
Submitted:
24 November 2023
Posted:
25 November 2023
You are already at the latest version
Abstract
Celiac disease is an autoimmune disease triggered by oral ingestion of gluten, with certain gluten residues resistant to digestive tract enzymes. Within the duodenum, the remaining peptides incite immunogenic responses, including the generation of autoantibodies and inflammation, leading to irreversible damage. Our previous exploration unveiled a glutenase called Bga1903 derived from the Gram-negative bacterium Burkholderia gladioli. The cleavage pattern of Bga1903 indicated its moderate ability to mitigate the toxicity of pro-immunogenic peptides. The crystal structure of Bga1903, along with the identification of subsites within its active site, was determined. To improve its substrate specificity toward prevalent motifs like QPQ within gluten peptides, the active site of Bga1903 underwent site-directed mutagenesis according to structural insights and enzymatic kinetics. Among the double-site mutants, E380Q/S387L exhibits an approximately 38-fold increase in its specificity constant toward the QPQ sequence, favoring glutamines at the P1 and P3 positions compared to the wild type. Not only does the heightened specificity of E380Q/S387L improve the hydrolytic activity against pro-immunogenic peptides, but also holds this version of the enzyme as a promising candidate as an oral therapy enzyme for celiac disease.
Keywords:
celiac disease
; glutenase
; gliadinase
; gluten-free diet
; gluten-derived pro-immunogenic peptides
; oral enzyme therapy
; enzyme active-site modification.
1. Introduction
Celiac disease is an autoimmune disorder triggered by the consumption of gluten and is characterized by various gastrointestinal and extraintestinal symptoms [1]. Human leukocyte antigen (HLA)-DQ2 is associated with >90% of patients, while the remaining patients possess HLA-DQ8 [2]. It is noteworthy that additional unknown factors influence the development of the disease [3]. The diagnosis of celiac disease can be accomplished through the utilization of duodenal biopsy or serological testing [4,5]. Traditionally, celiac disease is confirmed via endoscopic biopsy, revealing villous atrophy and crypt hyperplasia in the small intestine [4]. However, owing to the invasive nature of biopsy procedures, serologic tests, such as anti-transglutaminase 2 (TG2) antibodies and anti-endomysial antibody assays, are typically conducted in advance when there is suspicion of the disease [5].
The manifestation of the disease is primarily driven by gliadin, the ethanol-soluble component of wheat gluten [6]. The proline-rich composition renders gliadin partially resistant to proteolysis by human digestive enzymes. Some of the recalcitrant peptides, such as the 33-mer peptide from α2-gliadin and 26-mer peptide from γ5-gliadin, might enter the lamina propria of duodenum via the transepithelial pathway [7,8], where these pro-immunogenic peptides are subsequently subjected to deamination by TG2, leading to the conversion of specific glutamine residues into glutamic acids [9]. Epitopes on the deaminated peptides are presented to gluten-specific CD4+ T cells by antigen-presenting cells possessing HLA-DQ2 and/or HLA-DQ8 [2]. The activated gluten-specific CD4+ T cells secrete inflammatory cytokines and stimulate specific B cells, leading to the production of anti-gliadin antibodies and anti-TG2 autoantibodies [10,11]. Since TG2 is implicated in cell-adhesive cytoskeletal cross-linking throughout the body [12], the assault on tissues by anti-TG2 autoantibodies would compromise intestinal barrier integrity, further increasing the influx of gluten peptides and exacerbating the systemic autoimmune response and malabsorption of nutrients [13].
At present, there is no cure for patients; therefore, a strict gluten-free diet is the only prescription to mitigate the onset of celiac disease. However, maintaining this diet for life is difficult because it is costly, inconvenient, and sometimes impossible due to inadvertent contamination during food processing [14,15]. Given the substantial burden that celiac disease imposes on public health and individual well-being, drugs with diverse mechanisms have been under development by pharmaceutical companies and academia [16,17]. Oral enzyme therapy has been suggested as one of the adjunctive treatments for celiac disease, involving the enzymatic hydrolysis of pro-immunogenic peptides derived from gluten within the stomach and duodenum [18,19,20]. In recent years, the potential of a couple of peptidases has been assessed through clinical trials. Kuma-062 (also called TAK-062), an iteratively engineered acid-tolerant S53 peptidase originally from Alicyclobacillus sendaiensis, has recently progressed to the phase 2 clinical trial (clinical trial number: NCT05353985) [21]. Latiglutenase (formerly ALV003 or IMGX003), a cocktail formulation containing peptidases sourced from germinating barley seeds (EP-B2) and from Sphingomonas capsulate (SC-PEP), reduced gluten-induced intestinal mucosal damage in the phase 2 clinical trial (clinical trial number: NCT03585478) [22,23].
In previous studies, we identified a serine peptidase, named Bga1903, with gliadin hydrolytic activity from Burkholderia gladioli [24]. Bga1903 has the capability of being secreted into the culture medium by Escherichia coli BL21(DE3). The structure of Bga1903 was determined through X-ray crystallography, and the architecture of the substrate-binding subsites was defined [25]. In the present study, an endeavor was made to further enhance its hydrolytic activity toward the 33-mer and 26-mer peptides by rationally modifying the S1 subsite of Bga1903 through mutagenesis. The alterations in catalytic constants were investigated to verify their respective effects.
2. Results
2.1. The substrate-binding subsites of Bga1903
Bga1903 preferentially cut the bond at the carbonyl side of glutamine when it acted on the 26- and 33-mer peptides; nonetheless, it showed relaxed selectivity at the P1 residue, with a preference for lysine, followed by phenylalanine, leucine, and glutamine when bovine serum albumin served as the substrate [24]. These observations imply that elevating the catalytic specificity of Bga1903 toward pro-immunogenic peptides could be potentially achieved through strategic alterations of subsite S1 within the peptidyl-binding pocket. The crystal structure of Bga1903, highlighting residues constituting the S1-S3 subsites (Figure 1), indicates that the broader specificity for P1 residues may stem from the larger cavity of S1 compared to S2 and S3. An earlier study investigated the importance of residues E380 and S387, which line the wall of S1, in influencing the peptidase’s catalytic efficiency toward specific chromogenic peptidyl substrates [25]. The present study conducts a more detailed examination of these two residues, anticipating that tailoring S1 will result in changes in selectivity for acceptable P1 residues.
2.2. The specific activity of Bga1903 toward various chromogenic tripeptidyl substrates
To assess the substrate specificity of Bga1903, we employed several tripeptidyl chromogenic substrates, namely Z-HPX-pNA and Z-QPQ-pNA (where Z stands for benzyloxycarbonyl, pNA for p-nitroanilide, and X for an amino acid). The design of Z-HPX-pNA was informed by Bga1903's cleavage patterns on bovine serum albumins, revealing a preference for proline and histidine at the P2 and P3 positions, respectively [24]. Meanwhile, Z-QPQ-pNA represents a common motif found in the 26- and 33-mer peptides. The specific activity toward various tripeptidyl substrates is shown in Figure 2.
Within the Z-HPX-pNA group, the reactivity order is HPK >> HPQ > HPL > HPY ≈ HPF > HPE, affirming that the preferred order at the P1 position is K >> Q > L > Y ≈ F > E. Notably, Bga1903 exhibits reduced activity toward Z-QPQ-pNA compared to Z-HPQ-pNA, indicating a preference for histidine over glutamine at the P3 position. The high preference for Z-HPK-pNA and low preference for Z-HPE-pNA may be directly linked to the negatively charged potential in S1, particularly contributed by E380. Moreover, the S1 cavity appears to inadequately accommodate bulky hydrophobic groups, as evidenced by the disfavoring of substrates Z-HPY-pNA and Z-HPF-pNA.
2.3. Effect of E380 substitutions on the enzymatic activity of Bga1903
To probe the function of E380 and explore the potential for altering the substrate specificity of Bga1903, E380 was substituted with eight alternatives, including four nonpolar amino acid residues (F, I, M, and W) and four polar ones (K, N, Q, and T). The catalytic constants of the mutants toward Z-QPQ-pNA, Z-HPQ-pNA, Z-HPL-pNA, and Z-HPK-pNA were then determined. The turnover number (kcat) and Michaelis constant (KM) which reflects the binding affinity to the ground-state substrate, are detailed in Table 1. The specificity constants (kcat/KM), providing insight into the binding affinity to the transition-state substrate, are illustrated in Figure 3.
