
Submitted:
24 November 2023
Posted:
26 November 2023
You are already at the latest version
Abstract
High-performance properties of interpenetration polymer network (IPN) hydrogels, based on physically crosslinked chitosan (CS) and chemically crosslinked poly(N-isopropylacrylamide) (PNiPAM), have been successfully developed. The IPN of CS/PNiPAM is proposed to overcome the limited mechanical properties of the single CS network. In this study, the viscoelastic behaviors of prepared materials in both solution and gel states have been extensively examined, considering the UV exposure time and crosslinker concentration as key factors. The effect of these factors on gel formation, hydrogel structures, thermal stabilities of networks, and HeLa cell adhesion were studied sequentially. The sol-gel transition was effectively demonstrated through the scaling law, which agrees well with Winter and Chambon's theory. By subjecting the CS hydrogel to the process operation in an ethanol solution, its properties can be significantly enhanced with increased crosslinker concentration, including the shear modulus, crosslinking degree, gel strength, and thermal stability in its swollen state. The IPN samples exhibit a smooth and dense surface with irregular pores, allowing for much water absorption. The HeLa cells were adhered to and killed using the CS surface cationic derivative, then released through hydrolysis by utilizing the hydrophilic/hydrophobic switchable property or thermo-reversible gelation of the PNiPAM network. The results demonstrated that IPN is a highly attractive candidate for anti-fouling materials.
Keywords:
chitosan
; poly(N-isopropylacrylamide)
; thermo-reversible gelation
; interpenetration polymer network
; viscoelastic property
; anti-fouling materials
1. Introduction
Hydrogels are hydrophilic polymer networks that can retain a large amount of water within their structure without dissolving or losing their three-dimensional (3D) shape. This is possible due to their chemically or physically crosslinked networks. Both natural and synthetic polymers have gained significant attention in various biomedical applications due to their biocompatibility, making them similar to living tissues [1,2,3]. Additionally, hydrogels have been utilized in fields such as 3D printing [4], wearable devices [5], actuators [5,6], and energy storage devices [7,8]. However, many applications frequently encounter issues with the mechanical properties of hydrogels, both in dry and swollen states. This is particularly true for single-network hydrogels. In a recent study, we presented a simple yet effective approach to enhance the mechanical properties of single-network hydrogels. This was achieved by introducing a second network structure using the interpenetrating polymer networks (IPNs) method [9]. The IPNs involve the creation of two or more intertwined polymer networks that are not chemically bonded to each other [9,10]. Numerous studies have been conducted on preparing IPN hydrogels using various hydrophilic polymers or their precursors. The main classes of these polymers include natural polymers and their derivatives (such as polysaccharides and proteins), as well as synthetic polymers that contain hydrophilic functional groups like –COOH, –OH, –CONH2, -SO3H, amines, and -R4N+, and ether [10].
Chitosan (CS) is a polysaccharide obtained through the deacetylation process of chitin. Chitin is a linear copolymer consisting of linked β(1→4) glucosamine (2-amino-2-deoxy-D-glucose) and N-acetyl-D-glucosamine (2-acetamido-2-deoxy-D-glucose). The remarkable characteristics of excellent biocompatibility and admirable biodegradability, coupled with ecological safety and low toxicity, along with versatile biological activities like antimicrobial activity and low immunogenicity, have opened up numerous opportunities for further development [11]. CS hydrogels can be chemically constructed using crosslinking reagents, such as glutaraldehyde. Alternatively, CS gels can be formed simply through physical crosslinking, which occurs through H-bonds, ionic bonds, dipole interactions, or hydrophobic associations with hydrophobic substituted glucose units, similar to acetylated substituted dextran-based polysaccharides [12,13]. Although these physical gels might dissociate when deformed, they still provide an energy dissipative mechanism that helps prevent sudden shocks or impacts that could harm materials in the application device [14].
Poly(N-isopropylacrylamide) (PNiPAM) is a widely recognized thermo-responsive polymer that undergoes a reversible conformational transition from a coil to a globule state at a lower critical solution temperature (LCST) of approximately 32.0 °C in an aqueous solution. This temperature is relatively close to the human body temperature [15]. Similarly, the PNiPAM hydrogel is a well-known temperature-sensitive gel that exhibits a volume phase transition temperature (VPTT) at around 34°C [16]. Below this temperature, the gel undergoes swelling due to the formation of strong hydrogen bonds between the hydrophilic amide groups and water. As the temperature is raised, the hydrogel undergoes a shrinkage due to the disruption of hydrogen bonds and the significant strengthening of hydrophobic interactions among the hydrophobic groups. This polymer chain collapse results in the hydrogel network’s phase transition [17,18]. Both linear and hydrogel structures possess attractive features that make them suitable biomedical polymers. These features include biocompatibility and non-toxicity, and have garnered significant attention in diverse applications [19,20,21,22,23].
CS/PNiPAM hydrogels have garnered significant attention due to their exceptional properties for biomedical and related applications [20,21,24,25,26,27,28,29]. However, it is worth noting that these applications have typically been explored without thoroughly examining the hydrogels’ mechanical properties [20,21,24,25,26,27]. For instance, a temperature-sensitive polymer consisting of palmatine (PA)-loaded cysteine (Cys) modified chitosan (Cs) grafted with PNIPAM (Cs-Cys-PN/PA) was prepared. This polymer was developed as a fluorescent probe for living cell temperature sensors and for its antibacterial application. The materials have successfully polymerized, exhibiting aggregation-induced emission enhancement (AIEE) properties, resulting in a reversible hydrogel formation in an aqueous solution. These hydrogels demonstrate low cytotoxicity and do not require any mechanical reinforcement [24]. Excellent mechanical properties are crucial in biomedical fields. However, the inherent mechanical weakness of hydrogels is a significant drawback that limits their applications. Liu, M. et al. evaluated the mechanical properties of the hydrogels through rheological analysis. The addition of chitosan to alginate-g-poly(N-isopropylacrylamide) (Alg-PN31-77%) resulted in an enhancement of the elastic modulus, making it approximately 3.5 times larger than that of the Alg-PN31-77% copolymer hydrogel. However, the magnitudes were reported unsatisfactorily to be about 180 Pa [20]. To fine-tune the mechanical behavior, crosslinking agents such as glutaraldehyde were commonly used to crosslink chitosan chemically. Additionally, linear PNiPAM was initially functionalized before grafting onto the chitosan structure. As mentioned in previous reports, the hydrogel formation was undoubtedly complex.
We recently demonstrated a facile and versatile approach to constructing the sequential IPN of CS and PNiPAM. This is typically achieved by swelling the chitosan hydrogel in a solution containing a mixture of N-isopropylacrylamide monomer (NiPAM), N,N-methylenebisacrylamide cross-linker (BIS), and 2-hydroxyl-4-(2-hydroxyethoxy)-2-methylpropiophenone photo-initiator (I2965). These components are dissolved in two different solvents: deionized water and ethanol. The CS hydrogel is formed through physical crosslinking. The 3D network of PNiPAM is crosslinked using ultraviolet light (UV), as shown in the supplementary information (Figure S1). Theoretically, the preparation method, polymer concentration, and concentration of crosslinking agent can significantly contribute to developing new hydrogel systems and increase the practical value [30]. Therefore, controlling the degree of the cross-linking agent is expected to enhance the mechanical properties of the IPN structure. Further studies were conducted to analyze the rheological and viscoelastic properties, network structure formation, microstructure, thermal properties, and swelling properties. Additionally, HeLa cell adhesion was examined to ensure that the designed material can be used as an anti-fouling material.
2. Results and Discussion
2.1. UV-induced sol to gel transition of PNiPAM solution
Before constructing the IPN network, the conditions for PNiPAM gel formation were optimized. Unlike other systems where temperatures varied from around 50°C to as high as 75 °C [17,31], the IPN preparation was operated explicitly at room temperature. Therefore, the gelation time needed to be identified to ensure the formation of a three-dimensional network. Several techniques, such as light scattering (LS) [12,32], beam diffraction [13], and rheology [33,34,35], have been widely used to elucidate the mechanism of material gelation and its molecular structure. In this work, the rheological technique was used to investigate the crosslinking behavior due to its direct correlation with the evolving physical and mechanical properties of the system during the crosslinking process.
Figure 1(a) illustrates the relationship between zero-shear viscosity (η0) and UV irradiation time (t) for the mixture of NiPAM (1 M) and BIS (0.01 M) containing I2965 (0.02 M). η0 gradually increased with increasing UV irradiation time within 0 - 60 minutes. Subsequently, it exhibited a rapid increase after exposure to UV radiation for more than 60 minutes. The relationship between and t has intermittently changed within a UV irradiation time of 120 – 240 minutes, exhibiting linearity. The gel point (tg) is tentatively identified as the intersection at 120 minutes. In another experiment, the gelation point has also been associated with the parallel lines of shear and loss modulus (G′ and G″), as shown in Figure 1(b). As gelation proceeds beyond the sol-gel transition, log–log plots of G′ and G″ versus angular frequency produce a parallel line at t = 120 minutes, with the corresponding relaxation exponent, n, for G′ ~ G″~ ωn [36]. The slope, n, was found to be 0.29, which corresponds to the chemical gel in previous work (n < 0.5) [37]. Therefore, a UV irradiation time of 120 minutes is considered to be an appropriate condition for initiating the formation of the gel network of PNiPAM in our experimental condition. However, to achieve a fully developed three-dimensional network of PNiPAM, subsequent material construction was carried out using a UV irradiation time of 180 minutes.
2.2. Effect of crosslinker concentration on viscoelastic and mechanical behaviors of single and interpenetrating polymer networks
The composition of the starting materials and the abbreviations for the resulting hydrogels are listed in Table 1. The viscoelastic properties of the hydrogels were analyzed by studying the frequency-dependent behavior of the shear modulus, G′, loss modulus, G″, and tan δ at room temperature. As shown in Figure 2, the G′ was significantly higher than G″, and both G′ and G″ exhibited slight variations with increasing frequency for all IPN hydrogels. This indicated that the hydrogels have a stable network with elastic characteristics owing to the high molecular intra- and/or inter-actions level. The results demonstrate that the viscoelastic properties of chitosan are significantly enhanced by incorporating the PNiPAM chain structure.
