
Submitted:
26 November 2023
Posted:
27 November 2023
You are already at the latest version
Abstract
Synthesis of heterodinuclear 3d transition metal complexes in which two metal ions are introduced in equivalent coordination sites of a ligand is quite uncommon. We have previously shown that the application of the cis-trans isomerization of the oxamido group was able to furnish such a possibility. In the present paper we demonstrate that this synthetic pathway, relying on the possibility of changing the conformation (cis vs trans) of the oxamido group, can be extended to other ligands. Although we have not been able to structurally characterize these complexes, EPR and magnetic data do confirm the efficiency of this reaction pathway. We also demonstrate that the cis-trans isomerization of the oxamido group is not due to a particular property of the ion involved in the reaction. In fact, isomerization is governed by the basicity of the reaction medium. According to the reaction condition, it is possible to favor or not this phenomenon, and to increase the accessibility to a larger number of novel genuine complexes. But the oxophilic character of the lanthanide ions impedes this isomerization in 3d-4f complexes. The very weak magnetic interactions observed in these Cu-Gd complexes can be explained thanks to spin polarization.
Keywords:
coordination chemistry
; oxamide ligands
; magnetism
; EPR
; 3d-3d complexes
; 3d-4f complexes
1. Introduction
The reaction of diamines with dicarboxylic acids or their derivatives yields condensation polymers called polyamides, molecules made of repetitive units linked by amide functions [1]. These polyamides (nylon for example) have known a huge industrial development, going from textiles to a lot of different applications [2,3]. A few decades ago, as we were interested in the syntheses of transition metal complexes with a limited number of metal atoms, we decided to prepare ligands involving a limited number of amide functions [4]. And we described synthetic processes able to give ligands possessing two amide functions only by use of diethyloxalate, a derivative of the simplest diacid molecule, oxalic acid, reacting it with diverse substituted diamine molecules (Scheme 1) [5,6,7,8]. Note that one amide function can be replaced by an oxamate or an acid function [9,10,11]. So, starting with a symmetrical ligand such as L1H4 with two equivalent coordination sites, we could isolate a mixed metal complex involving CuII and NiII ions, a result that was at first sight really unexpected [4]. It relies on the possible cis vs. trans conformation change of these oxamide ligands. In the present paper we demonstrate that this property is not limited to the L1H4 ligand, but that it can extended to other ligands. We also show that this conformation change can be avoided to yield again heteronuclear transition metal complexes or even heteronuclear transition metal-lanthanide complexes.
2. Results
