirtuin 1
SRA = SET- and RING-associated domain proteins
SUMO = small ubiquitin-like modifier
TNFα = tumor necrosis factor α
TSA = trichostatin A
UHRF1 = ubiquitin-like containing PHD ring finger 1
UHRF1 = ubiquitin-like containing PHD ring finger 1
ZBTB4 = zinc finger and BTB domain-containing protein 4
ZBTB33 = zinc finger and BTB domain-containing protein 33
ZBTB 38 = zinc finger and BTB domain-containing protein 38
References
- Ingram, N. Waddington, Holmyard and Alchemy: Perspectives on the Epigenetic Landscape. Endeavour 2019, 43, 100690. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of aging. Nat Rev Genet 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Sun, J.; Yang, J.; Miao, X.; Loh, H. H.; Pei, D.; Zheng, H. Proteins in DNA methylation and their role in neural stem cell proliferation and differentiation. Cell Regeneration 2021, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends in genetics: TIG 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat Rev Genet 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Moore, L. D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? Embo j 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Roth, S. Y.; Denu, J. M.; Allis, C. D. Histone acetyltransferases. Annu Rev Biochem 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C. D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Greer, E. L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat Rev Genet 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Rossetto, D.; Truman, A. W.; Kron, S. J.; Côté, J. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clinical cancer research: An official journal of the American Association for Cancer Research 2010, 16, 4543–4552. [Google Scholar] [CrossRef]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev 2001, 15, 2343–2360. [Google Scholar] [CrossRef]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev 2004, 18, 2046–2059. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, H.; Liu, M.; Zhou, T.; Cheng, X.; Huang, W.; Cao, L. The role and mechanism of histone lactylation in health and diseases. Front Genet 2022, 13, 949252. [Google Scholar] [CrossRef]
- Jiang, G.; Li, C.; Lu, M.; Lu, K.; Li, H. Protein lysine crotonylation: Past, present, perspective. Cell Death & Disease 2021, 12, 703. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanović, O. Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol 2018, 19, 774–790. [Google Scholar] [CrossRef]
- Wei, J. W.; Huang, K.; Yang, C.; Kang, C. S. Non-coding RNAs as regulators in epigenetics (Review). Oncol Rep 2017, 37, 3–9. [Google Scholar] [CrossRef]
- Reichard, J.; Zimmer-Bensch, G. The Epigenome in Neurodevelopmental Disorders. Front Neurosci 2021, 15, 776809. [Google Scholar] [CrossRef]
- Zhubi, A.; Chen, Y.; Guidotti, A.; Grayson, D. R. Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. International Journal of Developmental Neuroscience 2017, 62, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.; Lee, K. H.; Ming, G. L.; Yoon, K. J.; Song, H. Setting the clock of neural progenitor cells during mammalian corticogenesis. Semin Cell Dev Biol 2023, 142, 43–53. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E. A.; Nery, J. R.; Urich, M.; Puddifoot, C. A.; Johnson, N. D.; Lucero, J.; Huang, Y.; Dwork, A. J.; Schultz, M. D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Xie, J.; Xie, L.; Wei, H.; Li, X. J.; Lin, L. Dynamic Regulation of DNA Methylation and Brain Functions. Biology (Basel) 2023, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.; Moyon, S. DNA methylation in Schwann cells and in oligodendrocytes. Glia 2020, 68, 1568–1583. [Google Scholar] [CrossRef]
- Lubin, F. D.; Roth, T. L.; Sweatt, J. D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- Maegawa, S.; Hinkal, G.; Kim, H. S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L. A.; Issa, J. P. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 2010, 20, 332–340. [Google Scholar] [CrossRef]