At a single frequency of 1 Hz (6.28 rad s-1), the G′ of all the hydrogels was compared as a function of the crosslinker content, as shown in Figure 3(a). The G′ of CS hydrogel increased significantly with an increase in the crosslinking agent. This can be attributed to the inherent structure of pure PNiPAM, as illustrated in the inset of Figure 3(b), where the G′ of pure PNiPAM prepared in an ethanol solution also showed a significant increase with an increase in the crosslinking agent. These hydrogels prepared in ethanol (IPNE systems) provided a higher value of G′ compared to those prepared in deionized water (IPNw systems). Interestingly, the G′ of the IPNE system showed a substantial concentration dependency for the crosslinker content, whereas that of the IPNw system remained almost constant value even when the crosslinker increased. This might be due to the excess crosslinker content above 0.01 M (1.45wt%) in the aqueous solution, where the conversion of NiPAM to PNiPAM remained almost constant regardless of the crosslinking agent used in this polymerization condition. The G′ of the IPN hydrogels (0.6–3.37 MPa) was approximately 70 to 400 times higher than that of the CS hydrogel. Previous studies have shown that the G′ value for the hydrogel containing polyvinyl pyrrolidone/carboxymethyl cellulose at a 1:4 ratio was approximately 0.43 MPa [38]. On the other hand, the G′ of Salecan and poly(2-acrylamido-2-methylpropanesulfonic acid-co-N-hydroxymethyl acrylamide) semi-IPN hydrogels ranged from 0.27 to1.14 kPa [39]. This indicates a relatively high modulus for hydrogels prepared in this study.
The theory of rubber elasticity states that the elastic modulus of a soft polymer network is directly proportional to the density of the crosslink points. In the case of a phantom network, the G′ of gels is determined by the crosslinking density (ve) and the extent of solvent swelling, as depicted in Equation (1) and Equation (2) [39]:
where v2 is the volume fraction of cross-linked polymer in the hydrogel; R and T are the gas constant and absolute temperature, respectively; ϕ is the functionality of the crosslinks. The term qF represents the mass of swollen gel in equilibrium divided by the constant weight of the hydrogel after solvent evaporation. This value increases as the crosslinker content increases and reaches its maximum at 2.65 wt% crosslinker. The details of this phenomenon are discussed in Section 2.5, specifically referred to as the equilibrium swelling ratio (SRe). The term ρ represents the polymer density and d represents the density of the solvent [39]. The crosslink densities ve were calculated from Equation (1) and Equation (2) and they are depicted in Figure 3(b) for the IPNE system. At the concentration of 21.4wt% of BIS content (IPNE4), ve increased further, but the G′ decreased, suggesting a high degree of brittleness in the IPNE4 structures. It has been reported adding an excess amount of crosslinking agent enhances the density of the CS network and decreases the flexibility of the chains due to the reinforced chain entanglements [40].
To gain insights into the influence of BIS concentration on the viscoelastic properties of liquid and solid gels, we have scrutinized important parameters such as the relaxation exponent and gel strength of the materials. Following the Winter-Chambon criterion [41], the power law relation is also evident in dynamic mechanical experiments, as expressed below:
where Γ(1 - n) represents the gamma function, n denotes the relaxation exponent, δ is the phase angle that remains independent of frequency but is proportional to the relaxation exponent, given by δ = nπ/2. Additionally, S represents the gel strength parameter, which relies on the crosslinking density and molecular chain flexibility.
The power law of mechanical behavior represents the self-similar (fractal) structure of clusters at the gel point (GP) [36,42]. The statistical self-similarity of a polymer is quantitatively represented by a fractal dimension, df, which describes the relation between the mass of a molecular cluster (M) in the network to its radius (R) through the expression of Rdf ~ M. Muthukumar established an expression for the relaxation exponent, n in terms of the df for polydisperse material, where the excluded volume effect of the polymer chain is completely screened. The equation is given as follows [42]:
where d (= 3) is the spatial dimension. As mentioned earlier, the values of relaxation exponent, n are determined by analyzing a log-log plot and the scaling relation of G′ and G″. The df values are obtained in the range of 2.22-2.44 for IPNE, which is close to 2.50 of complete screening of excluded volume interactions. It indicates a tight and compact network structure develops [43].
The effect of different amounts of BIS on gel strength S in Equation (3) is shown in Figure 4. When ethanol was used as a solvent, a clear dependence of the S value on the crosslinker concentration was observed. The gel strength S of CS hydrogel was outstandingly enhanced with the crosslinker content. Interestingly, the IPNE3 showed a synergistic character resulting from the crosslinking density. At low concentrations of BIS, the gel network becomes fragile and susceptible to additional cross-links, resulting in an increase in the strength parameter S. However, at the highest concentration of BIS, the strength of the network is primarily influenced by polymer entanglements, and the impact of additional cross-links on the gel strength parameter is not as significant as it is at lower levels of polymer cross-linking. These investigations have shown that the parameter S is sensitive to changes in the strand length between crosslinks. When the strand length shrinks (e.g., due to increased BIS concentration), the cross-linking density increases, resulting in a “harder” gel with a higher value of S.
2.3. Temperature dependence on mechanical stability of swollen hydrogel networks
Hydrogels typically contain a significant amount of moisture, which results in poor mechanical properties. This is due to the high degree of swelling and low density of the polymer chains. Consequently, this poses a significant challenge for their practical application. We manipulated the water content (%) within the hydrogels to assess their mechanical stability in their swollen state. Figure 5 (and Figure S7 in details) illustrates the temperature influence on G′ and G″ for the chitosan hydrogel, comparing it with IPNE3 hydrogel in the 30 – 80 oC temperature range. In the case of the chitosan hydrogel, the slope of log G′ vs. log ω increased with the temperature rise. For example, G′ decreases more rapidly as the frequency decreases at 80 °C, compared to the other temperatures. The difference between G′ and G″ at low frequency became smaller as temperature increased from 30 to 80 oC. At 80 oC, a crossover between G′ and G″ occurred at approximately 0.06 Hz. On the other hand, the crossover of the G′ and G″ was not observed for IPNE3 hydrogel. The network structure of chitosan relies on several key interactions: hydrophobic forces, molecular entanglements, and secondary forces such as ionic and hydrogen bonding. These interactions become more time-sensitive under stress. When we decrease the testing frequency, which corresponds to longer experimental durations, the elastic property of the gel, represented as G′, decreases, and its viscous nature, G", increases. At high temperatures and low frequencies, G′ and G″ become more similar, eventually crossing over. At this point, G" takes over, indicating a shift from a solid-like to a liquid-like behavior due to a thermal disruption in the physical network.
The IPNE3 exhibits a stable network when subjected to stress at varying temperatures. This characteristic of our system can be utilized in the development of shape memory hydrogels. Applying temperature or stress can cause the permanent shape of IPNE3 to disengage, transforming it into a temporary shape. This temporary shape can be restored by cooling the deformed sample while under load or reducing stress. Shape recovery occurs when the gel in its temporary shape is reheated and/or when a sufficiently large stress is applied to break the physical network. From a molecular perspective, the shape memory cycle regulates the relaxation times of the temporary shape through the amplitude of stress and/or temperature.
2.4. Chemical structure of single and IPN hydrogels
We assessed the chemical structures of the materials using Fourier-transform infrared spectroscopy (FT-IR). Figure 6 shows the FT-IR spectra of the NiPAM monomer, PNiPAME hydrogel, CS hydrogels, and IPNE3. Several changes became apparent in the FT-IR spectrum of the PNiPAME hydrogel compared to the NiPAM monomer, as illustrated in Figure 6(a) and 6(b). The sharp peak at 3274 cm-1 in the NiPAM monomer, attributed to the N-H stretching vibrations, became significantly broader in the PNiPAME hydrogel. This broadening was due to overlap with O-H vibrations from alcohol residues in the solvent. The distinctive spectral peaks of the NiPAM monomer at 1618 and 960 cm-1, associated with C=C bending and the vinyl group, vanished in the PNiPAME hydrogel, indicating polymerization proceeds. Moreover, specific peaks related to the isopropyl group vibrations appeared at 1386 and 1367 cm−1, shifting to a lower wavelength than the monomer (1400 cm-1), suggesting that these isopropyl groups act as crosslinking points. This formation of crosslinking points was supported by interactions between the tert-C atom of the side isopropyl group and the main chain isopropyl group, as explained in a previous study [44].
For the CS hydrogel presented in Figure 6(c), we found overlapping peaks at 3356 and 3288 cm-1, indicating -OH stretching, symmetric N-H vibration, and intermolecular H-bonds between polysaccharide chains. The carbonyl stretching vibration (amide-I), N-H stretching vibration (amide-II), and C-N stretching vibration (amide-III) of CS were observed at 1643, 1556, and 1311 cm-1, respectively. Additionally, the symmetrical deformation of the methyl (CH3) groups of CS was identified at 1373 cm-1, consistent with prior research [45].
The characteristic peaks of the IPN hydrogel indicate the presence of functional groups from both neat hydrogels. Some shifts in peak positions were observed as shown in Figure 6(d). These shifts suggest that the initial reactants could penetrate the CS hydrogel, and upon exposure to UV light, the PNiPAM structure was formed. Further shifts in wavenumber and band broadening indicate the formation of intermolecular associations between these pure hydrogels, which display compatible characteristics of both CS and PNiPAM hydrogels.
2.5. Equilibrium swelling ratio (SRe) of hydrogel networks
Figure 7 shows the swelling ratio SRe of the CS and IPN hydrogels with varying crosslinker contents, calculated using Equation (5) [46].
where wd and we represent the weight of dried hydrogels before being immersed in deionized water and the swollen sample reaching its equilibrium state at room temperature, respectively.
The interpenetration of the PNiPAM network resulted in an increased SRe for the CS hydrogel. The CS hydrogel is formed through physical crosslinking, primarily held together by hydrophobic interactions of the N-acetyl group and inter- and/or intra-molecular hydrogen bondings. This results in a polymer network that can easily deform, leading to disintegration and dissolution in water. Moreover, the CS network lacks stable crosslinking points, making it capable of absorbing less water than the IPNs, which have a stable network with chemically crosslinked structures.