2.1. 3d-3d Complexes
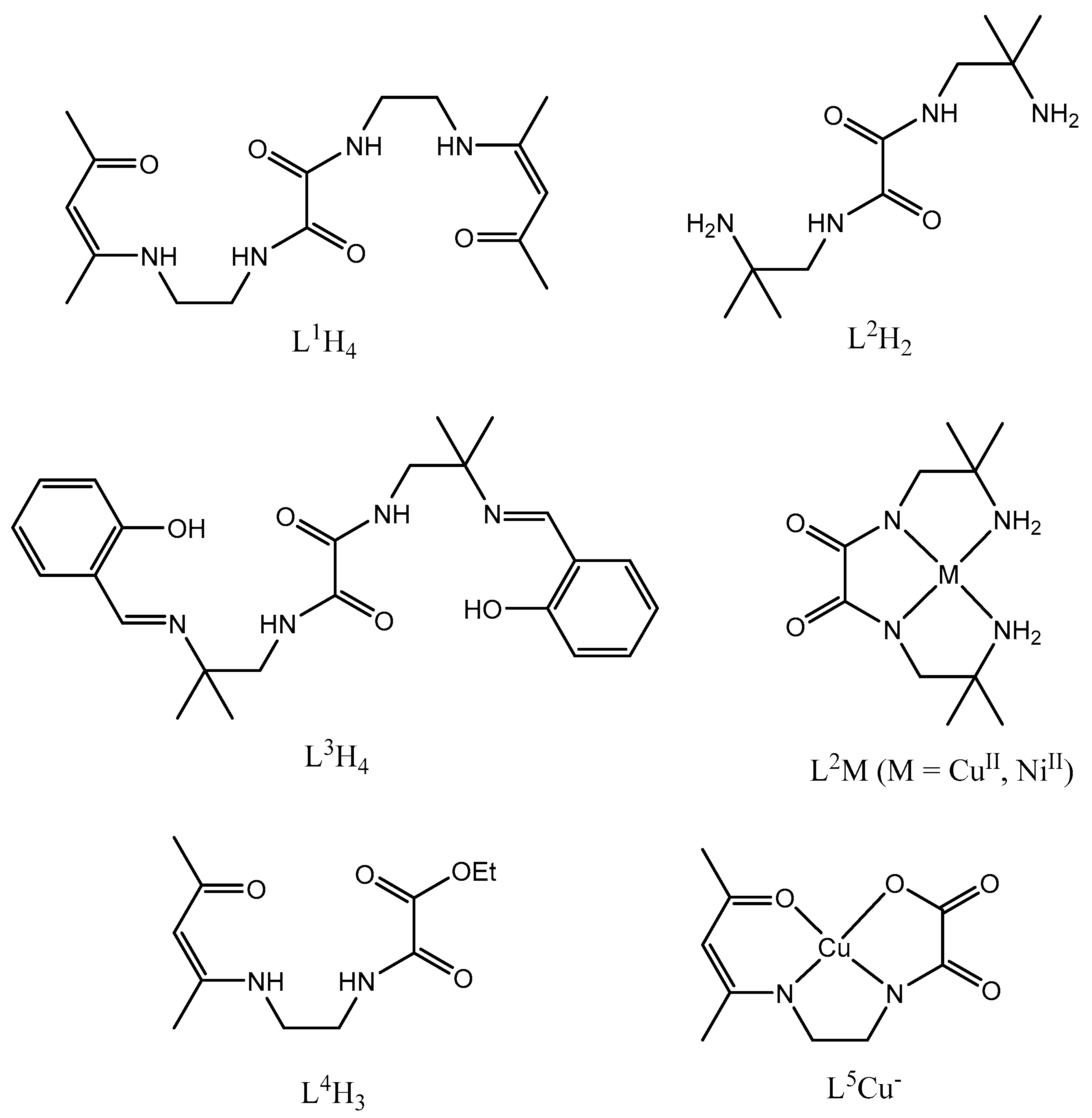
In previous work we showed that symmetrical oxamide ligands such as N-N’-bis(4-methyl-5-aza-3-hepten-2-on-7-yl) oxamide (L1H4, Scheme 1) or N-N’-bis[2,2-dimethyl-4-(2-hydroxyphenyl)-3-aza-3-buten] oxamide (L3H4, Scheme 1) are able to coordinate transition metal ions to yield neutral homodinuclear complexes such as CuL1Cu, NiL1Ni [4] or CuL3Cu, NiL3Ni, VOL3VO [5], as demonstrated by the structural determination of the [CuL3Cu](H2O) complex [7]. More surprisingly, thanks to the cis-trans isomerization of the oxamide function, the L1H4 can react with copper ions to give an anionic species formulated (L1HCuNa).2H2O. The EPR spectrum confirms that three nitrogen atoms are involved in the copper coordination sphere. In a following step this species acts as a ligand toward a nickel ion and yields the neutral heterodinuclear CuL1Ni complex [4]. Note that the L1H4 ligand is prepared by reacting the half-unit ligand 7-amino-4-methyl-5-aza-3-hepten-2-one with diethyl oxalate in a 2:1 ratio while the reaction of this half-unit ligand with diethyl oxalate in a 1/3 ratio allows isolation of the L4H3 ligand (Scheme 1) [9]. The preparation of this L3H4 ligand necessitates a two-step reaction. The reaction of diethyl oxalate with 1,2-diamino-2-methylpropane in a 1:2 ratio first yields the L2H2 ligand while addition of salicylaldehyde to the aforementioned L2H2 ligand in a 2:1 ratio furnishes the L3H4 ligand [5]. According to the cis-trans isomerization of the oxamide function, it is possible to isolate L2M complexes (M = CuII, NiII). The EPR spectrum of L2Cu does confirm that the copper ion is bound to two pairs of inequivalent nitrogen atoms (AN = 15 G and A’N = 8 G) giving a superhyperfine structure of 13 lines in addition to the hyperfine structure (Aiso = 95 G, giso = 2.08) [8]. Addition of copper ions to this CuL2 complex induces a trans isomerization of the oxamide ligand and formation of an infinite chain compound in which the copper ions are alternately bridged by the dideprotonated L2 ligand and two nitrite anions, as demonstrated by the structural determination of the [Cu(NO2)2CuL2]n compound [8].
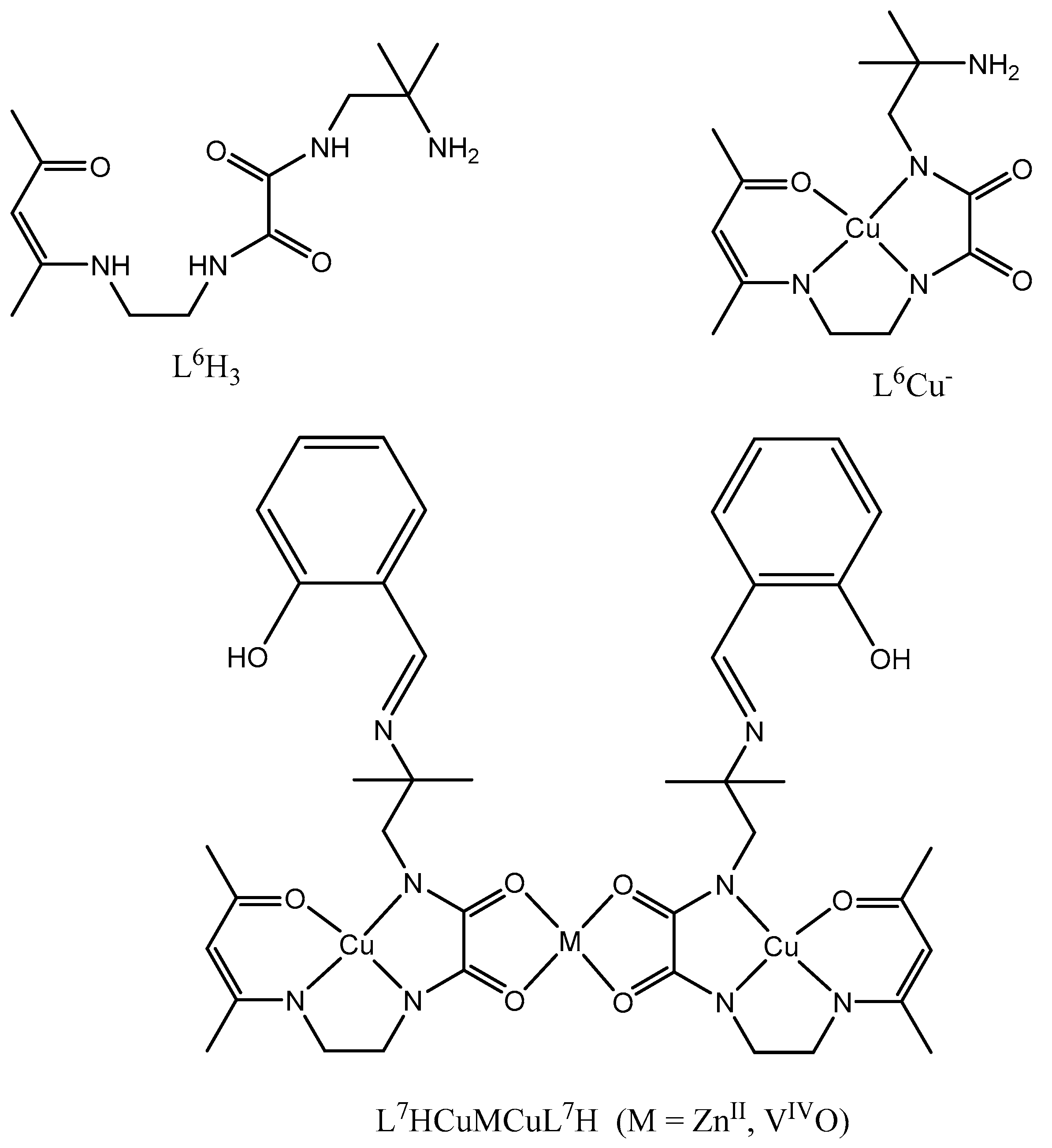
In the present work we have built a new L6H3 ligand possessing two different coordination sites. In a first step the L4H3 ligand is reacted in a 1:1 ratio with 1,2-diamino-2-methylpropane, thus allowing isolation of the L6H3 ligand. Reaction of copper ions in a basic medium gives the anionic L6Cu- entity in which the copper ion presents a N3O coordination sphere. Surprisingly, reaction with Zn(sal)2.2H2O or VO(Sal)2 (sal = deprotonated salicylaldehyde) has not allowed to isolate the expected CuL7Zn or CuL7VO neutral complexes. From the analytical, FAB+, magnetic data, it becomes clear that the isolated complexes correspond to the (L7HCu)2Zn·2CH3OH or (L7HCu)2VO·2CH3OH formulation, the phenol function remaining protonated. As