In the context of wild-type Bga1903, KM values range from 1.48 to 4.31 mM, while kcat/KM values range from 33.91 to 1.18 mM−1·sec−1. Notably, the peptidase exhibits a stronger binding affinity for Z-HPK-pNA, surpassing other tested substrates, in both ground-state and transition-state interactions. The substantial difference in kcat/KM values indicates that the enhanced catalysis toward Z-HPK-pNA stems from an overwhelming affinity to the transition state of the substrate. Conversely, Z-QPQ-pNA emerges as the least preferable substrate, suggesting room for improvement if Bga1903 is considered for use in celiac disease treatment. The significant difference in kcat/KM values between Z-QPQ-pNA (1.18 ± 0.07 mM−1·sec−1) and Z-HPQ-pNA (5.51 ± 0.91 mM−1·sec−1), aligning with specific activity data, primarily arises from their distinct kcat values. Accordingly, S3 also plays a pivotal role in the catalytic hydrolysis of the 26- and 33-mer peptides.
No detectable proteolytic activity toward Z-HPK-pNA was observed in any of the E380-substituted variants, underscoring the vital role of the negative charge associated with E380 in favoring lysine at the P1 position. Intriguingly, except for E380K, none of the mutants displays detectable proteolytic activity toward Z-HPL-pNA. The kcat/KM value of E380K dropped from 3.03 ± 0.68 to 1.44 ± 0.07 mM−1·sec−1 toward Z-HPL-pNA.
When Z-QPQ-pNA serves as the substrate, three mutations—E380W, E380K, and E380Q—significantly heighten the proteolytic activity of Bga1903. This enhancement is evident in the reduction of the KM value from 4.31 ± 0.21 to 2.31 ± 0.11, 2.02 ± 0.09, and 1.02 ± 0.02 mM, accompanied by respective increases in the kcat/KM value from 1.18 ± 0.07 to 4.38 ± 0.48, 7.35 ± 0.35, and 12.19 ± 0.48 mM−1·sec−1.
Conversely, with Z-HPQ-pNA as the substrate, most mutants display either reduced or similar kcat/KM values compared to the wild type. Notably, E380Q stands out by exhibiting the lowest KM value, i.e., 0.64 mM, among the variants. This change confers a 2.67-fold increase in kcat/KM on E380Q, compared to the wild type.
2.4. Cumulative effect of S387L with different E380 mutations on the enzymatic activity
A prior investigation uncovered that the S387L mutation augmented the specific constant, kcat/KM, toward Z-HPQ-pNA and significantly enhanced the breakdown of the 26-mer peptide. To assess whether the beneficial effects are accumulative, the impact of S387L mutation was re-evaluated in the current study on the background of E380K, E380W, and E380Q mutants as well as the wild-type enzyme. The resulting kcat and KM values are listed in Table 2, and the corresponding kcat/KM values for each variant are illustrated in Figure 4.
Without interfering with the specificity constant toward Z-HPK-pNA, S387L mutation increases the kcat/KM value by approximately 9, 2, and 3 folds toward Z-QPQ-pNA, Z-HPQ-pNA, and Z-HPL-pNA, respectively. It's worth noting that the increase in kcat/KM is primarily driven by reductions in the KM value rather than increases in the kcat value.
The mutant E380K/S387L has the kcat/KM value of 12.2 mM−1·sec−1 toward Z-QPQ-pNA, which only increases slightly (~15%) compared to the mutant S387L. The kcat/KM value reduced from 10.82 ± 0.51 to 4.47 ± 0.50 mM−1·sec−1 if the substrate was replaced with Z-HPQ-pNA. From the perspective of eliminating QPQ motifs, the introduction of E380K substitution to the mutant S387L is not beneficial, since there is no obvious cumulative effect. However, it retains one of the properties from mutant E380K, in which no activity was detected utilizing Z-HPK-pNA and Z-HPL-pNA as substrates.
The mutant E380W/S387L exhibits the kcat/KM value of 20.40 ± 0.92 mM−1·sec−1 toward Z-QPQ-pNA, which is an approximately 2-fold and 5-fold increase compared to mutants S387L and E380W, respectively. As for Z-HPQ-pNA, the kcat/KM value increases from 10.82 ± 0.51 to 17.09 ± 1.91 when the E380W mutation is introduced to mutant S387L. The rise in kcat/KM values for Z-QPQ-pNA and Z-HPQ-pNA is contributed by their individual KM values, both of which decrease by nearly half in contrast with their S387L counterparts. Same as mutant E380K/S387L, no hydrolytic activity toward HPK-pNA and Z-HPL-pNA is detected.
The mutant E380Q/S387L displays the highest kcat/KM value toward Z-QPQ-pNA, i.e., 44.34 ± 1.86 mM−1·sec−1, an approximately 4-fold increase when compared to either mutants S387L or E380Q. When the substrate is Z-HPQ-pNA, the kcat/KM value changes to 35.78 ± 3.93, which is two to four times higher than those of E380Q and S387L, respectively. The exceptionally high kcat/KM values for both Z-QPQ-pNA and Z-HPQ-pNA are attributed to the small KM values among all variants. Although the mutant can hydrolyze Z-HPL-pNA, the kcat/KM value is low compared to that of using Z-HPQ-pNA and Z-QPQ-pNA.
In summary, mutants E380W/S387L and E380Q/S387L have much higher specificity constant against Z-QPQ-pNA and Z-HPQ-pNA, demonstrating the combined impact of double-site mutations. Moreover, the pronounced preference for Z-HPK-pNA over Z-QPQ-pNA, which was originally seen in the wild type, has shifted in the opposite direction due to the double mutation E380Q/S387L.
2.5. Molecular docking models of Bga1903s and QPQ peptides
To elucidate the underlying mechanism responsible for the increased specificity constant resulting from double mutations (E380W/S387L or E380Q/S387L), computational docking models of peptidase Bga1903 with a QPQ-peptide substrate were constructed using HADDOCK software. Interactions between the QPQ peptide and different versions of Bga1903 were plotted according to the simulation provided by LigPlot+ software, as depicted in Figure 5.
In the wild-type Bga1903, as depicted in Figure 5A, a charged hydrogen bond is formed between the OE2 atom of E380 and the NE2 atom of glutamine at the P1 position. Additional hydrogen bonds are also established between the backbones of the QPQ peptide and the P3 subsites (S348 and G350), acting as anchor points to further stabilize the QPQ substrate.
In the case of mutant S387L, as illustrated in Figure 5B, the presence of L387 pushes the side chain of glutamine (P1) closer to the backbone of G378-N379-E380; consequently, two more hydrogen bonds are formed between the P1 residue and the subsites when compared to the wild-type Bga1903. This may account for the improved binding affinity for the QPQ substrate in both the ground state and transition state.
For mutant E380W/S387L, as shown in Figure 5C, there's a noteworthy rotation in the side chain of glutamine (P1) when compared to the wild-type scenario. This rotation results in the formation of two hydrogen bonds. One of these bonds is directed toward the NE1 atom on the side chain of W380, while the other connects with the N atom on the backbone of N379. Additionally, the carboxyl end of glutamine (P1) establishes two more hydrogen bonds, one with the ND2 atom of N379 and the other with the N atom on the backbone of S449. These interactions further enhance the substrate's affinity.
As for mutant E380Q/S387L, as presented in Figure 5D, the rotation of the glutamine side chain (P1) is similar to that of the E380W/S387L mutant, and a hydrogen bond forms between Q380 and the glutamine at the P1 position of the QPQ substrate. An additional hydrogen bond is formed between Q262 and the P3 residue of the QPQ substrate, compared to the E380W/S387L scenario. This could explain why mutant E380Q/S387L exhibits the highest specificity constant toward the QPQ substrate among various mutated versions of Bga1903.
2.6. Hydrolysis of pro-immunogenic peptides by mutant E380W/S387L and E380Q/S387L.
To verify their hydrolytic activity toward the 26-mer or 33-mer peptide, wild-type Bga1903, mutant E380W/S387L, and mutant E380Q/S387L were incubated with the peptide at pH 6.0. After incubation at 37 °C for 2 hours, the hydrolysate was subjected to RP-HPLC for analysis. Overall, the 26-mer peptide is more susceptible to hydrolysis compared to the 33-mer peptide. Both the double-mutant variants exhibit stronger proteolytic activity than the wild type toward both peptides. Particularly noteworthy is the superior efficiency of mutant E380Q/S387L in hydrolyzing the 33-mer peptide as compared to the wild type and mutant E380W/S387L.
