
Submitted:
24 November 2023
Posted:
24 November 2023
You are already at the latest version
Abstract
As humans age, aberrancies in several signaling pathways occur. In the aging population, some patients display abnormal bone morphogenetic protein (BMP) signaling. This can lead to osteoporosis (OP), a debilitating bone disorder caused by an imbalance between osteoblasts and osteoclasts. In 2002, the Food and Drug Administration (FDA) approved usage of recombinant human BMP-2 (rhBMP-2) during spinal fusion surgery as it is crucial for bone formation. However, complications have been reported with rhBMP-2 and primary osteoblasts isolated from OP patients are unresponsive to BMP-2. While patient samples are available, previous medical history may alter results. Therefore, C57BL/6 mice, an aging model that displays aberrant BMP-signaling, can be utilized to investigate OP and aging. We show that BMP receptor type Ia (BMPRIa) is upregulated in the bone marrow stromal cells (BMSCs) of 15-month mice similar to previous data. We show that a conjugation between BMP-2 and Quantum Dots (QDot®s) effectively binds to BMPRIa and can investigate BMP-2 activity. After incubating BMSCs with methyl-β-cyclodextrin (MβCD), BMP-signaling is restored in 15-month mice as shown by western blotting and the von Kossa assay. MβCD may be used to rescue BMPRIa and the BMP-signaling pathway can be utilized by future therapeutics to treat OP.
Keywords:
osteoporosis
; osteoblasts
; BMP-2
; BMPRIa
; C57BL/6 mice
; MβCD
; QDot®s
1. Introduction
During the natural aging process of humans, the body undergoes senescence and alterations in signaling pathways. As such, the development of several diseases, including metabolic disorders, neurocognitive disorders, and bone disorders such as osteoporosis (OP) can occur [1,2,3]. While the process of aging has been investigated, the precise molecular and cellular aberrancies that are presented during this process are unclear. As the bone morphogenetic protein (BMP) signaling pathway is altered in patients diagnosed with OP, it can be explored to determine the role of BMP-2 in aging and if these effects can be reversed in bone.
OP is a devastating bone disorder that affects more than 10 million Americans, and 80% of those diagnosed are women [4,5,6]. OP is a drastic decrease in bone mineral density (BMD) that is likely caused by an imbalance between osteoblasts (bone forming cells) and osteoclasts (bone resorbing cells) [7,8]. Further, OP is extremely costly, decreases the quality of life of patients, is associated with increased mortality, and is currently incurable [4,9,10].
Current treatment options are commonly anabolic, which restores bone loss, or anti-resorptive, which reduces bone degradation [11,12,13,14,15,16,17,18]. However, each therapeutic has been associated with adverse side-effects which include osteolysis, hematoma formation, necrosis, and others [5,19,20,21,22,23,24]. In addition, there is currently only one drug approved by the Food and Drug Administration (FDA) that targets osteoblasts and osteoclasts simultaneously, and this is known as Romosozumab [25,26,27,28,29,30]. This therapeutic is a sclerostin antibody and induces osteogenesis while limiting bone degradation. Its entire mechanism is currently being deciphered, especially as its long-term effects are unclear. Further, side-effects of Romosozumab have been reported and include hepatitis and osteonecrosis [25,26,27,28,29,30]. Moreover, a treatment option that safely treats OP without adverse side-effects and targets osteoblast and osteoclast activity is desperately needed.
A pathway that may be targeted for the treatment of OP is bone morphogenetic protein (BMP) signaling. BMPs are members of the transforming growth factor-β (TGF-β) superfamily and regulate many cellular and molecular processes [31,32,33,34]. Specifically, BMP-2 was identified in 1965 as a crucial growth factor that regulates osteogenesis during development and in adulthood [35,36]. BMP-2 also regulates many other pathways such as chondrogenesis, adipogenesis, cardiogenesis, somite formation, and limb development [37,38,39,40,41,42,43,44,45,46]. To activate these pathways, BMP-2 binds with high affinity to BMP Receptor Type Ia (BMPRIa) localized in caveolae enriched with caveolin-1 alpha (Cav1 α) isoforms or within clathrin coated pits (CCPs) [47,48,49,50,51,52]. After, BMP Receptor Type II (BMPRII) is recruited to the membrane or is already localized to the membrane domains with BMPRIa as pre-formed complexes containing the Cav1 αβ isoform [47,48,51,52]. BMPRII phosphorylates BMPRIa at its glycine-serine (GS) box and causes the release of protein kinase CK2 (CK2) [53,54,55,56]. The release of CK2 from BMPRIa allows the receptor to phosphorylate downstream proteins including SMAD1, 5, and/or 8 in the canonical signaling pathway [44,49,50,52,53,54,57,58,59,60]. The p-SMADs recruit SMAD4 and the entire complex translocates to the nucleus to serve as transcription factors for osteogenesis. In the non-canonical pathway, BMPRIa activates the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) signaling pathways to increase cellular survival and proliferation. [60,61,62,63,64,65]
As BMP-2 is essential for osteogenesis, the FDA approved it as a bone regeneration therapeutic during anterior lumbar interbody fusion (ALIF) surgery and facial reconstruction in 2002 [66]. Shortly after its appearance in the clinic, several side-effects were reported and the osteoblasts isolated from patients diagnosed with OP do not respond positively to this treatment, even though these cells overexpress BMPRIa [67,68,69,70,71]. As such, the usefulness of BMP-2 is contradictory and must be used with extreme caution. However, the BMP-signaling may still be utilized in future medical intervention without exogenous BMP-2.
As receptor localization determines BMP-signaling, it is plausible that BMPRIa and/or BMPRII may be mis-localized in patients diagnosed with OP. Recent data demonstrate that after primary osteoblasts isolated from OP patients are stimulated with BMP-2 treatment, there is not an increase in canonical or non-canonical signaling and mineralization is not induced [58,67]. This lack of signaling may be caused by improper localization or shuttling of BMPRs, which prevents BMP-2 activity. However, the precise role and localization of BMP-2 in these cells is currently unknown. A current pharmacological agent that can disrupt receptor localization and prevent endocytosis for short time-frames is methyl-β-cyclodextrin (MβCD) [72,73]. It is possible that after treating cells with MβCD and then BMP-2, BMPRIa and/or BMPRII can be re-localized to their proper location to restore signaling. To uncover the role of BMP-2 in these cells, this protein can be conjugated to Quantum Dot®s (QDot®s) to determine its mode of action. QDot®s are photostable molecules that photo-bleach less than other traditionally used dyes [74,75,76,77,78]. Further, QDot®s can be carboxylated to allow for bond formation with peptides or proteins [79,80]. In addition, while primary osteoblasts isolated from OP patients is an effective method to study the disease, several complications arise including previous medical history, genetic differences, and comorbidities. Thus, a model that eliminates these factors must be explored. One model that can be utilized to overcome these obstacles is the C57BL/6 (B6) mouse. These mice display low peak BMD and recent data demonstrates that they are a comparable model to study human OP [81,82,83]. Further, the localization of BMP-2 in the bone marrow stromal cells (BMSCs) within these mice is unknown and requires further attention.
Here, we first confirm that the 15-month female B6 mice overexpress BMPRIa compared to the 6-month mice via Western Blotting. These data are representative of previously collected results. Next, we demonstrate that BMP-2 conjugated to QDot®s colocalizes with BMPRIa in the BMSCs of 6-month mice and binds with a lesser extent to BMPRIa in 15-month mice. However, after treating the BMSCs isolated from 15-month mice with MβCD, the binding of BMP-2-QDot®s with BMPRIa was increased. After, Western Blotting was conducted and we determined that in the 15-month BMSCs, pSmad1 concentration was lower after BMP-2 stimulated compared to 6-month mice. However, after MβCD treatment, pSmad1 was activated in 15-month BMSCs, indicating the signaling pathway was restored. Finally, we observed that BMSCs from 6-month mice produced an increased mineralization area after BMP-2 stimulation compared to the control groups, but this effect was not observed in 15-month mice. However, upon treatment with MβCD and then BMP-2, the BMSCs isolated from 15-month mice displayed an increase in mineralization. Altogether, these data demonstrate that MβCD is able to rescue BMP-signaling, suggesting that the receptors are mis-localized on the plasma membrane in aged-mice that are reflective of OP patients. These results can inform future therapeutics that are desperately needed to treat OP.
2. Materials and Methods
2.1. Mice Acquisition and Ethical Approval
C57BL/6 (B6) female mice aged 6- (N = 20) and 15-months (N = 20) were obtained from Charles River Laboratories (Horsham, PA, USA). The mice were housed at the University of Delaware in the Life Sciences Research Facility (University of Delaware, Newark, DE, USA) and acclimated to the new environment for one week. Each mouse had access to food and water, and they were housed 4-5 mice of the same age per cage. All research conducted in this study was approved by the Institutional Animal Care and Use Committee (University of Delaware, Newark, DE, USA): AUP #1194.
2.2. Organ and Cell Isolation
The B6 mice were euthanized with CO2 and their femurs were extracted. Each femur was sliced at distal and proximal femoral heads and then flushed three times with alpha minimum essential media (αMEM; Caisson Labs, Smithfield, UT, USA) to isolate bone marrow stromal cells (BMSCs). After, the flushed solutions were filtered with 70 μM cell strainers (Stellar Scientific, Baltimore, MD, USA) into 50 mL conical tubes (Cole-Palmer, Vernon Hills, IL, USA) and centrifuged at 1,500 rotations per minute (RPM) for 5 min at 4 °C. The supernatant was aspirated and the cell pellets were resuspended with αMEM supplemented with 10% fetal bovine serum (FBS; Gemini Bioproducts, West Sacramento, CA), 1% penicillin/streptomycin (pen/strep; Fisher Scientific, Pittsburg, PA, USA), and 1% antibiotic/antimycotic (anti/anti; Gemini Bioproducts, West Sacramento, CA, USA). The cell densities were counted and cells were plated as noted by each method.