an example the FAB+ data for (L7HCu)2Zn·2CH3OH gives a first signal corresponding to the [(CuL7H)2Zn +1]+ cation with a maximum peak at m/z = 963 uma and a second one for the [(CuL7H)2Zn + Na]+ cation at m/z = 985 uma. The theoretical and experimental isotopic patterns of these cations are in complete agreement (Figures S1 and S2). In order to explain such a behavior, we can expect that the free amine function reacts with the aldehyde function of the salicylaldehydato ligand coordinated to the ZnII ion, which induces a decomplexation of the salicylaldehydato ligand coordinated to the ZnII ion along with a concomitant protonation of the free phenol function by a proton coming from the reacting nitrogen atom. Then the oxygen atoms of the two oxamide functions that are in the vicinity of the ZnII ion can enter into coordination with this ion. According to this observation, it appears that the Zn ion is not able to induce the cis-trans isomerization of the oxamide function. A similar observation is made for complex (L7HCu)2VO·2CH3OH. The χMT product for this complex is constant and equal to 1.12 cm3mol-1K from 100 to 4 K (Figure S3). Such a value corresponding to three non interacting S = ½ ions does confirm that the vanadyl ion is again not able to induce the cis-trans isomerization of the oxamide function. Contrary to our previous results showing that Cu and Ni ions are able to induce the cis-trans isomerization [4,8], it seems that Zn and VO ions are not able to favor such an isomerization.
Scheme 2.
Schematic representation of genuine ligands and complexes.
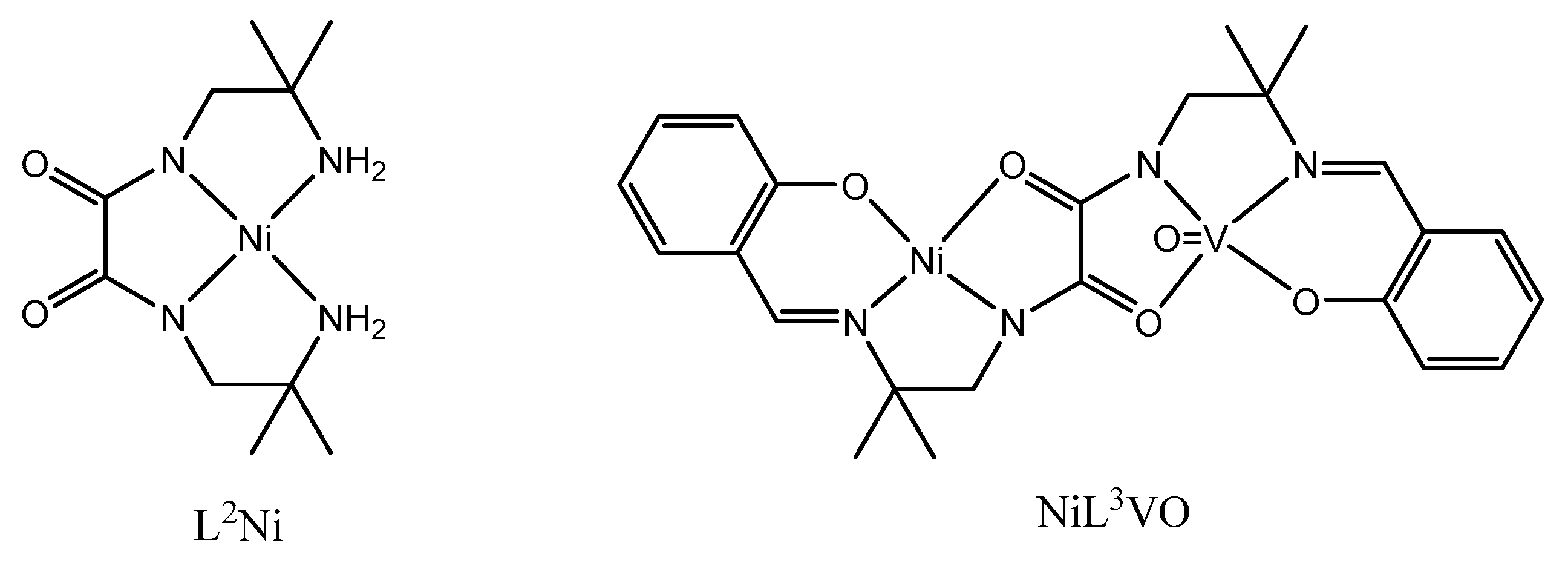
Reacting first the NiL2 complex with salicylaldehyde in a basic medium, followed by addition of vanadyl ions allows isolation of the NiL3VO complex in which the two 3d ions are coordinated to the symmetrical ligand possessing two N2O2 coordination sites. As the Ni ion is diamagnetic, we observe a nice EPR spectrum with 8 lines at room temperature and in solution (CH2Cl2), characteristic of the vanadyl ion (Figure S4). In the present case we must conclude that the reaction of the amine functions with salicylaldehyde induces the cis-trans isomerization, thus yielding the L3 ligand in which only one N2O2 coordination site is occupied by the Ni ion while the vanadyl ion is introduced in the other N2O2 coordination site to give the NiL3VO compound. The eight lines EPR spectrum (Figure S4) confirms the isolation of a heterodinuclear complex, with absence of scrambling of the Ni and VO ions. Its magnetic behavior follows a Curie law with C equal to 0.40 cm3mol-1K. In order to understand the absence of scrambling, the reaction of the primary amine functions with the aldehyde functions must yield a [NiL3H]- anionic species in a first stage, in which the NiII ion is coordinated to three nitrogen atoms and to the oxygen atom of a deprotonated phenol function. Then the cis-trans isomerization is favored by the basic