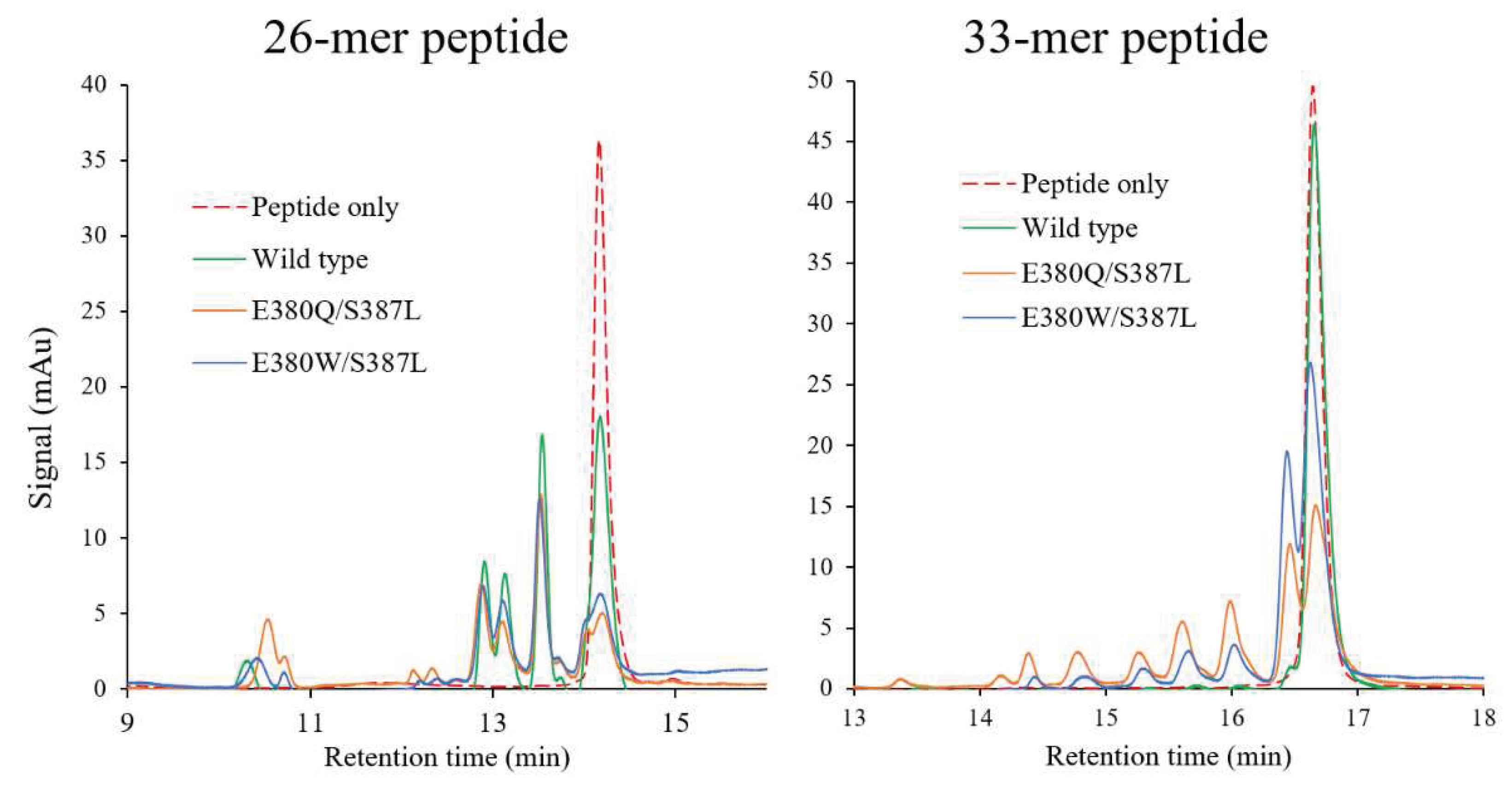
Figure 6.
Hydrolysis of pro-immunogenic peptides by wild-type Bga1903, mutant E380Q/S387L, and mutant E380W/S387L. The 26-mer or 33-mer peptide (1 mg/mL) was incubated with or without the peptidase (50 μg/mL) at pH 6.0 for 2 hours, and the digested fragments were analyzed by RP-HPLC.
Figure 6.
Hydrolysis of pro-immunogenic peptides by wild-type Bga1903, mutant E380Q/S387L, and mutant E380W/S387L. The 26-mer or 33-mer peptide (1 mg/mL) was incubated with or without the peptidase (50 μg/mL) at pH 6.0 for 2 hours, and the digested fragments were analyzed by RP-HPLC.
3. Discussion
Given their potential as supporting therapeutic agents for celiac disease, proteases that can effectively degrade toxic peptides derived from gluten have been extensively investigated [18,26]. These gluten-degrading proteases have been sourced from diverse origins and are classified such as aspartic protease, cysteine protease, glutamate protease, or serine protease [24]. Notable examples include nepenthesin (an aspartic protease from Nepenthes × ventrata) [27], neprosin (a glutamate protease from Nepenthes × ventrata) [29], RmuAP1 (an aspartic protease from Rhodotorula mucilaginosa) [30], EP-B2 (a cysteine protease from barley) [31], AN-PEP (a serine protease of the S28 family from Aspergillus niger) [32], SC-PEP (a serine protease of the S9 family from Sphigomonas capsulate) [33], subtilisin Carlsberg (a serine protease of the S8 family from Bacillus licheniformis) [34], Rmep (a serine protease of the S8 family from Rothia mucilaginosa) [35], Bga1903 (a serine protease of the S8 family from Burkholderia gladioli) [24,25], and Kuma (serine proteases of the S53 family serially engineered from kumamolisin of Alicyclobacillus sendaiensis) [36,37].
Among these, Latiglutenase, a combination of recombinant EP-B2 and SC-PEP, stands out as it has undergone the most extensive investigation in human clinical trials up to the present [23,38,39,40]. The phase II clinical trials for this drug yielded cautiously optimistic results, suggesting promising therapeutic potential. EP-B2, when presented as the zymogen form, exhibits optimal activity at pH 4.5 and displays a notable specificity for glutamine at the P1 position [31]. In contrast, SC-PEP functions as a prolyl endopeptidase and remains active in the pH range of 6-7 [33]. Consequently, these two glutenases complement each other not only in their substrate preferences but also in their action sites, encompassing both the stomach and duodenum where the proteolytic reactions occur.
In the present study, site-directed mutagenesis was employed to modify the S1 subsite of Bga1903. The importance of E380 in determining the peptidase’s preferential activity for the peptidyl substrate with lysine at the P1 position was established. The peptidase’s activity towards Z-HPK-pNA is not supported by any substitution for E380 in this study. However, a notable improvement in the peptidase’s activity towards Z-QPQ-pNA is observed upon the replacement of E380 with glutamine. When compared to the wild type, the substrate preference of the E380Q mutant undergoes a reversal from the HPK peptide, which is the most favored substrate by the wild type, to the QPQ and HPQ peptides, which are disfavored by the wild type. The introduction of the S387L mutation further heightens the peptidase’s preference for QPQ (with the kcat/KM value of 44.34 ± 1.86 mM−1·sec−1) and HPQ (with the kcat/KM value of 35.78 ± 3.93 mM−1·sec−1). The approximately 44-fold increase in the kcat/KM value towards Z-QPQ-pNA should account for the enhanced activity of E380Q/S387L in the hydrolysis of the 22- and 33-mer peptides. In summary, the approach of reshaping the catalytic subsites of Bga1903 has proven to be useful in enhancing the peptidase’s activity for the removal of gluten-derived toxic peptides. In combination with other peptidases with complementary properties, E380Q/S387L can serve as part of a potential glutenase preparation for the treatment of celiac disease.
4. Materials and Methods
4.1. Chemicals and reagents
Chromogenic peptidyl substrates, including Z-HPK-pNA, Z-HPQ-pNA, Z-HPL-pNA, Z-HPY-pNA, Z-HPF-pNA, Z-HPE-pNA, and Z-QPQ-pNA were synthesized by Kelowna International Scientific (Taipei, Taiwan). Q5 site-directed mutagenesis kit was purchased from New England Biolabs (Ipswich, MA). Primers for PCR used in mutagenesis were synthesized by Genomics (New Taipei, Taiwan), and listed in Table 3. The 26- and 33-mer immunogenic peptides were chemically synthesized by Mission Biotech (Taipei, Taiwan).
4.2. E. coli strains and site-directed mutagenesis
E. coli BL21(DE3) pLysS was purchased from Merck (Rahway, NJ), and utilized as the host for recombinant peptidase production. The process of obtaining plasmid pET-Bga1903+ was mentioned previously [24]. In brief, the Bga1903 gene cloned from Burkholderia gladioli was inserted into plasmid pET-Duet1, and the resulting plasmid pET-Bga1903 was chemically resynthesized to a codon-optimized version, pET-Bga1903+. The substitutions of amino acids were performed through PCR-based site-directed mutagenesis on plasmid pET-Bga1903+ utilizing a Q5 site-directed mutagenesis kit.