2.3. Immunofluorescent Staining
2.3.1. Staining of BMPRIa of Unstimulated (US) B6 Mice
The BMSCs isolated from the femurs of 6- and 15-month were plated at 1x106 cells/mL in 12-well plates (Nest Scientific, Woodbridge Township, NJ, USA) on 18 mm diameter rounded coverslips (Catalog #CS-R18-100, Amscope, Irvine, CA, USA). The cells were grown in αMEM supplemented with 10% FBS, 1% pen/strep, and 1% anti/anti for 7-10 days. Every third day, fresh media was replenished. On the final day of cell culture, the media was aspirated and the cells were washed with ice-cold 1X phosphate buffered saline (PBS) and fixed with 4.4% paraformaldehyde (PFA, pH 7.2; Sigma Aldrich, St. Louis, MO, USA for 15 min at room temperature. After, the cells were washed three times with ice-cold 1X PBS. The cells were permeabilized with 0.1% Saponin (Sigma-Aldrich, St. Louis, MO, USA) dissolved in 1X PBS for 10 min and then blocked for 1 hr on ice with 3% bovine serum albumin (BSA, Fisher Scientific, Pittsburgh, PA, USA) and 0.1% Saponin diluted in 1X PBS. The cells, except for those in the secondary control condition, were incubated with 1:100 dilutions of a primary rabbit polyclonal BMPRIa antibody (Catalog #100743-T08; Sino Biological, Beijing, China) in 1X PBS, 3% BSA, and 0.1% Saponin for 1 hr on ice. The cells were washed three times with ice-cold 1X PBS and all experimental groups were incubated with 1:500 solutions of a secondary chicken-anti-rabbit antibody (Alexa FluorTM594, Catalog #A21442; Invitrogen, Eugene, OR, USA) in 1X PBS, 3% BSA, and 0.1% Saponin for 1 h on ice away from light. The cells were washed three times with ice-cold 1X PBS and the nuclei were stained with Hoechst 33,342 (Catalog #AR0039, Bolster Bio, Pleasanton, CA, USA) for 7.5 min. The cells were washed once with 1X PBS and the coverslips were mounted onto glass slides with Airvol. After drying, the slides were imaged using the Zeiss LSM880 confocal microscope with Airyscan (Wolf Hall, University of Delaware, Newark, DE, USA) with the 63x objective lens. At least 10 images were obtained and analyzed in ImageJ. All data were normalized to the secondary control.
2.3.2. Staining of Cells Treated with Methyl-β-Cyclodextrin and BMP-2-QDot®s
Similar to above, the BMSCs were isolated from the femurs of B6 mice. The cells were plated and on day 6, they were treated with 100 µM of methyl-β-cyclodextrin (MβCD; Catalog# M1356, TCI America, Montgomeryville, PA, USA) or left US for 24 hr. The cells were then treated with 40 nM of a BMP-2-QDot®s conjugation for 6 hr or left US as previously described [79]. The cells were subjected to immunostaining as described in Section 2.3.1. The cells were imaged using confocal microscopy using the 63X objective lens and an additional magnification of 10 to focus on BMPRIa.
2.4. Lysate Collection
As described before, BMSCs were obtained from the femurs of 6- and 15-month B6 mice. The cells were seeded into 6-well plates at a density of 1x107 cells/mL and grown in αMEM. On day 6, the cells were treated with 100 µM of MβCD for 24 hr or left US. After, the cells were treated with 40 nM BMP-2 or left US for 6 hr. The cells were washed three times with ice-cold 1X PBS and scraped into 300 µL of Radioimmunoprecipitation Assay (RIPA) lysis buffer that contained .44 g NaCl, 1 mL TritonX-100 (Acros Organics, Geel, Belgium), 0.5 g Sodium Deoxycholate (Thermo Scientific, Rockford, IL, USA), 12.5 mL 0.5 M Tris Buffer pH 6.8, 0.05 g Sodium Dodecyl Sulfate (SDS, Fisher Scientific, Geel, Belgium), and 36.5 mL sterile H2O. Further, 1 mM (PMSF, MP Biomedicals, Solon, Ohio, USA) and 1 mM protease inhibitor (PI) cocktail (Thermo Scientific, Rockford, IL, USA) were added the RIPA buffer. After incubation in RIPA for 10-minutes on ice, the lysates were transferred to 1.5 mL microcentrifuge tubes and sonicated (Cleanosonic, Branson Digital Sonifier, Richmond, VA, USA) at 21% amperes for 20-seconds, three times. The lysates were centrifuged at 12,700 RPM for 10-minutes at 4 °C and the supernatants were collected. Protein estimation was conducted with the PierceTM BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA) and analyzed with the NanoDropTM One/OneC Microvolume Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, US). All lysates were normalized to the lowest protein concentration before loading into the gels.
2.5. Western Blotting
2.5.1. Western Blotting of BMPRIa of Unstimulated (US) B6 Mice
The lysates were boiled for 10-minutes at 95 °C and 20 µL of each were loaded into a 10% SDS-polyacrylamide (SDS-PAGE) gel. Gel electrophoresis was carried out at 120 V for 1 hr and 30 min and the proteins were transferred to a PVDF (Immobilon, Darmstadt, Germany) membrane for 1 hr and 10 min at 15 V using the semi-dry transfer machine (BioRad, Hercules, CA, USA). The membrane was blocked with 5% BSA diluted in 1X Tris-Buffered Saline-Tween (TBST) for 1-hour on ice. The blot was incubated with a 1:1000 dilution of BMPRIa (same as above) and 1:1000 dilution of glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Lot# 14C10, Rabbit monoclonal, Cell Signaling Technology, Beverly, MA, USA diluted in 1X TBST supplemented with 1% BSA overnight at 4 °C. On the following day, the blot was washed three times with 1X TBST and incubated with a 1:5000 dilution of goat-anti-rabbit IgG-HRP (ab6721, Abcam, Cambridge, UK) secondary antibody diluted in 1X TBST with 1% BSA for 1 hr. The blots were washed three more times with 1X TBST and treated with SuperSignalTM West Pico Plus Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA) for five-minutes. All protein bands were detected with the Invitrogen iBright1500 machine (Thermo Fisher Scientific, Waltham, MA, USA).
2.5.2. Western Blotting of Cells Treated with Methyl-β-Cyclodextrin and BMP-2-QDot®s
Similar to above, the lysates were boiled and loaded onto gels. After gel electrophoresis and transferring, the blots were incubated with the following primary antibodies: BMPRIa (same as above), pSMAD 1 (STJ90743, Rabbit polyclonal IgG, St John’s Laboratory Ltd, London, UK), SMAD 1/5/8 (Lot# D2110, Rabbit polyclonal, Santa Cruz Biotechnology, Dallas, TX, USA), and GAPDH (Lot# 14C10, Rabbit monoclonal, Cell Signaling Technology, Beverly, MA, USA). After washing the following day, the blots were incubated with the following secondary antibodies: goat-anti-rabbit IgG-HRP (same as above) or rabbit-anti-mouse IgG-HRP (ab6728, Abcam, Cambridge, UK). The bands were detected as described above.
2.6. Von Kossa Assay
BMSCs were isolated from the femurs of 6- and 15-month B6 female mice. The cells were plated in 24-well plates at a density of 1x105 cells/mL in αMEM for six days. On day 6, the cells were treated with 100 µM MβCD for 24 hr or left US. On the following day, the cells were treated with 40 nM BMP-2 or left US for 5 days. The media was changed and stimulations were replaced on day 3. On day 5, the media was aspirated, the plates were washed with 1X PBS, and the cells were fixed with 4.4% PFA at room temperature for 15 min. The wells were washed three times with 1X PBS and incubated with 5% silver nitrate (Science Company, Lakewood, CO, USA) dissolved in DiH2O for 1 hr under UV light. The silver nitrate was removed and the cells were washed with DiH2O until excess solute was removed. After drying for two days, random images were acquired and data were analyzed in ImageJ. All data were normalized to the US groups for both ages.
2.7. Statistical Analysis
The Chauvenet’s criterion test was employed to remove any outliers from all experimentation. The data were statistically analyzed using the single factor analysis of variance (ANOVA) and the Tukey-Kramer HSD statistical tests. The error bars above the bars in the graph are representative of standard error of the mean (SEM). Statistical significance was set to p < 0.05 and is displayed as lettering; here the letters “a, b, c” etc. represent group one as the letter “a,” group two corresponds to the letter “b,” and so on. For example, if there is a letter “a” above a bar, that means this group is statistically different from group one.
3. Results
3.1. 15-Month B6 Mice Overexpress BMPRIa compared to 6-Month Mice via Western Blotting
It was recently demonstrated that primary osteoblasts isolated from OP patients overexpress BMPRIa compared to control patients via immunofluorescent staining and western blotting [67]. Additionally, this overexpression of BMPRIa is also observed in the BMSCs isolated from 15-month mice compared to 6-month mice as illustrated by confocal microscopy [81]. However, the total protein concentration of BMPRIa from whole cell lysates has never been determined and should be explored to confirm this overexpression. Here, we isolated lysates from US BMSCs of 6- and 15-month mice and detected for BMPRIa. As noted in Figure 1, the 15-month mice overexpress BMPRIa compared to 6-month mice, confirming the results obtained in previous studies. Next, we determined if BMP-2 -QDot®s were able to bind to the overexpressed receptor, or if treatment with MβCD was necessary to re-establish proper signaling.