medium, that allows the VO ion to enter into coordination in the newly formed N2O2 site, as in the case of the CuL1Ni complex [4]. This result is surprising in view of our previous assertion in the above paragraph, where we were telling that the VO ion was not able to favor the cis-trans isomerization. We must conclude that the cis-trans isomerization is possible in a basic medium and is not observed in a neutral medium, whatever the concerned ions. So we again show that a symmetrical ligand possessing two identical N2O2 coordination sites allows formation of heterodinuclear complexes, what is quite surprising and original.
Scheme 3.
Schematic representation of the starting and final complexes.
We have previously shown that the free L3H4 ligand reacts with vanadyl ions in basic medium to yield the VOL3VO complex [5]. We have also demonstrated that the thermal dependence of the magnetic susceptibility follows a Curie law, with C = 0.69 cm3mol-1K, so that the magnetic interaction of the two vanadyl ions is absent or very weak. The EPR spectra (CH2Cl2 solution, 295 K) are more informative but they can be slightly different on going from a sample to another one, depending on the reaction time. It appears that two spectra are superimposed with a g parameter equal to 1.98, one with 8 lines and the other one with 15 lines, the 8 lines species appearing as a minor species giving shoulders in some lines of the main EPR spectrum after a prolongated heating, while the 8 lines species is favored before the thermal equilibrium is reached. (Figure S5). Such a result can be explained by the presence of two isomers, with a cis or trans arrangement of the V=O groups. The hyperfine splittings A are respectively equal to 94.6 x 10-4 cm-1 (8 lines spectrum) and 47.3 x 10-4 cm-1 (15 lines spectrum). From these data we can estimate the interaction parameters J. For the eight lines spectrum J must be lower than A, so that a J value lower than 10-3 cm-1 is expected, while for the fifteen lines spectrum the J parameter must be higher than A. It is expected to be comprised in between 10-3 and 10-2 cm-1. These J values are very weak, in complete agreement with previous literature data [12]. In absence of structural determination, we can expect that the main compound should be the most symmetrical complex, with the apical VO groups in a trans arrangement. The previously published structural determination of the [(terpy)Cu(ox)VO(ox)(H2O)]H2O complex indicates that the Cu and VO ions are linked by an oxalate bridge while a very weak Cu-VO magnetic interaction, expected to be ferromagnetic, is observed thanks to the magnetic study. Furthermore it is told that the magnetic susceptibility of the trinuclear [(terpy)Cu(ox)VO(ox)(H2O)Cu(terpy](ClO4) ·2H2O complex follows a Curie law in the 4-100 K temperature domain, with a χMT value of 1.20 cm3mol-1K [12]. This result does agree with our observation for the complex (L7HCu)2VO·2CH3OH described above.