4.3. Protein expression and purification
Recombinant Bga1903 was produced and purified as described previously [24]. In brief, E. coli BL21(DE3) pLysS was cultured overnight with Lysogeny broth (LB, containing 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl). Fresh LB medium containing 100 μg/mL ampicillin inside a shake flask was inoculated with overnight bacterial culture, and placed inside a shaking incubator at 37 °C at 200 rpm until the OD600 reached approximately 1.0. Administrated with 1 mM final concentration of isopropyl β-D-1-thiogalactopyranoside, the cell culture was incubated for 18 hours at 28 °C at 200 rpm. To harvest secreted Bga1903 within the medium, the bacterial broth was centrifuged at 10,000× g for 15 minutes at 4 °C, and the cell pellet was discarded afterward. Bga1903 inside the culture medium was harvested and subsequently purified by using a column packed with Qiagen Ni-NTA agarose resin (Venlo, Netherlands). The purified protein was obtained by concentrating elution fractions using a 10 kDa amicon ultra-4 centrifugal filter unit (Merck, Darmstadt, Germany), and preserved in the buffer containing 20 mM Tris [pH 8.0] and 500 mM NaCl. The concentration of the purified protein was measured using Bradford protein assay reagent (Thermo Fisher Scientific, Waltham, MA) with bovine serum albumin as the standard.
4.4. Enzyme activity assay utilizing chromogenic peptidyl substrates
The enzymatic activity toward various chromogenic peptidyl substrates was measured according to the release of p-nitroaniline from the substrates. One activity unit (U) was defined as the release of 1 nmol p-nitroaniline per second at the indicated conditions. Unless specified, the reaction containing 0.5 mM peptidyl substrates, 12.5 μg/mL of the purified enzyme, 1.5% (v/v) Triton X-100, and 40 mM citrate-phosphate buffer (pH 6.0) was performed at 37 °C. The catalytic rate was calculated following the time-course increment of optical density at 405 nm and based on the molar extinction coefficient of p-nitroaniline, 9960 M−1 ·cm−1. For steady-state kinetics, the concentration of chromogenic peptidyl substrates was subject to change when the reaction was performed. All reactions were performed in triplicate.
4.5. Computational evaluation of the interaction between Bga1903s and QPQ peptide
The tertiary structure determination of Bga1903 by X-ray crystallography was described previously [25]. Bga1903’s atomic coordinates and structure factors were deposited in the Protein Data Bank with PDB ID code 7W2A. Interactions of the representative motif of 26-mer peptide, QPQ, and mutated variants of Bga1903 were built according to the suggestion of FoldX software [41], and their computational dockings were performed with HADDOCK2.4 website [42,43]. The interactions between QPQ peptides and Bga1903 molecules were predicted by Ligplot+ [44].
4.6. Hydrolysis of the 26- and 33-mer pro-immunogenic peptides
The hydrolysis of the 26- and 33-mer pro-immunogenic peptides was carried out by incubating the peptide (1 mg/mL) and the purified peptidase (50 μg/mL) in 50 mM citrate-phosphate buffer, pH 6.0 at 37 °C for 2 hours, followed by incubation at 95 °C for 5 min to deactivate the peptidase. The degree of the peptide degradation was analyzed with a Gilson HPLC system (Middleton, WI) using a C18 column (Ascentis Express 25 cm × 4.6 mm, 5 µm, Supelco, Bellefonte, PA). After sample injection, the C18 column was subjected to H2O with 0.1% (v/v) trifluoroacetic acid for 5 minutes and then proceeded with linear gradient elution mode from 0–80% acetonitrile with 0.1% (v/v) trifluoroacetic acid for 20 minutes. The flow rate was set at a constant 1 mL/min, and the elution was monitored at 230 nm.
Author Contributions
Conceptualization, M. M.; methodology, Y.-Y. L.; software, Y.-Y. L.; validation, M. M. and Y.-Y. L.; formal analysis, Y.-Y. L. and R.-L. Y.; investigation, Y.-Y. L. and R.-L. Y.; resources, M. M.; data curation, Y.-Y. L.; writing—original draft preparation, Y.-Y. L.; writing—review and editing, M. M.; visualization, Y.-Y. L.; supervision, M. M.; project administration, M. M.; funding acquisition, M. M.
Funding
This research was funded by the National Science and Technology Council, Taiwan, ROC, grant number 109-2313-B-005-018-MY3.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dewar, D. H.; Ciclitira, P. J. Clinical features and diagnosis of celiac disease. Gastroenterology 2005, 128, S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Tollefsen, S.; Arentz-Hansen, H.; Fleckenstein, B.; Molberg, Ø.; Ráki, M.; Kwok, W. W.; Jung, G.; Lundin, K. E. A.; Sollid, L. M. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J. Clin. Invest. 2006, 116, 2226–2236. [Google Scholar] [CrossRef]
- Singh, P.; Arora, S.; Lal, S.; Strand, T. A.; Makharia, G. K. Risk of Celiac Disease in the First- and Second-Degree Relatives of Patients With Celiac Disease: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2015, 110, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Gasbarrini, A.; Cammarota, G. Endoscopic tools for the diagnosis and evaluation of celiac disease. World J. Gastroenterol. 2013, 19, 8562–8570. [Google Scholar] [CrossRef]
- Schwertz, E.; Kahlenberg, F.; Sack, U.; Richter, T.; Stern, M.; Conrad, K.; Zimmer, K. P.; Mothes, T. Serologic assay based on gliadin-related nonapeptides as a highly sensitive and specific diagnostic aid in celiac disease. Clin. Chem. 2004, 50, 2370–2375. [Google Scholar] [CrossRef]
- Shewry, P. R.; Tatham, A. S.; Forde, J.; Kreis, M.; Miflin, B. J. The classification and nomenclature of wheat gluten proteins: A reassessment. J. Cereal Sci. 1986, 4, 97–106. [Google Scholar] [CrossRef]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G. M.; Sollid, L. M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Qiao, S. W.; Arentz-Hansen, H.; Molberg, Ø.; Gray, G. M.; Sollid, L. M.; Khosla, C. Identification and analysis of multivalent proteolytically resistant peptides from gluten: implications for celiac sprue. J. Proteome Res. 2005, 4, 1732–1741. [Google Scholar] [CrossRef]
- Molberg, Ø.; McAdam, S. N.; Körner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Norén, O.; Roepstorff, P.; Lundin, K. E.; Sjöström, H.; Sollid, L. M. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef]
- Unsworth, D. J.; Kieffer, M.; Holborow, E. J.; Coombs, R. R.; Walker-Smith, J. A. IgA anti-gliadin antibodies in coeliac disease. Clin. Exp. Immunol. 1981, 46, 286–293. [Google Scholar]
- Mankai, A.; Sakly, W.; Landolsi, H.; Gueddah, L.; Sriha, B.; Ayadi, A.; Sfar, M. T.; Skandrani, K.; Harbi, A.; Essoussi, A. S.; Korbi, S.; Fabien, N.; Jeddi, M.; Ghedira, I. Tissue transglutaminase antibodies in celiac disease, comparison of an enzyme linked immunosorbent assay and a dot blot assay. Pathol. Biol. (Paris) 2005, 53, 204–209. [Google Scholar] [CrossRef]
- Nurminskaya, M. V.; Belkin, A. M. Cellular Functions of Tissue Transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar]
- Uhde, M.; Ajamian, M.; Caio, G.; De Giorgio, R.; Indart, A.; Green, P. H.; Verna, E. C.; Volta, U.; Alaedini, A. Intestinal cell damage and systemic immune activation in individuals reporting sensitivity to wheat in the absence of coeliac disease. Gut. 2016, 65, 1930–1937. [Google Scholar] [CrossRef]
- Singh, J.; Whelan, K. Limited availability and higher cost of gluten-free foods. J. Hum. Nutr. Diet. 2011, 24, 479–486. [Google Scholar] [CrossRef]
- Lerner, B. A.; Phan Vo, L. T.; Yates, S.; Rundle, A. G.; Green, P. H. R.; Lebwohl, B. Detection of gluten in gluten-free labeled restaurant food: Analysis of crowd-sourced data. Am. J. Gastroenterol. 2019, 114, 792–797. [Google Scholar] [CrossRef]