3.2. MβCD Increases the Binding of BMP-2-QDot®s to BMPRIa in BMSCs Isolated from 15-Month B6 Mice
To further understand the biological function of BMP-2 in BMSCs of 15-month female B6 mice, we conjugated this protein to Quantum Dots (QDot®s). As stated previously, the QDot®s are carboxylated and in the presence of DCC, and then this fluorescent probe can form bonds with proteins or peptides, such as BMP-2. This powerful technique will provide critical fluorescent details regarding the binding activity of BMP-2 within the BMSCs. As noted, in 15- and 20-month mice, BMP-2 is not functioning properly in the BMSCs. However, whether BMP-2 can bind to BMPRs or endocytosed into the cells is unclear and must be elucidated. Here, we isolated BMSCs from both 6-momth and 15-month B6 mice. These cells were treated with 100 µM MβCD for 24-hours, an agent that depletes membranous cholesterol to prevent endocytosis, followed by stimulation with BMP-2-QDot®s for 6-hours or left US. The cells were then immunostained for the nuclei, BMPRIa, and BMP-2-QDot®s, which will fluoresce green when present. As demonstrated in Figure 2 and Figure 3, all BMSCs of 6-month mice express BMPRIa, whereas only the BMP-2-QDot®s treated cells display green. Further, the green is confined to BMPRIa, suggesting that the conjugation specifically targets and binds only to this receptor. Next, it is visually notably that within all treatment groups, there is an overexpression of BMPRIa in 15-month cells compared to the 6-month cells, except for in the BMP-2-QDot®s + MβCD treated group. Further, the BMSCs of 15-month mice did not display effective BMP-2-QDot®s binding; however, when these cells were treated with MβCD, the binding of BMP-2-QDot®s to BMPRIa significantly increased.
Next, we semi-quantified these data by obtaining 10 random images and measuring the fluorescence of red and green within all groups (Figure 4). As shown, 15-month US cells and BMP-2-QDot®s only treated cells overexpressed BMPRIa when compared to the 6-month US BMSCs. Interestingly, treatment with MβCD and BMP-2-QDot®s in 15-momth cells slightly decreased the expression of BMPRIa compared to the other conditions in this age group. In addition, as shown in Figure 4B, treatment with MβCD increased the fluorescence of BMP-2-QDot®s when compared to the other 15-month conditions. Thus, these data suggest that BMPRIa is mis-localized and may be rescued by a cellular uptake inhibitor, such as MβCD.
3.3. MβCD Treated Cells Isolated from 15-Month Mice Display an Upregulation in pSMAD1
Stated above, MβCD improved the binding of BMP-2 to its receptor, BMPRIa, in the BMSCs isolated from 15-month female B6 mice. This increase in binding is likely to upregulate the BMP-signaling pathways and downstream proteins. Directly after BMP-2 binds to BMPRs, SMAD 1, 5, and/or 8 are phosphorylated by BMPRIa. Therefore, if BMP-2 binds to BMPRs after MβCD treatment, it is expected that there will be an increase in SMAD phosphorylation. To assess pathway activation, lysates were collected from BMSCs isolated from 6- and 15-month mice treated with MβCD and 40 nM BMP-2 or left US. The protein concentration was normalized and the samples were subjected to SDS-PAGE electrophoresis followed by transfer to a PVDF membrane. As demonstrated in Figure 5, the BMP-2 and BMP-2 + MβCD upregulated pSMAD1 compared to the US group in 6-month mice. However, as depicted in Figure 3, BMP-2 did not increase pSMAD1, but an upregulation was demonstrated in the BMP-2 + MβCD in 15-month mice. In addition, the total SMAD1/5/8 expression was similar across all groups, suggesting that the total SMAD level remains constant while pSMAD1 is upregulated. As suggested by these data, BMSCs isolated from 15-month mice remain unresponsive to BMP-2 unless they are treated with MβCD. Further, it is possible that MβCD disrupts BMPRIa mis-localization and allows these receptors to function properly after treatment.
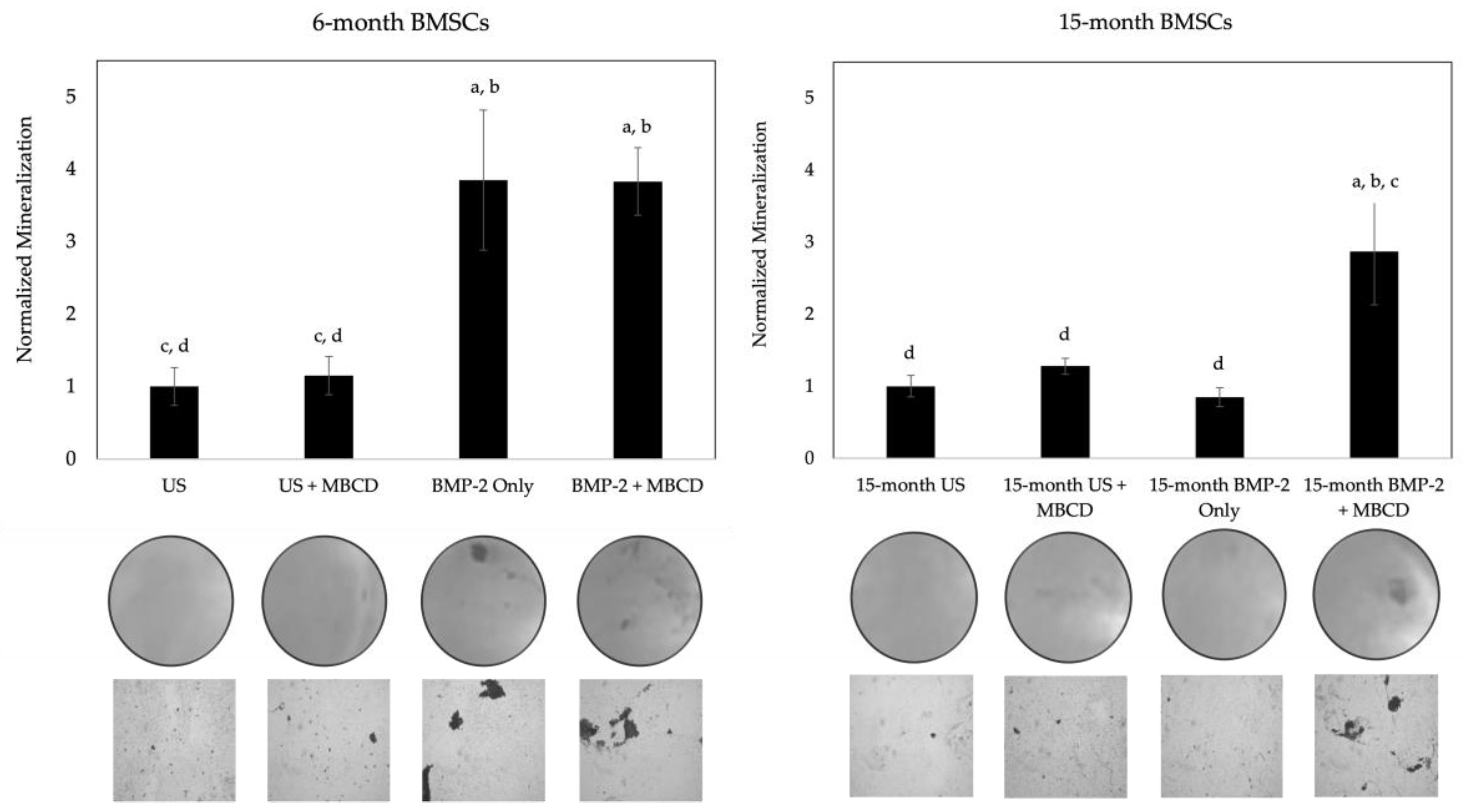
3.4. MβCD Treated Cells Displayed an Increase in Mineralization After BMP-2 Stimulation Compared to Untreated Cells
As noted in the previous section, MβCD was able to disrupt the endocytosis of membrane domains, allowing for the possible rescue of BMPRIa mis-localization. Specifically, we unraveled that BMP-2 conjugated to QDot®s is successfully able to bind to BMPRIa in the BMSCs of both 6- and 15-month mice, and this colocalization increased in 15-month mice when the cells were treated with MβCD. While this colocalization was observed, it remains unclear whether this increased binding will also lead to the activation of BMP-signaling. For this next experimentation, the BMSCs isolated from 6- and 15-month female B6 mice were obtained and seeded into 24-well plates. The cells were treated with MβCD or left untreated for 24-hours, and then stimulated with BMP-2 or left US for 5-days. After, the cells were fixed and subjected to the von Kossa assay as described in the Methods section. As shown in Figure 6, the BMSCs of 6-month mice displayed a significant increase in mineralization after BMP-2 treatment in both MβCD and no MβCD conditions compared to the US group. However, the 15-month mice only responded positively to BMP-2 when the cells were also treated with MβCD. Representative images are displayed under each bar. These data further affirm that BMPRIa may be mis-localized, and MβCD rescues these receptors to allow them to bind to BMP-2 and activate downstream signaling pathways.
4. Discussion
Natural aging and the effects of senescence are becoming increasingly prevalent as the world’s aging population is growing. This process poses many disorders and causes aberrancies in several signaling pathways. These disrupted signaling pathways can cause metabolic, neurocognitive, and bone or cartilage disorders, such as OP. OP is a debilitating bone disorder that affects one in four women and one in five men over the age of 50 [58]. This disease is characterized by a lower-than-normal BMD that likely arises from an imbalance between osteoblasts and osteoclasts. The activation of osteogenic signaling pathways in osteoblasts is mediated by BMP-2, which was approved by the FDA in 2002 to be used as a therapeutic [84]. However, several side-effects were reported after usage of BMP-2, such as hematoma formation, radiculitis, and increased osteolysis, as BMP-2 also enhances osteoclastogenesis [67,68,85,86]. Further, it was demonstrated that primary osteoblasts isolated from OP patients and BMSCs obtained from aged (15- and 20-month) B6 mice are unresponsive to BMP-2 stimulation [58,67]. Interestingly, there was an increase in BMPRIa expression, even though BMP-2 binding is aberrant. This unresponsiveness and upregulation of BMPRIa could contribute partly to OP and may arise from mis-localization or improper shuttling of BMPRs on the surface of cells. Thus, we explored whether these receptors could be shuttled from incorrect domains and into proper regions of the cells.
To ensure B6 mice are a reliable model to investigate aberrant BMP-signaling, the expression of BMPRIa must be further established. We first confirmed that the BMSCs of US 15-month B6 mice overexpress BMPRIa compared to US 6-month mice via western blotting and confocal microscopy, confirming previous data (Figure 1) [67,81]. This overexpression may be caused by aberrant BMP-signaling, and the cells are compensating for the lack of signaling and thereby upregulating BMPRIa expression. Further, while the receptor may be upregulated, it is plausible that the receptors themselves are mis-localized and restrict pathway activation by BMP-2. As such, the binding dynamics of BMP-2 to BMPRIa and the activation of the SMAD signaling pathway were explored.