2.2. 3d-4f Complexes
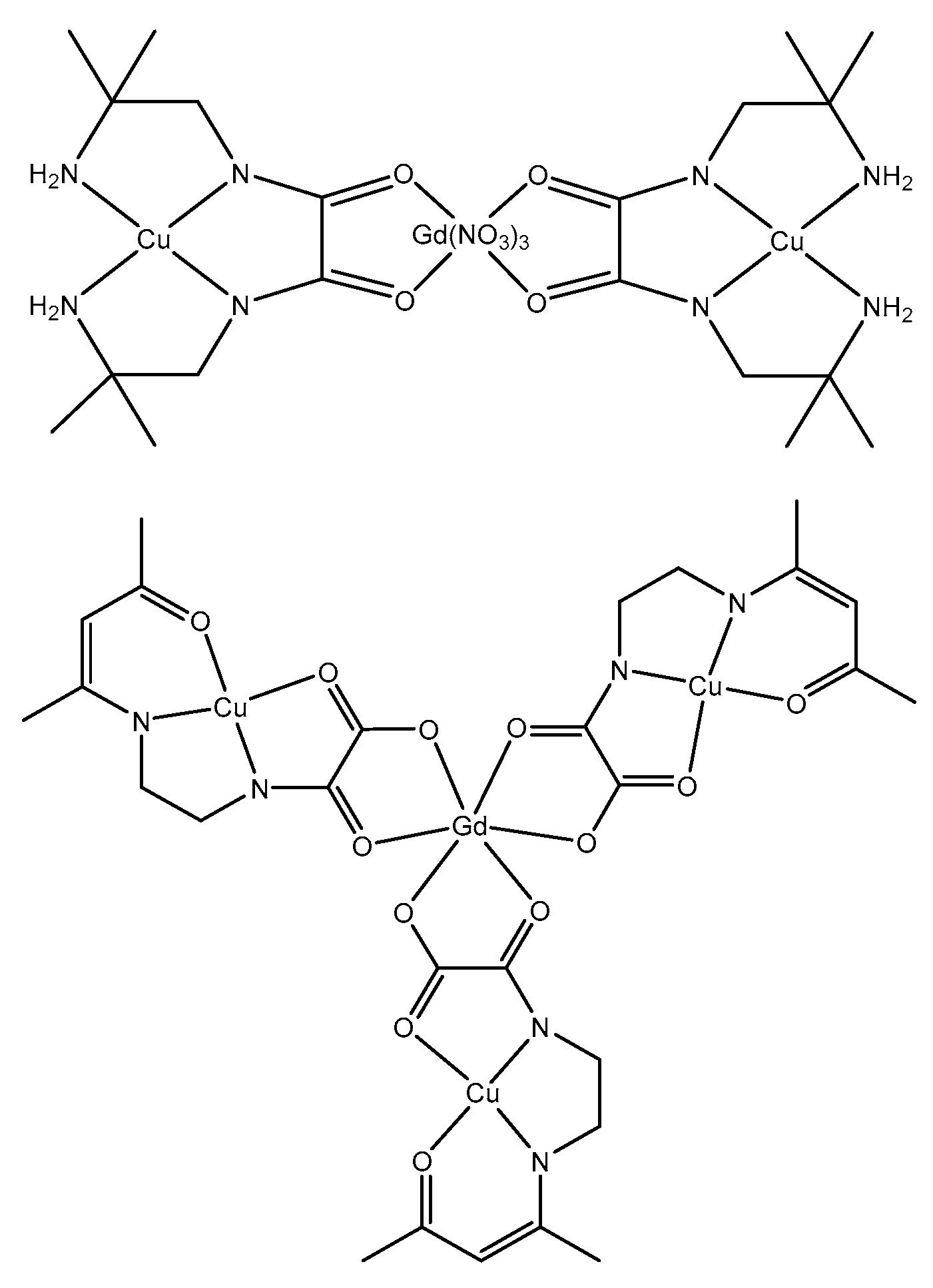
It is possible to prepare 3d-4f entities by reaction of some of these mononuclear 3d complexes with lanthanide ions. So the L2Cu complex reacts with gadolinium nitrate to yield a compound formulated (L2Cu)2Gd(NO3)3·5H2O while the [L5Cu]- anionic species gives a neutral complex formulated (L5Cu)3Gd·5H2O. In these compounds, the oxophilic character of the Ln ions impedes any cis-trans isomerization. A look at the magnetic data is of interest. In the first complex the Gd ion is surrounded by two Cu ions and the χMT product is equal to 8.30 cm3mol-1K at 300 K. It remains constant till 10 K and then slightly decreases to 8.14 cm3mol-1K at 2K (Figure S6). In the second compound the Gd ion is surrounded by three Cu ions. The χMT product is equal to 9.15 cm3mol-1K at 300 K, does not change in the 20-300 K temperature domain while a slight increase at 9.80 cm3mol-1K is observed at very low temperature (Figure S7). We have previously shown that spin polarization is the mechanism that allows to understand the strength and sign of the magnetic Cu–Gd interactions in complexes assembling Cu and Gd ions [13]. When spin polarization is active an odd number of bridging atoms in between the ions bearing the spins favors a parallel alignment of the spins, whereas an even number of bridging atoms favors an antiparallel alignment. In the present complexes we have a mixture of even and odd numbers of atoms in the ligand bridging the metal ions, so that the resulting magnetic interaction is expected to be very weak. Furthermore we have to remember that the presence of singly occupied 3dx2-y2 orbitals is needed for the observation of ferromagnetic 3d-Gd interactions, the 3d ions with unoccupied 3dx2-y2 orbitals yielding antiferromagnetic 3d-Gd interactions [14]. In the trinuclear CuGdCu complex (L2Cu)2Gd(NO3)3·5H2O, the Cu and Gd ions are interacting through two three-atom OCN bridges and two four-atom OCCN bridges. It has been previously shown that such OCN bridges [13] and OCO bridges [15] give weak ferromagnetic interactions. Observation of a weak antiferromagnetic interaction in the trinuclear complex means that the interaction through the four-atom OCCN bridge overcomes the one coming from the three-atom OCN bridge. In the tetranuclear complex (L5Cu)3Gd·5H2O, the Cu and Gd ions are interacting through two three-atom OCO and OCN bridges, and two four-atom OCCO and OCCN bridges. Observation of a weak ferromagnetic interaction implies that, according to the magnetic behavior of the trinuclear (L2Cu)2Gd(NO3)3·5H2O complex, the magnetic interaction through the three-atom OCO bridges is slightly larger than the one coming from the four-atom bridges and becomes the predominant magnetic interaction. Eventually we can conclude that the magnetic behavior of these two complexes assembling Cu and Gd ions can be understood by the presence of an active spin polarization interaction. But we cannot forget that these interactions are very weak. Structural determinations of similar complexes along with theoretical calculations will be necessary to confirm such an assertion.