- Hsieh, J.; Gage, F. H. Chromatin remodeling in neural development and plasticity. Curr Opin Cell Biol 2005, 17, 664–671. [Google Scholar] [CrossRef]
- Maze, I.; Nestler, E. J. The epigenetic landscape of addiction. Ann N Y Acad Sci 2011, 1216, 99–113. [Google Scholar] [CrossRef]
- Peixoto, L.; Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013, 38, 62–76. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P. D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef]
- Reul, J. M. Making memories of stressful events: A journey along epigenetic, gene transcription, and signaling pathways. Front Psychiatry 2014, 5, 5. [Google Scholar] [CrossRef]
- Jarome, T. J.; Perez, G. A.; Webb, W. M.; Hatch, K. M.; Navabpour, S.; Musaus, M.; Farrell, K.; Hauser, R. M.; McFadden, T.; Martin, K.; et al. Ubiquitination of Histone H2B by Proteasome Subunit RPT6 Controls Histone Methylation Chromatin Dynamics During Memory Formation. Biol Psychiatry 2021, 89, 1176–1187. [Google Scholar] [CrossRef]
- Hou, S. T. The regulatory and enzymatic functions of CRMPs in neuritogenesis, synaptic plasticity, and gene transcription. Neurochem Int 2020, 139, 104795. [Google Scholar] [CrossRef]
- Rajasethupathy, P.; Antonov, I.; Sheridan, R.; Frey, S.; Sander, C.; Tuschl, T.; Kandel, E. R. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 2012, 149, 693–707. [Google Scholar] [CrossRef]
- Giacoman-Lozano, M.; Meléndez-Ramírez, C.; Martinez-Ledesma, E.; Cuevas-Diaz Duran, R.; Velasco, I. Epigenetics of neural differentiation: Spotlight on enhancers. Front Cell Dev Biol 2022, 10, 1001701. [Google Scholar] [CrossRef]
- Champagne, F. A. Epigenetic influence of social experiences across the lifespan. Dev Psychobiol 2010, 52, 299–311. [Google Scholar] [CrossRef]
- Samblas, M.; Milagro, F. I.; Martínez, A. DNA methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics 2019, 14, 421–444. [Google Scholar] [CrossRef]
- Xu, H.; Li, B.; Li, L.; Fan, Z.; Gong, X.; Wu, L.; Yan, C. Environmental enrichment mitigates PTSD-like behaviors in adult male rats exposed to early life stress by regulating histone acetylation in the hippocampus and amygdala. J Psychiatr Res 2022, 155, 120–136. [Google Scholar] [CrossRef]
- Renthal, W.; Nestler, E. J. Epigenetic mechanisms in drug addiction. Trends Mol Med 2008, 14, 341–350. [Google Scholar] [CrossRef]
- González, B.; Jayanthi, S.; Gomez, N.; Torres, O. V.; Sosa, M. H.; Bernardi, A.; Urbano, F. J.; García-Rill, E.; Cadet, J. L.; Bisagno, V. Repeated methamphetamine and modafinil induce differential cognitive effects and specific histone acetylation and DNA methylation profiles in the mouse medial prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry 2018, 82, 1–11. [Google Scholar] [CrossRef]
- Hunter, R. G.; McEwen, B. S. Stress and anxiety across the lifespan: Structural plasticity and epigenetic regulation. Epigenomics 2013, 5, 177–194. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduction and Targeted Therapy 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J. H.; Baxter, M. G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat Rev Neurosci 2012, 13, 240–250. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front Aging Neurosci 2021, 13, 646924. [Google Scholar] [CrossRef]
- Madden, D. J.; Bennett, I. J.; Burzynska, A.; Potter, G. G.; Chen, N. K.; Song, A. W. Diffusion tensor imaging of cerebral white matter integrity in cognitive aging. Biochim Biophys Acta 2012, 1822, 386–400. [Google Scholar] [CrossRef]
- Borella, E.; Carretti, B.; Riboldi, F.; De Beni, R. Working memory training in older adults: Evidence of transfer and maintenance effects. Psychology and aging 2010, 25, 767–778. [Google Scholar] [CrossRef]
- Nyberg, L.; Lövdén, M.; Riklund, K.; Lindenberger, U.; Bäckman, L. Memory aging and brain maintenance. Trends Cogn Sci 2012, 16, 292–305. [Google Scholar] [CrossRef]
- Gazzaley, A.; D‘Esposito, M. Top-down modulation and normal aging. Ann N Y Acad Sci 2007, 1097, 67–83. [Google Scholar] [CrossRef]