Next, we determined the binding affinity of BMP-2 to BMPRIa with QDot®s. BMP-2 was conjugated to QDot®s and added to BMSCs that were treated or untreated with MβCD. MβCD is a pharmacological agent capable of preventing endocytosis and disrupting membrane domains, which may allow BMPRIa and/or BMPRII to shuttle out and resume proper signaling. As depicted in Figure 2 and Figure 3, BMP-2-QDot®s effectively bound to BMPRIa in both the MβCD treated and untreated groups compared to the US and secondary control groups. However, as shown, while BMP-2-QDot®s co-localized with BMPRIa, this effect was drastically increased after MβCD treatment in both age groups. The fluorescence was analyzed and quantified, including statistical significance between the groups (Figure 4A,B). Further, it is possible that because BMPRIa is overexpressed in 15-month BMSCs, there is an increase of substrates that BMP-2-QDot®s can bind. Therefore, the activation of SMAD signaling pathways must be elucidated to determine BMP-2 activity.
As BMP-2-QDot®s are able to bind to BMPRIa in both 6- and 15-month B6 mice, we investigated the activation of canonical SMAD signaling by BMP-2 with or without MβCD. After BMP-2 binds to BMPRIa, CK2 is released and BMPRIa is phosphorylated by BMPRII. This allows BMPRIa to freely phosphorylate downstream signaling proteins, including SMAD1, 5, and/or 8. The pSMADs will recruit SMAD4 and the complex will translocate to the nucleus and serve as an osteogenic transcription factor. Thus, after incubating BMSCs with BMP-2 and MβCD, we detected for pSMAD1, total SMAD1/5/8, and GAPDH. As illustrated above, BMP-2 effectively activated pSMAD1 signaling in 6-month mice with or without MβCD (Figure 5). However, in 15-months, pSMAD1 was only upregulated when the BMSCs were incubate with both BMP-2 and MβCD, but not BMP-2 alone. Further, the expression of SMAD1/5/8 was consistent across all samples. These data illustrate aberrant BMP-signaling in 15-month mice that may be rescued by MβCD treatment or other therapeutics that disrupt membrane domains and transiently allow for re-shuttling.
Finally, we investigated the signaling output of BMP-2 with or without MβCD via the von Kossa assay for mineralization. The von Kossa assay visualizes mineralization deposition by first binding silver nitrate to phosphate or calcium, and then exposing the cells to UV light. As such, the mineralization deposits are representative of osteogenesis, as ALP cleaves pyrophosphate molecules to yield phosphate [87]. Similar to the western blotting data, BMP-2 was able to induce mineralization in both BMP-2 alone and BMP-2 + MβCD treated cells (Figure 6). However, mineralization only occurred in 15-month BMSCs when the cells were exposed to both BMP-2 and MβCD (Figure 6). This indicates that while BMP-2 may bind to BMPRIa in 15-month BMSCs (Figure 3), it is unable to activate SMAD signaling to induce mineralization, unless the cells are simultaneously treated with MβCD (Figure 4 and Figure 5).
Taken together, these data demonstrate that BMPRIa is mis-localized on the plasma membrane of BMSCs isolated from 15-month B6 mice and contributes to aberrant BMP-signaling as a result of aging. Further, the receptors may be shuttled out of the improper domains after treatment with MβCD treatment. These data confirm aberrant BMP-signaling in aged-B6 mice that are reflective of OP patients and propose a potential therapeutic to restore BMP-signaling. In addition, while BMP-2 itself is ineffective, the BMP-signaling pathway may still be implicated in future therapeutics to treat OP.
Author Contributions
Conceptualization, D.H. and A.N.; methodology, D.H.; software, D.H. and A.N.; validation, D.H., V.P., K.C., and A.N.; formal analysis, D.H. and A.N.; investigation, A.N.; resources, A.N.; data curation, D.H., V.P., K.C., and A.N.; writing—original draft preparation, D.H.; writing—review and editing, D.H., V.P., K.C., and A.N.; visualization, D.H. and A.N.; supervision, A.N.; project administration, A.N.; funding acquisition, A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, grant number 1R21AR076689-01A1; the Nathan Shock Center Pilot Award; and the Institutional Development Award (IDeA) from the National Institute of Health’s General Medical Sciences under grant number P20GM103446.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of University of Delaware (AUP #1194; date of approval: 3 October 2020).
Informed Consent Statement
Not applicable.
Data Availability Statement
Any data or material that support the findings of this study can be made available by the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moerman, E.J.; Teng, K.; Lipschitz, D.A.; Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 2004, 3, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Bae, Y.S. CK2 Down-Regulation Increases the Expression of Senescence-Associated Secretory Phenotype Factors through NF-κB Activation. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Khosla, S. Skeletal changes through the lifespan--from growth to senescence. Nat Rev Endocrinol 2015, 11, 513–521. [Google Scholar] [CrossRef]
- Wright, N.C.; Looker, A.C.; Saag, K.G.; Curtis, J.R.; Delzell, E.S.; Randall, S.; Dawson-Hughes, B. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res 2014, 29, 2520–2526. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011, 3, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.X.; Yu, Q. Primary osteoporosis in postmenopausal women. Chronic Dis Transl Med 2015, 1, 9–13. [Google Scholar] [CrossRef]
- Ponnapakkam, T.; Katikaneni, R.; Sakon, J.; Stratford, R.; Gensure, R.C. Treating osteoporosis by targeting parathyroid hormone to bone. Drug Discov Today 2014, 19, 204–208. [Google Scholar] [CrossRef]
- Wippert, P.M.; Rector, M.; Kuhn, G.; Wuertz-Kozak, K. Stress and Alterations in Bones: An Interdisciplinary Perspective. Front Endocrinol (Lausanne) 2017, 8, 96. [Google Scholar] [CrossRef] [PubMed]
- Rashki Kemmak, A.; Rezapour, A.; Jahangiri, R.; Nikjoo, S.; Farabi, H.; Soleimanpour, S. Economic burden of osteoporosis in the world: A systematic review. Med J Islam Repub Iran 2020, 34, 154. [Google Scholar] [CrossRef]
- Weaver, C.M.; Gordon, C.M.; Janz, K.F.; Kalkwarf, H.J.; Lappe, J.M.; Lewis, R.; O’Karma, M.; Wallace, T.C.; Zemel, B.S. The National Osteoporosis Foundation’s position statement on peak bone mass development and lifestyle factors: a systematic review and implementation recommendations. Osteoporos Int 2016, 27, 1281–1386. [Google Scholar] [CrossRef]
- Reeve, J.; Meunier, P.J.; Parsons, J.A.; Bernat, M.; Bijvoet, O.L.; Courpron, P.; Edouard, C.; Klenerman, L.; Neer, R.M.; Renier, J.C.; et al. Anabolic effect of human parathyroid hormone fragment on trabecular bone in involutional osteoporosis: a multicentre trial. Br Med J 1980, 280, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Cyriac, M.; Kyhos, J.; Iweala, U.; Lee, D.; Mantell, M.; Yu, W.; O’Brien, J.R. Anterior Lumbar Interbody Fusion With Cement Augmentation Without Posterior Fixation to Treat Isthmic Spondylolisthesis in an Osteopenic Patient-A Surgical Technique. Int J Spine Surg 2018, 12, 322–327. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Bisphosphonates for the treatment of osteoporosis: insights for clinicians. Ther Adv Chronic Dis 2010, 1, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Lv, H.; Yin, P.; Li, Z.; Tang, P.; Wang, Y. Combination therapy with parathyroid hormone analogs and antiresorptive agents for osteoporosis: a systematic review and meta-analysis of randomized controlled trials. Osteoporos Int 2019, 30, 59–70. [Google Scholar] [CrossRef]