The hydrophilicity of CS improved when the PNiPAM network was introduced, leading to an increased SRe with a rise in crosslinker content. At lower crosslinker contents, a less stable network was formed, allowing polymer chains to partially dissolve in water and easily diffuse from the polymer structure. The network structure exhibited high hydrophilicity, interacting with water molecules most effectively at a crosslinker content of 2.65 wt%, resulting in the highest water uptake. However, SRe decreased at crosslinking concentrations exceeding 2.65 wt% due to the increased density of crosslinks and the entanglement of polymer chains within the gel network. This led to a dense structure that hindered further swelling, consistent with our crosslink density findings. As a result, water molecules diffused more slowly into the network, limiting the relaxation of network chains in the hydrogels [47].
The IPNE system exhibited a higher magnitude of SRe than the IPNw system. This is because less water could penetrate the dense IPNw structure, whereas the IPNE networks had a more porous structure, illustrating the morphology in FE-SEM results. The mechanism of water absorption will be thoroughly investigated in our future research.
2.6. Microstructure of the CS and IPN hydrogels
We used FE-SEM to examine the surface and cross-sectional structures of the fresh hydrogels. Figure 8 illustrates the morphologies of CS and IPN hydrogels with different BIS concentrations. The CS hydrogel exhibited a network of interconnected fibers with both coarse and smooth surfaces, consistent with previous findings [48]. Such surface structures are typical of polyelectrolyte components like CS and sodium alginate, as well as their mixtures [49]. When the PNiPAM network was added, the surface of the CS hydrogel became smoother and more uniform as the BIS crosslinker content increased. The roughness of the hydrogel surface played a crucial role in altering its morphology. A smoother surface enhances the antifouling properties of hydrogel materials, making them more resistant to unwanted adhesion.
Cross-sectional images of the samples revealed different structures based on the BIS concentration. The CS hydrogel exhibited a well-defined lamellar structure due to the system’s homogeneity, allowing the NiPAM solution to penetrate easily. In the IPNs, a smooth and dense structure with irregular pores was observed, indicating excellent compatibility between the two polymers. The lamellar layer of the CS hydrogel was replaced by the PNiPAM network, creating a porous structure ideal for absorbing wound fluid and facilitating oxygen supply, which promotes faster wound healing [50]. Furthermore, it is a useful material for loading and releasing preservative substances in food processing applications [51]. The IPN structure becomes denser with a higher crosslinker content, resulting from the increased crosslink density of PNiPAM within the CS network. Moreover, this material finds application in food processing for loading and releasing preservatives [51]. With a higher crosslinker content, the IPN structure became denser due to increased crosslink density of PNiPAM within the CS network. This homogeneity confirmed the compatibility between CS and PNiPAM, contributing to the high mechanical properties observed in the G′ results.
2.7. Wettability of hydrogel surface
Water contact angle (WCA) is a common metric used to gauge the hydrophobicity or hydrophilicity of a material’s surface. Hydrophilicity is a key factor in developing anti-biofouling materials [52]. On the tested hydrogel surfaces, WCAs ranged from 56.2° to 61.2° (Table 1), which is notably lower than the 79° observed on the surface of PS (polystyrene) [53]. The hydrogels contain various hydrophilic functional groups, including -NH2 and -OH groups from CS and amide (-CONH-) groups from PNiPAM. These groups contribute to the hydrogels having a more hydrophilic surface. Greater surface hydrophilicity enhances the material’s antifouling properties by reducing its affinity for hydrophobic fouling agents and forming a hydrated surface layer [54].
2.8. HeLa cell adhesion and proliferation on the hydrogels’ surface
The biocompatibility of biomaterials can be assessed by examining how cells adhere to them. In this study, HeLa cells, a commonly used immortalized human cancer cell line in research labs worldwide, were used [55]. The morphology of HeLa cells on both the CS and IPNE hydrogels was observed and compared to those on the PS control surface after 1 hour of cell seeding, as shown in Figure 9(a) to 9(c). No significant differences were observed in cell morphology between the control and hydrogel surfaces. However, it was evident that the hydrogel surfaces had more cell colonies than the control surface. The positively charged sites in the CS structure likely enhanced electrostatic interactions with the negatively charged cell membranes and proteins, facilitating cell adhesion to the CS surface [56]. Additionally, the hydrophobic nature of PNiPAM-based IPN might contribute to its cell adhesion properties, as it can switch from a hydrophilic to a hydrophobic state above its VPTT (37°C), as shown in Figure S9. The appearance of the swollen IPNE3 in the PBS solution changed from transparent at room temperature to opaque at the incubation temperature (37°C).
After 24 hours of cell culture initiation, HeLa cell proliferation was observed. Cells on the polystyrene (PS) control surface displayed a flattened morphology, indicating strong cell adhesion to the surface, as shown in Figure 9(a′). In contrast, cells on the hydrogel surfaces exhibited a rounder morphology, with lower adhesion and less proliferation, as shown in Figure 9(b′) to 9(c′). Cell populations were counted after detaching from the material surface, and revealing that more cells had grown and spread on the PS control surface (3.6 ± 0.10 × 106 cells/cm3) compared to the hydrogels (Table 1). The hydrogels likely had a significant impact on the strength of interactions with HeLa cells, leading to improved resistance against fouling [57]. The number of adhered cells on the IPN surfaces was slightly lower than on the CS surface, possibly due to the smoother surface, as indicated by the FE-SEM results. In general, cells adhere well to stiff surfaces [58,59], and materials with higher roughness are more prone to fouling as contaminants tend to accumulate in the "valleys" of rough surfaces [60,61]. Furthermore, the surface of IPNE3 underwent a thermal reversal, transitioning from a hydrophobic state at the incubation temperature (37°C) to its original hydrophilic state at room temperature. This demonstrated the detachment or release of HeLa cells from the surface. In other words, the excellent thermo-reversible gelation of this material offers numerous benefits for various applications. The gel’s strength is significantly enhanced when the IPNE3 is swollen and used above the VPTT, as described in Section 2.3 and in the Supplement Information. Conversely, it can be reversed to its original transparent state when cooled down below the VPTT.
3. Conclusions
We have explored a straightforward and versatile method for crafting hydrogel-based interpenetrating polymer networks (IPNs) with several notable advantages, such as enhanced mechanical properties, multifunctionality, and anti-fouling attributes. The process involves a sequential approach in which a CS hydrogel is swollen in a solution containing NiPAM monomer in ethanol or an aqueous solution of a BIS crosslinking agent, employing the I2965 photo-initiator. The PNiPAM network is then established as a cross-linked structure through UV polymerization at room temperature. The gel formation is evident through the sol-gel transition, as confirmed by the scaling law of the Winter and Chambon hypothesis.
Furthermore, we investigated the impact of BIS concentration on the resulting hydrogels’ properties. Increasing the BIS concentration from 1.45 to 21.72 wt% led to improvements in all analyzed rheological and viscoelastic properties of the CS hydrogel. However, exceeding the maximum BIS concentration had a detrimental effect. Higher degrees of crosslinking resulted in the formation of stiffer modules with reduced wettability. Among all BIS concentrations, intermediate values of the crosslinking agent (5.16 wt%) demonstrated the best performance.
The incorporation of the PNiPAM network into the CS hydrogel induced the formation of a porous structure, increasing water uptake within the hydrogel networks. Additionally, these materials exhibited significantly greater thermal and mechanical stabilities when exposed to varying temperatures and applied forces, compared to the CS hydrogel, which was vulnerable to thermal disruption. These materials are anticipated to demonstrate exceptional performance characteristics. They can effectively adhere to and eliminate living organisms through the cationic properties of the CS surface while also releasing undesirable molecules through hydrolysis, thanks to the hydrophilic/hydrophobic switchable property of the PNiPAM network.
4. Materials and Methods
4.1. Materials
All reagents used in this study were analytical grade. Medium molecular weight CS of 75–85% deacetylation, N-isopropylacrylamide, N,N-methylenebisacrylamide (BIS), and 2-hydroxyl-4-(2-hydroxyethoxy)-2-methylpropiophenone (I2965) were purchased from Sigma-Aldrich. BIS was utilized as a crosslinker, while I2965 served as a photo-initiator. All solvents, including acetic acid, 1, 3-propanediol, and ethanol, were purchased from Wako Pure Chemicals, Osaka, Japan.
4.2. Characterization of the samples
4.2.1. Fourier-transform infrared spectroscopy (FT-IR)
The chemical structure of the single network and IPN hydrogels was characterized using FT-IR analysis, which was conducted on a Horiba FT-IR 720 spectrometer equipped with an attenuated total reflectance accessory. The obtained spectra were averaged from 64 scans at a resolution of 4 cm-1 within the spectral range of 650–4000 cm-1.
4.2.2. Rheological test
The viscoelastic behavior of fresh and equilibrated samples was evaluated using a TA instrument TRIOS, employing a parallel plate of 25 mm diameter. The measurements were conducted within the linear viscoelasticity region. The shear modulus (G′), loss modulus (G″), and loss tangent (tan δ) were measured as a function of the frequency within a frequency range of 0.1–100 rad s-1 with 0.01% strain at 25 oC. Each hydrogel was automatically loaded at the normal force of 1.0 N. Thermal and mechanical stabilities of the hydrogels in fresh and swollen states were investigated by measurement of G′ as a function of frequency over the temperature range of 30–80oC.
4.2.3. Field emission-SEM (FE-SEM)
The surface and cross-section morphology of the CS and IPNE samples were examined using FE-SEM (Hitachi SU-4800) operated with an accelerating voltage of 3.0 kV and emission current of 10 mA. Before measurement, the fracture surfaces of the materials were sputter-coated with gold.
4.2.4. Measurement of equilibrium swelling ratio (SRe)
The swelling characteristics of the hydrogels were measured gravimetrically. The dried hydrogels were weighed before being immersed in deionized water at room temperature (Wd). The swollen hydrogels were then removed from the water at regular intervals and weighed after excess water on the hydrogel surfaces was removed using filter paper (Wt). The average value of three measurements was taken for each hydrogel to minimize errors. The water absorption of the hydrogel was continuously measured to allow it to reach its equilibrium swelling value (We). The equilibrium swelling ratio (SRe) was calculated using Equation (5) [46].
4.2.5. Wettability test; water contact angle (WCA)
The wettability of samples was observed by measuring the WCA on the hydrogel surface using a contact angle meter (DM-300; Kyowa Interface Science, Saitama, Japan).