- Christophersen, A.; Risnes, L. F.; Dahal-Koirala, S.; Sollid, L. M. Therapeutic and Diagnostic Implications of T Cell Scarring in Celiac Disease and Beyond. Trends Mol. Med. 2019, 25, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A. New therapeutic strategies for celiac disease. Autoimmun. Rev. 2010, 9, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M. T.; Khosla, C. Oral enzyme therapy for celiac sprue. Methods Enzymol. 2012, 502, 241–271. [Google Scholar]
- Scherf, K. A.; Wieser, H.; Koehler, P. Novel approaches for enzymatic gluten degradation to create high-quality gluten-free products. Food Res. Int. 2018, 110, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Caputo, I.; Lepretti, M.; Martucciello, S.; Esposito, C. Enzymatic strategies to detoxify gluten: Implications for celiac disease. Enzyme Res. 2010, 2010, 174354. [Google Scholar] [CrossRef]
- Pultz, I. S.; Hill, M.; Vitanza, J. M.; Wolf, C.; Saaby, L.; Liu, T.; Winkle, P.; Leffler, D. A. Gluten Degradation, Pharmacokinetics, Safety, and Tolerability of TAK-062, an Engineered Enzyme to Treat Celiac Disease. Gastroenterology 2021, 161, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.; Garber, M. E.; Spencer, A. G.; Botwick, W.; Kumar, P.; Williams, R. N.; Kozuka, K.; Shreeniwas, R.; Pratha, V.; Adelman, D. C. Safety, tolerability, and activity of ALV003: results from two phase 1 single, escalating-dose clinical trials. Dig. Dis. Sci. 2012, 57, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Murray, J. A.; Syage, J. A.; Wu, T. T.; Dickason, M. A.; Ramos, A. G.; Van Dyke, C.; Horwath, I.; Lavin, P. T.; Mäki, M.; Hujoel, I.; Papadakis, K. A.; Bledsoe, A. C.; Khosla, C.; Sealey-Voyksner, J. A. Latiglutenase Protects the Mucosa and Attenuates Symptom Severity in Patients With Celiac Disease Exposed to a Gluten Challenge. Gastroenterology 2022, 163, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Y.; Lee, C. C.; Hsu, J. H.; Leu, W. M.; Meng, M. Efficient hydrolysis of gluten-derived celiac disease-triggering immunogenic peptides by a bacterial serine protease from Burkholderia gladioli. Biomolecules 2021, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Lin, I. C.; Chen, P.-C.; Lee, C.-C.; Meng, M. Crystal structure of a Burkholderia peptidase and modification of the substrate-binding site for enhanced hydrolytic activity toward gluten-derived pro-immunogenic peptides. Int. J. Biol. Macromol. 2022, 222, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Helmerhorst, E. J.; Darwish, G.; Blumenkranz, G.; Schuppan, D. Gluten degrading enzymes for treatment of celiac disease. Nutrients 2020, 12, 2095. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Yang, M.; Lee, L.; Zhang, Y.; Sheff, J. G.; Sensen, C. W.; Mrazek, H.; Halada, P.; Man, P.; McCarville, J. L.; Verdu, E. F.; Schriemer, D. C. Addressing proteolytic efficiency in enzymatic degradation therapy for celiac disease. Sci. Rep. 2016, 6, 30980. [Google Scholar] [CrossRef] [PubMed]
- Ting, T.-Y.; Baharin, A.; Ramzi, A. B.; Ng, C.-L.; Goh, H.-H. Neprosin belongs to a new family of glutamic peptidase based on in silico evidence. Plant Physiol. Biochem. 2022, 183, 23–35. [Google Scholar] [CrossRef] [PubMed]
- del Amo-Maestro, L.; Mendes, S. R.; Rodríguez-Banqueri, A.; Garzon-Flores, L.; Girbal, M.; Rodríguez-Lagunas, M. J.; Guevara, T.; Franch, À.; Pérez-Cano, F. J.; Eckhard, U.; Gomis-Rüth, F. X. Molecular and in vivo studies of a glutamate-class prolyl-endopeptidase for coeliac disease therapy. Nat. Commun. 2022, 13, 4446. [Google Scholar] [CrossRef]
- Zhang, Y. H.; Leu, W. M.; Meng, M. Hydrolysis of Gluten-Derived Celiac Disease-Triggering Peptides across a Broad pH Range by RmuAP1: A Novel Aspartic Peptidase Isolated from Rhodotorula mucilaginosa. J. Agric. Food Chem. 2023, 71, 17202–17213. [Google Scholar] [CrossRef]
- Bethune, M. T.; Strop, P.; Tang, Y.; Sollid, L. M.; Khosla, C. Heterologous expression, purification, refolding, and structural-functional characterization of EP-B2, a self-activating barley cysteine endoprotease. Chem. Biol. 2006, 13, 637–647. [Google Scholar] [CrossRef]
- Akeroyd, M.; van Zandycke, S.; den Hartog, J.; Mutsaers, J.; Edens, L.; van den Berg, M.; Christis, C. AN-PEP, Proline-Specific Endopeptidase, Degrades All Known Immunostimulatory Gluten Peptides in Beer Made from Barley Malt. J. Am. Soc. Brew. Chem. 2018, 74, 91–99. [Google Scholar] [CrossRef]
- Shan, L.; Marti, T.; Sollid, L. M.; Gray, G. M.; Khosla, C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: implications for coeliac sprue. Biochem. J. 2004, 383, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Darwish, G.; Helmerhorst, E. J.; Schuppan, D.; Oppenheim, F. G.; Wei, G. Pharmaceutically modified subtilisins withstand acidic conditions and effectively degrade gluten in vivo. Sci. Rep. 2019, 9, 7505. [Google Scholar] [CrossRef]
- Wei, G.; Tian, N.; Siezen, R.; Schuppan, D.; Helmerhorst, E. J. Identification of food-grade subtilisins as gluten-degrading enzymes to treat celiac disease. Am. J. Physiol.: Gastrointest. Liver Physiol. 2016, 311, G571–G580. [Google Scholar] [CrossRef]
- Okubo, A.; Li, M.; Ashida, M.; Oyama, H.; Gustchina, A.; Oda, K.; Dunn, B. M.; Wlodawer, A.; Nakayama, T. Processing, catalytic activity and crystal structures of kumamolisin-As with an engineered active site. FEBS J. 2006, 273, 2563–2576. [Google Scholar] [CrossRef]
- Wolf, C.; Siegel, J. B.; Tinberg, C.; Camarca, A.; Gianfrani, C.; Paski, S.; Guan, R.; Montelione, G.; Baker, D.; Pultz, I. S. Engineering of Kuma030: A Gliadin Peptidase That Rapidly Degrades Immunogenic Gliadin Peptides in Gastric Conditions. J. Am. Chem. Soc. 2015, 137, 13106–13113. [Google Scholar] [CrossRef] [PubMed]
- Lähdeaho, M. L.; Kaukinen, K.; Laurila, K.; Vuotikka, P.; Koivurova, O. P.; Kärjä-Lahdensuu, T.; Marcantonio, A.; Adelman, D. C.; Mäki, M. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 2014, 146, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Murray, J. A.; Kelly, C. P.; Green, P. H. R.; Marcantonio, A.; Wu, T.-T.; Mäki, M.; Adelman, D. C.; Ansari, S.; Ayub, K.; Basile, A.; et al. No Difference Between Latiglutenase and Placebo in Reducing Villous Atrophy or Improving Symptoms in Patients With Symptomatic Celiac Disease. Gastroenterology 2017, 152, 787–798. [Google Scholar] [CrossRef]
- Syage, J. A.; Murray, J. A.; Green, P. H. R.; Khosla, C. Latiglutenase Improves Symptoms in Seropositive Celiac Disease Patients While on a Gluten-Free Diet. Dig. Dis. Sci. 2017, 62, 2428–2432. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R. V.; Koukos, P. I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. M. J. J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G. C. P.; Rodrigues, J. P. G. L. M.; Trellet, M.; Schmitz, C.; Kastritis, P. L.; Karaca, E.; Melquiond, A. S. J.; van Dijk, M.; de Vries, S. J.; Bonvin, A. M. J. J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R. A.; Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
Figure 1.
The docking model featuring the active site of Bga1903 complexed with QPQ tripeptide in stereo view. The coloring scheme designates the surface of the S1, S2, S3, and S1’ sites as cyan, magenta, orange, and wheat, respectively. Residues E380 and S387, targeted for mutagenesis in the present study, are colored elementally (C: cyan; N: blue; O: red). The catalytic triad composed of D203, H277, and S449 is colored by elements (C: yellow; N: blue; O: red).
Figure 1.
The docking model featuring the active site of Bga1903 complexed with QPQ tripeptide in stereo view. The coloring scheme designates the surface of the S1, S2, S3, and S1’ sites as cyan, magenta, orange, and wheat, respectively. Residues E380 and S387, targeted for mutagenesis in the present study, are colored elementally (C: cyan; N: blue; O: red). The catalytic triad composed of D203, H277, and S449 is colored by elements (C: yellow; N: blue; O: red).