- Delmas, P.D.; Recker, R.R.; Chesnut, C.H.; Skag, A.; Stakkestad, J.A.; Emkey, R.; Gilbride, J.; Schimmer, R.C.; Christiansen, C. Daily and intermittent oral ibandronate normalize bone turnover and provide significant reduction in vertebral fracture risk: results from the BONE study. Osteoporos Int 2004, 15, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Chesnut, C.H.; Skag, A.; Christiansen, C.; Recker, R.; Stakkestad, J.A.; Hoiseth, A.; Felsenberg, D.; Huss, H.; Gilbride, J.; Schimmer, R.C.; et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res 2004, 19, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.T.; Watts, N.B.; Genant, H.K.; McKeever, C.D.; Hangartner, T.; Keller, M.; Chesnut, C.H.; Brown, J.; Eriksen, E.F.; Hoseyni, M.S.; et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA 1999, 282, 1344–1352. [Google Scholar] [CrossRef]
- Chen, L.R.; Ko, N.Y.; Chen, K.H. Medical Treatment for Osteoporosis: From Molecular to Clinical Opinions. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Miller, P.D.; Bilezikian, J.P.; Diaz-Curiel, M.; Chen, P.; Marin, F.; Krege, J.H.; Wong, M.; Marcus, R. Occurrence of hypercalciuria in patients with osteoporosis treated with teriparatide. J Clin Endocrinol Metab 2007, 92, 3535–3541. [Google Scholar] [CrossRef]
- Marx, R.E.; Cillo, J.E.; Ulloa, J.J. Oral bisphosphonate-induced osteonecrosis: risk factors, prediction of risk using serum CTX testing, prevention, and treatment. J Oral Maxillofac Surg 2007, 65, 2397–2410. [Google Scholar] [CrossRef]
- Charopoulos, I.; Orme, S.; Giannoudis, P.V. The role and efficacy of denosumab in the treatment of osteoporosis: an update. Expert Opin Drug Saf 2011, 10, 205–217. [Google Scholar] [CrossRef]
- Bone, H.G.; Hosking, D.; Devogelaer, J.P.; Tucci, J.R.; Emkey, R.D.; Tonino, R.P.; Rodriguez-Portales, J.A.; Downs, R.W.; Gupta, J.; Santora, A.C.; et al. Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Engl J Med 2004, 350, 1189–1199. [Google Scholar] [CrossRef]
- Miller, P.D.; Schwartz, E.N.; Chen, P.; Misurski, D.A.; Krege, J.H. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporos Int 2007, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Thiruchelvam, N.; Randhawa, J.; Sadiek, H.; Kistangari, G. Teriparatide induced delayed persistent hypercalcemia. Case Rep Endocrinol 2014, 2014, 802473. [Google Scholar] [CrossRef] [PubMed]
- Sølling, A.S.K.; Harsløf, T.; Langdahl, B. The clinical potential of romosozumab for the prevention of fractures in postmenopausal women with osteoporosis. Ther Adv Musculoskelet Dis 2018, 10, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Geusens, P.; Oates, M.; Miyauchi, A.; Adachi, J.D.; Lazaretti-Castro, M.; Ebeling, P.R.; Perez Niño, C.A.; Milmont, C.E.; Grauer, A.; Libanati, C. The Effect of 1 Year of Romosozumab on the Incidence of Clinical Vertebral Fractures in Postmenopausal Women With Osteoporosis: Results From the FRAME Study. JBMR Plus 2019, 3, e10211. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Adanty, C. Romosozumab (sclerostin monoclonal antibody) for the treatment of osteoporosis in postmenopausal women: A review. J Popul Ther Clin Pharmacol 2020, 27, e25–e31. [Google Scholar] [CrossRef]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Lewiecki, E.M.; Jaller-Raad, J.; Zerbini, C.; Milmont, C.E.; Meisner, P.D.; Libanati, C.; Grauer, A. Romosozumab FRAME Study: A Post Hoc Analysis of the Role of Regional Background Fracture Risk on Nonvertebral Fracture Outcome. J Bone Miner Res 2018, 33, 1407–1416. [Google Scholar] [CrossRef]
- Kerschan-Schindl, K. Romosozumab: a novel bone anabolic treatment option for osteoporosis? Wien Med Wochenschr 2019. [Google Scholar] [CrossRef]
- Chang, C. Agonists and Antagonists of TGF-β Family Ligands. Cold Spring Harb Perspect Biol 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, M.M.; Kim, S.K.; Barron, L.; Kodali, R.; Baardsnes, J.; Hinck, C.S.; Krzysiak, T.C.; Henen, M.A.; Pakhomova, O.; Mendoza, V.; et al. Binding Properties of the Transforming Growth Factor-β Coreceptor Betaglycan: Proposed Mechanism for Potentiation of Receptor Complex Assembly and Signaling. Biochemistry 2016, 55, 6880–6896. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Halloran, D.; Durbano, H.W.; Nohe, A. Bone Morphogenetic Protein-2 in Development and Bone Homeostasis. J Dev Biol 2020, 8. [Google Scholar] [CrossRef]
- Sountoulidis, A.; Stavropoulos, A.; Giaglis, S.; Apostolou, E.; Monteiro, R.; Chuva de Sousa Lopes, S.M.; Chen, H.; Stripp, B.R.; Mummery, C.; Andreakos, E.; et al. Activation of the canonical bone morphogenetic protein (BMP) pathway during lung morphogenesis and adult lung tissue repair. PLoS One 2012, 7, e41460. [Google Scholar] [CrossRef]
- Gaussin, V.; Morley, G.E.; Cox, L.; Zwijsen, A.; Vance, K.M.; Emile, L.; Tian, Y.; Liu, J.; Hong, C.; Myers, D.; et al. Alk3/Bmpr1a receptor is required for development of the atrioventricular canal into valves and annulus fibrosus. Circ Res 2005, 97, 219–226. [Google Scholar] [CrossRef]
- Angello, J.C.; Kaestner, S.; Welikson, R.E.; Buskin, J.N.; Hauschka, S.D. BMP induction of cardiogenesis in P19 cells requires prior cell-cell interaction(s). Dev Dyn 2006, 235, 2122–2133. [Google Scholar] [CrossRef]
- Gámez, B.; Rodriguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front Cell Neurosci 2013, 7, 87. [Google Scholar] [CrossRef]
- Pajni-Underwood, S.; Wilson, C.P.; Elder, C.; Mishina, Y.; Lewandoski, M. BMP signals control limb bud interdigital programmed cell death by regulating FGF signaling. Development 2007, 134, 2359–2368. [Google Scholar] [CrossRef]
- Huang, P.; Chen, A.; He, W.; Li, Z.; Zhang, G.; Liu, Z.; Liu, G.; Liu, X.; He, S.; Xiao, G.; et al. BMP-2 induces EMT and breast cancer stemness through Rb and CD44. Cell Death Discov 2017, 3, 17039. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef]
- Bragdon, B.; Bonor, J.; Shultz, K.L.; Beamer, W.G.; Rosen, C.J.; Nohe, A. Bone morphogenetic protein receptor type Ia localization causes increased BMP2 signaling in mice exhibiting increased peak bone mass phenotype. J Cell Physiol 2012, 227, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Heubel, B.; Nohe, A. The Role of BMP Signaling in Osteoclast Regulation. J Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: a critical review. Cell Signal 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Nohe, A.; Keating, E.; Underhill, T.M.; Knaus, P.; Petersen, N.O. Dynamics and interaction of caveolin-1 isoforms with BMP-receptors. J Cell Sci 2005, 118, 643–650. [Google Scholar] [CrossRef]
- Nohe, A.; Keating, E.; Underhill, T.M.; Knaus, P.; Petersen, N.O. Effect of the distribution and clustering of the type I A BMP receptor (ALK3) with the type II BMP receptor on the activation of signalling pathways. J Cell Sci 2003, 116, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Bragdon, B.; Thinakaran, S.; Bonor, J.; Underhill, T.M.; Petersen, N.O.; Nohe, A. FRET reveals novel protein-receptor interaction of bone morphogenetic proteins receptors and adaptor protein 2 at the cell surface. Biophys J 2009, 97, 1428–1435. [Google Scholar] [CrossRef]
- Bonor, J.; Adams, E.L.; Bragdon, B.; Moseychuk, O.; Czymmek, K.J.; Nohe, A. Initiation of BMP2 signaling in domains on the plasma membrane. J Cell Physiol 2012, 227, 2880–2888. [Google Scholar] [CrossRef]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 2002, 277, 5330–5338. [Google Scholar] [CrossRef] [PubMed]
- Nohe, A.; Keating, E.; Knaus, P.; Petersen, N.O. Signal transduction of bone morphogenetic protein receptors. Cell Signal 2004, 16, 291–299. [Google Scholar] [CrossRef]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; King, D.; Young, K.; Litchfield, D.W.; Petersen, N.O.; Nohe, A. Casein kinase 2 beta-subunit is a regulator of bone morphogenetic protein 2 signaling. Biophys J 2010, 99, 897–904. [Google Scholar] [CrossRef]
- Bragdon, B.; Thinakaran, S.; Moseychuk, O.; Gurski, L.; Bonor, J.; Price, C.; Wang, L.; Beamer, W.G.; Nohe, A. Casein kinase 2 regulates in vivo bone formation through its interaction with bone morphogenetic protein receptor type Ia. Bone 2011, 49, 944–954. [Google Scholar] [CrossRef]