Scheme 4.
Schematic representation of trinuclear and tetranuclear Cu-Gd complexes.
3. Discussion
Although we have not been able to structurally characterize these genuine complexes, EPR and magnetic data do confirm the efficiency of this reaction pathway. The example of the heterodinuclear NiL3VO complex is quite informative. If scrambling was active, a mixture of NiL3Ni and VOL3VO complexes in a 1/1 ratio would be present. The magnetic data corresponding to such a mixture could agree with the data of the genuine NiL3VO complex, as we demonstrate that the magnetic interaction in the dinuclear VOL3VO complex is very weak and that de dinuclear NiL3Ni complex is diamagnetic. On the contrary, the EPR data become informative. Observation of an eight-lines spectrum for the NiL3VO complex does not agree with the fifteen-lines spectrum corresponding the VOL3VO complex. This spectrographic result does confirm the existence of a genuine NiL3VO entity. In addition the study of the homodinuclear VOL3VO complex indicates that this entity is not unique, with the axial VO groups in a trans or cis conformation. The EPR study shows that the fifteen-lines species becomes the main product after a prolongated heating, so that we can expect that the most symmetrical species is the main species and that it corresponds to the trans conformation of the VO groups. It is not surprising to think that the eight-lines species corresponding to the cis conformation is favored before the thermal equilibrium is reached. The strength of the magnetic interaction in these two conformational VOL3VO species can also be deduced from the EPR spectrum, thus confirming the weakness of these interactions.
We also demonstrate that the cis-trans isomerization of the oxamido group is not due to a particular property of the ion involved in the reaction. Indeed isomerization is governed by the basicity of the reaction medium. According to the reaction condition, it is possible to favor or not this phenomenon, and to increase the accessibility to a larger number of novel genuine complexes. In that case, the positive FAB mass spectra data are of prime interest for the complexes resulting from an absence of cis trans isomerization. Due to the isotopic pleiade of the 3d and 4f ions, the corresponding cationic signals give a direct information on the molecular mass of these species and the number of metal ions involved in a compound, at the exception of the solvent molecules (coordinated or not). This is the case for the neutral complexes (L7HCu)2Zn·2CH3OH, (L7HCu)2VO·2CH3OH, (L5Cu)3Gd·5H2O for which the main peaks appearing at 963, 966 and 983 uma, respectively, correspond to the [(CuL7H)2Zn +1]+ , [(CuL7H)2VO +1]+ and [(L5Cu)3Gd + 1]+ cationic species. For the (L2Cu)2Gd(NO3)3·5H2O complex possessing three nitrato anions, the main signal appears at 866 uma, which corresponds to the [(L2Cu)2Gd(NO3)2]+ formulation, with a loss of a nitrato anion. The magnetic data of these different entities do agree with these formulae and confirm the presence or absence of very weak magnetic interactions. Eventually the oxophilic character of the lanthanide ions limits the use of this synthetic pathway because it impedes this isomerization. As the magnetic interaction in 3d-4f complexes is dominated by spin polarization, very weak magnetic interactions can be expected in these complexes, due to the antagonist effects coming from the presence of even and odd numbers of bridging atoms in the ligand transmitting the interaction between the Cu and Gd ions.
4. Materials and Methods
2-methyl-1,2-diaminopropane, Cu(Ac)2·H2O, Ni(Ac)2·4H2O, Gd(NO3)3·5H2O, (Aldrich) were used as purchased. The ligands L1H4 [4], L4H3 [9], L2H2 [5], L3H4 [5] and the complexes L2Cu [5], VOL3VO [5], L5CuNa·1.33H2O [9] were prepared as previously described. High-grade solvents (dichloromethane, dimethylformamide, diethyl oxide, diisopropyl oxide, acetone, ethanol and methanol) were used for the syntheses of ligands and complexes.
4.1. Complexes
L2Ni. To L2H2 (1 g, 4.35 mmol) in ethanol (20 cm3) was first added an aqueous solution (5 cm3) of NaOH (0.35 g, 8.7 mmol). Then, nickel acetate (1.08 g, 4.35 mmol) in water (10 cm3) was added dropwise under stirring. Three hours later, the orange precipitate which appeared progressively was filtered off, washed with cold ethanol and diethyl ether and air-dried. Yield: 0.85 g (68%). Anal. Calcd for C10H20N4NiO2 (287.0): C, 41.85; H, 7.02 ; N, 19.52. Found : C, 41.52 ; H, 6.89 ; N, 19.23 %.