- Volkow, N. D.; Koob, G.; Baler, R. Biomarkers in Substance Use Disorders. ACS Chemical Neuroscience 2015, 6, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Damoiseaux, J. S.; Beckmann, C. F.; Arigita, E. J.; Barkhof, F.; Scheltens, P.; Stam, C. J.; Smith, S. M.; Rombouts, S. A. Reduced resting-state brain activity in the “default network” in normal aging. Cereb Cortex 2008, 18, 1856–1864. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S. L.; Bonini, N. M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Wallin, A.; Kapaki, E.; Boban, M.; Engelborghs, S.; Hermann, D. M.; Huisa, B.; Jonsson, M.; Kramberger, M. G.; Lossi, L.; Malojcic, B.; et al. Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease - A consensus report. BMC neurology 2017, 17, 102. [Google Scholar] [CrossRef]
- Penner, M. R.; Roth, T. L.; Barnes, C. A.; Sweatt, J. D. An epigenetic hypothesis of aging-related cognitive dysfunction. Front Aging Neurosci 2010, 2, 9. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Xu, N.; Zhang, S.; Wang, S.; Mao, Y.; Zhu, Y.; Li, B.; Jiang, Y.; Tan, Y.; et al. NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Cell Mol Life Sci 2019, 76, 3005–3018. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Villa, C.; Stoccoro, A. Epigenetic Peripheral Biomarkers for Early Diagnosis of Alzheimer’s Disease. Genes (Basel) 2022, 13, 1308. [Google Scholar] [CrossRef] [PubMed]
- Higgins-Chen, A. T.; Thrush, K. L.; Levine, M. E. Aging biomarkers and the brain. Semin Cell Dev Biol 2021, 116, 180–193. [Google Scholar] [CrossRef]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A. T.; Levine, M. E.; Boks, M. P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res Rev 2021, 69, 101348. [Google Scholar] [CrossRef]
- Levine, M. E.; Lu, A. T.; Quach, A.; Chen, B. H.; Assimes, T. L.; Bandinelli, S.; Hou, L.; Baccarelli, A. A.; Stewart, J. D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Hwang, J. Y.; Aromolaran, K. A.; Zukin, R. S. Author Correction: The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci 2018, 19, 771. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes (Basel) 2023, 14, 873. [Google Scholar] [CrossRef]
- Raj, K.; Horvath, S. Current perspectives on the cellular and molecular features of epigenetic ageing. Exp Biol Med (Maywood) 2020, 245, 1532–1542. [Google Scholar] [CrossRef]
- Hwang, I.; Tang, D.; Paik, J. Oxidative stress sensing and response in neural stem cell fate. Free Radic Biol Med 2021, 169, 74–83. [Google Scholar] [CrossRef]
- Yu, J.; Wu, Y.; Yang, P. High glucose-induced oxidative stress represses sirtuin deacetylase expression and increases histone acetylation leading to neural tube defects. J Neurochem 2016, 137, 371–383. [Google Scholar] [CrossRef]
- Karpova, N. N.; Sales, A. J.; Joca, S. R. Epigenetic Basis of Neuronal and Synaptic Plasticity. Curr Top Med Chem 2017, 17, 771–793. [Google Scholar] [CrossRef]
- Dai, D. F.; Santana, L. F.; Vermulst, M.; Tomazela, D. M.; Emond, M. J.; MacCoss, M. J.; Gollahon, K.; Martin, G. M.; Loeb, L. A.; Ladiges, W. C.; Rabinovitch, P. S. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathology and Applied Neurobiology 2010, 36, 320–330. [Google Scholar] [CrossRef]
- Saito, T.; Braun, P. R.; Daniel, S.; Jellison, S. S.; Hellman, M.; Shinozaki, E.; Lee, S.; Cho, H. R.; Yoshino, A.; Toda, H.; Shinozaki, G. The relationship between DNA methylation in neurotrophic genes and age as evidenced from three independent cohorts: Differences by delirium status. Neurobiol Aging 2020, 94, 227–235. [Google Scholar] [CrossRef]
- Dhar, G. A.; Saha, S.; Mitra, P.; Nag Chaudhuri, R. DNA methylation and regulation of gene expression: Guardian of our health. The Nucleus 2021, 64, 259–270. [Google Scholar] [CrossRef]
- Yang, Y. N.; Su, Y. T.; Wu, P. L.; Yang, C. H.; Yang, Y. S. H.; Suen, J. L.; Yang, S. N. Granulocyte Colony-Stimulating Factor Alleviates Bacterial-Induced Neuronal Apoptotic Damage in the Neonatal Rat Brain through Epigenetic Histone Modification. Oxid Med Cell Longev 2018, 2018, 9797146. [Google Scholar] [CrossRef]