- Akkiraju, H.; Bonor, J.; Nohe, A. CK2.1, a novel peptide, induces articular cartilage formation in vivo. J Orthop Res 2017, 35, 876–885. [Google Scholar] [CrossRef]
- Halloran, D.; Pandit, V.; Nohe, A. The Role of Protein Kinase CK2 in Development and Disease Progression: A Critical Review. J Dev Biol 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ho, C.C.; Bang, E.; Rejon, C.A.; Libasci, V.; Pertchenko, P.; Hébert, T.E.; Bernard, D.J. Bone morphogenetic protein 2 stimulates noncanonical SMAD2/3 signaling via the BMP type 1A receptor in gonadotrope-like cells: implications for FSH synthesis. Endocrinology 2014, 155, 1970–1981. [Google Scholar] [CrossRef]
- Weidner, H.; Yuan Gao, V.; Dibert, D.; McTague, S.; Eskander, M.; Duncan, R.; Wang, L.; Nohe, A. CK2.3, a Mimetic Peptide of the BMP Type I Receptor, Increases Activity in Osteoblasts over BMP2. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Vrathasha, V.; Weidner, H.; Nohe, A. Mechanism of CK2.3, a Novel Mimetic Peptide of Bone Morphogenetic Protein Receptor Type IA, Mediated Osteogenesis. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Akkiraju, H.; Bonor, J.; Olli, K.; Bowen, C.; Bragdon, B.; Coombs, H.; Donahue, L.R.; Duncan, R.; Nohe, A. Systemic injection of CK2.3, a novel peptide acting downstream of bone morphogenetic protein receptor BMPRIa, leads to increased trabecular bone mass. J Orthop Res 2015, 33, 208–215. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J Biochem 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb Perspect Biol 2017, 9. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb Perspect Biol 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Kelly, S.; Wood, R.; Heubel, B.; Nohe, A. A Synthetic Peptide, CK2.3, Inhibits RANKL-Induced Osteoclastogenesis through BMPRIa and ERK Signaling Pathway. J Dev Biol 2020, 8. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Burkus, J.K.; Gornet, M.F.; Dickman, C.A.; Zdeblick, T.A. Anterior lumbar interbody fusion using rhBMP-2 with tapered interbody cages. J Spinal Disord Tech 2002, 15, 337–349. [Google Scholar] [CrossRef]
- Durbano, H.W.; Halloran, D.; Nguyen, J.; Stone, V.; McTague, S.; Eskander, M.; Nohe, A. Aberrant BMP2 Signaling in Patients Diagnosed with Osteoporosis. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- James, A.W.; LaChaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng Part B Rev 2016, 22, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio, A.T.; Burneikiene, S. RhBMP-2-induced radiculitis in patients undergoing transforaminal lumbar interbody fusion: relationship to dose. Spine J 2016, 16, 1208–1213. [Google Scholar] [CrossRef]
- McClellan, J.W.; Mulconrey, D.S.; Forbes, R.J.; Fullmer, N. Vertebral bone resorption after transforaminal lumbar interbody fusion with bone morphogenetic protein (rhBMP-2). J Spinal Disord Tech 2006, 19, 483–486. [Google Scholar] [CrossRef]
- Lewandrowski, K.U.; Nanson, C.; Calderon, R. Vertebral osteolysis after posterior interbody lumbar fusion with recombinant human bone morphogenetic protein 2: a report of five cases. Spine J 2007, 7, 609–614. [Google Scholar] [CrossRef]
- Chen, X.; Resh, M.D. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J Biol Chem 2002, 277, 49631–49637. [Google Scholar] [CrossRef] [PubMed]
- Rodal, S.K.; Skretting, G.; Garred, O.; Vilhardt, F.; van Deurs, B.; Sandvig, K. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell 1999, 10, 961–974. [Google Scholar] [CrossRef]
- Tabatabaei-Panah, A.S.; Jeddi-Tehrani, M.; Ghods, R.; Akhondi, M.M.; Mojtabavi, N.; Mahmoudi, A.R.; Mirzadegan, E.; Shojaeian, S.; Zarnani, A.H. Accurate sensitivity of quantum dots for detection of HER2 expression in breast cancer cells and tissues. J Fluoresc 2013, 23, 293–302. [Google Scholar] [CrossRef]
- Fang, M.; Chen, M.; Liu, L.; Li, Y. Applications of Quantum Dots in Cancer Detection and Diagnosis: A Review. J Biomed Nanotechnol 2017, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Bakhshi, R.; Petrova, D.; Pocock, R.; Imani, M.; Seifalian, A.M. Biological applications of quantum dots. Biomaterials 2007, 28, 4717–4732. [Google Scholar] [CrossRef]
- Forder, J.; Smith, M.; Wagner, M.; Schaefer, R.J.; Gorky, J.; van Golen, K.L.; Nohe, A.; Dhurjati, P. A Physiologically-Based Pharmacokinetic Model for Targeting Calcitriol-Conjugated Quantum Dots to Inflammatory Breast Cancer Cells. Clin Transl Sci 2019, 12, 617–624. [Google Scholar] [CrossRef]
- Geng, X.F.; Fang, M.; Liu, S.P.; Li, Y. Quantum dot-based molecular imaging of cancer cell growth using a clone formation assay. Mol Med Rep 2016, 14, 3007–3012. [Google Scholar] [CrossRef] [PubMed]
- Halloran, D.; Vrathasha, V.; Durbano, H.W.; Nohe, A. Bone Morphogenetic Protein-2 Conjugated to Quantum Dot. Nanomaterials (Basel) 2020, 10. [Google Scholar] [CrossRef]
- Vrathasha, V.; Booksh, K.; Duncan, R.L.; Nohe, A. Mechanisms of Cellular Internalization of Quantum Dot® Conjugated Bone Formation Mimetic Peptide CK2.3. Nanomaterials (Basel) 2018, 8. [Google Scholar] [CrossRef]
- Halloran, D.; Pandit, V.; MacMurray, C.; Stone, V.; DeGeorge, K.; Eskander, M.; Root, D.; McTague, S.; Pelkey, H.; Nohe, A. Age-Related Low Bone Mineral Density in C57BL/6 Mice Is Reflective of Aberrant Bone Morphogenetic Protein-2 Signaling Observed in Human Patients Diagnosed with Osteoporosis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Piotrowska, K.; Zgutka, K.; Kupnicka, P.; Chlubek, D.; Pawlik, A.; Baranowska-Bosiacka, I. Analysis of Bone Mineral Profile After Prolonged Every-Other-Day Feeding in C57BL/6J Male and Female Mice. Biol Trace Elem Res 2020, 194, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Kerschan-Schindl, K.; Papageorgiou, M.; Föger-Samwald, U.; Butylina, M.; Weber, M.; Pietschmann, P. Assessment of Bone Microstructure by Micro CT in C57BL/6J Mice for Sex-Specific Differentiation. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.F.; Jones, C.B.; Sietsema, D.L. Complications of rhBMP-2 utilization for posterolateral lumbar fusions requiring reoperation: a single practice, retrospective case series report. Spine J 2013, 13, 1244–1252. [Google Scholar] [CrossRef]
- Faundez, A.; Tournier, C.; Garcia, M.; Aunoble, S.; Le Huec, J.C. Bone morphogenetic protein use in spine surgery-complications and outcomes: a systematic review. Int Orthop 2016, 40, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Halloran, D.R.; Heubel, B.; MacMurray, C.; Root, D.; Eskander, M.; McTague, S.P.; Pelkey, H.; Nohe, A. Differentiation of Cells Isolated from Human Femoral Heads into Functional Osteoclasts. J Dev Biol 2022, 10. [Google Scholar] [CrossRef]
- Pujari-Palmer, M.; Pujari-Palmer, S.; Lu, X.; Lind, T.; Melhus, H.; Engstrand, T.; Karlsson-Ott, M.; Engqvist, H. Pyrophosphate Stimulates Differentiation, Matrix Gene Expression and Alkaline Phosphatase Activity in Osteoblasts. PLoS One 2016, 11, e0163530. [Google Scholar] [CrossRef]
Figure 1.
Overexpression of BMPRIa in BMSCs isolated from US 6- and 15-month B6 mice. BMSCs were isolated from the femurs of 6- and 15-month B6 mice. The cells were grown, without additional stimulation, for 10-12 days. (A) lysates were collected and probed for BMPRIa. Protein concentration was normalized and GAPDH was used as the loading control. (B) BMPRIa was detected via immunofluorescent staining and images were acquired with confocal microscopy. Representative images are displayed and scalebars are set to 10 µm.
Figure 1.
Overexpression of BMPRIa in BMSCs isolated from US 6- and 15-month B6 mice. BMSCs were isolated from the femurs of 6- and 15-month B6 mice. The cells were grown, without additional stimulation, for 10-12 days. (A) lysates were collected and probed for BMPRIa. Protein concentration was normalized and GAPDH was used as the loading control. (B) BMPRIa was detected via immunofluorescent staining and images were acquired with confocal microscopy. Representative images are displayed and scalebars are set to 10 µm.
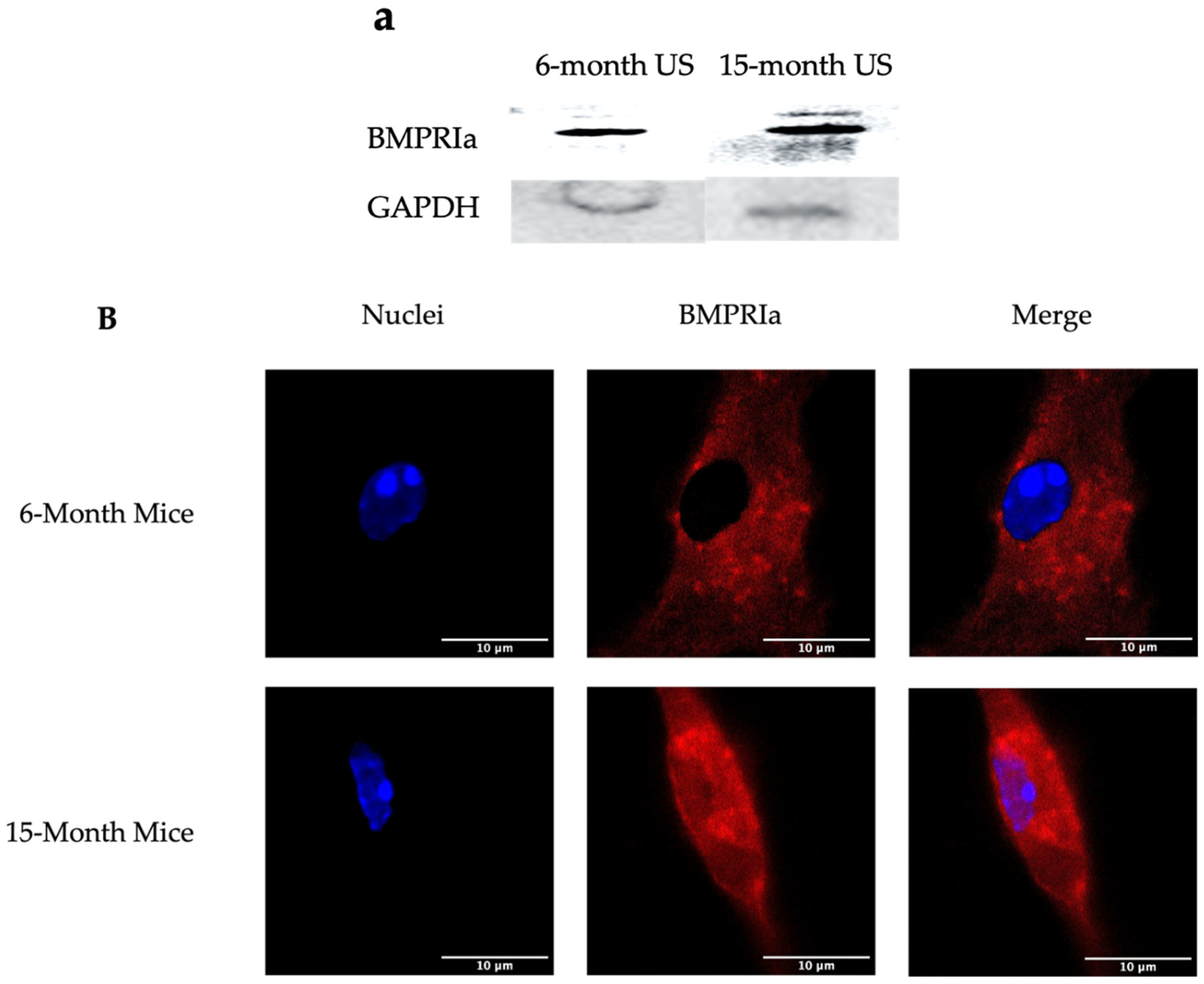
Figure 2.