L6CuNa·2H2O. A dichloromethane solution (30 mL) of L4H3 (1.7 g, 7.0 mmol) and 2-methyl-1,2-diaminopropane (0.62 g, 7.0 mmol) was stirred overnight. To the oil obtained by solvent evaporation was added ethanol (20 mL) with stirring, followed by a water solution (3 mL) of NaOH (0.84 g, 21.0 mmol) and then a dropwise addition of an ethanol solution (20 mL) of Cu(ClO4)2·6H2O (2.6 g, 7.0 mmol). The resulting solution was stirred for three hours and concentrated to 15mL. Addition of acetone (60 mL) induced formation of lilac precipitate that was filtered off, washed with acetone and dried. Yield : 0.85g (30 %). Anal. Calcd for C13H25CuN4NaO5 (403.5): C, 38.66; H, 6.24; N, 13.87. Found : C, 38.45 ; H, 6.16 ; N, 13.53.
(L7HCu)2VO·2CH3OH. A methanol solution (15 mL) containing L6CuNa·2H2O (0.20 g, 0.5 mmol) and VO(Sal)2·H2O (0.16 g, 0.5 mmol) was heated and stirred for 30 min. The resulting solution was cooled and evaporated. Addition of CH2CL2 (20 mL) give a solution that was filtered and concentrated to a few mL. Addition of isopropyl ether induced formation of a violet precipitate that was filtered off by suction and dried. Yield : 0.08 g (31 %). Anal. Calcd for C42H58Cu2N8O11V (1029.0): C, 49.02; H, 5.68 ; N, 10.89. Found : C, 48.60 ; H, 5.36 ; N, 10.65. FAB+ (DMSO, mnba): m/z = 966 (100), [L7HCu)2VO + 1]+.
(L7HCu)2Zn·2CH3OH. A methanol solution (15 mL) containing L6CuNa·2H2O (0.20 g, 0.5 mmol) and Zn(Sal)2·2H2O (0.17 g, 0.5 mmol) was heated and stirred for 30 min. The resulting solution was cooled and evaporated. Addition of CH2CL2 (20 mL) give a solution that was filtered and concentrated to a few mL. Addition of isopropyl ether induced formation of a red precipitate that was filtered off by suction and dried. Yield : 0.09 g (35 %). Anal. Calcd for C42H58Cu2N8O10Zn (1027.5): C, 49.10; H, 5.69 ; N, 10.91. Found : C, 48.65 ; H, 5.32 ; N, 10.55. FAB+ (DMSO, mnba): m/z = 963 (87), [L7HCu)2Zn + 1]+, 985 (55) [L7HCu)2Zn + Na]+.
NiL3VO. A mixture of L2Ni (0.15 g, 0.5 mmol), sodium acetate (0.08 g, 1 mmol) in methanol (15 mL) was first stirred, followed by addition of VO(Sal)2·H2O (0.16 g, 0.5 mmol) and then heated for thirty minutes and left to cool under stirring. The maroon precipitate that appeared was filtered off, washed with diethyl ether and dried . Yield : 0.12 g (41 %). Anal. Calcd for C24H28N4NiO6V (578.1): C, 49.86; H, 4.88; N, 9.69. Found: C, 49.38; H, 4.86; N, 9.59.
(L2Cu)2Gd(NO3)3·5H2O. A mixture of L2Cu (0.15 g, 0.5 mmol) and Gd(NO3)3·5H2O (0.11 g, 0.25 mmol) in methanol (5 mL) was stirred for 1 hour. Addition of acetone (20 mL) and isopropylether (50 mL) induced formation of a violet precipitate that was filtered off, washed with acetone and diethyl ether. Yield: (0.23 g, 90 %). Anal. Calcd for C20H50Cu2GdN11O18 (1017.0): C, 23.62 ; H, 4.96 ; N, 15.15. Found : C, 23.34 ; H, 4.76 ; N, 14.82. . FAB+ (DMSO, mnba): m/z = 866 (100), [(L2Cu)2Gd(NO3)2]+.
(L5Cu)3Gd·5H2O. To L5CuNa·1.33H2O (0.2 g, 0.62 mmol) in water (5 mL) was added with stirring Gd(NO3)3·5H2O (0.09 g, 0.20 mmol) dissolved in methanol (10 mL). The violet precipitate that appeared quickly was filtered off, washed with water, acetone and diethyl ether. Yield: (0.11 g, 50 %). Anal. Calcd for C27H43Cu3GdN6O17 (1071.5): C, 30.26 ; H, 4.04 ; N, 7.84. Found : C, 30.04 ; H, 4.06 ; N, 7.76. FAB+ (DMSO, mnba): m/z = 983 (100), [(L5Cu)3Gd + 1]+.
4.2. Physical Measurements
C, H, and N elemental analyses were carried out at the Laboratoire de Chimie de Coordination Microanalytical Laboratory in Toulouse, France. Mass spectra (FAB+) were recorded in dmf as solvent and 3-nitrobenzyl alcohol matrix with a Nermag R10–10 spectrometer. Magnetic data were obtained with a Quantum Design MPMS SQUID susceptometer. Magnetic susceptibility measurements were performed in the 2-300 K temperature range under a 0.1 T applied magnetic field, and diamagnetic corrections were applied by using Pascal’s constants [16].
5. Conclusions
Synthesis of heterodinuclear 3d transition metal complexes in which two metal ions are introduced in equivalent coordination sites of a ligand is quite uncommon. In the present paper we shown that the application of the cis-trans isomerization of the oxamido group, that was observed previously, can be extended to other ligands. Although we have not been able to structurally characterize these genuine complexes, EPR and magnetic data do confirm the efficiency of this reaction pathway. The EPR spectrum of the homodinuclear VOL3VO complex does agree with the presence of two conformers, with a cis or trans arrangement of the axial VO groups. We also demonstrate that the cis-trans isomerization of the oxamido group is not due to a particular property of the 3d ion involved in the reaction. Indeed isomerization is governed by the basicity of the reaction medium. According to the reaction condition, it is possible to favor or not this phenomenon, and to increase the accessibility to a larger number of novel genuine complexes. But the oxophilic character of the lanthanide ions limits the use of this synthetic pathway because it impedes this isomerization in 3d-4f complexes. As the magnetic interaction in Cu-Gd complexes is dominated by spin polarization, very weak magnetic interactions can be expected in these complexes, due to the antagonist effects coming from the presence of even and odd numbers of bridging atoms in the ligand transmitting the interaction between the Cu and Gd ions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The authors are grateful to A. Mari and J.-F. Meunier (Laboratoire de Chimie de Coordination) and C. Claparols (UPS) for their technical assistance in recording magnetic, EPR and mass spectrometry data.