- Yang, K.; Zeng, L.; Ge, A.; Wang, S.; Zeng, J.; Yuan, X.; Mei, Z.; Wang, G.; Ge, J. A systematic review of the research progress of non-coding RNA in neuroinflammation and immune regulation in cerebral infarction/ischemia-reperfusion injury. Front Immunol 2022, 13, 930171. [Google Scholar] [CrossRef]
- Rachmian, N.; Krizhanovsky, V. Senescent cells in the brain and where to find them. Febs j 2023, 290, 1256–1266. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S. A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B. M.; Patel, A. D.; Pirtskhalava, T.; Inman, C. L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K. F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Kanduri, C. Long noncoding RNAs: Lessons from genomic imprinting. Biochim Biophys Acta 2016, 1859, 102–111. [Google Scholar] [CrossRef]
- Huang, C. H.; Chang, M. C.; Lai, Y. C.; Lin, C. Y.; Hsu, C. H.; Tseng, B. Y.; Hsiao, C. K.; Lu, T. P.; Yu, S. L.; Hsieh, S. T.; Chen, W. J. Mitochondrial DNA methylation profiling of the human prefrontal cortex and nucleus accumbens: Correlations with aging and drug use. Clin Epigenetics 2022, 14, 79. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M. R.; Bentivoglio, A. R.; Coelho-Júnior, H. J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int J Mol Sci 2019, 20, 805. [Google Scholar] [CrossRef]
- Wood, J. G.; Hillenmeyer, S.; Lawrence, C.; Chang, C.; Hosier, S.; Lightfoot, W.; Mukherjee, E.; Jiang, N.; Schorl, C.; Brodsky, A. S.; et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 2010, 9, 971–978. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P. P.; Nativio, R.; Berger, S. L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Itoh, Y.; Takada, Y.; Yamashita, Y.; Suzuki, T. Recent progress on small molecules targeting epigenetic complexes. Curr Opin Chem Biol 2022, 67, 102130. [Google Scholar] [CrossRef]
- Azzalin, C. M.; Lingner, J. Telomere functions grounding on TERRA firma. Trends Cell Biol 2015, 25, 29–36. [Google Scholar] [CrossRef]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R. C.; Cota, P.; Wittnam, J. L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M. F. The role of epigenetics in aging and age-related diseases. Ageing Res Rev 2009, 8, 268–276. [Google Scholar] [CrossRef]
- Gionchiglia, N.; Granato, A.; Merighi, A.; Lossi, L. Association of Caspase 3 Activation and H2AX γ Phosphorylation in the Aging Brain: Studies on Untreated and Irradiated Mice. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Merighi, A.; Gionchiglia, N.; Granato, A.; Lossi, L. The Phosphorylated Form of the Histone H2AX (γH2AX) in the Brain from Embryonic Life to Old Age. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Zhang, D.; Deng, Y.; Kukanja, P.; Agirre, E.; Bartosovic, M.; Dong, M.; Ma, C.; Ma, S.; Su, G.; Bao, S.; et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 2023, 616, 113–122. [Google Scholar] [CrossRef]
- Urdinguio, R. G.; Sanchez-Mut, J. V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Kilgore, M.; Miller, C. A.; Fass, D. M.; Hennig, K. M.; Haggarty, S. J.; Sweatt, J. D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef]
- Sung, Y. M.; Lee, T.; Yoon, H.; DiBattista, A. M.; Song, J. M.; Sohn, Y.; Moffat, E. I.; Turner, R. S.; Jung, M.; Kim, J.; Hoe, H. S. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp Neurol 2013, 239, 192–201. [Google Scholar] [CrossRef]
- Williams, J. B.; Cao, Q.; Wang, W.; Lee, Y. H.; Qin, L.; Zhong, P.; Ren, Y.; Ma, K.; Yan, Z. Inhibition of histone methyltransferase Smyd3 rescues NMDAR and cognitive deficits in a tauopathy mouse model. Nat Commun 2023, 14, 91. [Google Scholar] [CrossRef]
- Zhao, Y.; Nomiyama, T.; Findeisen, H. M.; Qing, H.; Aono, J.; Jones, K. L.; Heywood, E. B.; Bruemmer, D. Epigenetic regulation of the NR4A orphan nuclear receptor NOR1 by histone acetylation. FEBS Lett 2014, 588, 4825–4830. [Google Scholar] [CrossRef]