Immunostaining of BMSCs isolated from 6- and 15-month female B6 mice. Cells were obtained from 6-month and 15-month mice and stimulated with BMP-2-QDot®s or left US. The cells were immunostained and observed with confocal microscopy. Representative images are displayed and scale bars are set to 1 or 5 μm. As displayed in the images, the expression of BMPRIa is increased in the 15-month mice. Meanwhile, BMP-2-QDot®s less effectively bind to BMPRIa in 15-month mice compared to 6-month mice as indicated by less green fluorescence.
Figure 2.
Immunostaining of BMSCs isolated from 6- and 15-month female B6 mice. Cells were obtained from 6-month and 15-month mice and stimulated with BMP-2-QDot®s or left US. The cells were immunostained and observed with confocal microscopy. Representative images are displayed and scale bars are set to 1 or 5 μm. As displayed in the images, the expression of BMPRIa is increased in the 15-month mice. Meanwhile, BMP-2-QDot®s less effectively bind to BMPRIa in 15-month mice compared to 6-month mice as indicated by less green fluorescence.
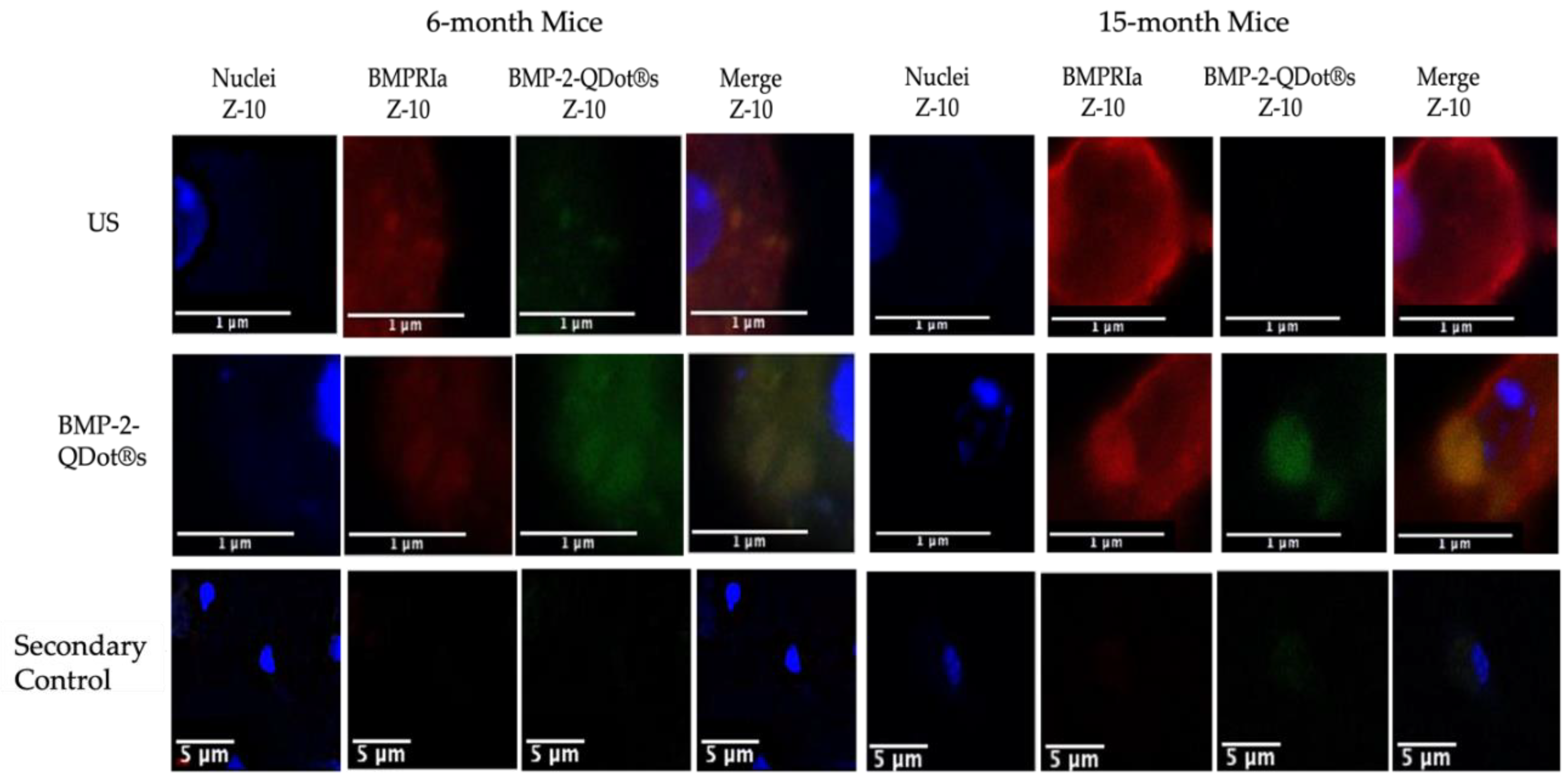
Figure 3.
Immunostaining of BMSCs isolated from 6- and 15-month female B6 mice. Cells were obtained from 6-month and 15-month mice and stimulated with BMP-2-QDot®s or left US after treatment with MβCD. The cells were immunostained and observed with confocal microscopy. Representative images are displayed and scale bars are set to 1 or 5 μm. As displayed in the images, the expression of BMPRIa is similar between the two age groups. Further, BMP-2-QDot®s effectively binds to BMPRIa in 15-month mice compared to 6-month mice as indicated by an increase in green fluorescence.
Figure 3.
Immunostaining of BMSCs isolated from 6- and 15-month female B6 mice. Cells were obtained from 6-month and 15-month mice and stimulated with BMP-2-QDot®s or left US after treatment with MβCD. The cells were immunostained and observed with confocal microscopy. Representative images are displayed and scale bars are set to 1 or 5 μm. As displayed in the images, the expression of BMPRIa is similar between the two age groups. Further, BMP-2-QDot®s effectively binds to BMPRIa in 15-month mice compared to 6-month mice as indicated by an increase in green fluorescence.
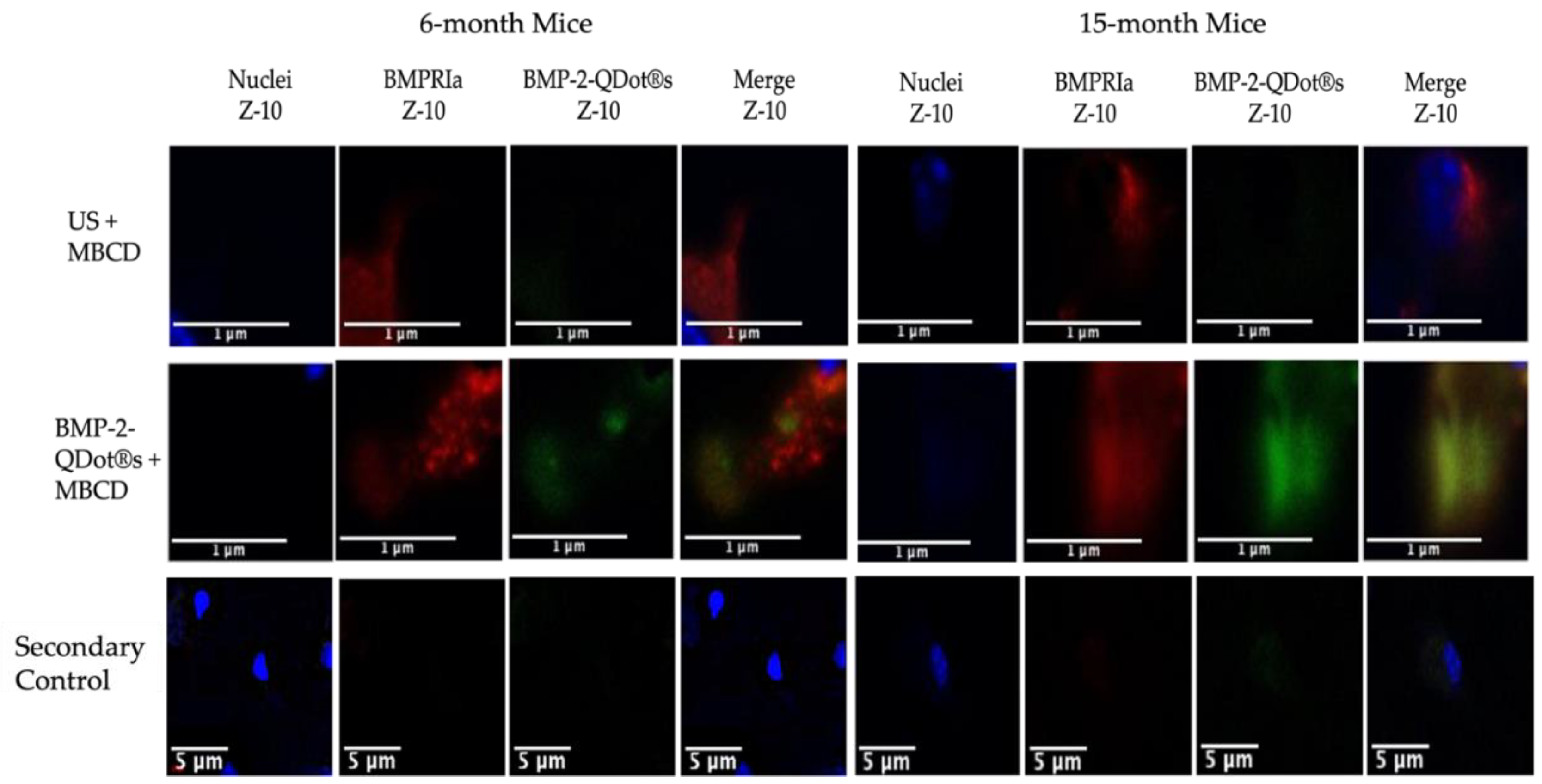
Figure 4.
Quantification of immunostaining conducted on BMSCs. To further assess the increase or decrease of BMPRIa and BMP-2 binding, the images acquired from confocal microscopy were semi-quantified. (A) The red fluorescence of BMPRIa was measured across all conditions of both 6- and 15-month B6 mice. (B) The green fluorescence of BMPRIa was measured across all conditions of both 6- and 15-month B6 mice. SEM bars are displayed above each bar, and lettering indicates significance. All data were analyzed in ImageJ and statistical analyses were performed with the Tukey-Kramer HSD test.