4.2.6. Cell adhesion test
HeLa cells (JCRB, #JCRB9004), an immortal cell line derived from human cervical cancer cells, were cultured in a complete medium (CM). The CM consisted of Dulbecco’s Modified Eagle’s medium supplemented with 2 mM L-glutamine and 10% (v/v) fetal bovine serum (Biowest, France). After rinsing with phosphate-buffered saline (PBS) at pH 7.2, the sub-cultured cells were harvested from a tissue culture polystyrene dish using a 0.25% (w/v) trypsin/1 mM-EDTA solution. The cells were then recovered by centrifugation at 500 ×g for 5 minutes. Subsequently, the cells were seeded on polymer sheets at a density of 1.0 × 104 cells/cm2. The polymer sheets, free from any solvent contaminants and unreactive components, were pre-equilibrated in PBS for 2 hours before being cultured in complete medium (CM) for 24 hours at 37 oC under 5% (v/v) CO2. After being washed with PBS, the medium was replaced with fresh CM, and the cells were imaged using a Nikon BW-S507 fluorescence microscope at Tokai University Imaging Center for Advanced Research. The cell numbers were counted after adding trypsin blue solution to a final 0.2% (v/v).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, D.I.; methodology, D.I.; software, D.I; validation, D.I.; formal analysis, D.I.; investigation, D.I., T.F., S.N., Y.S., and K.R.; writing-original draft preparation, D.I.; writing-review and editing, D.I., T.F., S.N., Y.S., and K.R.; visualization, D.I., T.F., S.N., Y.S., and K.R.; supervision, D.I. and K.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the Micro/Nano Technology Center, Tokai University, Kanagawa, Japan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors can provide any necessary information on the study upon request.
Acknowledgments
The FT-IR and WCA measurements were guided by Prof. Osamu Kanie (Department of Applied Biochemistry) and Prof. Yosuke Okamura (Department of Applied Chemistry), Tokai University. The authors thank Prof. Hiroshi Kimura’s assistant researcher for the cell adhesion test (Bio-Microfluidic System Laboratory, Department of Mechanical Engineering, Tokai University).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jiang, S.Y.; Cao, Z.Q. Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications. Adv Mater 2010, 22, 920–932. [Google Scholar] [CrossRef] [PubMed]
- DeFlorio, W.; Liu, S.; White, A.R.; Taylor, T.M.; Cisneros-Zevallos, L.; Min, Y.; Scholar, E.M.A. Recent developments in antimicrobial and antifouling coatings to reduce or prevent contamination and cross-contamination of food contact surfaces by bacteria. Compr. Rev. Food Sci. Food Saf. 2021, 20, 3093–3134. [Google Scholar] [CrossRef]
- Dueramae, I.; Nishida, M.; Nakaji-Hirabayashi, T.; Matsumura, K.; Kitano, H. Biodegradable shape memory polymers functionalized with anti-biofouling interpenetrating polymer networks. J. Mater. Chem. B 2016, 4, 5394–5404. [Google Scholar] [CrossRef]
- Bauman, L.; Zhao, B. Multi-thermo responsive double network composite hydrogel for 3D printing medical hydrogel mask. J. Colloid Interface Sci. 2023, 638, 882–892. [Google Scholar] [CrossRef]
- Du, J.; Ma, Q.; Wang, B.; Sun, L.; Liu, L. Hydrogel fibers for wearable sensors and soft actuators. iScience 2023, 26, 106796. [Google Scholar] [CrossRef]
- Jian, Y.; Wu, B.; Yang, X.; Peng, Y.; Zhang, D.; Yang, Y.; Qiu, H.; Lu, H.; Zhang, J.; Chen, T. Stimuli-responsive hydrogel sponge for ultrafast responsive actuator. Supramol. Mater. 2022, 1, 100002. [Google Scholar] [CrossRef]
- Dueramae, I.; Okhawilai, M.; Kasemsiri, P.; Uyama, H. High electrochemical and mechanical performance of zinc conducting-based gel polymer electrolytes. Sci. Rep. 2021, 11, 13268. [Google Scholar] [CrossRef]
- Aruchamy, K.; Ramasundaram, S.; Divya, S.; Chandran, M.; Yun, K.; Oh, T.W. Gel polymer electrolytes: Advancing solid-state batteries for high-performance applications. Gel 2023, 9, 585. [Google Scholar] [CrossRef]
- Creton, C. 50th Anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 2017, 50, 8297–8316. [Google Scholar] [CrossRef]
- Dragan, E.S. Design and applications of interpenetrating polymer network hydrogels. A review. Chem. Eng. J. 2014, 243, 572–590. [Google Scholar] [CrossRef]
- Jayakumar, R.; Prabaharan, M.; Sudheesh, K.P.T.; Nair, S.V.; Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef]
- Dueramae, I.; Yoneyama, N.; Shinyashiki, S.; Yagihara, S.; Kita, R. Self-assembly of acetylated dextran with various acetylation degrees in aqueous solutions: Studied by light scattering. Carbohydr. Polym. 2017, 159, 171–177. [Google Scholar] [CrossRef]
- Dueramae, I.; Yoneyama, N.; Shinyashiki, S.; Yagihara, S.; Kita, R. Thermal diffusion of aqueous solution of acetylated dextran: The effect of hydrophobicity using optical beam deflection technique. Int. J. Heat Mass Transf. 2019, 132, 997–1003. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, A.; Galaev, I.Y.; Mattiassaon, B. Smart polymers: physical forms and bioengineering applications. Prog. Poly. Sci. 2007, 32, 1205–1237. [Google Scholar] [CrossRef]
- Wei, M.; Gao, Y.; Li, X.; Serpe, M.J. Stimuli-responsive polymers and their applications. Polym. Chem. 2017, 8, 127–143. [Google Scholar] [CrossRef]
- Hirokawa, Y.; Tanaka, T. Volume phase transition in a nonionic gel. J. Chem. Phys. 1984, 81, 6379–6380. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuo, R.; Cui, J.; Zhang, J. A novel thermo-responsive drug delivery System with positive controlled release. Int. J. Pharm. 2002, 235, 43–50. [Google Scholar] [CrossRef]
- Dueramae, I.; Okhawilai, M.; Kasemsiri, P.; Uyama, H.; Kita, R. Properties enhancement of carboxymethyl cellulose with thermo-responsive polymer as solid polymer electrolyte for zinc ion battery. Sci. Rep. 2020, 10, 12587. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhu, J.; Song, X.; Wen, Y.; Li, J. Smart hydrogel formed by alginate-g-poly(N-isopropylacrylamide) and chitosan through polyelectrolyte complexation and its controlled release properties. Gels 2022, 8, 441. [Google Scholar] [CrossRef] [PubMed]
- Afloarea, O.-T.; Cheaburu Yilmaz, C.N.; Verestiuc, L.; Bibire, N. Development of vaginal carriers based on chitosan-grafted-PNIPAAm for progesterone administration. Gels 2022, 8, 596. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, B.; Takatsuka, M.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Rathinasabapathy, S. Dynamics of the poly(N-Isopropylacrylamide) microgel aqueous suspension investigated by dielectric relaxation spectroscopy. Macromolecules 2022, 55, 1218–1229. [Google Scholar] [CrossRef]
- Fukai, T.; Shinyashiki, N.; Yagihara, S.; Kita, R.; Tanaka, F. Phase behavior of co-nonsolvent systems: Poly(N-isopropylacrylamide) in mixed solvents of water and methanol. Langmuir 2018, 34, 3003–3009. [Google Scholar] [CrossRef]
- Xu, L.; Liang, X.; You, L.; Yang, Y.; Fen, G.; Gao, Y.; Cui, X. Temperature-sensitive poly(N-isopropylacrylamide)-chitosan hydrogel for fluorescence sensors in living cells and its antibacterial application. Int. J. Biol. Macromol. 2021, 189, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.T.; Jo, S.-H.; Phan, Q.-T.; Park, H.; Park, S.-H.; Oh, C.-W.; Lim, K.T. Dual pH-/thermo-responsive chitosan-based hydrogels prepared using "click" chemistry for colon-targeted drug delivery applications. Carbohydr. Polym. 2021, 260, 117812. [Google Scholar] [CrossRef]
- Romero, M.R.; Coser, N.A.; P´erez, M.A.; Gomez, C.G. Poly(N-isopropylacrylamide)-interpenetrated chitosan coils working as nanoreactors for controlled silver nanoparticle growth. Carbohydr. Polym. 2022, 288, 119374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Chen, W.; Liao, L.; Xie, P.; Zhang, T.C.; Wu, X.; Feng, X. Comparative adsorption of emerging contaminants in water by functional designed magnetic poly(N-isopropylacrylamide)/chitosan hydrogels. Sci. Total Environ. 2019, 671, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-T.; Zeng, K.; Fuhrmann, B.; Woelk, C.; Zhang, K.; Groth, T. Engineering of stable cross-linked multilayers based on thermo-responsive PNIPAM-grafted chitosan/heparin to tailor their physiochemical properties and biocompatibility. ACS Appl. Mater. Interfaces 2022, 14, 29550–29562. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-Y.; Luo, L.-J. Chitosan-g-poly(N-isopropylacrylamide) copolymers as delivery carriers for intracameral pilocarpine administration. Eur J Pharm Biopharm 2017, 113, 140–148. [Google Scholar] [CrossRef]
- Kopač, T.; Ručigaj, A.; Krajnc, M. The mutual effect of the crosslinker and biopolymer concentration on the desired hydrogel properties. Int. J. Biol. Macromol. 2020, 159, 557–569. [Google Scholar] [CrossRef]
- Dueramae, I.; Ishiyama, T.; Torigaki, A.; Nakano, S.; Sasaki, K.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Katsumoto, Y.; Yoneyama, M. Separation of micro-Brownian motion and side-group rotational motion for poly(N-isopropylacrylamide) in 1,4-dioxane studied by dielectric relaxation spectroscopy. Macromolecules 2023, 56, 4041–4048. [Google Scholar] [CrossRef]
- Dueramae, I.; Fukuzawa, S.; Shinyashiki, N.; Yagihara, S.; Kita, R. Dynamics of amyloid -like aggregation and gel formation of hen egg-white lysozyme in highly concentrated ethanol solution. J. Biorheology 2017, 31, 21–28. [Google Scholar] [CrossRef]
- Rimdusit, S.; Punson, K.; Dueramae, I.; Somwangthanaroj, A.; Tiptipakorn, S. Rheological and thermomechanical characterizations of fumed silica-filled polybenzoxazine nanocomposites. Eng. J. 2011, 15, 27–38. [Google Scholar] [CrossRef]
- Carmona, P.; Tasici, A.M.; Sande, S.A.; Knudsen, K.D.; Nyström, B. Glyceraldehyde as an efficient chemical crosslinker agent for the formation of chitosan hydrogels. Gels 2021, 7, 186. [Google Scholar] [CrossRef]
- Joly, J.-P.; Aricov, Ludmila.; Balan, G.-A.; Popescu, E.I.; Mocanu, S.; Leonties, A.R.; Matei, I.; Marque, S.R.A.; Ionita, G. Formation of alginate/chitosan interpenetrated networks revealed by EPR spectroscopy. Gels 2023, 9, 231.