- Jakubowski, J. L.; Labrie, V. Epigenetic Biomarkers for Parkinson’s Disease: From Diagnostics to Therapeutics. J Parkinsons Dis 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Porcu, M.; Chiarugi, A. The emerging therapeutic potential of sirtuin-interacting drugs: From cell death to lifespan extension. Trends Pharmacol Sci 2005, 26, 94–103. [Google Scholar] [CrossRef]
- Francelle, L.; Outeiro, T. F.; Rappold, G. A. Inhibition of HDAC6 activity protects dopaminergic neurons from alpha-synuclein toxicity. Scientific Reports 2020, 10, 6064. [Google Scholar] [CrossRef] [PubMed]
- Godena, V. K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R. M.; Miller, C. C.; Whitworth, A. J.; De Vos, K. J. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun 2014, 5, 5245. [Google Scholar] [CrossRef]
- Whittle, N.; Singewald, N. HDAC inhibitors as cognitive enhancers in fear, anxiety and trauma therapy: Where do we stand? Biochem Soc Trans 2014, 42, 569–581. [Google Scholar] [CrossRef]
- Horvath, S.; Mah, V.; Lu, A. T.; Woo, J. S.; Choi, O. W.; Jasinska, A. J.; Riancho, J. A.; Tung, S.; Coles, N. S.; Braun, J.; et al. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY) 2015, 7, 294–306. [Google Scholar] [CrossRef]
- Marioni, R. E.; Shah, S.; McRae, A. F.; Chen, B. H.; Colicino, E.; Harris, S. E.; Gibson, J.; Henders, A. K.; Redmond, P.; Cox, S. R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 2015, 16, 25. [Google Scholar] [CrossRef]
- Fahy, G. M.; Brooke, R. T.; Watson, J. P.; Good, Z.; Vasanawala, S. S.; Maecker, H.; Leipold, M. D.; Lin, D. T. S.; Kobor, M. S.; Horvath, S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef]
- Le Clercq, L. S.; Kotzé, A.; Grobler, J. P.; Dalton, D. L. Biological clocks as age estimation markers in animals: A systematic review and meta-analysis. Biol Rev Camb Philos Soc 2023. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the three main groups of epigenetic modifications in mammalian cells: DNA methylation. Histone epigenetic modifications, and non-coding RNAs. DNA methylation that acts as an off-switch to block translation occurs at CpG sites that are observed across the genome. Methylation can occur in intergenic regions, CpG islands, and the gene body. Nevertheless, CpG islands that are considered normal exhibit a lack of methylation throughout all stages of development. This lack of methylation enables the transcription of the specific gene, provided that the necessary transcription factors are present, and the chromatin structure is accessible to these factors. Histone modifications are chemical alterations, which can have profound effects on gene expression and, consequently, various cellular processes. These modifications form an epigenetic code that imparts a distinct feature on chromatin architecture. The enzymes that catalyze these modifications can be classified as writers, readers, and erasers. Writers are enzymes that are responsible for the acetylation, methylation, phosphorylation, ubiquitination, sumoylation, lactylation, and crotonylation of histones. Among them, KMTs and HATs are of relevance. Readers are responsible for the recognition of the epigenetic marks on histones. Among readers, are the readers of methyl- and acetyl-lysine residues. Among the erasers are histone demethylases and deacetylases. Non-coding RNAs are divided into short and long non-coding RNAs. For simplicity, only the miRNA generation pathway is represented. Non-coding RNAs can interact with DNA, RNA, and protein molecules to modulate gene transcription, contribute to RNA inhibition or degradation, or serve as molecular guides, scaffolds, or decoys for specific proteins, such as transcription factors. These many functions occur either in the nucleus or the cell cytoplasm. Abbreviations: AC =acetylation; Ex = exon; lncRNA = long non-coding RNA; Me = methylation; NXF1 = nuclear RNA export factor 1; Su = sumoylation; XPO5 = exportin 5. Created with BioRender.com.
Figure 1.