Figure 4.
Quantification of immunostaining conducted on BMSCs. To further assess the increase or decrease of BMPRIa and BMP-2 binding, the images acquired from confocal microscopy were semi-quantified. (A) The red fluorescence of BMPRIa was measured across all conditions of both 6- and 15-month B6 mice. (B) The green fluorescence of BMPRIa was measured across all conditions of both 6- and 15-month B6 mice. SEM bars are displayed above each bar, and lettering indicates significance. All data were analyzed in ImageJ and statistical analyses were performed with the Tukey-Kramer HSD test.
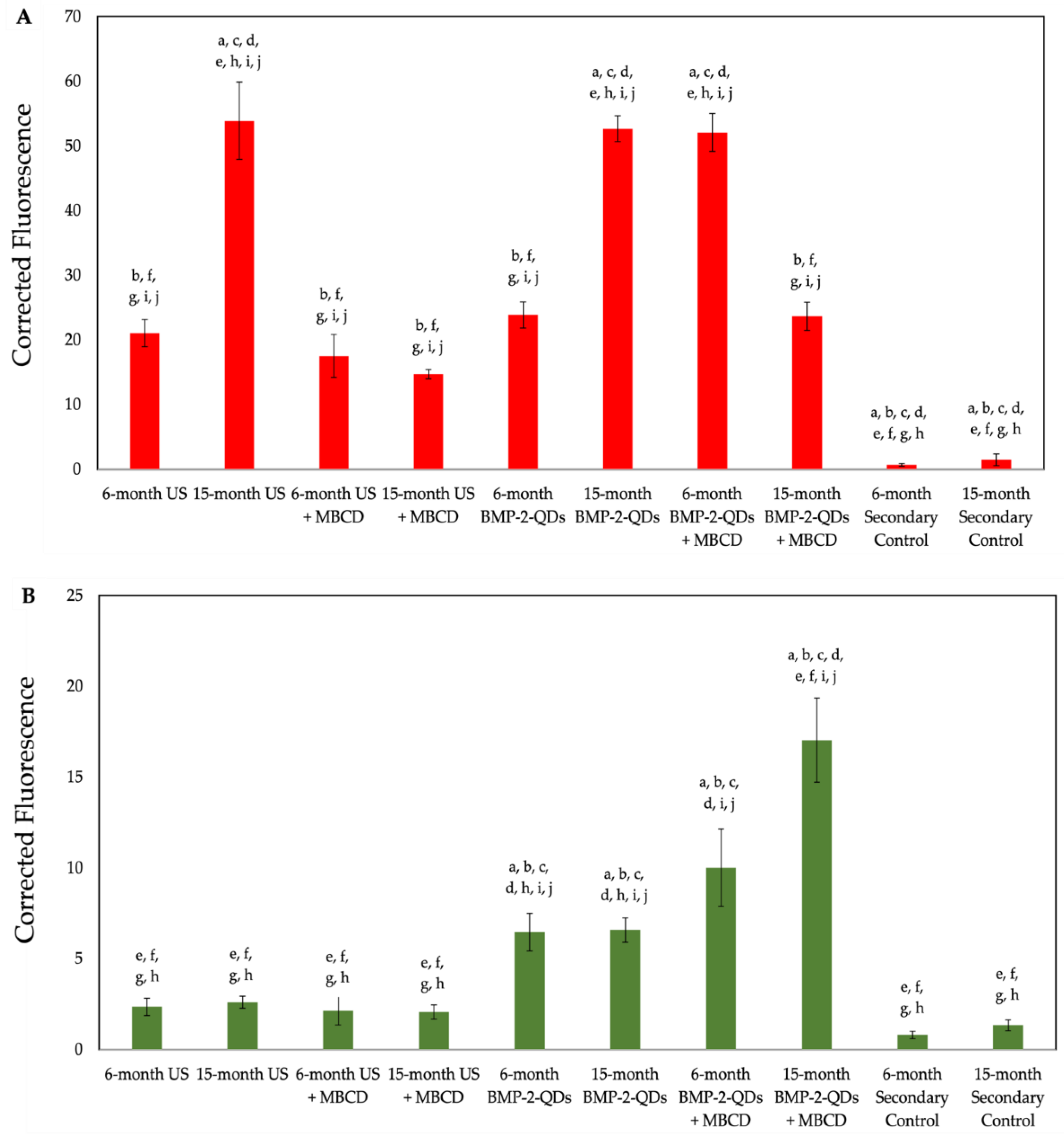
Figure 5.
Western blotting of lysates collected from 6- and 15-month BMSCs after treatment with BMP-2 and MβCD. BMSCs were obtained from 6- and 15-month female B6 mice and treated with MβCD or left untreated. After, cells were stimulated with BMP-2 or left US. BMP-2 enhanced pSMAD1 phosphorylation in 6-month cells in both MβCD treated and untreated cells, whereas in 15-months, only BMP-2 + MβCD led to an upregulation in pSMAD1. Total SMAD1/5/8 detection was similar across all samples. GAPDH is detected as the loading control. All protein concentration was normalized.
Figure 5.
Western blotting of lysates collected from 6- and 15-month BMSCs after treatment with BMP-2 and MβCD. BMSCs were obtained from 6- and 15-month female B6 mice and treated with MβCD or left untreated. After, cells were stimulated with BMP-2 or left US. BMP-2 enhanced pSMAD1 phosphorylation in 6-month cells in both MβCD treated and untreated cells, whereas in 15-months, only BMP-2 + MβCD led to an upregulation in pSMAD1. Total SMAD1/5/8 detection was similar across all samples. GAPDH is detected as the loading control. All protein concentration was normalized.
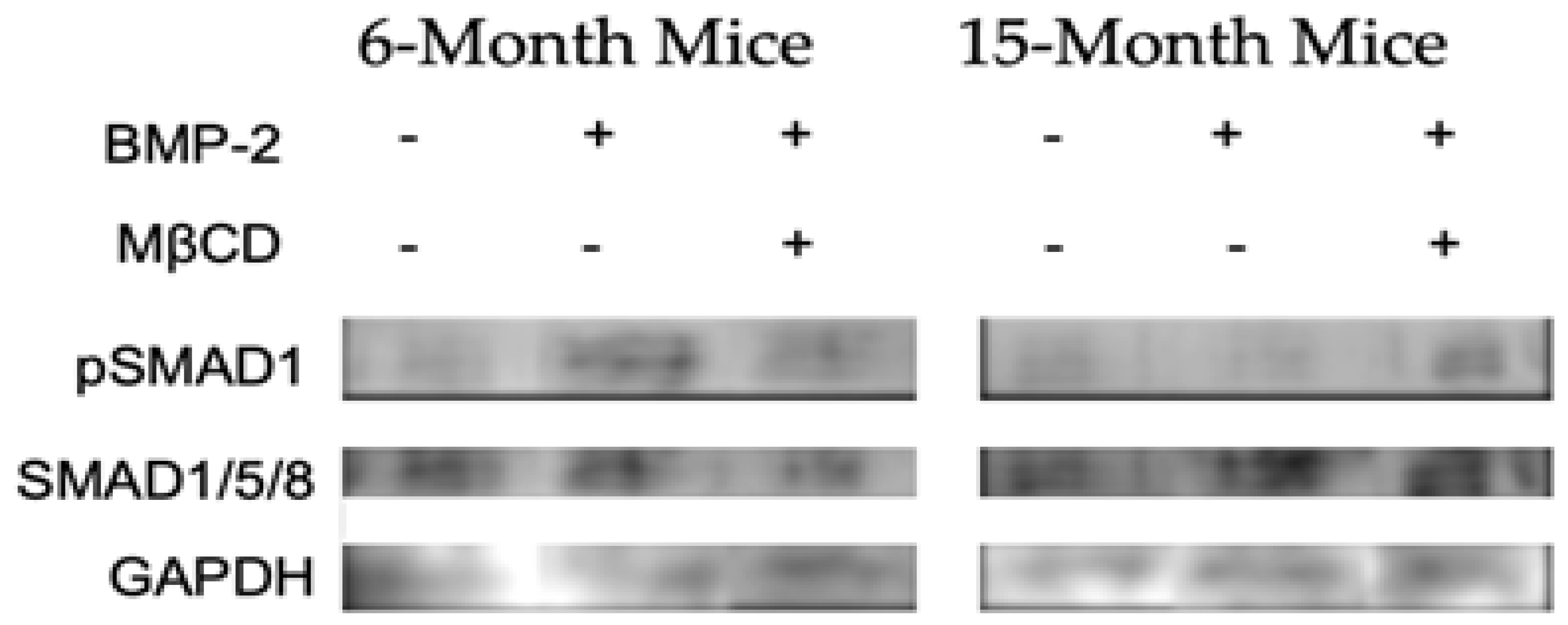
Figure 6.
von Kossa assay of MβCD treated BMSCs isolated from 6- and 15-month mice. BMSCs were obtained from 6- and 15-month female B6 mice and treated with MβCD or left untreated. After, cells were stimulated with BMP-2 or left US. BMP-2 enhanced mineralization in 6-month cells in both MβCD treated and untreated cells, whereas in 15-months, only BMP-2 + MβCD led to mineralization. Random images were obtained from each condition and analyzed in ImageJ. Representative images are displayed underneath each bar. Error bars represent SEM and significance was determined using the Tukey Kramer-HSD test.
Figure 6.
von Kossa assay of MβCD treated BMSCs isolated from 6- and 15-month mice. BMSCs were obtained from 6- and 15-month female B6 mice and treated with MβCD or left untreated. After, cells were stimulated with BMP-2 or left US. BMP-2 enhanced mineralization in 6-month cells in both MβCD treated and untreated cells, whereas in 15-months, only BMP-2 + MβCD led to mineralization. Random images were obtained from each condition and analyzed in ImageJ. Representative images are displayed underneath each bar. Error bars represent SEM and significance was determined using the Tukey Kramer-HSD test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