Conflicts of Interest
The author declares no conflict of interest.
References
- Carothers, W.H. Studies on polymerization and ring formation. An introduction to the general theory of condensation polymers. J. Am. Chem. Soc. 1929, 51, 2548–2559. [Google Scholar] [CrossRef]
- Moncrieff, R.W. Man-made fibers; John Wiley: New-York, USA, 1963; pp. 335–355, John Wiley: New-York, USA, 1963; pp. 335-355. [Google Scholar]
- Ndiaye, P. Du nylon et des bombes; Belin: Paris, France, 2001. [Google Scholar]
- Costes, J.P.; Laurent, J.P. New pathway to heterodinuclear complexes with equivalent coordination sites: application of the cis-trans isomerization of the oxamido group. Inorg. Chem. 1989, 28, 2234–2236. [Google Scholar] [CrossRef]
- Laurent, J.P.; Costes, J.P.; Pradié, G. Dinuclear (Cu/Cu, Ni/Ni, V/V, Fe/Fe) complexes of a novel oxamide-type ligand coordinating in the trans conformation. Inorg. Chim. Acta. 1994, 213, 57–63. [Google Scholar] [CrossRef]
- Costes, J.P.; Commenges, G.; Dominguez-Vera, J.M.; Laurent, J.P. Geometrical and optical isomers of the dinickel complexes of two chiral ligands of the N,N’-disubstituted oxamide type : 1D and 2D NMR study. Inorg. Chim. Acta. 1994, 216, 237–243. [Google Scholar] [CrossRef]
- Costes, J.P.; Dahan, F.; Laurent, J.P. A Dicopper Complex of N,N’bis[2,2-dimethyl-4-(2-hydroxyphenyl)-3-aza-3-buten] oxamide. Structure and Magnetic Behaviour of the mono hydrated form. Inorg. Chim. Acta. 1995, 230, 199–203. [Google Scholar] [CrossRef]
- Costes, J.P.; Dahan, F.; Laurent, J.P.; Drillon, M. An alternating copper(II) chain with bridging oxamidato and nitrito ligands: crystal structure and magnetic properties of [Cu(NO2)2CuL]n. Inorg. Chim. Acta. 1998, 294, 8–13. [Google Scholar] [CrossRef]
- Costes, J.P.; Dahan, F.; Laurent, J.P. Magnetic properties of mono-, bi-, and tri-nuclear Copper (II) complexes of novel oxamato and oxamido ligands. Crystal structure of a mononuclear precursor. Dalton Trans. 1989, 1017–1025. [Google Scholar] [CrossRef]
- Costes, J.P.; Laurent, J.P.; Moreno Sanchez, J.M.; Suarez Varela, J.; Alhgren, M.; Sundberg, M. Magnetic properties of a series of trinuclear complexes (CuL)2Mn.xB (L representing the deprotonated form of N-(4-Methyl-6-oxo-3-azahept-4-enyl)oxamic acid and B representing respectively H2O (x = 5, 4,5, 3, 1), (CH3)2SO (2 = 2), and C5H5N (x = 4)). Crystal and molecular structure of (CuL)2Mn.2(CH3)2SO. Inorg. Chem. 1997, 36, 4641–4646. [Google Scholar] [PubMed]
- Cador, O.; Mathonière, C.; Kahn, O.; Costes, J.P.; Verelst, M.; Lecante, P. Spectroscopic determination of Magnetic Exchange Parameters and Structural Geometry for Trinuclear Compounds: (CuL)2Mn.xB (L = N-(4-Methyl-6-oxo-3-azahept-4-enyl)oxamato and B = (CH3)2SO (x = 2) or H2O (x = 5)). Inorg. Chem. 1999, 38, 2643–2648. [Google Scholar] [CrossRef]
- Cortes, R.; Karmele Urtiaga, M.; Lezama, L.; Arriortua, M.I.; Rojo, T. A ferromagnetic copper(II)-vanadium(IV) oxide m-oxalato complex: crystallographic structure and spectroscopic and magnetic properties. Inorg. Chem. 1994, 33, 829–832. [Google Scholar] [CrossRef]
- Costes, J.P.; Duhayon, C.; Mallet-Ladeira, S.; Shova, S.; Vendier, L. Does the sign of the Cu-Gd magnetic interaction depend on the number of atoms in the bridge? Chem. Eur. J. 2016, 22, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Gomez, V.; Vendier, L.; Corbella, M.; Costes, J.P. Tetranuclear [Co-Gd]2 complexes: aiming at a better understanding of the 3d-Gd magnetic interaction. Inorg. Chem. 2012, 51, 6396–6404. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.E.; Hughbanks, T. Magnetic coupling in dinuclear Gd complexes. J. Am. Chem. Soc. 2006, 128, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Pascal, P. Magnetochemical studies. Annales de Chimie et de Physique, 1910, 19, 5–70. [Google Scholar]
Scheme 1.
Schematic representation of previously used ligands and complexes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