- Winter, H.H. Can the gel point of a cross-linking polymer be detected by the G′ – G″ crossover? Polym. Eng. Sci. 1987, 27, 698–1702. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Chen, L.A.; McDowell, C.; Khan, S.A.; Hwang, R.; White, S. Rheological study of crosslinking and gelation in chlorobutyl elastomer systems. Polymer 1996, 37, 5869–5875. [Google Scholar] [CrossRef]
- Hassan, A.; Niazi, M.B.K.; Hussain, A.; Earrukh, S.; Ahmad, T. Development of anti-bacterial PVA/starch based hydrogel emmbrane for wound dressing. J. Polym. Environ. 2018, 26, 235–243. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Y.; Zhang, L.; Xu, M.; Dong, W.; Zhang, J. Fabrication of Salecan/poly(AMPS-co-HMAA) semi-IPN hydrogels for cell adhesion. Carbohydr. Polym. 2017, 174, 171–181. [Google Scholar] [CrossRef]
- Nie, W.; Yuan, X.; Zhao, J.; Zhou, Y.; Bao, H. Rapidly in situ forming chitosan/ɛ-polylysine hydrogels for adhesive sealants and hemostatic material. Carbohydr. Polym. 2013, 96, 342–348. [Google Scholar] [CrossRef]
- Chambon, F.; Winter, H.H. Linear viscoelasticity at the gel point of a crosslinking PDMS with imbalanced stoichiometry. J. Rheol. 1987, 31, 683–697. [Google Scholar] [CrossRef]
- Muthukumar, M. Screening effect on viscoelasticity near the gel point. Macromolecules 1989, 22, 4656–4658. [Google Scholar] [CrossRef]
- Scanlan, J.C.; Winter, H.H. Composition dependence of the viscoelasticity of end-linked poly(dimethylsiloxane) at the gel point. Macromolecules 1991, 24, 47–54. [Google Scholar] [CrossRef]
- Kurecic, M.; Sfiligoj-Smole, M.; Stana-Kleinschek, K. UV polymerization of poly(N-isopropylacrylamide) hydrogel. Mater. Technol. 2012, 46, 87–91. [Google Scholar]
- Maity, J.; Ray, S.K. Enhanced adsorption of methyl violet and congo red by using semi and full IPN of polymethacrylic acid and chitosan. Carbohydr. Polym. 2014, 104, 8–16. [Google Scholar] [CrossRef]
- Mandal, B.; Ray, S.K. Synthesis of interpenetrating network hydrogel from poly(acrylic acid-co-hydroxyethyl methacrylate) and sodium alginate: Modeling and kinetics study for removal of synthetic dyes from water. Carbohyd. Polym. 2013, 98, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, H. Controlled release of diclofenac sodium from pH-responsive carrageenan-g-poly(acrylic acid) superabsorbent hydrogel. J. Chem. Sci. 2010, 122, 651–659. [Google Scholar] [CrossRef]
- Kaya, M.; Baran, T. Description of a new surface morphology for chitin extracted from wings of cockroach (Periplaneta americana). Int. J. Biol. Macromol. 2015, 75, 7–12. [Google Scholar] [CrossRef]
- Kulig, D.; Zimoch-Korzycka, A.; Jarmoluk, A.; Marycz, K. Study on alginate–chitosan complex formed with different polymers ratio. Polymers. 2016, 8, 167. [Google Scholar] [CrossRef]
- Natarajan, N.; Shashirekha, V.; Noorjahan, S.E.; Rameshkumar, M.; Rose, C.; Sastry, T.P.J. Fibrin–chitosan–gelatin composite film: preparation and characterization. J. Macromol. Sci. A 2005, 42, 945–953. [Google Scholar] [CrossRef]
- Fuciños, C.; Fuciños, P.; Pastrana, L.M.; Rúa, M.L. Functional characterization of poly(N-isopropylacrylamide) nanohydrogels for the controlled release of food preservatives. Food Bioprocess Technol. 2014, 7, 3429–3441. [Google Scholar] [CrossRef]
- Pasmore, M.; Todd, P.; Smith, S.; Baker, D.; Silverstein, J.; Coons, D.; Bowman, C.N. Effects of ultrafiltration membrane surface properties on Pseudomonas aeruginosa biofilm initiation for the purpose of reducing biofouling. J. Membr. Sci. 2001, 194, 15–32. [Google Scholar] [CrossRef]
- Kolodin, A.N.; Bulavchenko, A.I. Contact angle and free surface energy of CdS films on polystyrene substrate. Appl. Surf. Sci. 2019, 463, 820–828. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Xiong, S.; Shi, Y.S.; Zhu, J.; Hu, Q.L.; Liu, J.; Wang, Y. Antifouling enhancement of polyimide membrane by grafting DEDA-PS zwitterions. Chemosphere 2018, 198, 30–39. [Google Scholar] [CrossRef]
- Santos, P.A.; Rocha, C.S.; Baptista, M.S. Adhesion and proliferation of HeLa and fibroblast cells onchemically-modified gold surfaces. Colloids. Surf. B. 2014, 123, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.P.; Fang, N. Adhesion dynamics, morphology, and organization of 3T3 fibroblast on chitosan and its derivative: the effect of O-carboxymethylation. Biomacromolecules 2005, 6, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wei, J. Hydrogel brushes grafted from stainless steel via surface-initiated atom transfer radical polymerization for marine antifouling. Appl. Surf. Sci. 2016, 382, 202–216. [Google Scholar] [CrossRef]
- Pelham, R.J.; Wang, Y.L. Cell locomotion and focal adhesions are regulated bysubstrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef]
- Hadjipanayi, E.; Mudera, V.; Brown, R.A. Close dependence of fibroblast proliferation on collagen scaffold matrix stiffness. J. Tissue Eng. Regen. Med. 2009, 3, 277–284. [Google Scholar] [CrossRef]
- Yuliwati, E.; Ismail, A.F. Effect of additives concentration on the surface properties and performance of PVDF ultrafiltration membranes for refinery produced wastewater treatment. Desalination 2011, 273, 226–234. [Google Scholar] [CrossRef]
- Bae, T.H.; Kim, I.C.; Tak, T.M. Preparation and characterization of fouling-resistant TiO2 self-assembled nanocomposite membranes. J. Membr. Sci. 2006, 275, 1–5. [Google Scholar] [CrossRef]
Figure 1.
Investigation of sol-gel transition for PNIPAM solutions, containing a BIS concentration of 1.45wt% (0.01 M) measured at room temperature; (a) Zero-shear viscosity η0 vs. UV irradiation time, t, (b) the frequency-dependent behavior of the moduli, i.e. G′~ G″ ~ ωn at t = 120 min.
Figure 1.
Investigation of sol-gel transition for PNIPAM solutions, containing a BIS concentration of 1.45wt% (0.01 M) measured at room temperature; (a) Zero-shear viscosity η0 vs. UV irradiation time, t, (b) the frequency-dependent behavior of the moduli, i.e. G′~ G″ ~ ωn at t = 120 min.
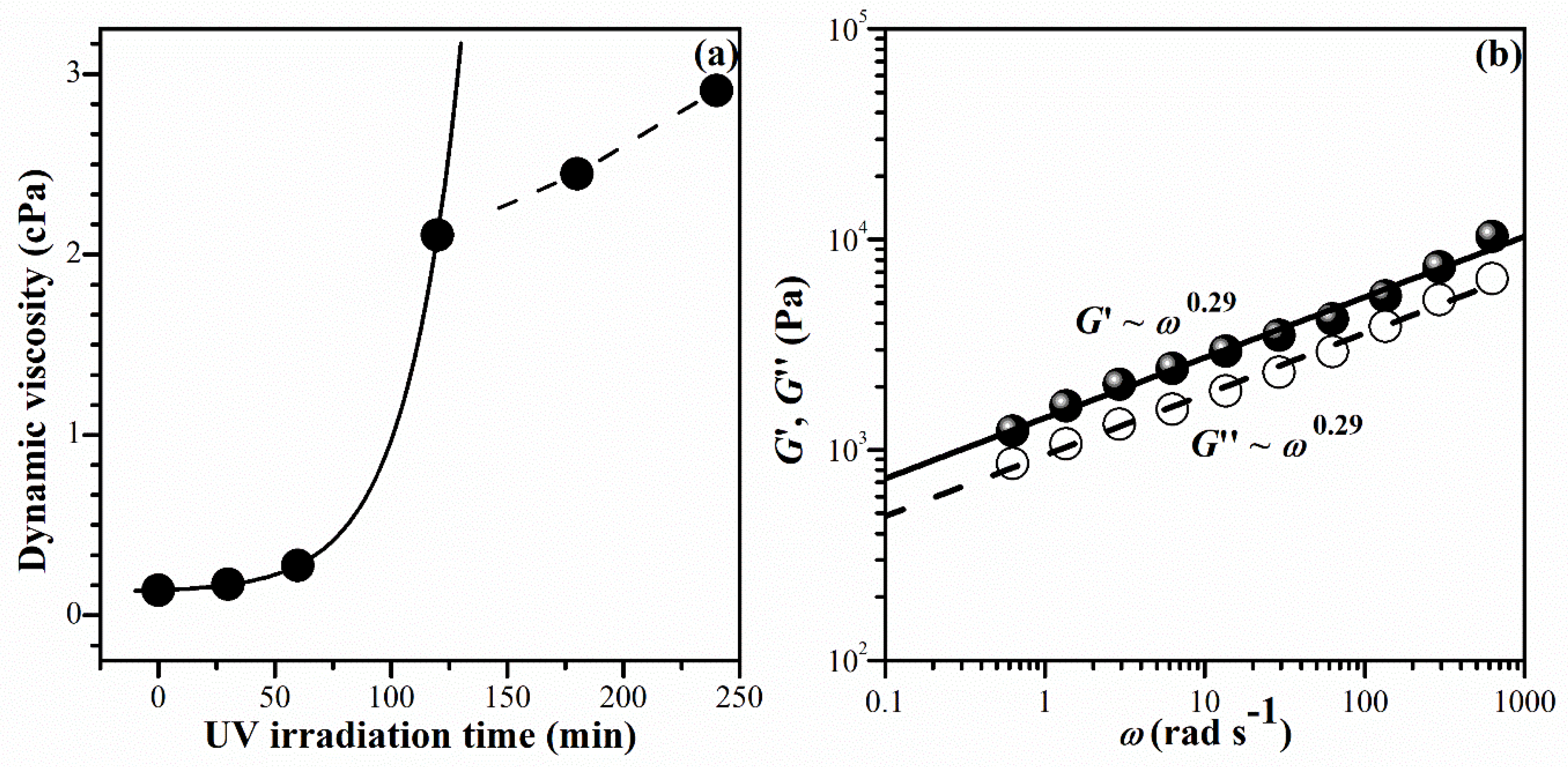
Figure 2.