Schematic representation of the three main groups of epigenetic modifications in mammalian cells: DNA methylation. Histone epigenetic modifications, and non-coding RNAs. DNA methylation that acts as an off-switch to block translation occurs at CpG sites that are observed across the genome. Methylation can occur in intergenic regions, CpG islands, and the gene body. Nevertheless, CpG islands that are considered normal exhibit a lack of methylation throughout all stages of development. This lack of methylation enables the transcription of the specific gene, provided that the necessary transcription factors are present, and the chromatin structure is accessible to these factors. Histone modifications are chemical alterations, which can have profound effects on gene expression and, consequently, various cellular processes. These modifications form an epigenetic code that imparts a distinct feature on chromatin architecture. The enzymes that catalyze these modifications can be classified as writers, readers, and erasers. Writers are enzymes that are responsible for the acetylation, methylation, phosphorylation, ubiquitination, sumoylation, lactylation, and crotonylation of histones. Among them, KMTs and HATs are of relevance. Readers are responsible for the recognition of the epigenetic marks on histones. Among readers, are the readers of methyl- and acetyl-lysine residues. Among the erasers are histone demethylases and deacetylases. Non-coding RNAs are divided into short and long non-coding RNAs. For simplicity, only the miRNA generation pathway is represented. Non-coding RNAs can interact with DNA, RNA, and protein molecules to modulate gene transcription, contribute to RNA inhibition or degradation, or serve as molecular guides, scaffolds, or decoys for specific proteins, such as transcription factors. These many functions occur either in the nucleus or the cell cytoplasm. Abbreviations: AC =acetylation; Ex = exon; lncRNA = long non-coding RNA; Me = methylation; NXF1 = nuclear RNA export factor 1; Su = sumoylation; XPO5 = exportin 5. Created with BioRender.com.
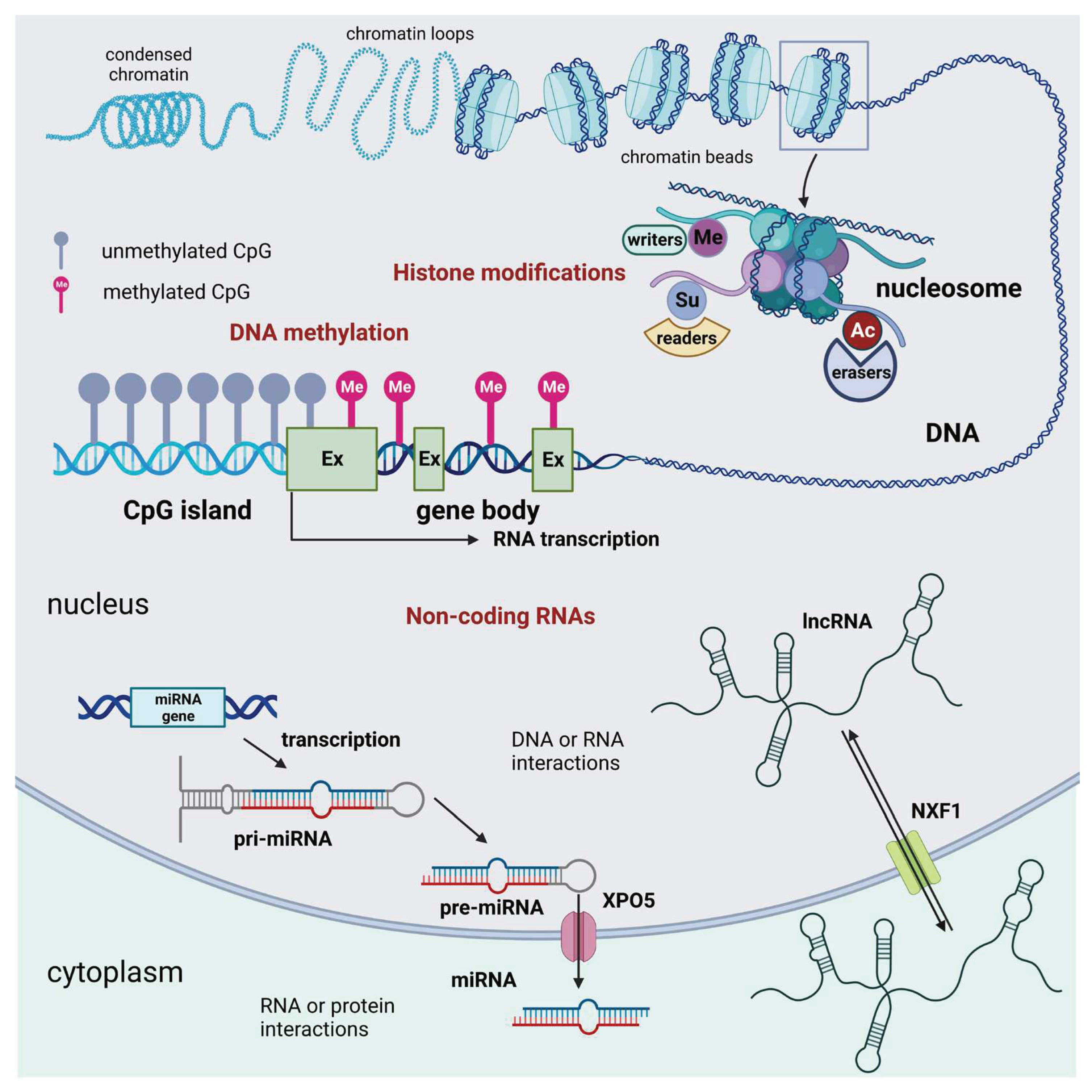
Figure 2.