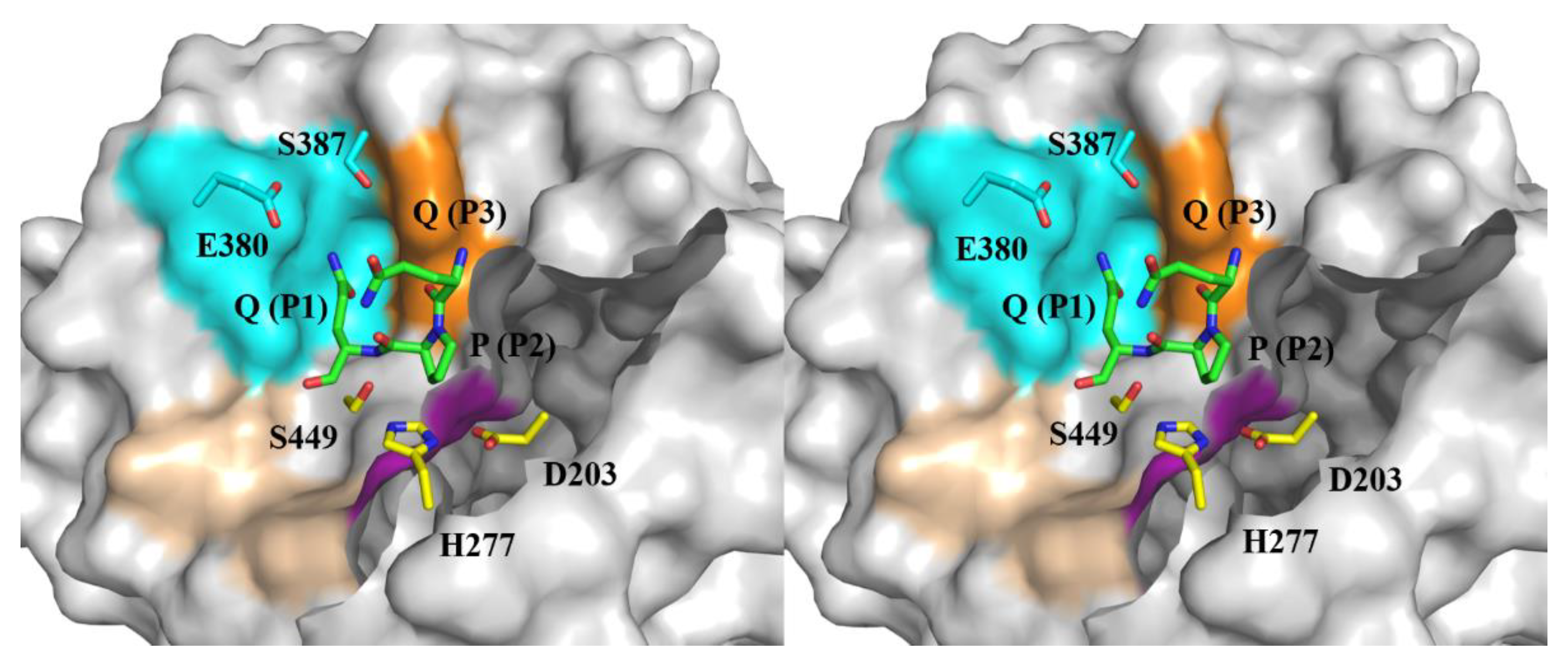
Figure 2.
The specific activity of Bga1903 toward chromogenic tripeptidyl substrates. The reactions were performed in triplicate at 37 °C, pH 6.0. The activity rate was calculated based on the release of p-nitroaniline as described in Materials and Methods.
Figure 2.
The specific activity of Bga1903 toward chromogenic tripeptidyl substrates. The reactions were performed in triplicate at 37 °C, pH 6.0. The activity rate was calculated based on the release of p-nitroaniline as described in Materials and Methods.
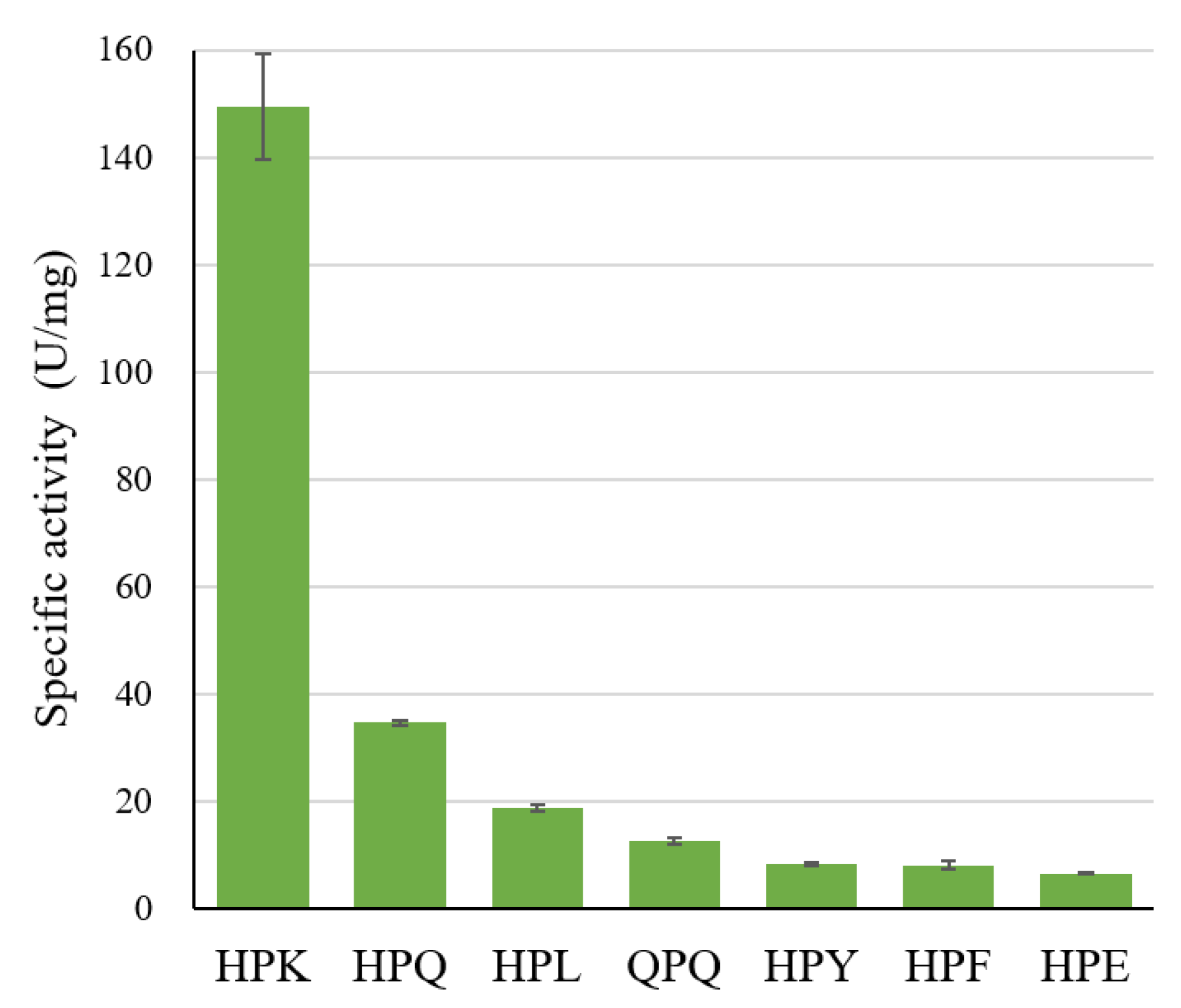
Figure 3.
The specificity constants of Bga1903 and E380 substituted mutants toward selected substrates. The specificity constants (kcat/KM) toward Z-QPQ-pNA (green), Z-HPQ-pNA (orange), Z-HPL-pNA (blue), and Z-HPK-pNA (gray) were determined according to the Michaelis-Menten model.
Figure 3.
The specificity constants of Bga1903 and E380 substituted mutants toward selected substrates. The specificity constants (kcat/KM) toward Z-QPQ-pNA (green), Z-HPQ-pNA (orange), Z-HPL-pNA (blue), and Z-HPK-pNA (gray) were determined according to the Michaelis-Menten model.