Frequency dependence of the (a) G′, and (b) G″ at 25 oC of the hydrogels with a BIS crosslinker content of 0 wt.% (CS hydrogel) (○), 1.45 wt.% IPNE1(●), 2.65 wt.% IPNE2(■), 5.16 wt.% IPNE3(▲), and 21.42 wt.% IPNE4 (♦).
Figure 2.
Frequency dependence of the (a) G′, and (b) G″ at 25 oC of the hydrogels with a BIS crosslinker content of 0 wt.% (CS hydrogel) (○), 1.45 wt.% IPNE1(●), 2.65 wt.% IPNE2(■), 5.16 wt.% IPNE3(▲), and 21.42 wt.% IPNE4 (♦).
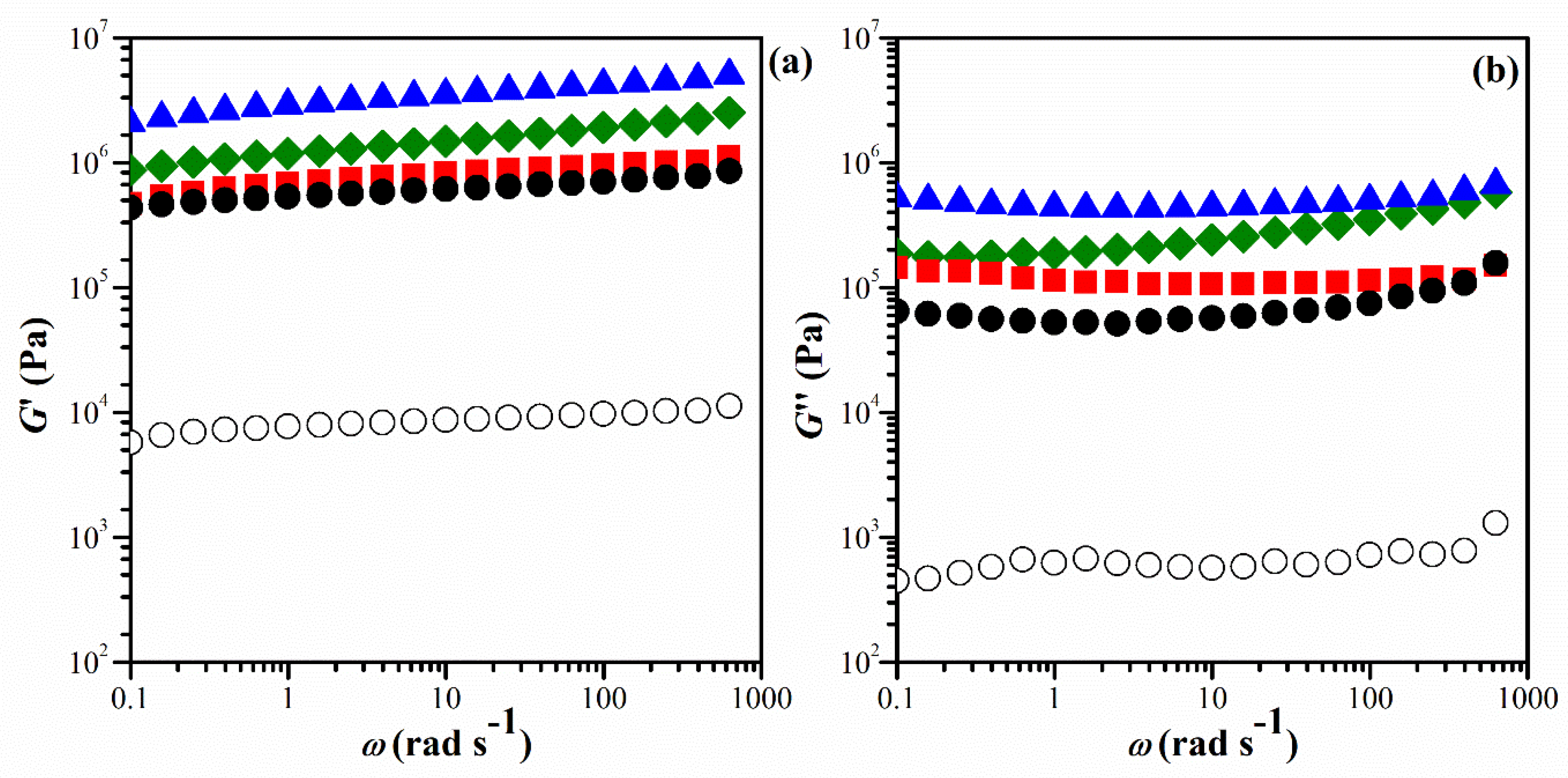
Figure 3.
(a) Shear modulus, G′, obtained at 1 Hz (6.28 rad s-1) with various crosslinker contents. G′ of IPNs prepared in ethanol solution (IPNE system) (●) and IPN prepared in aqueous solution (IPNW system) (●). The inset shows G′ of PNiPAM hydrogels prepared in ethanol solution (PNiPAME) (●) and in aqueous solution (PNiPAMW) (●) as a function of crosslinker content (Table 1 involves 5.16 wt% BIS only for PNiPAME and PNiPAMW systems). (b) The crosslinking density ve as a function of crosslinker content for IPNE system. The inset shows a schematic IPN network model in which grey and blue networks represented the CS and PNiPAM chains, respectively. Yellow circles are the crosslinking points. Curves are drawn to guide eyes.
Figure 3.
(a) Shear modulus, G′, obtained at 1 Hz (6.28 rad s-1) with various crosslinker contents. G′ of IPNs prepared in ethanol solution (IPNE system) (●) and IPN prepared in aqueous solution (IPNW system) (●). The inset shows G′ of PNiPAM hydrogels prepared in ethanol solution (PNiPAME) (●) and in aqueous solution (PNiPAMW) (●) as a function of crosslinker content (Table 1 involves 5.16 wt% BIS only for PNiPAME and PNiPAMW systems). (b) The crosslinking density ve as a function of crosslinker content for IPNE system. The inset shows a schematic IPN network model in which grey and blue networks represented the CS and PNiPAM chains, respectively. Yellow circles are the crosslinking points. Curves are drawn to guide eyes.
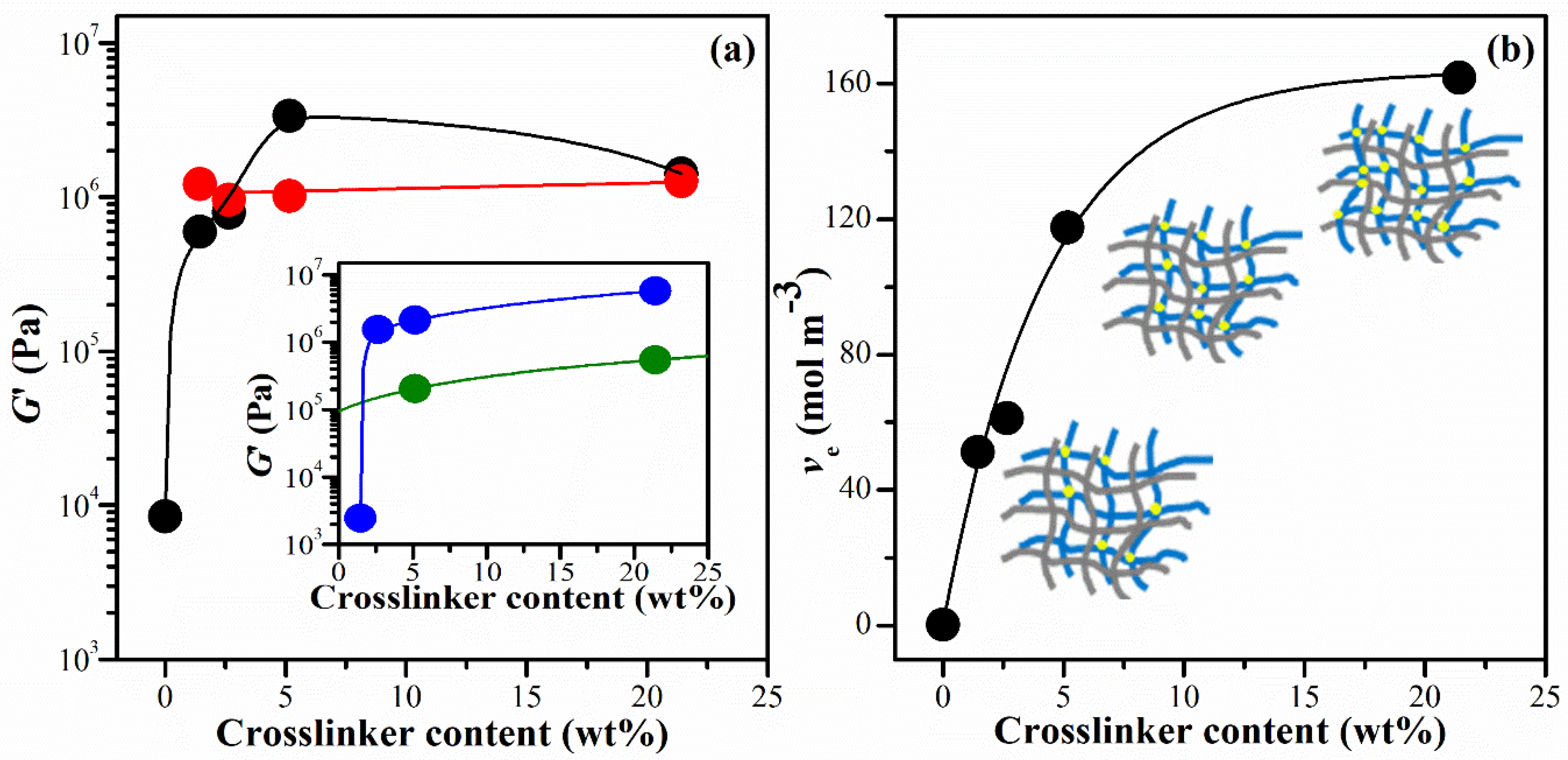
Figure 4.