Histone tail epigenetic modifications described in the brain. All core histone proteins contain intrinsically disordered tail regions that protrude from the DNA-enveloped core and are known to play critical roles in chromatin regulation (see text). Aminoacidic residues are indicated by one-letter notation. Amino lysine acid residues (K) undergoing epigenetic changes so far described in the old brain are indicated in red font. The sequences specific to H2AX are indicated by yellow circles. Abbreviations: AC =acetylation; Cr = crotonylation; Las =lactylation; Me = methylation; P = phosphorylation; Su = sumoylation; Ub = ubiquitination. Created with BioRender.com.
Figure 2.
Histone tail epigenetic modifications described in the brain. All core histone proteins contain intrinsically disordered tail regions that protrude from the DNA-enveloped core and are known to play critical roles in chromatin regulation (see text). Aminoacidic residues are indicated by one-letter notation. Amino lysine acid residues (K) undergoing epigenetic changes so far described in the old brain are indicated in red font. The sequences specific to H2AX are indicated by yellow circles. Abbreviations: AC =acetylation; Cr = crotonylation; Las =lactylation; Me = methylation; P = phosphorylation; Su = sumoylation; Ub = ubiquitination. Created with BioRender.com.
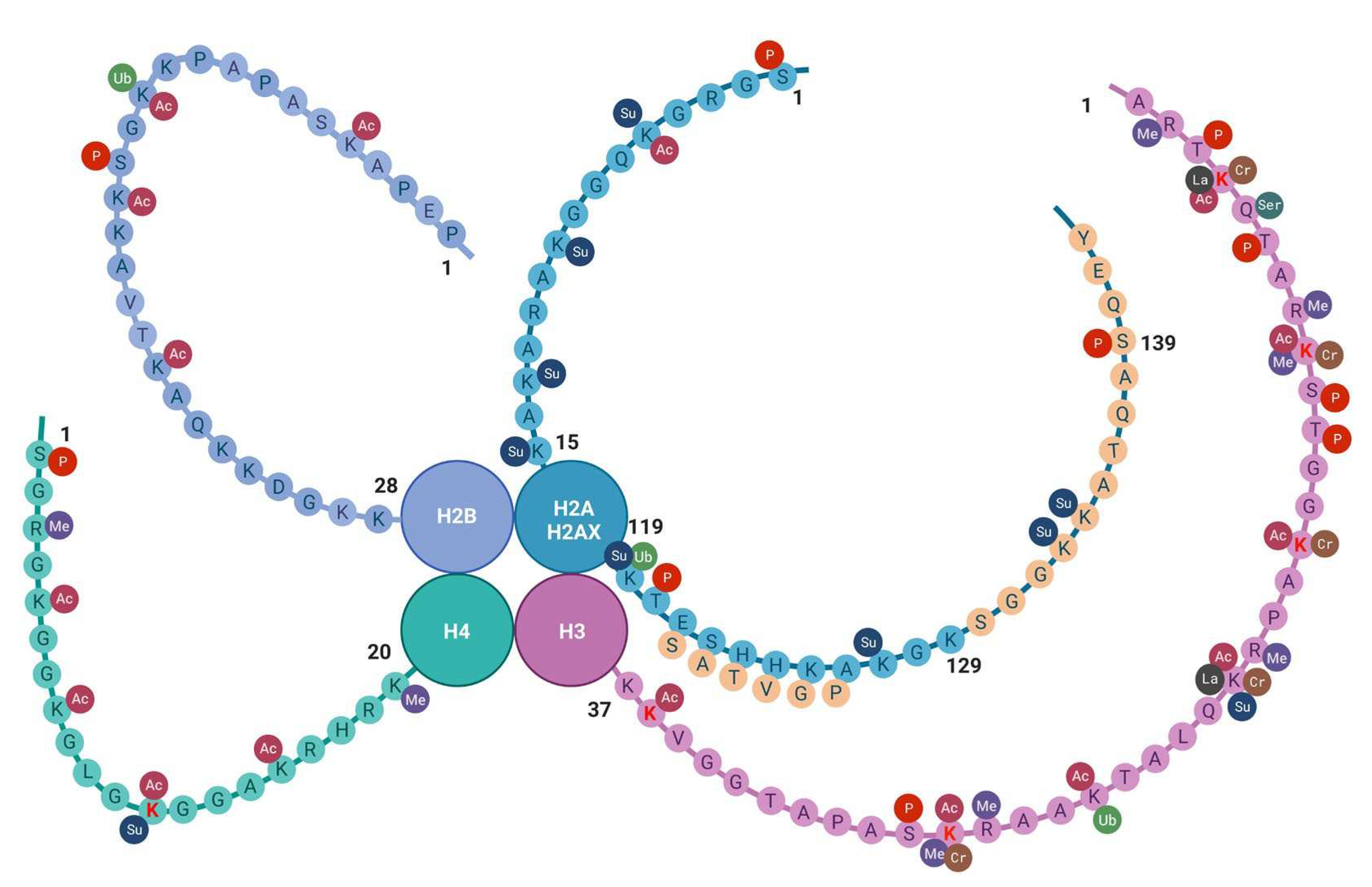
Table 1.
Proteins acting on DNA methylation. For abbreviations see the list at the end of the paper.
Table 1.
Proteins acting on DNA methylation. For abbreviations see the list at the end of the paper.
| Protein family | Family members | Function |
|---|---|---|
| DNA methyltransferases (DNMTs) | DNMT1 DNMT3A DNMT3B |
DNMT1 is the switch from neurogenesis to gliogenesis during NSC differentiation. DNMT3A regulates NSC proliferation and differentiation. |
| Methyl-CpG-binding proteins (MBPs) |
MBD proteins MBD1–5 2MeCP2 |