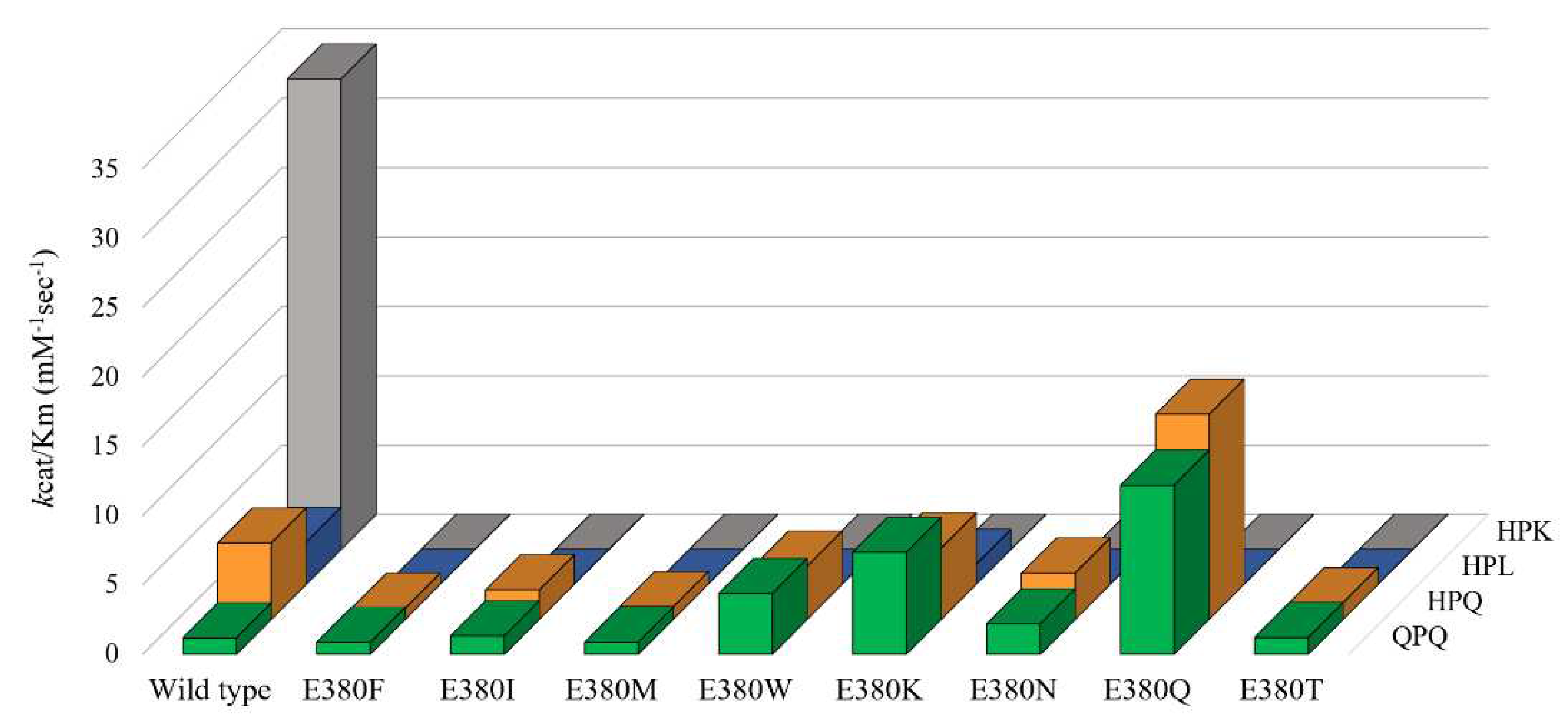
Figure 4.
Cumulative effects of mutations at S387 and E380. The specificity constants of Bga1903 and the indicated variants toward selected peptidyl substrates, including Z-QPQ-pNA (green), Z-HPQ-pNA (orange), Z-HPL-pNA (blue), and Z-HPK-pNA (gray), were determined according to the Michaelis-Menten model.
Figure 4.
Cumulative effects of mutations at S387 and E380. The specificity constants of Bga1903 and the indicated variants toward selected peptidyl substrates, including Z-QPQ-pNA (green), Z-HPQ-pNA (orange), Z-HPL-pNA (blue), and Z-HPK-pNA (gray), were determined according to the Michaelis-Menten model.
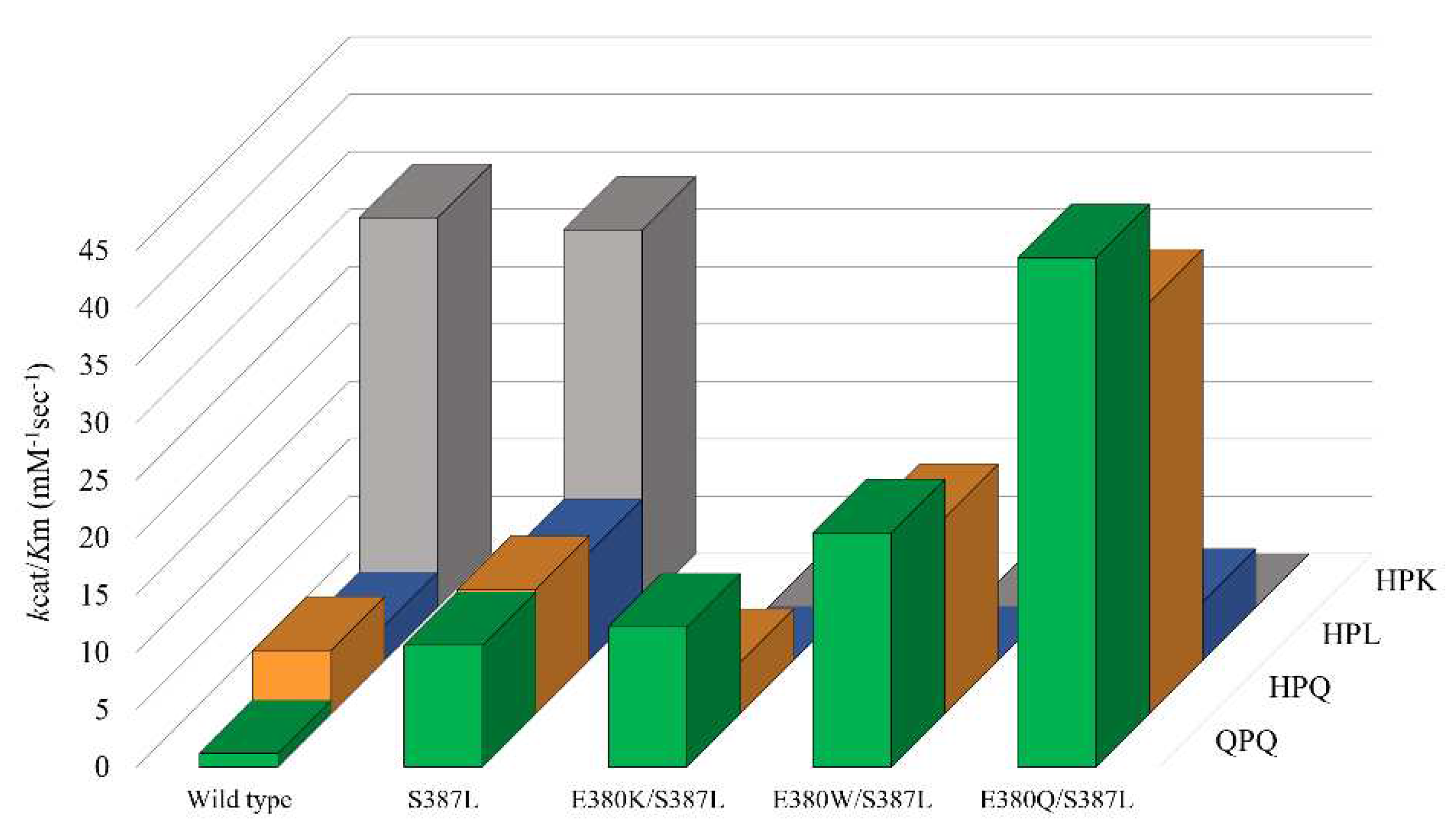
Figure 5.
The molecular docking models of the QPQ peptides with different variants of Bga1903s, including (A) wild-type Bga1903, (B) mutant S387L, (C) mutant E380W/S387L, and (D) E380Q/S387L. The QPQ peptides located at S1-S3 positions are colored elementally (C: green; N: blue; O: red), and spatially neighboring residues from the peptidase are colored by elements (C: white; N: blue; O: red). The catalytic triad (D203, H277, S449) is colored by elements (C: magenta; N: blue; O: red). H-bonds (hydrogen bonds) between N atoms and C atoms are illustrated as yellow-dotted lines.
Figure 5.
The molecular docking models of the QPQ peptides with different variants of Bga1903s, including (A) wild-type Bga1903, (B) mutant S387L, (C) mutant E380W/S387L, and (D) E380Q/S387L. The QPQ peptides located at S1-S3 positions are colored elementally (C: green; N: blue; O: red), and spatially neighboring residues from the peptidase are colored by elements (C: white; N: blue; O: red). The catalytic triad (D203, H277, S449) is colored by elements (C: magenta; N: blue; O: red). H-bonds (hydrogen bonds) between N atoms and C atoms are illustrated as yellow-dotted lines.