Gel strength S as a function of BIS concentration of hydrogels for IPNE system (●), IPNw system (●), and PNiPAME system (●). Curves are drawn to guide eyes.
Figure 4.
Gel strength S as a function of BIS concentration of hydrogels for IPNE system (●), IPNw system (●), and PNiPAME system (●). Curves are drawn to guide eyes.
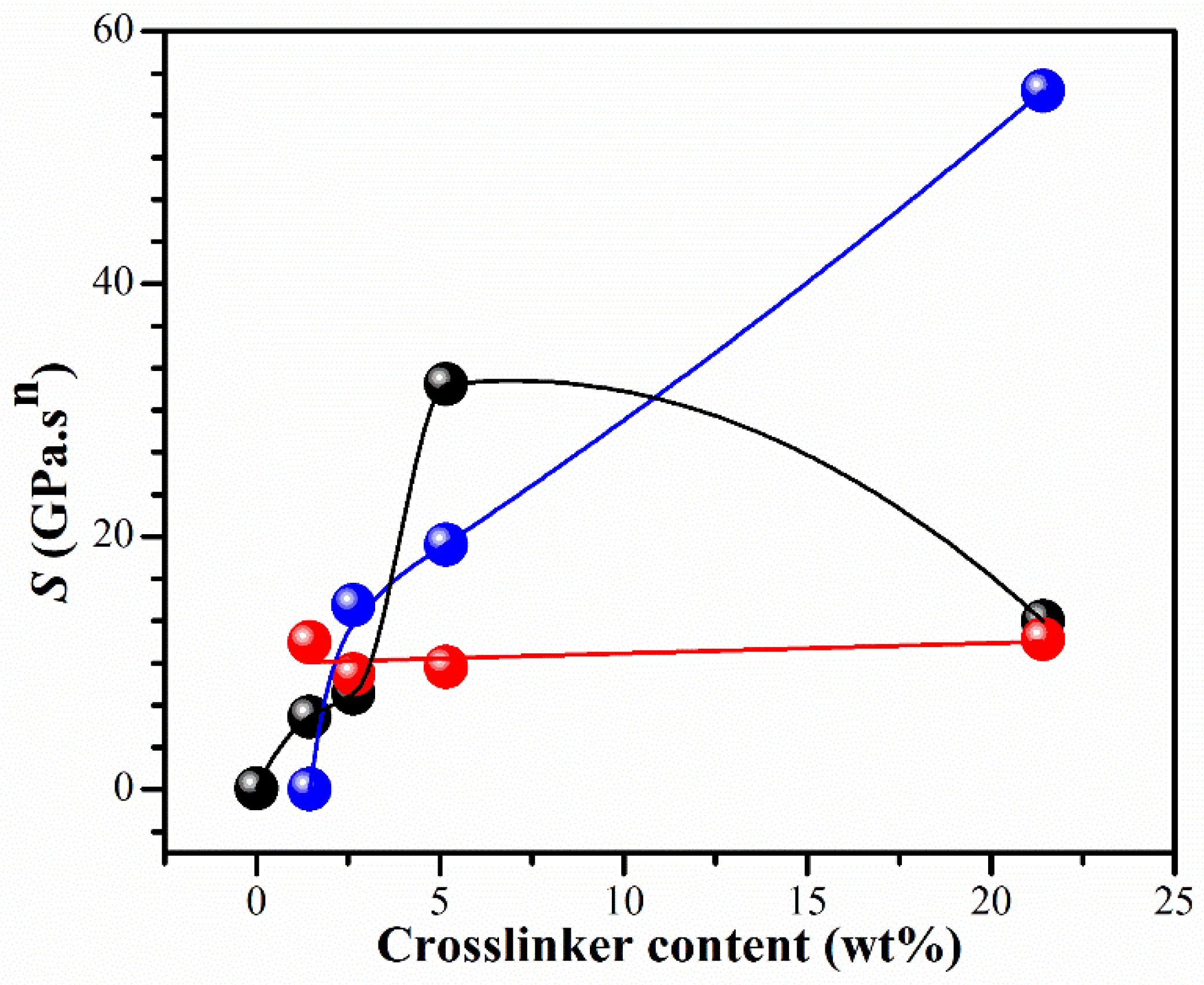
Figure 5.
The frequency dependence on the G′ and G″ of (a) CS (chitosan) and (b) IPNE3 hydrogels: G′ (filled circles) and G″ (hollow circles) at different temperatures.
Figure 5.
The frequency dependence on the G′ and G″ of (a) CS (chitosan) and (b) IPNE3 hydrogels: G′ (filled circles) and G″ (hollow circles) at different temperatures.
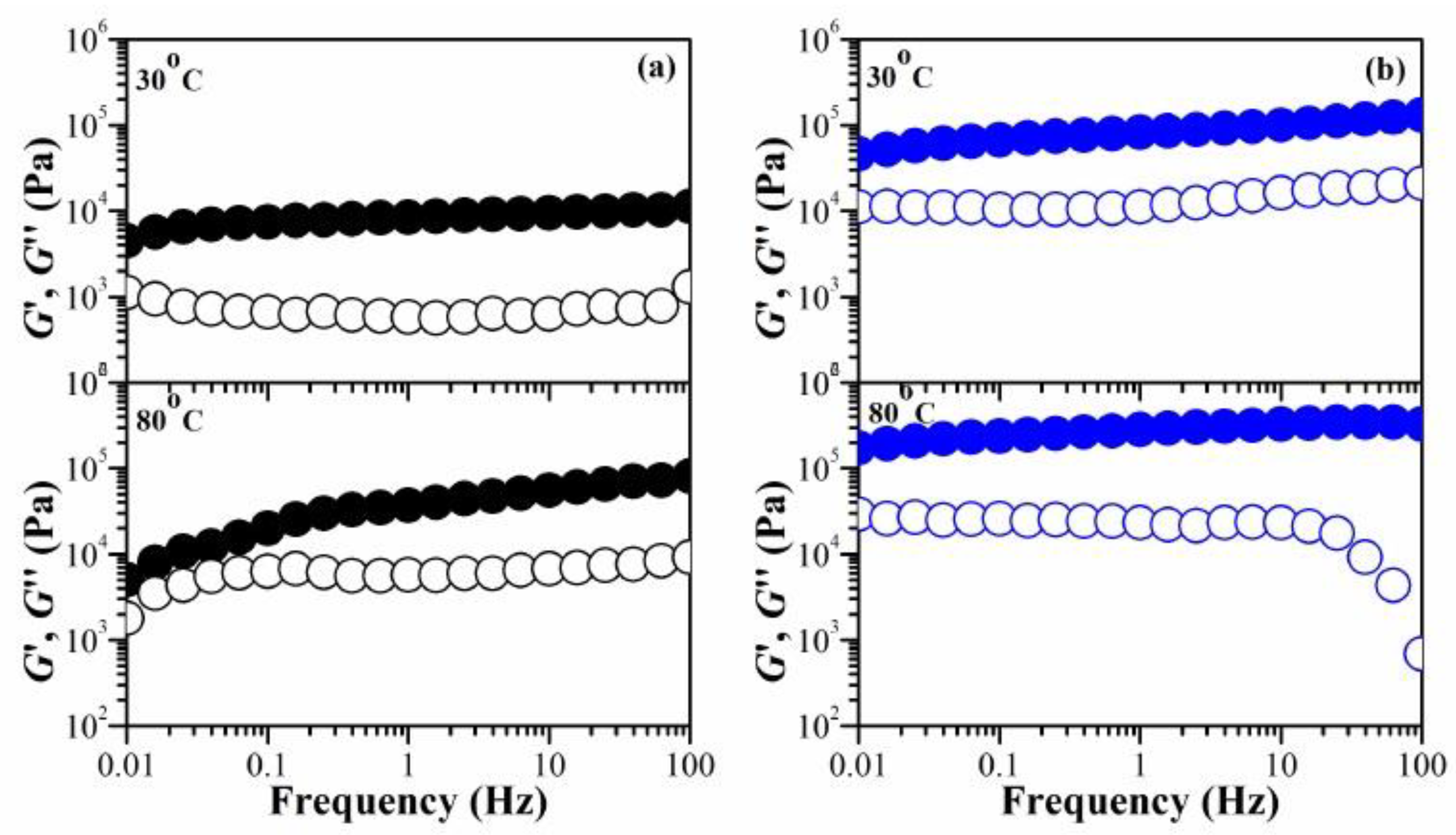
Figure 6.
Representative FT-IR spectra of the (a) NiPAM monomer, (b) PNiPAME hydrogel, (c) CS hydrogel, and (d) IPNE3 hydrogel.
Figure 6.
Representative FT-IR spectra of the (a) NiPAM monomer, (b) PNiPAME hydrogel, (c) CS hydrogel, and (d) IPNE3 hydrogel.