MBD1 deficiency causes the accumulation of NSCs and the impairment of neuronal lineage differentiation. 2MeCP2 controls neuronal maturation and dendritic arborization in both developing and adult brains |
|
Zinc finger/Kaiso proteins Kaiso/ZBTB33 ZBTB4 ZBTB38 |
ZBTB4 and ZBTB38 exhibited high expression levels in the brain | |
|
SRA proteins UHRF1 UHRF2 |
UHRF1 is critical for the maintenance of DNA methylation through cell division and is also involved in DNA damage repair. UHRF2 is involved in cell cycle progression. |
Table 3.
Epigenetic marks in the old brain. Aminoacidic residues are indicated by one-letter notation. For abbreviations see the list at the end of the paper.
Table 3.
Epigenetic marks in the old brain. Aminoacidic residues are indicated by one-letter notation. For abbreviations see the list at the end of the paper.
| Epigenetic mark | Biological effects | Brain region | Target |
|---|---|---|---|
| Reduction of H3K9ac | Lowered expression of key genes to neuronal and synaptic development Decrease in age-related memory and learning capacity |
Hippocampus | IEGs |
| Reduction of H3K14acc | Hippocampus | IEGs | |
| Reduction of H3K27ac | Prefrontal cortex Hippocampus |
GATA3, BDNF | |
| Reduction of H4K12a | Hippocampus | Synaptic function-related genes | |
| Increase in H3K9me2 | Aging | Cerebral cortex Hippocampus |
_ |
| Increase of H3K9me3 | Reduction of dendritic growth and stability | Cerebral cortex Hippocampus |
BDNF |
| Memory deficit | Hippocampus | BDNF IEGs |
|
| Learning and memory ability decline | Brain tissue from AD patients and mouse models | Mitochondrial function-related genes | |
| Increase of H3K4me2 | Increased expression of related stress-response proteins and inducing cognitive impairment | Prefrontal cortex | Stress-related genes |
| Increase of H3K27me3 | Activation of stress and immune inflammation | Brain | Stress-related genes |
| Reduction of H3K36me3 | Impaired memory function | Cerebral cortex Hippocampus |
_ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