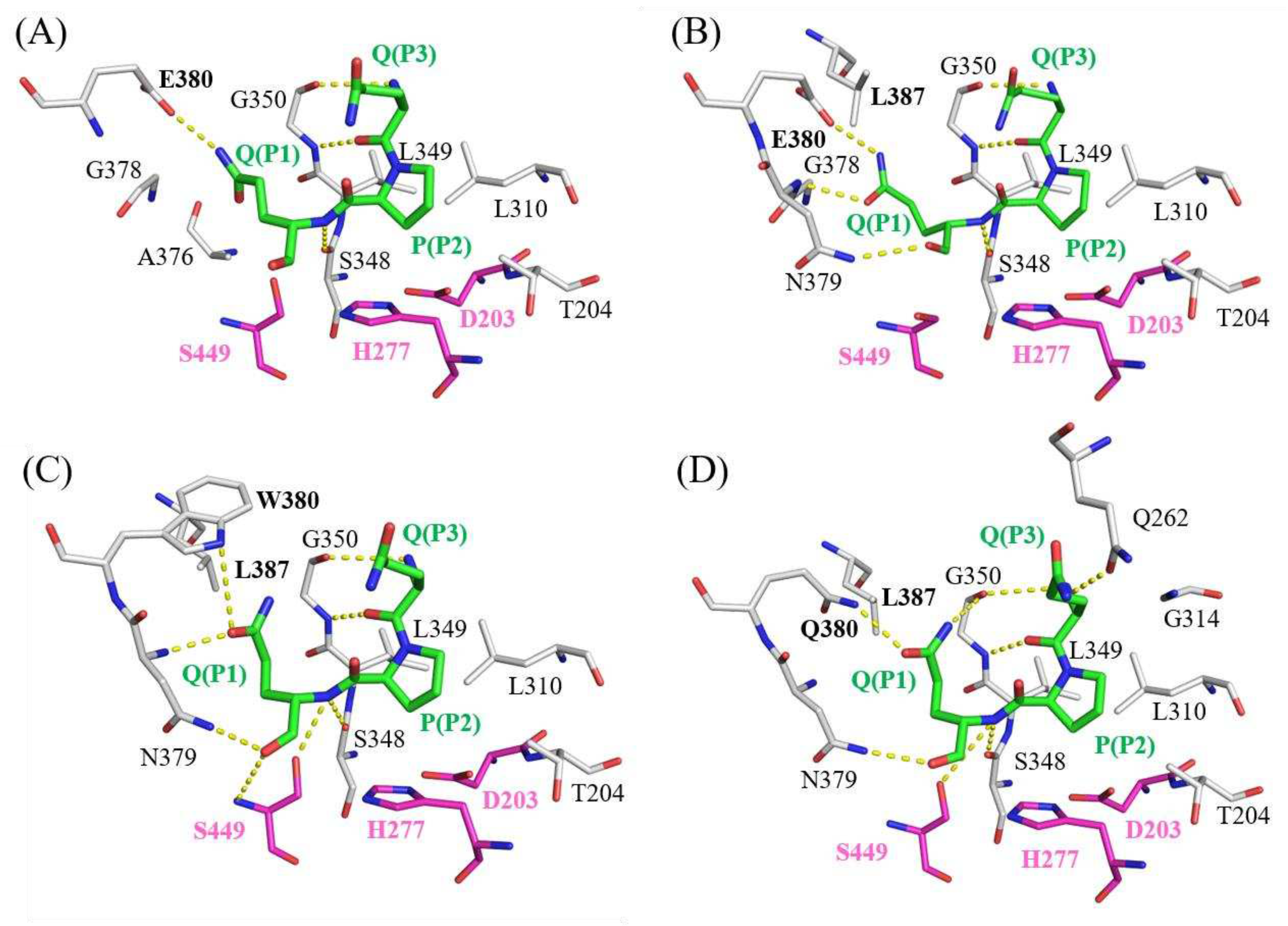

Table 1.
Mutational effects of E380 substitution on the kinetic constants of selected peptidyl substrates.
Table 1.
Mutational effects of E380 substitution on the kinetic constants of selected peptidyl substrates.
| Variants | QPQ | HPQ | HPL | HPK | ||||
|---|---|---|---|---|---|---|---|---|
| kcat | KM | kcat | KM | kcat | KM | kcat | KM | |
| (sec−1) | (mM) | (sec−1) | (mM) | (sec−1) | (mM) | (sec−1) | (mM) | |
| Wild type | 5.08 ± 0.15 | 4.31 ± 0.21 | 22.60 ± 0.50 | 4.10 ± 0.11 | 9.20 ± 1.22 | 3.04 ± 0.55 | 50.18 ± 2.96 | 1.48 ± 0.17 |
| E380F | 3.61 ± 0.13 | 4.15 ± 0.30 | 4.45 ± 0.54 | 5.64 ± 1.11 | BDL* | BDL* | BDL* | BDL* |
| E380I | 10.18 ± 0.96 | 7.71 ± 1.03 | 5.81 ± 0.46 | 2.75 ± 0.37 | BDL* | BDL* | BDL* | BDL* |
| E380M | 6.93 ± 1.15 | 8.14 ± 2.16 | 7.54 ± 0.73 | 8.38 ± 0.97 | BDL* | BDL* | BDL* | BDL* |
| E380W | 10.19 ± 0.41 | 2.31 ± 0.11 | 8.65 ± 0.92 | 2.27 ± 0.38 | BDL* | BDL* | BDL* | BDL* |
| E380K | 14.81 ± 0.25 | 2.02 ± 0.09 | 9.79 ± 0.15 | 1.91 ± 0.04 | 0.95 ± 0.02 | 0.66 ± 0.03 | BDL* | BDL* |
| E380N | 18.44 ± 1.85 | 8.42 ± 1.05 | 9.51 ± 0.55 | 2.85 ± 0.24 | BDL* | BDL* | BDL* | BDL* |
| E380Q | 12.50 ± 0.42 | 1.02 ± 0.02 | 9.43 ± 0.38 | 0.64 ± 0.03 | BDL* | BDL* | BDL* | BDL* |
| E380T | 16.67 ± 2.76 | 13.76 ± 2.58 | 35.33 ± 2.64 | 29.72 ± 2.26 | BDL* | BDL* | BDL* | BDL* |
* BDL stands for below detection limit.
Table 2.
Mutational effects of double mutations at E380 and S387 on selected peptidyl substrates.
| Variants | QPQ | HPQ | HPL | HPK | ||||
|---|---|---|---|---|---|---|---|---|
| kcat | KM | kcat | KM | kcat | KM | kcat | KM | |
| (sec−1) | (mM) | (sec−1) | (mM) | (sec−1) | (mM) | (sec−1) | (mM) | |
| Wild type | 5.08 ± 0.15 | 4.31 ± 0.21 | 22.60 ± 0.50 | 4.10 ± 0.11 | 9.20 ± 1.22 | 3.04 ± 0.55 | 50.18 ± 2.96 | 1.48 ± 0.17 |
| S387L | 9.87 ± 0.16 | 0.93 ± 0.02 | 9.96 ± 0.18 | 0.92 ± 0.04 | 6.74 ± 0.19 | 0.72 ± 0.06 | 22.78 ± 0.47 | 0.69 ± 0.02 |
| E380K/S387L | 7.21 ± 0.06 | 0.59 ± 0.01 | 6.40 ± 0.37 | 1.44 ± 0.14 | BDL* | BDL* | BDL* | BDL* |
| E380W/S387L | 9.25 ± 0.04 | 0.45 ± 0.02 | 7.82 ± 0.23 | 0.46 ± 0.05 | BDL* | BDL* | BDL* | BDL* |
| E380Q/S387L | 10.49 ± 0.09 | 0.24 ± 0.01 | 9.92 ± 0.28 | 0.28 ± 0.03 | 6.14 ± 0.37 | 1.21 ± 0.04 | BDL* | BDL* |
* BDL stands for below detection limit.
Table 3.
Primers utilized in site-directed mutagenesis.
| Primer name | Sequence (5′→3′) |
|---|---|
| 1903+ E380-R | GGCCACAACCACGGTGGCACCTTTGGC |
| 1903+ E380I-F | GCCGGTAATATTGCGAGCCCGGTTAGC |
| 1903+ E380K-F | GCCGGTAATAAAGCGAGCCCGGTTAGC |
| 1903+ E380M-F | GCCGGTAATATGGCGAGCCCGGTTAGC |
| 1903+ E380N-F | GCCGGTAATAACGCGAGCCCGGTTAGC |
| 1903+ E380Q-F | GCCGGTAATCAGGCGAGCCCGGTTAGC |
| 1903+ E380T-F | GCCGGTAATACCGCGAGCCCGGTTAGC |
| 1903+ E380W-F | GCCGGTAATTGGGCGAGCCCGGTTAGC |
| 1903+ E380Y-F | GCCGGTAATTATGCGAGCCCGGTTAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