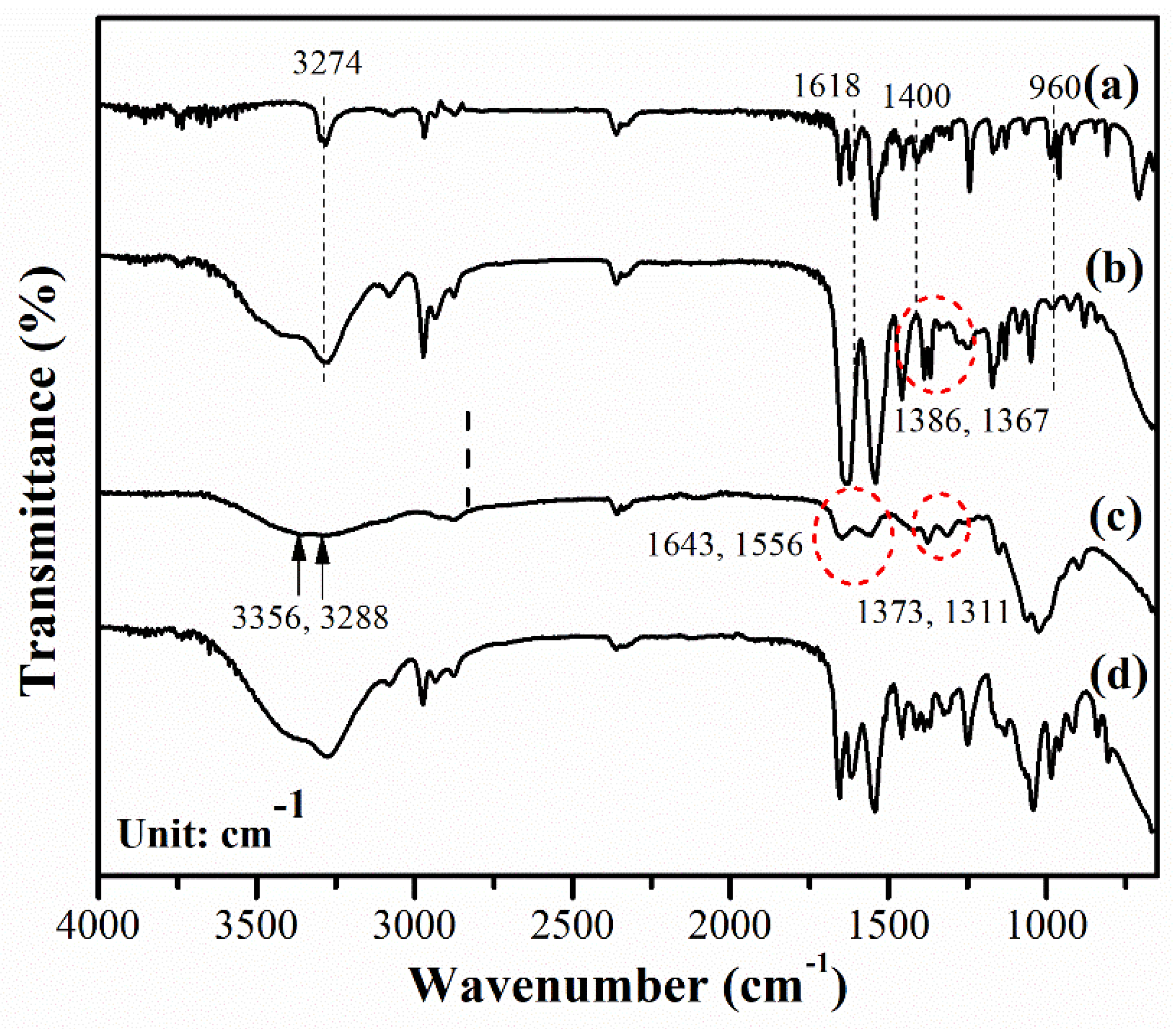
Figure 7.
Equilibrium swelling ratio (SRe) of IPNE system (gray), IPNw system (wine) with various crosslinker contents. Data are shown as the mean, derived from 3 repeats.
Figure 7.
Equilibrium swelling ratio (SRe) of IPNE system (gray), IPNw system (wine) with various crosslinker contents. Data are shown as the mean, derived from 3 repeats.
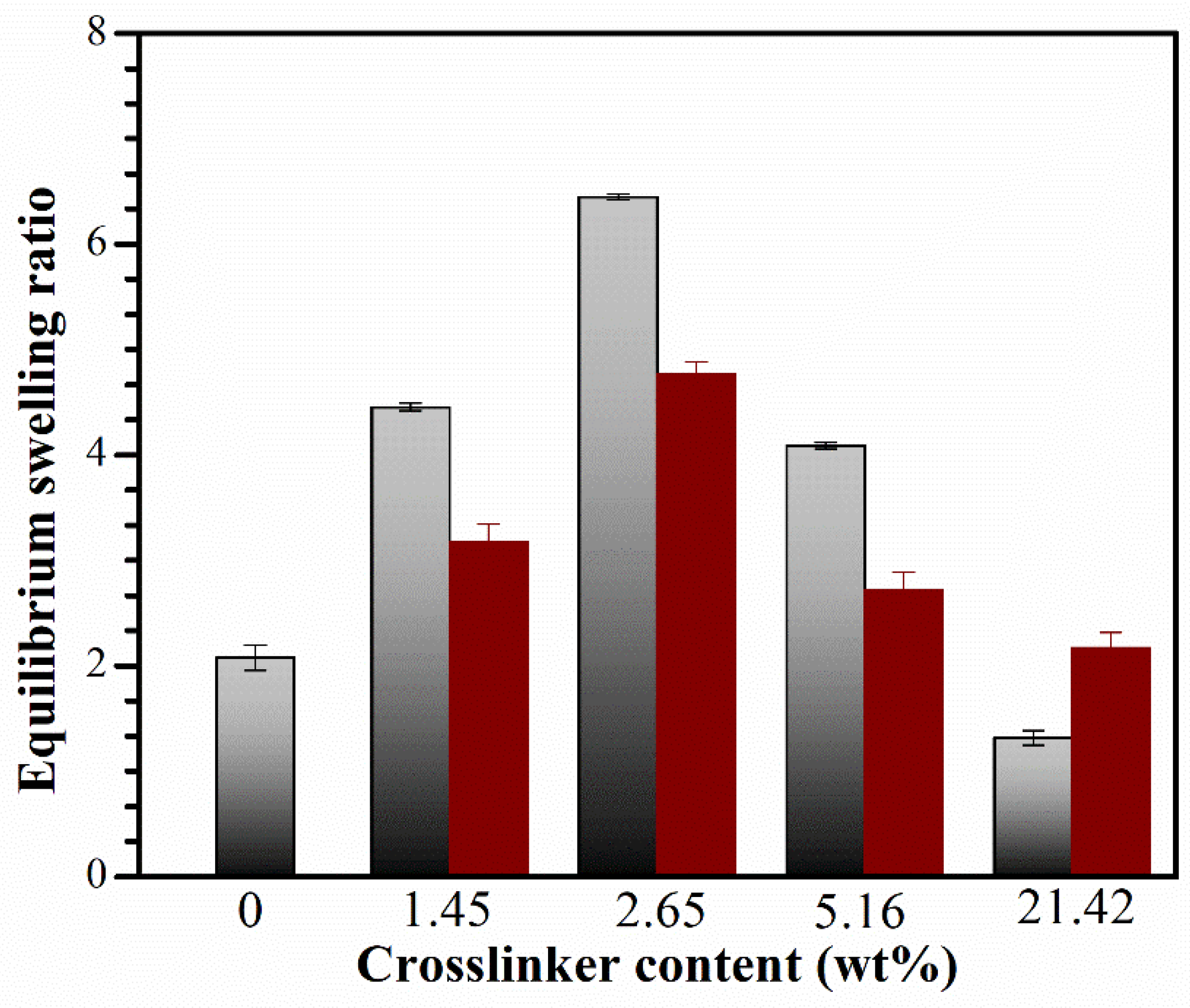
Figure 8.
Representative SEM images showing the (a-d) surface and (a′-d′) cross-sectional morphology of IPN hydrogels with different crosslinking agents as: (a, a′) 0 wt.% CS hydrogel, (b, b′) 1.45 wt.% IPNE1, (c, c′) 5.16 wt.% IPNE2, and (d, d′) 21.42 wt.% IPNE4.
Figure 8.
Representative SEM images showing the (a-d) surface and (a′-d′) cross-sectional morphology of IPN hydrogels with different crosslinking agents as: (a, a′) 0 wt.% CS hydrogel, (b, b′) 1.45 wt.% IPNE1, (c, c′) 5.16 wt.% IPNE2, and (d, d′) 21.42 wt.% IPNE4.

Figure 9.
Representative fluorescence images of cells cultured for (a - c, top) 1 h and (a′- c′, bottom) 24 h on the controlled surface of (a, a′) PS, (b, b′) CS, and (c, c′) IPNE3.
Figure 9.
Representative fluorescence images of cells cultured for (a - c, top) 1 h and (a′- c′, bottom) 24 h on the controlled surface of (a, a′) PS, (b, b′) CS, and (c, c′) IPNE3.


Table 1.
Characteristics of the single gels (chitosan and PNiPAM) and IPN hydrogels.
| Name | NiPAM (M) |
BIS (M, wt%) |
photo-initiator /I2959 (M) |
Prepared solvent | Cell content (103 cell/cm2)a | Contact angle (degree) |
|---|---|---|---|---|---|---|
| Single polymer network | ||||||
|
CS (Chitosan) PNiPAME PNiPAMW |
0 1 1 |
0 0.04(5.16) 0.04(5.16) |
0 0.02 0.02 |
Ethanol Water |
4.50 ± 0.54 N/A N/A |
62.7 ± 5.9 |
| Interpenetrated polymer network of CS/PNiPAM prepared in ethanol | ||||||
| IPNE1 | 1 | 0.01 (1.45) | 0.02 | Ethanol | 3.70 ± 0.29 | 58 ± 11 |
| IPNE2 | 1 | 0.02 (2.65) | 0.02 | 3.30 ± 0.50 | 56 ± 7.3 | |
| IPNE3 | 1 | 0.04 (5.16) | 0.02 | 2.40 ± 0.52 | 61 ± 5.3 | |
| IPNE4 | 1 | 0.20 (21.4) | 0.02 | 2.40 ± 0.43 | 61 ± 8.2 | |
| Interpenetrated polymer network of CS/PNiPAM prepared in water | ||||||
| IPNw1 | 1 | 0.01 (1.45) | 0.02 | Water (DI) |
N/A | |
| IPNw2 | 1 | 0.02 (2.65) | 0.02 | |||
| IPNw3 | 1 | 0.04 (5.16) | 0.02 | |||
| IPNw4 | 1 | 0.20 (21.4) | 0.02 | |||
*The subscribed E and W represent the ethanol and deionized water, respectively, as solvents. aData are shown as the mean, derived from 3 repeats.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



